
Submitted:
12 May 2023
Posted:
15 May 2023
You are already at the latest version
Abstract
In 2021, amidst the COVID-19 pandemic and global food insecurity, the Nigerian poultry sector was yet exposed to highly pathogenic avian influenza (HPAI) virus and its economic challenges. Between 2021 and 2022, HPAI caused 467 outbreaks reported in 31 of the 37 administrative regions in Nigeria. In this study, we characterized the genome of 97 influenza A viruses of the subtypes H5N1, H5N2 and H5N8 identified in different agro-ecological zones and farms during the 2021-2022 epidemic. The phylogenetic analysis of the HA genes showed widespread distribution of the H5Nx clade 2.3.4.4b and similarity with the HPAI H5Nx viruses detected in Europe since late 2020. Topology of the phylogenetic trees indicates the occurrence of several independent introductions of the virus into the country followed by a regional evolution of the virus most probably linked to its persistent circulation in West African territories. An additional evidence of the evolutionary potential of HPAI viruses circulating in this region is the identification in this study of a putative H5N1/H9N2 reassortant virus in a mixed-species commercial poultry farm. Our data confirm Nigeria as a crucial hotspot for HPAI virus introduction from the Eurasian territories and reveal a dynamic pattern of avian influenza virus evolution within the Nigerian poultry population.
Keywords:
Highly pathogenic avian influenza
; H5N1/H9N2 reassortant virus
; Nigeria
1. Introduction
Poultry operations in Nigeria faced many challenges amidst the Coronavirus disease (COVID-19) pandemic and global food insecurity and economic downturn [1]. Though the problems associated with highly pathogenic avian influenza (HPAI) in Nigeria and many countries predate COVID-19 pandemic, yet persistent outbreaks, morbidity and mortality in chickens and other birds have continued unabated and has worsen the economic and public health impacts. The fear that HPAI, a zoonotic disease, could be a progenitor of another pandemic is rife driven by ecological, behavioural, and socioeconomic changes, as well as by the ability of this virus to rapidly evolve through both genetic reassortment and acquisition of mutations [2,3].
The HPAI phenotype is caused by H5 and H7 subtypes of influenza A virus (IAV) of the Orthomyxoviridae family; the virus occurs naturally in wild waterfowls as low pathogenic avian influenza (LPAI) virus and can mutate into HPAI [4] when transmitted to poultry. Influenza A viruses (IAV) are divided into subtypes based on the combinations of the two surface glycoproteins HA and NA; in birds 16 HA and 9 NA types have been identified [5].
In the last two decades, the world has witnessed multiple intercontinental epidemic waves of HPAI of the H5Nx subtype from Asia, Europe to Africa [6] as a consequence of the spread of viruses descendent of the H5N1 virus A/goose/Guangdong/1/1996 (Gs/GD), which was first detected in China in 1996. Since their emergence, these viruses have dramatically expanded their geographical distribution spreading to distinct continents and remaining well entrenched in a number of countries. They have been acquiring a broader host range with a constantly expanding list of free-living wild bird species, which can potentially be affected by the virus and spillover events in mammals have been increasingly reported [7,8].
Since 2006, multiple Gs/GD/96 H5Nx lineages have been introduced in Nigeria, which have negatively impacted on poultry productivity, food security and threaten public health. The first introduction of the Gs/Gd lineage of the HPAI clade 2.2 to Nigeria happened during the harmattan (winter) season of 2006 [9,10]. Between 2006-2008, when the epidemic lasted, millions of poultry birds either died or were culled to contain further spread and economic losses to farmers [11]. These control efforts paid off and the epizootic was declared over by a self-declaration of disease-free status at the World Organization for Animal Health (WOAH, formerly OIE) General Assembly in 2013 (https://www.woah.org/app/uploads/2022/06/eng-archive-2000-may-20222.pdf) [12].
Not long afterwards, a more devastating resurgence of another strain of HPAI caused by clade 2.3.2.1c was witnessed in 2015/2016 [13,14]. Thereafter, a series of re-introductions of various subtypes of HPAI into the Nigerian agro-ecological space, including subtype H5N8, became more frequent [15] and H5N6 was detected in live bird markets (LBMs) in 2019 [16]. Added to these plethora of outbreaks and detections of H5Nx in Nigeria, the low pathogenic avian influenza (LPAI) subtype, H9N2, has been added to the mix and frequently and widely isolated in poultry, especially at LBMs [17].
Apparently, Nigeria has emerged as a regional hotspot of HPAI in sub-Saharan Africa [6] and accounting for a significant proportion of cases reported to the WOAH from the African region in the last two decades (https://www.woah.org/en/disease/avian-influenza/#ui-id-2). More intriguing is the diversity of HPAI and LPAI that has been detected in the Nigerian-agro-ecological space since 2006. HPAI of the H5N2 subtype has previously been detected in healthy wild waterfowl in Nigeria [18]. By 2011, Snoeck et al., [19] again reported the detection of reassortant LPAI H5N2 viruses in African wild birds sampled in Hadejia-Nguru wetlands in northern Nigeria. Similarly, in 2013, Coker et al., [12] recovered genetically similar strains of the H5N2 in commercial waterfowls at a LBM in southwestern Nigeria. These previous H5N2 strains were limited in their spread, as subsequent surveillance activities did not detect the H5N2 even in LBM [16,17].
The agro-ecological source of the introduction of AIV into Nigeria is not fully understood and poorly investigated [10]. We can merely theorize based on available epidemiological and genetic data that AIV is most likely introduced seasonally to Nigeria through activities and contact with migratory waterfowls from Eurasia [20]. Nigeria remained a country at risk of the continuous new introduction of AIV with chances that an endemic situation may result in co-circulation, co-infection and the emergence of possible new reassortant viruses.
Since early January 2021, new outbreaks of HPAI H5Nx have caused multiple outbreaks across the country and have been reported in 30 out of the 37 administrative territories (Pers Comm: Federal Department of Veterinary and Pest Control Services) resulting in 467 positive cases as of December 2022. The transmission of these new HPAIVs in poultry flocks where the H9N2 subtype is also entrenched in the poultry population is a matter of great concern given the proclivity for reassortment of AIV [17]. Using the wide collection of samples gathered over the 2021 and 2022 epidemic events in Nigeria, we investigated the genetic diversity of the AIVs circulating across the Nigerian states and provided evidences of the emergence of a putative H5N1/H9N2 reassortant in a mixed-species commercial poultry farm.
2. Materials and Methods
2.1. Sample collection and necropsy
Diagnostic outbreak investigations were carried out following suspicion of HPAI outbreaks in Nigeria. From previous experience, poultry farmers are sensitive to abnormal mortality patterns and are quick to raise the index of suspicion by contacting the nearest private and state veterinarians. Attending veterinarians’ initial observations with respect to clinical manifestations are documented. Fresh carcasses were selected and shipped under cold-chain to the Regional Laboratory for Animal Influenza, National Veterinary Research Institute (NVRI) in Vom for diagnosis and necropsy. Parenchymatous organs and tissues were harvested from the carcasses, pooling several tissues from the same flock and species for further processing.
2.2. Virus identification and isolation
Tissues were homogenized and supernatant fluid collected for nucleic acid extraction using the Qiagen Viral RNA kit (Qiagen, Hilden, Germany) following the manufacturer’s instructions. Total RNA was extracted from a pool of combined tissues and first screened for IAV matrix gene (M-gene), H5, H7, H9 and Nx IAV subtypes by RT-qPCR using QuantiTect Multiplex (Qiagen, Hilden, Germany) and OneStep RT-PCR (Qiagen, Hilden, Germany) kit as previously described [21,22,23,24,25]. Influenza A positive samples were isolated and typed according to OIE Terrestrial Manual (https://www.woah.org/fileadmin/Home/eng/Animal_Health_in_the_World/docs/pdf/2.03.04_AI.pdf).
