
Submitted:
08 May 2023
Posted:
08 May 2023
You are already at the latest version
Abstract
In this paper we propose a simple procedure to obtain multi-walled carbon nanotubes (MWCNT) decorated with TiO2-Ag nanoparticles (MWCNT-TiO2-Ag). MWCNT were decorated with TiO2-Ag via combined functionalisation with -OH and –COOH groups and polymer wrapping technique using poly(allylamine hydrochloride) (PAH). TiO2 modified Ag nanoparticles were synthesized through Pechini method using mixture of acetylacetonate-modified titanium (IV) isopropoxide with silver nitrate (the Ag+/Ti4+ atomic ratio, is 0.5, 1,0, 1.5, 2.0 and 2.5 %, respectively) and the L(+)-ascorbic acid, as reducing agents. XRD analysis revealed the formation of nanocomposite containing CNT, TiO2 (anatase and rutile) and Ag. By SEM was evidenced the presence of the nanoparticles on MWCNT surface. The increases of the ratio between Ag+ and Ti4+ decrease the band gap energy of the samples. In Raman spectra the characteristic vibration of the TiO2, Ag and C atoms of graphite are identified. The photocatalytic activity of the MWCNT-TiO2-Ag nanocomposite was assessed by examining the degradation of Allura red (E129) aqueous solution under UV irradiation. The dye photodegradation process follows a pseudo – first – order kinetic with respect to the Langmuir – Hinshelwood reaction mechanism.
Keywords:
Ag doped TiO2
; MWCNT
; Allura Red
1. Introduction
Currently, semiconductor photocatalysis has been considered as a cost-effective and eco-friendly depolution technology for the removal of different organic pollutants in wastewater [1]. Titanium dioxide (TiO2) are amongst the most promising photocatalyst due to it intrinsic properties, high chemical and thermal stability, biocompatibility and low cost, which is widely used for many industrial application such as in environmental purification, pigments, decomposition of carbonic acid gas, catalyst supports coatings, solar cells, biomaterial, etc [2,3,4,5,6,7,8]. However, TiO2 posseses same disadvantages like specificity to UV light and to rapid recombination of charge carriers produced under irradiation with light in specific wavelength [9,10]. Thus, different strategies were achieved to improve the electron-hole separation of TiO2 and prolong the visible light absortion. Noble metals, such as Ag, Au, Pt and Pd deposited on a TiO2 surface could enhance the photocatalytic efficiency because they act as an electron trap promoting interfacial charge transfer processes in the composite systems [11,12,13,14]. Additionally, the excellent electronic properties of MWCNT provide continuous electronic state in the conduction band for transferring electrons, in the same time favoring the migration of excited electron into MWCNT with result in increasing the photocatalytic activity under visible light. Thanks to the unique properties of composite materials resulting from the combination of two entities with different functions, many systems based on MWCNTs and semiconductors have been studied [15,16,17].
Due to the rapid industrialization, numerous pollutants, like dyes, antibiotics, pesticides were released in surface water causing devastating effects on the water environment. Among them, the dyes have a carcinogenic and mutagenic effect, affecting aquatic organisms [18,19,20].
This study present the synthesis of the MWCNTs – TiO2 modified by Ag photocatalyst obtained the decoration of MWCNT with TiO2 modified Ag mediated by specific functionalisation with -OH and –COOH groups and poly(allyamine)hydrochloride (PAH) as polymer binder. The specific experimental conditions and complex investigation were performed to evidence the properties of new nanocomposite materials for improve photocatalytic efficiency. The photocatalytic degradation of Allura Red (E129) was investigated under visible – light irradiation.
2. Results
2.1.1. Precursors investigation
Thermal analyses of dried precursors were performed in order to establish the optimum crystallization temperature for the corresponding TiO2 oxides. Figure 1 show the thermogravimetric (TG) and differential thermal analysis (DTA) and derivative thermogravimetric (DTG) curves of dried (at room temperature, for 10 days) titanium precursor’s solution in the temperature range 20°C – 1200°C at a heating rate of 10°C/min. in an airflow.
Considering that the quantity of Ag doping does not have a significant influence on the thermal transformations and the total mass loss of samples, we chose for the thermal investigation the sample with the highest percentage of Ag in the TiO2 matrix (Ag+ /Ti4+ = 2.5%).
Figure 1.
Thermogravimetric(TG), differential thermal analysis (DTA) derivative thermogravimetric (DTG) curves of the dried (100°C) titania precursors (Ag+ /Ti4+ = 2.5%).
Figure 1.
Thermogravimetric(TG), differential thermal analysis (DTA) derivative thermogravimetric (DTG) curves of the dried (100°C) titania precursors (Ag+ /Ti4+ = 2.5%).
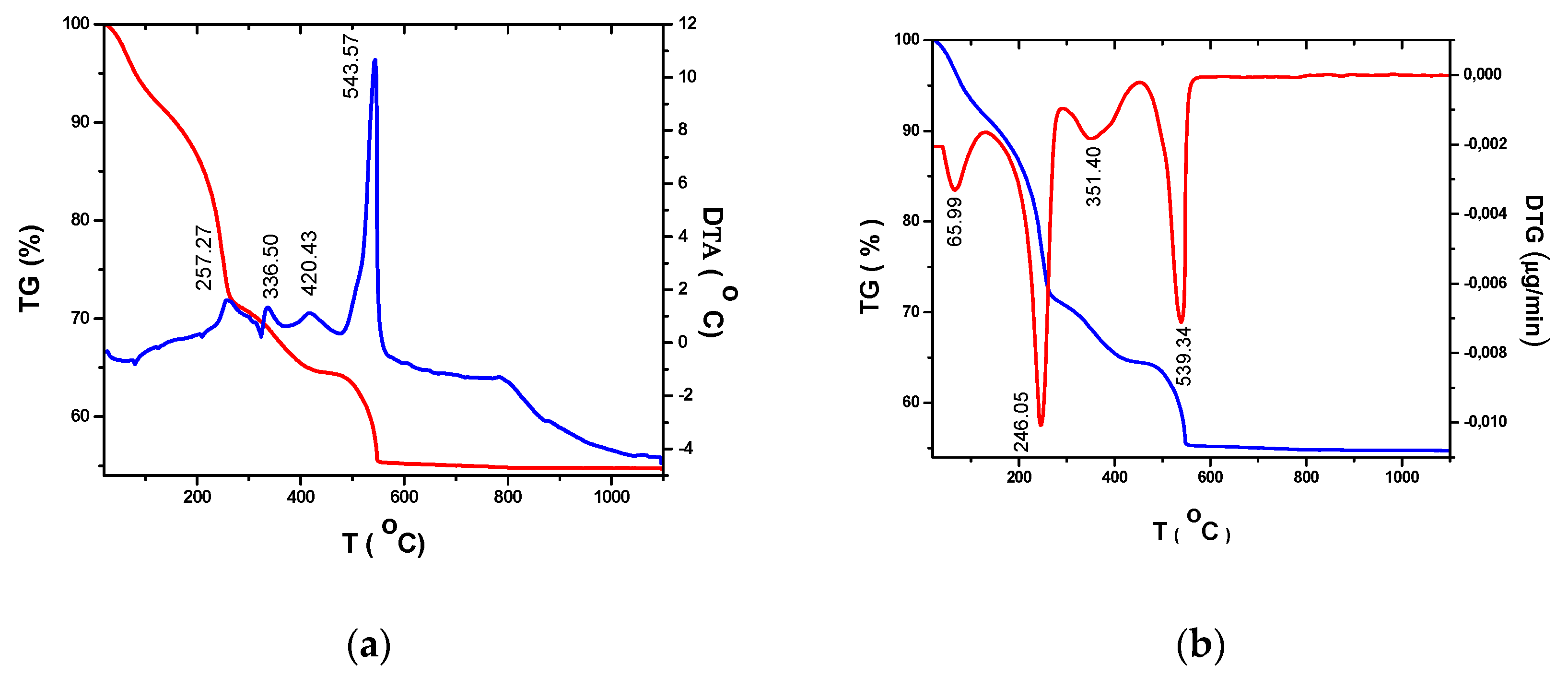
The thermal decomposition of TiO2 precursors take place in different progressive steps of temperature (TG) depending on physical and chemical processes that occur with increasing the temperature. The total weight loss (44,75%) associated to transformation of precursors into Ag+ / Ti4+ = 2.5% is the results of physical and chemical adsorbed water (8.07%), volatilization and combustion of residual water and organic parts from polymeric gels (20.63%gr), combustions of residual carbon (6.45%), Ag-crystallization of amorphous anatase (9.6%). The corresponding thermal effects are shown in DTA curve. Thus, a significant effects corresponding to the exothermic peak at ~ 420.43°C in the DTA curve are related to the beginning of crystallization of TiO2 anatas, whereas that around 543.57°C indicates the phase transformation anatase – rutile. These transformations are confirmed by the transformations corresponding to the DTG curves.
