
Submitted:
15 May 2023
Posted:
16 May 2023
You are already at the latest version
Abstract
The Virus HIV-1 infection still represents a serious disease even if actually it is transformed in chronic pathology. Considering the crucial role of the enzyme Protease in life cycle of HIV many efforts have been made in the research of new organic compounds showing inhibitory activity. After development of several series of non peptidic inhibitors we report here the synthesis of novel simple HIV-Protease inhibitors containing heteroaryl carboxamides and their antiviral activity in vitro and in HEK293 cells. Benzofuryl- benzothienyl- and indolyl rings as well as aryl sulfonamides with different electronic properties have been introduced by efficient synthetic procedures. All compounds showed inhibitory activity similar to the commercial drug Darunavir, effective against both wild-type HIV-1 protease and that containing the V32I or V82A mutations. ADME properties were also evaluated, showing the potential of such compounds to be developed as drugs.
Keywords:
mutate HIV protease inhibitors
; carboxamides
; heteroarenes
; mammalian cells essay
; ADME
1. Introduction
In the last two decades great efforts have been dedicated to the treatment of human immunodeficiency virus (HIV-1) infection, transforming it from a fatal disease into a manageable chronic pathology. Nevertheless, over the last three years, the multiple and overlapping world crises have had a devastating impact on people living with and affected by HIV, and they have knocked back the global response to the AIDS pandemic [1].
HIV-1 Protease (HIV-1 PR) is essential to the life cycle of the virus and many inhibitors have been developed and introduced into combination-therapy regimens. Taking advantage of the detailed structure of HIV protease and its substrate, many commercially available drugs have been based on the tetrahedral transition state mimetic concept, in which a not hydrolysable hydroxyethylamine moiety has been used as the central core of the molecule [2,3].
Despite the commercial Protease Inhibitors (PIs) have proved their role in the major advances in HIV/AIDS therapies, there are still many drawbacks, mainly in terms of toxicity and systemic complications involving several organs [4]. Moreover, the emergence of multiple drug resistance mutations remains a challenge [5,6], reducing long-term viral inhibition.
During our investigation on new peptidomimetics and non peptidic inhibitors, we found beneficial effect of heteroaryl rings. In this respect we successfully modified first generation PIs nelfinavir and saquinavir preparing new thienyl analogues which showed the same or even higher activity against both wild type and mutant HIV protease [7].
Recently, the concept of targeting the protein backbone in structure-based drug design prompted the preparation of new non-peptidic templates, which can maximize interactions in the HIV-protease active site, particularly with the enzyme backbone atoms. This approach has led to many potent inhibitors such as FDA-approved Darunavir and several related compounds [8,9,10,11,12,13].
Following this concept, we developed a systematic study on simple substituted stereodefined isopropanolamine derivatives, in which the high effect of the moiety between the heteroaryl group and the core, as well as the type of heteroaryl group, were evident [14,15,16]. Easily synthesized benzothienyl-, benzofuryl- and indolyl derivatives bearing either a carboxyamide or a carbamoyl spacer in general showed high in vitro activity against native protease. They also confirmed their inhibition activity in mammalian cells, showing a general low citotoxicity and great metabolic stability, thus demonstrating their promising potential [17,18,19,20].
In particular, regarding carboxyamide derivatives we found critical the presence of benzyl fragment in the core, reaching an IC50 value of 1 nM in vitro for compound A (Figure 1)[17]. Hence, with the aim of checking the effect of different arylsulfonamide groups (in terms of electronic properties) and heteroaryl rings as P2 ligand, we run a systematic study on the synthesis and on the inhibition activity of new simple derivatives of structure B.
2. Materials and Methods
2.1. Chemistry
Preparative chromatography was carried out on Merck silica gel (0.063–0.200 mm particle size) by progressive elution with suitable solvent mixtures. 1H and 13C NMR spectra were carried out in CDCl3 solutions on a VARIAN INOVA 500 MHz or Bruker 400 MHz and referenced to CDCl3. Mass spectra were obtained with a Hewlett-Packard 5971 mass-selective detector on a Hewlett–Packard 5890 gas chromatograph ((OV-1 capillary column between 70 and 250 °C (20 °C min-1)). The optical rotation was evaluated by using a polarimeter JASCO Mod Dip-370. CH2Cl2 was dried by distillation over anhydrous CaCl2 in an inert atmosphere. Dry THF and DMF were commercially available,
All 1H and 13C NMR spectra for the following compounds were consistent to literature data: (1R,2S)-(1-Benzyl-2-hydroxy-3-iso-butylamino-propyl)-carbamic acid tert-butyl ester (2) [17], (1S,2R)-[1-Benzyl-2-hydroxy-3-iso-butyl-(4-methoxy-benzenesulfonyl)amino-propyl]-carbamic acid tert-butyl ester (3a) [18], (1S,2R)-[1-Benzyl-2-hydroxy-3-iso-butyl-(4-nitro-benzenesulfonyl)amino-propyl]-carbamic acid tert-butyl ester (3b) [21], (2R,3S)-N-(3-Amino-2-hydroxy-4-phenyl-butyl)-N-isobutyl-4-methoxy-benzenesulfonamide (4a) [18], (2R,3S)-N-(3-Amino-2-hydroxy-4-phenyl-butyl)-N-isobutyl-4-nitro-benzenesulfonamide (4b) [20].