To ascertain the presence of an H5N2 reassortant virus in samples potentially co-infected with H9N2 and HPAI H5N1 viruses, homogenate supernatants were subjected in parallel to cloning by multiple rounds of limiting dilution in embryonated chicken eggs and to plaque purification in MDCK cells. Briefly, for the limiting dilution cloning in eggs, the original sample was serially diluted in phosphate buffer solution with antibiotics and incubated for 2 hours at room temperature with chicken polyclonal antiserum raised against a G1 lineage H9N2 virus before inoculation in eggs. To plaque purify the virus, homogenate supernatant was serially diluted in cell culture medium and plated onto MDCK cells for 1 hour at 37 °C in 5% CO2. After removal of the inoculum, a 0.9% agar overlay was added and plates incubated for 72 hours. The allantoic fluid and several plaques collected from eggs/cells infected with the highest dilution underwent further diagnostic screening and NGS analyses.
2.3. Genomic sequencing and phylogenetic analysis
Ninety-seven HPAI H5 positive clinical specimens were selected based on epidemiological locations, species, time of outbreak and sent to the EURL, WOAH and FAO reference laboratory for avian influenza (AI) in Italy (Istituto Zooprofilattico Sperimentale delle Venezie) for sequencing and genetic data analysis.
A target RT-PCR approach was used to amplify influenza A virus whole genomes as previously described [6]. Sequencing libraries were obtained using Nextera XT DNA Sample preparation kit (Illumina, San Diego, CA, USA) and sequenced on MiSeq instrument using a 2 x 250 bp PairedEnd [PE] mode.
Raw sequencing reads produced by MiSeq instrument were cleaned with Trimmomatic v0.32 [26] with minimum quality 20. Illumina Nextera XT adapters sequences were clipped from reads using scythe v0.991 (https://github.com/vsbuffalo/scythe) and amplification primers were removed using sickle v1.33 (https://github.com/najoshi/sickle). Read shorter than 80bp or unpaired were discarded. The cleaned reads were aligned against a reference genome using the MEM algorithm from BWA v0.7.1229. Picard tools v2.1.0 (http://broadinstitute.github.io/picard/) and GATK v3.530-32 [27] where used to improve alignment quality, correct potential errors and recalibrate base quality score. LoFreq v2.1.233 [28]was used to call single nucleotide polymorphisms (SNPs).
The consensus sequence was created with an in-house script. Consensus sequences were aligned using MAFFT v7 online server (https://mafft.cbrc.jp/alignment/server/) [29] and compared with sequences of most related virus strains available in GISAID. Maximum likelihood phylogenetic trees of each gene segment were obtained by using IQTREE v1.6.6 (https://github.com/iqtree/iqtree1) and its robustness was determined through an ultrafast bootstrap resampling analysis of 1000 replications [30,31]. Phylogenetic trees were visualized using FigTree v1.4.4 software (http://tree.bio.ed.ac.uk/software/figtree/).
FluSurver tool (https://flusurver.bii.a-star.edu.sg/) was used to identify amino acid mutations in the influenza A virus proteins, that could affect biological functions such as virulence, host adaptation, drugs resistance.
3. Results
3.1. Virus identification and pathological findings
Depression, somnolence, drooling fluid from the mouth, diarrhea, hock sitting, inappetence, drop in egg production and sudden/high mortality were the most frequent clinical signs raising the suspicion of HPAI infection. Laboratory investigations applied on samples collected from farms reporting the HPAI suspicions allowed to confirm 467 outbreaks of HPAI H5Nx across 31 administrative regions in Nigeria from January 2021 to December 2022 (Figure 1).
More specifically, 454 A(H5N1), 12 A(H5N8) and one A(H5N2) outbreaks were reported. Of note, the unique A (H5N2) subtype was identified in tissue sample collected in a mixed species poultry flock of 20,792 birds (8,271 ducks and 12,521 layer chickens) in Toro LGA of Bauchi State, northeastern Nigeria (Longitude:10.056813 and Latitude:09.0685465) (Figure 1) where a sudden increase in the mortality was registered (2000 birds within 24 hours).
The HPAI H5N1 and H5N8 outbreaks were reported in a mixture of backyard, semi-intensive and intensive commercial operations with mixed species mainly including chickens (broiler, layers, growers) and ducks (Table S1). Necropsy revealed gross lesions that are typical of HPAI including diffuse and ecchymotic haemorrhages which is an evidence of capillary damage. Congestion observed in carcasses from commercial chickens were subtle and did not involve multiple organ damages that is known with HPAI. The gross lesions observed among the peacocks, geese and broilers/indigenous chickens were classical and include cyanoses of combs, beaks and wattles, subcutaneous haemorrhages on the shank, hock joints and breast muscles, ecchymotic haemorrhages in the proventriculus and ventriculus. In addition, there were hepatic congestion with friable texture and streaks of peripheral pallor, petechial haemorrhages in thigh and breast muscles, enlarged and congested spleen, severe peritonitis and adhesion of visceral organs, haemorrhagic enteritis, severe haemorrhagic tracheitis as well as haemorrhages in the ceca and cecal tonsils. These major pathological presentations are shown in Figure 2.
3.2. Phylogenetic analyses
Whole genome sequences of ninety viruses (eleven H5N8, seventy-eight H5N1 and one H5N2 virus) and partial genomes of five H5N1 and two H5N8 viruses were obtained (Table S1).
The phylogenetic analysis of the HA genes confirmed that all the HPAI H5N1, H5N2 and H5N8 Nigerian viruses belong to clade 2.3.4.4b. In depth analysis of the HA phylogeny revealed the existence of two major genetic groups in Nigeria, namely 1 and 2 (Figure 3). Group 1 comprises the H5N1 (N=83) and the H5N2 (N=1) Nigerian strains as well as H5N1 viruses collected in Europe (2020-2021) and in other West African countries (2020-2022). In particular, the Nigerian H5N1 viruses form different genetic clusters within Group 1, suggesting the occurrence of multiple virus introductions at the beginning of 2021 followed by a local spread. Only one of these cluster (Group 1-A), which includes most of the characterized H5N1 viruses (72/83), persistently circulated in the country up to 2022. Group 2 comprises only Nigerian viruses of the H5N8 subtype, which showed the highest identity with H5N8 viruses identified in Central Asia and Europe between October 2020 and January 2021, suggesting a separate virus incursions, likely through a different route. Of note, the H5N8 subtype has been detected in the country only for few months after its introduction at the beginning of 2021 and has not been extensively circulating in other West African countries.
The tree topology of N1 (Figure S1), N8 (Figure S2) and the other six internal gene segments confirms the genetic clustering similar to that shown for the haemagglutinin gene for the H5N1 and the H5N8 viruses respectively. Differently, the complete genome analyses of the H5N2 virus indicate that it emerged from reassortment events between the H5N1 and the low pathogenic H9N2 viruses circulating in poultry in Nigeria. Specifically, six genes (PB2, PB1, PA, HA, NP and NS) are highly related to the Nigerian H5N1 viruses, while two genes (NA and M) show the highest identity with of the H9N2 viruses of G1 lineage identified in Nigeria between 2019 and 2021 (Figure 1, Figures S3–S9) [17]. As for the endemic circulation of the H9N2 subtype in Nigeria and the weak positivity detected by real time RT-PCR for the H9 subtype [25], an ultradeep sequencing approach was applied to characterize the population diversity within the H5N2 positive sample and to exclude a H5N1/H9N2 coinfection. No reads ascribable to the N1 subtype were identified suggesting the existence of a H5N2/H9N2 coinfection. No reads ascribable to the N1 subtype were identified. Therefore, clonal purification by limiting dilution in embryonated eggs and MDCK cells of the isolate obtained from the original clinical material was performed and the clone was genetically characterized. Genetic analyses of the eight gene segments confirmed the presence of a H5N2 reassortant subtype.
3.3. Genetic characterization
Molecular assessment of the consensus sequences of all the Nigerian viruses revealed the presence of several mutations that are likely associated with an increased zoonotic potential or adaptation to poultry. In details analysis of the HA gene shows in all the viruses, except for one (A/Avian/Nigeria/VRD-21-019_21RS744-61/2021), the mutation S137A (H3 numbering) and the mutation S158N, whereas the mutation T160A is present in all the H5N8 strains, and in eighteen H5N1 viruses. All these mutations has been demonstrated to cause an increased alpha2,6-SA binding [32,33]. Glycosylation on hemagglutinin protein need particular attention considering its role on viral properties involved in virus attachment to target cell receptors, virulence, receptor-binding specificity and host adaptation [34].