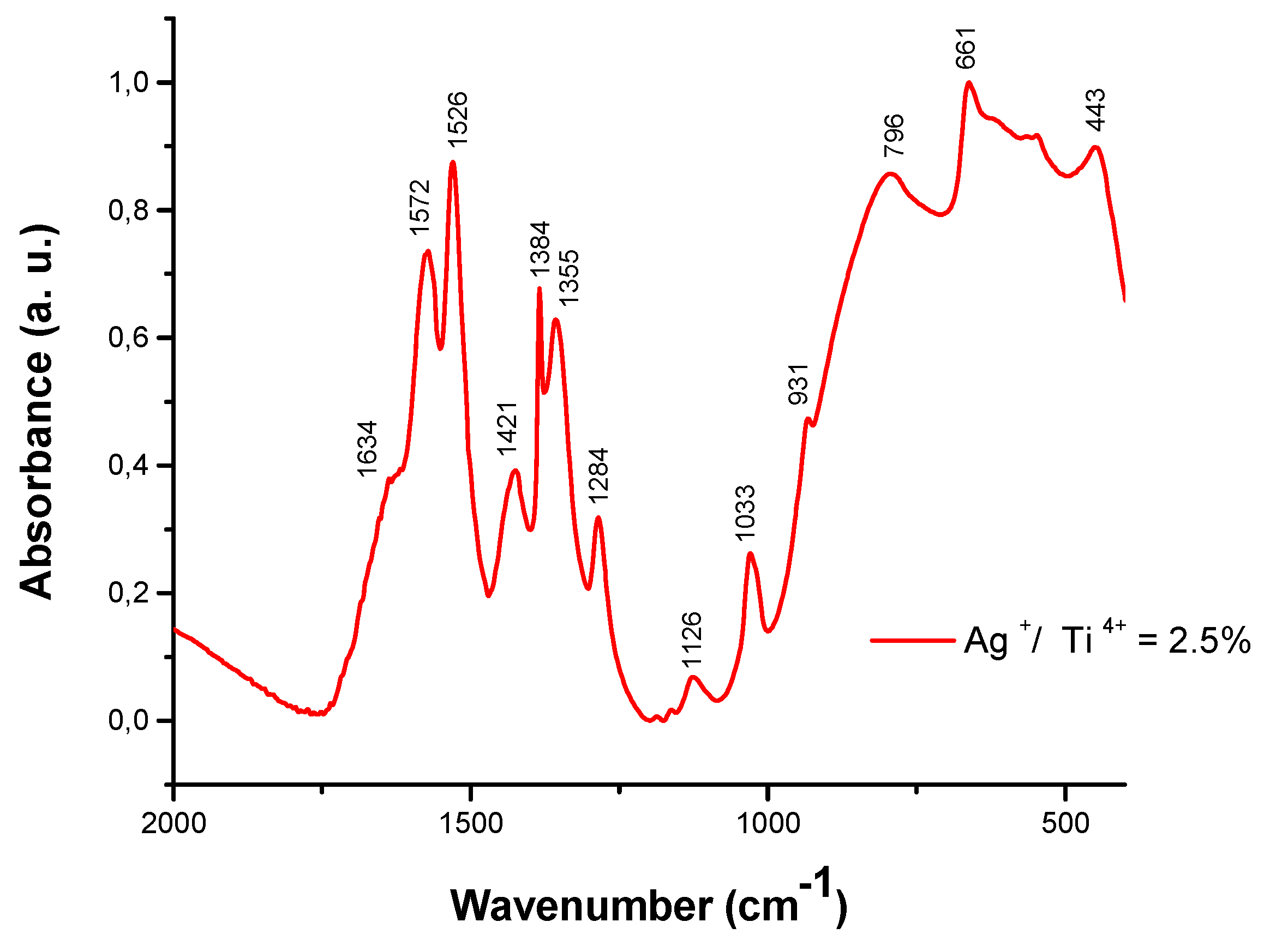
To further investigate the TiO2 precursors, the FT-IR spectra of the precursor solution containing Ag+ / Ti4+ = 2.5% are presented in Figure 2. The absorption shoulders at 1634cm-1 is attributed to bending vibrations of the –OH groups, and the bands centered at 1580, 1530, 1430 and 1025cm-1 corresponded to conjugated C – O vibrations [C = C - (C = O)] and [C = C - (C-) O-] of the acac ligand. The peak at 1355 and 931cm-1 is related to the CH2 symmetric vibration and CH3 – C – CH3 stretching modes of the isopropoxy group. The band at 1126cm-1 is due to Ti-O-C stretching vibration, the broad peaks in the range 653 – 550cm-1 and 495 - 436cm-1 indicated the presence of νTi-O and νTi-O-Ti bond respectively, and the peak located at 443cm-1 is correlated with asymmetric stretching vibration of Ti-O-Ag [21].
Figure 2.
The FT - IR spectra of precursor solution containing Ag+/Ti4+ = 2.5%.
2.2. Structural, morphologic and optical properties
Crystalline structure and structural parameters of MWCNT-TiO2-Ag nanocomposites were confirmed by XRD as shown in figure 3a. The indexed diffraction peaks correspond to tetragonal anatase crystalline phase (PDF card 01-089-4921) and Ag (PDF card 00-002-1098). No other peaks related to secondary phases was observed in the diffractograms. Devi et al. [22] reported that Ag deposited nitrogen doped TiO2 lead to the same effects. The peak at la 2θ=26.4° is typical for the 002 direction of graphite [23] (file PDF 01-075-0444) is probably superposed with (101) at 25.38° of anatase [24,25]. The small right - shifting to higher 2θ values of diffraction peaks of anatase (101) with the increse in Ag content (Figure 3b) less than 0.1°, was reported by Yuliati et al. [26] and was assigned to the presence of Ag on TiO2 with minimal crystal distorsion. The presence of MWCNT is also underlined by the elemental chemical analysis (Figure 3).
Figure 3.
XRD diffraction patterns of MWCNT - TiO2 - Ag nanocomposites. a. 2θ = 20 - 80° and b. 2θ = 24.8 - 26°.
Figure 3.
XRD diffraction patterns of MWCNT - TiO2 - Ag nanocomposites. a. 2θ = 20 - 80° and b. 2θ = 24.8 - 26°.
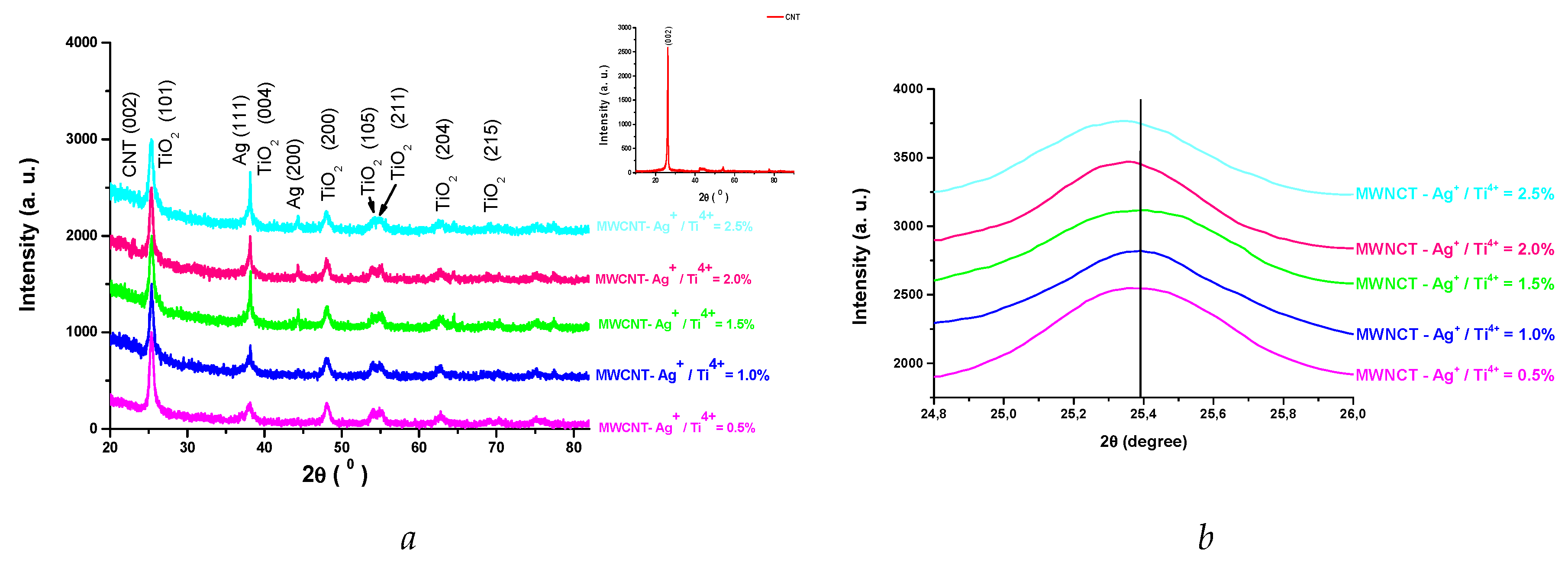
The Warren - Averbach X-ray profile Fourier analysis of the (101) (2θ=28.31°), (200) (2θ= 48.05) and (215) (2θ=75.058°) anatase peak profiles were processed by the XRLINE [27] computer program in order to determine the effective crystallite mean size (Deff). The crystallite size distribution function was determined from the second derivative of the strain corrected Fourier coefficients [28]. Also, from this profile were obtained the root mean square (rms) of the microstrains averaged along the real space distance, [29].
The elementary parameters of the cell was determined by Rietveld type refinement using the Powder Cell program [30], developed by Werner Kraus & Gert Nolze (BAM Berlin).
Table 1 summarizes the microstructural parameters of TiO2 anatase nanoparticles from the MWCNT-TiO2-Ag.

Table 1.
The effective crystalline mean size, (nm), the root mean square (rms) of the microstrains averaged along the real space,
Table 1.
The effective crystalline mean size, (nm), the root mean square (rms) of the microstrains averaged along the real space,
| Sample | Unit cell parameter |
Cell volume [Å3] |
(nm) | x 103 | |
| a [Å] | c [Å] | ||||
| MWCNT - Ag+/ Ti4+ = 0.5% | 3.7717 | 9.4603 | 134.576 | 14.21 | 11.3 |
| MWCNT - Ag+/ Ti4+ = 1.0% | 3.7824 | 9.4821 | 135.656 | 14.08 | 12.44 |
| MWCNT - Ag+/ Ti4+ = 1.5% | 3.7892 | 9.4888 | 136.240 | 13.86 | 13.86 |
| MWCNT - Ag+/ Ti4+ = 2.0% | 3.7920 | 9.4842 | 136.375 | 13.35 | 14.2 |
| MWCNT - Ag+/ Ti4 += 2.5% | 3.7951 | 9.5046 | 136.892 | 13.18 | 15.6 |
From microstructural data, it can be observed that as the dopant concentration increases the average crystallite decreases from 14.21 to 13.18nm.The decrease of crystallite size is due to the effects of Ag at the boundary of TiO2 [31].