General procedure for carboxyamides 5a-c and 6a-c
To a solution of 5-heteroaryl acid (0.13 mmol), EDCI (0.20 mmol), HOBt (0.20 mmol) in anhydrous CH2Cl2, a solution of amine 4a-b (0.13 mmol) and diisopropylethylamine (0.78 mmol) in anhydrous CH2Cl2 was added at 0°C under argon atmosphere and it was allowed to stir for 16h at room temperature. The reaction mixture was quenched with water and extracted with CH2Cl2. The organic layers were dried on Na2SO4, filtered and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (CH2Cl2/AcOEt 9/1) to furnish 5a-c and 6 a-c .
N-((2S,3R)-3-hydroxy-4-(N-isobutyl-4-methoxyphenylsulfonamido)-1-phenylbutan-2-yl)benzo[b]thiophene -5-carboxamide (5a). Following the general procedure compound 5a was obtained as a white solid, in 42% yield. [α] D20=+3.7 (c 0.2, CHCl3) 1H NMR (400 MHz, CDCl3): δ 8.09 (s, 1H), 7.89 (d, J = 8.4 Hz, 1H), 7.68 (d, J = 8.8 Hz, 2H), 7.55 (m, 2H), 7.30 (m, 6H), 6.93 (d, J = 8.8 Hz, 2H), 6.55 (d, J = 8.4 Hz, 1H), 4.44 (m, 2H), 4.03 (m, 1H), 3.85 (s, 3H), 3.17 (m, 4H), 2.89 (m, 2H), 1.88 (m, 1H), 0.89 (d, J = 6.2 Hz, 6H). 13C NMR (100 MHz, CDCl3): δ 168.3, 163.0, 142.9, 139.4, 137.8, 130.3, 129.8, 129.4, 128.7, 128.0 126.7, 124.2, 122.7, 122.6, 122.3, 114.3, 72.9, 58.9, 55.6, 54.7, 53.6, 35.0, 27.2, 20.1, 20.0.
N-((2S,3R)-3-hydroxy-4-(N-isobutyl-4-methoxyphenylsulfonamido)-1-phenylbutan-2-yl)benzo[b]furan-5-carboxamide (5b). Following the general procedure, the compound 5b was obtained as a white solid, in 44% yield.[α]D20=+9.6 (c 0.5, CHCl3). 1H NMR (400 MHz, CDCl3): δ 7.89 (s, 1H), 7.67 (m, 3H), 7.56 (d, J = 8.6 Hz, 1H), 7.47 (d, J = 8.6Hz, 1H), 7.28 (m, 4H), 7.22 (m, 1H), 6.91 (d, J = 8.8 Hz, 2H), 6.78 (brs, 1H), 6.58 (d, J = 8.0 Hz, 1H), 4.48 (brs, 1H), 4.42 (m, 1H), 4.05 (m, 1H), 3.84 (s, 3H), 3.16 (m, 5H), 2.88 (m, 2H), 1.88 (m, 1H), 0.87 (d, J = 6.5 Hz, 6H). 13C NMR (100 MHz, CDCl3): δ 168.3, 163.0, 156.6, 146.3, 137.9, 129.8, 129.41, 129.40, 129.1, 128.6, 127.5, 126.6, 123.3, 120.7, 114.2, 111.4, 106.9, 72.9, 58.8, 55.6, 54.7, 53.5, 35.0, 27.2, 20.1, 20.0
N-((2S,3R)-3-hydroxy-4-(N-iso-butyl-4-methoxyphenylsulfonamido)-1-phenylbutan-2-yl)indol-5-carboxamide (5c). Following the general procedure, the compound 5c was obtained as a white solid, in 45% yield. [α] D20=+22.2° (c 1.1, CHCl3). 1H NMR (400 MHz, CDCl3): δ 8.94 (d, J = 13.0 Hz, 1H), 7.94 (s, 1H), 7.66 (d, J = 8.7 Hz, 2H), 7.43 (d, J = 8.6 Hz, 1H), 7.28 (m, 7H), 6.88 (d, J = 8.7 Hz, 2H), 6.55 (brs, 2H), 4.69 (brs, 1H), 4.40 (m, 1H), 4.04 (m, 1H), 3.81 (s, 3H), 3.31 (dd, J = 15.2 Hz, J = 4.8 Hz, 1H), 3.15 (m, 2H), 3.05 (dd, J = 15.2 Hz, J = 7.6 Hz, 1H), 2.92 (dd, J = 13.3 Hz, J = 7.2 Hz, 1H), 2.82 (dd, J = 13.3 Hz, J = 7.2 Hz, 1H), 1.88 (m, 1H), 0.85 (m, 6H). 13C NMR (100 MHz, CDCl3): δ 169.5, 162.9, 138.0, 137.7, 129.7, 129.5, 129.4, 128.6, 127.5, 126.6, 126.0, 125.4, 120.7, 120.3, 114.3, 111.2, 103.4, 73.0, 58.8, 55.6, 54.9, 53.5, 35.1, 27.2, 20.1, 20.0.
