
Submitted:
13 May 2023
Posted:
15 May 2023
You are already at the latest version
Abstract
A controllable synthesis of trisubstituted imidazoles and pyrroles has been developed through rhodium(II)-catalyzed regioselective annulation of N-sulfonyl-1,2,3-trizaoles with β-enaminones. The imidazole ring was formed through 1,1-insertion of N-H bond to α-imino rhodium carbene and subsequential intramolecular 1,4-conjugate addition when α-carbon atom of amino group bearing with methyl, whereas utilizing phenyl substituent constructed pyrrole ring via intramolecular nucleophilic addition. Features such as mild conditions, good functional groups tolerance, gram-scale synthesis, and valuable transformations of the products qualified this unique protocol as an efficient tool for the synthesis of N-heterocycles.
Keywords:
annulation
; β-enaminones
; imidazoles
; pyrroles
; N-sulfonyl trizaoles
1. Introduction
Nitrogen-containing heterocycles are privileged structural motifs in various of natural products and bioactive compounds.[1,2] Among them, imidazole and pyrrole framework are very common structural units widely distributed in natural products, pharmaceutics, agrochemicals, and other functional materials.[3,4] For this reason, the synthesis of such compounds continues to be a hot topic in modern synthetic chemistry.[5,6,7,8] Consequently, a large number of new reactions have been developed to construct structurally diverse imidazole and pyrrole derivatives, such as multicomponent reactions,[9,10] [3+2] cycloaddition,[11,12,13,14] as well as both metal-catalyzed inter-[15,16] and intramolecular[17,18] cyclization strategies. Despite all the achievements, the development of efficient methods for their synthesis, particularly regiocontrolled synthesis of those containing multiple substituents, from readily accessible compounds is of ever-increasing importance.
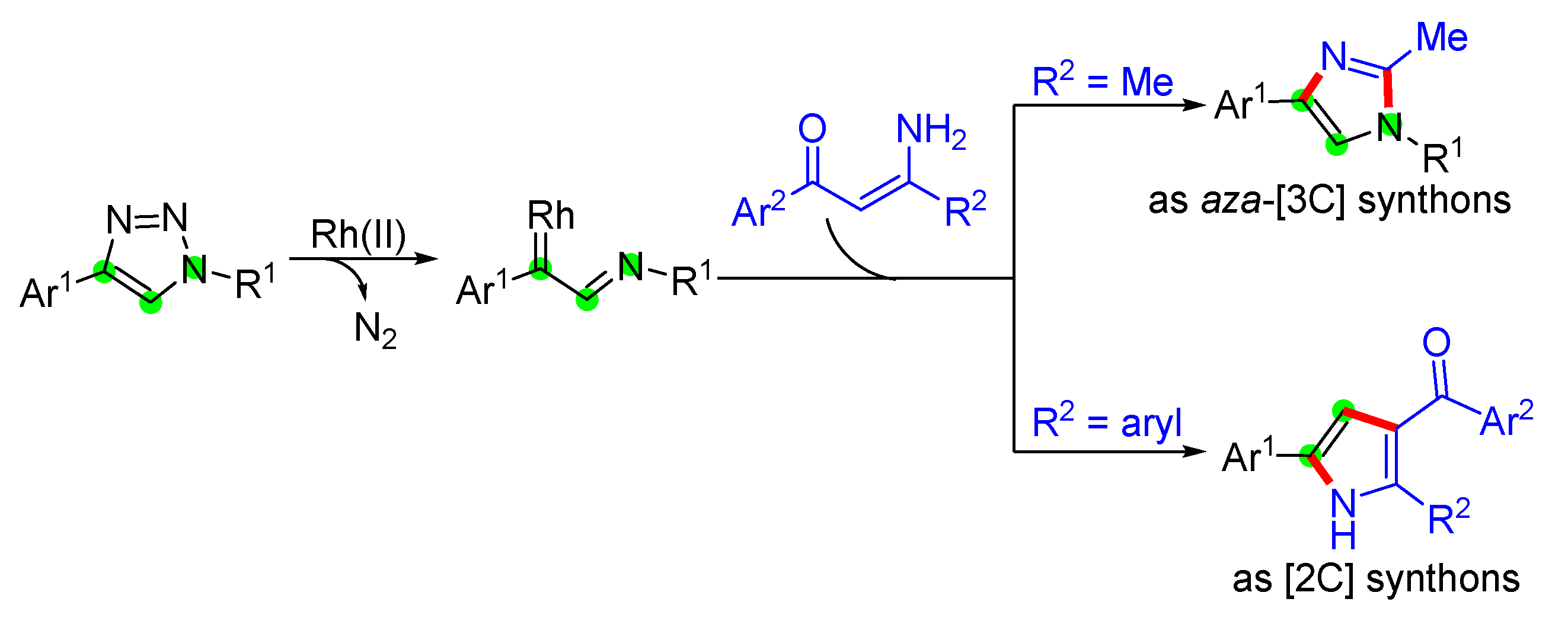
In the past decades, 1,2,3-triazoles have emerged as capable precursors for the synthesis of various nitrogen heterocycles.[19,20] Upon treatment with rhodium(II) catalysts, N-sulfonyl-1,2,3-triazoles readily undergo denitrogenative reaction to form α-imino rhodium carbenes, a versatile intermediate that could promote a wide range of transformations.[21,22] Besides the common reactivity, such as cyclopropanation,[23,24] X-H insertion,[25,26,27,28] and ylide formation,[29,30,31] α-imino rhodium carbenes can be employed as [1C]- or aza-[3C]-synthons to participate in stepwise cycloadditions, giving various N-heterocycles.[32,33,34,35] As the major part of our research efforts in developing new methodologies for the construction of heterocycles,[36,37,38,39] we herein describe an efficient strategy for regioselective synthesis of trisubstituted imidazoles and pyrroles involving the cascade N-H insertion to α-imino rhodium carbene followed by substituent-controllable intramolecular annulation (Scheme 1), in which the α-imino rhodium carbene acted as a [2C] and aza-[3C]-synthons, respectively.
2. Results and Discussion
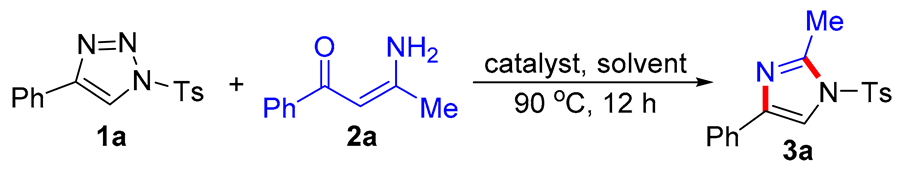
The optimization of a one-pot procedure for the formation of imidazole 3a from triazole 1a and β-enaminone 2a was undertaken (Table 1). Screening of various transition-metal catalysts revealed that dirhodium catalyst Rh2(OAc)4 and Rh2(oct)4 were demonstrated more efficient than other metal catalysts for this reaction (Table 1, entries 1-6). Further investigation showed that at a lower catalytic loading (2 mol%) had a positive effect on the reaction (Table 1, entries 7-10). Other solvents, including toluene and chlorobenzene, could better promote this transformation and then utilization chlorobenzene for further optimization (Table 1, entries 11-16). Further variation of reaction temperatures revealed that 80 °C was the optimal condition (Table 1, entries 17-19). Extension of reaction time did not find beneficial to the product yield (Table 1, entries 20-22). Thus, the optimal reaction conditions were found to be Rh2(oct)4 in chlorobenzene at 90 °C for 12 h (Table 1, entry 13).
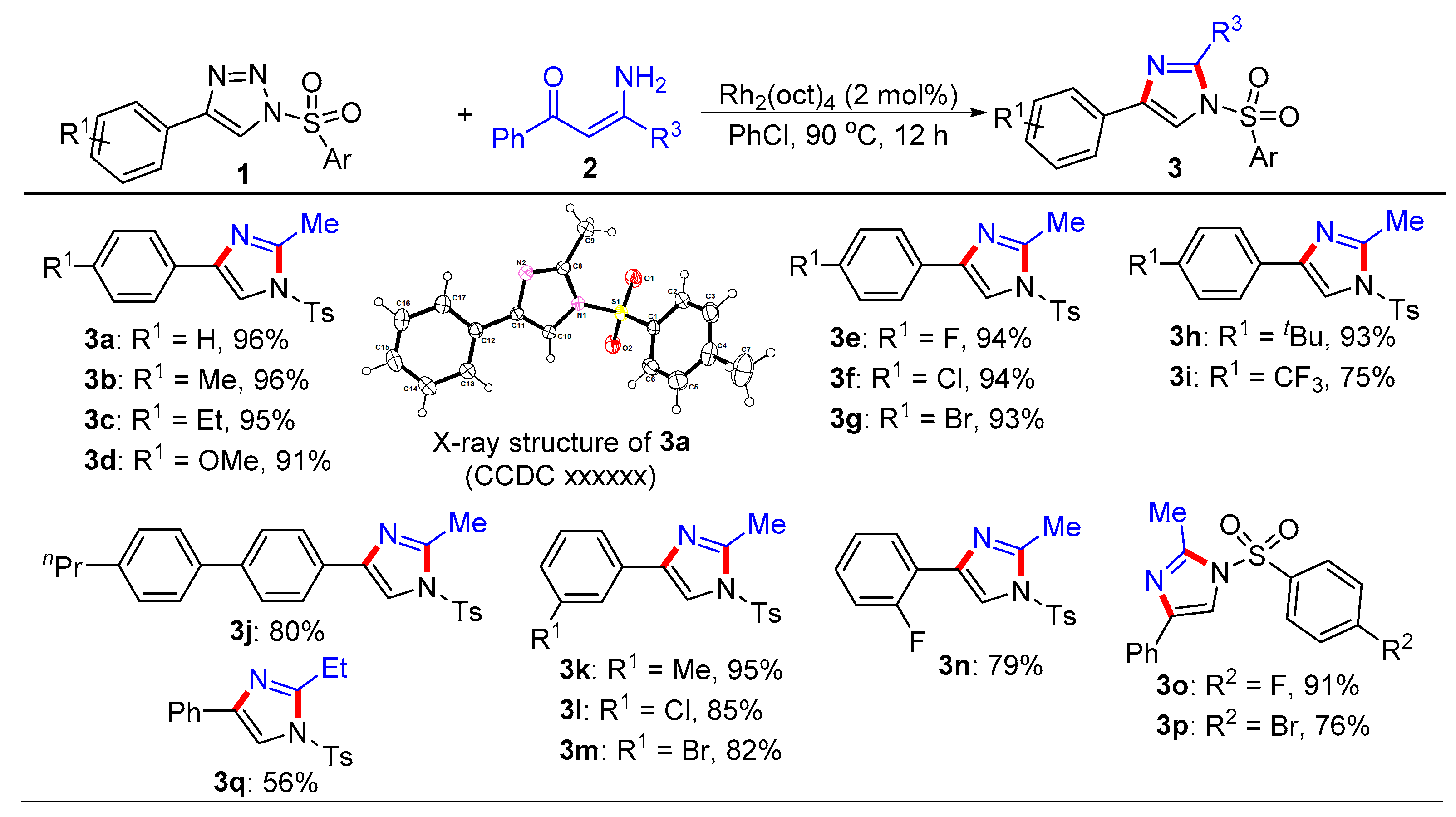
With the optimal conditions in hand, we set out to explore the scope and generality of this [3+2] annulation with a combination of various substituted N-sulfonyl-1,2,3-triazoles 1 and β-enamino ketones 2 (Scheme 2). We firstly evaluated the effect of substituents of R1 group on the phenyl of N-sulfonyl-1,2,3-triazoles, and the results indicated that the introduction of electron-neutral (-Me, -Et), electron-rich (-OMe), and electron-deficient (-F, -Cl, -Br) substituents at the para-positions were tolerated to this transformation and gave the desired imidazoles (products 3b-3g) in 91-96% yields. Notably, the triazoles 1 bearing a bulky tert-butyl or a strong electron-withdrawing trifluoromethyl at the para-position of benzene ring led to a smooth reaction process, generating the corresponding products 3h and 3i in 93% and 75% yields, respectively. Moreover, the extended π structure did not show an influence, and the desired product 3j was successfully obtained in 80% yield. Additionally, substituents variations on the meta- and ortho-position could work well to produce the corresponding products 3k-3n in 79-95% yields. Furthermore, the N-arylsulfonyl groups of the triazole substrates were also examined. The reactions of fluoro- and bromo-substituted phenylsulfonyl triazoles proceeded well, giving the desired products 3o and 3p in 91% and 76% yields, respectively. In addition, (Z)-3-amino-1-phenylpent-2-en-1-one was also viable substrate for the transformation, generating the product 3q in 56% yield.