2.3. FT – IR spectroscopy
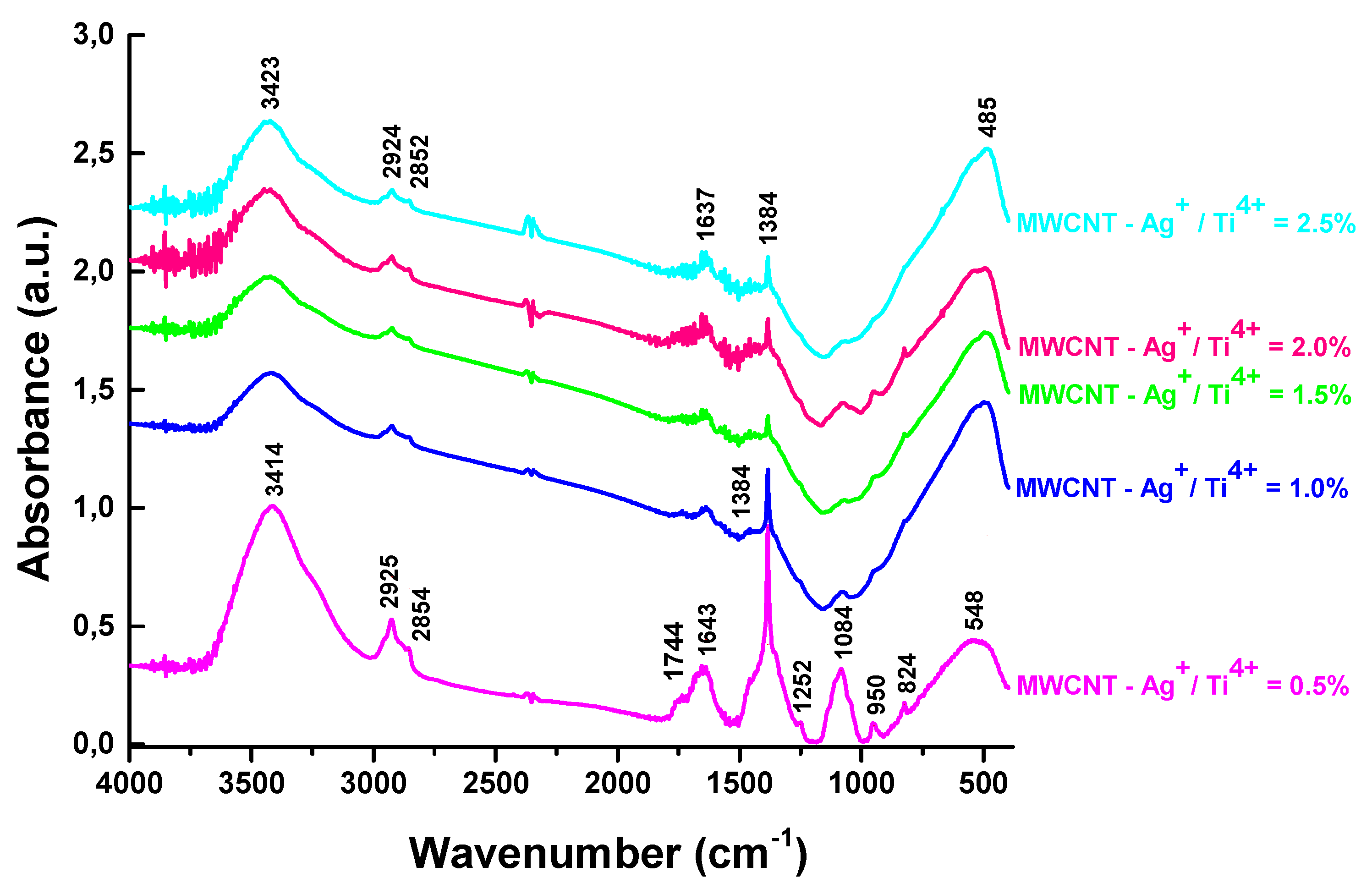
Figure 4 presents the FT-IR spectra obtained for MWCNT - TiO2-Ag nanocomposites with various Ag+/Ti4+ ratios. In the FT- IR spectra, the peak from 3414 cm-1 can be associated to the -OH stretching vibrations. The C-H stretching mode from the MWCNT structure [32] are evidenced in all the MWCNT - TiO2 - Ag samples at about 2852 and 2924cm-1 (figure 4.). The bending mode of the adsorbed water molecules on the nanocomposites are responsible for the 1637 cm-1 peak [33]. The band associated with Ti-O-Ti lattice vibration at 1384cm-1 can be observed [33]. The broad band centered at 480 cm-1 and 548 cm-1 could be associated with the Ti-O-C stretching vibrations and to Ti–O vibration, respectively [34]. In the spectrum of MWCNT-Ag+/Ti4+= 0.5%, more bands can be identified and associated with the presence of the COOH (peak from 1744 cm-1) or C-O-C vibrations (peak from 1084 cm-1) [34,35]. They can be also seen in less intensity in the other FTIR spectra of MWCNT- TiO2 - Ag samples, suggesting the presence of the MWCNT structures in the composition of all these hybrid materials.
Figure 4.
The FT- IR spectra of MWCNT- TiO2 -Ag.
2.4. Raman spectroscopy
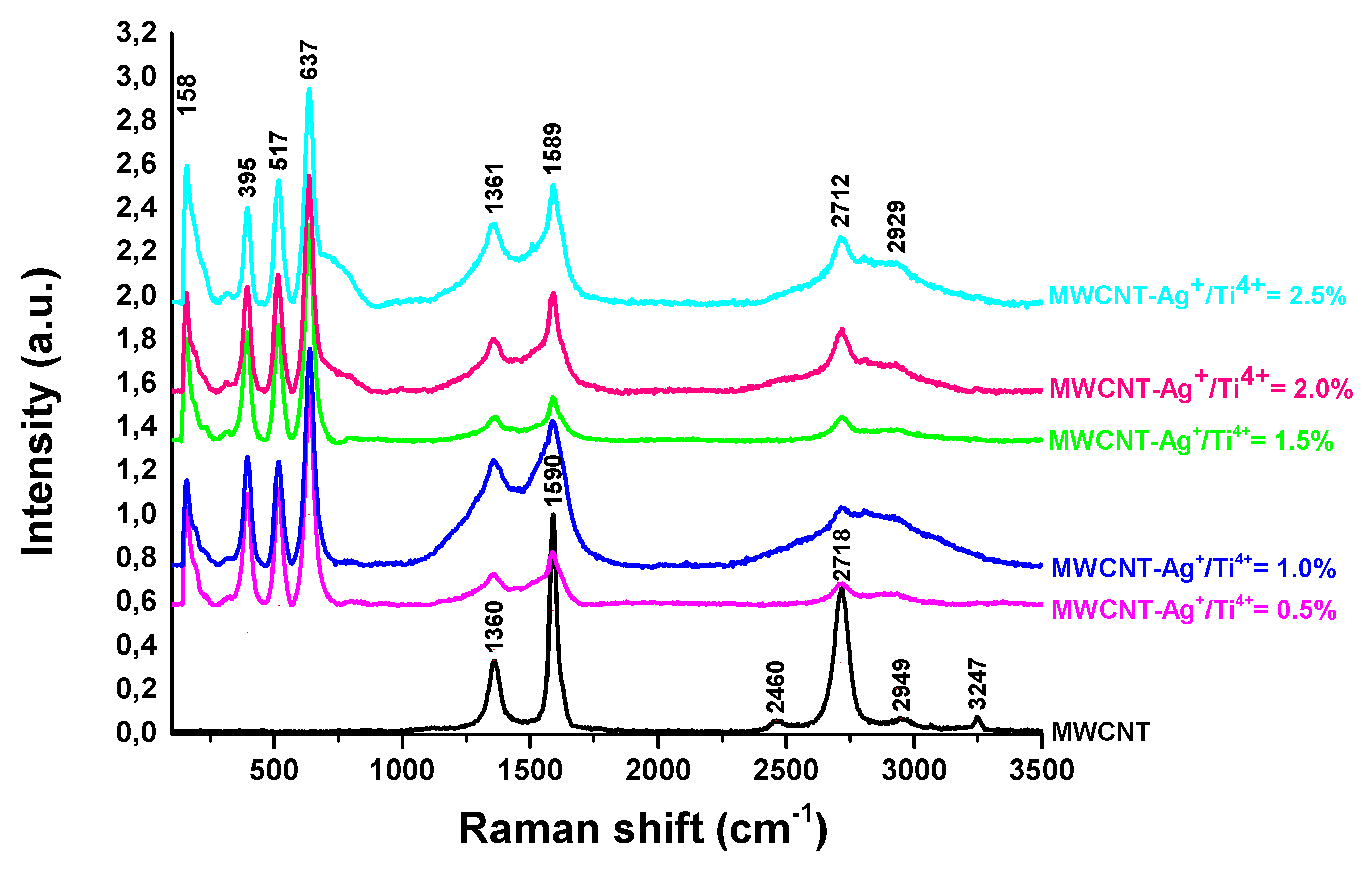
The typical Raman active modes of anatase TiO2 are composed of four main peaks around 152 (Eg), 389 (B1g), 510 (A1 ) and 633 cm-1 (Eg ). In addition to these bands, a small peak, assigned to the Eg mode, is localized at about 189 cm-1 [36]. The MWCNT - TiO2 - Ag nanocomposites contain supplementary bands which are characteristic to the carbon nanostructures, Figure 5. Thus, a D band appeared at about 1361 cm-1, being specific to the presence of defects in the carbon system, while the G peak from about 1589 cm-1 is related to the vibration of the sp2 – bonded carbon atoms [37]. A 2D band (also called G’) is present at about 2712 cm-1 in the spectrum of the MWCNT-Ag+/Ti4+ =2.5% sample and at 2718 cm-1 in the CNT precursor sample.
Figure 5.
Raman spectra of MWCNTs – TiO2 with different amounts of Ag nanoparticles recorded with 514 nm excitation laser line.
Figure 5.
Raman spectra of MWCNTs – TiO2 with different amounts of Ag nanoparticles recorded with 514 nm excitation laser line.
2.5. Electron spin resonance measurements
EPR spectra are shown in the Figure. The analysis of EPR spectra reveal a narrow resonance signal centered at g = 2.004 due to an oxygen vacancy with trapped electron named F - center [38]. By increasing the Ag content in TiO2 lattice, this signal gradually diminishes. Besides this signal, at higher and lower field side there are two low resonance signal centered at g = 1.97 and g = 2.03 shown in the inset of the Figure 6. The signal at g = 1.97 was attributed to Ti3+ ion in anatase crystalline phase, and the signal at g = 2.03 show the presence of surface O- ions [38].
Figure 6.
a. EPR spectra of MWCNT - TiO2 - Ag.
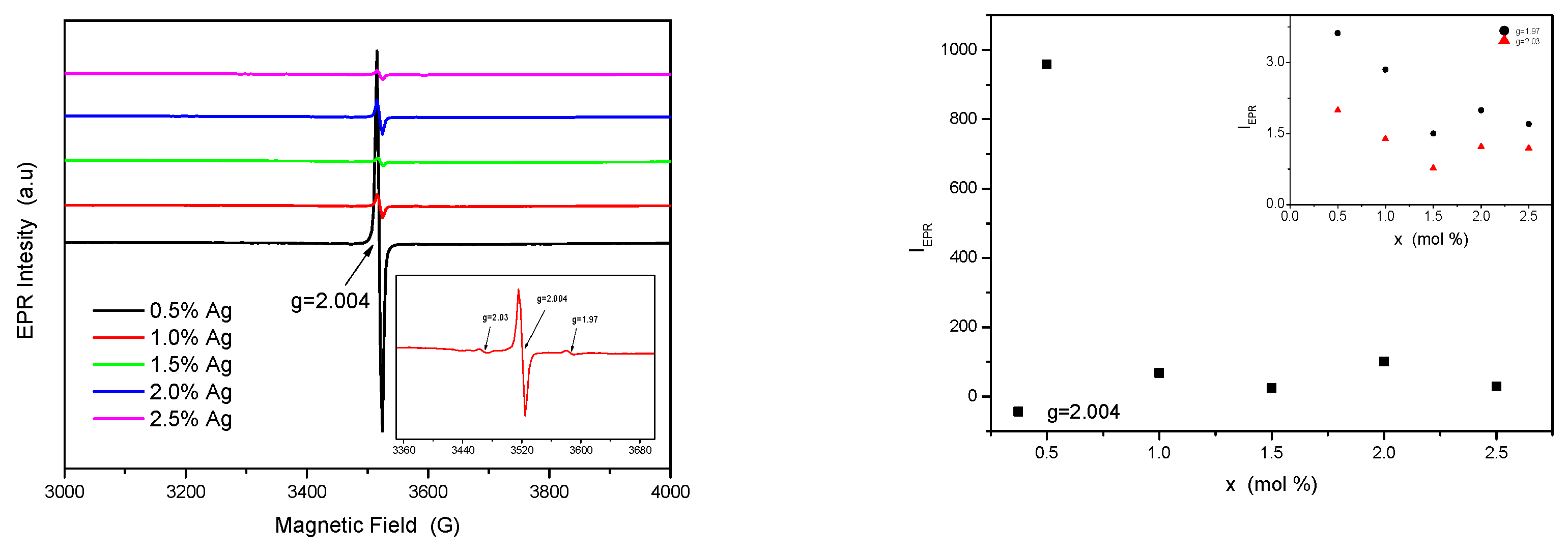
The EPR integral intensity of the resonance signal, which is proportional with the number of the electron spins from sample is illustrated in Figure 6b. It could be observed that all resonance signals show same behavior, decreasing with the increase of dopant concentration up to 1.5 % mol Ag, followed by a slightly increase. The oxygen vacancies concentration shows a significant decrease for sample with high Ag content (≥1.5 mol% ).