For the NA protein, most of the H5N1 Nigerian viruses (seventy-three out of eighty-two) present a twenty-two amino acid deletion in the NA stalk region. It has been shown that this feature can increase the virulence of the virus [35] and it is a marker of virus adaptation from wild aquatic birds to poultry [36,37,38].
The investigation of the polymerase complex protein, revealed that the PB2 protein of the A/chicken/Nigeria/743A_22VIR3286-80/2021 virus possesses the mutation D701N which has been recognized as an important mammalian adaptive marker, related to increased replication and virulence in mammals [39,40,41,42,43]. Furthermore, the H5N1 virus A/Avian/Nigeria/271PT_22VIR3286-71/2021 presents K482R, which has been reported to increase polymerase activity in mammalian cell line [44].
Eighty-two out of eighty-tree viruses show Y52H mutation in NP protein, which seems to confer resistance to butyrophilin subfamily 3 member A3 (BTN3A3), a potent avian influenza virus inhibitor in humans [45]. Mutations detected in Matrix-2 (M2) protein and associated to amantadine resistance have been found in three viruses; in details the mutation S31N [46,47] in the H5N2 A/chicken/Nigeria/743A_22VIR3286-800/2021 and the V27I [48] in the H5N1 A/Avian/Nigeria/741_22VIR3286-29/2021 and A/Chicken/Nigeria/751_22VIR3286-35/2022 viruses.
All the viruses show in the NS1 protein the mutation P42S which has been described to increase virulence in mice and it was also demonstrated to be critical for the H5N1 virus to antagonize the interferon induction [49]. Furthermore the mutations L103F and I106M that can increase virulence in mice, have been detected in all viruses here analysed [50].
All H5N1 and the H5N2 viruses show the mutations N205S in NS1 and T48A in NS2, that cause decreased antiviral response in host and correspond to N200S and T47A in the study previously published by Imai et al. [51] (Table 1). Further mutations supposed to be host specificity markers according to statistical inferences are shown in Table 1.
4. Discussion
Nigeria has been identified as a regional hotspot for HPAI since 2006 [6,9]. How the maiden outbreak was introduced into the agro-ecological space is debatable, but introduction through migratory waterfowls has been widely acclaimed [6,8,14].
In the current study, clade 2.3.4.4b H5N1, H5N2 and H5N8 AIVs were confirmed in commercial poultry farms across Nigeria where there is concurrent endemic circulation of H9N2. Longitudinal outbreak investigation in the last decade revealed frequent seeding and reassortments of HPAI H5Nx from external sources to poultry in Nigeria [14].
In the past, AIV subtype H5N2 has been detected in Nigeria under different agro-ecological situations and conditions, but none of those involved chickens in commercial poultry farms [12,18,19]. Molecular and phylogenetic analysis of these earlier H5N2 strains that were detected in Nigeria showed some degree of relationship, especially in the ecology and origin as it relates to waterfowls from Europe [20]. None of these viral strains have become established in the local poultry or other bird population despite sustained active and passive surveillance for AIV in Nigeria [17].
In the last 10 years, other subtypes and clades of AIV including H5N1, H5N8, H5N6 and H9N2 subtypes detected in Nigeria were mostly the consequence of direct introduction through migratory waterfowls expect for the H9N2 subtype which most probably was introduced through poultry trade [6]. However, in the present study, apart from re-introductions at different times of the H5Nx, there appears to be evidence of genetic reassortment between circulating H5N1 and H9N2 viruses. This is not surprising as Asian countries as well as Egypt have had experiences similar to Nigeria where the introduction and spread of HPAI H5Nx resulted in inter-subtype reassortment with H9N2 viruses endemically circulating in local poultry [56,57]. However, the identification of a new reassortant H5N1/H9N2 avian influenza virus in Nigeria suggests that the emergence of reassortant highly pathogenic H5N1/H9N2 avian influenza viruses has recently become a more frequent event in West Africa. Indeed, another H5N1/H9N2 virus containing H9N2 PA gene was reported in Burkina Faso in late 2021 [58].
Genomic and virological evidences suggests that the reassortment and emergence of H5N2 virus in Nigeria may have taken place in Nigerian agro-ecology. Indeed, the combination of H5N1 and H9N2, where the HA and NA segments were donated by the circulating H5N1 and H9N2 parent virus to the new progeny appears to be unique to Nigeria. An interesting dimension to this reassortment event is also the co-existence of an entire H9N2 subtype in the same cocktail of sample where the reassortment genes were detected, providing further proof that the N2 was donated from the H9N2 progenitor and that the emergence of the reassortant strain was most probably identified at its source. As no further outbreaks caused by this reassortant strain has been identified in the region so far, it may be suggested that the early detection of the virus prevented its further spread. However, additional experimental studies are needed to exclude that the lack of viral dissemination was the consequence of a reduced viral fitness of the emerged H5N2 virus. This also lend credence to the necessity of implementing active surveillance and virus search in the region in order to detect these and other circulating IAV subtypes for an improved control of the disease.
The emergence of a H9N2/H5N1 reassortant virus was not the unique evidence of the extensive diversity and evolution characterizing the HPAI H5 viruses identified in Nigeria. Analyses of the phylogenetic topologies revealed that the sequences found in Nigeria were dispersed throughout the trees forming multiple genetic clusters, indicating the occurrence of several independent introductions of the virus into the country as occurred in previous epidemic waves [6,15]. Two main clusters can be identified both finding their potential precursors in viruses identified in Europe and Central Asia: a cluster provisionally named here Group 1 which includes most of the Nigerian H5N1 viruses and viruses circulating in Niger, Benin, Mali, Burkina Faso and Ghana from June 2021 to January 2022 and one genetic group formed by all the Nigerian H5N8 viruses (Group 2) characterized in this study. The high genetic similarity existing between the Nigerian viruses identified within Group 1 and 2 respectively, indicates that lateral transmission events most likely happened, determining a high number of secondary cases in the poultry sector after virus introduction. Despite sources of introduction are far from being conclusively identified, the phylogenies indicate that the country has been experiencing the co-circulation of viruses newly introduced most probably from Europe through wild birds in late 2020. Since then, the viruses have been then evolving in the Western African areas. Indeed, what raise up as distinct from previous epidemic seasons is that most of the 2021-2022 H5N1 viruses characterized here are highly related to the HPAI H5N1 viruses identified in West Africa since early 2021 suggesting a persistent circulation of the virus in the region.
Whole genome analyses revealed the presence of molecular mutations in the HPAI H5N1 and H5N2 viruses that could be associated with an increased zoonotic potential. Particularly interesting is the presence of the mammalian adaptive markers D701N found in the PB2 protein of the H5N2 virus A/chicken/Nigeria/743A_22VIR3286-80/2021; it was experimentally shown by Yang et al. [59], that in a mouse-adapted H9N2 virus this mutations conferred an higher virulence and more efficient viral replication in mouse lung and liver. Furthermore the H5N1 virus A/avian/Nigeria/271PT_22VIR3286-71/2021 isolated in Kano presents K482R, which has been reported to increase polymerase activity in mammalian cell line [44]. All the H5N1 described here present the mutation S137A (H3 numbering)(position 149 from the initial methionine) which has been demonstrated to cause an increased alpha2,6-SA binding and has been already described as characterizing the HA gene of all the H5 viruses identified in Europe since early 2021 [7,32].
5. Conclusions
This study confirms Nigeria as a hotspot for virus introduction and evolution of HPAI H5Nx viruses. Despite the lack of efficient surveillance plans in wild birds across the Western African region, the results of our investigations suggest that HPAI H5Nx viruses were introduced from Eurasian territories most likely via migratory birds. The occurrence of lateral spread transmission events between distinct poultry farms indicate biosecurity breach in the poultry system in the country although a regional spread through wild bird movements cannot be excluded. The relatedness between the Nigerian viruses and the ones identified in other Western African countries suggest the possible existence of inter border livestock movement and poultry trade in these territories.
