
Submitted:
17 May 2023
Posted:
18 May 2023
You are already at the latest version
Abstract
Alcoholic Liver Disease (ALD) and Non-Alcoholic Fatty Liver Disease (NAFLD) are the most common causes of chronic liver disease and are increasingly emerging as a global health problem. Such disorders can lead to liver damage, resulting in the release of pro-inflammatory cytokines and the activation of infiltrating immune cells. These are some of the common features of ALD progression in ASH (alcoholic steatohepatitis) and NAFLD to NASH (non-alcoholic steatohepa-titis). Hepatic steatosis and subsequent fibrosis, whose continuous progression is accompanied by angiogenesis, lead to hypoxia, inducing the activation of vascular factors, which in turn triggers pathological angiogenesis and subsequent fibrosis, resulting in a vicious cycle. This condition further exacerbates liver injury and may contribute to the development of comorbidities, such as metabolic syndrome as well as hepatocellular carcinoma. Increasing evidence suggests that antiangiogenic therapy may have beneficial effects on these hepatic disorders and their exacerbation. Therefore, there is a great interest to deepen the knowledge of the molecular mechanisms of natural antiangiogenic products that could both prevent and control liver diseases. In this review, we focus on the role of major natural antian-giogenic compounds against steatohepatitis and determine their potential therapeutic benefits in the treatment of liver inflammation.
Keywords:
liver disease
; inflammation
; steatosis
; ALD
; NAFLD
; ASH
; NASH
; angiogenesis
; hepatocellular carcinoma
; natural compounds
1.1. The Liver: Function and Diseases
Liver is an essential organ that plays a critical role in maintaining the metabolic and biochemical balance of the body. It is involved in nutrient metabolism, regulation of blood volume, storage of vitamins and minerals, support of the immune system, endocrine control of growth signaling pathways, lipid and cholesterol homeostasis, and degradation of xenobiotic compounds. In addition, its ability to oxidize lipids and package their excess for secretion and storage in other tissues makes it the major player in fat accumulation in various diseases [1,2].
The liver is composed of several cell types, mainly including hepatocytes, cholangiocytes, stellate cells, sinusoidal cells and Kupffer cells. Hepatocytes are the most abundant cells in the liver volume, and they perform many of the functions attributed to the liver; cholangiocytes are polarized cells that line the bile ducts within the liver and play a crucial role in the secretion and modification of bile, which is essential for digestion and absorption of fats in the small intestine, in addition to their immunological functions; hepatic stellate cells (HSCs) are a dynamic cell population that can exist in a quiescent state and can be activated when damage is induced to promote wound healing; sinusoidal endothelial cells are a specialized endothelial population that coat the hepatic sinusoids and allow the exchange of nutrients, hormones, and waste products between the blood and the liver cells; finally, Kupffer cells are the resident macrophage population of the liver that help to remove foreign particles such as bacteria and viruses [3,4].
1.1.1. ALD and NAFLD
In the industrial city, increasing alcohol abuse and excessive eating are responsible for alcoholic liver disease (ALD) and non-alcoholic liver disease (NAFLD), respectively; both diseases have hepatic steatosis in common and range from a mild condition to more aggressive forms of the disease called alcoholic steatohepatitis (ASH) and non-alcoholic steatohepatitis (NASH), where steatosis, hepatocyte apoptosis, inflammation and fibrogenesis coexist [5,6].
Hepatic steatosis represents the first step of these pathologies and is characterized by the accumulation of lipids, especially triglycerides, in the cytoplasm of hepatocytes, yet in the absence of inflammation, cell death or hepatic fibrosis. In this regard, two forms of steatosis can be distinguished: a macrovesicular form, where there is an arrangement of fat in a single large droplet that dislodges the nucleus in a peripheral location of the hepatocyte; and a microvesicular form, where fat collects in small droplets that do not dislodge the nucleus of the cells. These two aspects often reflect different stages of steatosis, and indeed clinical cases in which the two forms are associated are not uncommon [7,8].
Many hypotheses have been proposed to explain the pathogenetic mechanisms of ALD and NAFLD.
In ALD, as the liver is the main organ responsible for ethanol metabolism, ethanol and its metabolites (acetaldehyde-acetate, fatty acid ethanol esters (FAEE), ethanol-protein adducts) may exert a direct cytotoxic effect as hepatotoxins [9].
Hepatic ethanol metabolism involves oxidative and non-oxidative pathways. The main steps of the oxidative pathway are mediated by alcohol dehydrogenase (ADH) and acetaldehyde dehydrogenase (ALDH). ADH and ALDH convert ethanol to acetaldehyde and acetaldehyde to acetate, respectively [10]. The end products of these reactions are acetaldehyde, acetate, and high levels of NADH. Acetaldehyde injures the liver by directly inducing inflammation, extracellular matrix remodeling, and fibrogenesis [11]. In addition, it covalently binds to proteins and DNA, leading to lipid peroxidation and the subsequent production of toxic metabolites (e.g., malondialdehyde, 4-hydroxy-2,3-trans-nonenal), which are commonly analyzed under oxidative stress conditions [12,13]. Finally, acetaldehyde stimulates transforming growth factor (TGF)-β signaling in HSCs, which acquire a pro-fibrogenic and pro-inflammatory profile [14].
Changes in the NADH/NAD+ ratio can affect biochemical reactions in the mitochondria and gene expression in the nucleus. In particular, the re-oxidation of NADH requires additional oxygen in the mitochondria. To this end, hepatocytes absorb more than their normal share of oxygen from the arterial blood, but not enough to supply all regions of the liver adequately. Therefore, alcohol consumption causes significant hypoxia in perivenous hepatocytes, which are the first to show evidence of chronic alcohol-induced damage [15].
Cytochrome P450, in particular cytochrome P450 2E1 (CYP2E1), is upregulated in chronic alcohol abuse and assists ADH in the conversion of alcohol to acetaldehyde [16] (Figure 1). Reactive oxygen species (ROS), such as hydrogen peroxide and superoxide ions, generated by CYP2E1 are responsible for the pro-inflammatory profile of alcohol-induced liver injury by:
(1) activation of redox-sensitive transcription factors (e.g., nuclear factor kappa B (NF-κB), nuclear factor erythroid 2-related factor 2 (Nrf2), activating transcription factor 4 (ATF4), etc.) [17,18,19];
(2) recruitment of neutrophils and other immune cells [20];
(3) increasing levels of circulating inflammatory cytokines (e.g., tumor necrosis factor-α (TNF-α) and interleukin-1β (IL-1β)) [21];
(4) exacerbated lipid peroxidation associated with alcoholic liver injury [22].
Finally, the last player in the inflammatory and fibrogenic phenomena is catalase, a peroxisomal enzyme involved in the regulation of non-oxidative alcohol metabolism, the product by which FAEE are responsible for alcoholic steatosis and useful as biomarkers of chronic alcohol consumption [16] (Figure 1). Obesity, type 2 diabetes, hyperlipidemia and other conditions associated with insulin resistance are commonly present in patients with metabolic syndrome (MS). A large body of evidence suggests that NAFLD is associated with MS. The latter is a clinical entity characterized by a cluster of metabolic changes, including glucose intolerance, dyslipidemia, and hypertension [23,24,25]. Approximately 90% of NAFLD patients have one or more diagnostic criteria for MS, allowing NAFLD to be defined as the "hepatic representation" of MS [26].
We can distinguish two types of NAFLD: primary (associated with MS) and secondary (associated with other metabolic or iatrogenic conditions, distinct from MS). The pathophysiology of primary NAFLD is not fully understood.
Since 1998, the two-hit pathogenetic model of liver injury proposed by Day and James has been widely accepted [27]. The first hit leads to the initial accumulation of triglycerides within the hepatocyte, caused by increased hepatic lipid uptake. These metabolic imbalances correlate with insulin resistance, a characteristic feature of MS. Such an event is, among others, bidirectional to steatosis: on one hand, insulin resistance promotes the progression of NAFLD, but on the other hand, NAFLD triggers the development of insulin resistance [28]. Later, the condition of steatosis would thus predispose the liver to the second event (second hit), after which inflammation, fibrosis and liver damage would develop. The factors determining the second hit included several biomolecules capable of interfering with the insulin-induced signal transduction mechanism, including inflammatory cytokines (e.g., TNF-α, IL-6, IL-1β, hormones produced by adipose tissue such as adipokines, leptins, and adiponectins), oxidative stress and lipid peroxidation markers, fatty acids, and even ceramides.
In fact, inhibition of ceramide synthesis relieves NAFLD and fibrosis. Moreover, increased hepatic steatosis is associated with ceramide-rich hepatic lipids [29,30].
In addition, insulin resistance results in increased hyperglycemia-induced endoplasmic reticulum (ER) stress, peripheral lipolysis, hepatic fatty acid (FA) uptake, and hepatic synthesis of de novo lipids [31,32,33]. Therefore, the oxidation and disposal of fatty acids results in a defect compared to their accumulation and neo-synthesis, leading to an accumulation of fats within hepatocytes with important consequences, such as dysregulated ketogenesis [34,35]. In addition, fatty acids negatively affect the intracellular insulin signaling mechanism and cause hepatic insulin resistance through multiple pathways mediating activation of protein kinase C (PKC-3), c-Jun N-terminal kinase (JNK), I-kappaB (I-κB) kinase β, and NF-κB [36,37,38].
Furthermore, insulin resistance has been found to be associated in increased mitochondrial fatty acid oxidation. This process, both mitochondrial and peroxisomal, can produce ROS which are hepatotoxic and contribute to the development of oxidative stress. In addition, fatty acids and their metabolites are ligands of PPARα (peroxisomal proliferator-activated receptor-α), a transcription factor that regulates the expression of several genes encoding enzymes involved in fatty acid oxidation at the mitochondrial, peroxisomal and microsomal levels. In support of this, deletion of PPARα in hepatocytes impaired fatty acid catabolism, resulting in hepatic lipid accumulation in steatosis models [39,40].
Regarding the role of inflammatory cytokines in the pathogenesis of NAFLD, they could cause hepatic and systemic insulin resistance, as well as promote liver injury, apoptosis, neutrophil chemotaxis, and HSC activation [41,42,43]. Studies have reported several cytokines involved in the development and progression of NAFLD, such as IL-1β, IL-6, TNF-α, C-reactive protein (CRP), and NOD-like receptor protein 3 (NLRP3) inflammasome activation. Some of these inflammatory mediators can be used as biomarkers to assess the severity and prognosis of NAFLD [44,45] (Figure 1).
In addition, fatty acids accumulated in hepatocytes can stimulate cytokine production via the NF-κB-dependent pathway [46]. An additional source of pro-inflammatory cytokines is the infiltrating macrophages in the adipose tissue of obese subjects; cytokines are increased in addition to promoting the onset of a state of insulin resistance. They appear to reduce the production of certain peptides produced by visceral adipose tissue, such as leptins, resistins and adiponectins, which are involved in the pathogenesis of NAFLD [47].
1.1.2. ASH and NASH
Alcoholic steatohepatitis (ASH) and non-alcoholic steatohepatitis (NASH) are two of the most common causes of chronic liver disease worldwide. ASH is caused by chronic alcohol consumption, while NASH is associated with poor dietary habits and obesity, and is associated with insulin resistance and type 2 diabetes [48]. Although ASH and NASH differ in their etiology, both diseases are characterized by excessive fat deposition and lipotoxicity, leading to inflammation, hepatocellular swelling, and fibrogenesis [49].
Between 90 and 100% of individuals who consume more than 40 g of alcohol per day will eventually develop alcoholic fatty liver. Approximately 10-35% of these individuals develop ASH, which is characterized by a severe inflammatory state of the liver, hepatocyte swelling, neutrophil infiltration, and/or hepatic fibrosis [50]. In approximately 8-20% of patients with ASH, the risk of developing hepatocellular carcinoma (HCC) increases by approximately 2% per year [51]. Factors involved in the pathophysiology of ASH include hepatic steatosis, oxidative stress, acetaldehyde-mediated toxicity, and cytokine- and chemokine-induced inflammation, but hepatic inflammation is the critical prerequisite for the development of liver fibrosis, cirrhosis, and HCC [52].
Recent studies indicate that 5-20% of patients with NAFLD develop NASH during their clinical course, 10-20% develop higher grade fibrosis and <5% progress to cirrhosis [53]. Thus, ASH and NASH share a common pathogenesis and are mediated by different mechanisms [54]. The "two-hit" theory suggests that oxidative stress and cytokines lead to the progression of necroinflammation and ultimately to fibrosis and cirrhosis [55]. Indeed, hepatic macrophages and Kupffer cells are able to activate HSCs through the production of IL-6 and TNF-β, while HSCs in turn produce pro-inflammatory cytokines, such as TNF-α and IL-8 [56]. Fat-derived products can also cause inflammation and injury in the liver by activating the inflammasome and inducing the production of IL-1 and IL-18. Fat products can also cause inflammation and injury in the liver by activating the NLRP3 inflammasome, which induces the production of IL-1β and IL-18; evidence for the critical role of NLRP3 activation is the marked protection from disease in knockout mice following acute or chronic alcohol administration [57]. In addition, oxidative stress strains antioxidant defenses, which become inadequate. This results in liver injury through direct cellular damage and NF-κB-mediated cellular signaling [58]. Several studies also show that oxidative stress and the increase in ER stress-mediated Nrf2 are induced, which appears to be some common features of ASH and NASH (Figure 1); accordingly, altered markers of ER stress are recorded in numerous experimental preclinical models whose diets are administered alcohol or HFD [59].
As previously reported, the mechanisms that drive disease progression also induce steatosis. Therefore, steatosis could be considered as an "adaptive" response of the liver to stress, and continuous insult leads to ASH and NASH, in which we find not only steatosis but also inflammation and fibrosis [61,62].
In addition to oxidative stress and cytokines production, insulin resistance and hyperglycemia are active players in the progression of the pathology by directly inducing fibrosis or by regulating connective tissue synthesis [63,64].
Unfortunately, although significant progress has been made in identifying the key players that mediate the transition from steatosis to steatohepatitis, treatment options for ASH and NASH are limited [65]. Exercise and dietary interventions remain the main recommendations for patients with NAFLD and NASH. However, it is difficult for patients to adhere to such lifestyle changes for a variety of social, psychological, physical, genetic and epigenetic reasons; therefore, pharmacotherapy is essential [66,67].
Figure 1.
Role of alcohol consumption and inadequate diet on liver disease.