2.6. UV – ViS spectroscopy
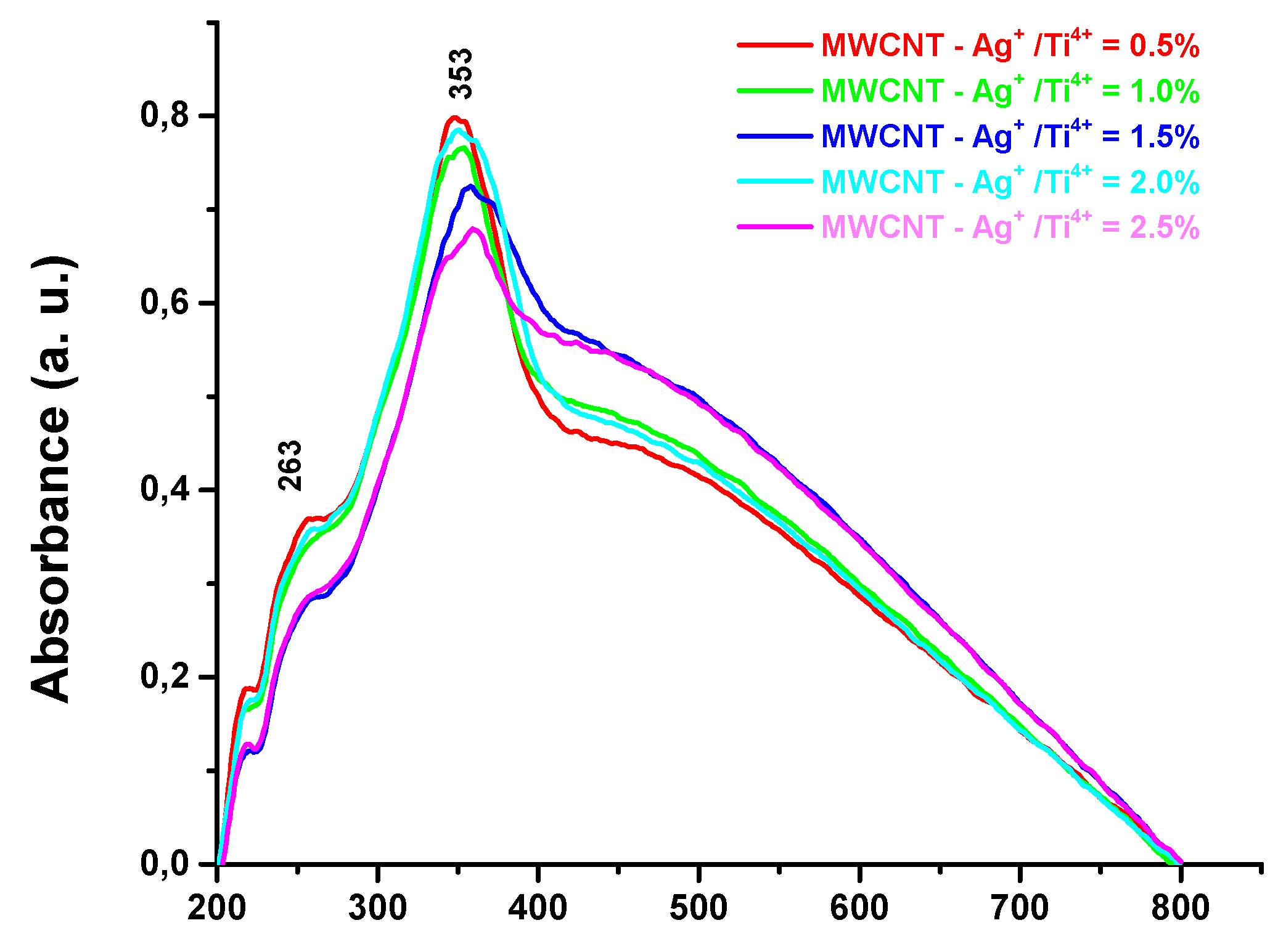
Figure 7 indicates the UV – VIS spectra of MWCNTs – TiO2 with different Ag nanoparticles amounts. An intense absorption is observed at 353 nm specific to intrinsic band gap of TiO2 nanoparticles [39]. The broad absorption which covers the whole range of visible range, is caused by the addition of MWCNTs [40]. Moreover, the strong absorption peak around 470 – 580nm arise from the surface plasmon absorption due to Ag particles [41]. By increasing the Ag concentration a red shift to a higher wavelength is observed in the absorption edge of the nanocomposites, due to electronic interaction between MWCNT and TiO2 modified Ag [42] . Supplementary, the spectrum contain a very low absorption peak at 263 nm attributes to the transition to Ag higher energy state of valence electrons [43].
Figure 7.
UV – Figure 7. UV - VIS spectrum of MWCNTs – TiO2 -Ag.
2.7. Optical band gap energy, Eg determination
The Eg of the nanopowder was estimated by using Tauc's plot [44,45]. The band gap values for direct transitions is 2.81-3.02eV (Figure 8) (Table 2). The redshift to the longer wavelengths originates, as we mention and was obsreved, aslo, by Li et al. [46], from the band gap narrowing.
Figure 8.
Extrapolation of the band gap energy for direct transition.
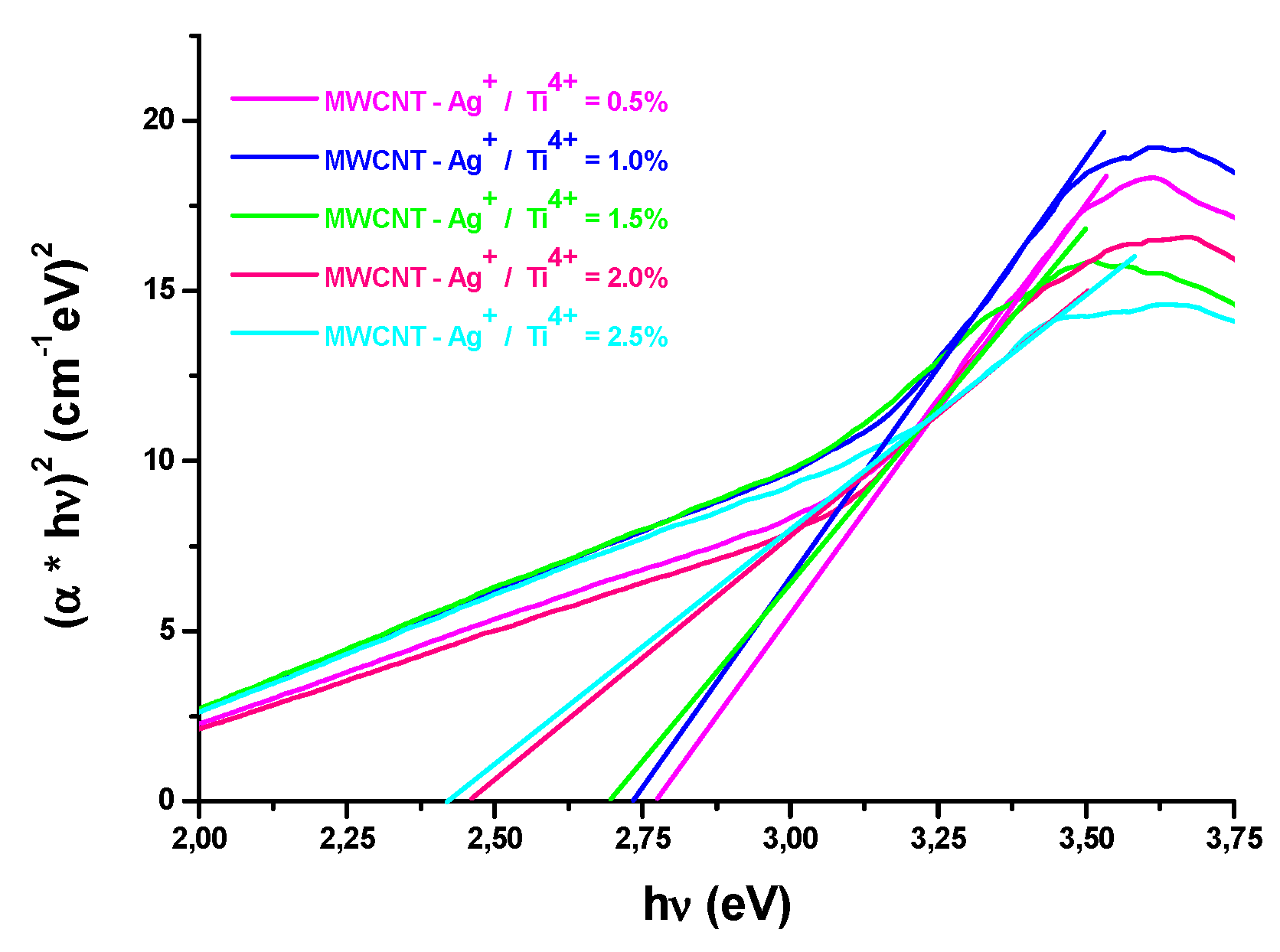
Table 2.
Optical band gap energy of MWCNT -TiO2-Ag nanocomposites.
| Sample | Eg – direct transition |
|---|---|
| MWCNT - Ag+/ Ti4+ = 0.5% | 2.77 |
| MWCNT - Ag+ / Ti4+ = 1.0% | 2.73 |
| MWCNT - Ag+ / Ti4+ = 1.5% | 2.69 |
| MWCNT - Ag+ / Ti4+ = 2.0% | 2.46 |
| MWCNT - Ag+ / Ti4+ = 2.5% | 2.41 |
2.8. Morphology of nanocomposites
Conventional TEM images revealed the presence of polyhedral nanopartices over the nanotube surface having a tendency to agglomerate.
Figure 9.
TEM images of MWCNTs - TiO2 - Ag . a. Ag+ /Ti4+ = 0.5% and b. Ag+ /Ti4+ = 2.5%.

In addition, the EDX shown in Figure 10 reveals the presence of elements including C, Ti, O and Ag in the MWCNTs –TiO2 – Ag samples.
Figure 10.
EDX mapping of MWCNTs – Ag+/Ti 4+ = 1.5%.