The emergence of a further putative reassortant virus in West Africa, with the presence of a mutation with zoonotic potential (D701N), added to the identification of other H5N1/H9N2 reassortants in Burkina Faso during late 2021 [58] , raises concerns about the risk that western African countries may be a new source of emerging avian influenza viruses with increased zoonotic potential. Indeed, the role of H9N2 subtype as a main donor of viral genes that resulted in zoonotic infections is well documented [60]. The diversified biosecurity measures applied across the country, the sales of poultry through poorly regulated live markets and the limited financial resources available to implement appropriate HPAI viruses surveillance and control measures are all features that make the eradication of the disease in this area an extremely challenging goal, besides increasing the chance for the virus to evolve as expected. Given the diversity and pattern of introduction and spread of IAV in Nigeria, the importance of pre-emptive bio-surveillance of wild migratory birds at major wetlands and Live Bird Markets, combined with stringent biosecurity measures cannot be overemphasised. Future discussion on additional control measure such as vaccination may be an appropriate tool in some circumstances, but its use may sometimes have negative impacts. Therefore, the government must first carry out risk/benefit assessment of the situation. The economic and public health threat of endemicity of AIV in Nigeria as portends in this study may have far-reaching consequences for sub-Saharan Africa and the rest of the world.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org., Table S1: Nigerian influenza A viruses sequenced in this study, Table S2: Acknowledgment table of the authors, originating and submitting laboratories of the sequences from GISAID’s EpiFlu™ Database on which this research is based in part. Figure S1: Maximum likelihood phylogenetic tree of the NA (N1) gene; Figure S2: Maximum likelihood phylogenetic tree of the NA (N8) gene; Figure S3: Maximum likelihood phylogenetic tree of the NA (N2) gene; Figure S4: Maximum likelihood phylogenetic tree of the PB2 gene; Figure S5: Maximum likelihood phylogenetic tree of the PB1 gene; Figure S6: Maximum likelihood phylogenetic tree of the PA gene; Figure S7: Maximum likelihood phylogenetic tree of the NP gene; Figure S8: Maximum likelihood phylogenetic tree of the M gene; Figure S9: Maximum likelihood phylogenetic tree of the NS gene.
Author Contributions
C.M., I.M., A.M. I.S., M.M.- involved in study design, conceptualization, laboratory analysis, manuscript writing. B.I., C.C.- involved in lab analysis, manuscript writing. J.A.- involved in sampling, lab analyses, manuscript writing. B.Z., F.B., A.F., C.T., F.G. - involved in laboratory analysis, manuscript writing. S.M.- involved in analyses, manuscript writing. A.A, M.N. – involved in manuscript writing. T.O.- involved in sampling and data analysis. All authors read and approved the final manuscript..
Funding
Partial support for this work was provided by the United Nations Food and Agriculture Organization (UN-FAO) with funding from USAID under the OSRO/GLO/507/USA project within the “Global Health Security Agenda” (GHSA) program. The work was partially funded by the Italian Ministry of Health—Ricerca Corrente IZSVE IZS VE 10/19 RC “Sequenziamento di terza generazione: sviluppo di strategie innovative per l’identificazione e la caratterizzazione di virus prioritari per il settore avicolo”. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The consensus sequences of the viruses analyzed in this study were submitted to GISAID’s EpiFlu™ Database
Acknowledgments
We acknowledge Dr. Christianah Ronke Odita for constructing the outbreak map. We appreciate State and Federal Surveillance Officers of the Ministry of Agriculture for outbreak samples collection. We would also like to thank Dr. Morgane Gourlaouen (FAO-UN) for her expert advice and technical support.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Omotayo, A.O.; Omotoso, A.B.; Daud, S.A.; Omotayo, O.P.; Adeniyi, B.A. Rising Food Prices and Farming Households Food Insecurity during the COVID-19 Pandemic: Policy Implications from SouthWest Nigeria. Agric. 2022, 12. [Google Scholar] [CrossRef]
- Morse, S.S.; Mazet, J.A.K.; Woolhouse, M.; Parrish, C.R.; Carroll, D.; Karesh, W.B.; Zambrana-Torrelio, C.; Lipkin, W.I.; Daszak, P. Prediction and Prevention of the next Pandemic Zoonosis. Lancet (London, England) 2012, 380, 1956–1965. [Google Scholar] [CrossRef]
- Lutz, M.M.; Dunagan, M.M.; Kurebayashi, Y.; Takimoto, T. Key Role of the Influenza A Virus PA Gene Segment in the Emergence of Pandemic Viruses. Viruses 2020, 12. [Google Scholar] [CrossRef] [PubMed]
- de Bruin, A.C.M.; Funk, M.; Spronken, M.I.; Gultyaev, A.P.; Fouchier, R.A.M.; Richard, M. Hemagglutinin Subtype Specificity and Mechanisms of Highly Pathogenic Avian Influenza Virus Genesis. Viruses 2022, 14. [Google Scholar] [CrossRef] [PubMed]
- Webster, R.G.; Bean, W.J.; Gorman, O.T.; Chambers, T.M.; Kawaoka, Y. Evolution and Ecology of Influenza A Viruses. Microbiol. Rev. 1992, 56, 152–179. [Google Scholar] [CrossRef] [PubMed]
- Fusaro, A.; Zecchin, B.; Vrancken, B.; Abolnik, C.; Ademun, R.; Alassane, A.; Arafa, A.; Awuni, J.A.; Couacy-Hymann, E.; Coulibaly, M. ’. B.; et al. Disentangling the Role of Africa in the Global Spread of H5 Highly Pathogenic Avian Influenza. Nat. Commun. 2019, 10. [Google Scholar] [CrossRef]
- Adlhoch, C.; Fusaro, A.; Gonzales, J.L.; Kuiken, T.; Marangon, S.; Niqueux, É.; Staubach, C.; Terregino, C.; Guajardo, I.M.; Chuzhakina, K.; et al. Avian Influenza Overview June - September 2022. EFSA journal. Eur. Food Saf. Auth. 2022, 20. [Google Scholar] [CrossRef]
- Meseko, C.; Globig, A.; Ijomanta, J.; Joannis, T.; Nwosuh, C.; Shamaki, D.; Harder, T.; Hoffman, D.; Pohlmann, A.; Beer, M.; et al. Evidence of Exposure of Domestic Pigs to Highly Pathogenic Avian Influenza H5N1 in Nigeria. Sci. Rep. 2018, 8. [Google Scholar] [CrossRef]
- Joannis, T.M.; Meseko, C.A.; Oladokun, A.T.; Ularamu, H.G.; Egbuji, A.N.; Solomon, P.; Nyam, D.C.; Gado, D.A.; Luka, P.; Ogedengbe, M.E.; et al. Serologic and Virologic Surveillance of Avian Influenza in Nigeria, 2006-7. Euro Surveill. Bull. Eur. sur les Mal. Transm. = Eur. Commun. Dis. Bull. 2008, 13. [Google Scholar] [CrossRef]
