
Submitted:
05 May 2023
Posted:
09 May 2023
You are already at the latest version
Abstract
Biomimetic N-acetylcysteamine thioesters are essential for the study of polyketide synthases, non-ribosomal peptide synthetases and fatty acid synthases. The chemistry for their preparation is however limited by their specific functionalization and their susceptibility to undesired side reactions. This is especially detrimental to transition metal-catalyzed reactions. Here we report a method for the rapid preparation of N-acetylcysteamine (SNAC) 7-hydroxy-2-enethioates, which are suitable for the study of various enzymatic domains of megasynthase enzymes, particularly oxygen heterocycle-forming cyclase domains. The method is based on a one-pot sequence of hy-droboration and Suzuki-Miyaura reaction. Optimization of the reaction conditions made it possi-ble to suppress potential side reactions and to introduce the highly functionalized SNAC meth-acrylate unit in high yield. The versatility of the sequence was demonstrated on a dienal precur-sor, which was subjected to Brown crotylation followed by the hydroboration-Suzuki-Miyaura reaction sequence and deprotection, finally giving a complex polyketide SNAC thioester. Back-bone extension by six carbons and a terminal SNAC enethioate was achieved, introducing an E-configured double bond and two adjacent stereocenters in a highly selective manner. The pre-sented method allows for the synthesis of the target motif in significantly fewer steps and with higher overall yield than previously described approaches, while maintaining higher flexibility and control over the stereogenic elements. It is also the first reported example of a transition met-al-catalyzed cross-coupling reaction in the presence of an SNAC thioester.
Keywords:
Suzuki-Miyaura reaction
; biomimetic thioesters
; polyketide synthases
; enzymes
; cyclases
1. Introduction
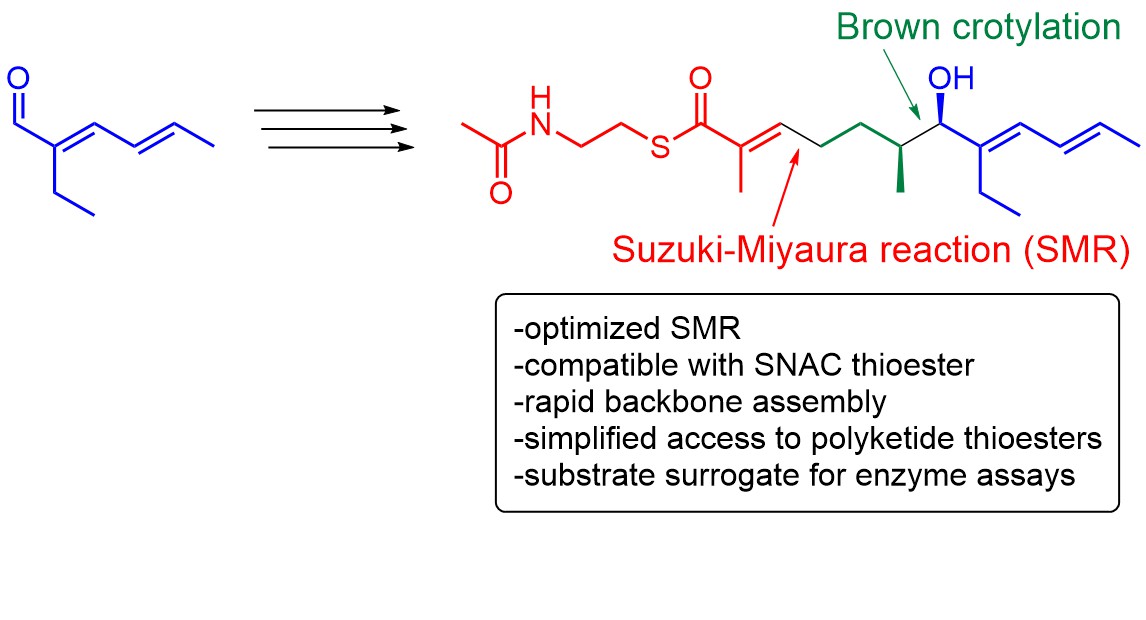
Thioesters are an important functional group in many biosynthetic systems. They often serve to link biosynthetic acyl intermediates to carrier thiols, which can be free molecules such as coenzyme A (CoA) or proteins. Important systems working with protein-bound metabolites are so-called megasynthase enyzmes like fatty acid synthases, polyketide synthases (PKS) and non-ribosomal peptide synthetases (NRPS) and their hybrids [1,2]. They are responsible for the formation of polyketide and peptide natural products, including some of the most important small-molecule drugs in clinical use, such as erythromycin, rapamycin or epothilone. The availability of suitable substrate surrogates is essential for the functional study of these biosynthetic systems. N-Acetylcysteamine (SNAC) thioesters are of particular importance for this as they effectively mimic protein attachment of the substrate via the 4’-phosphopantetheine arm and thus allow simplified studies with active enzymes (Figure 1A) [3,4,5,6,7,8,9,10].