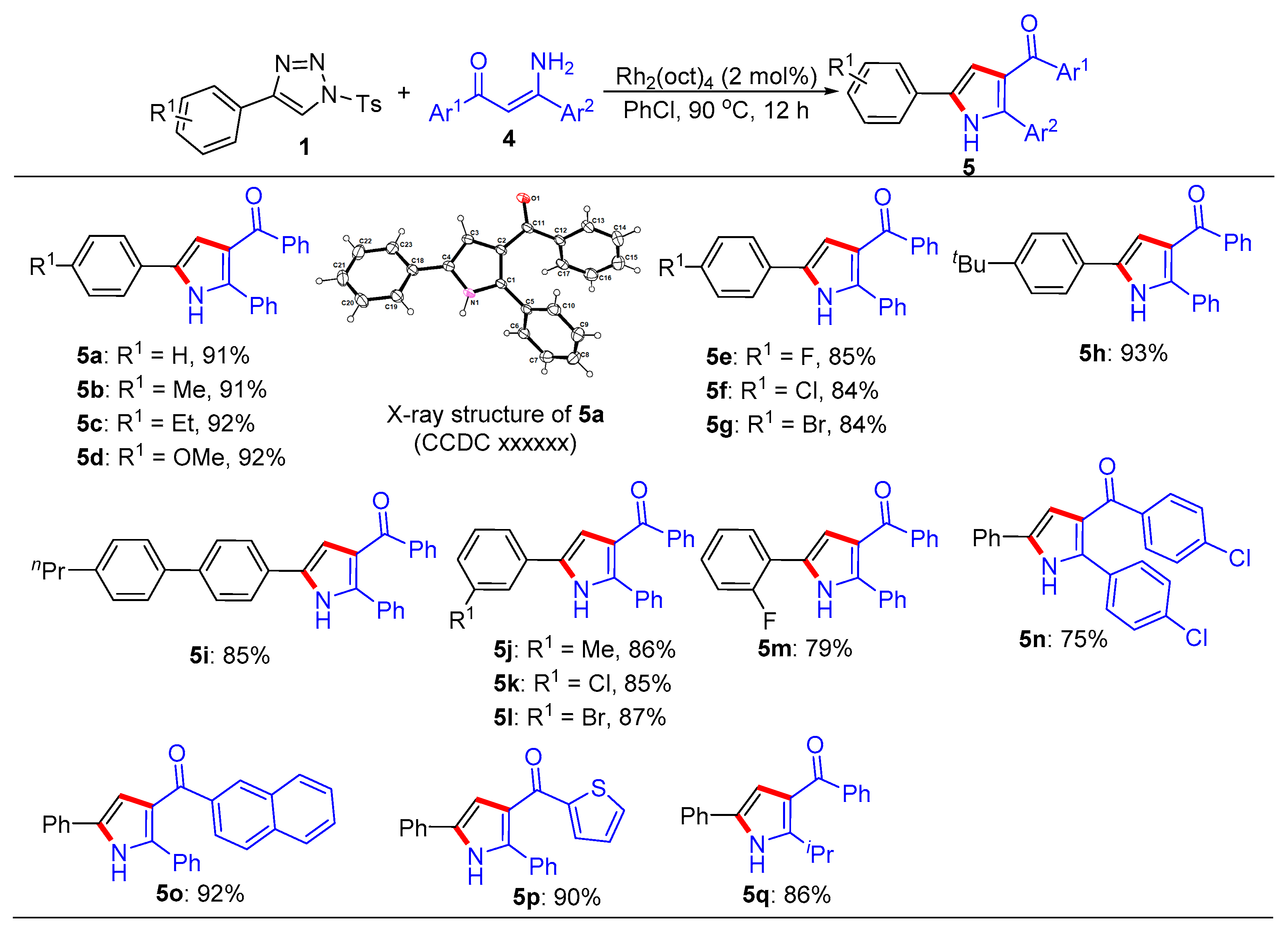
Subsequently, an unexpected pyrrole product 5a was obtained in 91% yield under the standard conditions when the phenyl group (4a) replace of methyl group of β-enaminones. We further evaluated the feasibility by using the 2,5-diaryl β-enaminones as starting materials (Scheme 3). As expected, a wide range of electronically different substituents, including alkyl, methoxy, halogen, and bulky tert-butyl groups, were successfully installed into the products 5a-5h. Also, an extended π-system was implemented on the pyrrole structure (product 5i). Particularly noteworthy is that the halogen groups (e.g. -F, -Cl, -Br) remained intact during the course of the reaction, which makes this transformation particularly attractive in terms of increasing the molecular complexity via transition metal-catalyzed coupling reactions (5e-5g and 5k-5m). Additionally, we turned our attention to investigating the suitability of the substrate 1,3-diaryl β-enaminones 4, and the desired products 5n–5q were successfully obtained in 76-92% yields. It was gratifying that the introduction of a naphthyl and thienyl group also proceeded smoothly to produce the desired products 5o and 5p in a yield of 92% and 90%, respectively. Likewise, changing the phenyl group to a bulky isopropyl was also tolerated in the reaction to give the desired product 5r in 86% yield.
Based on the above results, we hypothesis that the reaction of N-sulfonyl-1,2,3-triazoles and β-enaminones might be controlled by the steric hinder of the substituent on the α-position of amino. When a moderate steric group, such as n-propyl and n-butyl, was subjected to the amino α-position of β-enaminones under the standard conditions, the corresponding products imidazoles (3r and 3s) and pyrroles (5s and 5t) were obtained (Scheme 4a). In the synthesis of pyrroles, the presence of a methyl p-tolyl on the amino group resulted in a satisfactory yield of compounds 7a and 7b (72% and 85%), respectively (Scheme 4b).
Notably, the reaction could be easily scaled up. As shown in Scheme 3, imidazole 3a could be obtained in a satisfactory yield of 77% (1.44 g) when the scale of the reaction was increased to 5 mmol, and the 3,5-disubstituted pyrrole 5a was obtained in an 75% yield (1.21 g) on the same scale. Subsequently, several transformations were performed to demonstrate the utility of the target products. The desulfonation of imidazole 3a afforded the unprotected imidazole 8 in 96% yield (Scheme 5a). In addition, treating pyrrole 5a with hydroxylamine hydrochloride and iodomethane successfully realized the formation of the pyrrolyl oxime 9 in 94% yield (Scheme 5b). In the presence of sodium hydride, N-methylation between compound 5a and iodomethane easily generated N-methylpyrrole derivative 10 in 95% yield (Scheme 5c), highlighting the synthetic utility of the current protocol.
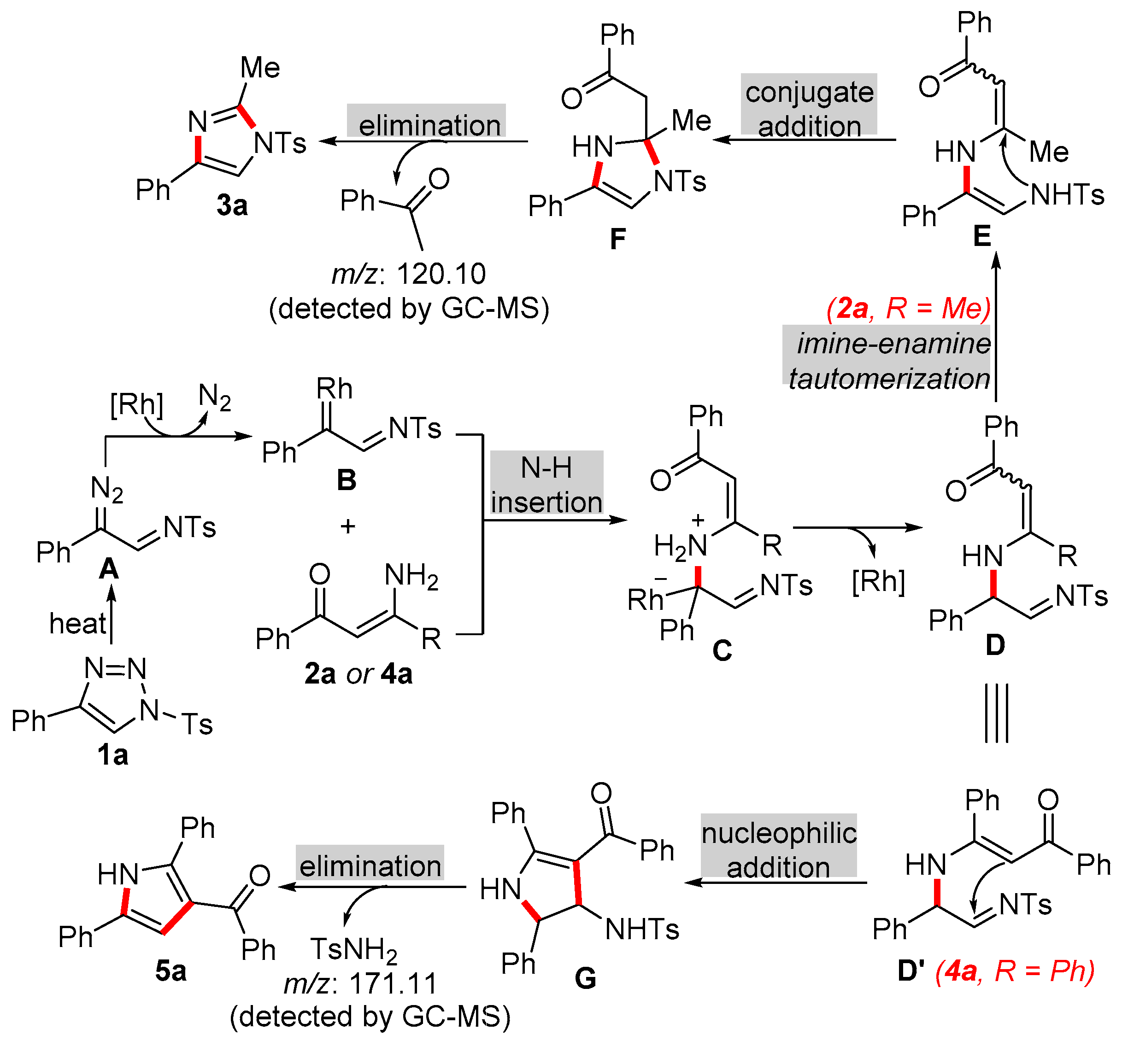
The mechanism of this reaction was proposed as shown in Scheme 4 on the basis of the above experimental results and previous reports.[40,41,42] α-Diazo imino intermediate A, which was generated from the ring-chain tautomerization of triazole 1a, could be decomposed by the rhodium(II) catalyst efficiently to form α-imino rhodium carbene intermediate B along with the release of nitrogen gas. β-Enaminones (2a or 4a) attacked the electrophilic carbene center of intermediate B, and 1,1-insertion occurred to convert intermediate D with the regeneration of the rhodium(II) catalyst. When R was methyl group, an imino-enamine taumerization could be induced to give more stable intermediate E, which could undergo an intramolecular 1,4-conjugate addition affording the intermediate F. The elimination of the intermediate F results the desired product 3a. In the case of 4a, the phenyl group was bulky to form the intermediate D'. Therefore, after the subsequential intramolecular nucleophilic addition and elimination process, the corresponding product 5a was obtained.
Scheme 4.
Proposed mechanism for the formation of NH-free isoquinolones.
3. Materials and Methods
The detailed procedures for the synthesis and characterization of the products are given in Appendix A section.