N-((2S,3R)-3-hydroxy-4-(N-iso-butyl-4-nitrophenylsulfonamido)-1-phenylbutan-2-yl)benzo[b]thiophene-5-carboxamide (6a). Following the general procedure, the compound 6a was obtained as a white solid, in 54 % yield. [α] D20= +6.8 (c 0.3, CHCl3). 1H NMR (400 MHz, CDCl3): δ 8.30 (d, J = 8.6 Hz, 2H), 8.09 (s, 1H), 8.03 (brs, 1H), 7.92 (d, J = 8.6 Hz, 2H), 7.83 (d, J = 8.8 Hz, 1H), 7.53 (m, 2H), 7.36 (m, 1H), 7.30 (m, 5H), 4.35 (m, 1H), 4.02 (m, 1H), 3.35 (brd, J = 15.2 Hz, 1H), 3.19 (m, 1H), 3.13 (brd, J = 7.1 Hz, 2H), 3.00 (m, 2H), 1.91 (m, 1H), 0.86 (d, J = 5.7 Hz, 3H), 0.85 (d, J = 5.7 Hz, 3H). 13C NMR (100 MHz, CDCl3): δ 169.7, 150.0, 145.1, 137.7, 137.21, 135.1, 129.3, 128.8 (3C), 128.4, 128.3, 126.8, 125.8, 124.3, 120.9, 120.4, 71.8, 55.1, 53.4, 51.3, 35.6, 31.4, 30.2, 29.7
N-((2S,3R)-3-hydroxy-4-(N-iso-butyl-4-nitrophenylsulfonamido)-1-phenylbutan-2-yl)benzofuran-5-carboxamide (6b). Following the general procedure, the compound 6b was obtained as a white solid, in 56% yield. [α]D20= -3.0° (c 0.2, CHCl3). 1H NMR (400 MHz, CDCl3): δ 8.30 (d, J =8.8 Hz, 2H), 7.92 (d, J =8.8 Hz, 2H), 7.87 (s, 1H), 7.69 (s,1H), 7.53 (A part of AB system, JAB =8.8 Hz, 1H), 7.49 (B part of AB system, JAB = 8.8 Hz, 1H), 7.32 (m, 4H), 6.81 (s, 1H), 6.41 (d, J = 7.5 Hz, 1H), 4.45 (brs, 1H), 4.34 (brs, 1H), 4.02 (brs, 1H), 3.35 (brd, J = 15.2 Hz, 1H), 3.20 (dd, J = 15.2Hz, J = 8.4 Hz, 1H), 3.13 (d, J = 6.8 Hz, 2H), 3.03 (dd, J = 13.7, J = 7.2 Hz, 1H), 2.95 (dd, J = 13.7, J = 7.2 Hz, 1H), 1.91 (m, 1H), 0.87 (d, J = 6.8 Hz, 3H), 0.85 (d, J = 6.8 Hz, 3H). 13C NMR (100 MHz, CDCl3): δ 168.7, 156.7, 150.0, 146.5, 144.7, 137.5, 129.3, 129.2, 128.8, 128.5, 127.6, 126.9, 124.3, 123.2, 120.7, 111.5, 106.9, 72.4, 57.7, 55.3, 52.5, 35.2, 26.9, 19.9.
N-((2S,3R)-3-hydroxy-4-(N-iso-butyl-4-nitrophenylsulfonamido)-1-phenylbutan-2-yl)indol-5-carboxamide (6c). Following the general procedure, the compound 6c was obtained as a white solid, in 83% yield.[α]D20= + 26.7 (c 1.2, CHCl3). 1H NMR (400 MHz, CDCl3): δ 8.66 (brs, 1H), 8.22 (d, J =8.8 Hz, 2H), 7.89 (s, 1H), 7.88 (d, J =8.8 Hz, 2H), 7..44 (d , J = 8.6 Hz, 1H), 7.34 (d, J = 8.6 Hz, 1H) 7.31 (m, 4H), 7.26 (m, 2H), 6.58 (brs, 1H), 6.43 (d, J = 7.4 Hz, 1H), 4.35 (m, 1H), 4.00 (m, 1H), 3.39 (dd, J = 15.1 Hz, J = 4.0 Hz, 1H), 3.19 (dd, J = 15.1 Hz, J = 8.4 Hz, 1H), 3.13 (d, J = 7.1 Hz, 2H), 3.07 (dd, J = 13.6 Hz, J = 8.0 Hz, 1H), 2.92 (dd, J = 13.6 Hz, J = 7.2 Hz, 1H), 1.91 (m, 1H), 0.87 (d, J = 6.8 Hz, 3H), 0.84 (d, J = 6.8 Hz, 3H). 13C NMR (100 MHz, CDCl3): δ 169.7, 149.8, 144.7, 137.7, 137.5, 129.3, 128.8, 128.5, 127.5, 126.9, 125.9, 125.2, 124.2, 120.8, 120.3, 111.2, 103.7, 72.4, 57.6, 55.4, 52.3, 35.5, 26.9, 19.9.
2.2. Biology
2.2.1. Materials
Dulbecco’s Modification of Eagle’s Medium (DMEM) was purchased from Corning. Dimethyl sulfoxide (DMSO), Trypsin–EDTA solution and Darunavir were purchased from Sigma Aldrich-Merck. Dulbecco’s Phosphate Buffered Saline (DPBS), L–Glutamine, Penicillin-Streptomycin solution and Fetal Bovine Serum were obtained from EuroClone. Lipofectamine 3000 was purchased from Thermofisher.
2.2.2. Cell culture and drug treatment
Human embryonic kidney (HEK293) cells were cultured in DMEM supplemented with 10% FBS, 2 mM L-glutamine, 100 U/ml penicillin and 100 µg/ml streptomycin at 37 °C in a humidified incubator with 5% CO2. HIV-protease inhibitors were solubilized in DMSO as a 5 mM stock solution and then diluted in complete DMEM to 10 µM as working concentration. Cells treated only with 0.2% DMSO (vehicle) were used as control.