- Fusaro, A.; Joannis, T.; Monne, I.; Salviato, A.; Yakubu, B.; Meseko, C.; Oladokun, T.; Fassina, S.; Capua, I.; Cattoli, G. Introduction into Nigeria of a Distinct Genotype of Avian Influenza Virus (H5N1). Emerg. Infect. Dis. 2009, 15, 445–447. [Google Scholar] [CrossRef]
- Oladokun, A.T.; Meseko, C.A.; Ighodalo, E.; John, B.; Ekong, P.S. Effect of Intervention on the Control of Highly Pathogenic Avian Influenza in Nigeria. Pan Afr. Med. J. 2012, 13, 14. [Google Scholar] [PubMed]
- Coker, T.; Meseko, C.; Odaibo, G.; Olaleye, D. Circulation of the Low Pathogenic Avian Influenza Subtype H5N2 Virus in Ducks at a Live Bird Market in Ibadan, Nigeria. Infect. Dis. poverty 2014, 3. [Google Scholar] [CrossRef] [PubMed]
- Monne, I.; Meseko, C.; Joannis, T.; Shittu, I.; Ahmed, M.; Tassoni, L.; Fusaro, A.; Cattoli, G. Highly Pathogenic Avian Influenza A(H5N1) Virus in Poultry, Nigeria, 2015. Emerg. Infect. Dis. 2015, 21, 1275–1277. [Google Scholar] [CrossRef]
- Tassoni, L.; Fusaro, A.; Milani, A.; Lemey, P.; Awuni, J.A.; Sedor, V.B.; Dogbey, O.; Commey, A.N.O.; Meseko, C.; Joannis, T.; et al. Genetically Different Highly Pathogenic Avian Influenza A(H5N1) Viruses in West Africa, 2015. Emerg. Infect. Dis. 2016, 22, 2132–2136. [Google Scholar] [CrossRef] [PubMed]
- Laleye, A.T.; Bianco, A.; Shittu, I.; Sulaiman, L.; Fusaro, A.; Inuwa, B.; Oyetunde, J.; Zecchin, B.; Bakam, J.; Pastori, A.; et al. Genetic Characterization of Highly Pathogenic Avian Influenza H5Nx Clade 2.3.4.4b Reveals Independent Introductions in Nigeria. Transbound. Emerg. Dis. 2022, 69, 423–433. [Google Scholar] [CrossRef] [PubMed]
- Shittu, I.; Bianco, A.; Gado, D.; Mkpuma, N.; Sulaiman, L.; Laleye, A.; Gobbo, F.; Bortolami, A.; Bonfante, F.; Vakuru, C.; et al. First Detection of Highly Pathogenic H5N6 Avian Influenza Virus on the African Continent. Emerg. Microbes Infect. 2020, 9, 886–888. [Google Scholar] [CrossRef]
- Sulaiman, L.; Shittu, I.; Fusaro, A.; Inuwa, B.; Zecchin, B.; Gado, D.; Schivo, A.; Bianco, A.; Laleye, A.; Gobbo, F.; et al. Live Bird Markets in Nigeria: A Potential Reservoir for H9N2 Avian Influenza Viruses. Viruses 2021, 13. [Google Scholar] [CrossRef]
- Gaidet, N.; Cattoli, G.; Hammoumi, S.; Newman, S.H.; Hagemeijer, W.; Takekawa, J.Y.; Cappelle, J.; Dodman, T.; Joannis, T.; Gil, P.; et al. Evidence of Infection by H5N2 Highly Pathogenic Avian Influenza Viruses in Healthy Wild Waterfowl. PLoS Pathog. 2008, 4. [Google Scholar] [CrossRef]
- Snoeck, C.J.; Adeyanju, A.T.; de Landtsheer, S.; Ottosson, U.; Manu, S.; Hagemeijer, W.; Mundkur, T.; Muller, C.P. Reassortant Low-Pathogenic Avian Influenza H5N2 Viruses in African Wild Birds. J. Gen. Virol. 2011, 92, 1172–1183. [Google Scholar] [CrossRef]
- Meseko, C.A.; Ehizibolo, D.O.; Vakuru, C. Migratory Waterfowls from Europe as Potential Source of Highly Pathogenic Avian Influenza Infection to Nigeria Poultry. Niger. Vet. J. 2018, 39, 1. [Google Scholar] [CrossRef]
- Spackman, E.; Senne, D.A.; Myers, T.J.; Bulaga, L.L.; Garber, L.P.; Perdue, M.L.; Lohman, K.; Daum, L.T.; Suarez, D.L. Development of a Real-Time Reverse Transcriptase PCR Assay for Type A Influenza Virus and the Avian H5 and H7 Hemagglutinin Subtypes. J. Clin. Microbiol. 2002, 40, 3256–3260. [Google Scholar] [CrossRef] [PubMed]
- Slomka, M.J.; Pavlidis, T.; Banks, J.; Shell, W.; McNally, A.; Essen, S.; Brown, I.H. Validated H5 Eurasian Real-Time Reverse Transcriptase-Polymerase Chain Reaction and Its Application in H5N1 Outbreaks in 2005-2006. Avian Dis. 2007, 51, 373–377. [Google Scholar] [CrossRef] [PubMed]
- Monne, I.; Ormelli, S.; Salviato, A.; De Battisti, C.; Bettini, F.; Salomoni, A.; Drago, A.; Zecchin, B.; Capua, I.; Cattoli, G. Development and Validation of a One-Step Real-Time PCR Assay for Simultaneous Detection of Subtype H5, H7, and H9 Avian Influenza Viruses. J. Clin. Microbiol. 2008, 46, 1769–1773. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, B.; Hoffmann, D.; Henritzi, D.; Beer, M.; Harder, T.C. Riems Influenza a Typing Array (RITA): An RT-QPCR-Based Low Density Array for Subtyping Avian and Mammalian Influenza a Viruses. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Panzarin, V.; Marciano, S.; Fortin, A.; Brian, I.; D’amico, V.; Gobbo, F.; Bonfante, F.; Palumbo, E.; Sakoda, Y.; Le, K.T.; et al. Redesign and Validation of a Real-Time RT-PCR to Improve Surveillance for Avian Influenza Viruses of the H9 Subtype. Viruses 2022, 14. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Van der Auwera, G.A.; Carneiro, M.O.; Hartl, C.; Poplin, R.; Del Angel, G.; Levy-Moonshine, A.; Jordan, T.; Shakir, K.; Roazen, D.; Thibault, J.; et al. From FastQ Data to High Confidence Variant Calls: The Genome Analysis Toolkit Best Practices Pipeline. Curr. Protoc. Bioinformatics 2013, 43, 11–10. [Google Scholar] [CrossRef]
- Wilm, A.; Aw, P.P.K.; Bertrand, D.; Yeo, G.H.T.; Ong, S.H.; Wong, C.H.; Khor, C.C.; Petric, R.; Hibberd, M.L.; Nagarajan, N. LoFreq: A Sequence-Quality Aware, Ultra-Sensitive Variant Caller for Uncovering Cell-Population Heterogeneity from High-Throughput Sequencing Datasets. Nucleic Acids Res. 2012, 40, 11189–11201. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Nguyen, L.T.; Schmidt, H.A.; Von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Hoang, D.T.; Chernomor, O.; Von Haeseler, A.; Minh, B.Q.; Vinh, L.S. UFBoot2: Improving the Ultrafast Bootstrap Approximation. Mol. Biol. Evol. 2018, 35, 518–522. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.-Y.; Wei, C.-J.; Kong, W.-P.; Wu, L.; Xu, L.; Smith, D.F.; Nabel, G.J. Immunization by Avian H5 Influenza Hemagglutinin Mutants with Altered Receptor Binding Specificity. Science 2007, 317, 825–828. [Google Scholar] [CrossRef] [PubMed]
- Suttie, A.; Deng, Y.M.; Greenhill, A.R.; Dussart, P.; Horwood, P.F.; Karlsson, E.A. Inventory of Molecular Markers Affecting Biological Characteristics of Avian Influenza A Viruses. Virus Genes 2019, 55, 739–768. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Deng, G.; Sun, H.; Song, J.; Zhang, W.; Li, H.; Wei, X.; Li, F.; Zhang, X.; Liu, J.; et al. N-Linked Glycosylation Enhances Hemagglutinin Stability in Avian H5N6 Influenza Virus to Promote Adaptation in Mammals. PNAS nexus 2022, 1. [Google Scholar] [CrossRef]
- Munier, S.; Larcher, T.; Cormier-Aline, F.; Soubieux, D.; Su, B.; Guigand, L.; Labrosse, B.; Cherel, Y.; Quéré, P.; Marc, D.; et al. A Genetically Engineered Waterfowl Influenza Virus with a Deletion in the Stalk of the Neuraminidase Has Increased Virulence for Chickens. J. Virol. 2010, 84, 940–952. [Google Scholar] [CrossRef]