4. Conclusions
In conclusion, we have demonstrated that the Rh(II)-catalyzed substituent-controllable regioselective annulations provide a new synthetic strategy to trisubstituted imidazoles and pyrroles. The highlight of the current reaction is the substituent-dependent product selectivity. The imidazole skeleton was formed via N-H insertion to α-imino rhodium carbene, followed by intramolecular 1,4-conjugate addition when α-carbon atom of amino group bearing with methyl. Switching methyl to phenyl group, the pyrrole framework was generated through N-H insertion and intramolecular nucleophilic addition process. The large-scale reactions and transformations of the products further demonstrated the potential synthetic value of this strategy.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Characterization data for product 3 and 4, include 1H- and 13C-NMR spectroscopies are available online. CCDC 2260879 and 2260880 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif, or by emailing data_request@ccdc.cam.ac.uk, or by contacting The Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; Fax: +44 1223 336033.
Author Contributions
Conceptualization, X.H., Q.Y.; methodology, H.W. and T.Z.; formal analysis, T.Z.; data curation, H.W.; writing—original draft preparation, H.W. and Y.Q.; writing—review and editing, X.H. and Q.Y.; supervision, Q.Y. and X.H.; project administration, X.H. All authors have read and agreed to the published version of the manuscript.
Funding
We thank the Natural Scientific Research Foundations of Anhui Provincial Universities (2022AH050210), the Excellent Scientific Research and Innovation Team of Anhui Provincial Universities (No. 2022AH010011), the University Synergy Innovation Program of Anhui Province (No. GXXT-2020-074), and the Excellent Young Talents Support Program of Education Administration of Anhui Province (No. gxyq2022233) for financial support.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available in this article.
Conflicts of Interest
The authors declare no conflict of interest.
Sample Availability
Samples of the compounds are available from the authors.
Appendix A. Experimental Section
Unless otherwise specified, all reagents and starting materials were purchased from commercial sources and used as received, and the solvents were purified and dried using standard procedures. The chromatography solvents were technical grade and distilled prior to use. The NMR spectra were recorded with a Bruker Avance 500 spectrometer (500 MHz for 1H and 125 MHz for 13C) with CDCl3 as solvent with tetramethylsilane (TMS) as the internal standard at room temperature. Chemical shifts are given in δ relative to TMS, the coupling constants J are given in Hz. HRMS spectra were obtained with an Agilent 6200 using a quadrupole time-of-flight mass spectrometer equipped with an ESI source. The melting points were measured using SGWX-4 melting point apparatus and not corrected. The X-ray source used for the single crystal X-ray diffraction analysis of compound 3a and 5a was Mo Kα (λ = 0.71073 Å), and the thermal ellipsoid was drawn at the 30% probability level.
General procedure for the synthesis of trisubstituted imidazoles 3 and pyrroles 5. N-Sulfonyl-1H-1,2,3-triazoles 1 (0.2 mmol), β-enaminones 2 (0.2 mmol), and Rh2(oct)4 (2 mol%) were successively added to a Schlenk reaction tube. The reaction set was evacuated and backfilled with argon atmosphere for three times. Then chlorobenzene (2.0 mL) was added into the reaction tube through a syringe. The reaction mixture was stirred vigorously in an oil bath preheated to 90 °C for 12 hours. After the reaction was complete, the reaction mixture was cooled to room temperature, extracted with CH2Cl2 (3 × 10 mL), and washed with brine. The organic layers were combined, dried over Na2SO4 and then evaporated under vacuum. The residue was purified by flash column chromatography on silica gel (200-300 mesh) using ethyl acetate and petroleum ether (1:8, v/v) as the elution solvent to give desired products 3 or 5.
General procedure for the synthesis of compound 8. 2-Methyl-4-phenyl-1-tosyl-1H-imidazole 3a (0.15 mmol) and NaOH (2.25 mmol) were successively added to a Schlenk reaction tube. The reaction set was evacuated and backfilled with argon atmosphere for three times. Then methanol (2.0 mL) was added into the reaction tube through a syringe. The reaction mixture was stirred vigorously in an oil bath preheated to 70 °C for 30 minutes. After the reaction was complete, the reaction mixture was cooled to room temperature, extracted with CH2Cl2 (3 × 10 mL), and washed with brine. The organic layers were combined, dried over Na2SO4 and then evaporated under vacuum. The residue was purified by flash column chromatography on silica gel (200-300 mesh) using ethyl acetate and petroleum ether (1:3, v/v) as the elution solvent to give desired product 8 in 96% yield.
General procedure for the synthesis of compound 9. A mixture of (2,5-diphenyl-1H-pyrrol-3-yl)(phenyl)methanone 5a (0.2 mmol), hydroxylamine hydrochloride (0.4 mmol), and sodium acetate (0.5 mmol) were added to a round-bottomed flask with reflux condenser. Ethanol (4 mL) was then added and the reaction mixture was stirred vigorously at reflux in oil bath with stirring 12 hours. After quenching with water, the residue was extracted with ethyl acetate twice. The combined layer was washed with brine, dried over Na2SO4 and then evaporated under vacuum. The residue was purified by flash column chromatography on silica gel (200-300 mesh) using ethyl acetate and petroleum ether (1:8, v/v) as the elution solvent to give desired product 9 in 94% yield.
General procedure for the synthesis of compound 10. NaH (60% in mineral oil, 0.5 mmol, 1.7 equiv.) was added to a solution of the 5a (0.25 mmol) in DCM (4 mL) at 0 °C in portions. After stirring for 5 min at 0 °C, MeI (0.22 mmol, 1.1 equiv.) was added drop-wise and the reaction mixture was allowed to warm to room temperature and stirred for another 19 h. After quenching with water, the residue was extracted with ethyl acetate twice. The combined organic layer was washed with brine, dried over Na2SO4, filtrated and concentrated, and purified by column chromatography to afford 10 in 95% yield.
2-Methyl-4-phenyl-1-tosyl-1H-imidazole (3a). This compound was purified by column chromatography (ethyl acetate/petroleum ether = 1: 8) to afford a white solid in 96% yield (60 mg); mp 122-124 °C; 1H NMR (500 MHz, CDCl3) δ 7.81 (d, J = 8.5 Hz, 2H), 7.73 (d, J = 7.0 Hz, 2H), 7.67 (s, 1H), 7.39-7.35 (m, 4H), 7.25 (d, J = 7.5 Hz, 1H), 2.57 (s, 3H), 2.44 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 146.5, 146.4, 141.0, 135.5, 132.7, 130.8, 129.1, 128.2, 127.8, 125.6, 114.4, 22.1, 15.6; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C17H17N2O2S 313.1005; Found 313.1006.
2-Methyl-4-(p-tolyl)-1-tosyl-1H-imidazole (3b). This compound was purified by column chromatography (ethyl acetate/petroleum ether = 1: 8) to afford a white solid in 96% yield (62 mg); mp 60-62 °C; 1H NMR (500 MHz, CDCl3) δ 7.80 (d, J = 8.5 Hz, 2H), 7.62 (d, J = 8.0 Hz, 3H), 7.35 (d, J = 8.0 Hz, 2H), 7.18 (d, J = 8.0 Hz, 2H), 2.57 (s, 3H), 2.44 (s, 3H), 2.35 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 146.4, 146.3, 141.0, 138.0, 135.4, 130.7, 129.9, 129.8, 127.7, 125.5, 113.9, 22.1, 21.7, 15.6; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C18H19N2O2S 327.1162; Found 327.1170.
4-(4-Ethylphenyl)-2-methyl-1-tosyl-1H-imidazole (3c). This compound was purified by column chromatography (ethyl acetate/petroleum ether = 1: 8) to afford a white solid in 95% yield (64 mg); mp 77-79 °C; 1H NMR (500 MHz, CDCl3) δ 7.80 (d, J = 8.5 Hz, 2H), 7.65 (d, J = 8.0 Hz, 3H), 7.34 (d, J = 8.0 Hz, 2H), 7.21 (d, J = 8.0 Hz, 2H), 2.64 (q, J = 7.5 Hz, 2H), 2.57 (s, 3H), 2.42 (s, 3H), 1.23 (t, J = 7.5 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 146.4, 146.3, 144.4, 141.0, 135.4, 130.8, 130.1, 128.6, 127.7, 125.5, 113.9, 29.1, 22.1, 15.9, 15.6; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C19H21N2O2S 341.1318; Found 341.1319.
4-(4-Methoxyphenyl)-2-methyl-1-tosyl-1H-imidazole (3d). This compound was purified by column chromatography (ethyl acetate/petroleum ether = 1: 8) to afford a white solid in 91% yield (62 mg); mp 66-68 °C; 1H NMR (500 MHz, CDCl3) δ 7.80 (d, J = 8.5 Hz, 2H), 7.65 (d, J = 9.0 Hz, 2H), 7.57 (s, 1H), 7.34 (d, J = 8.0 Hz, 2H), 6.90 (d, J = 9.0 Hz, 2H), 3.81 (s, 3H), 2.56 (s, 3H), 2.42 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 159.8, 146.4, 146.3, 140.8, 135.4, 130.7, 127.7, 126.9, 125.5, 114.5, 113.2, 55.7, 22.1, 15.6; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C18H19N2O3S 343.1111; Found 343.1112.
4-(4-Fluorophenyl)-2-methyl-1-tosyl-1H-imidazole (3e). This compound was purified by column chromatography (ethyl acetate/petroleum ether = 1: 8) to afford a white solid in 94% yield (62 mg); mp 107-109 °C; 1H NMR (500 MHz, CDCl3) δ 7.81 (d, J = 8.5 Hz, 2H), 7.69 (dd, J = 8.5, 5.0 Hz, 2H), 7.61 (s, 1H), 7.36 (d, J = 8.0 Hz, 2H), 7.06 (t, J = 8.5 Hz, 2H), 2.56 (s, 3H), 2.44 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 162.9 (d, JC-F = 246.3 Hz), 146.6, 146.5, 140.1, 135.3, 130.8, 129.0, 127.8, 127.3 (d, JC-F = 8.0 Hz), 116.0 (d, JC-F = 21.6 Hz), 114.0, 22.1, 15.6; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C17H16FN2O2S 331.0911; Found 331.0909.
4-(4-Chlorophenyl)-2-methyl-1-tosyl-1H-imidazole (3f). This compound was purified by column chromatography (ethyl acetate/petroleum ether = 1: 8) to afford a white solid in 94% yield (65 mg); mp 107-109 °C; 1H NMR (500 MHz, CDCl3) δ 7.80 (d, J = 7.5 Hz, 2H), 7.66-7.65 (m, 3H), 7.36-7.32 (m, 4H), 2.56 (s, 3H), 2.43 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 146.6, 146.6, 139.9, 135.2, 133.8, 131.3, 130.8, 129.2, 127.8, 126.8, 114.5, 22.1, 15.5; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C17H16ClN2O2S 347.0616; Found 347.0612.
4-(4-Bromophenyl)-2-methyl-1-tosyl-1H-imidazole (3g). This compound was purified by column chromatography (ethyl acetate/petroleum ether = 1: 8) to afford a white solid in 93% yield (72 mg); mp 104-106 °C; 1H NMR (500 MHz, CDCl3) δ 7.80 (d, J = 8.5 Hz, 2H), 7.67 (s, 1H), 7.60 (d, J = 8.5 Hz, 2H), 7.48 (d, J = 8.5 Hz, 2H), 7.35 (d, J = 8.5 Hz, 2H), 2.56 (s, 3H), 2.42 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 146.7, 146.6, 139.9, 135.2, 132.2, 131.7, 130.8, 127.8, 127.1, 122.0, 114.6, 22.1, 15.6; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C17H16BrN2O2S 391.0110; Found 391.0109.
4-(4-(tert-Butyl)phenyl)-2-methyl-1-tosyl-1H-imidazole (3h). This compound was purified by column chromatography (ethyl acetate/petroleum ether = 1: 8) to afford a white solid in 93% yield (68 mg); mp 69-71 °C; 1H NMR (500 MHz, CDCl3) δ 7.79 (d, J = 7.0 Hz, 2H), 7.69-7.64 (m, 3H), 7.39 (d, J = 7.0 Hz, 2H), 7.34 (d, J = 8.0 Hz, 2H), 2.57 (s, 3H), 2.42 (s, 3H), 1.32 (s, 9H); 13C NMR (125 MHz, CDCl3) δ 151.3, 146.4, 146.4, 141.0, 135.5, 130.7, 129.9, 127.7, 126.0, 125.3, 114.0, 35.0, 31.7, 22.1, 15.6; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C21H25N2O2S 369.1631; Found 369.1634.