Acylated SNACs contain an acetamide and a thioester as conserved reactive functional groups that afford them with problematic properties (Figure 1B) [7,11]. The thioester can undergo side reactions with external or internal nucleophiles, resulting in irreversible loss of substance. Due to its polarity, the acetamide can cause problems during substance purification and can, as a nucleophilic/protic group, cause undesired side reactions. The functionalization distance between acetamide and thioester carries the risk that they act as a chelate ligand and interact with metals. The synthesis of SNAC thioester surrogates of late-stage biosynthetic intermediates is as challenging as the synthesis of natural products of similar structural complexity but, for the above-mentioned reasons, has the challenge of an additional problematic functional group. A useful strategy to overcome this problem would be to introduce the SNAC moiety at a late stage of synthesis along with a larger fraction of the polyketide moiety.
Improving the specific methodology for the synthesis of complex polyketide-SNAC thioesters is therefore of great interest to the biosynthetic research community. Transition metal-mediated reactions are well suited for late-stage attachment in the convergent synthesis of complex biosynthetic thioester surrogates but have only very rarely been described in the presence of SNAC thioesters. To the best of our knowledge, the literature currently only contains a report about olefin cross metathesis between SNAC-acrylates and hydroxyolefins catalyzed by the 2nd generation Grubbs catalyst [12].
The Suzuki-Miyaura reaction (SMR) is a highly versatile Pd-catalyzed cross-coupling reaction. It allows couplings between halides and non-toxic boronic acid derivatives under relatively mild conditions (Figure 2) [13,14]. In addition to sp2-sp2 bond formations, it is now possible to carry out couplings between sp2 and sp3 centers as well as between two sp3 centers. Two aspects of the SMR could be problematic when applied to SNAC thioesters. On the one hand, the use of base is necessary to accelerate the essential group transfer from the boronic acid to the Pd during the catalytic cycle (step 3). Moreover, Pd can also insert into the C-S bond of the thioester instead of the C-halide bond (step 1) [15]. This reactivity is so pronounced that it forms the basis of the Liebeskind-Srogl reaction, a modification of the SMR for the direct synthesis of ketones from thioesters [16,17].
Among the diverse enzymatic PKS domains, cyclases that form saturated oxygen heterocycles by intramolecular oxa-Michael addition (IMOMA) stand out for the synthetical value of this transformation (Figure 1C) [18,19]. It has been shown that they catalyze ring formation with exceptional stereoselectivity and therefore represent a potential new type of biocatalyst [20,21,22,23,24,25,26,27,28,29]. For the study of such enzymes, SNAC-7-hydroxy-2-enethioates are required as substrate surrogates. The synthetic methodology for the selective installation of this structural motif is however not well developed, making the generation of precursor libraries a difficult task. The multi-step routes described in the literature are either highly elaborate, are not stereoselective or lack flexibility and are therefore narrow in their applicability [20,21,22,23]. For example, the synthesis of the SNAC surrogate of 2 in stereochemical pure form was accomplished in eight steps and required multiple purification procedures [21,22]. A lack of convergence furthermore makes it necessary to carry out the largest part of this sequence from different starter building blocks to access derivatives with variations in the eastern part of the molecule. Other reported routes are shorter, but also less flexible due to the choice of larger starting building blocks or the choice of the introduction reaction for the SNAC thioester. Olefin cross-metathesis for example is only possible with SNAC-acrylthioates and not with SNAC-methacrylthioates. Therefore, we set out to develop a flexible, straightforward and broadly applicable method for the preparation of SNAC-7-hydroxy-2-enethioates.
As a solution, we turned to a sequence of hydroboration and SMR to assemble the backbone and directly introduce the SNAC moiety. The specific challenge was to effectively perform the SMR in the presence of the SNAC thioester, which has not been achieved before to the best of our knowledge. The versatility of the method should be shown on the example of the synthesis of 4 (Figure 1D). This compound was on the one hand specifically required for our enzymatic studies on new IMOMA cyclases. On the other hand, it represents a particularly challenging substrate during the preparation of which various detrimental side reactions could occur and thus a reasonable benchmark.
2. Results and Discussion
Thioester-halides are rare substrates in SMRs. The literature however contains an example of coupling reactions of simple 4-bromothiophenols with 4-tolyl-boronic acid in which the bulky acyl unit of the thioesters served as a protecting group for the thiol [15]. We used the reported conditions for the synthesis of an ethylenoate sensitive towards hydrolysis and base described by Suzuki et al. as the starting point for our studies [30]. These were carried out using the SNAC (E)-3-bromo-2-methylprop-2-enethioate 5 and the OTBS-protected 3-hydroxyolefin 6 (1.0 equiv. of 5, 1.1 equiv. of 6, 1.1 equiv. 9-BBN, 5mol% PdCl2(dppf) und 2.0 equiv. K2CO3). Fortunately, a basic coupling reactivity was observed. The yields of the reactions however varied hardly reproducibly over a wide range and showed a strong dependence on even small variations of the amounts of thioester, alkene, borane and Pd catalyst. This suggests that several side reactions might proceed at rates similar to the desired pathway. We therefore carried out a systematic optimization study (Table 1).