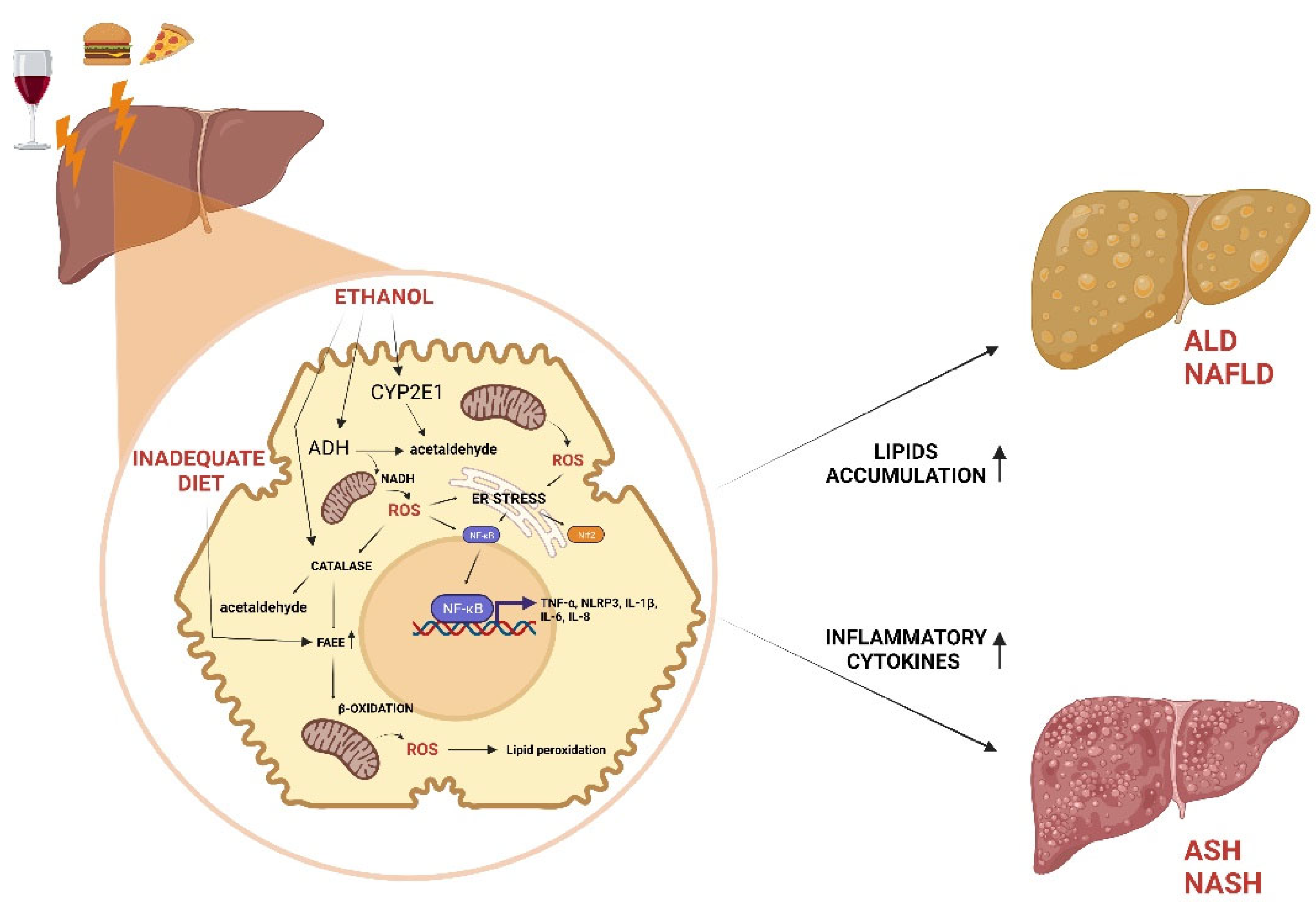
ADH, catalase and CYP2E1 contribute to oxidative metabolism of ethanol to generate acetaldehyde. Alcohol metabolism, high fat and high carbohydrate diet induce NAFLD and ALD by FAEE accumulating in lipid droplets. Accumulation of ROS resulting from ethanol or β-oxidation leads to formation of lipid peroxides and ER stress, which induce NF-kB activation and consequent inflammatory state at the base of ASH and NASH.
2.1. Angiogenesis and Hepatic Inflammation
In this review, we focus on the role of the main natural compounds with health beneficial properties, including their role in angiogenesis within the pathophysiology of liver fibrosis.
Angiogenesis is a widely described and studied composite process that leads to the development of new vessels from pre-existing vasculature [68,69]. The angiogenic phenomenon occurs physiologically during normal tissue growth and wound healing, but also in pathological contexts such as liver disease and tumorigenesis [70,71].
In the liver, angiogenesis proceeds with molecular mechanisms similar to those demonstrated in other parts of the body. However, several differences in this organ make angiogenesis more complex, such as a more complex parenchyma with two distinct microvascular structures (portal vein and hepatic sinusoids), the presence of liver-derived angiopoietin-like peptide 3 (ANGPTL3), and the activity of HSCs, which play an active role in liver fibrosis by proliferating and becoming myofibroblasts that can modulate angiogenesis [72,73,74,75].
Multiple factors trigger angiogenesis in liver disease, including tissue inflammation and fibrosis. These lead to progressive tissue hypoxia, which in turn causes capillarization of sinusoids and initiation of the angiogenic process that leads to progression of NAFLD and ALD to NASH and ASH, respectively. Hypoxia activates angiogenesis through signaling mediated by transcription factors called hypoxia-inducible factors (HIFs), such as HIF-1 (Figure 2), which, among other things, stimulates inflammation via the NF-kB pathway [76].
All this occurs because the intrahepatic vascular remodeling, which causes the deposition of fibrillar collagen (type I) instead of sinusoidal collagen (type IV), generates a continuous capillarization of the sinusoids and the development of intrahepatic shunts, leading to decreased effective hepatocyte perfusion and consequent loss of specific endothelial fenestrations. This process, together with the accumulation of fibrotic tissue, creates vascular resistance and decreases oxygen transport to liver parenchyma, resulting in the upregulation of pro-angiogenic mechanisms via hypoxia, establishing a vicious cycle [77,78,79].
In pathological liver angiogenesis, there is a strong dialogue between different cell populations: indeed, wound healing generates an increase in the expression of some cytokines and factors with pro-angiogenic and inflammatory actions. Kupffer cells may contribute to angiogenesis by releasing ROS and inducing new vessel formation by stimulating the expression of TNF-α, HIF-1 and VEGF (vascular endothelial growth factor) [80]. Upon liver injury, these cells further produce TGF-β, the major regulator in chronic liver disease, contributing to all stages of disease progression from initial insult, through inflammation and fibrosis, to cirrhosis and HCC [81] (Figure 2). HSCs express several chemokines capable of stimulating angiogenesis and respond to hypoxia in a HIF-1α-related pathway through the production of IL-8 and the increase of VEGF, Ang-1 (angiopoietin-1) and their corresponding receptors VEGFR-2 and Tie-2 [82,83] (Figure 2). In addition, other cells not present in the liver are activated: leukocytes and mast cells are involved in the regulation of angiogenesis by releasing histamine, heparin, cytokines and other angiogenic factors (Ang-2, EGF (epidermal growth factor), PlGF (placental growth factor), TGF-β1, VEGF, etc.) [84,85].
Therefore, angiogenesis represents a key stage of inflammation and fibrosis [86] in liver disease. For these reasons, several antiangiogenic molecules (e.g., the anti-vascular endothelial growth factor (VEGF) monoclonal antibodies bevacizumab, brivanib, dovitinib, nintedanib, etc.) are currently used in the treatment of various cancers, including HCC, suggesting that angiogenesis is a promising therapeutic target [87,88,89]. Therefore, it is of great importance to determine the potential therapeutic benefits of various natural compounds in the prevention and treatment of liver inflammation. In this scenario, there are several natural compounds active in this field.
Figure 2.
Vicious circle generated from hypoxia, fibrosis, and angiogenesis in liver disease.
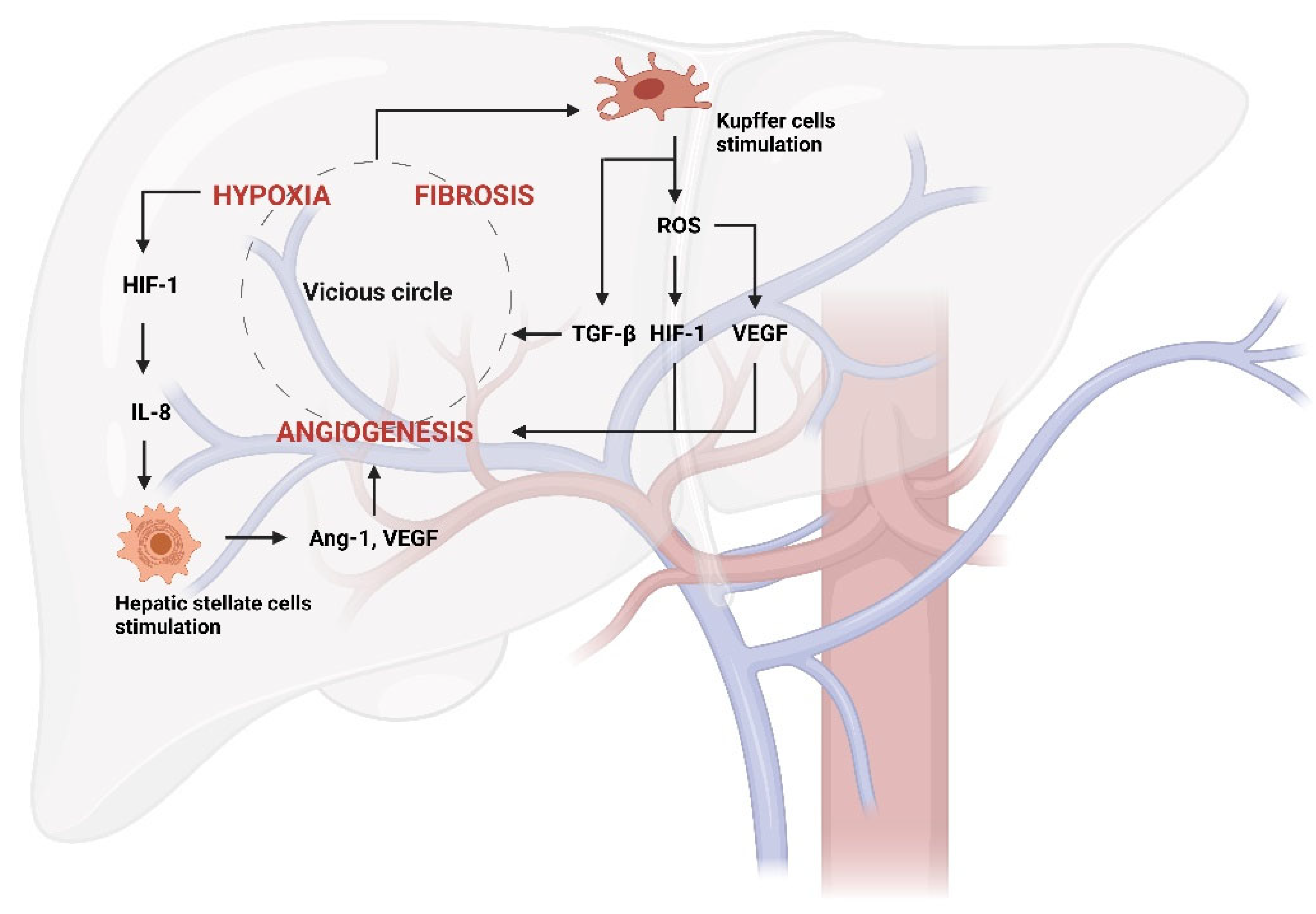
Hypoxia activates hypoxia-inducible factors (HIFs), such as HIF-1, which among other things stimulates hepatic stellate cells (HSCs) activation via IL-8 production. They consequently produce angiogenic factors, such as Ang-1 (Angiopoietin-1) and VEGF (Vascular Endothelial Growth Factor). The stimulation of Kupffer cells by the vicious circle further aggravates this pathological process through TGF-β release. Furthermore, Kupffer cells can produce ROS inducing new vessel formation by the stimulation of HIF-1 and VEGF.
3.1. Phytochemicals to Counteract the Stages of Liver Disease via Angiogenesis Inhibition
3.1.1. Quercetin
Quercetin is a flavonoid found in fruits such as apples, red grapes, citrus fruits, tomatoes, onions, and other green leafy vegetables. Quercetin possesses various biological and pharmacological activities, including antioxidant, antiviral, anti-inflammatory, antiproliferative, and antifibrotic effects [90,91]. In recent studies, quercetin inhibited hepatic inflammation and fibrosis in mice by downregulating the HMGB1-TLR2/4-NF-κB signaling pathway [92]. Moreover, it also limited hepatic fibrosis by inhibiting HSCs activation and reducing autophagy by regulating crosstalk between TGF-β1/Smads and PI3K/Akt pathways [93].
Nevertheless, recent data have shown that macrophages play a complex role in liver fibrogenesis and are directly involved in the progression and resolution of liver fibrosis [94,95]. Indeed, inflammatory cytokines released by macrophages perpetuate inflammation and activate HSCs; quercetin showed direct effects by reducing the number of hepatic macrophages and ameliorating liver fibrosis after treatment of mice with CCl4 [96] (Table 1). In conclusion, quercetin is a flavonoid with numerous beneficial biological and since its anti-inflammatory and antifibrotic effects it may hold promise as a potential therapeutic agent for liver fibrosis.
3.1.2. Silybin
Silybin is a flavonolignan that exists as a mixture of two diastereomers, silybin A and silybin B, in a roughly equimolar ratio [97]. It is usually extracted together with other compounds from milk thistle (Silybum marianum) in a mixture called silymarin, consisting of 50-60% silybin and the rest of silydianin, silycristin, taxifolin and other polyphenolic components [98]; known compounds with antioxidant activity [99]. Several pharmacological actions of silybin have been identified, including antioxidant and anti-inflammatory properties, antifibrotic effects, and modulation of insulin resistance. Specifically, in chronic inflammation, it underlies liver fibrosis and the development of cirrhosis [100].
The common mechanism underlying the initiation and progression of liver inflammation is oxidative stress [101]. NF-κB is a key transcriptional regulator of the inflammatory response and plays an essential role in the regulation of inflammatory signaling pathways in the liver [102]. Furthermore, NF-κB is activated in virtually all chronic liver diseases, including ALD, NAFLD, viral hepatitis, and biliary liver disease [103,104,105]. There is increasing evidence for a general inhibition of inflammatory mediators such as NF-κB and inflammatory metabolites (e.g. prostaglandin E2 (PGE 2) and leukotriene B4 (LTB 4)) by silymarin [106].
In isolated rat Kupffer cells, silymarin weakly inhibited the formation of PGE 2 but strongly blocked the biosynthesis of LTB 4 [107]. This selective inhibition of LTB 4 formation by Kupffer cells and possibly other cell types may explain its anti-inflammatory potential.
In addition, it can reduce or normalize liver function parameters, such as transaminase levels, and improve ultrasound parameters of liver anatomy [108]. Therefore, silymarin (formulation derived from Eurosil 85®) has been investigated as a therapeutic option for NAFLD and NASH (Table 1).
Therefore, silybin a has antioxidant and anti-inflammatory properties and may inhibit NF-κB and inflammatory metabolites, having potential as a therapy for NAFLD and NASH, since it improves liver function and multiple ultrasound parameters used in liver ultrasonography. Silybin holds promise as a therapeutic agent for liver inflammation and fibrosis.