2-Methyl-1-tosyl-4-(4-(trifluoromethyl)phenyl)-1H-imidazole (3i). This compound was purified by column chromatography (ethyl acetate/petroleum ether = 1: 8) to afford a white solid in 75% yield (57 mg); mp 76-78 °C; 1H NMR (500 MHz, CDCl3) δ 7.84-7.82 (m, 4H), 7.76 (s, 1H), 7.62 (d, J = 8.0 Hz, 2H), 7.38 (d, J = 8.0 Hz, 2H), 2.58 (s, 3H), 2.45 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 146.8, 139.5, 136.2, 135.1, 130.9, 129.9 (q, JC-F = 32.5 Hz), 128.5, 127.9, 126.6, 126.1(q, JC-F = 3.8 Hz), 124.6(q, JC-F = 270.0 Hz), 115.6, 22.1, 15.5.; HRMS (ESI-TOF) m/z: [M + H] + Calcd for C18H16F3N2O2S 381.0879; Found 381.0878.
2-Methyl-4-(4'-propyl-[1,1'-biphenyl]-4-yl)-1-tosyl-1H-imidazole (3j). This compound was purified by column chromatography (ethyl acetate/petroleum ether = 1: 8) to afford a white solid in 80% yield (69 mg); mp 94-96 °C; 1H NMR (500 MHz, CDCl3) δ 7.83-7.79 (m, 4H), 7.71(s, 1H), 7.61 (d, J = 8.5 Hz, 2H), 7.54 (d, J = 8.0 Hz, 2H), 7.36 (d, J = 8.0 Hz, 2H), 7.26 (d, J = 8.5 Hz, 2H), 2.63 (t, J = 7.5 Hz, 2H), 2.59 (s, 3H), 1.72-1.64 (m, 2H), 0.98 (t, J = 7.5 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 146.5, 146.5, 142.4, 140.9, 140.7, 138.4, 135.4, 131.4, 130.8, 129.3, 127.8, 127.6, 127.1, 125.9, 114.3, 38.1, 25.0, 22.1, 15.6, 14.3; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C26H27N2O2S 431.1788; Found 431.1781.
2-Methyl-4-(m-tolyl)-1-tosyl-1H-imidazole (3k). This compound was purified by column chromatography (ethyl acetate/petroleum ether = 1: 8) to afford a white solid in 95% yield (62 mg); mp 107-109 °C; 1H NMR (500 MHz, CDCl3) δ 7.80 (d, J = 8.5 Hz, 2H), 7.67 (s, 1H), 7.58 (s, 1H), 7.51 (d, J = 7.5 Hz, 1H), 7.35 (d, J = 8.0 Hz, 2H), 7.26 (t, J = 7.5 Hz, 1H), 7.09 (d, J = 7.5 Hz, 1H), 2.57 (s, 3H), 2.43 (s, 3H), 2.37 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 146.5, 146.4, 141.0, 138.8, 135.4, 132.5, 130.8, 129.0, 127.8, 126.2, 122.6, 114.3, 22.1, 21.8, 15.6; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C18H19N2O2S 327.1162; Found 327.1163.
4-(3-Chlorophenyl)-2-methyl-1-tosyl-1H-imidazole (3l). This compound was purified by column chromatography (ethyl acetate/petroleum ether = 1: 8) to afford a white solid in 85% yield (59 mg); mp 83-85 °C; 1H NMR (500 MHz, CDCl3) δ 7.81 (d, J = 8.0 Hz, 2H), 7.73 (s, 1H), 7.68 (s, 1H), 7.59 (d, J = 7.5 Hz, 1H), 7.37 (d, J = 8.5 Hz, 2H), 7.29 (t, J = 8.0 Hz, 1H), 7.24 (d, J = 8.0 Hz, 1H), 2.56 (s, 3H), 2.44 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 146.7, 139.7, 135.1, 134.6, 130.8, 130.3, 128.1, 127.8, 125.7, 123.6, 115.0, 22.1, 15.5; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C17H16ClN2O2S 347.0616; Found 347.0625.
4-(3-Bromophenyl)-2-methyl-1-tosyl-1H-imidazole (3m). This compound was purified by column chromatography (ethyl acetate/petroleum ether = 1: 8) to afford a white solid in 82% yield (64 mg); mp 79-81 °C; 1H NMR (500 MHz, CDCl3) δ 7.89 (s, 1H), 7.80 (d, J = 8.5 Hz, 2H), 7.67 (s, 1H), 7.64 (d, J = 7.5 Hz, 1H), 7.39-7.35 (m, 3H), 7.22 (t, J = 8.0 Hz, 1H), 2.56 (s, 3H), 2.43 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 146.7, 146.6, 139.5, 135.2, 134.8, 131.0, 130.8, 130.6, 128.6, 127.8, 124.1, 123.3, 115.0, 22.1, 15.5; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C17H16BrN2O2S 391.0110; Found 391.0119.
4-(2-Fluorophenyl)-2-methyl-1-tosyl-1H-imidazole (3n). This compound was purified by column chromatography (ethyl acetate/petroleum ether = 1: 8) to afford a white solid in 79% yield (52 mg); mp 57-59 °C; 1H NMR (500 MHz, CDCl3) δ 8.03 (t, J = 7.5 Hz, 1H), 7.85 (d, J = 4.0 Hz, 1H), 7.82 (d, J = 8.0 Hz, 2H), 7.35 (d, J = 8.0 Hz, 2H), 7.23 (t, J = 7.0 Hz, 1H), 7.17 (t, J = 7.5 Hz, 1H), 7.10 (t, J = 10 Hz, 1H), 2.58 (s, 3H), 2.43 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 160.2 (d, JC-F = 247.9 Hz), 146.5, 145.9, 135.4, 134.5, 130.8, 129.1 (d, JC-F = 8.5 Hz), 128.2 (d, JC-F = 3.6 Hz), 127.8, 124.7 (d, JC-F = 3.6 Hz), 120.6 (d, JC-F = 12.5 Hz), 118.4 (d, JC-F = 15.4 Hz), 116.0 (d, JC-F = 21.5 Hz), 22.1, 15.5; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C17H16FN2O2S 331.0911; Found 331.0914.
1-((4-Fluorophenyl)sulfonyl)-2-methyl-4-phenyl-1H-imidazole (3o). This compound was purified by column chromatography (ethyl acetate/petroleum ether = 1: 8) to afford a white solid in 91% yield (57 mg); mp 107-109 °C; 1H NMR (500 MHz, CDCl3) δ 7.97-7.94 (m, 2H), 7.73 (d, J = 7.5 Hz, 2H), 7.67 (s, 1H), 7.38 (t, J = 8.0 Hz, 2H), 7.29 (t, J = 7.5 Hz, 1H), 7.24 (t, J = 8.0 Hz, 2H), 2.58 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 166.6 (d, JC-F = 257.5 Hz), 146.4, 141.3, 134.4 (d, JC-F = 2.8 Hz), 132.5, 130.7 (d, JC-F = 9.8 Hz), 129.1, 128.4, 125.6, 117.7 (d, JC-F = 22.9 Hz), 114.2, 15.7; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C16H14FN2O2S 317.0755; Found 317.0760.
1-((4-Bromophenyl)sulfonyl)-2-methyl-4-phenyl-1H-imidazole (3p). This compound was purified by column chromatography (ethyl acetate/petroleum ether = 1: 8) to afford a white solid in 76% yield (57 mg); mp 97-99 ℃; 1H NMR (500 MHz, CDCl3) δ 7.78 (d, J = 9.0 Hz, 2H), 7.72 (t, J = 9.0 Hz, 4H), 7.65 (s, 1H), 7.38 (d, J = 7.5 Hz, 2H), 7.29 (d, J = 7.5 Hz, 1H), 2.58 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 146.5, 141.4, 137.3, 133.6, 132.4, 130.6, 129.1, 129.1, 128.4, 125.6, 114.2, 15.7; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C16H14BrN2O2S 376.9954; Found 376.9952.
2-Ethyl-4-phenyl-1-tosyl-1H-imidazole (3q). This compound was purified by column chromatography (ethyl acetate/petroleum ether = 1: 8) to afford a white solid in 56% yield (37 mg); mp 49-51 °C; 1H NMR (500 MHz, CDCl3) δ 7.79 (d, J = 8.5 Hz, 2H), 7.75 (d, J = 7.0 Hz, 2H), 7.67 (s, 1H), 7.39-7.34 (m, 4H), 7.28 (d, J = 7.5 Hz, 1H), 2.90 (q, J = 7.5 Hz, 2H), 2.43 (s, 3H), 1.32 (t, J = 7.5 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 151.4, 146.4, 140.9, 135.7, 132.9, 130.7, 129.0, 128.1, 127.7, 125.6, 114.3, 22.4, 22.1, 12.5; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C18H19N2O2S 327.1162; Found 327.1163.
4-Phenyl-2-propyl-1-tosyl-1H-imidazole (3r). This compound was purified by column chromatography (ethyl acetate/petroleum ether = 1: 8) to afford a colorless oil in 45% yield (31 mg); 1H NMR (500 MHz, CDCl3) δ 7.79 (d, J = 8.5 Hz, 2H), 7.75 (d, J = 7.0 Hz, 2H), 7.66 (s, 1H), 7.38-7.33 (m, 4H), 7.27 (t, J = 7.5 Hz, 1H), 2.85 (t, J = 7.5 Hz, 2H), 2.43 (s, 3H), 1.81-1.74 (m, 2H), 0.98 (t, J = 7.5 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 150.4, 146.4, 141.0, 135.8, 132.9, 130.7, 129.0, 128.1, 127.6, 125.6, 114.3, 30.8, 22.1, 21.9, 14.3; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C19H21N2O2S 341.1318; Found 341.1312.
2-Butyl-4-phenyl-1-tosyl-1H-imidazole (3s). This compound was purified by column chromatography (ethyl acetate/petroleum ether = 1: 8) to afford a colorless oil in 44% yield (31 mg); 1H NMR (500 MHz, CDCl3) δ 7.79 (d, J = 8.5 Hz, 2H), 7.75 (d, J = 7.5 Hz, 2H), 7.67 (s, 1H), 7.38-7.33 (m, 4H), 7.28 (d, J = 7.5 Hz, 1H), 2.87 (t, J = 8 Hz, 2H), 2.43 (s, 3H), 1.72-1.69 (m, 2H), 1.42-1.37 (m, 2H), 0.91 (t, J = 7.5 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 150.5, 146.4, 140.9, 135.7, 132.9, 130.7, 129.1, 128.2, 127.7, 125.6, 114.3, 30.5, 28.6, 22.9, 22.1, 14.2; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C20H23N2O2S 355.1475; Found 355.1482.
(2,5-Diphenyl-1H-pyrrol-3-yl)(phenyl)methanone (5a). This compound was purified by column chromatography (ethyl acetate/petroleum ether = 1: 8) to afford a yellow solid in 91% yield (59 mg); mp 81-83 °C; 1H NMR (500 MHz, CDCl3) δ 8.98 (s, 1H), 7.80 (d, J = 7.0 Hz, 2H), 7.54 (d, J = 7.5 Hz, 2H), 7.45-7.42 (m, 3H), 7.39 (t, J = 7.5 Hz, 2H), 7.32 (t, J = 7.5 Hz, 2H), 7.29-7.24 (m, 4H), 6.84 (d, J = 3.0 Hz, 1H); 13C NMR (125 MHz, CDCl3) δ 192.9, 139.8, 138.3, 132.3, 132.1, 131.8, 130.1, 129.5, 128.9, 128.8, 128.5, 128.3, 127.5, 124.5, 122.3, 110.9; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C23H18NO 324.1383; Found 324.1382.