2.2.3. Cell transfection and FACS analysis
HEK293 cells were seeded in 24-well plates at the density of 2.5 x 105 cells/well. After 24 hours, cells were transfected with pcDNA3/GFP-PR plasmid (gift from Nico Dantuma, Addgene plasmid # 20253) or with mutant plasmids carrying V32I or V82A mutation, generated as previously described [18], using Lipofectamine 3000 according to the manufacturer’s instructions.
Where indicated, immediately after transfection, cells were treated with HIV-1 protease inhibitors at 10 µM. Cells treated with Darunavir (DRV) were used as positive control. GFP fluorescence was quantified in HEK293 cells harvested 24 hours after trasfection and resuspended in DPBS, using a BD FACS Canto II flow cytometer (Ex/Em: 480/510 nm) [17].
2.2.4. Statistical analysis
Data were presented as means ± Standard Error (SE) of three independent experiments, each performed in triplicate and were analyzed by GraphPad Prism software (version 8, GraphPad Software, San Diego, California, USA), using one-way analysis of variance (ANOVA) followed by Dunnett's post hoc test (p-values <0.05 were considered as statistically significant).
3. Results
3.1. Chemistry
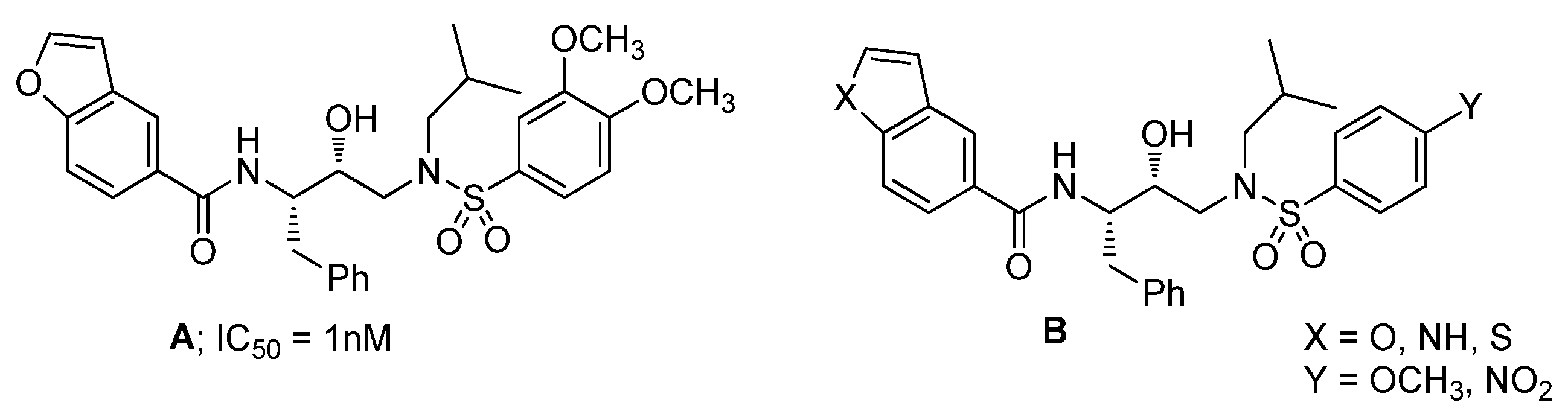
The synthetic approach for the preparation of compounds of general structure B is well established [17] and uses a four step reactions sequence (Scheme 1).
In particular, if a benzyl group is present in the central core (Figure 1), synthesis started from commercially available homochiral N-Boc protected amino epoxide 1 (Scheme 1). The epoxide was firstly opened with iso-butyl amine to afford the monoprotected diaminoalcohol 2, 4-OMe- and 4-NO2-phenylsulfonyl moieties were alternatively introduced on secondary amine, affording 3a and 3b, respectively. N-Boc group was then efficiently displaced by TFA and the crude ammonium trifluoroacetate derivatives were treated with Et3N, affording the free amines 4a-b [18,20].
From these common intermediates, we were easily able to achieve two class of compounds: 5a-c and 6a-c, in which heteroaryl group is spaced from core by carboxyamide functionality. This was inserted reacting amines 4a-b with the suitable 5-heteroarylcarboxylic acid, previously activated with 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC) and hydroxybenzotriazole (HOBt).
3.2. Biology
All these new compounds were tested as HIV Protease inhibitors firstly in vitro against the wild type enzyme and the results are reported in Table 1. All the tested compounds were powerful inhibitors of the native enzyme in vitro. The IC50 of three derivatives, namely 5a, 5b and 6a, could not be measured under our experimental conditions, as the enzyme was fully inhibited even at 0.1 nM concentration of the inhibitors. However, also 5c, 6b and 6c were extremely active, with IC50 ranging from 0.6 to 95 nM.
The ability of these compounds to inhibit HIV-protease in HEK293 cells was also evaluated, using the assay developed by Lindsten et al. [22], as described in our previous work [17,18].
Briefly, HEK293 cells transiently expressing a nontoxic HIV-protease precursor fused to GFP protein were treated with 10 µM of each inhibitor and GFP fluorescence was quantified by FACS.
As shown in Figure 2, cells treated without any inhibitor showed a low fluorescence, because the recombinant protein is toxic by autocatalytic cleavage; conversely, in presence of proper inhibitors, cells were able to express intact chimera and GFP fluorescence accordingly increased. The tested compounds (5a, 5b, 5c, 6a, 6b, 6c) were able to inhibit wild type HIV-1 protease (Figure 2A) similarly to Darunavir (no significant differences were detected), encouraging to test their inhibitory capacity also towards HIV-protease carrying mutation V32I or V82A.