2.11. Photoluminescence spectroscopy.
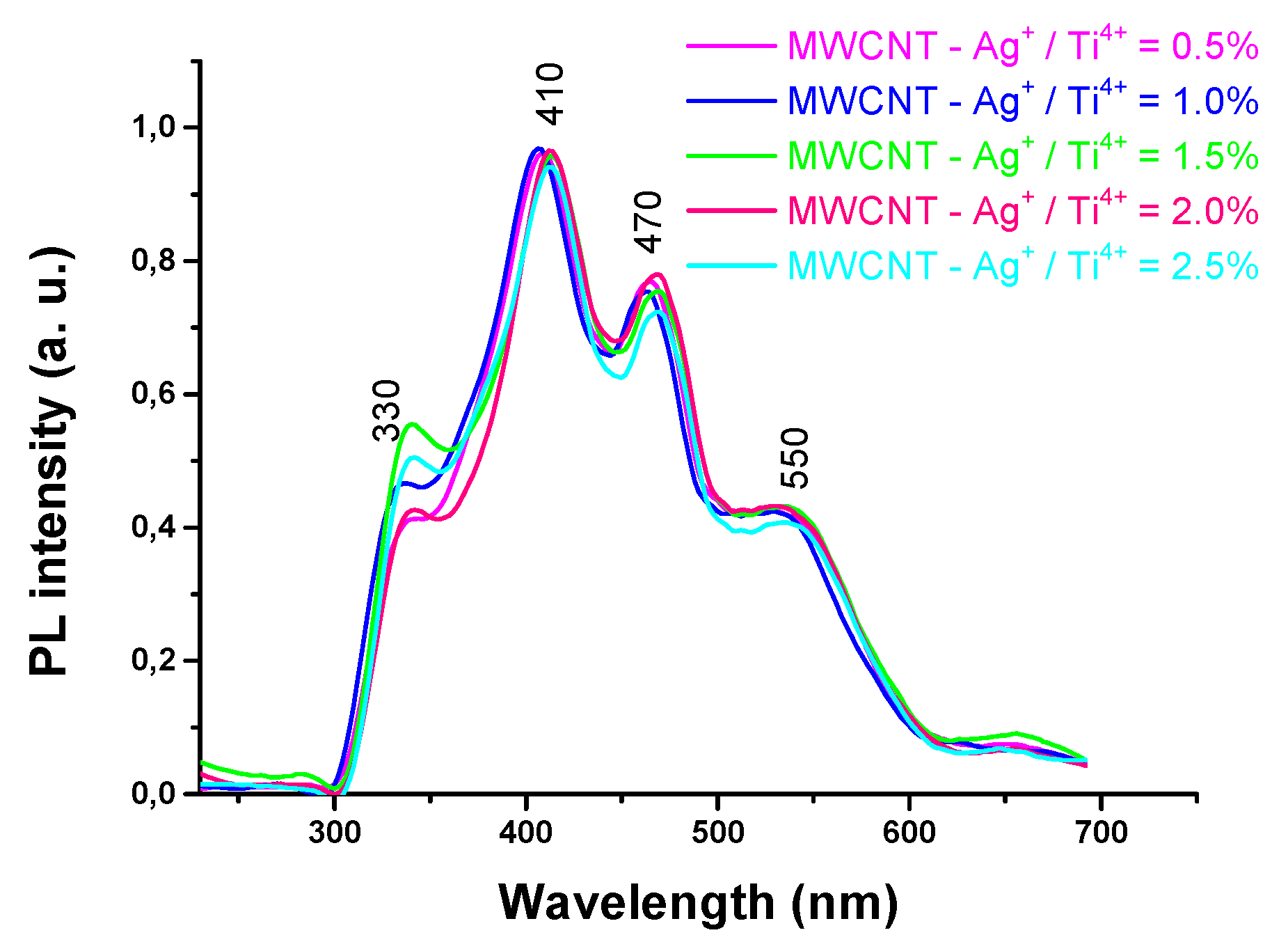
The PL spectra of MWCNTs – TiO2 with different amount of Ag nanoparticles is illustrated in Figure 11. As can be seen in the PL spectra, the intensity of the PL bands decreases with the incorporation and increasing of the content of Ag+ ions in the TiO2 the content of Ag+ ions in the TiO2 lattice, because Ag nanoparticles act as trapes to capture the photoreduced electrons and thus inhibit recombination of electron – hole pair. Presence of peak at ~ 470 nm is due defect states formed by oxygen vacancies [47], and peak from 330nm is associated with band – gap of titania [48]. Emissions around ~550 nm are associated to electron transition from to O on the surface [49].
Figure 11.
PL of MWCNTs – TiO2 – Ag nanocomposites.
2.12. Photocatalytic activity.
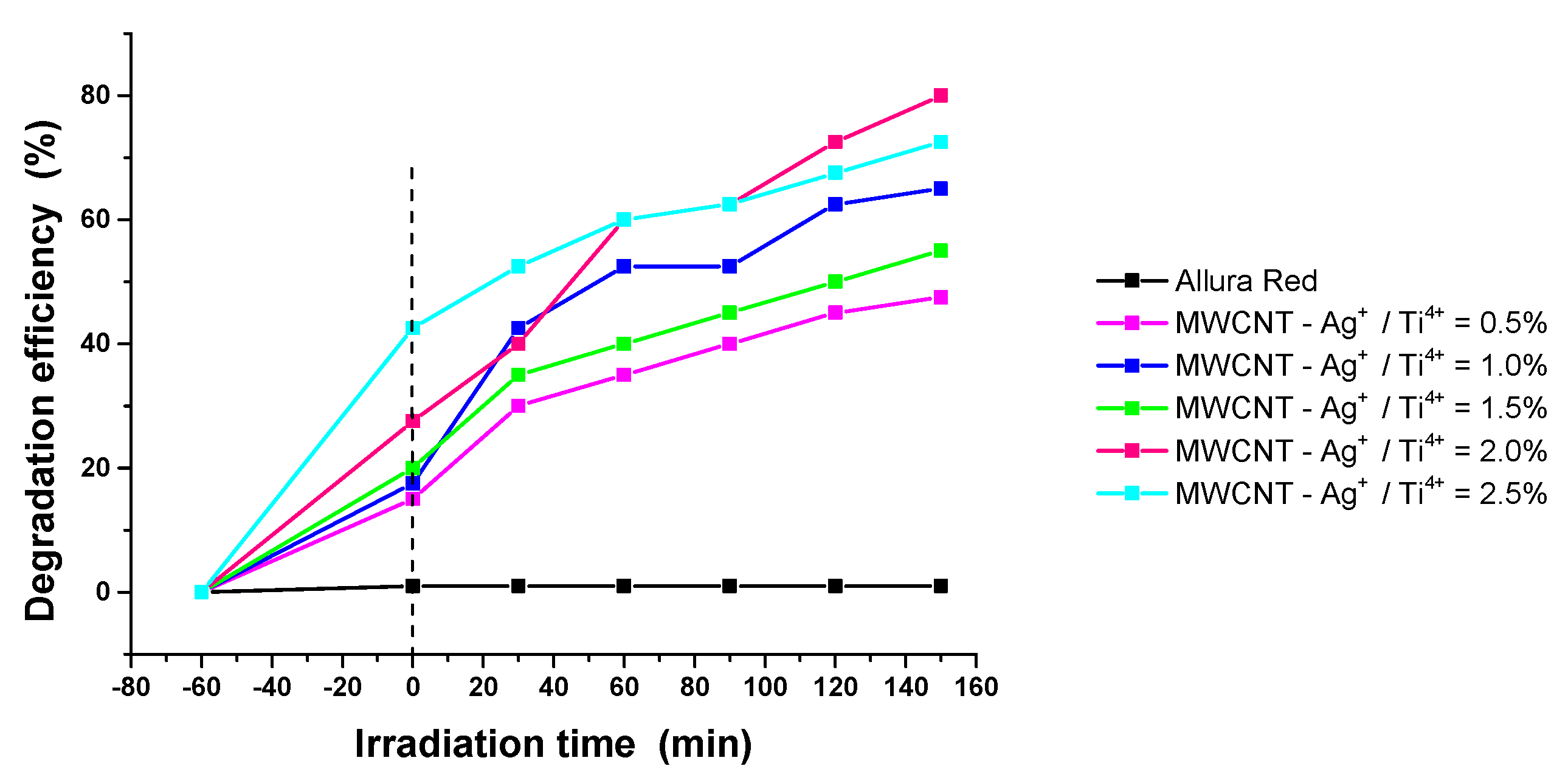
Photocatalytic activity of the samples was investigated using a synthetic solution of Allura red-E129 under UV irradiation. Since the efficiency of the degradation process depends on both the concentration of the pollutants and the amount of the photocatalyst in the studied systems, minimal amounts of each component were chosen for the study.Prior to irradiation, the samples were kept in dark for 60 min to achieve adsorption-desorption equilibrium. The testing of the photocatalytic activity of MWCNT decorated with TiO2-Ag nanoparticles was carried out by tracking the spectrophotometric decrease, over time, of the concentration of pigment E129, (λ = 502nm), following adsorption and photodecomposition of nanostructures. Figure 12 shows degradation efficiency of the samples. The adsorption capacity of the samples increases with Ag content.
The efficiency of the photocatalysts for selected Allura Red degradation was calculated according to Eq. (1):
where Co is the initial concentration of dye, and Ct is the concentration of colorant at time t. The nanostructures with 2.0% Ag have the best photocatalytic activity for the degradation of Allura Red under UV irradiation demonstrating degradation efficiency of 79.99% (Figure 12) .According to literature data the photocatalytic activity of Ag doped titania is dependent on the concentration of noble metal particles [50,51] and the diminish of the photoreaction at higher loading is due to so-called “screening effect” [52]. The presence of Ag nanoparticles stimulates the separation of charges but too high concentration of Ag particles shields the active centers of the semiconductor, thus reducing its photocatalytic activity.
Figure 12.
Photocatalytic degradation of Allura Red by MWCNT nanostructures.
According to Langmuir – Hinselwood expression:
, unde and are the dye concentration at time 0 and t (after irradiation), k (min-1) is the pseudo - first order rate constant, the best photocatalyst is the one with 2.0% Ag (Figure 13), as mentioned above. The obtained results are shown in Table 3.
Figure 13.
Kinetics of Allura Red degradation by nanostructures.