For this, we varied the individual reaction parameters. Since we assumed that side reactions of the 3-bromoacryl thioate 5a were be a particular problem, we worked with an excess of 1.5 equiv. of alkene 6 and 9-BBN. Different thioester halides (Br and I), bases (K2CO3 and Cs2CO3), additives (P(o-furyl)3 and AsPh3) and temperatures (20 °C, 50 °C and 65°C) were tested. All reactions were carried out on a scale of 90–100 µmol of 5a/5b and compared based on the isolated yield after column chromatography. The yields in the basic experiment with an excess of 1.5 equiv. of 6 and 9-BBN (entry a) were fortunately stable upon repetition in a range slightly above 50%. The variation of individual parameters did not lead to a marked increase of this value, whereas the addition of P(o-furyl)3 and the decrease of the reaction temperature even significantly reduced the yield (entries b and e). Fortunately, the combined change of several parameters led to a significantly improved result (entry i). Using 3-iodoacryl thioate 5b, Cs2CO3, AsPh3 and carrying out the reaction at room temperature resulted in a yield of 78%.
Side products were regularly detected in the low-yielding reactions that could not be isolated and fully analyzed. According to TLC, these were highly polar compounds whose migration behavior suggests that they are derived from SNAC. We assume that a major part of this is the homocoupling product of the thioester acrylates 5a/5b and the 2-(N-acetamidyl)-ethylketone resulting after C–S bond insertion of the Pd, a side reaction described previously for low-functionalized thioesters. [15] The yield improvement observed in the optimization study would be consistent with the suppression of these side reactions. Cs2CO3 is much better soluble in DMF than K2CO3 leading to a much higher effective concentration of carbonate. This should significantly improve the activation of the ate complex for alkyl group transfer to the Pd (step 3 in Figure 2) and accelerate the heterocoupling reaction. The iodoacrylate is more reactive towards Pd insertion than the bromoacrylate, thus favoring this productive reaction (step 1) compared to insertion of the Pd into the C–S bond. This selectivity is expected to be even more pronounced at room temperature than at 50 °C. The addition of AsPh3 supports these effects by accelerating both, the formation of the active Pd(0) from the Pd(II) species and the transmetallation due to its lower σ-donor effect than PPh3. [31,32]
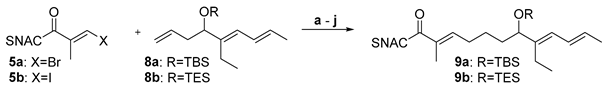
We shifted to the coupling between the thioester 5 with the olefins 8a and 8b that resemble the sensitive 5-hydroxy-tri-1,3,7-ene in target molecule 4 (Table 2). The higher degree of functionalization makes them more susceptible to side reactions during the introduction and removal of the protecting group and during the coupling cascade. Beside screening the same thioester halides (5a/5b) as in Table 1, a broader panel of bases (Cs2CO3, K2CO3 and K3PO4) and hydroxyl protection groups (TBS and TES) on the olefinic coupling partner were examined. Due to the superiority in the previous optimization, only AsPh3 was applied as an additive and only 20 °C and 50 °C were tested as reaction temperature.
Compared to the basic experiments (entries a and c, Table 2), a decrease in yield was observed when the reaction was carried out at 20 °C instead of 50 °C or when K3PO4 was employed as a base (entries a–e). In contrast to the experiments with the simpler coupling partner 6 (Table 1), the change of one or two reaction parameters already led to an increase of the yield up to 80% (entries f–i). When these measures were combined and the reaction was carried out at 20 °C, a further increase to 87% yield was achieved (entry j). The TES group is expected to be easier removable than the TBS group (vide infra). As it showed to be stable under the conditions tested and as both protecting groups gave similar yields in the comparable entries a-d, the optimization in entries f–j was conducted with this silyl protection group. The results summarized in Table 2 are in agreement with those observed in Table 1 and confirm the conclusions / interpretations drawn from them.
Numerous side reactions are conceivable during the deprotection of silyl ethers 9a and 9b, such as eliminations, intramolecular oxa-Michael additions or interference with the thioester. The slightly acidic conditions in the presence of PPTS resulted in elimination of the alcohol / silylether (entry a, Table 3). With TBAF, the formation of the desired product was also not observed in any case. No reaction of the TBS ether 9a was found after 1 h at 0 °C (entry b). Decomposition occurred for the TBS ether 9b after overnight reaction at 20 °C and for the TES ether already after 1 h at 0 °C (entries c and d). Standard HF*pyridine treatment also resulted in decomposition (entry e). A successful procedure was finally adopted from a protocol previously reported by Carreira et al., which relied on using a premixed stock solution of HF*pyridine in THF supplemented with additional pyridine at 0 °C [33]. Deprotection was successful for both silylethers and led to the desired alcohol 10 in pure form after column chromatography (entries f and g). The reactions were continuously monitored by TLC and stopped before noticeable decomposition occurred. The yield for the TES ether was significantly better than that of the TBS ether, suggesting the former to be the preferable protecting group for the synthesis of 4.