All compounds inhibited the viral protease carrying V32I mutation, although less efficiently than commercial inhibitor used as positive control, as demonstrated by significant differences revealed between our molecules and Darunavir (Figure 2B).
As shown in Figure 2C, cells transfected with mutant plasmid carrying V82A mutation and treated with 6a showed poor fluorescence, comparable to fluorescence measured in control cells, suggesting that the V82A mutation compromised 6a inhibitory activity. Compounds 5a and 6b were able to inhibit mutant protease, but the levels of fluorescence detected were lower than those measured in presence of Darunavir; conversely, significant accumulation of GFP reporter was detected in cells treated with 5b, 5c and 6c (no significant differences were revealed respect to Darunavir), indicating that V82A mutation do not affect in any way their activity.
4. Discussion
This paper is the last of a long series, as our work in the design, synthesis and optimization of inhibitors bearing heterocyclic systems at P1/P2 positions started in 2012 and has led to over 30 selected molecules tested on the wt HIVpro [14,15,17,18,19,23,24,25,26].
During our research, we started from initial IC50 values of ten millimolar obtained for a few molecules among large pools of inactive compounds and arrived at pools containing only active molecules with sub-nanomolar affinities.
In our previous papers, we have carried out extensive molecular modelling to explain the observed structure-activity relationships and we have discussed in detail the modelled interactions involving indole and benzofuran side chains, and the eventual entropic effects connected to less counterproductive desolvation in benzothiophene. This analysis has been helpful to drive our design to more focused structures, and in the first works we have been able to correct the most unfavorable features of the first sets of inhibitors (such as the length of the chain connecting the interacting groups as in the case of carbamates) [27].
The most powerful inhibitors found in our previous works are reported for reference in Figure 3.
With the reported molecules in this paper, and those reported in the previous ones we reached a limit in which most of the molecules show IC50 values that are under the limit of measure of our experimental systems (< 0.6 nM, as we work at 1.2 nM enzyme). In this condition, molecular modelling is no longer useful to obtain further insights in the binding mode of the inhibitors, as we cannot compare calculated energies or scores with experimental values.
Thus, rather than report modeling explanations for the results found in this paper, we attempted to carry out an overall analysis of the results of the whole of our work.
We have selected a set of the 27 best inhibitors from this and our previous works, with affinities ranging from 15 μM to < 0.6 nM. (Figure S1) and we have calculated the values of commonly used molecular descriptors for all the compounds, including molar refractivity, total polar surface area, logP, logS (water solubility), log Kp (skin permeability) (see Table S2) [28] .
We have then carried out a covariance analysis of our experimental log (IC50) and the descriptors, and we have found that activity is slightly (but suggestively) correlated directly with logP (0.758) and inversely with molar refractivity and logS (-0.771, -0.757). This suggests that hydrophobic interactions are more important than polar ones to increase the affinity for the catalytic site.
Such outcome is not unexpected due to the nature of the HIVpr site, however it seems to confirm that we have driven hydrophobic interactions to a limit.
We have also carried out an evaluation of the ADME properties in comparison with the predicted properties of Darunavir on the SwissADME facility: the results are reported in Table 2S, where compounds performing better or equally than Darunavir in several rule systems are highlighted in green, while worse compounds are highlighted in red. 12 out 27 compounds (including 5b and 5c described in this work) perform better or equally than Darunavir in all the ADME prediction models. The greater inhibitory activity of 5b and 5c molecules, respect to other molecules tested, was also highlighted in the assays conducted in HEK293 cells. The efficacy of these compounds was comparable to that of darunavir both towards HIV-1 protease wild type and towards V82A mutated variant. Mutation V32I, instead, seems to weakly compromise their activity.
5. Conclusions
In conclusion, in this work we have reported the synthesis of a novel series of carboxamidic compounds with a high inhibitory activity against both the native HIV-1 protease and a mutant one; the inhibitory activity has been measured both in vitro and in mammalian cells, by using methodologies just reported in our previous papers. The obtained results can open new perspectives in the research for new inhibitors to overcome the problem of drug resistance. Furthermore, the ADME evaluations show that not only the novel inhibitors reported in this paper, but all the compounds in the Table 1 are competitive with Darunavir structure.
Work is in progress in the attempt to understand the structure-activity relationships for a develop of the research.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Figure S1: Structures of the inhibitors used in the analysis of ADME properties; Table S1: Collection of molecular descriptors for the set of inhibitors; Figure S2: ADME predictions; Copies of 1H and 13C NMR spectra of compounds 5a-c and 6a-c recorded in CDCl3 with a Varian 400 MZ instrument.
Author Contributions
“Conceptualization, M.F.A., P.L., M.F. and L.C..; methodology, R.M., I.N., A.S. and R.D.; validation, F.B. (units), L.C., P.L. and M.F.A; investigation, F.B. (units); resources, F.B. (unibas); writing—original draft preparation, F.B. (units); writing—review and editing, L.C., P.L. and R.M.; supervision, M.F., M.F.A., P.L. and L.C. All authors have read and agreed to the published version of the manuscript.”
Funding
“This research was funded by INBIOMED PROJECT (National Operational Plan (PON) of the Italian Ministry of Education, University and Research (MIUR), ARS01_01081).
Data Availability Statement
The data presented in this study are available within this article.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Global Report: UNAIDS report on the global AIDS epidemic 2022. WHO Library Cataloguing-in-Publication Data, Joint United Nations Programme on HIV/AIDS (UNAIDS), 2022.