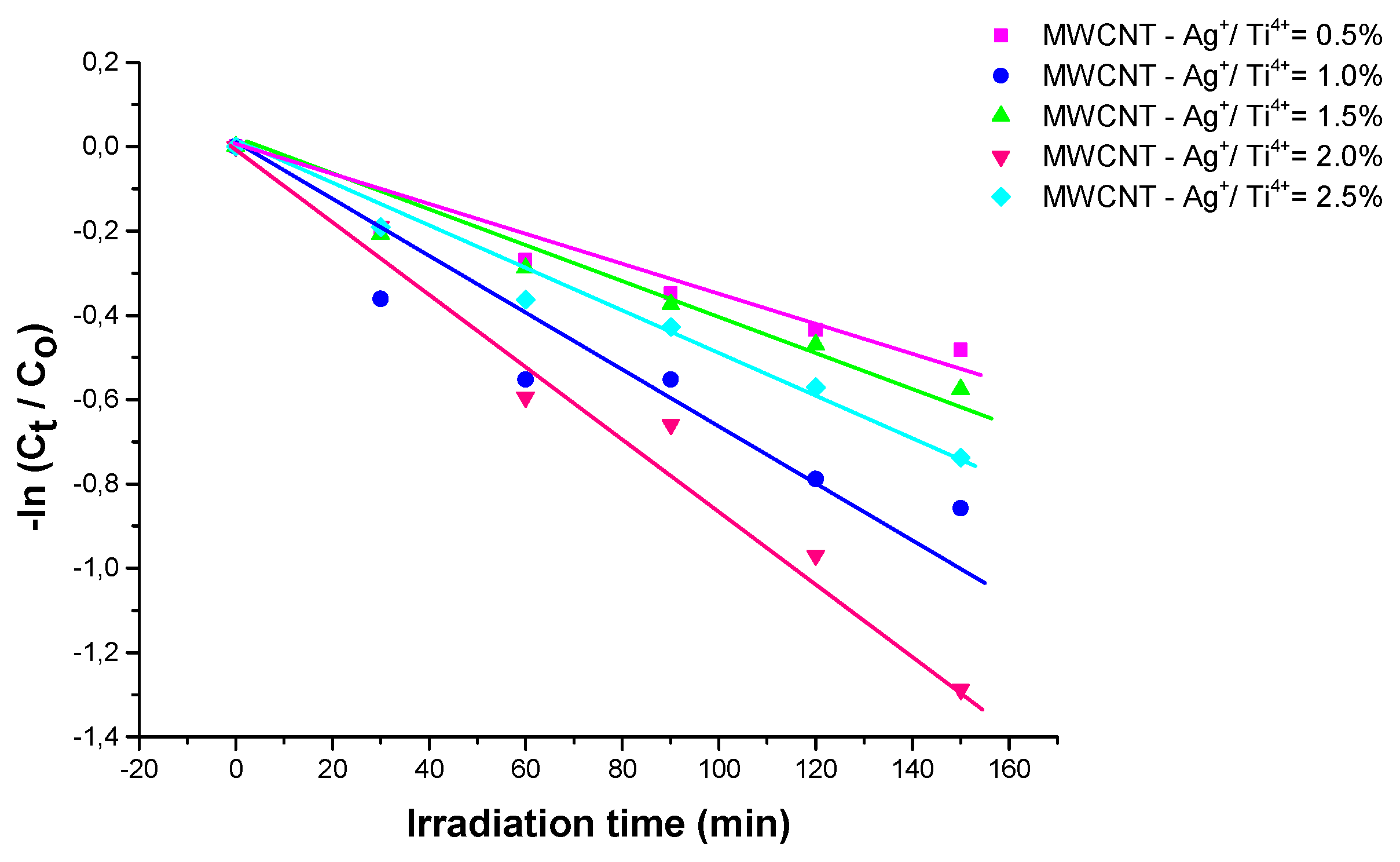
Table 3.
Photodegradation rate, apparent first-order rate constant (ki) of photocatalytic degradation and correlation coefficient (R2).
Table 3.
Photodegradation rate, apparent first-order rate constant (ki) of photocatalytic degradation and correlation coefficient (R2).
| Sample | Photodegradation rate (%) |
|
R2 | |
| MWCNT - Ag+/ Ti4+ = 0.5% | 47.5 | 3. 06 | 0.95097 | |
| MWCNT - Ag+/ Ti4+ = 1.0% | 65 | 5.30 | 0.91623 | |
| MWCNT - Ag+/ Ti4+ = 1.5% | 55 | 3.57 | 0.97206 | |
| MWCNT - Ag+/ Ti4+ = 2.0% | 80 | 8.42 | 0.98006 | |
| MWCNT - Ag+/ Ti4 += 2.5% | 72.5 | 4. 66 | 0.98538 |
To identify the reactive oxygen species involved in the photocatalytic process of MWCNT-TiO2-Ag, ESR coupled with spin-trapping method was used. As spin trapping agent was used DMPO.
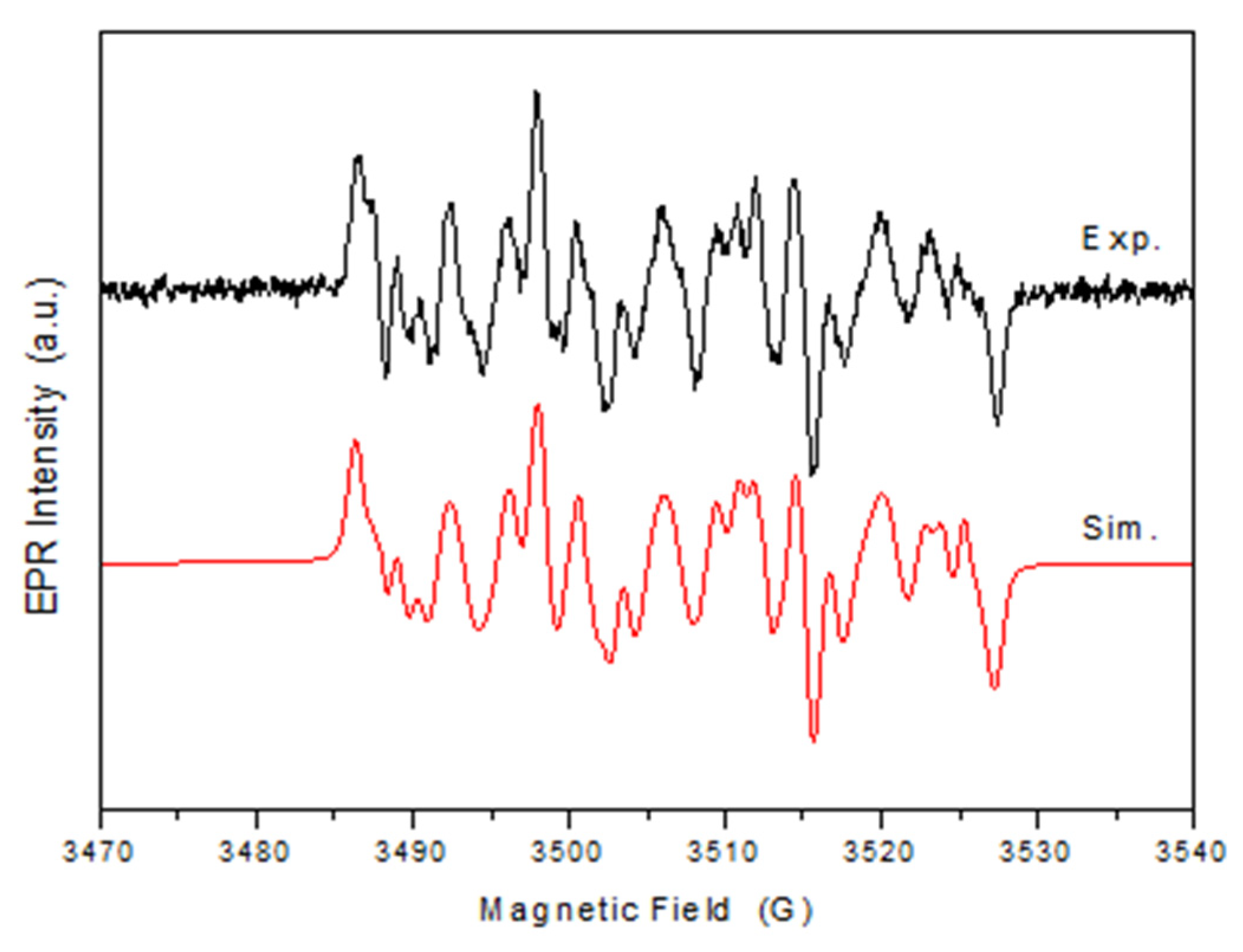
The experimental spectrum obtained after 30 min UV irradiation of DMPO-MWCNT-TiO2-Ag solution is shown in Figure 15. The obtained spectrum is a complex one consisting of many resonance lines due to different spin adducts generated in the time of irradiation. To identify and extract the contribution of the spin adducts, the simulation of the spectrum was performed. The simulated spectrum represents the linear contribution of the following spin adducts: •DMPO-OCH3 ( = 13.2 G, = 8.3 G, = 2.0 G, relative concentration 23.8%), •DMPO-O2- ( = 13.0 G, = 11.3 G, = 2.6 G, relative concentration 37.9%), •DMPO-OOH ( = 13.5 G, = 11.0 G, = 0.6 G, relative concentration 17.2%), nitroxide-like radical ( = 13.9 G, relative concentration 21%). The presence of •DMPO-OCH3 spin adducts is the proof of the presence of •OH radicals as a results of the interaction between the DMSO solvent and •OH radical [53]. •OOH radical is obtained by protonation of superoxide radical •O2- [54]. Nitroxide-like radical appears by cleavage the N‒C bond and ring opening of DMPO [55]. These results attest the presence of both •OH and •O2- radicals, relative concentration•O2- radicals have been more than twice than hydroxyl radicals concentration.
Figure 15.
Experimental and spectrum simulation of DMPO spin adducts generated by MWCNT-TiO2-Ag sample after 30 min of irradiation.
Figure 15.
Experimental and spectrum simulation of DMPO spin adducts generated by MWCNT-TiO2-Ag sample after 30 min of irradiation.
4. Materials and Methods
4.1. Materials.
Materials and reagents used for the preparation of MWCNT-TiO2:AgX% were: multi-walled carbon nanotubes (MWCNTs) with a 99% purity were purchased commercially (Sigma-Aldrich, Merck, KGaA, Darmstadt, Germany), poly-allylamine hydrochloride (PAH) (Alfa –Aesar , Thermo Fisher (Kandel) GmbH, Germany), sodium chloride, NaCl-( Alpha Aesar, Bio Aqua Group, Targu Mures, Romania), titanium tetraisopropoxide Ti[OCH(CH3)2]4, note Ti(OPri)4 (Fluka Chemie, GmBH, Sigma – Aldrich Chemie, Steinheim, UK), acetylacetone, C5H8O, note AcAc (Merck, KGaA, Darmstadt, Germany), AgNO3 (VWR Chemicals, UK), L+ ascorbic acid (Merck, KGaA, Darmstadt, Germany), absolute ethanol (C2H5OH-EtOH) (Alpha Aesar, Bio Aqua Group, Targu Mures, Romania). All chemicals were of analytical grade and used without further purification. The aqueous solutions were prepared with Milli-Q water obtained from Direct-Q 3UV system (Millipore, Bedford, MA, USA).
4.2. Sample preparation.
To obtain carbon nanotubes (MWCNT) decorated with TiO2 - Ag (MWCNT - TiO2:AgX%) several synthesis steps were performed, as follows: in the first stage MWCNT were functionalized with -OH and –COOH groups and in the second stage the TiO2:AgX% nanoparticles previously prepared by modified Pechini method were attached on functionalized MWCNT through polyallylamine hidrochloride (PAH).
4.2.1. Functionalization of MWCNTs
Functionalization of MWCNTs was performed by acid treatment of commercial MWCNTs (D x L 110-170nm x 5-9 μm, Aldrich) in 200 ml mixture of H2SO4:HNO3 (3:1 vol ratio) in ultrasound bath for 4 h. Then, the content was cooled, centrifuged and washed with distilled water several times to maintain its neutralization. Further, it was dried at 65oC in the oven for obtaining its functionalized MWCNTs.