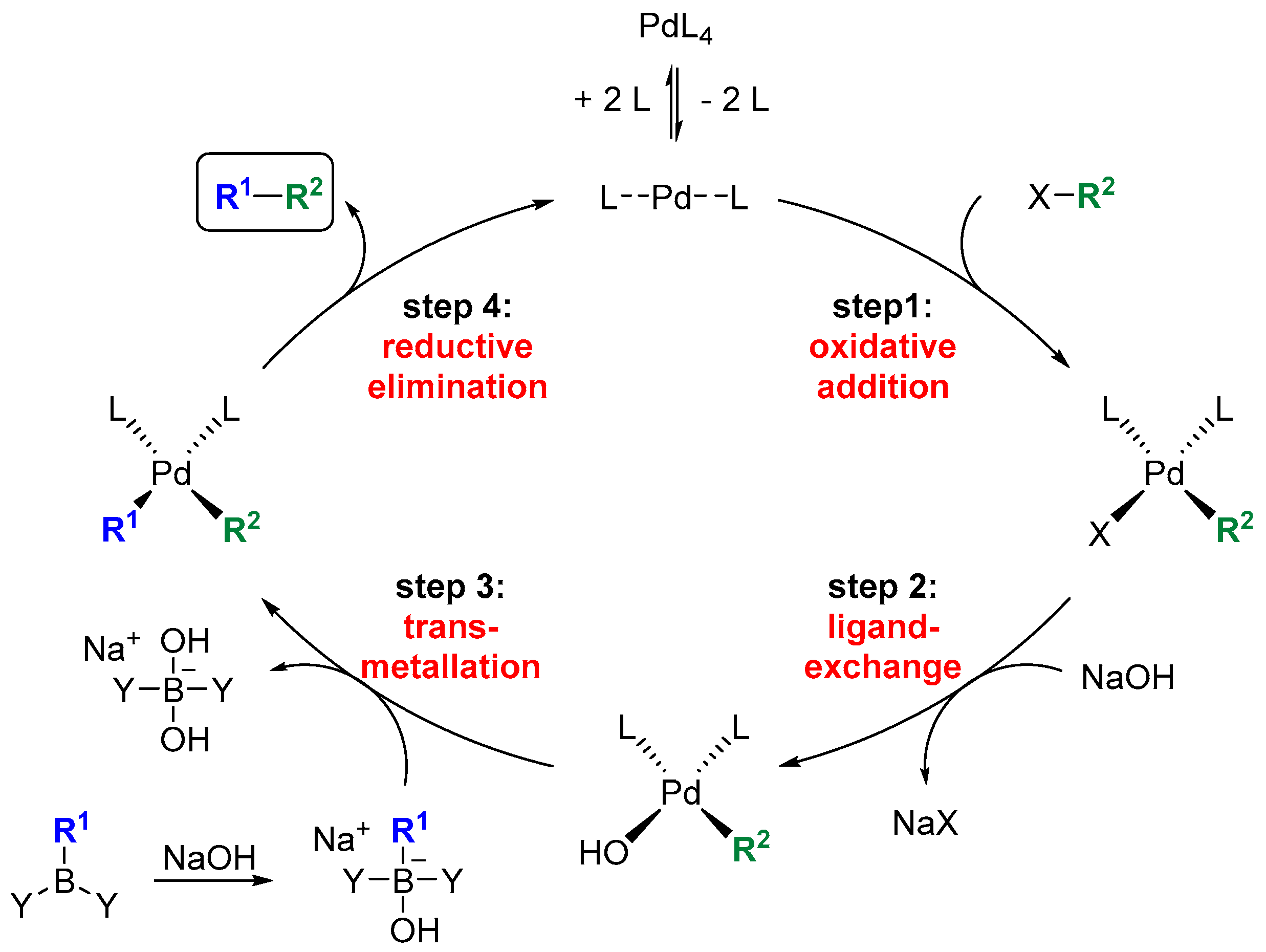
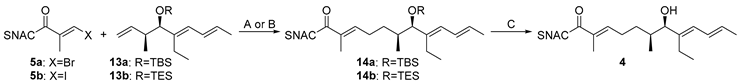
The reaction sequences to 4 were carried out starting from aldehyde 11. Brown crotylation first afforded the highly sensitive hydroxytriene 12 in 71% yield with an e.e. of 86%, which was immediately transformed into the isolatable TBS and TES ethers 13a and 13b in 91% and 84% yield (Scheme 1). This was followed by the established one-pot-two-step-cascade of hydroboration and SMR to give 14a and 14b, which were deprotected to give 7-hydroxy-2-ene-SNAC thioate 4 in overall yields of 16–60% (Table 4). These results confirm on the one hand that TES is the preferable protecting group compared to TBS (entries a and b). On the other hand, they show the positive effect of optimizing the SMR conditions, which led to a yield improvement from 51% to 74% in the coupling step (entries b and c).
In total, product 4 was obtained in four synthetic operations from aldehyde 11 with an overall yield of 36% in high stereoisomeric purity. This represents a significant improvement over previously described routes to similar compounds, which either required significant more steps and gave lower overall yields (8 steps, 10% overall yield for the SNAC thioester analog of 2). Other routes gave 7-hydroxy-2-ene SNAC thioates in five steps from TBS-protected 1,5-hexanediol with a total yield of 23%. The latter however only gave access to racemic products, which were also not branched in the α-position, and did not offer the flexibility in backbone installation that the presented method does.
5. Conclusions
The SMR-based coupling method presented here is compatible with the presence of SNAC thioesters and can be used in the future for the flexible and efficient preparation of substrate surrogates for studies of IMOMA cyclases. Such 2-ene thioates can also be used for studies of other catalytic megasynthase domains that act on similar functionalization patterns as present at C-1–C-6 in 4. The method should also be of interest for the synthesis of precursors of non-enzymatic IMOMA reactions. It has been shown for chemically-catalyzed IMOMA reactions that cis-THP stereoselectivity can be more reliably achieved with enethioates than with enoates so that the former are attractive precursors.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, The Supplementary Materials contain detailed synthetic procedures and analytical data including 1H and 13C NMR spectra.