- World Health Organization. Consolidated guidelines on the use of antiretroviral drugs for treating and preventing HIV infection: recommendations for a public health approach. 2nd ed. Geneva: World Health Organization; 2016.
- Weber, I. T.; Wang, Y-F; Harrison, R. W. HIV Protease: Historical Perspective and Current Research Viruses, 2021, 13, 839. [CrossRef]
- Lv, Z.; Chu, Y.; Wang, Y. HIV protease inhibitors: a review of molecular selectivity and toxicity Research and Palliative Care, 2015, 7, 95-104. [CrossRef]
- Wong-Sam, A.; Wang, Y-F.; Zhang, Y.; Ghosh, A. K.; Harrison, R. W.; Weber, I. T. Drug Resistance Mutation L76V Alters Nonpolar Interactions at the Flap-Core Interface of HIV-1 Protease ACS Omega, 2018, 3, 121132-121140. [CrossRef]
- Wang, B.; He, Y.; Wen, X.; Xi, Z. Prediction and molecular field view of drug resistance in HIV-1 protease mutants, Nature Portfolio Scientific Reports, 2022, 12, 2913. [CrossRef]
- Bonini, C.; Chiummiento, L.; De Bonis, M.; Di Blasio, N.; Funicello, M.; Lupattelli, P.; Pandolfo, R.; Tramutola, F.; Berti, F. Synthesis of New Thienyl Ring Containing HIV-1 Protease Inhibitors: Promising Preliminary Pharmacological Evaluation against Recombinant HIV-1 Proteases. J. Med. Chem. 2010, 53, 1451-1457. [CrossRef]
- Ghosh, A. K.; Shahabi, D.; Kipfmiller, M.; Ghosh, Ajay K.; Johnson, M.; Wang, Y.-F.; Agniswamy, J.; Amano, M.; Weber, I. T.; Mitsuya, H., Evaluation of darunavir-derived HIV-1 protease inhibitors incorporating P2’ amide-derivatives: Synthesis, biological evaluatio and structural studies. Bioorg. Med. Chem. Lett. 2023, 83, 129168. [CrossRef]
- Ghosh, A. K.; Weber, I. T.; Mitsuya, H. Beyond darunavir: recent development of next generation HIV-1 protease inhibitors to combat drug resistance Chem. Commun. 2022, 58, 11762-11782.
- Ghosh, A. K.; Fyvie, W. S.; Brindisi, M.; Steffey, M.; Agniswami, J.; Wang, Y.-F.; Aoki, M.; Amano, M.; Weber, I. T.; Mitsuya, H. Design, Synthesis, Biological Evaluation, and X-ray Studies of HIV-1 Protease Inhibitors with Modified P2’ Ligands of Darunavir ChemMedChem 2017, 12, 1942-1952. [CrossRef]
- Zhang, Y.; Chang, Y.-C. E.; Lousi, J. M.; Wang, Y.-F.; Harrison, R. W.; Weber, I. T. Structures of Darunavir-Resistant HIV-1 Protease Mutant Reveal Atypical Binding of Darunavir to Wide Open Flaps ACS Chem. Biol. 2014, 9, 1351-1358. [CrossRef]
- Ghosh, A. K.; Anderson, D. D.; Weber, I. T.; Mitsuya, H. Enhancing Protein Backbone Binding—A Fruitful Concept for Combating Drug-Resistant HIV. Angew. Chem. Int. Ed. 2012, 51, 1778-1802. [CrossRef]
- Ghosh, A. K.; Chapsal, B. D.; Weber, I. T.; Mitsuya, H, Design of HIV Protease Inhibitors Targeting Protein Backbone: An Effective Strategy for Combating Drug Resistance. Acc. Chem. Res. 2008, 41, 78-86. [CrossRef]
- Bonini, C.; Chiummiento, L.; Di Blasio, N.; Funicello, M.; Lupattelli, P.; Tramutola, F.; Berti, F.; Ostric, A.; Miertus, S.; Frecer, V.; Kong, D.-X. Synthesis and biological evaluation of new simple indolic non peptidic HIV Protease inhibitors: The effect of different substitution patterns. Bioorg. Med. Chem. 2014, 22, 4792-4802. [CrossRef]
- Chiummiento, L.; Funicello, M.; Lupattelli, P.; Tramutola, F.; Berti, F.; Marino-Merlo, F., Synthesis and biological evaluation of novel small non-peptidic HIV-1 PIs: The benzothiophene ring as an effective moiety. Bioorg. Med. Chem. Lett. 2012, 22, 2948-2950. [CrossRef]