- Banks, J.; Speidel, E.S.; Moore, E.; Plowright, L.; Piccirillo, A.; Capua, I.; Cordioli, P.; Fioretti, A.; Alexander, D.J. Changes in the Haemagglutinin and the Neuraminidase Genes Prior to the Emergence of Highly Pathogenic H7N1 Avian Influenza Viruses in Italy. Arch. Virol. 2001, 146, 963–973. [Google Scholar] [CrossRef]
- Croville, G.; Soubies, S.M.; Barbieri, J.; Klopp, C.; Mariette, J.; Bouchez, O.; Camus-Bouclainville, C.; Guérin, J.L. Field Monitoring of Avian Influenza Viruses: Whole-Genome Sequencing and Tracking of Neuraminidase Evolution Using 454 Pyrosequencing. J. Clin. Microbiol. 2012, 50, 2881–2887. [Google Scholar] [CrossRef]
- Li, J.; Dohna, H.Z.; Cardona, C.J.; Miller, J.; Carpenter, T.E. Emergence and Genetic Variation of Neuraminidase Stalk Deletions in Avian Influenza Viruses. PLoS One 2011, 6. [Google Scholar] [CrossRef]
- Gao, Y.; Zhang, Y.; Shinya, K.; Deng, G.; Jiang, Y.; Li, Z.; Guan, Y.; Tian, G.; Li, Y.; Shi, J.; et al. Identification of Amino Acids in HA and PB2 Critical for the Transmission of H5N1 Avian Influenza Viruses in a Mammalian Host. PLoS Pathog. 2009, 5. [Google Scholar] [CrossRef]
- Le, Q.M.; Sakai-Tagawa, Y.; Ozawa, M.; Ito, M.; Kawaoka, Y. Selection of H5N1 Influenza Virus PB2 during Replication in Humans. J. Virol. 2009, 83, 5278–5281. [Google Scholar] [CrossRef]
- Li, Z.; Chen, H.; Jiao, P.; Deng, G.; Tian, G.; Li, Y.; Hoffmann, E.; Webster, R.G.; Matsuoka, Y.; Yu, K. Molecular Basis of Replication of Duck H5N1 Influenza Viruses in a Mammalian Mouse Model. J. Virol. 2005, 79, 12058–12064. [Google Scholar] [CrossRef] [PubMed]
- Steel, J.; Lowen, A.C.; Mubareka, S.; Palese, P. Transmission of Influenza Virus in a Mammalian Host Is Increased by PB2 Amino Acids 627K or 627E/701N. PLoS Pathog. 2009, 5. [Google Scholar] [CrossRef] [PubMed]
- Taft, A.S.; Ozawa, M.; Fitch, A.; Depasse, J. V.; Halfmann, P.J.; Hill-Batorski, L.; Hatta, M.; Friedrich, T.C.; Lopes, T.J.S.; Maher, E.A.; et al. Identification of Mammalian-Adapting Mutations in the Polymerase Complex of an Avian H5N1 Influenza Virus. Nat. Commun. 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Yamayoshi, S.; Kiso, M.; Yasuhara, A.; Ito, M.; Shu, Y.; Kawaoka, Y. Enhanced Replication of Highly Pathogenic Influenza A(H7N9) Virus in Humans. Emerg. Infect. Dis. 2018, 24, 746–750. [Google Scholar] [CrossRef] [PubMed]
- Pinto, R.M.; Bakshi, S.; Lytras, S.; Zakaria, M.K.; Swingler, S.; Worrell, J.C.; Herder, V.; Varjak, M.; Cameron-Ruiz, N.; Rodriguez, M.C.; et al. Zoonotic Avian Influenza Viruses Evade Human BTN3A3 Restriction. bioRxiv, 4961. [Google Scholar] [CrossRef]
- Cheung, C.L.; Rayner, J.M.; Smith, G.J.D.; Wang, P.; Naipospos, T.S.P.; Zhang, J.; Yuen, K.Y.; Webster, R.G.; Peiris, J.S.M.; Guan, Y.; et al. Distribution of Amantadine-Resistant H5N1 Avian Influenza Variants in Asia. J. Infect. Dis. 2006, 193, 1626–1629. [Google Scholar] [CrossRef]
- Lan, Y.; Zhang, Y.; Dong, L.; Wang, D.; Huang, W.; Xin, L.; Yang, L.; Zhao, X.; Li, Z.; Wang, W.; et al. A Comprehensive Surveillance of Adamantane Resistance among Human Influenza A Virus Isolated from Mainland China between 1956 and 2009. Antivir. Ther. 2010, 15, 853–859. [Google Scholar] [CrossRef]
- Lee, J.; Young, J.S.; Jeung, H.P.; Lee, J.H.; Yun, H.B.; Song, M.S.; Oh, T.K.; Han, H.S.; Pascua, P.N.Q.; Choi, Y.K. Emergence of Amantadine-Resistant H3N2 Avian Influenza A Virus in South Korea. J. Clin. Microbiol. 2008, 46, 3788–3790. [Google Scholar] [CrossRef]
- Jiao, P.; Tian, G.; Li, Y.; Deng, G.; Jiang, Y.; Liu, C.; Liu, W.; Bu, Z.; Kawaoka, Y.; Chen, H. A Single-Amino-Acid Substitution in the NS1 Protein Changes the Pathogenicity of H5N1 Avian Influenza Viruses in Mice. J. Virol. 2008, 82, 1146–1154. [Google Scholar] [CrossRef]
- Kuo, R.-L.; Krug, R.M. Influenza a Virus Polymerase Is an Integral Component of the CPSF30-NS1A Protein Complex in Infected Cells. J. Virol. 2009, 83, 1611–1616. [Google Scholar] [CrossRef]
- Imai, H.; Shinya, K.; Takano, R.; Kiso, M.; Muramoto, Y.; Sakabe, S.; Murakami, S.; Ito, M.; Yamada, S.; thi Quynh Le, M.; et al. The HA and NS Genes of Human H5N1 Influenza A Virus Contribute to High Virulence in Ferrets. PLoS Pathog. 2010, 6. [Google Scholar] [CrossRef]
- Miotto, O.; Heiny, A.T.; Tan, T.W.; Thomas, J.T.; Brusic, V. Identification of Human-to-Human Transmissibility Factors in PB2 Proteins of Influenza A by Large-Scale Mutual Information Analysis. BMC Bioinformatics. [CrossRef]
- Finkelstein, D.B.; Mukatira, S.; Mehta, P.K.; Obenauer, J.C.; Su, X.; Webster, R.G.; Naeve, C.W. Persistent Host Markers in Pandemic and H5N1 Influenza Viruses. J. Virol. 2007, 81, 10292–10299. [Google Scholar] [CrossRef] [PubMed]
- Allen, J.E.; Gardner, S.N.; Vitalis, E.A.; Slezak, T.R. Conserved Amino Acid Markers from Past Influenza Pandemic Strains. BMC Microbiol. 2009, 9. [Google Scholar] [CrossRef] [PubMed]
- Heikkinen, L.S.; Kazlauskas, A.; Melén, K.; Wagner, R.; Ziegler, T.; Julkunen, I.; Saksela, K. Avian and 1918 Spanish Influenza a Virus NS1 Proteins Bind to Crk/CrkL Src Homology 3 Domains to Activate Host Cell Signaling. J. Biol. Chem. 2008, 283, 5719–5727. [Google Scholar] [CrossRef] [PubMed]
- Hassan, K.E.; Saad, N.; Abozeid, H.H.; Shany, S.; El-Kady, M.F.; Arafa, A.; EL-Sawah, A.A.A.; Pfaff, F.; Hafez, H.M.; Beer, M.; et al. Genotyping and Reassortment Analysis of Highly Pathogenic Avian Influenza Viruses H5N8 and H5N2 from Egypt Reveals Successive Annual Replacement of Genotypes. Infect. Genet. Evol. 2020, 84. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Xu, L.; Liu, T.; Xie, S.; Li, K.; Li, X.; Zhang, M.; Wu, Y.; Wang, X.; Wang, J.; et al. Novel Reassortant Avian Influenza A(H5N6) Virus, China, 2021. Emerg. Infect. Dis. 2022, 28, 1701–1707. [Google Scholar] [CrossRef]
- Ouoba, L.B.; Habibata-Zerbo, L.; Zecchin, B.; Barbierato, G.; Hamidou-Ouandaogo, S.; Palumbo, E.; Giussani, E.; Bortolami, A.; Niang, M.; Traore-Kam, A.; et al. Emergence of a Reassortant 2.3.4.4b Highly Pathogenic H5N1 Avian Influenza Virus Containing H9N2 PA Gene in Burkina Faso, West Africa, in 2021. Viruses 2022, 14. [Google Scholar] [CrossRef]
- Yang, F.; Zhang, X.; Liu, F.; Yao, H.; Wu, N.; Wu, H. Rapid Emergence of a PB2 D701N Substitution during Adaptation of an H9N2 Avian Influenza Virus in Mice. Arch. Virol. 2022, 167, 2299–2303. [Google Scholar] [CrossRef]
- Peacock, T.P.; James, J.; Sealy, J.E.; Iqbal, M. A Global Perspective on H9N2 Avian Influenza Virus. Viruses 2019, 11. [Google Scholar] [CrossRef]
Figure 1.