4.2.2. Synthesis of TiO2 modified by Ag
In order to obtain TiO2 modified by Ag nanopowder by Pechini method, titanium tetraisopropoxide Ti[OCH(CH3)2]4, and acetylacetone, C5H8O, is used as precursor. The mixture, as obtained, a different Ag+/Ti4+ atomic ratio, respectively 0.5, 1.0, 1.5, 2.0 and 2.5% of AgNO3 () (note as Ag+ /Ti4+ = 0.5%, Ag+ /Ti4+ = 1.0%, Ag+ /Ti4+ = 1.5%, Ag+ /Ti4+ = 2.0% and Ag+ /Ti4+ = 2.5%) is added in solution, after 2h. By continuous stirring, the L(+)-ascorbic acid was added. The AgNO3 : L(+)-ascorbic acid was 1:1 (%wt). This mixture was dried at room temperature, for 10 days, and then calcinated, in air, at 455°C, for 4h.
4.3. Decoration of MWCNT with TiO2 modified by Ag
To ensure a good attaching, the functionalized MWCNTs were modified by polymer wrapping with poly(allylamine hydrochloride) (PAH). The nanotubes (8 mg) were dispersed in a 0.5 wt% PAH salt solution (0.5M NaCl, 500 ml) and sonicated for 4 h, then stirred overnight at 80°C. The excess polymer was removed by repeated centrifugation and redispersed in water, until a stable, homogenous MWCNT suspension was obtained. The amine functionalities on the MWCNTs surface (MWNT-PAH) ensure good separation and stability due to electrostatic interactions (repulsions) in an aqueous solution. Before the decoration process, the functionalized MWCNT and TiO2 :Ag nanoparticles were dispersed separately by sonication in ethanol for 1h and then were mixed together, the process is continued for another 4h. The obtained MWCNT decorated with TiO2: Ag (MWCNT-TiO2:Ag) were separated by centrifugation, washed with distilled water several times and then were dried in an oven at 70°C.
4.4. Characterisation
Thermal analysis (TG–DTA–DTG) was recorded with a Mettler-Toledo Thermogravimeter 851e (Columbus, USA) equipment. The TG–DTA–DTG was performed in air, in the temperature range 20–1000°C using upgraded computer controlled equipment. About 38,679 mg of sample was heated in Pt-holder with another Pt-holder containing a-alumina as reference material. The sample was heated at a rate of 10°C/min from ambient temperature to 1000°C in static air.
The XRD was recorded on the BRUKER D8 Advance X-ray diffractometer (Rheinstetten, Baden-Württemberg, Germany), working at 45kV and 45mA. The CuKα radiation, Ni filtered was collimated with Soller slits. A germanium monochromator was used. The data of the X-ray diffraction patterns were collected in a step-scanning mode with steps of Δ2θ = 0.01°. Pure silicon powder (standard sample) was used to correct the data for instrumental broadening. Crystallographic identification was accomplished by comparing the experimental XRD patterns with those of MATCH software (Kreuzherrenstr, Germany) version 1.11.
The Warren - Averbach X-ray profile Fourier analysis of the (101), (004) (200) and (204) anatase peak profiles were processed by the XRLINE [27] computer program in order to determine the effective crystallite mean size (Deff). The crystallite size distribution function was determined from the second derivative of the strain corrected Fourier coefficients [28].
FT-IR spectra of the powder samples using KBr pellet technique, in the absorbance mode have been recorded using JASCO FT/IR-6100 Fourier Transform Infrared Spectrometer (JASCO International Co, LTD. Tokyo, Japan) in the 400 - 4000cm-1 wavenumber range with a resolution of 4cm-1.
The Raman spectra have been recorded at room temperature with a JASCO NRS 3300 spectrophotometer (JASCO International Co, LTD. Tokyo, Japan ) equipped with a CCD detector (-69°C) using the laser excitation wavelength of 515 nm line of an Ar-ion laser was used as the excitation source and a laser power of 0.7mW. An 100x Olympus objective, an exposure time of 60s and 3 accumulations were used for each spectral measurement. The spectrometer was calibrated using the Si Raman peak from 521cm-1.
EPR measurements of powder samples were carried out on a Bruker E-500 ELEXSYS X-band (9.52GHz) spectrometer (Rheinstetten, Baden-Württemberg, Germany) at room temperature under identical conditions: microwave frequency of 9.5248GHz, microwave power 2mW, modulation frequency of 100kHz and modulation amplitude 10G.
For morphology of samples, a Scanning Electron Microscope Hitachi SU8230 scanning electronic microscope (Tokyo, Japan), using 30kV, 15mm working distance. The instrumental analysis of the sample composition was determined by Oxford Instruments EDS System (Oxford, UK) and AZtech software (Greeley, CO, USA).
Optical UV-VIS absorption spectra were recorded on the PERKIN - ELMER LAMBDA 45 spectrophotometer (JASCO International Co, LTD. Tokyo, Japan), equipped with an integrating sphere assembly in the range 200-900nm.
The fluorescence spectra were obtained using an ABL&JASCO V 6500 spectrofluorometer (Tokyo, Japan) with xenon lamp of 150W.
Photocatalytic measurements were performed by immersing the 0.5mg samples in the Allura Red solution were irradiated with UV lamp (15 W) emitting at 365 nm. The absorbance of Allura Red solution was measured using a T80+ UV – VIS, Pro In-struments Ltd. Spectrophotometer (Leicestershire, UK).
ESR coupled with the spin trapping probe technique was employed. 5,5-dimethyl- 1-pyrroline N-oxide (DMPO, Sigma-Aldrich, Merck, KGaA, Darmstadt, Germany) was used as a spin trapping reagent. The nanoparticles (10 mg) were dispersed in DMSO (1 mL) and homogenized in an ultrasound bath (30 min) before use. DMPO of 0.2 mol/L concentration was added to the suspension. The samples were prepared immediately before measurements and transferred into the quartz flat cell optimized for liquid measurements.
5. Conclusions
The present paper reported the synthesis and characterization of MWCNT-TiO2-Ag nanocomposites for photocatalytic oxidation of Allura Red. The investigation results show that samples have crystalline structures containing anatase phase with average crystallite size of 14 nm.The nanocomposites have a polyhedral shape and are distributed over the nanotube surface with a tendency to agglomerate as evidenced by TEM. FT – IR spectra suggest the presence of the vibrations corresponding to MWCNT – TiO2 - Ag structures in the nanocomposites. The existance of active modes of anatase TiO2 and vibration of the sp2 bonded carbon atoms characteristic of graphite are evidenced by Raman characterization. The presence of oxygen vacancies was evidenced by EPR spectroscopy, and their concentration decrease with the increasing of dopant concentration up to 1.5% mol, followed by a slightly increase in accordance with the PL results. Moreover, a narrowing of the band gap by doping was observed. The highest photocatalytic activity (79.99%) was achieved for the sample containing 2%Ag. A higher concentration of Ag particles shields the active centers of the semiconductor, thus reducing its catalytic activity. Moreover, the generation under UV light irradiation of both active radicals OH• and •O2- have been highlighted by ESR spin trapping technique. The •O2- are the main species generated and responsible for Allura Red degradation.
Aknowlegments: The authors acknowledge financial support from the MCID through the "Nucleu" Programe within the National Plan for Research, Development and Innovation 2022-2027, project PN 23 24 01 03 and through Program 1—Development of the national research and development system, Subprogram 1.2—Institutional performance—Projects that finance the RDI excellence, Contracts no. 37PFE/30.12.2021.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Zhao, D.X.; Cai, C. Cerium-based UiO-66 metal-organic framework for synergistic dye adsorption and photodegradation: A discussion of the mechanism. Dyes Pigments 2021 185 (3) 108957. [CrossRef]
- Xiang, C.; Wang, W.; Liu, S; Huang, Y., Li; M.; Wang, D. Humidity-stimulated film actuator with dual-responsive of bending deformation and discoloration. Sens Actuators B Chem 2023 380 133344. [CrossRef]
- Rajakani, V.; Sahaya Shajan, X., Arulgnanam, A.; Sumithraj Premkumar, P. Photovoltaic studies on iodine incorporated titania aerogel nanocomposites. Results in Optics 2023, 10, 100346. [CrossRef]
- Mandić, V.; Panžić, I.; Brnardić, I.; Jajčinović, I.; Mičetić, M. Lateral and vertical evolution of the compositional and morphological profiles in nanostructured photocatalytic titania thin films. Appl Surf Sci 2023 613 56047. [CrossRef]
- Kausar, F.; Varghese A.; Pinheiro, D.; Sunaja, D. K. R. Recent trends in photocatalytic water splitting using titania based ternary photocatalysts-A review. Int J Hydrog Energy 2022 47 (53) 22371-22402. [CrossRef]
- Sadia, M.; Naz, R.; Khan, J.; Zahoor, M.; Ullah, R.; Khan, R.; Naz, S.; Almoallim, H. S.; Alharbi, S. A. Metal doped titania nanoparticles as efficient photocatalyst for dyes degradation. J King Saud Univ Sci. 2021 33 (2) 101312. [CrossRef]
- Ahmed, D. S.; Mohammed, M. K. A.; Mohammad, M. R. Sol–gel synthesis of Ag-doped titania-coated carbon nanotubes and study their biomedical applications. Chem Zvesti 2020 74 197–208. [CrossRef]
- Arun, J.; Nachiappan, S.; Rangarajan, G.; Alagappan, R. P.; Gopinath, K. P.; Lichtfouse, E. Synthesis and application of titanium dioxide photocatalysis for energy, decontamination and viral disinfection: A review. Environ Chem Lett 2023 21 339 – 362. [CrossRef]
- Cringoli, M. C.; Perathoner, S.; Fornasiero, P.; Marchesan, S. Carbon nanostructures decorated with titania: Morphological control and applications. Appl Sci 2021 11 6814. [CrossRef]
- Rajakani, V.; Sahaya, X.; Shajan, A.; Arulgnanam, Sumithraj Premkumar, P. Studies on the silver incorporated titania aerogel nanostructure as a photoanode in quasi solid dye- sensitized solar cells. Mater Today Proc 2022 65 (5) 2473-2479. [CrossRef]