- Chiummiento, L.; Funicello, M.; Lupattelli, P.; Tramutola, F.; Campaner, P., New indolic non-peptidic HIV protease inhibitors from (S)-glycidol: synthesis and preliminary biological activity. Tetrahedron, 2009, 65, 5984-5989. [CrossRef]
- Funicello, M.; Chiummiento, L.; Tramutola, F.; Armentano, M. F.; Bisaccia, F.; Miglionico, R.; Milella, L.; Benedetti, F.; Berti, F.; Lupattelli, P. Synthesis and biological evaluation in vitro and in mammalian cells of new heteroaryl carboxyamides as HIV-protease inhibitors Bioorg. Med. Chem. 2017, 25, 4715-4722. [CrossRef]
- Tramutola, F.; Armentano, M. F.; Berti, F.; Chiummiento, L.; Lupattelli, P.; D’Orsi, R.; Miglionico, R.; Milella, L.; Bisaccia, F.; Funicello, M., New heteroaryl carbamates: Synthesis and biological screening in vitro and in mammalian cells of wild-type and mutant HIV protease inhibitors. Bioorg. Med. Chem. 2019, 27, 1863-1870. [CrossRef]
- D’Orsi, R.; Funicello, M.; Laurita, T.; Lupattelli, P.; Berti, F.; Chiummiento, L. The Pseudo-Symmetric N-benzyl Hydroxyethylamine Core in a New Series of Heteroarylcarboxyamide HIV-1 Pr Inhibitors: Synthesis, Molecular Modelling and Biological Evaluation. Biomolecules, 2021, 11, 1584. [CrossRef]
- Rinaldi, R.; Miglionico, R.; Nigro, I.; D’Orsi, R.; Chiummiento, L.; Funicello, M.; Lupattelli, P.; Laurenzana, I.; Sgambato, A.; Monné, M.; Bisaccia, F.; Armentano, M. F., Two Novel Precursors of the HIV-1 Protease Inhibitor Darunavir Target the UPR/Proteasome System in Human Hepatocellular Carcinoma Cell Line HepG2. Cells, 2021, 10, 3052. [CrossRef]
- Kanemitsu, T.; Inoue, M.; Yoshimura, N.; Yoneyama, K.; Watarai, R.; Miyazaki, M.; Odanaka, Y.; Nagata, K.; Itoh, T. A Concise One-Pot Organo- and Biocatalyzed Preparation of Enantiopure Hexahydrofuro[2,3-b]furan-3-ol: An Approach to the Synthesis of HIV Protease Inhibitors. Eur. J. Org. Chem. 2016, 1874-1880. [CrossRef]
- Lindsten, K.; Uhlikova, T.; Konvalinka, J.; Masucci, M.G.; Dantuma, N.P. Cell-based fluorescence assay for human immunodeficiency virus type 1 protease activity. Antimicrob Agents Chemother. 2001, 45, 2616–2622. [CrossRef]
- Chiummiento, L.; Funicello, M.; Lupattelli, P.; Tramutola, F.; Berti, F.; Marino-Merlo, F. Synthesis and biological evaluation of novel small non-peptidic HIV-1 PIs: the benzothiophene ring as an effective moiety. Bioorg. Med. Chem. Lett. 2012, 22, 2948-2950. [CrossRef]
- Cerminara, I.; Chiummiento, L.; Funicello, M.; Guarnaccio, A.; Lupattelli, P. Heterocycles in Peptidomimetics and Pseudopeptides: Design and Synthesis. Pharmaceutical 2012, 5, 297. [CrossRef]
- Chiummiento, L.; Funicello, M.; Lupattelli, P.; Tramutola, F. Ligand-Free Suzuki Coupling of Arylboronic Acids with Methyl (E)-4-Bromobut-2-enoate: Synthesis of Unconventional Cores of HIV-1 Protease Inhibitors. Org. Lett. 2012, 14, 3928-3931. [CrossRef]
- Tramutola, F.; Chiummiento, L.; Funicello, M.; Lupattelli, P. Practical and efficient ipso-iodination of arylboronic acids via KF/I2 system. Tetrahedron Lett. 2015, 56, 1122-1123. [CrossRef]