3.1.3. Breviscapine
Breviscapine is a crude mixture of several flavonoids from the traditional Chinese herb Erigeron breviscapus. It consists of more than 90% scutellarin and also contains baicalein, naringenin, scopoletin, kaempferol, apigenin, scutellarin, luteolin, caffeic acid, and protocatechuic acid [109]. It has multiple pharmacological activities, including anti-inflammatory, antioxidant, antiapoptotic, vasorelaxant, antiplatelet, anticoagulant, and myocardial protective activities [110,111]. Recent studies show that breviscapine protects against CCl4-induced liver injury by reducing proinflammatory cytokine secretion and oxidative stress [112]. In addition, scutellarin, the main component of breviscapine, has been shown to regulate lipid metabolism and reduce oxidative stress in NAFLD [113].
Further studies show that in mice fed a high-fat diet (HFD), high-fat/high-cholesterol diet (HFHC), or methionine- and choline-deficient diet (MCD), breviscapine attenuates hepatic lipid accumulation, inflammation, and fibrosis by mediating effects via direct inhibition of TGF-β-activated kinase 1 (TAK1) MAPKKK (mitogen-activated protein kinase kinase kinase).
In addition to being an upstream protein kinase of MAPK signaling, TAK1 also activates NF-κB during NAFLD progression [114]. Because of the pivotal role of TAK1 in many physiological processes, such as inflammation, cell differentiation, and apoptosis, inhibition of metabolic stress-induced TAK1 activation may provide a potent hepatoprotective effect [115]. Indeed, recent studies have highlighted the important role of TAK1 in regulating hepatocyte lipid metabolism and reducing hepatocyte inflammation in NAFLD and NASH [116,117] (Table 1).
3.1.4. ALS-L1023
ALS-L1023 is an extract isolated from Melissa officinalis, known in natural medicine as an antiangiogenic agent [118,119,120,121]. ALS-L1023 modulated the mRNA expression of angiogenic factors (VEGF-A and fibroblast growth factor 2 (FGF-2)), metalloproteinases (MMPs) MMP-2 and MMP-9, and their inhibitors (TIMP-1, TIMP-2, and thrombospondin (TSP-1)) [122,123]. It also reduces visceral adipose tissue (VAT) production in mice with HFD-induced NAFLD by suppressing lipid synthesis and steatosis, inflammatory cell infiltration, and collagen accumulation in the liver; thus, supporting the idea that ALS-L1023 may have a great contribution to regulate the development and progression of NAFLD and consequent NASH [124,125]. In addition, this extract reduced inflammation in diet-induced NAFLD in female ovariectomized mice and rescued from oxidative stress through Akt activation. For these reasons, ALS-L1023 is in a Phase IIa clinical trial in patients with NAFLD. Recent studies are also evaluating whether ALS-L1023 could alleviate liver fibrosis, as anti-fibrotic effects are essential properties of NASH drugs. In this regard, biochemical analysis showed that the ALS-L1023 mouse group had significantly decreased alanine transaminase and aspartate transaminase. In addition, the area of fibrosis was significantly reduced after administration of ALS-L1023, and its anti-fibrotic effect was greater than that of the reference compound OCA (obeticholic acid) [126] (Table 1). ALS-L1023 has antiangiogenic and anti-inflammatory properties, reduces VAT production and inflammation in NAFLD as well as to alleviate liver fibrosis, with a significative antifibrotic effect.
3.1.5. Curcumin
Curcumin, along with demethoxycurcumin, bisdemethoxycurcumin, and cyclocurcumin, is the major phenylpropanoid compound found in the roots of the perennial plant Curcuma longa. It is unclear whether all four analogs have the same activity. Although curcumin was found to be the most potent in most systems [127,128,129,130], bisdemethoxycurcumin and cyclocurcumin were found to have higher activity in some studies [131,132]. There is also evidence that the mixture of all three is more potent than just one [132]. Curcumin has shown anti-angiogenic and other beneficial properties in several experimental models of liver injury by inhibiting the NF-κB pathway [133]. It prevents aflatoxin-induced liver injury [134] and regresses cirrhosis [135].
In acute CCl4 intoxication, oxidative stress and expression of pro-inflammatory cytokines, such as TNF-α, IL-1β and IL-6, and NF-κB activation are associated with some increases in several markers of liver injury and distortion of hepatic microscopic structure; pretreatment with curcumin prevented oxidative stress, NF-κB activation, and liver injury. Similarly, in biliary cirrhosis, curcumin showed anti-fibrogenic properties associated with downregulation of the cytokine TGF-β [136] (Table 1).
NAFLD induced in methionine and choline-deficient (MCD) mice significantly upregulates the hexosamine biosynthetic pathway and O-GlcNAc transferase via ER stress. This model has been shown to be a useful experimental model for NASH. Curcumin treatment alleviates the severity of hepatic steatosis by blocking the NF-κB signaling pathway through inhibition of O-GlcNAcylation and enhancing antioxidant systems, including SOD1 (superoxide dismutase 1), GPx (glutathione peroxidase), and CAT (catalase) [137] (Table 1).
3.1.6. Sulforaphane
Sulforaphane (SFN) is an organosulfur compound in the isothiocyanate group that is found in high concentrations in cruciferous vegetables such as broccoli and cauliflower [138]. It has been reported to exert a variety of bioactive effects, including antioxidant, anti-inflammatory, cytotoxic, and cytoprotective effects [139]. In tumor angiogenesis, SFN inhibits NF-κB-regulated VEGF expression in human prostate cancer cells [140]; Peng liu et al. have shown that SFN interferes with the proliferation, migration and tube formation of endothelial cells, but it is also capable of inhibiting the pro-angiogenic effect of HepG2 cells both in vitro, ex vivo and in vivo. SFN significantly inhibited the viability and migration of HUVECs (human umbilical vein endothelial cells), consistent with previous findings [141].
SFN has been shown to inhibit STAT3 (signal transducer and activator of transcription 3), and this is consistent with reduced expression of HIF-1α and VEGF in HepG2, suggesting that STAT3/HIF-1α/VEGF may be responsible for the anti-angiogenic effects of SFN. This latter also inhibits TGF-β-induced epithelial-mesenchymal transition of HCC via ROS-dependent pathway [142]. SFN-induced Nrf2 activation was indeed confirmed by mRNA upregulation of redox genes such as HO-1 (heme oxygenase-1), MRP2 (multidrug resistance-associated protein 2), and NQO1 (NAD(P)H quinone dehydrogenase 1) in several cell lines [143,144] (Table 1). Essentially, SFN is a bioactive compound with a range of beneficial effects, including antioxidant, anti-inflammatory, cytotoxic, and cytoprotective effects, which has been shown to inhibit angiogenesis in human prostate and liver cancer cells by inhibiting NF-κB-regulated VEGF expression, and through inhibiting the STAT3/HIF-1α/VEGF pathway. SFN also induces Nrf2 activation, which may contribute to its antitumor effects.
3.1.7. Cordycepin
Cordycepin is an adenosine derivative, antimetabolite and antibiotic from the fungus Cordyceps militaris [145]. Cordycepin has a broad spectrum of biological activities, including anti-inflammatory, antioxidant, antifibrotic, antiadipogenic and antitumor activities [146,147]. Guo Z. et al. have shown that it suppresses the migration and invasion of human liver cancer cells by downregulating CXCR4 [148]. A hallmark of NASH pathogenesis is the inflammatory response, which initially triggers the release of numerous types of proinflammatory cytokines. Cordycepin suppresses the production of proinflammatory cytokines in LPS-stimulated macrophages and exerts anti-inflammatory effects by suppressing NLRP3 activity, an important regulator of the inflammatory process in macrophages [149]. Furthermore, NLRP3 deficiency in HCC cells enhances the activity of natural killer cells to delay tumor development in the xenograft mouse model [150].
Tian Lan et al. also demonstrate in their study how cordycepin attenuates metabolic stress-induced NASH by preventing steatosis, inflammation, and fibrosis. Specifically, cordycepin inhibited the activation of the NF-κB signaling pathway and attenuated the secretion of proinflammatory cytokines in hepatocytes under metabolic stress. This is due to an increase in AMPK phosphorylation through an interaction with the α-subunit of AMPK. The latter acts in mice treated with NASH-inducing diets as a central regulator of fatty acid, cholesterol and glucose homeostasis through phosphorylation of enzymes that regulate metabolism, including ACCα (acetyl-CoA carboxylase), glycogen synthase, glucose transporter 4, HMG-CoA (3-hydroxy-3-methyl-glutaryl-CoA) reductase [151] (Table 1). In conclusion, cordycepin, an adenosine derivative, has been shown to exhibit various biological activities, including anti-inflammatory, antioxidant, antifibrotic, anti-adipogenic, and antitumor effects. Cordycepin also enhances natural killer cell activity to delay tumor development in xenograft mice models. These findings suggest that cordycepin may have potential therapeutic applications in the treatment of liver diseases, including NASH and liver cancer.
3.1.7. Methoxyeugenol
Methoxyeugenol has been identified in Brazilian red propolis, produced by bees of the species Apis mellifera, and the therapeutic effects of propolis have been known in folk medicine for centuries and have been used to treat infections, gastric disorders and to improve wound healing [152,153]. Another source of methoxyeugenol is nutmeg (Myristica fragrans Houtt.), which is used as a spice and whose use in folk medicine is reported mainly for the treatment of gastrointestinal disorders [154]. However, studies support its use in the treatment of nonalcoholic hepatic steatosis [155], as it appears to promote lipid metabolism regulation and exert anti-inflammatory effects. Bruno de Souza Basso et al. have shown that methoxyeugenol promotes HSC deactivation without evidence of cell death, suggesting a reduction in proliferative rate [156]. In addition, the cytokine TGF-β, a growth factor that plays an important role in the development of liver fibrosis, particularly by activating quiescent HSCs, is reduced in the same cells treated with methoxyeugenol.
The maintenance of lipid metabolism is mediated by nuclear receptors of the PPAR family, mainly PPARα/ɣ, and several studies have shown that PPARγ activation can inhibit macrophage activation and NF-κβ-mediated inflammatory pathways [157,158]. In light of this, PPARγ activation may enhance phenotypic modulation of activated HSCs by restoring lipid metabolism and inhibiting inflammatory signaling. In this regard, methoxyeugenol increases PPARɣ mRNA expression. In vivo studies on the use of methoxyeugenol, administered in the CCl4-induced liver fibrosis model, attenuate the inflammatory process and fibrosis, reducing intralobular inflammation by decreasing the gene expression of TNF-α, IL-6 and IL-8, as well as NF-κB protein expression [156] (Table 1). Overall, methoxyeugenol shows promising potential as a natural compound for the treatment of liver diseases.
3.1.8. Naringenin
Naringenin is a flavone found in various plants and is particularly abundant in citrus fruits [159]. Several studies show that it exerts multiple biological effects, including anti-inflammatory, antioxidant, hypolipidemic effects: protective factors for NAFLD [160,161].
The study by Wang et al. showed that the mRNA expression levels of NF-κB, IL-1β, IL-18 and NLRP3 were improved in mice fed an MCD diet. In vivo experiments showed that naringenin significantly reduced the mRNA and protein levels of these factors. In addition, hepatic triglyceride levels were not further reduced in NLRP3-/- mice fed an MCD diet treated with naringenin. For further confirmation, primary hepatocytes from wild-type and NLRP3 -/- mice were isolated and NLRP3 -/- cells were stimulated with LPS and oleic acid. It was found that naringenin is highly effective in preventing lipid deposition in WT primary hepatocytes, but much less effective in NLRP3 -/- primary hepatocytes, demonstrating that this flavonoid acts precisely by reducing the levels of the NLRP3 inflammasome [162] (Table 1).
3.1.9. Ferulic Acid
Ferulic acid (FA) is a phenolic acid widely found in grains, vegetables, and plants such as Angelica sinensis. FA exhibits several biological activities, including antioxidant, anticancer, antidiabetic, and immune function-enhancing effects [163,164].
Several recent studies have demonstrated the hepatoprotective effects of FA [165]. Indeed, its administration improves lipid metabolism and liver inflammation in apolipoprotein E-deficient mice fed a HFD by upregulating AMPKα and downregulating lipogenic genes [166]. In addition, FA showed beneficial effects against oxidative stress and liver damage by activating Nrf2/HO-1 and PPARγ pathways [167].
Jianzhi Wu et al. reported FA as an agent capable of preventing all histological changes of CCl4-induced liver fibrosis in mice by suppressing hepatic oxidative stress, inflammatory response, macrophage activation, and HSCs activation through phosphorylation of AMPK by direct binding and consequent inhibition of PTP1B (protein tyrosine phosphatase 1B) [168]. Therefore, activated AMPK in hepatocytes not only suppressed apoptosis and NOX2 (NADPH oxidase 2)-derived ROS production, but also inhibited the production of endothelial pro-inflammatory cytokines [169]. FA is also able to prevent the nuclear translocation of NF-κB. All this makes FA a good player in the treatment of liver fibrosis and its advanced complications (Table 1).
3.1.10. Betaine
Betaine is an essential biochemical modulator of the methionine/homocysteine cycle that is originally extracted from the juice of sugar beet (Beta vulgaris) [170] and is also found in various microorganisms, plants, and animals [171]. Aqueous extracts of betaine are used in traditional Eastern medicine to treat liver disease [172]. Recent studies show that betaine suppresses inflammation through activation of NF-κB during aging [173]. Eui-Yeun Yi et al. found that betaine suppressed angiogenesis in the in vivo Matrigel plug assay, in addition to in vitro inhibition in tube formation, migration, and invasion assays performed on HUVECs. Betaine also inhibits NF-κB and Akt activation [174].
VEGF and FGF-2 are known to be key stimuli for angiogenesis [175,176]. When investigating the involvement of betaine in the expression of key angiogenic factors, it was found that the mRNA expression of FGF-2 was significantly reduced. Betaine also shows beneficial effects by downregulating the expression of MMP-2 and MMP-9, key regulators of extracellular matrix turnover through the degradation of a variety of extracellular matrix proteins. Extracellular proteolytic activity is important in the process of endothelial cell migration and invasion, focal events in angiogenesis [177,178] (Table 1). Therefore, betain suppresses inflammation and angiogenesis by inhibiting NF-κB and Akt activation, reducing the expression of key angiogenic factors and regulating the extracellular matrix turnover.
3.1.11. Catechins
Catechins are a group of flavanols that are abundant in plant fruits, vegetables, and beverages. Fruits such as grapes, apple, cocoa, and green tea are considered the major sources of catechins, which represent some promising candidates in the field of biomedicine [179,180,181,182]. Green tea is one of the most popular beverages in the world and has been studied for its health-promoting properties in several diseases such as cancer, obesity, diabetes, and cardiovascular disease [183]. Many of the biological effects of green tea are mediated by its polyphenolic catechins, particularly (-)-epigallocatechin-3-gallate (EGCG), which accounts for 50-85% of total catechins. Several studies show how green tea in animals restores gene and protein levels of proinflammatory cytokines in galactosamine-induced hepatitis [184]. Similar observations have indeed been reported in CCl4-induced liver injury in rodents [185], where mRNA expression levels of TNF-α, COX-2, iNOS, smooth muscle actin, TGF-1, procollagen-I, MMP-2, MMP-9, and TIMPs are increased along with an increase in NF-κB activity. Treatment with green tea extract significantly reduced liver damage, oxidative stress, inflammatory response, and expression of all pro-fibrogenic markers analyzed except TIMP-2 and MMP-9. Other studies show that treatment with such extract inhibits the levels of IL-1, IL-6, TNF-α [186]. Furthermore, vascular endothelial (VE) cadherin and Akt, known downstream proteins in the VEGFR-2-mediated cascade, are other target proteins by which EGCG inhibits angiogenesis [187]. It has also been observed that pentameric procyanidins isolated from cocoa inhibit gene expression of the tyrosine kinase ErbB2, thereby slowing down in vitro angiogenesis of human aortic endothelial cells [188] (Table 1).