Phenyl(2-phenyl-5-(p-tolyl)-1H-pyrrol-3-yl)methanone (5b). This compound was purified by column chromatography (ethyl acetate/petroleum ether = 1: 8) to afford a yellow solid in 91% yield (61 mg); mp 81-83 °C; 1H NMR (500 MHz, CDCl3) δ 9.12 (s, 1H), 7.79 (d, J = 8.0 Hz, 2H), 7.47 – 7.38 (m, 5H), 7.31 (t, J = 7.7 Hz, 2H), 7.21 - 7.17 (m, 5H), 6.78 (s, 1H), 2.36 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 193.1, 139.8, 138.1, 137.3, 132.5, 132.2, 132.1, 130.1, 130.1, 129.1, 128.9, 128.7, 128.4, 128.3, 124.6, 122.2, 110.4, 21.6; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C24H20NO 338.1539; Found 338.1545.
(5-(4-Ethylphenyl)-2-phenyl-1H-pyrrol-3-yl)(phenyl)methanone (5c). This compound was purified by column chromatography (ethyl acetate/petroleum ether = 1: 8) to afford a yellow solid in 92% yield (64 mg); mp 71-73 °C; 1H NMR (500 MHz, CDCl3) δ 8.92 (s, 1H), 7.80 (d, J = 7.0 Hz, 2H), 7.47-7.42 (m, 5H), 7.32 (t, J = 7.5 Hz, 2H), 7.26-7.22 (m, 5H), 6.81 (d, J = 3.0 Hz, 1H), 2.67 (q, J = 7.5 Hz, 2H), 1.26 (t, J = 7.5 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 193.0, 143.8, 139.8, 138.0, 132.5, 132.2, 132.1, 130.1, 129.3, 128.9, 128.9, 128.7, 128.4, 128.3, 124.6, 122.2, 110.4, 29.0, 15.9; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C25H22NO 352.1693; Found 352.1689.
(5-(4-Methoxyphenyl)-2-phenyl-1H-pyrrol-3-yl)(phenyl)methanone (5d). This compound was purified by column chromatography (ethyl acetate/petroleum ether = 1: 8) to afford a yellow solid in 92% yield (65 mg); mp 111-113 °C; 1H NMR (500 MHz, CDCl3) δ 8.96 (s, 1H), 7.79 (d, J = 7.5 Hz, 2H), 7.46 (d, J = 8.5 Hz, 2H), 7.41 (d, J = 7.0 Hz, 3H), 7.31 (t, J = 7.5 Hz, 2H), 7.22 (d, J = 7.5 Hz, 3H), 6.92 (d, J = 8.5 Hz, 2H), 6.72 (d, J = 3.0 Hz, 1H), 3.82 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 193.1, 159.3, 139.9, 137.9, 132.4, 132.3, 132.1, 130.1, 128.9, 128.7, 128.3, 128.3, 126.0, 124.8, 122.2, 114.9, 109.8, 55.8; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C24H20NO 354.1489; Found 354.1489.
(5-(4-Fluorophenyl)-2-phenyl-1H-pyrrol-3-yl)(phenyl)methanone (5e). This compound was purified by column chromatography (ethyl acetate/petroleum ether = 1: 8) to afford a yellow solid in 85% yield (58 mg); mp 104-106 °C; 1H NMR (500 MHz, CDCl3) δ 8.93 (s, 1H), 7.78 (d, J = 7.0 Hz, 2H), 7.50 (dd, J = 9.0, 5.0 Hz, 2H), 7.45-7.41 (m, 3H), 7.31 (t, J = 7.5 Hz, 2H), 7.24-7.22 (m, 3H), 7.08 (t, J = 8.5 Hz, 2H), 6.76 (d, J = 7.5 Hz, 1H); 13C NMR (125 MHz, CDCl3) δ 192.9, 162.4 (d, JC-F = 245.0 Hz), 139.7, 138.3, 132.2, 132.1, 131.5, 130.1, 128.8 (d, JC-F = 6.3 Hz), 128.6, 128.3, 128.2, 126.4, 126.3, 122.4, 116.5 (d, JC-F = 22.5 Hz), 110.7; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C23H17FNO 342.1289; Found 342.1287.
(5-(4-Chlorophenyl)-2-phenyl-1H-pyrrol-3-yl)(phenyl)methanone (5f). This compound was purified by column chromatography (ethyl acetate/petroleum ether = 1: 8) to afford a yellow solid in 84% yield (60 mg); mp 112-114 ℃; 1H NMR (500 MHz, CDCl3) δ 9.04 (s, 1H), 7.77 (d, J = 7.0Hz, 2H), 7.46 – 7.42 (m, 3H), 7.39 (dd, J = 6.5, 3.0 Hz, 2H), 7.35-7.30 (m, 4H), 7.23 – 7.19 (m, 3H), 6.79 (d, J = 3.0 Hz, 1H); 13C NMR (125 MHz, CDCl3) δ 192.9, 139.6, 138.6, 133.1, 132.2, 131.9, 131.3, 130.4, 130.1, 129.6, 128.9, 128.8, 128.6, 128.3, 125.8, 122.4, 111.2; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C23H17ClNO 358.0993; Found 358.1002.
(5-(4-Bromophenyl)-2-phenyl-1H-pyrrol-3-yl)(phenyl)methanone (5g). This compound was purified by column chromatography (ethyl acetate/petroleum ether = 1: 8) to afford a yellow solid in 84% yield (67 mg); mp 127-129 °C; 1H NMR (500 MHz, CDCl3) δ 8.77 (s, 1H), 7.79 (d, J = 8.5 Hz, 2H), 7.52 (d, J = 8.5 Hz, 2H), 7.46-7.43 (m, 3H), 7.40 (d, J = 8.5 Hz, 2H), 7.33 (t, J = 7.5 Hz, 2H), 7.29-7.27 (m, 3H), 6.85 (d, J = 7.5 Hz, 1H); 13C NMR (125 MHz, CDCl3) δ 193.0, 139.6, 138.7, 132.5, 132.3, 131.9, 131.3, 130.8, 130.1, 128.9, 128.7, 128.6, 128.3, 126.1, 122.4, 121.1, 111.3; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C23H17BrNO 402.0488; Found 402.0489.
(5-(4-(tert-Butyl)phenyl)-2-phenyl-1H-pyrrol-3-yl)(phenyl)methanone (5h). This compound was purified by column chromatography (ethyl acetate/petroleum ether = 1: 8) to afford a yellow solid in 93% yield (70 mg); mp 98-100 °C; 1H NMR (500 MHz, CDCl3) δ 8.89 (s, 1H), 7.80 (d, J = 8.0 Hz, 2H), 7.49 – 7.40 (m, 7H), 7.32 (t, J = 8.0 Hz, 2H), 7.28 – 7.23 (m, 3H), 6.82 (s, 1H), 1.34 (s, 9H); 13C NMR (125 MHz, CDCl3) δ 192.9, 150.7, 139.8, 138.0, 132.4, 132.3, 132.1, 130.1, 129.1, 128.9, 128.8, 128.4, 128.3, 126.4, 124.3, 122.3, 110.5, 35.0, 31.7; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C27H26NO 380.2009; Found 380.2008.
Phenyl(2-phenyl-5-(4'-propyl-[1,1'-biphenyl]-4-yl)-1H-pyrrol-3-yl)methanone (5i). This compound was purified by column chromatography (ethyl acetate/petroleum ether = 1: 8) to afford a yellow solid in 85% yield (72 mg); mp 149-151 °C; 1H NMR (500 MHz, CDCl3) δ 9.43 (s, 1H), 7.80 (d, J = 7.5 Hz, 2H), 7.59 (s, 4H), 7.52 (d, J = 8.0 Hz, 2H), 7.44 (t, J = 7.5 Hz, 1H), 7.41-7.36 (m, 2H), 7.32 (t, J = 7.5 Hz, 2H), 7.25 (d, J = 8.0 Hz, 2H), 7.20 – 7.15 (m, 3H), 6.85 (d, J = 3.0 Hz, 1H), 2.64 (d, J = 8.0 Hz, 2H), 1.69 (m, 2H), 0.99 (t, J = 7.5 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 193.3, 142.5, 140.0, 139.8, 138.7, 138.2, 132.3, 132.2, 132.1, 130.5, 130.2, 129.4, 129.0, 128.7, 128.4, 128.3, 127.8, 127.1, 125.0, 122.3, 110.9, 38.1, 25.0, 14.3; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C32H28NO 442.2165; Found 442.2170.
Phenyl(2-phenyl-5-(m-tolyl)-1H-pyrrol-3-yl)methanone (5j). This compound was purified by column chromatography (ethyl acetate/petroleum ether = 1: 8) to afford a yellow solid in 86% yield (58 mg); mp 118-120 °C; 1H NMR (500 MHz, CDCl3) δ 9.08 (s, 1H), 7.80 (d, J = 7.5 Hz, 2H), 7.43 (d, J = 6.0 Hz, 3H), 7.37-7.31 (m, 4H), 7.27 (d, J = 7.5 Hz, 1H), 7.22 (t, J = 6.0 Hz, 3H), 7.08 (d, J = 7.5 Hz, 1H), 6.82 (d, J = 2.5 Hz, 1H), 2.38 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 193.1, 139.8, 139.0, 138.4, 132.5, 132.2, 132.1, 131.8, 130.1, 129.3, 128.9, 128.7, 128.4, 128.3, 125.4, 122.2, 121.7, 110.8, 21.9; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C24H20NO 338.1539; Found 338.1531.
(5-(3-Chlorophenyl)-2-phenyl-1H-pyrrol-3-yl)(phenyl)methanone (5k). This compound was purified by column chromatography (ethyl acetate/petroleum ether = 1: 8) to afford a yellow solid in 85% yield (60 mg); mp 83-85 °C; 1H NMR (500 MHz, CDCl3) δ 9.19 (s, 1H), 7.78 (d, J = 7.5 Hz, 2H), 7.51 (s, 1H), 7.44 (t, J = 7.5 Hz, 1H), 7.41-7.38 (m, 3H), 7.34-7.27 (m, 3H), 7.23 – 7.18 (m, 4H), 6.80 (d, J = 2.5 Hz, 1H); 13C NMR (125 MHz, CDCl3) δ 193.0, 139.6, 138.9, 135.4, 133.6, 132.3, 131.8, 130.9, 130.6, 130.1, 128.9, 128.7, 128.6, 128.4, 127.3, 124.6, 122.6, 122.3, 111.7; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C23H17ClNO 358.0993; Found 358.0992.
(5-(3-Bromophenyl)-2-phenyl-1H-pyrrol-3-yl)(phenyl)methanone (5l). This compound was purified by column chromatography (ethyl acetate/petroleum ether = 1: 8) to afford a yellow solid in 87% yield (69 mg); mp 99-101 °C; 1H NMR (500 MHz, CDCl3) δ 9.10 (s, 1H), 7.78 (d, J = 7.0 Hz, 2H), 7.67 (s, 1H), 7.46 – 7.40 (m, 4H), 7.37 (d, J = 8.0 Hz, 1H), 7.33 (t, J = 8.0 Hz, 2H), 7.25 – 7.21 (m, 4H), 6.81 (d, J = 2.5 Hz, 1H); 13C NMR (125 MHz, CDCl3) δ 192.9, 139.6, 138.9, 133.9, 132.3, 131.8, 130.9, 130.7, 130.2, 130.1, 128.9, 128.8, 128.7, 128.4, 127.4, 123.6, 123.1, 122.4, 111.7; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C23H17BrNO 402.0488; Found 402.0489.
(5-(2-Fluorophenyl)-2-phenyl-1H-pyrrol-3-yl)(phenyl)methanone (5m). This compound was purified by column chromatography (ethyl acetate/petroleum ether = 1: 8) to afford a yellow solid in 79% yield (54 mg); mp 77-79 °C; 1H NMR (500 MHz, CDCl3) δ 9.39 (s, 1H), 7.81 (d, J = 7.0 Hz, 2H), 7.64 (t, J = 8.0 Hz, 1H), 7.48 - 7.44 (m, 3H), 7.34 (t, J = 8.0 Hz, 2H), 7.29 - 7.26 (m, 3H), 7.23 – 7.13 (m, 3H), 6.98 (d, J = 3.0 Hz, 1H); 13C NMR (125 MHz, CDCl3) δ192.8, 159.2 (d, JC-F = 124.1 Hz), 139.7, 138.3, 132.2, 132.0, 130.1, 128.8, 128.6, 128.57 (d, JC-F = 8.5 Hz), 128.4, 127.3 (d, JC-F = 4.0 Hz), 127.1, 125.3 (d, JC-F = 3.0 Hz), 121.7, 119.4, 119.3, 116.8 (d, JC-F = 23.8 Hz), 112.7; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C23H17FNO 342.1289; Found 342.1281.