Author Contributions
Conceptualization, S.D. and F.H.; investigation, S.D. and L.S.; resources, F.H.; writing—original draft preparation, F.H.; writing—review and editing, S.D., L.S. and F.H.; supervision, F.H.; project administration, F.H.; funding acquisition, F.H. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Deutsche Forschungsgemeinschaft (DFG), grant number HA 5841/5-1.
Data Availability Statement
The data presented in this study are available on request from the corresponding author (Prof. Frank Hahn).
Acknowledgments
We thank Central Analytics of the Department of Chemistry as well as the North Bavarian NMR Centre (NBNC) at the University of Bayreuth.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Weissman, K.J.; Müller, R. Protein–Protein Interactions in Multienzyme Megasynthetases. ChemBioChem 2008, 9, 826–848. [Google Scholar] [CrossRef]
- Grininger, M. Enzymology of Assembly Line Synthesis by Modular Polyketide Synthases. Nat. Chem. Biol. 2023, 19, 401–415. [Google Scholar] [CrossRef]
- Ge, H.-M.; Huang, T.; Rudolf, J.D.; Lohman, J.R.; Huang, S.-X.; Guo, X.; Shen, B. Enediyne Polyketide Synthases Stereoselectively Reduce the β-Ketoacyl Intermediates to β-d-Hydroxyacyl Intermediates in Enediyne Core Biosynthesis. Org. Lett. 2014, 16, 3958–3961. [Google Scholar] [CrossRef]
- Sahner, J.H.; Sucipto, H.; Wenzel, S.C.; Groh, M.; Hartmann, R.W.; Müller, R. Advanced Mutasynthesis Studies on the Natural α-Pyrone Antibiotic Myxopyronin from Myxococcus Fulvus. ChemBioChem 2015, 16, 946–953. [Google Scholar] [CrossRef]
- Pinto, A.; Wang, M.; Horsman, M.; Boddy, C.N. 6-Deoxyerythronolide B Synthase Thioesterase-Catalyzed Macrocyclization Is Highly Stereoselective. Org. Lett. 2012, 14, 2278–2281. [Google Scholar] [CrossRef]
- Hansen, D.A.; Rath, C.M.; Eisman, E.B.; Narayan, A.R. H.; Kittendorf, J.D.; Mortison, J.D.; Yoon, Y.J.; Sherman, D.H. Biocatalytic Synthesis of Pikromycin, Methymycin, Neomethymycin, Novamethymycin, and Ketomethymycin. J. Am. Chem. Soc. 2013, 135, 11232–11238. [Google Scholar] [CrossRef]
- Franke, J.; Hertweck, C. Biomimetic Thioesters as Probes for Enzymatic Assembly Lines: Synthesis, Applications, and Challenges. Cell Chem. Biol. 2016, 23, 1179–1192. [Google Scholar] [CrossRef]
- Hahn, F.; Kandziora, N.; Friedrich, S.; Leadlay, P.F. Synthesis of Complex Intermediates for the Study of a Dehydratase from Borrelidin Biosynthesis. Beilstein J. Org. Chem. 2014, 10, 634–640. [Google Scholar] [CrossRef]
- Berkhan, G.; Merten, C.; Holec, C.; Hahn, F. The Interplay between a Multifunctional Dehydratase Domain and a C-Methyltransferase Effects Olefin Shift in Ambruticin Biosynthesis. Angew. Chem. Int. Ed. 2016, 55, 13589–13592. [Google Scholar] [CrossRef]
- Schröder, M.; Roß, T.; Hemmerling, F.; Hahn, F. Studying a Bottleneck of Multimodular Polyketide Synthase Processing: The Polyketide Structure-Dependent Performance of Ketoreductase Domains. ACS Chem. Biol. 2022, 17, 1030–1037. [Google Scholar] [CrossRef]
- Wunderlich, J.; Roß, T.; Schröder, M.; Hahn, F. Step-Economic Synthesis of Biomimetic β-Ketopolyene Thioesters and Demonstration of Their Usefulness in Enzymatic Biosynthesis Studies. Org. Lett. 2020, 22, 4955–4959. [Google Scholar] [CrossRef]
- Sundaram, S.; Kim, H.J.; Bauer, R.; Thongkongkaew, T.; Heine, D.; Hertweck, C. On-Line Polyketide Cyclization into Diverse Medium-Sized Lactones by a Specialized Ketosynthase Domain. Angew. Chem. Int. Ed. 2018, 57, 11223–11227. [Google Scholar] [CrossRef]
- Hooshmand, S.E.; Heidari, B.; Sedghi, R.; Varma, R.S. Recent Advances in the Suzuki–Miyaura Cross-Coupling Reaction Using Efficient Catalysts in Eco-Friendly Media. Green Chem. 2019, 21, 381–405. [Google Scholar] [CrossRef]
- Miyaura, Norio. ; Suzuki, Akira. Palladium-Catalyzed Cross-Coupling Reactions of Organoboron Compounds. Chem. Rev. 1995, 95, 2457–2483. [Google Scholar] [CrossRef]
- Zeysing, B.; Gosch, C.; Terfort, A. Protecting Groups for Thiols Suitable for Suzuki Conditions. Org. Lett. 2000, 2, 1843–1845. [Google Scholar] [CrossRef]