3.1.12. Puerarin
Puerarin, a natural compound extracted from Pueraria lobata, has antioxidant and anti-inflammatory effects [189]. Various studies show that puerarin may regulate leptin signaling through the Janus kinase 2 (JAK2)/STAT3 pathway, thereby ameliorating hepatic steatosis [190]. Jingxuan Zhou et al. showed that in a rat model of HFD-induced NAFLD, puerarin administration reduced the abdominal fat coefficient [191]. In addition, puerarin significantly reduces inflammation caused by lipid accumulation. This induces ROS overproduction and consequent activation of the NF-κB pathway, as well as a variety of inflammatory factors, such as interleukins (IL-1β and IL-18) and TNF-α [192]. In addition, puerarin decreased thiobarbituric acid reactive substances and protein carbonyl content in the liver of CCl4-treated mice by regulating the expression of phosphorylated JNK, phosphorylated c-Jun protein, and cholesterol 7a hydroxylase (CYP7A1) in the liver [193] (Table 1).
3.1.13. Resveratrol
Resveratrol (RSV) is a stilbene found in grape skins, blueberries, raspberries, mulberries, and peanuts, and its high concentration in red wine partly explains the relatively low incidence of cardiovascular disease despite the prevalence of HFD in populations using it [194]. Resveratrol has antioxidant activity in a wide range of liver diseases [194,195]. In particular, its effect is mainly exerted through ROS reduction and enhancement of endogenous antioxidant enzyme activity (e.g., SOD, CAT). In addition, RSV promotes the synthesis of antioxidant molecules and the expression of related genes involved in mitochondrial energy biogenesis, particularly through the AMPK/SIRT1/Nrf2, ERK/p38 (extracellular signal-regulated kinase), and PTEN/Akt (phosphatase and tensin homologue/protein kinase B) signaling pathways [196]. Resveratrol acts as an antioxidant by inhibiting the ubiquitination of Nrf2, thereby preserving its critical function. Nrf2, in turn, translocates to the nucleus and induces the transcription of various antioxidant genes, such as SOD and CAT, thereby enhancing antioxidant defense [197].
In NAFLD, RSV seems to play an interesting role in regulating the deposition of de novo fibrogenesis by acting on several key pathways. Indeed, RSV administration reduces portal pressure, HSC activation and improves hepatic endothelial function in cirrhotic rats, with an overall beneficial effect on cirrhosis and portal hypertension [198,199]. Again, it is well known that RSV administration in hepatocytes causes inhibition of mRNA expression of inflammatory mediators, including TNF-α, IL-1β, as well as TNF-α and IL-6 in cultured hepatocyte cells, and reduces the number of Kupffer CD68(+) cells recruited to the liver [200,201] (Table 1).
3.1.14. Fucoidan
Fucoidan, a sulfated polysaccharide containing substantial amounts of L-fucose and sulfate ester groups, is readily present in edible brown seaweeds, which are widely consumed in Asian countries due to their extensive health benefits [202,203]. An ex vivo angiogenesis assay showed that fucoidan caused a significant reduction in microvessel outgrowth and significantly reduced the expression of the angiogenesis factor VEGF-A [204]. Fucoidan was also evaluated for its activity in combination with sorafenib and bevacizumab in HCC. In vitro, on Huh-7 cells, fucoidan showed a potent synergistic effect with anti-angiogenic drugs and significantly reduced HCC cell line viability in a dose-dependent manner via inhibition of pro-angiogenic PI3K/AKT/mTOR and KRAS/BRAF/MAPK pathways [205] (Table 1).
3.1.15. Carnosol and Carnosic Acid
Carnosol and carnosic acid are two major components of rosemary extracts that contribute to the chemopreventive, anti-inflammatory, antitumor, and antimetastatic activities of Rosmarinus officinalis [206,207]. Lopez-Jimenez et al. demonstrate for the first time how these diterpenes modulate different relevant steps of the angiogenic process, inhibiting cytokine-induced adhesion molecule expression, monocyte adhesion to endothelial cells through a mechanism related to NF-κB, and capillary tube formation by endothelial cells, in addition to inducing apoptosis in endothelial and tumor cells in vitro. These results suggest their potential use in the treatment of angiogenesis-related malignancies [208] (Table 1).

Table 1.
Antiangiogenic effects of natural compounds and their molecular mechanism.
| PHYTOCHEMICALS | MAIN DIETARY SOURCES | DESIGNED STUDY | ANTIANGIOGENIC EFFECTS |
REF. |
|---|---|---|---|---|
| Quercetin | apples, red grapes, citrus fruits, tomatoes, onions and green leafy vegetables | Mice | HMGB1/TLR2/4-NF-κB signaling pathway downregulation; TGF-β1/Smads and PI3K/Akt crosstalk regulation |
92; 93 |
| Silybin | Silybum marianum | Rats | NF-κB, PGE 2 and LTB 4 inhibition | 102; 106 |
| Breviscapine | Erigeron breviscapus | Mice | TGF-β-activated kinase 1 (TAK1)/ NF-κB signaling pathway Inhibition | 114 |
| ALS-L1023 | Melissa officinalis | Mice | mRNA modulation of VEGF-A, FGF-2, MMP-2, MMP-9 TIMP-1, TIMP-2, TSP-1 | 122; 123 |
| Curcumin | Curcuma longa | Mice | NF-κB inhibition; TGF-β downregulation | 133; 136 |
| Sulforaphane | Broccoli, cauliflowers | Hep G2 and HUVEC cells | NF-κB/VEGF, STAT3/HIF-1α/VEGF signaling pathway inhibition; HO-1, NQO1, MRP2 genes upregulation |
140; 142-144 |
| Cordycepin | Cordyceps militaris | HCC cells | CXCR4 downregulation; NLRP3 suppression; NF-κB inhibition |
148-150 |
| Methoxyeugenol | Brazilian red propolis | Mice | mRNA up-regulation of PPARγ; TNF-α, IL-6 e IL-8 genes downregulation |
156 |
| Naringenin | citrus fruits | Mice | mRNA downregulation of NF-κB, IL-1β, IL-18 NLRP3 | 162 |
| Ferulic acid | cereals, vegetables, and plants such as Angelica sinensis | Mice | AMPKα up-regulation; lipogenic genes downregulation; Nrf2/HO-1 and PPARγ pathways activation; nuclear translocation block of NF-κB; PTP1B inhibition |
166-169 |
| Betaine | Beta vulgaris | HUVEC cells | NF-κB and Akt inhibition; FGF-2 MMP-2 MMP-downregulation |
174; 177; 178 |
| Catechins | grapes, apple and cocoa | Haec cells; Mice |
IL-1, IL-6, TNF-α inhibition; ErbB2 gene downregulation |
186; 188 |
| Puerarin | Pueraria lobata | Rats | (JAK2)/STAT3 signaling pathway regulation; JNK activity regulation, | 190; 193 |
| Resveratrol | grape skins, blueberries, raspberries, mulberries, and peanuts | Primary hepatocyte; Mice |
Nrf2 ubiquitination inhibition; TNF-α, IL-1β, IL-6 mRNA inhibition |
197; 200; 201 |
| Fucoidan | edible brown seaweeds | HCC; Huh-7 cells | VEGF-A gene downregulation | 204; 205 |
|
Carnosol and Carnosic Acid |
Rosmarinus officinalis |
BAECs and HUVEC cells | NF-κB inhibition | 208 |
4.1. Conclusions and Perspectives
Angiogenesis plays an important role in the development of liver disease, contributing to the progression of fibrosis and remodeling leading to capillarization of the sinusoids with the formation of intrahepatic shunts. As a result, vascular resistance increases and hepatocyte perfusion decreases, leading to hypoxia.
Many anti-angiogenic drugs, mainly tyrosine kinase inhibitors and humanized monoclonal antibodies, are currently used, but they are often expensive or toxic, limiting their use in many cases. Therefore, it is useful to identify less toxic, inexpensive, novel and effective anti-angiogenic compounds. In this regard, several natural plant products have shown prominent anti-angiogenic effects. However, the bioavailability of natural products is a critical issue since many of them have poor aqueous solubility and low absorption rate.
On the positive side, a large number of natural anti-angiogenic substances are being discovered and the study of their complex mechanisms has not yet been fully investigated.
Therefore, it is of great interest to study natural compounds and their derivatives to gain new insights into the biochemical mechanisms involved in signaling pathways in liver diseases. Accordingly, research on new natural products with anti-angiogenic effects seems very promising and may lead to the discovery of new drugs, which would be a significant achievement in this field. Furthermore, this sets the stage has the potential to identify new targets for more selective and effective synthetic anti-angiogenic agents.
Author Contributions
Writing—original draft preparation, S.N. and V.V.; writing—review and editing, N.T., A.B., M.F.T.; supervision, P.C. and M.F.T. All authors have read and agreed to the published version of the manuscript.
Funding
Not applicable.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Trefts, E.; Gannon, M.; Wasserman, D. H. The liver. Current biology. 2017, CB, 27, R1147–R1151. [CrossRef]
- Racanelli, V.; Rehermann, B. The liver as an immunological organ. Hepatology (Baltimore, Md.). 2006, 43, S54–S62. [CrossRef]
- Ko, S.; Russell, J. O.; Molina, L. M.; Monga, S. P. Liver Progenitors and Adult Cell Plasticity in Hepatic Injury and Repair: Knowns and Unknowns. Annual review of pathology. 2020, 15, 23–50. [CrossRef]
- Tsuchida, T.; Friedman, S. L. Mechanisms of hepatic stellate cell activation. Nature reviews. Gastroenterology & hepatology. 2017, 14, 397–411. [CrossRef]
- Friedman, S. L.; Neuschwander-Tetri, B. A.; Rinella, M.; Sanyal, A. J.Mechanisms of NAFLD development and therapeutic strategies. Nature medicine. 2018, 24, 908–922. [CrossRef]
- Gilgenkrantz, H.; Collin de l'Hortet, A. Understanding Liver Regeneration: From Mechanisms to Regenerative Medicine. The American journal of pathology. 2018, 188, 1316–1327. [CrossRef]
- Nassir, F.; Rector, R. S.; Hammoud, G. M.; Ibdah, J. A. Pathogenesis and Prevention of Hepatic Steatosis. Gastroenterology & hepatology. 2015, 11, 167–175.