(4-Chlorophenyl)(2-(4-chlorophenyl)-5-phenyl-1H-pyrrol-3-yl) methanone (5n). This compound was purified by column chromatography (ethyl acetate/petroleum ether = 1: 8) to afford a yellow solid in 75% yield (58 mg); mp 94-96 °C; 1H NMR (500 MHz, CDCl3) δ 8.75 (s, 1H), 7.77 (d, J = 9.0 Hz, 2H), 7.53 (d, J = 7.0 Hz, 2H), 7.46 (d, J = 8.5 Hz, 2H), 7.42 (t, J = 8.0 Hz, 2H), 7.34 (m, 4H), 6.80 (d, J = 2.5 Hz, 1H); 13C NMR (125 MHz, CDCl3) δ 191.2, 138.7, 138.0, 136.8, 134.8, 132.7, 131.4, 130.5, 130.0, 129.6, 129.2, 128.8, 127.9, 124.6, 122.3, 110.8; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C23H16Cl2NO 392.0603; Found 392.0612.
(2,5-Diphenyl-1H-pyrrol-3-yl) (naphthalen-2-yl) methanone (5o). This compound was purified by column chromatography (ethyl acetate/petroleum ether = 1: 8) to afford a yellow solid in 92% yield (68 mg); mp 107-109 °C; 1H NMR (500 MHz, CDCl3) δ 8.81 (s, 1H), 8.34 (s, 1H), 7.94 (d, J = 8.5 Hz, 1H), 7.83 (t, J = 8.0 Hz, 3H), 7.60 – 7.52 (m, 5H), 7.49 (d, J = 7.0 Hz, 1H), 7.42 (t, J = 8.0 Hz, 2H), 7.31 – 7.26 (m, 3H), 7.22 (d, J = 7.5 Hz, 1H), 6.92 (d, J = 2.5 Hz, 1H); 13C NMR (125 MHz, CDCl3) δ 192.7, 138.1, 137.0, 135.4, 132.7, 132.3, 132.2, 131.8, 131.7, 129.7, 129.5, 128.9, 128.8, 128.6, 128.2, 128.2, 128.1, 127.6, 126.8, 126.1, 124.5, 122.6, 111.0; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C27H20NO 374.1539; Found 374.1537.
(2,5-Diphenyl-1H-pyrrol-3-yl) (thiophen-2-yl) methanone (5p). This compound was purified by column chromatography (ethyl acetate/petroleum ether = 1: 8) to afford a yellow solid in 90% yield (59 mg); mp 86-88 °C; 1H NMR (500 MHz, CDCl3) δ 8.77 (s, 1H), 7.66 (d, J = 3.5 Hz, 1H), 7.60 – 7.54 (m, 5H), 7.42 (t, J = 8.0 Hz, 2H), 7.36 (t, J = 7.0 Hz, 2H), 7.33 – 7.28 (m, 2H), 7.04 (dd, J = 5.0, 4.0 Hz, 1H), 6.99 (d, J = 3.0 Hz, 1H); 13C NMR (125 MHz, CDCl3) δ 184.1, 145.9, 137.4, 134.1, 133.2, 132.4, 132.1, 131.8, 129.5, 129.0, 128.7, 128.6, 128.0, 127.6, 124.6, 122.3, 110.2; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C21H16NOS 330.0947; Found 330.0955.
(2-Isopropyl-5-phenyl-1H-pyrrol-3-yl) (phenyl)methanone (5q). This compound was purified by column chromatography (ethyl acetate/petroleum ether = 1: 8) to afford a yellow solid in 86% yield (50 mg); mp 85-87 °C; 1H NMR (500 MHz, CDCl3) δ 8.79 (s, 1H), 7.84 (d, J = 7.0 Hz, 2H), 7.53 (t, J = 7.0 Hz, 1H), 7.48 – 7.44 (m, 4H), 7.36 (t, J = 7.5 Hz, 2H), 7.23 (t, J = 7.5 Hz, 1H), 6.64 (d, J = 3.0 Hz, 1H), 3.87 (m, 1H), 1.37 (d, J = 7.0 Hz, 6H); 13C NMR (125 MHz, CDCl3) δ 192.8, 147.8, 141.1, 132.2, 131.6, 129.8, 129.5, 129.4, 128.5, 127.2, 124.3, 120.2, 110.1, 26.8, 22.4; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C20H20NO 290.1539; Found 290.1530.
Phenyl(5-phenyl-2-propyl-1H-pyrrol-3-yl) methanone (5r). This compound was purified by column chromatography (ethyl acetate/petroleum ether = 1: 8) to afford a yellow solid in 42% yield (24 mg); mp 74-76 °C; 1H NMR (500 MHz, CDCl3) δ 8.68 (s, 1H), 7.87 – 7.82 (m, 2H), 7.53 (t, J = 7.5 Hz, 1H), 7.49 – 7.44 (m, 4H), 7.39 – 7.34 (m, 2H), 7.23 (t, J = 7.5 Hz, 1H), 6.66 (d, J = 3.0 Hz, 1H), 3.02 (t, J = 7.5 Hz, 2H), 1.76 (m, 2H), 1.60 (s, 3H), 1.01 (t, J = 7.5 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 192.7, 142.4, 141.0, 132.1, 131.6, 130.0, 129.5, 129.4, 128.5, 127.1, 124.2, 121.2, 109.8, 30.1, 23.1, 14.4; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C20H20NO 290.1539; Found 290.1542.
(2-Butyl-5-phenyl-1H-pyrrol-3-yl) ( phenyl)methanone (5s). This compound was purified by column chromatography (ethyl acetate/petroleum ether = 1: 8) to afford a yellow solid in 49% yield (30 mg); mp 74-76 °C; 1H NMR (500 MHz, CDCl3) δ 9.24 (s, 1H), 7.85 (d, J = 8.0 Hz, 2H), 7.55 - 7.45 (m, 5H), 7.34 (t, J = 7.0 Hz, 2H), 7.21 (t, J = 7.5 Hz, 1H), 6.67 (s, 1H), 3.01 (t, J = 7.5 Hz, 2H), 1.7.-1.64 (m, 2H), 1.39 - 1.32 (m, 2H), 0.89 (t, J = 7.0 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 192.7, 142.7, 140.7, 131.8, 131.3, 129.8, 129.1, 129.0, 128.1, 126.6, 123.9, 120.6, 109.4, 31.7, 27.5, 22.6, 14.0, 13.9; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C21H22NO 304.1696; Found 304.1702.
(1,2-Dimethyl-5-phenyl-1H-pyrrol-3-yl) (phenyl)methanone (7a). This compound was purified by column chromatography (ethyl acetate/petroleum ether = 1: 8) to afford a white solid in 72% yield (40 mg); mp 96-98 °C; 1H NMR (500 MHz, CDCl3) δ 7.64 (d, J = 7.0 Hz, 2H), 7.26 (t, J = 7.5Hz, 1H), 7.14 (t, J = 7.5 Hz, 2H), 7.04 (q, J = 8.0 Hz, 4H), 6.98 (d, J = 6.5 Hz, 1H), 6.64 (s, 1H), 3.62 (s, 3H), 2.38 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 194.4, 140.0, 135.8, 135.6, 131.9, 130.2, 128.8, 128.2, 128.0, 126.2, 125.9, 120.2, 120.1, 34.2, 11.7; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C19H18NO 276.1383; Found 276.1388.
1-(2-Methyl-5-phenyl-1-(p-tolyl)-1H-pyrrol-3-yl) ethan-1-one (7b). This compound was purified by column chromatography (ethyl acetate/petroleum ether = 1: 8) to afford a yellow solid in 85% yield (49 mg); mp 66-68 °C; 1H NMR (500 MHz, CDCl3) δ 7.38 (d, J = 4.0 Hz, 4H), 7.32 – 7.27 (m, 3H), 7.21 (d, J = 8.5 Hz, 2H), 6.64 (s, 1H), 2.42 (s, 3H), 2.39 (s, 3H), 2.07 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 198.1, 138.5, 136.6, 136.5, 135.8, 130.3, 129.7, 128.7, 127.2, 126.6, 126.4, 122.8, 121.1, 31.5, 21.5, 13.3; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C20H20NO 290.1539; Found 290.1538.
2-Methyl-4-phenyl-1H-imidazole (8). This compound was purified by column chromatography (ethyl acetate/petroleum ether = 1: 3) to afford a white solid in 96% yield (30 mg); mp 57-59 °C; 1H NMR (500 MHz, CDCl3) δ 7.67 (d, J = 7.0 Hz, 2H), 7.37 (s, 1H), 7.30 (t, J = 7.5Hz, 2H), 7.13 (t, J = 7.5 Hz, 1H), 2.29 (s, 3H); 13C NMR (125 MHz, DMSO-d6) δ 145.7, 138.2, 133.2, 129.1, 127.2, 125.1, 115.6, 14.2; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C10H11N2 159.0917; Found 159.091.
(E)-(2,5-Diphenyl-1H-pyrrol-3-yl) (phenyl)methanone oxime (9). This compound was purified by column chromatography (ethyl acetate/petroleum ether = 1: 8) to afford a white solid in 94% yield (63 mg); mp 95-97 °C; 1H NMR (500 MHz, DMSO-d6) δ 11.44 (s, 1H), 11.19 (s, 1H), 7.79 (d, J = 7.5 Hz, 2H), 7.49 (d, J = 7.5 Hz, 4H), 7.37 (t, J = 8.0 Hz, 2H), 7.26-7.23 (m, 5H), 7.19 (t, J = 7.5 Hz, 1H), 7.12 (t, J = 7.5 Hz, 1H), 6.52 (d, J = 3.0 Hz, 1H); 13C NMR (125 MHz, DMSO-d6) δ 153.7, 137.7, 133.2, 132.8, 131.6, 129.6, 129.5, 129.1, 129.0, 128.8, 127.4, 127.2, 127.0, 126.5, 124.8, 115.1, 109.2; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C23H19N2O 339.1492; Found 339.1496.
(1-Methyl-2,5-diphenyl-1H-pyrrol-3-yl) (phenyl)methanone (10). This compound was purified by column chromatography (ethyl acetate/petroleum ether = 1: 8) to afford a colorless oil in 98% yield (66 mg); 1H NMR (500 MHz, CDCl3) δ 7.76 (d, J = 7.0 Hz, 2H), 7.50 (d, J = 8.0 Hz, 2H), 7.45 (t, J = 8.0 Hz, 2H), 7.40 – 7.35 (m, 5H), 7.34 (s, 1H), 7.32 – 7.27 (m, 3H), 6.67 (s, 1H), 3.49 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 192.3, 140.8, 140.1, 135.7, 132.9, 132.3, 131.6, 131.2, 129.8, 129.4, 129.0, 128.6, 128.5, 128.1, 128.1, 122.3, 112.3, 34.3; HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C24H20NO 338.1539; Found 338.1544.