- Bonini, C.; Chiummiento, L.; De Bonis, M.; Funicello, M.; Lupattelli, P.; Suanno, G.; Berti, F.; Campaner, P. Synthesis, biological activity and modelling studies of two novel anti HIV PR inhibitors with a thiophene containing hydroxyethylamino core, Tetrahedron 2005, 61, 6580-6589. [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules.Sci. Rep. 2017, 7, 42717. [CrossRef]
Figure 1.
Active carboxamide inhibitor A and new simple derivatives B.
Scheme 1.
Preparation of compounds 5 and 6.
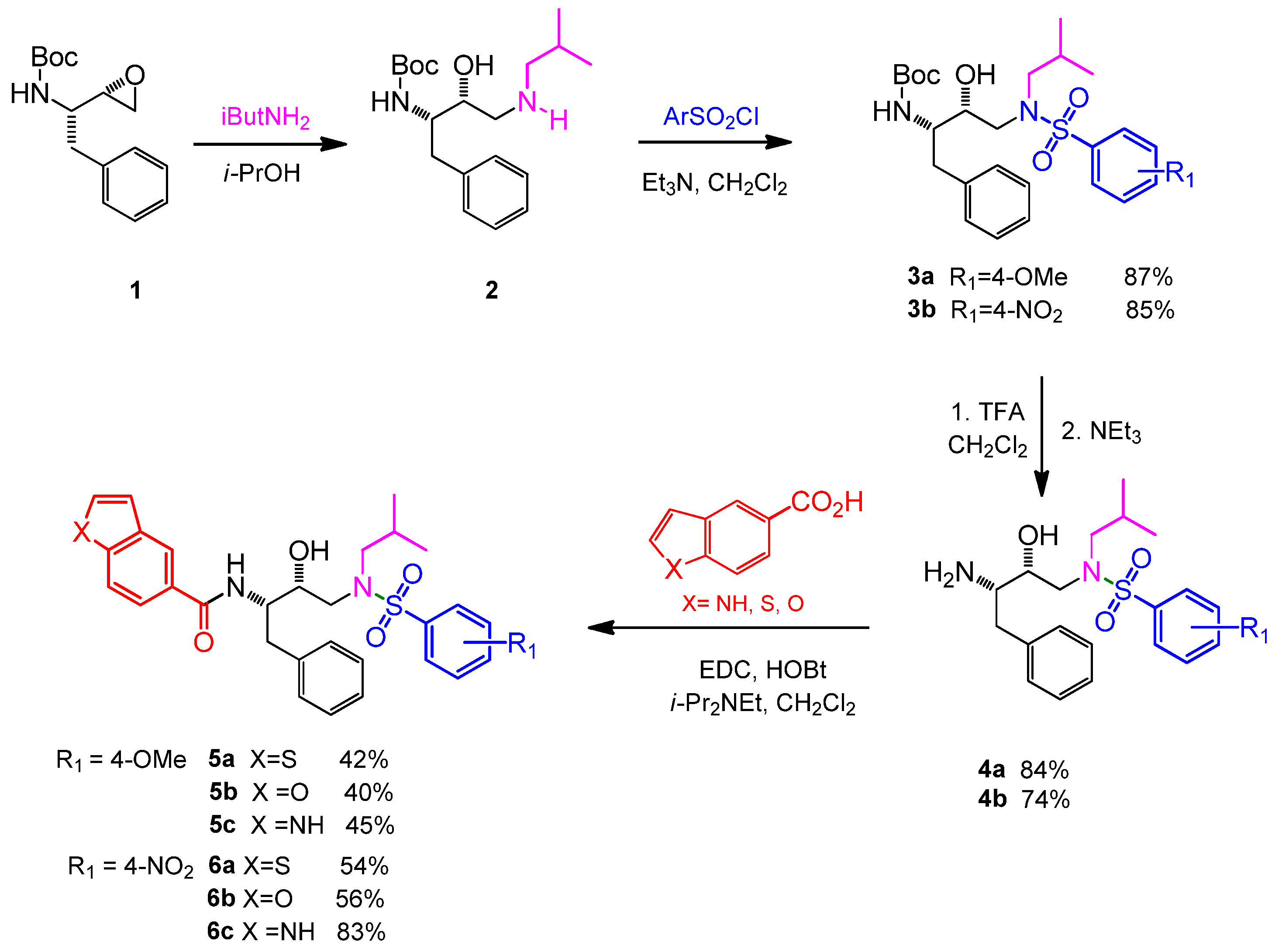
Figure 2.
Evaluation of HIV-1 protease inhibition. HIV-1 protease activity was detected measuring GFP fluorescence in HEK293 cells transiently transfected with pcDNA3/GFP-PR (wild type, wt) or with recombinant plasmids, expressing HIV-protease carrying mutation V32I or V82A, and treated for 24h with 10 µM HIV-1 protease inhibitors (5a, 5b, 5c, 6a, 6b, 6c). In each panel, the cells transfected with the plasmid (wt, V32I or V82A) and untreated with any inhibitor are to be considered as negative controls; cells transfected with the indicated plasmid and exposed to DRV are to be considered as positive controls. Relative fluorescence is expressed as fold change respect to transfected cells cultured without any inhibitor. All data are expressed as means ± Standard Error (SE) of three experiments, each performed in triplicate and statistical significance was evaluated using GraphPad Prism 8.4.2 software by one-way ANOVA followed by Dunnett's post hoc test, **p <0.01, ***p <0.001, ****p<0.0001 versus control [respectively wt (A), V32I (B) or V82A (C)]; $$p<0.01, $$$p <0.001, $$$$p<0.0001 versus DRV.
Figure 2.
Evaluation of HIV-1 protease inhibition. HIV-1 protease activity was detected measuring GFP fluorescence in HEK293 cells transiently transfected with pcDNA3/GFP-PR (wild type, wt) or with recombinant plasmids, expressing HIV-protease carrying mutation V32I or V82A, and treated for 24h with 10 µM HIV-1 protease inhibitors (5a, 5b, 5c, 6a, 6b, 6c). In each panel, the cells transfected with the plasmid (wt, V32I or V82A) and untreated with any inhibitor are to be considered as negative controls; cells transfected with the indicated plasmid and exposed to DRV are to be considered as positive controls. Relative fluorescence is expressed as fold change respect to transfected cells cultured without any inhibitor. All data are expressed as means ± Standard Error (SE) of three experiments, each performed in triplicate and statistical significance was evaluated using GraphPad Prism 8.4.2 software by one-way ANOVA followed by Dunnett's post hoc test, **p <0.01, ***p <0.001, ****p<0.0001 versus control [respectively wt (A), V32I (B) or V82A (C)]; $$p<0.01, $$$p <0.001, $$$$p<0.0001 versus DRV.
Figure 3.
inhibitors with IC50 < 0.6 nM described in our previous works; compounds are numbered according to the original schemes in refs. [18] and [19].

Table 1.
Structure and activity of compounds 5a-c and 6a-c.
| Entry | Inhibitor | IC50 (nM) | Entry | Inhibitor | IC50 (nM) |
|---|---|---|---|---|---|
| 1 |
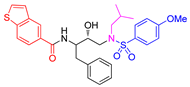 5a |
< 0.6 | 4 |
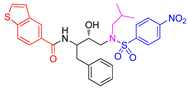 6a |
< 0.6 |
| 2 |
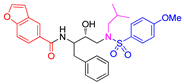 5b |
< 0.6 n | 5 |
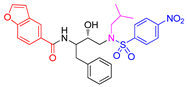 6b |
4.0 ± 0.5 |
| 3 |
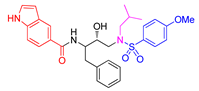 5c |
95 ± 7 | 6 |
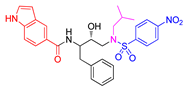 6c |
0.60 ± 0.03 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