- Forbes, S. J., Newsome, P. N. Liver regeneration - mechanisms and models to clinical application. Nature reviews. Gastroenterology & hepatology. 2016, 13, 473–485. [CrossRef]
- Seth, D.; Haber, P. S.; Syn, W. K.; Diehl, A. M.; Day, C. P. Pathogenesis of alcohol-induced liver disease: classical concepts and recent advances. Journal of gastroenterology and hepatology. 2011, 26, 1089–1105. [CrossRef]
- Li, S.; Tan, H. Y.; Wang, N.; Zhang, Z. J.; Lao, L.; Wong, C. W.; & Feng, Y. The Role of Oxidative Stress and Antioxidants in Liver Diseases. International journal of molecular sciences. 2015, 16, 26087–26124. [CrossRef]
- Ceni, E.; Mello, T.; & Galli, A. Pathogenesis of alcoholic liver disease: role of oxidative metabolism. World journal of gastroenterology. 2014, 20, 17756–17772. [CrossRef]
- Quagliariello, V.; Basilicata, M. G.; Pepe, G.; De Anseris, R.; Di Mauro, A.; Scognamiglio, G.; Palma, G.; Vestuto, V.; Buccolo, S.; Luciano, A.; Barbieri, M.; Bruzzese, F.; Maurea, C.; Pumpo, R.; Ostacolo, C.; Campiglia, P.; Berretta, M.; Maurea, N. Combination of Spirulina platensis, Ganoderma lucidum and Moringa oleifera Improves Cardiac Functions and Reduces Pro-Inflammatory Biomarkers in Preclinical Models of Short-Term Doxorubicin-Mediated Cardiotoxicity: New Frontiers in Cardioncology?. Journal of cardiovascular development and disease. 2022, 9, 423. [CrossRef]
- Cao, P.; Sun, J.; Sullivan, M. A.; Huang, X.; Wang, H.; Zhang, Y.; Wang, N.; Wang, K. Angelica sinensis polysaccharide protects against acetaminophen-induced acute liver injury and cell death by suppressing oxidative stress and hepatic apoptosis in vivo and in vitro. International journal of biological macromolecules. 2018, 111, 1133–1139. [CrossRef]
- Chen, A. Acetaldehyde stimulates the activation of latent transforming growth factor-beta1 and induces expression of the type II receptor of the cytokine in rat cultured hepatic stellate cells. The Biochemical journal. 2002, 368, 683–693. [CrossRef]
- Parola, M.; & Robino, G. Oxidative stress-related molecules and liver fibrosis. Journal of hepatology. 2001, 35, 297–306. [CrossRef]
- Kaphalia, B. S.; Cai, P.; Khan, M. F.; Okorodudu, A. O.; Ansari, G. A. Fatty acid ethyl esters: markers of alcohol abuse and alcoholism. Alcohol. 2004, 34, 151–158. [CrossRef]
- Cederbaum, A. I. Nrf2 and antioxidant defense against CYP2E1 toxicity. Sub-cellular biochemistry. 2013, 67, 105–130. [CrossRef]
- Lin, Q.; Kang, X.; Li, X.; Wang, T.; Liu, F.; Jia, J.; Jin, Z.; Xue, Y. NF-κB-mediated regulation of rat CYP2E1 by two independent signaling pathways. PloS one. 2019, 14, e0225531. [CrossRef]
- Magne, L.; Blanc, E.; Legrand, B.; Lucas, D.; Barouki, R.; Rouach, H.; Garlatti, M. ATF4 and the integrated stress response are induced by ethanol and cytochrome P450 2E1 in human hepatocytes. Journal of hepatology. 2011, 54, 729–737. [CrossRef]
- Hwang, S.; Yun, H.; Moon, S.; Cho, Y. E.; Gao, B. Role of Neutrophils in the Pathogenesis of Nonalcoholic Steatohepatitis. Frontiers in endocrinology. 2021, 12, 751802. [CrossRef]
- Lin, Q.; Kang, X.; Li, X.; Wang, T.; Liu, F.; Jia, J.; Jin, Z.; Xue, Y. NF-κB-mediated regulation of rat CYP2E1 by two independent signaling pathways. PloS one. 2019, 14, e0225531. [CrossRef]
- Ali, H.; Assiri, M. A.; Shearn, C. T.; Fritz, K. S. Lipid peroxidation derived reactive aldehydes in alcoholic liver disease. Current opinion in toxicology. 2019, 13, 110–117. [CrossRef]
- Abu-Freha, N.; Cohen, B.; Gordon, M.; Weissmann, S.; Fich, A.; Munteanu, D.; Yardeni, D.; Etzion, O. Comorbidities and Malignancy among NAFLD Patients Compared to the General Population, A Nation-Based Study. Biomedicines. 2023, 11, 1110. [CrossRef]
- Kosmalski, M.; Śliwińska, A.; Drzewoski, J. Non-Alcoholic Fatty Liver Disease or Type 2 Diabetes Mellitus—The Chicken or the Egg Dilemma. Biomedicines. 2023, 11, 1097. [CrossRef]
- Cusi K. Role of insulin resistance and lipotoxicity in non-alcoholic steatohepatitis. Clinics in liver disease. 2009, 13, 545–563. [CrossRef]
- Grønbaek, H.; Thomsen, K. L.; Rungby, J.; Schmitz, O.; Vilstrup, H. Role of nonalcoholic fatty liver disease in the development of insulin resistance and diabetes. Expert review of gastroenterology & hepatology. 2008, 2, 705–711. [CrossRef]
- Day, C. P.; James, O. F. Steatohepatitis: a tale of two "hits"?. Gastroenterology. 1998, 114, 842–845. [CrossRef]
- Mohammad, S.; Thiemermann, C. Role of Metabolic Endotoxemia in Systemic Inflammation and Potential Interventions. Frontiers in immunology. 2021, 11, 594150. [CrossRef]
- Luukkonen, P. K.; Zhou, Y.; Sädevirta, S.; Leivonen, M.; Arola, J.; Orešič, M.; Hyötyläinen, T.; Yki-Järvinen, H. Hepatic ceramides dissociate steatosis and insulin resistance in patients with non-alcoholic fatty liver disease. Journal of hepatology. 2016, 64, 1167–1175. [CrossRef]
- Adolph, T. E.; Grander, C.; Grabherr, F.; Tilg, H. Adipokines and Non-Alcoholic Fatty Liver Disease: Multiple Interactions. International journal of molecular sciences. 2017, 18, 1649. [CrossRef]
- Vestuto, V.; Di Sarno, V.; Musella, S.; Di Dona, G.; Moltedo, O.; Gomez-Monterrey, I.M.; Bertamino, A.; Ostacolo, C.; Campiglia, P.; Ciaglia, T. New Frontiers on ER Stress Modulation: Are TRP Channels the Leading Actors?.International journal of molecular sciences. 2023, 24, 185. [CrossRef]
- Fabbrini, E.; Magkos, F.; Mohammed, B. S.; Pietka, T.; Abumrad, N. A.; Patterson, B. W.; Okunade, A.; Klein, S. Intrahepatic fat, not visceral fat, is linked with metabolic complications of obesity. Proceedings of the National Academy of Sciences of the United States of America. 2009, 106, 15430–15435. [CrossRef]
- Baffy, G. Kupffer cells in non-alcoholic fatty liver disease: the emerging view. Journal of hepatology. 51, 212–223. [CrossRef]
- Grimaldi, M.; Palisi, A.; Marino, C.; Montoro, P.; Capasso, A.; Novi, S.; Tecce, M. F.; D'Ursi, A. M. NMR-based metabolomic profile of hypercholesterolemic human sera: Relationship with in vitro gene expression?. PloS one. 2020, 15(4), e0231506. [CrossRef]
- Asif, S.; Kim, R. Y.; Fatica, T.; Sim, J.; Zhao, X.; Oh, Y.; Denoncourt, A., Cheung, A. C., Downey, M., Mulvihill, E. E., & Kim, K. H. (). Hmgcs2-mediated ketogenesis modulates high-fat diet-induced hepatosteatosis. Molecular metabolism. 2022, 61, 101494. [CrossRef]
- Czaja M. J. JNK regulation of hepatic manifestations of the metabolic syndrome. Trends in endocrinology and metabolism: TEM. 2010, 21, 707–713. [CrossRef]
- Najjar, S.M.; Abdolahipour, R.; Ghadieh, H.E.; Jahromi, M.S.; Najjar, J.A.; Abuamreh, B.A.M.; Zaidi, S.; Kumarasamy, S.; Muturi, H.T. Regulation of Insulin Clearance by Non-Esterified Fatty Acids. Biomedicines. 2022, 10, 1899. [CrossRef]
- Polyzos, S. A.; Kountouras, J.; Zavos, C. Nonalcoholic fatty liver disease: the pathogenetic roles of insulin resistance and adipocytokines. Current molecular medicine. 2009, 9, 299–314. [CrossRef]
- Berger, J.; Moller, D. E. The mechanisms of action of PPARs. Annual review of medicine. 2002, 53, 409–435. [CrossRef]
- Montagner, A.; Polizzi, A.; Fouché, E.; Ducheix, S.; Lippi, Y.; Lasserre, F.; Barquissau, V.; Régnier, M.; Lukowicz, C.; Benhamed, F.; Iroz, A.; Bertrand-Michel, J.; Al Saati, T.; Cano, P.; Mselli-Lakhal, L.; Mithieux, G.; Rajas, F.; Lagarrigue, S.; Pineau, T.; Loiseau, N.; Guillou, H. Liver PPARα is crucial for whole-body fatty acid homeostasis and is protective against NAFLD. Gut. 2016, 65, 1202–1214. [CrossRef]
- Duan, Y.; Pan, X.; Luo, J.; Xiao, X.; Li, J.; Bestman, P. L.; Luo, M. Association of Inflammatory Cytokines With Non-Alcoholic Fatty Liver Disease. Frontiers in immunology. 2022, 13, 880298. [CrossRef]
- Khan, R. S.; Bril, F.; Cusi, K.; Newsome, P. N. Modulation of Insulin Resistance in Nonalcoholic Fatty Liver Disease. Hepatology (Baltimore, Md.). 2019, 70, 711–724. [CrossRef]
- Syn, W. K.; Choi, S. S.; Diehl, A. M. Apoptosis and cytokines in non-alcoholic steatohepatitis. Clinics in liver disease. 2009, 13, 565–580. [CrossRef]
- Auguet, T.; Bertran, L.; Binetti, J.; Aguilar, C.; Martínez, S.; Sabench, F.; Lopez-Dupla, J. M.; Porras, J. A.; Riesco, D.; Del Castillo, D.; Richart, C. Relationship between IL-8 Circulating Levels and TLR2 Hepatic Expression in Women with Morbid Obesity and Nonalcoholic Steatohepatitis. International journal of molecular sciences. 2020, 21, 4189. [CrossRef]
- Henao-Mejia, J.; Elinav, E.; Jin, C.; Hao, L.; Mehal, W. Z.; Strowig, T.; Thaiss, C. A.; Kau, A. L.; Eisenbarth, S. C.; Jurczak, M. J.; Camporez, J. P.; Shulman, G. I.; Gordon, J. I.; Hoffman, H. M.; Flavell, R. A. Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature. 2012, 482, 179–185. [CrossRef]
- Baker, R. G.; Hayden, M. S.; Ghosh, S. NF-κB, inflammation, and metabolic disease. Cell metabolism. 2011, 13, 11–22. [CrossRef]
- Oates, J. R.; McKell, M. C.; Moreno-Fernandez, M. E.; Damen, M. S. M. A.; Deepe, G. S.; Jr, Qualls, J. E.; Divanovic, S. Macrophage Function in the Pathogenesis of Non-alcoholic Fatty Liver Disease: The Mac Attack. Frontiers in immunology. 2019, 10, 2893. [CrossRef]
- Buzzetti, E.; Pinzani, M.; Tsochatzis, E. A. The multiple-hit pathogenesis of non-alcoholic fatty liver disease (NAFLD). Metabolism: clinical and experimental. 2016, 65, 1038–1048. [CrossRef]
- Seki, E.; Schwabe, R. F. Hepatic inflammation and fibrosis: functional links and key pathways. Hepatology (Baltimore, Md.). 2015. 61, 1066–1079. [CrossRef]
- Torres, S.; Segalés, P.; García-Ruiz, C.; Fernández-Checa, J. C. Mitochondria and the NLRP3 Inflammasome in Alcoholic and Nonalcoholic Steatohepatitis. Cells. 2022, 11, 1475. [CrossRef]
- Seitz, H. K.; Bataller, R.; Cortez-Pinto, H.; Gao, B.; Gual, A.; Lackner, C.; Mathurin, P.; Mueller, S.; Szabo, G.; Tsukamoto, H. Alcoholic liver disease. Nature reviews. Disease primers. 2018, 4, 16. [CrossRef]
- Llovet, J. M.; Kelley, R. K.; Villanueva, A.; Singal, A. G.; Pikarsky, E.; Roayaie, S.; Lencioni, R.; Koike, K.; Zucman-Rossi, J.; Finn, R. S. Hepatocellular carcinoma. Nature reviews. Disease primers. 2021, 7, 6. [CrossRef]
- Yang, Y. M., Cho, Y. E., & Hwang, S. Crosstalk between Oxidative Stress and Inflammatory Liver Injury in the Pathogenesis of Alcoholic Liver Disease. International journal of molecular sciences. 2022, 23, 774. [CrossRef]
- Dhar, D.; Baglieri, J.; Kisseleva, T.; Brenner, D. A. Mechanisms of liver fibrosis and its role in liver cancer. Experimental biology and medicine (Maywood, N.J.). 2020, 245, 96–108. [CrossRef]
- Pappachan, J. M.; Babu, S.; Krishnan, B.; Ravindran, N. C. Non-alcoholic Fatty Liver Disease: A Clinical Update. Journal of clinical and translational hepatology. 2017, 5, 384–393. [CrossRef]
- Ratziu, V., Bellentani, S., Cortez-Pinto, H., Day, C., & Marchesini, G. A position statement on NAFLD/NASH based on the EASL 2009 special conference. Journal of hepatology. 2010, 53, 372–384. [CrossRef]
- Shan, Z.; Ju, C. Hepatic Macrophages in Liver Injury. Frontiers in immunology. 2020, 11, 322. [CrossRef]
- Wree, A.; McGeough, M. D.; Peña, C. A.; Schlattjan, M.; Li, H.; Inzaugarat, M. E.; Messer, K.; Canbay, A.; Hoffman, H. M.; Feldstein, A. E. NLRP3 inflammasome activation is required for fibrosis development in NAFLD. Journal of molecular medicine (Berlin, Germany). 2014, 92, 1069–1082. [CrossRef]
- Papa, S.; Bubici, C.; Zazzeroni, F.; Franzoso, G. Mechanisms of liver disease: cross-talk between the NF-kappaB and JNK pathways. Biological chemistry, 2009, 390(10), 965–976. [CrossRef]
- Lee, J. S.; Zheng, Z.; Mendez, R.; Ha, S. W.; Xie, Y.; Zhang, K. Pharmacologic ER stress induces non-alcoholic steatohepatitis in an animal model. Toxicology letters. 2012, 211, 29–38. [CrossRef]
- Brunt, E. M.; Tiniakos, D. G. Pathology of steatohepatitis. Best practice & research. Clinical gastroenterology. 2002, 16, 691–707. [CrossRef]
- Tiniakos, D. G.; Vos, M. B.; Brunt, E. M. Nonalcoholic fatty liver disease: pathology and pathogenesis. Annual review of pathology. 2010, 5, 145–171. [CrossRef]
- Wong, V. W.; Wong, G. L.; Choi, P. C.; Chan, A. W.; Li, M. K.; Chan, H. Y.; Chim, A. M.; Yu, J.; Sung, J. J.; Chan, H. L. Disease progression of non-alcoholic fatty liver disease: a prospective study with paired liver biopsies at 3 years. Gut. 2010, 59, 969–974. [CrossRef]
- Loomba, R.; Lawitz, E.; Mantry, P. S.; Jayakumar, S.; Caldwell, S. H.; Arnold, H.; Diehl, A. M.; Djedjos, C. S.; Han, L.; Myers, R. P.; Subramanian, G. M.; McHutchison, J. G.; Goodman, Z. D.; Afdhal, N. H.; Charlton, M. R.; GS-US-384-1497 Investigators. The ASK1 inhibitor selonsertib in patients with nonalcoholic steatohepatitis: A randomized, phase 2 trial. Hepatology (Baltimore, Md.). 2018, 67, 549–559. [CrossRef]
- Bianco, C.; Casirati, E.; Malvestiti, F.; Valenti, L. Genetic predisposition similarities between NASH and ASH: Identification of new therapeutic targets. JHEP reports: innovation in hepatology. 2021, 3, 100284. [CrossRef]
- Neuschwander-Tetri B. A. Therapeutic Landscape for NAFLD in 2020. Gastroenterology. 2020, 158, 1984–1998.e3. [CrossRef]
- Philip Esteban, J.; Dinani, A. Lifestyle Interventions Beyond Diet and Exercise for Patients With Nonalcoholic Fatty Liver Disease. Gastroenterology & hepatology. 2020, 16, 119–130.
- Norata, G. D.; Pellegatta, F.; Catapano, A. L. "Peroxisome proliferator activated receptors" e patologie cardiovascolari [Peroxisome proliferator activated receptors and cardiovascular disorders]. Italian heart journal. Supplement: official journal of the Italian Federation of Cardiology. 2003, 4, 8–18.