References
- Weinreb, S.M. Some recent advances in the synthesis of polycyclic imidazole-containing marine natural products. Nat. Prod. Rep. 2007, 24, 931–948. [Google Scholar] [CrossRef] [PubMed]
- Robertson, J.; Stevens, K. Pyrrolizidine alkaloids. Nat. Prod. Rep. 2014, 31, 1721–1788. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, J.; Köck, M. Hybrid pyrrole-imidazole alkaloids from the sponge agelas sceptrum. J. Nat. Prod. 2016, 79, 434–437. [Google Scholar] [CrossRef] [PubMed]
- Clive, D.L.J.; Cheng, P. The marinopyrroles. Tetrahedron 2013, 69, 5067–5078. [Google Scholar] [CrossRef]
- Shabalin, D.A.; Camp, J.E. Recent advances in the synthesis of imidazoles. Org. Biomol. Chem. 2020, 18, 3950–3964. [Google Scholar] [CrossRef]
- Vessally, E.; Soleimani-Amiri, S.; Hosseinian, A.; Edjlali, L.; Bekhradnia, A. New protocols to access imidazoles and their ring fused analogues: synthesis from N-propargylamines. RSC Adv. 2017, 7, 7079–7091. [Google Scholar] [CrossRef]
- Khajuria, R.; Dham, S.; Kapoor, K.K. Active methylenes in the synthesis of a pyrrole motif: an imperative structural unit of pharmaceuticals, natural products and optoelectronic materials. RSC Adv. 2016, 6, 37039–37066. [Google Scholar] [CrossRef]
- Borah, B.; Dwivedi, K.D.; Chowhan, L.R. Recent approaches in the organocatalytic synthesis of pyrroles. RSC Adv. 2021, 11, 13585–13601. [Google Scholar] [CrossRef]
- Preeti; Singh, K. N. Multicomponent reactions: a sustainable tool to 1,2- and 1,3-azoles. Org. Biomol. Chem. 2018, 16, 9084–9116. [Google Scholar] [CrossRef]
- Estévez, V.; Villacampa, M.; Menéndez, J.C. Recent advances in the synthesis of pyrroles by multicomponent reactions. Chem. Soc. Rev. 2014, 43, 4633–4657. [Google Scholar] [CrossRef]
- Shi, S.; Xu, K.; Jiang, C.; Ding, Z. ZnCl2-Catalyzed [3+2] Cycloaddition of Benzimidates and 2H-Azirines for the Synthesis of Imidazoles. J. Org. Chem. 2018, 83, 14791–14796. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Huang, Y.; Song, H.; Liu, Y.; Wang, Q. Copper-Catalyzed Aerobic Oxidative [2+3] Cyclization/Aromatization Cascade Reaction: Atom-Economical Access to Tetrasubstituted 4,5-Biscarbonyl Imidazoles. Org. Lett. 2017, 19, 6056–6059. [Google Scholar] [CrossRef] [PubMed]
- Liao, J.-Y.; Shao, P.-L.; Zhao, Y. Catalytic Divergent Synthesis of 3H or 1H Pyrroles by [3 + 2] Cyclization of Allenoates with Activated Isocyanides. J. Am. Chem. Soc. 2015, 137, 628–631. [Google Scholar] [CrossRef] [PubMed]
- Vannada, J.; Sulthan, M.; Arun, D.; Dada, R.; Yaragorla, S. Regiodivergent Synthesis of Penta-Substituted Pyrroles through a Cascade [3 + 2] Cyclization of C-Acylimines with Activated Alkynes and Aromatic Nucleophiles. J. Org. Chem. 2020, 85, 6697–6708. [Google Scholar] [CrossRef]
- Dai, L.; Yu, S.; Lv, N.; Ye, X.; Shao, Y.; Chen, Z.; Chen, J. Synthesis of Imidazoles and Oxazoles via a Palladium-Catalyzed Decarboxylative Addition/Cyclization Reaction Sequence of Aromatic Carboxylic Acids with Functionalized Aliphatic Nitriles. Org. Lett. 2021, 23, 5664–5668. [Google Scholar] [CrossRef] [PubMed]
- Kurita, S.; Kiyota, S.; Komine, N.; Hirano, M. Ru(0)-Catalyzed Synthesis of Conjugated Iminotrienes and Subsequent Intramolecular Cyclization Giving Polysubstituted Pyrroles. Org. Lett. 2022, 24, 2973–2977. [Google Scholar] [CrossRef]
- Li, S.; Li, Z.; Yuan, Y.; Peng, D.; Li, Y.; Zhang, L.; Wu, Y. Au(I)-Catalyzed Intramolecular Hydroamination of the Fluorinated N′-Aryl-N-Propargyl Amidines: Mild Conditions for the Synthesis of 2-Fluoroalkyl Imidazole Derivatives. Org. Lett. 2012, 14, 1130–1133. [Google Scholar] [CrossRef]
- Li, W.; Shi, R.; Chen, S.; Zhang, X.; Peng, W.; Chen, S.; Li, J.; Xu, X.-M.; Zhu, Y.-P.; Wang, X. Synthesis of Diverse Pentasubstituted Pyrroles by a Gold(I)-Catalyzed Cascade Rearrangement-Cyclization of Tertiary Enamide. J. Org. Chem. 2022, 87, 3014–3024. [Google Scholar] [CrossRef]
- Akter, M.; Rupa, K.; Anbarasan, P. 1,2,3-Triazole and Its Analogues: New Surrogates for Diazo Compounds. Chem. Rev. 2022, 122, 13108–13205. [Google Scholar] [CrossRef]
- Huang, J.; Zhou, H.; Chen, Z. Advances on the Metallocarbene Formation Reactions Based on Triazole Derivatives. Chin. J. Org. Chem. 2016, 36, 1555–1563. [Google Scholar] [CrossRef]
- Davies, H.M.L.; Alford, J.S. Reactions of metallocarbenes derived from N-sulfonyl-1,2,3-triazoles. Chem. Soc. Rev. 2014, 43, 5151–5162. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Sun, R.; Tang, X.-Y.; Shi, M. Recent Advancesin the Synthesis of Heterocycles and RelatedSubstances Based on α-Imino Rhodium Carbene ComplexesDerived from N-Sulfonyl-1,2,3-triazoles. Chem. Eur. J. 2016, 22, 17910–17924. [Google Scholar] [CrossRef] [PubMed]
- Chuprakov, S.; Kwok, S.W.; Zhang, L.; Lercher, L.; Fokin, V.V. Rhodium-Catalyzed Enantioselective Cyclopropanation of Olefins with N-Sulfonyl 1,2,3-Triazoles. J. Am. Chem. Soc. 2009, 131, 18034–18035. [Google Scholar] [CrossRef] [PubMed]
- Miura, T.; Nakamuro, T.; Ishihara, Y.; Nagata, Y.; Murakami, M. Chiral Macrocycles Having C3 Symmetry Resulting from Orientation of Thiophene Rings. Angew. Chem., Int. Ed. 2020, 59, 20475–20479. [Google Scholar] [CrossRef] [PubMed]
- Chuprakov, S.; Worrell, B.T.; Selander, N.; Sit, R.K.; Fokin, V.V. Stereoselective 1,3-Insertions of Rhodium(II) Azavinyl Carbenes. J. Am. Chem. Soc. 2014, 136, 195–202. [Google Scholar] [CrossRef]
- He, X.; Wu, Y.; Zhou, T.; Zuo, Y.; Xie, M.; Li, R.; Duan, J.; Shang, Y. Rh-Catalyzed C−N Coupling of N-Sulfonyl-1,2,3-Trizales with Secondary Amines for Regioselective Synthesis of Phenylvinyl-1,2-Diamines. Synth. Commun. 2020, 50, 2685–2697. [Google Scholar] [CrossRef]
- Chen, Y.; Liu, Y.; Li, Z.; Dong, S.; Liu, X.; Feng, X. Tandem Insertion-[1,3]-Rearrangement: Highly Enantioselective Construction of α-Aminoketones. Angew. Chem., Int. Ed. 2020, 59, 8052–8056. [Google Scholar] [CrossRef]
- Miura, T.; Nakamuro, T.; Miyakawa, S.; Murakami, M. A Syn-Selective Aza-Aldol Reaction of Boron Aza-Enolates Generated fromNSulfonyl-1,2,3-Triazoles and 9-BBN-H. Angew. Chem. Int. Ed. 2016, 55, 8732–8735. [Google Scholar] [CrossRef]
- He, J.; Shi, Y.; Cheng, W.; Man, Z.; Yang, D.; Li, C.-Y. Rhodium-Catalyzed Synthesis of 4-Bromo-1,2-Dihydroisoquinolines: Access to Bromonium Ylides by the Intramolecular Reaction of a Benzyl Bromide and an α-Imino Carbene. Angew. Chem., Int. Ed. 2016, 55, 4557–4561. [Google Scholar] [CrossRef]
- Reddy, A.C.S.; Ramachandran, K.; Reddy, P.M.; Anbarasan, P. Rhodium-Catalyzed Sommelet Hauser Type Rearrangement of α-Diazoimines: Synthesis of Functionalized Enamides. Chem. Commun. 2020, 56, 5649–5652. [Google Scholar] [CrossRef]
- Yu, Y.; Zhu, L.; Liao, Y.; Mao, Z.; Huang, X. Rhodium(II)-Catalysed Skeletal Rearrangement of Ether Tethered N-Sulfonyl 1,2,3-Triazoles: A Rapid Approach to 2-Aminoindanone and Dihydroisoquinoline Derivatives. Adv. Synth. Catal. 2016, 358, 1059–1064. [Google Scholar] [CrossRef]
- Zhang, W.-B.; Xiu, S.-D.; Li, C.-Y. Rhodium-catalyzed synthesis of multi-substituted furans from N-sulfonyl-1,2,3-triazoles bearing a tethered carbonyl group. Org. Chem. Front. 2015, 2, 47–50. [Google Scholar] [CrossRef]
- Shi, Y.; Yu, X.; Li, C.-Y. Rhodium-Catalyzed Synthesis of 2,5-Epoxybenzo[f ][1,4]Oxazepines by Tandem Reaction of 1-Sulfonyl-1,2,3-Triazoles and Salicylaldehydes. Eur. J. Org. Chem. 2015, 2015, 6429–6433. [Google Scholar] [CrossRef]
- Yadagiri, D.; Reddy, A.C.S.; Anbarasan, P. Rhodium Catalyzed Diastereoselective Synthesis of 2,2,3,3-Tetrasubstituted Indolines from N-Sulfonyl-1,2,3-Triazoles and Ortho-Vinylanilines. Chem. Sci. 2016, 7, 5934–5938. [Google Scholar] [CrossRef] [PubMed]
- Miura, T.; Fujimoto, Y.; Funakoshi, Y.; Murakami, M. A Reaction of Triazoles with Thioesters to Produce β-Sulfanyl Enamides by Insertion of an Enamine Moiety into the Sulfur-Carbonyl Bond. Angew. Chem., Int. Ed. 2015, 54, 9967–9970. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Li, R.; Choy, P.Y.; Duan, J.; Yin, Z.; Xu, K.; Tang, Q.; Zhong, R.-L.; Shang, Y.; Kwong, F.Y. An expeditious FeCl3-catalyzed cascade 1,4-conjugate addition/annulation/1,5-H shift sequence for modular access of all-pyrano-moiety-substituted chromenes. Chem. Sci. 2022, 13, 13617–13622. [Google Scholar] [CrossRef] [PubMed]
- Zhou, T.; He, X.; Zuo, Y.; Wu, Y.; Hu, W.; Zhang, S.; Duan, J.; Shang, Y. Rh-catalyzed formal [3+2] cyclization for synthesis of 5-aryl-2-(quinolin-2-yl)oxazoles and its applications in metal ions probes. Chin. J. Chem. 2021, 39, 621–626. [Google Scholar] [CrossRef]
- He, X.; Xu, K.; Liu, Y.; Wang, D.; Tang, Q.; Hui, W.; Chen, H.; Shang, Y. Radical-Induced Cascade Annulation/Hydrocarbonylation for Construction of 2-Aryl-4H-chromen-4-ones. Molecules 2022, 27, 7412. [Google Scholar] [CrossRef]
- Fang, T.; Zhang, S.; Ye, Q.; Kong, S.; Yang, T.; Tang, K.; He, X.; Shang, Y. Rh-catalyzed Cascade C-H Activation/Annulation of N-Hydroxybenzamides and Propargylic Acetates for Modular Access to Isoquinolones. Molecules 2022, 27, 8553. [Google Scholar] [CrossRef]
- Jeon, H.J.; Jung, D.J.; Kim, J.H.; Kim, Y.; Bouffard, J.; Lee, S. From Triazoles to Imidazolines through the Sequential N−H Insertion of α-Imino Rhodium−Carbenes into β-Enamino Esters/Enamine−Imine Tautomerization/Conjugate Addition Cascade. J. Org. Chem. 2014, 79, 9865–9871. [Google Scholar] [CrossRef]
- Lei, X.; Li, L.; He, Y.-P.; Tang, Y. Rhodium(II)-Catalyzed Formal [3 + 2] Cycloaddition of N-Sulfonyl-1,2,3-Triazoles with Isoxazoles: Entry to Polysubstituted 3-Aminopyrroles. Org. Lett. 2015, 17, 5224–5227. [Google Scholar] [CrossRef] [PubMed]
- Rostovskii, N.V.; Ruvinskaya, J.O.; Novikov, M.S.; Khlebnikov, A.F.; Smetanin, I.A.; Agafonova, A.V. Switchable Synthesis of Pyrroles and Pyrazines via Rh(II)-Catalyzed Reaction of 1,2,3-Triazoles with Isoxazoles: Experimental and DFT Evidence for the 1,4-Diazahexatriene Intermediate. J. Org. Chem. 2017, 82, 256–268. [Google Scholar] [CrossRef] [PubMed]
Scheme 1.