Map of Nigeria showing location and distribution of HPAI in Nigeria, 2021-2022.
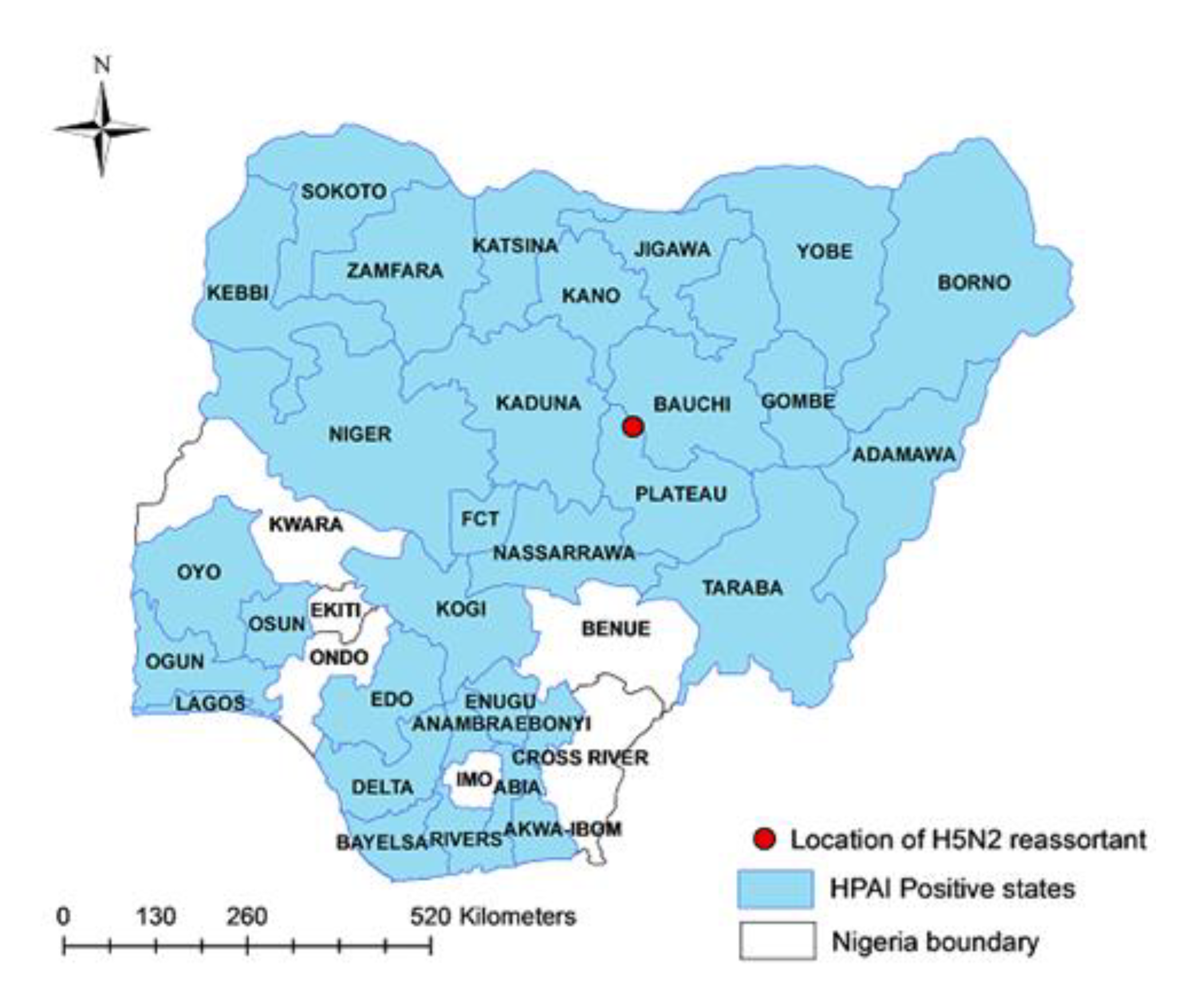
Figure 2.
Haemorrhages of organs and tissues caused by HPAI in Nigeria, 2021-2022.
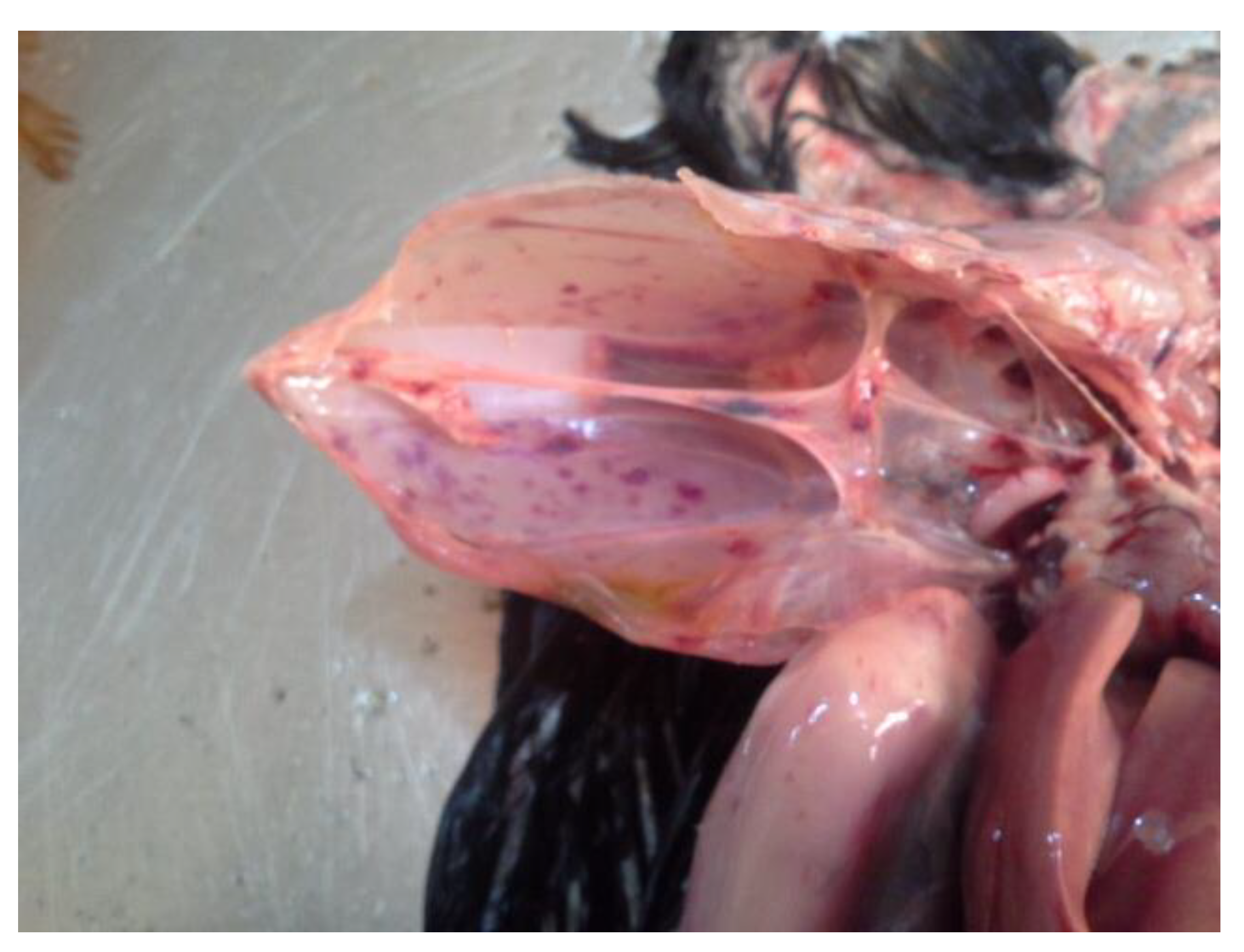
Figure 3.