- Loria, P.; Adinolfi, L. E.; Bellentani, S.; Bugianesi, E.; Grieco, A.; Fargion, S.; Gasbarrini, A.; Loguercio, C.; Lonardo, A.; Marchesini, G.; Marra, F.; Persico, M.; Prati, D.; Baroni, G. S.; NAFLD Expert Committee of the Associazione Italiana per lo studio del Fegato Practice guidelines for the diagnosis and management of nonalcoholic fatty liver disease. A decalogue from the Italian Association for the Study of the Liver (AISF) Expert Committee. Digestive and liver disease: official journal of the Italian Society of Gastroenterology and the Italian Association for the Study of the Liver. 2010, 42, 272–282. [CrossRef]
- Seaman, S.; Stevens, J.; Yang, M. Y.; Logsdon, D.; Graff-Cherry, C.; St Croix, B. Genes that distinguish physiological and pathological angiogenesis. Cancer cell. 2007, 11, 539–554. [CrossRef]
- Bocca, C.; Novo, E.; Miglietta, A.; Parola, M. Angiogenesis and Fibrogenesis in Chronic Liver Diseases. Cellular and molecular gastroenterology and hepatology. 2015, 1, 477–488. [CrossRef]
- Fernández, M.; Semela, D.; Bruix, J.; Colle, I.; Pinzani, M.; Bosch, J. Angiogenesis in liver disease. Journal of hepatology. 2009, 50, 604–620. [CrossRef]
- Camenisch, G.; Pisabarro, M. T.; Sherman, D.; Kowalski, J.; Nagel, M.; Hass, P.; Xie, M. H.; Gurney, A.; Bodary, S.; Liang, X. H.; Clark, K.; Beresini, M.; Ferrara, N.; Gerber, H. P. ANGPTL3 stimulates endothelial cell adhesion and migration via integrin alpha vbeta 3 and induces blood vessel formation in vivo. The Journal of biological chemistry. 2002, 277, 17281–17290. [CrossRef]
- Valfrè di Bonzo, L.; Novo, E.; Cannito, S.; Busletta, C.; Paternostro, C.; Povero, D.; Parola, M. Angiogenesis and liver fibrogenesis. Histology and histopathology. 2009, 24, 1323–1341. [CrossRef]
- Baumeister, S. E.; Völzke, H.; Marschall, P.; John, U.; Schmidt, C. O.; Flessa, S.; Alte, D. Impact of fatty liver disease on health care utilization and costs in a general population: a 5-year observation. Gastroenterology. 2008, 134, 85–94. [CrossRef]
- Nath, B.; Szabo, G. Hypoxia and hypoxia inducible factors: diverse roles in liver diseases. Hepatology (Baltimore, Md.). 2012, 55, 622–633. [CrossRef]
- Kukla M. Angiogenesis: a phenomenon which aggravates chronic liver disease progression. Hepatology international. 2013, 7, 4–12. [CrossRef]
- Parola, M.; Marra, F.; Pinzani, M. Myofibroblast - like cells and liver fibrogenesis: Emerging concepts in a rapidly moving scenario. Molecular aspects of medicine. 2008, 29:58–66 . [CrossRef]
- Lei, L.; Ei Mourabit, H.; Housset, C.; Cadoret, A.; Lemoinne, S. Role of Angiogenesis in the Pathogenesis of NAFLD. Journal of clinical medicine. 2021, 10, 1338. [CrossRef]
- Coulon, S.; Heindryckx, F.; Geerts, A.; Van Steenkiste, C.; Colle, I.; Van Vlierberghe, H. Angiogenesis in chronic liver disease and its complications. Liver international : official journal of the International Association for the Study of the Liver. 2011, 31, 146–162. [CrossRef]
- Dooley, S.; Ten Dijke, P. TGF-β in progression of liver disease. Cell and tissue research. 2012, 347, 245–256. [CrossRef]
- Zhu, B.; Lin, N.; Zhang, M.; Zhu, Y.; Cheng, H.; Chen, S.; Ling, Y.; Pan, W.; Xu, R. Activated hepatic stellate cells promote angiogenesis via interleukin-8 in hepatocellular carcinoma. Journal of translational medicine. 2015, 13, 365. [CrossRef]
- Copple, B. L.; Bai, S.; Burgoon, L. D.; Moon, J. O. Hypoxia-inducible factor-1α regulates the expression of genes in hypoxic hepatic stellate cells important for collagen deposition and angiogenesis. Liver international: official journal of the International Association for the Study of the Liver. 2011, 31, 230–244. [CrossRef]
- Weiskirchen, R.; Meurer, S. K.; Liedtke, C.; Huber, M. Mast Cells in Liver Fibrogenesis. Cells. 2019, 8, 1429. [CrossRef]
- Raevens, S.; Coulon, S.; Van Steenkiste, C.; Colman, R.; Verhelst, X.; Van Vlierberghe, H.; Geerts, A.; Perkmann, T.; Horvatits, T.; Fuhrmann, V.; Colle, I. Role of angiogenic factors/cell adhesion markers in serum of cirrhotic patients with hepatopulmonary syndrome. Liver international : official journal of the International Association for the Study of the Liver. 2015, 35, 1499–1507. [CrossRef]
- Bedogni, G.; Miglioli, L.; Masutti, F.; Castiglione, A.; Crocè, L. S.; Tiribelli, C.; Bellentani, S. Incidence and natural course of fatty liver in the general population: the Dionysos study. Hepatology (Baltimore, Md.). 2007, 46, 1387–1391. [CrossRef]
- Lee, C.K.; Lee, M.E.; Lee, W.S.; Kim, J.M.; Park, K.H.; Kim, T.S.; Lee, K.Y.; Ahn, J.B.; Chung, H.C.; Rha, S.Y. Dovitinib (TKI258), a Multi-Target Angiokinase Inhibitor, Is Effective Regardless of KRAS or BRAF Mutation Status in Colorectal Cancer. Am. J. Cancer Res. 2015, 5, 72–86.
- Lopes-Coelho, F.; Martins, F.; Pereira, S.A.; Serpa, J. Anti-Angiogenic Therapy: Current Challenges and Future Perspectives. Int. J. Mol. Sci. 2021, 22, 3765. [CrossRef]
- Zhu, X. D.; Tang, Z. Y.; Sun, H. C. Targeting angiogenesis for liver cancer: Past, present, and future. Genes & diseases. 2020, 7, 328–335. [CrossRef]
- Russo, M.; Spagnuolo, C.; Tedesco, I.; Bilotto, S.; Russo, G. L. The flavonoid quercetin in disease prevention and therapy: facts and fancies. Biochemical pharmacology. 2012, 83, 6–15. [CrossRef]
- Hisaka, T.; Sakai, H.; Sato, T.; Goto, Y.; Nomura, Y.; Fukutomi, S.; Fujita, F.; Mizobe, T.; Nakashima, O.; Tanigawa, M.; Naito, Y.; Akiba, J.; Ogasawara, S.; Nakashima, K.; Akagi, Y.; Okuda, K.; Yano, H. Quercetin Suppresses Proliferation of Liver Cancer Cell Lines In Vitro. Anticancer research. 2020, 40, 4695–4700. [CrossRef]
- Li, X.; Liu H. C.; Yao, Q. Y.; Xu, B. L.; Zhang, S. C.; Tu, C. T. Quercetin Protects Mice from ConA-Induced Hepatitis by Inhibiting HMGB1-TLR Expression and Down-Regulating the Nuclear Factor Kappa B Pathway. Inflammation. 2016, 96–106. [CrossRef]
- Wu, L.; Zhang, Q.; Mo, W.; Feng, J.; Li, S.; Li, J.; Liu, T.; Xu, S.; Wang, W.; Lu, X.; Yu, Q.; Chen, K.; Xia, Y.; Lu, J.; Xu, L.; Zhou, Y.; Fan, X.; Guo, C. Quercetin prevents hepatic fibrosis by inhibiting hepatic stellate cell activation and reducing autophagy via the TGF-β1/Smads and PI3K/Akt pathways. Scientific reports. 2017, 7(1), 9289. [CrossRef]
- Sica, A.; Invernizzi, P.; Mantovani, A. Macrophage plasticity and polarization in liver homeostasis and pathology. Hepatology (Baltimore, Md.). 2014, 59, 2034–2042. [CrossRef]
- Tacke, F.; Zimmermann, H. W. Macrophage heterogeneity in liver injury and fibrosis. Journal of hepatology. 2014, 60, 1090–1096. [CrossRef]
- Pradere, J. P.; Kluwe, J.; De Minicis, S.; Jiao, J. J.; Gwak, G. Y.; Dapito, D. H.; Jang, M. K.; Guenther, N. D.; Mederacke, I.; Friedman, R.; Dragomir, A. C.; Aloman, C.; Schwabe, R. F. Hepatic macrophages but not dendritic cells contribute to liver fibrosis by promoting the survival of activated hepatic stellate cells in mice. Hepatology (Baltimore, Md.). 2013, 58, 1461–1473. [CrossRef]
- Kren, V.; Walterová, D. Silybin and silymarin--new effects and applications. Biomedical papers of the Medical Faculty of the University Palacky, Olomouc, Czechoslovakia. 2005, 149, 29–41. [CrossRef]
- Javed, S.; Kohli, K.; Ali, M. Reassessing bioavailability of silymarin. Alternative medicine review : a journal of clinical therapeutic. 2011, 16, 239–249.
- Surai P. F. Silymarin as a Natural Antioxidant: An Overview of the Current Evidence and Perspectives. Antioxidants (Basel, Switzerland). 2015, 4, 204–247. [CrossRef]
- Younossi, Z. M.; Koenig, A. B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology (Baltimore, Md.). 2016, 64, 73–84. [CrossRef]
- Hahn, G.; Lehmann, H. D.; Kürten, M.; Uebel, H.; Vogel, G. Zur Pharmakologie und Toxikologie von Silymarin, des antihepatotoxischen Wirkprinzipes aus Silybum marianum (L.) Gaertn [On the pharmacology and toxicology of silymarin, an antihepatotoxic active principle from Silybum marianum (L.) Gaertn]. Arzneimittel-Forschung. 1968, 18, 698–704.
- Kondylis, V.; Kumari, S.; Vlantis, K.; Pasparakis, M. The interplay of IKK, NF-κB and RIPK1 signaling in the regulation of cell death, tissue homeostasis and inflammation. Immunological reviews. 2017, 277, 113–127. [CrossRef]
- Gobejishvili L, Barve S, Joshi-Barve S, Uriarte S, Gobejishvili, L.; Barve, S.; Joshi-Barve, S.; Uriarte, S.; Song, Z.; McClain, C. Chronic ethanol-mediated decrease in cAMP primes macrophages to enhanced LPS-inducible NF-kappaB activity and TNF expression: relevance to alcoholic liver disease. American journal of physiology. Gastrointestinal and liver physiology. 2006, 291, G681–G688. [CrossRef]
- Guan, Y. S.; He, Q.; Wang, M. Q.; Li, P. Nuclear factor kappa B and hepatitis viruses. Expert opinion on therapeutic targets. 2008, 12, 265–280. [CrossRef]
- Muriel P. NF-kappaB in liver diseases: a target for drug therapy. Journal of applied toxicology: JAT. 2009, 29, 91–100. [CrossRef]
- Lieber, C. S.; Leo, M. A.; Cao, Q.; Ren, C.; DeCarli; L. M. Silymarin retards the progression of alcohol-induced hepatic fibrosis in baboons. Journal of clinical gastroenterology. 2003, 37, 336–339. [CrossRef]
- Wenfeng, Z.; Yakun, W.; Di, M.; Jianping, G.; Chuanxin, W.; Chun, H. Kupffer cells: increasingly significant role in nonalcoholic fatty liver disease. Annals of hepatology. 2014, 13, 489–495. [CrossRef]
- Trubitsyna, I.E.; Chikunova, B.Z.; Tkachenko, E.V.; Tsaregorodtseva, T.M.; Vinokurova, L.V.; Varvanina, G.G. [Pathophysiology of hormonal, immune, metabolic changes in acute and chronic pancreatitis. Experimental and clinical studies]. Eksp Klin Gastroenterol. 2008, 7, 40-4.
- Pengyue, Z.; Tao, G.; Hongyun, H.; Liqiang, Y.; Yihao, D. Breviscapine confers a neuroprotective efficacy against transient focal cerebral ischemia by attenuating neuronal and astrocytic autophagy in the penumbra. Biomed Pharmacother. 2017,90, 69-76 . [CrossRef]
- Lin, L. L.; Liu, A. J.; Liu, J. G.; Yu, X. H; Qin, L. P.; Su, D. F. Protective effects of scutellarin and breviscapine on brain and heart ischemia in rats. Journal of cardiovascular pharmacology. 2007, 50, 327–332. [CrossRef]
- Wang, L.; Ma, Q. Clinical benefits and pharmacology of scutellarin: A comprehensive review. Pharmacology & therapeutics. 2018, 190, 105–127. [CrossRef]
- Liu, Y.; Wen, P. H.; Zhang, X. X.; Dai, Y.; He, Q. Breviscapine ameliorates CCl4-induced liver injury in mice through inhibiting inflammatory apoptotic response and ROS generation. International journal of molecular medicine. 2018, 42, 755–768. [CrossRef]
- Fan, H.; Ma, X.; Lin, P.; Kang, Q.; Zhao, Z.; Wang, L.; Sun, D.; Cheng, J.; Li, Y. Scutellarin Prevents Nonalcoholic Fatty Liver Disease (NAFLD) and Hyperlipidemia via PI3K/AKT-Dependent Activation of Nuclear Factor (Erythroid-Derived 2)-Like 2 (Nrf2) in Rats. Medical science monitor: international medical journal of experimental and clinical research. 2017, 23, 5599–5612. [CrossRef]
- Sun, H.; Wang, X.; Chen, J.; Song, K.; Gusdon, A. M.; Li, L.; Bu, L.; Qu, S. Melatonin improves non-alcoholic fatty liver disease via MAPK-JNK/P38 signaling in high-fat-diet-induced obese mice. Lipids in health and disease. 2016, 15, 202. [CrossRef]
- Wu, Y. K.; Hu, L. F.; Lou, D. S.; Wang, B. C.; Tan, J. Targeting DUSP16/TAK1 signaling alleviates hepatic dyslipidemia and inflammation in high fat diet (HFD)-challenged mice through suppressing JNK MAPK. Biochemical and biophysical research communications. 2020, 524, 142–149. [CrossRef]
- Wang, J.; Ma, J.; Nie, H.; Zhang, X. J.; Zhang, P.; She, Z. G.; Li, H.; Ji, Y. X.; Cai, J. Hepatic Regulator of G Protein Signaling 5 Ameliorates Nonalcoholic Fatty Liver Disease by Suppressing Transforming Growth Factor Beta-Activated Kinase 1-c-Jun-N-Terminal Kinase/p38 Signaling. Hepatology (Baltimore, Md.). 2021, 73, 104–125. [CrossRef]
- Shen, X.; Guo, H.; Xu, J.; Wang, J. Inhibition of lncRNA HULC improves hepatic fibrosis and hepatocyte apoptosis by inhibiting the MAPK signaling pathway in rats with nonalcoholic fatty liver disease. Journal of cellular physiology. 2019, 234, 18169–18179. [CrossRef]