- Liebeskind, L.S.; Srogl, J. Thiol Ester−Boronic Acid Coupling. A Mechanistically Unprecedented and General Ketone Synthesis. J. Am. Chem. Soc. 2000, 122, 11260–11261. [Google Scholar] [CrossRef]
- Cheng, H.-G.; Chen, H.; Liu, Y.; Zhou, Q. The Liebeskind–Srogl Cross-Coupling Reaction and Its Synthetic Applications. Asian J. Org. Chem. 2018, 7, 490–508. [Google Scholar] [CrossRef]
- Meng, S.; Tang, G.-L.; Pan, H.-X. Enzymatic Formation of Oxygen-Containing Heterocycles in Natural Product Biosynthesis. ChemBioChem 2018, 19, 2002–2022. [Google Scholar] [CrossRef]
- Hemmerling, F.; Hahn, F. Biosynthesis of Oxygen and Nitrogen-Containing Heterocycles in Polyketides. Beilstein J. Org. Chem. 2016, 12, 1512–1550. [Google Scholar] [CrossRef]
- Pöplau, P.; Frank, S.; Morinaka, B.I.; Piel, J. An Enzymatic Domain for the Formation of Cyclic Ethers in Complex Polyketides. Angew. Chem. Int. Ed. 2013, 52, 13215–13218. [Google Scholar] [CrossRef]
- Berkhan, G.; Hahn, F. A Dehydratase Domain in Ambruticin Biosynthesis Displays Additional Activity as a Pyran-Forming Cyclase. Angew. Chem. Int. Ed. 2014, 53, 14240–14244. [Google Scholar] [CrossRef]
- Hollmann, T.; Berkhan, G.; Wagner, L.; Sung, K.H.; Kolb, S.; Geise, H.; Hahn, F. Biocatalysts from Biosynthetic Pathways: Enabling Stereoselective, Enzymatic Cycloether Formation on a Gram Scale. ACS Catal. 2020, 10, 4973–4982. [Google Scholar] [CrossRef]
- Wagner, L.; Stang, J.; Derra, S.; Hollmann, T.; Hahn, F. Towards Understanding Oxygen Heterocycle-Forming Biocatalysts: A Selectivity Study of the Pyran Synthase PedPS7. Org. Biomol. Chem. 2022, 20, 9645–9649. [Google Scholar] [CrossRef]
- Sung, K.H.; Berkhan, G.; Hollmann, T.; Wagner, L.; Blankenfeldt, W.; Hahn, F. Insights into the Dual Activity of a Bifunctional Dehydratase-Cyclase Domain. Angew. Chem. Int. Ed. 2018, 57, 343–347. [Google Scholar] [CrossRef]
- Wagner, L.; Roß, T.; Hollmann, T.; Hahn, F. Cross-Linking of a Polyketide Synthase Domain Leads to a Recyclable Biocatalyst for Chiral Oxygen Heterocycle Synthesis. RSC Adv. 2021, 11, 20248–20251. [Google Scholar] [CrossRef]
- Wagner, D.T.; Zhang, Z.; Meoded, R.A.; Cepeda, A.J.; Piel, J.; Keatinge-Clay, A.T. Structural and Functional Studies of a Pyran Synthase Domain from a Trans-Acyltransferase Assembly Line. ACS Chem. Biol. 2018, 13, 975–983. [Google Scholar] [CrossRef]
- Ueoka, R.; Uria, A.R.; Reiter, S.; Mori, T.; Karbaum, P.; Peters, E.E.; Helfrich, E.J. N.; Morinaka, B.I.; Gugger, M.; Takeyama, H.; Matsunaga, S.; Piel, J. Metabolic and Evolutionary Origin of Actin-Binding Polyketides from Diverse Organisms. Nat. Chem. Biol. 2015, 11, 705–712. [Google Scholar] [CrossRef]
- Luhavaya, H.; Dias, M.V. B.; Williams, S.R.; Hong, H.; de Oliveira, L.G.; Leadlay, P.F. Enzymology of Pyran Ring A Formation in Salinomycin Biosynthesis. Angew. Chem. Int. Ed. 2015, 54, 13622–13625. [Google Scholar] [CrossRef]
- Woo, A.J.; Strohl, W.R.; Priestley, N.D. Nonactin Biosynthesis: The Product of NonS Catalyzes the Formation of the Furan Ring of Nonactic Acid. Antimicrob. Agents Chemother. 1999, 43, 1662–1668. [Google Scholar] [CrossRef]
- Miyaura, N.; Ishiyama, T.; Sasaki, H.; Ishikawa, M.; Sato, M.; Suzuki, A. Palladium-Catalyzed Inter- and Intramolecular Cross-Coupling Reactions of B-Alkyl-9-Borabicyclo [3.3.1]Nonane Derivatives with 1-Halo-1-Alkenes or Haloarenes. Syntheses of Functionalized Alkenes, Arenes, and Cycloalkenes via a Hydroboration-Coupling Sequence. J. Am. Chem. Soc. 1989, 111, 314–321. [Google Scholar] [CrossRef]
- Farina, V.; Krishnan, B. Large Rate Accelerations in the Stille Reaction with Tri-2-Furylphosphine and Triphenylarsine as Palladium Ligands: Mechanistic and Synthetic Implications. J. Am. Chem. Soc. 1991, 113, 9585–9595. [Google Scholar] [CrossRef]
- Chishiro, A.; Konishi, M.; Inaba, R.; Yumura, T.; Imoto, H.; Naka, K. Tertiary Arsine Ligands for the Stille Coupling Reaction. Dalton Trans. 2021, 51, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Carreira, E.M.; Du Bois, J. (+)-Zaragozic Acid C: Synthesis and Related Studies. J. Am. Chem. Soc. 1995, 117, 8106–8125. [Google Scholar] [CrossRef]
Figure 1.