Substituent-controllable cascade strategy for the synthesis of trisubstituted imidazoles and pyrroles.
Scheme 1.
Substituent-controllable cascade strategy for the synthesis of trisubstituted imidazoles and pyrroles.
Scheme 2.
Substrate scope of N-sulfonyl-1,2,3-triazoles and β-enaminones for the synthesis of trisubstituted imidazoles. Reaction conditions: N-sulfonyl-1,2,3-triazoles 1 (0.2 mmol), β-enaminones 2 (0.2 mmol), and Rh2(oct)4 (2 mol%) in PhCl (2 mL) at 90 ºC for 12 h under argon atmosphere. Isolated yields were reported.
Scheme 2.
Substrate scope of N-sulfonyl-1,2,3-triazoles and β-enaminones for the synthesis of trisubstituted imidazoles. Reaction conditions: N-sulfonyl-1,2,3-triazoles 1 (0.2 mmol), β-enaminones 2 (0.2 mmol), and Rh2(oct)4 (2 mol%) in PhCl (2 mL) at 90 ºC for 12 h under argon atmosphere. Isolated yields were reported.
Scheme 3.
Substrate scope of N-sulfonyl-1,2,3-triazoles and β-enaminones for the synthesis of trisubstituted pyrroles. Reaction conditions: N-sulfonyl-1,2,3-triazoles 1 (0.2 mmol), β-enaminones 2 (0.2 mmol), and Rh2(oct)4 (2 mol%) in PhCl (2 mL) at 90 ºC for 12 h under argon atmosphere. Isolated yields were reported.
Scheme 3.
Substrate scope of N-sulfonyl-1,2,3-triazoles and β-enaminones for the synthesis of trisubstituted pyrroles. Reaction conditions: N-sulfonyl-1,2,3-triazoles 1 (0.2 mmol), β-enaminones 2 (0.2 mmol), and Rh2(oct)4 (2 mol%) in PhCl (2 mL) at 90 ºC for 12 h under argon atmosphere. Isolated yields were reported.
Scheme 4.
Further studies.
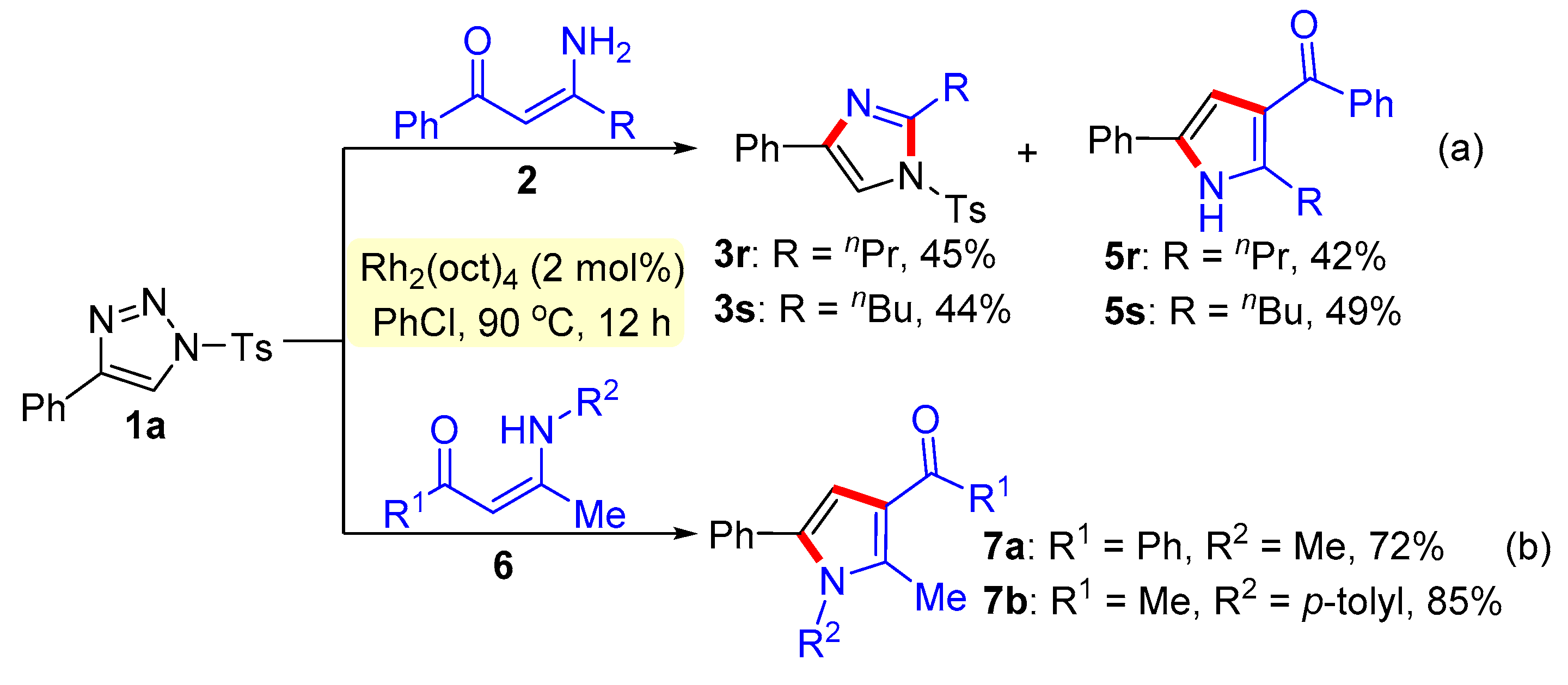
Scheme 5.
Gram-scale synthesis and further synthetic transformations.
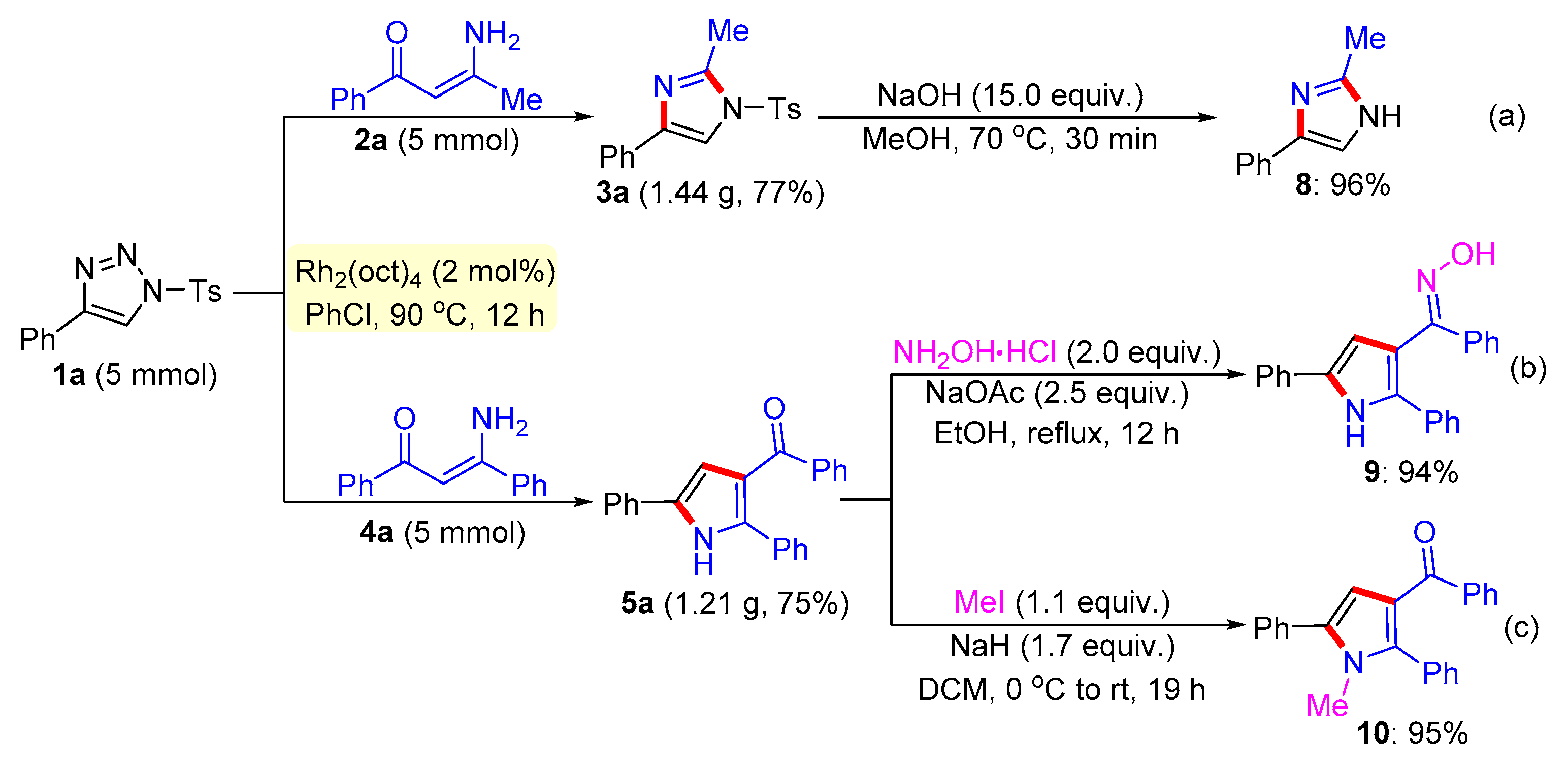

Table 1.
Optimization of the reaction conditionsa.
| Entry | Catalyst (x mol%) | Solvent | Yield (%) b |
|---|---|---|---|
| 1 | Rh2(OAc)4 (4) | DCM | 56 |
| 2 | Rh2(oct)4 (4) | DCM | 58 |
| 3 | CuI (4) | DCM | 44 |
| 4 | Sc(OTf)3 (4) | DCM | trace |
| 5 | Co2(CO)8 (4) | DCM | nr |
| 6 | Ni(acac)2 (4) | DCM | nr |
| 7 | Rh2(oct)4 (3) | DCM | 73 |
| 8 | Rh2(oct)4 (2) | DCM | 79 |
| 9 | Rh2(oct)4 (1) | DCM | 41 |
| 10 | / | DCM | nr |
| 11 | Rh2(oct)4 (2) | DCE | 55 |
| 12 | Rh2(oct)4 (2) | toluene | 84 |
| 13 | Rh2(oct)4 (2) | PhCl | 96 |
| 14 | Rh2(oct)4 (2) | CH3OH | trace |
| 15 | Rh2(oct)4 (2) | CH3NO2 | trace |
| 16 | Rh2(oct)4 (2) | DMF | nr |
| 17c | Rh2(oct)4 (2) | PhCl | nr |
| 18d | Rh2(oct)4 (2) | PhCl | 87 |
| 19e | Rh2(oct)4 (2) | PhCl | 94 |
| 20f | Rh2(oct)4 (2) | PhCl | 56 |
| 21g | Rh2(oct)4 (2) | PhCl | 78 |
| 22h | Rh2(oct)4 (2) | PhCl | 96 |
aReaction conditions: 4-phenyl-1-tosyl-1H-1,2,3-triazole 1a (0.2 mmol), 3-amino-1-phenylbut-2-en-1-one 2a (0.2 mmol), and catalyst in solvent (2 mL) at 90 ºC for 12 h under argon atmosphere. b Isolated yields. c At 60 ºC. d At 80 ºC. e At 100 ºC. f For 2 h. g For 6 h. h For 18 h. nr = no reaction.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