- Park, B. Y.; Lee, H.; Woo, S.; Yoon, M.; Kim, J.; Hong, Y.; Lee, H. S.; Park, E. K.; Hahm, J. C.; Kim, J. W.; Shin, S. S.; Kim, M. Y.; Yoon, M. Reduction of Adipose Tissue Mass by the Angiogenesis Inhibitor ALS-L1023 from Melissa officinalis. PloS one. 2015, 10, e0141612. [CrossRef]
- Kim, J.; Lee, H.; Lim, J.; Oh, J.; Shin, S. S.; Yoon, M. The Angiogenesis Inhibitor ALS-L1023 from Lemon-Balm Leaves Attenuates High-Fat Diet-Induced Nonalcoholic Fatty Liver Disease through Regulating the Visceral Adipose-Tissue Function. International journal of molecular sciences. 2017, 18, 846. [CrossRef]
- Lee, E. K.; Kim, Y. J.; Kim, J. Y.; Song, H. B.; Yu, H. G. Melissa officinalis extract inhibits laser-induced choroidal neovascularization in a rat model. PloS one. 2014, 9, e110109. [CrossRef]
- Sipos, S.; Moacă, E.-A.; Pavel, I.Z.; Avram, Ş.; Crețu, O.M.; Coricovac, D.; Racoviceanu, R.-M.; Ghiulai, R.; Pană, R.D.; Şoica, C.M.; Borcan, F.; Dehelean, C.A.; Crăiniceanu, Z. Melissa officinalis L. Aqueous Extract Exerts Antioxidant and Antiangiogenic Effects and Improves Physiological Skin Parameters. Molecules. 2021, 26, 2369. [CrossRef]
- Woo, S.; Yoon, M.; Kim, J.; Hong, Y.; Kim, M. Y.; Shin, S. S.; Yoon, M. The anti-angiogenic herbal extract from Melissa officinalis inhibits adipogenesis in 3T3-L1 adipocytes and suppresses adipocyte hypertrophy in high fat diet-induced obese C57BL/6J mice. Journal of ethnopharmacology. 2016, 178, 238–250. [CrossRef]
- Park, B. Y.; Lee, H.; Woo, S.; Yoon, M.; Kim, J.; Hong, Y.; Lee, H. S.; Park, E. K.; Hahm, J. C.; Kim, J. W.; Shin, S. S.; Kim, M. Y.; Yoon, M. Reduction of Adipose Tissue Mass by the Angiogenesis Inhibitor ALS-L1023 from Melissa officinalis. PloS one. 2015, 10, e0141612. [CrossRef]
- Kim, J.; Lee, H.; Lim, J.; Oh, J.; Shin, S. S.; Yoon, M. The Angiogenesis Inhibitor ALS-L1023 from Lemon-Balm Leaves Attenuates High-Fat Diet-Induced Nonalcoholic Fatty Liver Disease through Regulating the Visceral Adipose-Tissue Function. International journal of molecular sciences. 2017, 18, 846. [CrossRef]
- Kim, J.; Lee, H.; Lim, J.; Lee, H.; Yoon, S.; Shin, S. S.; Yoon, M. The lemon balm extract ALS-L1023 inhibits obesity and nonalcoholic fatty liver disease in female ovariectomized mice. Food and chemical toxicology: an international journal published for the British Industrial Biological Research Association. 2017, 106, 292–305. [CrossRef]
- Lee, E.J.; Kim, Y.; Kim, J.E.; Yoon, E.L.; Lee, S.R.; Jun, D.W. ALS-L1023 from Melissa officinalis Alleviates Liver Fibrosis in a Non-Alcoholic Fatty Liver Disease Model. Life. 2023, 13, 100. [CrossRef]
- Howells, L. M.; Iwuji, C. O. O.; Irving, G. R. B.; Barber, S.; Walter, H.; Sidat, Z.; Griffin-Teall, N.; Singh, R.; Foreman, N.; Patel, S. R.; Morgan, B.; Steward, W. P.; Gescher, A.; Thomas, A. L.; Brown, K. Curcumin Combined with FOLFOX Chemotherapy Is Safe and Tolerable in Patients with Metastatic Colorectal Cancer in a Randomized Phase IIa Trial. The Journal of nutrition. 2019, 149, 1133–1139. [CrossRef]
- Marino, P.; Pepe, G.; Basilicata, M.G.; Vestuto, V.; Marzocco, S.; Autore, G.; Procino, A.; Gomez-Monterrey, I.M.; Manfra, M.; Campiglia, P. Potential Role of Natural Antioxidant Products in Oncological Diseases. Antioxidants 2023, 12, 704. [CrossRef]
- Longobardi, C.; Damiano, S.; Andretta, E.; Prisco, F.; Russo, V.; Pagnini, F.; Florio, S.; Ciarcia, R. Curcumin Modulates Nitrosative Stress, Inflammation, and DNA Damage and Protects against Ochratoxin A-Induced Hepatotoxicity and Nephrotoxicity in Rats. Antioxidants (Basel, Switzerland). 2021, 10, 1239. [CrossRef]
- Damiano, S.; Longobardi, C.; Andretta, E.; Prisco, F.; Piegari, G.; Squillacioti, C.; Montagnaro, S.; Pagnini, F.; Badino, P.; Florio, S.; Ciarcia, R. Antioxidative Effects of Curcumin on the Hepatotoxicity Induced by Ochratoxin A in Rats. Antioxidants (Basel, Switzerland). 2021, 10, 125. [CrossRef]
- Randino, R.; Grimaldi, M.; Persico, M.; De Santis, A.; Cini, E.; Cabri, W.; Riva, A.; D'Errico, G.; Fattorusso, C.; D'Ursi, A. M.; & Rodriquez, M. Investigating the Neuroprotective Effects of Turmeric Extract: Structural Interactions of β-Amyloid Peptide with Single Curcuminoids. Scientific reports. 2016, 6, 38846. [CrossRef]
- Vera-Ramirez, L.; Pérez-Lopez, P.; Varela-Lopez, A.; Ramirez-Tortosa, M.; Battino, M.; Quiles, J.L. Curcumin and liver disease. BioFactors. 2013, 39, 88-100. [CrossRef]
- Bhandarkar, S. S.; Arbiser, J. L. Curcumin as an inhibitor of angiogenesis. Advances in experimental medicine and biology. 2007, 595, 185–195. [CrossRef]
- Damiano, S.; Jarriyawattanachaikul, W.; Girolami, F.; Longobardi, C.; Nebbia, C.; Andretta, E.; Lauritano, C.; Dabbou, S.; Avantaggiato, G.; Schiavone, A.; Badino, P.; Ciarcia, R. Curcumin Supplementation Protects Broiler Chickens Against the Renal Oxidative Stress Induced by the Dietary Exposure to Low Levels of Aflatoxin B1. Frontiers in veterinary science. 2022, 8, 822227. [CrossRef]
- Farzaei, M. H.; Zobeiri, M.; Parvizi, F.; El-Senduny, F. F.; Marmouzi, I.; Coy-Barrera, E.; Naseri, R.; Nabavi, S. M.; Rahimi, R.; Abdollahi, M. Curcumin in Liver Diseases: A Systematic Review of the Cellular Mechanisms of Oxidative Stress and Clinical Perspective. Nutrients. 2018, 10, 855. [CrossRef]
- Zhao, Y.; Ma, X., Wang, J.; He, X.; Hu, Y.; Zhang, P.; Wang, R.; Li, R.; Gong, M.; Luo, S.; Xiao, X. Curcumin protects against CCl4-induced liver fibrosis in rats by inhibiting HIF-1α through an ERK-dependent pathway. Molecules (Basel, Switzerland). 2014, 19, 18767–18780. [CrossRef]
- Lee, D. E.; Lee, S. J.; Kim, S. J.; Lee, H. S.; Kwon, O. S. Curcumin Ameliorates Nonalcoholic Fatty Liver Disease through Inhibition of O-GlcNAcylation. Nutrients. 2019, 11(11), 2702. [CrossRef]
- Nakagawa, K.; Umeda, T.; Higuchi, O.; Tsuzuki, T.; Suzuki, T.; Miyazawa, T. Evaporative light-scattering analysis of sulforaphane in broccoli samples: Quality of broccoli products regarding sulforaphane contents. Journal of agricultural and food chemistry. 2006, 54, 2479–2483. [CrossRef]
- Kaiser, A. E.; Baniasadi, M.; Giansiracusa, D.; Giansiracusa, M.; Garcia, M.; Fryda, Z.; Wong, T. L.; Bishayee, A. Sulforaphane: A Broccoli Bioactive Phytocompound with Cancer Preventive Potential. Cancers. 2021, 13, 4796. [CrossRef]
- Xu, C.; Shen, G.; Chen, C.; Gélinas, C.; Kong, A. N. Suppression of NF-kappaB and NF-kappaB-regulated gene expression by sulforaphane and PEITC through IkappaBalpha, IKK pathway in human prostate cancer PC-3 cells. Oncogene. 2005, 24, 4486–4495. [CrossRef]
- Davis, R.; Singh, K. P.; Kurzrock, R.; Shankar, S. Sulforaphane inhibits angiogenesis through activation of FOXO transcription factors. Oncology reports. 2009, 22, 1473–1478. [CrossRef]
- Hahm, E. R.; Singh, S. V. Sulforaphane inhibits constitutive and interleukin-6-induced activation of signal transducer and activator of transcription 3 in prostate cancer cells. Cancer prevention research (Philadelphia, Pa.). 2010, 3, 484–494. [CrossRef]
- Noh, J. R.; Kim, Y. H.; Hwang, J. H.; Choi, D. H.; Kim, K. S.; Oh, W. K.; Lee, C. H. Sulforaphane protects against acetaminophen-induced hepatotoxicity. Food and chemical toxicology: an international journal published for the British Industrial Biological Research Association. 2015, 80, 193–200. [CrossRef]
- S, Sato; K, Moriya; M, Furukawa; S, Saikawa; T, Namisaki; M, Kitade; H, Kawaratani; K, Kaji; H, Takaya; N, Shimozato; Y, Sawada; K, Seki; K, Kitagawa; T, Akahane; A, Mitoro; Y, Okura; H, Yoshiji; J, Yamao Sulforaphane Inhibits Liver Cancer Cell Growth and Angiogenesis. Annals of Behavioural Science. 2018, 4, – . [CrossRef]
- Tuli, H. S.; Sharma, A. K.; Sandhu, S. S.; Kashyap, D. Cordycepin: a bioactive metabolite with therapeutic potential. Life sciences. 2013, 93, 863–869. [CrossRef]
- Cao, T.; Xu, R.; Xu, Y.; Liu, Y.; Qi, D.; Wan, Q. The protective effect of Cordycepin on diabetic nephropathy through autophagy induction in vivo and in vitro. International urology and nephrology. 2019, 51, 1883–1892. [CrossRef]
- Yoon, S. Y.; Park, S. J.; Park, Y. J. The Anticancer Properties of Cordycepin and Their Underlying Mechanisms. International journal of molecular sciences. 2018, 19, 3027. [CrossRef]
- Guo, Z.; Chen, W.; Dai, G.; Huang, Y. Cordycepin suppresses the migration and invasion of human liver cancer cells by downregulating the expression of CXCR4. International journal of molecular medicine. 2020, 45, 141–150. [CrossRef]
- Yang, J.; Li, Y. Z.; Hylemon, P. B.; Zhang, L. Y.; Zhou, H. P. Cordycepin inhibits LPS-induced inflammatory responses by modulating NOD-Like Receptor Protein 3 inflammasome activation. Biomedicine & pharmacotherapy = Biomedecine & pharmacotherapie. 2017, 95, 1777–1788. [CrossRef]
- Lee, H. H.; Kim, D.; Jung, J.; Kang, H.; Cho, H. NLRP3 Deficiency in Hepatocellular Carcinoma Enhances Surveillance of NK-92 through a Modulation of MICA/B. International journal of molecular sciences. 2021, 22, 9285. [CrossRef]
- Lan, T.; Yu, Y.; Zhang, J.; Li, H.; Weng, Q.; Jiang, S.; Tian, S.; Xu, T.; Hu, S.; Yang, G.; Zhang, Y.; Wang, W.; Wang, L.; Zhu, Q.; Rong, X.; Guo, J. Cordycepin Ameliorates Nonalcoholic Steatohepatitis by Activation of the AMP-Activated Protein Kinase Signaling Pathway. Hepatology (Baltimore, Md.). 2021, 74, 686–703. [CrossRef]
- Costa, B. P.; Nassr, M. T., Diz; F. M., Fernandes; K. H. A., Antunes; G. L., Grun, L. K.; Barbé-Tuana, F. M.; Nunes, F. B.; Branchini, G.; de Oliveira, J. R. Methoxyeugenol regulates the p53/p21 pathway and suppresses human endometrial cancer cell proliferation. Journal of ethnopharmacology. 2021, 267, 113645. [CrossRef]
- Antunes, G. L.: Matzenbacher, L. S.; Costa, B. P.; de Sousa Basso, B.; Levorse, V. G. S.; Antunes, K. H.; Costa-Ferro, Z. S. M.; de Oliveira, J. R. Methoxyeugenol Protects Against Lung Inflammation and Suppresses Neutrophil Extracellular Trap Formation in an LPS-Induced Acute Lung Injury Model. Inflammation. 2022, 45, 1534–1547. [CrossRef]
- Hoda, S.; Vermani, M.; Joshi, R. K.; Shankar, J.; Vijayaraghavan, P. Anti-melanogenic activity of Myristica fragrans extract against Aspergillus fumigatus using phenotypic based screening. BMC complementary medicine and therapies. 2020, 20, 67. [CrossRef]
- Zhao, W.; Song, F.; Hu, D.; Chen, H.; Zhai, Q.; Lu, W.; Zhao, J.; Zhang, H.; Chen, W.; Gu, Z.; Wang, G. The Protective Effect of Myristica fragrans Houtt. Extracts Against Obesity and Inflammation by Regulating Free Fatty Acids Metabolism in Nonalcoholic Fatty Liver Disease. Nutrients. 2020, 12, 2507. [CrossRef]
- de Souza Basso, B.; Haute, G. V.; Ortega-Ribera, M.; Luft, C.; Antunes, G. L.; Bastos, M. S.; Carlessi, L. P.; Levorse, V. G.; Cassel, E.; Donadio, M. V. F.; Santarém, E. R.; Gracia-Sancho, J.; Rodrigues de Oliveira, J. Methoxyeugenol deactivates hepatic stellate cells and attenuates liver fibrosis and inflammation through a PPAR-ɣ and NF-kB mechanism. Journal of ethnopharmacology. 2021, 280, 114433. [CrossRef]
- Mirza, A. Z.; Althagafi, I. I.; Shamshad, H. Role of PPAR receptor in different diseases and their ligands: Physiological importance and clinical implications. European journal of medicinal chemistry. 2019, 166, 502–513. [CrossRef]
- Kim, J. H.; Song, J.; Park, K. W. The multifaceted factor peroxisome proliferator-activated receptor γ (PPARγ) in metabolism, immunity, and cancer. Archives of pharmacal research. 2015, 38, 302–312. [CrossRef]
- Orhan, I. E.; Nabavi, S. F.; Daglia, M.; Tenore, G. C.; Mansouri, K.; Nabavi, S. M. Naringenin and atherosclerosis: a review of literature. Current pharmaceutical biotechnology. 2015, 16, 245–251. [CrossRef]
- Wali, A. F., Rashid, S., Rashid, S. M., Ansari, M. A., Khan, M. R., Haq, N., Alhareth, D. Y., Ahmad, A., & Rehman, M. U. Naringenin Regulates Doxorubicin-Induced Liver Dysfunction: Impact on Oxidative Stress and Inflammation. Plants (Basel, Switzerland). 2020, 9(4), 550. [CrossRef]
- Zhao, M.; Li, C.; Shen, F.; Wang, M.; Jia, N.; Wang, C. Naringenin ameliorates LPS-induced acute lung injury through its anti-oxidative and anti-inflammatory activity and by inhibition of the PI3K/AKT pathway. Experimental and therapeutic medicine. 2017, 14, 2228–2234. [CrossRef]