(A) Structure of the 4’-phosphopantetheine prosthetic group of acyl carrier proteins and the partial structure that is mimicked by SNAC. (B) Susceptibilities and side reactions of acyl-SNACs. (C) IMOMA cyclases catalyze intramolecular oxa-Michael addition to oxygen heterocycles. The natural reaction of AmbDH3 is shown as an example. (D) Structure of the target compound required for our biosynthetic studies.
Figure 1.
(A) Structure of the 4’-phosphopantetheine prosthetic group of acyl carrier proteins and the partial structure that is mimicked by SNAC. (B) Susceptibilities and side reactions of acyl-SNACs. (C) IMOMA cyclases catalyze intramolecular oxa-Michael addition to oxygen heterocycles. The natural reaction of AmbDH3 is shown as an example. (D) Structure of the target compound required for our biosynthetic studies.
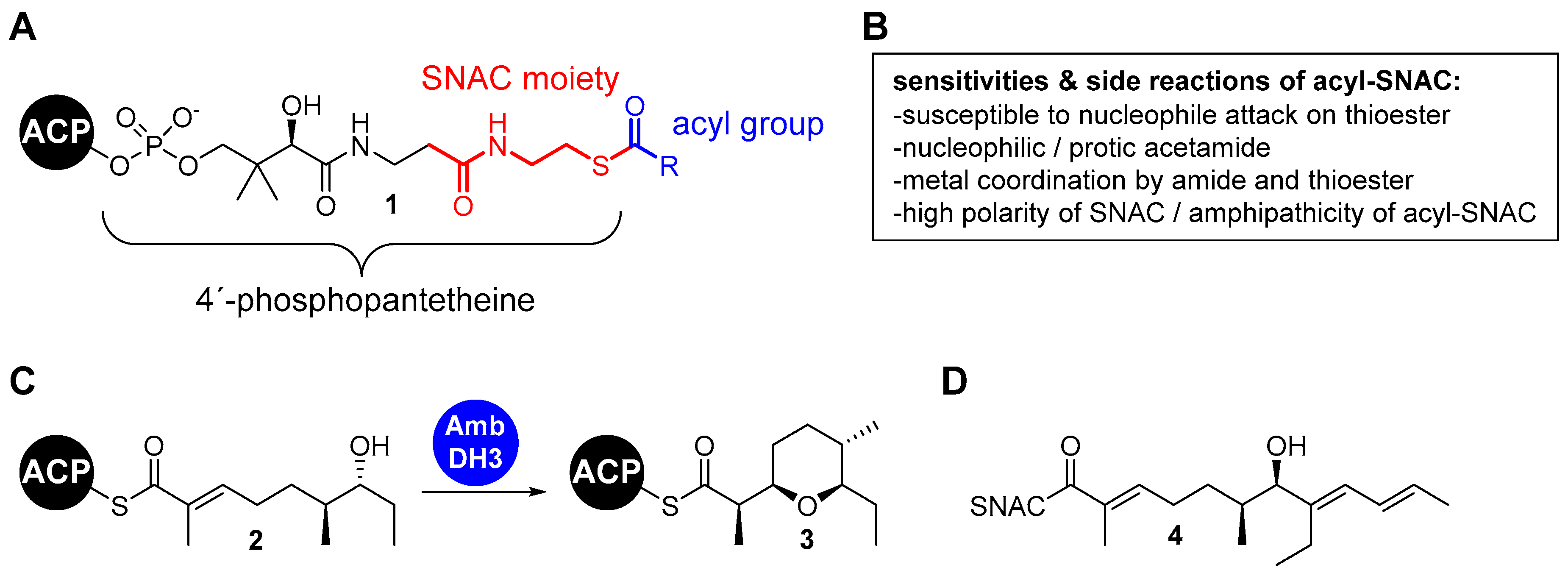
Figure 2.
Mechanism of the SMR.
Scheme 1.
Synthesis of protected hydroxytrienes 13a/13b from aldehyde 11.

Table 1.
Optimization of the conditions for the coupling of SNAC thioester halides 5a/5b and TBS-protected olefin 6.
Table 1.
Optimization of the conditions for the coupling of SNAC thioester halides 5a/5b and TBS-protected olefin 6.