- Topolski, A. Functionalization of titania nanotubes surface with platinum(II) complexes. Polyhedron 2023 230 116218. [CrossRef]
- Paszkiewicz, O.; Wang, K.; Rakoczy, R.; Kordas, M.; Leniec, G.; Kowalska, E.; Markowska-Szczupak, A. Antimicrobial properties of pristine and Pt-modified titania P25 in rotating magnetic field conditions. Chem Eng Process 2022 178 109010. [CrossRef]
- Chakhtouna, H.; Benzeid, H.; Zari, N.; Qaiss, A. el kacem; Bouhfid, R. Recent progress on Ag/TiO2 photocatalysts: Photocatalyticand bactericidal behaviors. Environ Sci Pollut Res 2021 28 44638–44666. [CrossRef]
- Sohrabi, L.; Taleshi, F.; Sohrabi, R. Effect of carbon nanotubes support on band gap energy of MgO nanoparticles. J Mater Sci: Mater Electron 2014 25 4110–4114. [CrossRef]
- Sameera, I.; Bhatia, R.; Prasad, V. Preparation, characterization and electrical conductivity studies of MWCNT/ZnO nanoparticles hybrid. Physica B: Condensed Matter 2010 405 1709-1714. [CrossRef]
- Younas, M.; Gondal, M.,A.; Dastageer, M.,A.; Harrabi, K. Efficient and cost-effective dye-sensitized solar cells using MWCNT-TiO2 nanocomposite as photoanode and MWCNT as Pt-free counter electrode. Solar Energy 2019 188 1178-1188. [CrossRef]
- Hosny, N. M.; Gomaa, I.; Elmahgary, M. G. Adsorption of polluted dyes from water by transition metal oxides: A review, Appl Surf Sci Adv 2023 15 100395. [CrossRef]
- Li, W.;, Wang, B.;Yuan, Y. ; Wang, S. Spatiotemporal distribution patterns and ecological risk of multi-pesticide residues in the surface water of a typical agriculture area in China, Sci Total Environ 2023 870 161872. [CrossRef]
- Jia, W.-L.; Song, C.; He, L. -Y.; Wang, B.; Gao, F. - Z.; Zhang, M.; Ying. G.- G. Antibiotics in soil and water: Occurrence, fate, and risk, Curr Opin Environ Sci Health 2023 32100437. [CrossRef]
- Barboux – Doeuff, S.; Sanchez, C. Synthesis and characterization of titanium oxide – based gels synthesized from acetate modified titanium butoxide precursors. Mat Res Bull 1990 29 41-13. [CrossRef]
- Devi Gamathi, L., Nagaraj, B., Eraiah Rajashekhar, K. Synergetic effect of Ag concentration and nitrogen doping in TiO2 for the degaradtion of phenol under solar irradiation in presence of electron acceptor. Chem Eng J 2012 181 – 182 259 -266. [CrossRef]
- Bouazza, N.; Ouzzine, M.; Lillo – Rόdenas, M. A.; Eder, D.; Linares – Solano, A. TiO2 nanotubes and CNT – TiO2 hybrid materials for the photocatalytic oxidation of propene at low concentration. Appl Catal B – Environ 2009 92 377 – 383. [CrossRef]
- Akbarzadeh, R.; Ghaedi; M., Kokhdan, S. N.;Vashaee, D. Remarkably improved electrochemical hydrogen storage by multi-walled carbon nanotubes decorated with nanoporous bimetallic Fe–Ag/TiO2 nanoparticles. Dalton Trans 2019 48 898. [CrossRef]
- Ashkarran, A. A.; Fakhari, M.; Hamidinezhad H.; Haddadi, H.; Nourani, M. R. TiO2 nanoparticles immobilized on carbon nanotubes for enhanced visible - light photo – induced activity. J Mater Res Technol 2015 4 (2) 126 – 132. [CrossRef]
- Yuliati, L.; Kimi, M.; Shamsuddin, M. High activity of Ag-doped Cd0.1Zn0.9S photocatalyst prepared by the hydrothermal method for hydrogen production under visible-light irradiation. Beilstein J Nanotechnol 2014 5 (587–595. [CrossRef]
- Aldea, N.; Indrea, E. XRLINE, a program to evaluate the crystallite size of supported metal-catalysts by single X-ray profile fourier-analysis. Comput Phys Commun 1990 60 155-159. [CrossRef]
- Indrea, E., Barbu, A. Indirect photon interaction in PbS photodetectors, Appl Surf Sci 1996 106 498. [CrossRef]
- Berkum, J. G. M. van; Vermeulen, A. C.; Delhez, R.; Keijser, T. H. de; Mittemeijer, E. M.. Applicabilities of the Warren-Averbach analysis and an alternative analysis for separation of size and strain broadening, J Appl Cryst 1994 27 345 - 357 . [CrossRef]
- Kraus, W.; Nolze, G. POWDER CELL — a Program for the Representation and Manipulation of Crystal Structures and Calculation of the Resulting X-ray Powder Patterns". Appl Cryst 1996 29 301—309. [CrossRef]
- Thiel, J.; Pakstis, L.; Buzby, S.; Raffi, M.; Ni, C.; Pochan, D. J.; Ismat Shah, S. Antibacterial properties of silver-doped titania, Small 2007 3 (5) 799–803. [CrossRef]
- Soler-Illia, G. J. de A. A.; Louis, A.; Sanchez, C. Synthesis and characterization of mesostructured titania-based materials through evaporation-induced self-assembly. Chem Mater 2002 14 (2) 750–759. [CrossRef]
- Chen, C.S.; Liu, T.G.; Lin, L.W.; Xie, X.D.; Chen, X.H.; Liu, Q.C.; Liang, B.; Yu, W.W.;. Qiu, C. Y. Multi-walled carbon nanotube-supported metal-doped ZnO nanoparticles and their photocatalytic property. J Nano Res 15 2013 1295–1304. [CrossRef]
- Alsharaeh, H. ; Bora, T.; Soliman, A.; Ahmed, F.; Bharath, G.; Ghoniem, M. G.; Abu-Salah, K. M.; Dutta, J. Sol-Gel-Assisted Microwave-Derived Synthesis of Anatase Ag/TiO2/GO Nanohybrids toward efficient visible light phenol degradation, Catalysts 2017 7 (5) 133. [CrossRef]
- Zhang, H.; Wang, X; Xia, J.; Meng, Q.; Ding, J.; Lu, J. Synthesis andcharcterisation of TiO2 / graphene oxide nanocomposites for photoreduction of heavy metal ions in reverse osmosis concentrate, RSC Adv 2018 8 (60) 34241-34251. [CrossRef]
- Abbasi, S.; Zebarjad, S. M.; Bogdan, S. H. Noe; Yousesfi, A. Synthesis of TiO2 nanoparticles and decorated multi-walled carbon nanotubes with various content of rutile titania. Synth React Inorg M 2015 45 10. [CrossRef]
- Ohsaka, T. Izumi, F., Fujiki, Y. Raman Spectrum of Anatase TiO2. J Raman Spectroscopy 1978 7 (6) 321-324. [CrossRef]
- Misra, K.; Andronenko, S. I.; Tipikin, D.; Freed, J. H.; Somani V.; Prakash, O. Study of paramagnetic defect centers in as-grown and annealed TiO2 anatase and rutile nanoparticles by a variable-temperature X-band and high-frequency (236GHz) EPR. J Magn Magn Mater 2016 401 495–505. [CrossRef]
- Caretti, I.; Keulemans, M.; Verbruggen, S. W.; Lenaerts, S.; Doorslaer, S. Van. Light-Induced Processes in Plasmonic Gold/TiO2 Photocatalysts Studied by Electron Paramagnetic Resonance. Top Catal 2015 58 776–782. [CrossRef]
- Grabowska, E., Marchelek,.M., Klimczuk, T., Trykowski, G., Zaleska-Medynska, A. Noble metal modified TiO2 microspheres: Surface properties and photocatalytic activity under UV–vis and visible light. J Mol Catal A Chem 423 191–206 . [CrossRef]
- Popa, A.; Stefan, M.; Macavei, S.; Muresan, L. E.; Leostean, C.; Floare-Avram, C. V.; Toloman, D. Photoluminescence and photocatalytic properties of MWNTs decorated with Fe-doped ZnO nanoparticles. Materials 2023 16(7) 2858. [CrossRef]
- He, J.; Ichinose, I.; Kunitake, T.; Nakao, A. In situ synthesis of noble metal nanoparticles in ultrathin TiO2-gel films by a combination of ion-exchange and reduction processes, Langmuir 2002 18(25) 10005-10010. [CrossRef]
- Nguyen, M. T.; Nguyen, C. K.; Vu, T. M. P.; Duong, Q.V.; Pham, T. L.; Nguyen. T. C.. A study on carbon nanotube titanium dioxide hybrids: Experiment and calculation. Nanosci Nanotechnol 2014 5 045018. [CrossRef]
- Siwach, O. P.; Sen, P. Fluorescence properties of Ag nanoparticles in water, methanol and hexane. J Lumin 2009 129 (1) 6 - 11. [CrossRef]
- Chrysicopoulou, P.; Davvazoglou, D.; Trapalis, Chr.; Kordas, L. G. Optical properties of very thin (<100 nm) sol–gel TiO2 films. Thin Solid Films 1998 323 188 -193. [CrossRef]
- Tauc, J.; Grigorovici, R.; Vancu, A. Optical properties and electronic structure of amorphous germanium, Phys Status Solidi 1966 15 627–637 . [CrossRef]
- Li, Y.; Ma, M.; Chen, W., Li. L.; Zen, M. Preparation of Ag – doped TiO2 nanoparticles by a miniemulsion method and their photoactivity in visible light illuminations. Mat Chem Phys 2011 129 501-505. [CrossRef]
- Chen, X.; Luo, W. Optical spectroscopy of rare earth ion-doped TiO2 nanophosphors. J Nanosci Nanotechno 2010 10 482–1494. [CrossRef]
- Selvam, K.; Swaminathan, M. Cost effective one-pot photocatalytic synthesis of quinaldines from nitroarenes by silver loaded. TiO2. J Mol Catal A - Chem 2011 351 52–61. [CrossRef]
- Lipping, W.; Baoshun, L.; Chao, L.; Xiujian, Z. Preparation, characterisation and photocatalytic property of Ag-loaded TiO2 powers using photodeposition method, J. Wuhan Univ Techno.-Mat Sci Edit 2009 24 258–263. [CrossRef]
- Zu, J.; Xiong J.; ChenB.; Liu, S. Fabrication and characterization of Ag–TiO2 multiphase nanocomposite thin films with enhanced photocatalytic activity. Applied Catal B 2005 60 211-221. [CrossRef]
- Falaras, P.; Arabatzis, I. M.; Stergiopoulos T.; Bernard, M. C. Enhanced activity of silver modified thin-film TiO2 photocatalysts. Int J Photoenergy 2003 5 123 – 130. [CrossRef]
- Sobczyński, A.. Photoassisted hydrogen production from a methanol-water mixture on platinized Cr2O3-doped TiO2. J Mol Cat 1987 39 45 – 53. [CrossRef]
- Klein, S. M.; Cohen, G.; Cederbaum, A. I. Production of formaldehyde during metabolism of dimethyl sulfoxide by hydroxyl radical-generating systems. Biochemistry 1981 20 6006–6012. [CrossRef]
- Alyani, S.J.; Pirbazari, A.E.; Khalilsaraei, F. E.; Kolur, N.A.; Gilani. N. Growing Co-doped TiO2 nanosheets on reduced graphene oxide for efficient photocatalytic removal of tetracycline antibiotic from aqueous solution and modeling the process by artificial neural network. J Alloy Compd 2019 799 169–182. [CrossRef]
- Diaz-Uribe, C. E.; Daza, M.; Martínez, C.; Páez-Mozo, F.; Guedes; E. A.; Mauro, C. L. B.; Di. E. Visible light superoxide radical anion generation by tetra(4-carboxyphenyl) porphyrin /TiO2: EPR characterization. J Photochem Photobiol A 2010 215 172–178. [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