Phylogenetic tree of the nucleotide sequence of the HA gene obtained with the Maximum Likelihood method by using IQ-TREE v.1.6.12. H5Nx Nigerian viruses analysed in this study are in bold. The orange box (Group-1) includes the 2021-2022 H5N1 (black) and the reassortant H5N2 (red) Nigerian viruses. The light blue box (Group-2) includes the H5N8 Nigerian viruses. The Ultra-fast bootstrap values higher than 80% are indicated next to the nodes. The ovals next to the tree show the genetic composition of the three different genotypes (H5N1, H5N8 and H5N2) characterized in Nigeria between 2021 and 2022. Horizontal bars within each oval represent the 8 gene segments, colored according to their origin (orange: H5N1; light blue: H5N8; red: H9N2).
Figure 3.
Phylogenetic tree of the nucleotide sequence of the HA gene obtained with the Maximum Likelihood method by using IQ-TREE v.1.6.12. H5Nx Nigerian viruses analysed in this study are in bold. The orange box (Group-1) includes the 2021-2022 H5N1 (black) and the reassortant H5N2 (red) Nigerian viruses. The light blue box (Group-2) includes the H5N8 Nigerian viruses. The Ultra-fast bootstrap values higher than 80% are indicated next to the nodes. The ovals next to the tree show the genetic composition of the three different genotypes (H5N1, H5N8 and H5N2) characterized in Nigeria between 2021 and 2022. Horizontal bars within each oval represent the 8 gene segments, colored according to their origin (orange: H5N1; light blue: H5N8; red: H9N2).
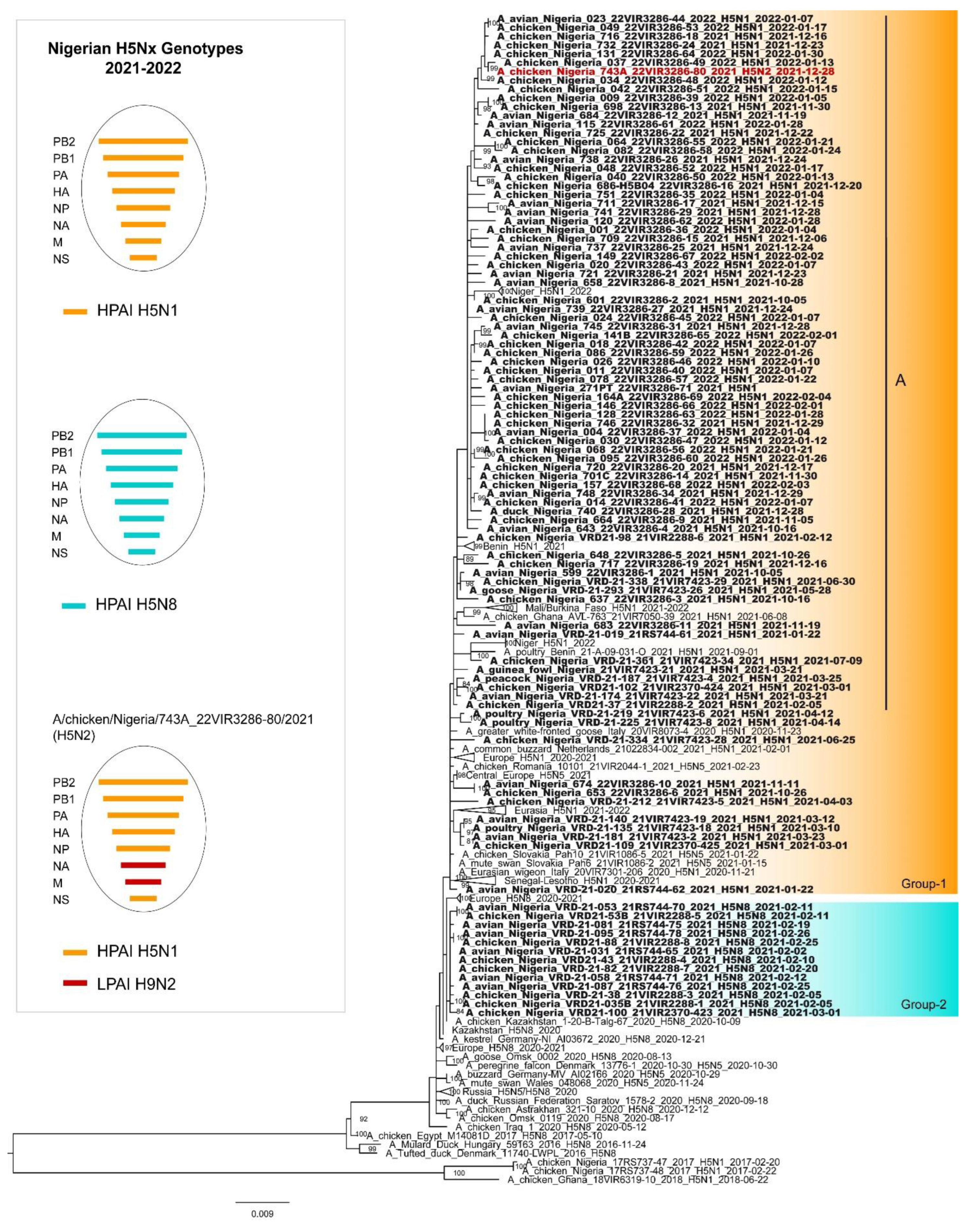

Table 1.
Amino acid markers associated with specific phenotypic effects found in the genome of HPAI H5Nx viruses from Nigeria sequenced in the current study.
Table 1.
Amino acid markers associated with specific phenotypic effects found in the genome of HPAI H5Nx viruses from Nigeria sequenced in the current study.
| Protein | Genetic marker | Phenotypic effect | Virus name | References |
|---|---|---|---|---|
| PB2 | T105V | May be associate to human adaptation (statistical analysis) | A/chicken/Nigeria/648_22VIR3286-5/2021 | [52] |
| D701N | Mammalian adaptive marker | A/chicken/Nigeria/743A_22VIR3286-80/2021 (H5N2) | [39,40,41,42,43] | |
| K482R | Increase polymerase activity in mammalian cell line | A/avian/Nigeria/271PT_22VIR3286-71/2021 | [44] | |
| PA | D55N | Host specificity marker through statistical methods (D in avian, N in human) | A/guinea_fowl/Nigeria/VRD-21-169_21VIR7423-21/2021 | [53] |
| S409N | Host specificity marker through statistical methods (S in avian, N in human) | A/chicken/Nigeria/040_22VIR3286-50/2022 | [53] | |
| HA | PLREKRRKR/GLF | HPAI cleavage site | All viruses | Not available |
| S137A | Increased α2-6 sialic acid virus binding | All viruses except for A/avian/Nigeria/VRD-21-019_21RS744-61/2021 | [32] | |
| S158N | Increased α2-6 sialic acid virus binding | All viruses | [33] | |
| T160A | Increased α2-6 sialic acid virus binding | In all the H5N8 strains, and in eighteen H5N1 viruses | [33] | |
| NA | 44-65 del in the stalk region | Enhanced virulence, marker of viral adaptation from wild bird to poultry | In 73 out of 82 H5N1 viruses | [35,36,37,38] |
| NP | Y52H | Confer resistance to BTN3A3 (butyrophilin subfamily 3 member A3) inhibitor | In 82 out of 83 H5N1 viruses | [45] |
| M2 | V27I | Associated to amantadine resistance | A/avian/Nigeria/741_22VIR3286-29/2021 A/chicken/Nigeria/751_22VIR3286-35/2022 | [48] |
| S31N | Associated to amantadine resistance | A/chicken/Nigeria/743A_22VIR3286-80/2021 (H5N2) | [46,47] | |
| NS1 | P42S | Increase virulence in mice and could be critical to antagonize the interferon induction | All viruses | [49] |
| P87S | host-specificity marker through statistical methods (S in human, P in avian) | All virus with exception of A/chicken/Nigeria/725_22VIR3286-22/2021 A/avian/Nigeria/115_22VIR3286-61/2022 | [54] | |
| L103F | Increase virulence in mice | All viruses | [50] | |
| I106M | Increase virulence in mice | All viruses | [50] | |
| K217E | Decrease replicative or pathogenic potential of the virus | A/chicken/Nigeria/157_22VIR3286-68/2022 | [55] | |
| N205S | Decreased antiviral response in host | All H5N1 and the H5N2 viruses | [51] | |
| NS2 | T48A | Decreased antiviral response in host | All H5N1 and the H5N2 viruses | [51] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