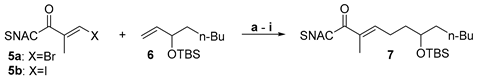 | |||||
|---|---|---|---|---|---|
| Entry | X | Base | Additive | Temperature [°C] | Isolated yield [%] |
| a | Br | K2CO3 | - | 50 | 54 |
| b | Br | K2CO3 | P(o-furyl)3 | 50 | 23 |
| c | Br | K2CO3 | AsPh3 | 50 | 55 |
| d | Br | Cs2CO3 | - | 50 | 55 |
| e | Br | K2CO3 | - | 20 | 13 |
| f | I | K2CO3 | - | 50 | 55 |
| g | I | Cs2CO3 | AsPh3 | 50 | 34 |
| h | I | Cs2CO3 | AsPh3 | 65 | - |
| i | I | Cs2CO3 | AsPh3 | 20 | 78 |
General reaction conditions: 1. 6 (1.5 eq., 1 m in THF), 9-BBN (1.5 eq., 0.5 m in THF), 0 °C to 20 °C, o.n.; 2. DMF (total 0.2 m), 5 (1.0 eq.), base (2.0 eq.), PdCl2dppf (5 mol%), additive (5 mol%), reaction control via TLC. Reaction scale: 90–100 µmol.
Table 2.
Optimization of the conditions for the coupling of 5 and protected trienes 8a/8b, varying protecting group, halogenide, base, additives and temperature.
Table 2.
Optimization of the conditions for the coupling of 5 and protected trienes 8a/8b, varying protecting group, halogenide, base, additives and temperature.
 | ||||||
|---|---|---|---|---|---|---|
| Entry | X | PG | Base | Additive | Temperature [° C] | Isolated yield [%] |
| a | Br | TBS | 2 eq. K2CO3 | - | 50 | 27 |
| b | Br | TBS | 3 eq. K3PO4 | - | 50 | 17 |
| c | Br | TES | 2 eq. K2CO3 | - | 50 | 25 |
| d | Br | TES | 3 eq. K3PO4 | - | 50 | 12 |
| e | Br | TES | 2 eq. K2CO3 | - | 20 | 15 |
| f | I | TES | 2 eq. K2CO3 | - | 50 | 49 |
| g | Br | TES | 2 eq. Cs2CO3 | - | 50 | 80 |
| h | Br | TES | 2 eq. K2CO3 | AsPh3 | 50 | 77 |
| i | I | TES | 2 eq. K2CO3 | - | 20 | 63 |
| j | I | TES | 2 eq. Cs2CO3 | AsPh3 | 20 | 87 |
Reaction conditions: 1. 8 (1.5 eq., 1 m in THF), 9-BBN (1.5 eq., 0.5 m in THF), 0 °C to 20 °C, o.n.; 2. DMF (total 0.2 m), 5 (1.0 eq.), Base (2.0 eq.), PdCl2dppf (5 mol%), additive (5 mol%), reaction control via TLC.
Table 3.
Testing conditions for silylether deprotection.
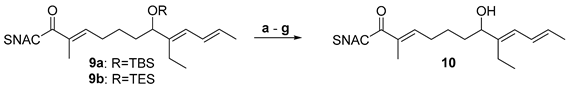 | ||||
|---|---|---|---|---|
| Entry | PG | Reagent | Conditions | Result |
| a | TBS | PPTS | DMSO, 50 °C, o.n. | Decomposition |
| b | TBS | TBAF | THF, 0 °C, 1 h | No reaction |
| c | TBS | TBAF | THF, 0 → 20 °C, o.n. | Decomposition |
| d | TES | TBAF | THF, 0 °C, 1 h | Decomposition |
| e | TBS | HF*pyridine | THF, 0 °C, 3 h | Decomposition |
| f | TBS | HF*pyridine, pyridine | THF, 0 → 20 °C, 3 h | 51% |
| g | TES | HF*pyridine, pyridine | THF, 0 → 20 °C, 3 h | 81% |
Table 4.
Two-pot-three-step reaction sequence to thioester 4.
 | ||||||
|---|---|---|---|---|---|---|
| Entry | X | PG | Conditions | Coupling yield [%] | Deprotection yield [%] | Overall yield [%] |
| a | Br | TBS | A | 30 | 53 | 16 |
| b | Br | TES | A | 51 | 86 | 44 |
| c | I | TES | B | 74 | 81 | 60 |
Reaction conditions: A. 13 (1.0 eq., 1 m in THF), 9-BBN (1.0 eq., 0.5 m in THF), 0 °C to 20 °C, o.n.; 2. DMF (total 0.2 m), 5 (1.5 eq.), K2CO3 (2.0 eq.), PdCl2dppf (5 mol%), 50 °C, reaction control via TLC; B. 13 (1.5 eq., 1 m in THF), 9-BBN (1.5 eq., 0.5 m in THF), 0 °C to 20 °C, o.n.; 2. DMF (total 0.2 m), 5 (1.0 eq.), Cs2CO3 (2.0 eq.), PdCl2dppf (5 mol%), AsPh3 (5 mol%), 20 °C, reaction control via TLC; C. 14 (10.0 mg, 1.0 eq.), 110 µL of HF-containing stock solution (1 part HF*pyridine, 2 parts pyridine, 8 parts THF).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