- Wang, Q.; Ou, Y.; Hu, G.; Wen, C.; Yue, S., Chen, C.; Xu, L.; Xie, J.; Dai, H.; Xiao, H.; Zhang, Y.; Qi, R. Naringenin attenuates non-alcoholic fatty liver disease by down-regulating the NLRP3/NF-κB pathway in mice. British journal of pharmacology. 2020, 177, 1806–1821. [CrossRef]
- Kohno, M.; Musashi, K.; Ikeda, H. O.; Horibe, T.; Matsumoto, A.; Kawakami, K. Oral administration of ferulic acid or ethyl ferulate attenuates retinal damage in sodium iodate-induced retinal degeneration mice. Scientific reports. 2020, 10, 8688. [CrossRef]
- Narasimhan, A.; Chinnaiyan, M.; Karundevi, B. Ferulic acid exerts its antidiabetic effect by modulating insulin-signalling molecules in the liver of high-fat diet and fructose-induced type-2 diabetic adult male rat. Applied physiology, nutrition, and metabolism = Physiologie appliquee, nutrition et metabolism. 2015, 40, 769–781. [CrossRef]
- Wang, J.M.; Sheng, Y.C.; Ji, L.L.; Wang, Z.T. Ferulic acid prevents liver injury and increases the anti-tumor effect of diosbulbin B in vivo. J Zhejiang Univ Sci B. 2014, 15, 540-7. [CrossRef]
- Hu, Y.; Li, J.; Chang, A. K.; Li, Y.; Tao, X.; Liu, W.; Wang, Z.; Su, W.; Li, Z.; Liang, X. Screening and tissue distribution of protein tyrosine phosphatase 1B inhibitors in mice following oral administration of Garcinia mangostana L. ethanolic extract. Food chemistry. 2021, 357, 129759. Advance online publication. [CrossRef]
- Mahmoud, A. M.; Hussein, O. E.; Hozayen, W. G.; Bin-Jumah, M.; Abd El-Twab, S. M. Ferulic acid prevents oxidative stress, inflammation, and liver injury via upregulation of Nrf2/HO-1 signaling in methotrexate-induced rats. Environmental science and pollution research international. 2020, 27, 7910–7921. [CrossRef]
- Wu, J.; Xue, X.; Fan, G.; Gu, Y.; Zhou, F.; Zheng, Q., Liu, R.; Li, Y.; Ma, B.; Li, S.; Huang, G.; Ma, L.; Li, X. Ferulic Acid Ameliorates Hepatic Inflammation and Fibrotic Liver Injury by Inhibiting PTP1B Activity and Subsequent Promoting AMPK Phosphorylation. Frontiers in pharmacology. 2021, 12, 754976. [CrossRef]
- Hawley, S. A.; Ford, R. J.; Smith, B. K.; Gowans, G. J.; Mancini, S. J.; Pitt, R. D.; Day, E. A.; Salt, I. P.; Steinberg, G. R.; Hardie, D. G. The Na+/Glucose Cotransporter Inhibitor Canagliflozin Activates AMPK by Inhibiting Mitochondrial Function and Increasing Cellular AMP Levels. Diabetes. 2016, 65, 2784–2794. [CrossRef]
- Craig S. A. Betaine in human nutrition. The American journal of clinical nutrition. 2004, 80, 539–549. [CrossRef]
- Holm, P. I.; Ueland, P. M.; Vollset, S. E.; Midttun, Ø.; Blom, H. J.; Keijzer, M. B.; den Heijer, M. Betaine and folate status as cooperative determinants of plasma homocysteine in humans. Arteriosclerosis, thrombosis, and vascular biology. 2005, 25, 379–385. [CrossRef]
- Kim, S. K.; Choi, K. H.; Kim, Y. C. Effect of acute betaine administration on hepatic metabolism of S-amino acids in rats and mice. Biochemical pharmacology. 2003, 65, 1565–1574. [CrossRef]
- Go, E. K.; Jung, K. J.; Kim, J. Y.; Yu, B. P.; Chung, H. Y. Betaine suppresses proinflammatory signaling during aging: the involvement of nuclear factor-kappaB via nuclear factor-inducing kinase/IkappaB kinase and mitogen-activated protein kinases. The journals of gerontology. Series A, Biological sciences and medical sciences. 2005, 60, 1252–1264. [CrossRef]
- Yi, E. Y.; Kim, Y. J. Betaine inhibits in vitro and in vivo angiogenesis through suppression of the NF-κB and Akt signaling pathways. International journal of oncology. 2012, 41, 1879–1885. [CrossRef]
- Ferrara, N.; Kerbel, R. S. Angiogenesis as a therapeutic target. Nature. 2005, 438, 967–974. [CrossRef]
- Muñoz-Chápuli, R., Quesada, A. R., & Angel Medina, M. Angiogenesis and signal transduction in endothelial cells. Cellular and molecular life sciences: CMLS. 2004, 61, 2224–2243. [CrossRef]
- Blavier, L.; Lazaryev, A.; Dorey, F.; Shackleford, G. M.; DeClerck, Y. A. Matrix metalloproteinases play an active role in Wnt1-induced mammary tumorigenesis. Cancer research. 2006, 66, 2691–2699. [CrossRef]
- Hussein, R. M.; Anwar, M. M.; Farghaly, H. S.; Kandeil, M. A. Gallic acid and ferulic acid protect the liver from thioacetamide-induced fibrosis in rats via differential expression of miR-21, miR-30 and miR-200 and impact on TGF-β1/Smad3 signaling. Chemico-biological interactions. 2020, 324, 109098. [CrossRef]
- Dos Santos, A. N.; de L Nascimento, T. R.; Gondim, B. L. C.; Velo, M. M. A. C.; de A Rêgo, R. I.; do C Neto, J. R.; Machado, J. R.; da Silva, M. V.; de Araújo, H. W. C.; Fonseca, M. G.; Castellano, L. R. C. Catechins as Model Bioactive Compounds for Biomedical Applications. Current pharmaceutical design. 2020, 26, 4032–4047. [CrossRef]
- Badolati, N.; Masselli, R.; Sommella, E.; Sagliocchi, S.; Di Minno, A.; Salviati, E.; Campiglia, P.; Dentice, M.; Tenore, G. C.; Stornaiuolo, M.; Novellino, E. The Hepatoprotective Effect of Taurisolo, a Nutraceutical Enriched in Resveratrol and Polyphenols, Involves Activation of Mitochondrial Metabolism in Mice Liver. Antioxidants (Basel, Switzerland). 2020, 9, 410. [CrossRef]
- Vestuto, V.; Amodio, G.; Pepe, G.; Basilicata, M.G.; Belvedere, R.; Napolitano, E.; Guarnieri, D.; Pagliara, V.; Paladino, S.; Rodriquez, M.; Bertamino, A.; Campiglia, P.; Remondelli, P.; Moltedo, O. Cocoa Extract Provides Protection against 6-OHDA Toxicity in SH-SY5Y Dopaminergic Neurons by Targeting PERK. Biomedicines 2022, 10, 2009. [CrossRef]
- Sugiyama, H.; Akazome, Y.; Shoji, T.; Yamaguchi, A.; Yasue, M.; Kanda, T.; Ohtake, Y. Oligomeric procyanidins in apple polyphenol are main active components for inhibition of pancreatic lipase and triglyceride absorption. Journal of agricultural and food chemistry. 2007, 55, 4604–4609. [CrossRef]
- Hayakawa, S.; Saito, K.; Miyoshi, N., Ohishi, T.; Oishi, Y.; Miyoshi, M.; Nakamura, Y. Anti-Cancer Effects of Green Tea by Either Anti- or Pro- Oxidative Mechanisms. Asian Pacific journal of cancer prevention: APJCP. 2016, 17, 1649–1654. [CrossRef]
- Abe, K.; Ijiri, M.; Suzuki, T.; Taguchi, K.;Koyama, Y.; Isemura, M. Green tea with a high catechin content suppresses inflammatory cytokine expression in the galactosamine-injured rat liver. Biomedical research (Tokyo, Japan). 2005,26, 187–192. [CrossRef]
- Tipoe, G. L.; Leung, T. M.; Liong, E. C.; Lau, T. Y.; Fung, M. L.; Nanji, A. A. Epigallocatechin-3-gallate (EGCG) reduces liver inflammation, oxidative stress and fibrosis in carbon tetrachloride (CCl4)-induced liver injury in mice. Toxicology. 2010, 273, 45–52. [CrossRef]
- Ding, Y.; Sun, X.; Chen, Y.; Deng, Y.; Qian, K. Epigallocatechin gallate attenuated non-alcoholic steatohepatitis induced by methionine- and choline-deficient diet. European journal of pharmacology. 2015, 761, 405–412. [CrossRef]
- Tang, F. Y.; Nguyen, N.; Meydani, M. Green tea catechins inhibit VEGF-induced angiogenesis in vitro through suppression of VE-cadherin phosphorylation and inactivation of Akt molecule. International journal of cancer. 2003, 106, 871–878. [CrossRef]
- Kenny, T. P.; Keen, C. L.; Jones, P.; Kung, H. J.; Schmitz, H. H.; Gershwin, M. E. Cocoa procyanidins inhibit proliferation and angiogenic signals in human dermal microvascular endothelial cells following stimulation by low-level H2O2. Experimental biology and medicine (Maywood, N.J.). 2004, 229, 765–771. [CrossRef]
- Zeng, X.; Feng, Q.; Zhao, F.; Sun, C.; Zhou, T.; Yang, J.; Zhan, X. Puerarin inhibits TRPM3/miR-204 to promote MC3T3-E1 cells proliferation, differentiation and mineralization. Phytotherapy research: PTR. 2018, 32, 996–1003. [CrossRef]
- Zheng, P.; Ji, G.; Ma, Z.; Liu, T.; Xin, L.; Wu, H.; Liang, X.; Liu, J. Therapeutic effect of puerarin on non-alcoholic rat fatty liver by improving leptin signal transduction through JAK2/STAT3 pathways. The American journal of Chinese medicine. 2009, 37, 69–83. [CrossRef]
- Zhou, J.; Zhang, N.; Aldhahrani, A.; Soliman, M. M.; Zhang, L.; Zhou, F. Puerarin ameliorates nonalcoholic fatty liver in rats by regulating hepatic lipid accumulation, oxidative stress, and inflammation. Frontiers in immunology. 2022, 13, 956688. [CrossRef]
- Park, J.; Min, J. S.; Kim, B.; Chae, U. B.; Yun, J. W.; Choi, M. S.; Kong, I. K.; Chang, K. T.; Lee, D. S. Mitochondrial ROS govern the LPS-induced pro-inflammatory response in microglia cells by regulating MAPK and NF-κB pathways. Neuroscience letters. 2015, 584, 191–196. [CrossRef]
- Ma, J. Q.; Ding, J.; Zhao, H.; Liu, C. M. Puerarin attenuates carbon tetrachloride-induced liver oxidative stress and hyperlipidaemia in mouse by JNK/c-Jun/CYP7A1 pathway. Basic & clinical pharmacology & toxicology. 2014, 115, 389–395. [CrossRef]
- BedÊ, T. P.; Jesuz, V. A.; Souza, V. R.; Elias, M. B.; Oliveira, F. L.; Dias, J. F.; Teodoro, A. J.; Azeredo, V. B. Effects of grape juice, red wine and resveratrol on liver parameters of rat submitted high-fat diet. Anais da Academia Brasileira de Ciencias. 2020, 92, e20191230. [CrossRef]
- Yun, H.; Park, S.; Kim, M. J.; Yang, W. K.; Im, D. U.; Yang, K. R.; Hong, J.; Choe, W.; Kang, I.; Kim, S. S.; Ha, J. AMP-activated protein kinase mediates the antioxidant effects of resveratrol through regulation of the transcription factor FoxO1. The FEBS journal. 2014, 281, 4421–4438. [CrossRef]
- Yun, H.; Park, S.; Kim, M. J.; Yang, W. K.; Im, D. U.; Yang, K. R.; Hong, J.; Choe, W.; Kang, I.; Kim, S. S.; Ha, J. AMP-activated protein kinase mediates the antioxidant effects of resveratrol through regulation of the transcription factor FoxO1. The FEBS journal. 2014, 281, 4421–4438. [CrossRef]
- Ahmed, S. M.; Luo, L.; Namani, A.; Wang, X. J.; Tang, X. Nrf2 signaling pathway: Pivotal roles in inflammation. Biochimica et biophysica acta. Molecular basis of disease. 2017, 1863, 585–597. [CrossRef]
- Bellezza, I.; Giambanco, I.; Minelli, A.; Donato, R. Nrf2-Keap1 signaling in oxidative and reductive stress. Biochimica et biophysica acta. Molecular cell research. 2018, 1865, 721–733. [CrossRef]
- Di Pascoli, M.; Diví, M.; Rodríguez-Vilarrupla, A.; Rosado, E.; Gracia-Sancho, J.; Vilaseca, M.; Bosch, J.; García-Pagán, J. C. Resveratrol improves intrahepatic endothelial dysfunction and reduces hepatic fibrosis and portal pressure in cirrhotic rats. Journal of hepatology. 2013, 58, 904–910. [CrossRef]
- de Oliveira, C. M.; Martins, L. A. M.; de Sousa, A. C.; Moraes, K. D. S.; Costa, B. P.; Vieira, M. Q.; Coelho, B. P.; Borojevic, R.; de Oliveira, J. R.; Guma, F. C. R. Resveratrol increases the activation markers and changes the release of inflammatory cytokines of hepatic stellate cells. Molecular and cellular biochemistry. 2021, 476, 649–661. [CrossRef]
- Lee, E. S.; Shin, M. O.; Yoon, S.; Moon, J. O. Resveratrol inhibits dimethylnitrosamine-induced hepatic fibrosis in rats. Archives of pharmacal research. 2010, 33, 925–932. [CrossRef]
- Li, B.; Lu, F.; Wei, X.; Zhao, R. Fucoidan: structure and bioactivity. Molecules (Basel, Switzerland). 2008, 13, 1671–1695. [CrossRef]
- Liu, F.; Wang, J.; Chang, A. K.; Liu, B.; Yang, L.; Li, Q.; Wang, P.; Zou, X. Fucoidan extract derived from Undaria pinnatifida inhibits angiogenesis by human umbilical vein endothelial cells. Phytomedicine: international journal of phytotherapy and phytopharmacology. 2012, 19, 797–803. [CrossRef]
- Abdollah, M. R. A.; Ali, A. A.; Elgohary, H. H.; Elmazar, M. M. Antiangiogenic drugs in combination with seaweed fucoidan: A mechanistic in vitro and in vivo study exploring the VEGF receptor and its downstream signaling molecules in hepatic cancer. Frontiers in pharmacology. 2023, 14, 1108992. [CrossRef]
- Lin, K. I.; Lin, C. C.; Kuo, S. M.; Lai, J. C.; Wang, Y. Q.; You, H. L.; Hsu, M. L.; Chen, C. H.; Shiu, L. Y. Carnosic acid impedes cell growth and enhances anticancer effects of carmustine and lomustine in melanoma. Bioscience reports. 2018, 38, BSR20180005. [CrossRef]
- Johnson J. J. Carnosol: a promising anti-cancer and anti-inflammatory agent. Cancer letters. 2011, 305, 1–7. [CrossRef]
- López-Jiménez, A.; García-Caballero, M.; Medina, M. Á.; Quesada, A. R. Anti-angiogenic properties of carnosol and carnosic acid, two major dietary compounds from rosemary. European journal of nutrition. 2013, 52, 85–95. [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



