
Submitted:
18 May 2023
Posted:
18 May 2023
You are already at the latest version
Abstract
Apoptosis under severe hypoxia is induced through p53 phosphorylation and HIF-1α-dependent p53 accumulation via ATR activation by DNA damage response (DDR) activation through replication stress. We previously demonstrated that the topoisomerase I catalytic inhibitor, 3EZ, 20Ac-ingenol, specifically induced apoptosis in Jeko-1 and Panc-1 cells, both of which are cell lines that show cyclin D1 overexpression. After progression to the S phase facilitated by nuclear cyclin D1, in the presence of 3EZ, 20Ac-ingenol, an intra S phase checkpoint was induced in ATR activation as part of replication stress-induced DDR. In this study, we examined whether 3EE, 20Ac-ingenol induces a higher degree of p53 phosphorylation and additional HIF-1α and p53 accumulation in response to replication stress-induced DDR activation under the hypoxic condition than under the normoxic condition by controlling ATR activation. 3EE, 20Ac-ingenol induced p53 activation and HIF-1α-dependent p53 accumulation through cooperative ATR activation via induced DDR with the hypoxia in Panc-1 cells. The Jeko-1 cells showed slight HIF-1α accumulation under hypoxia, but this was not decreased by 3EE, 20Ac-ingenol, so that the cells remained resistant to hypoxia. 3EE, 20Ac-ingenol induces an intricate interplay between p53 and HIF-1α accumulation via ATR activations that results in high p53 accumulation, which advanced transient expression and early disappearance of HIF-1?. The strong p53 accumulation and consequent PTEN activation also decreased HIF-1α accumulation and PD-L1 expression, which resulted in intense apoptosis.
Keywords:
Apoptosis
; PD-L1
; catalytic topo I inhibitor
; cyclin D1 overexpression
; ATR
; p53
1. Introduction
Upregulation of hypoxia-inducible factor 1, alpha subunit (HIF-1𝛼) under the hypoxic condition has been observed in a variety of solid cancers; HIF-1𝛼 serves as a direct master controller of the expressions of many genes related to tumor growth and vascularization [1,2]. Programmed death-ligand 1 (PD-L1) allows tumor cells to escape immune surveillance through interferon-𝛾 (INF-𝛾) activation in cancers [3,4]; PD-L1 expression is also known to be upregulated in a HIF-1𝛼- dependent manner [5]. Furthermore, HIF-1𝛼 has been shown to be capable of inducing tumor resistance to the topoisomerase (topo) I inhibitor SN-38 [6], gemcitabine [7], and various other cytotoxic agents [8]. However, under severe hypoxia, replication stress in regions of single-strand DNA leads to accumulation of HIF-1𝛼 and p53 through ataxia-telangiectasia mutated (ATM) and RAD3-related (ATR) activation [9,10]. In the presence of high p53 activation, p53 destroys the HIF-1𝛼 protein, and elimination of the HIF-1𝛼 promoter activity results in downregulated expressions of many genes, resulting in growth arrest/apoptosis [11,12,13]. Although HIF-1𝛼 serves as a major direct transcriptional regulator of the expressions of hypoxia-inducible genes under hypoxia, rat sarcoma viral oncogene-extracellular regulated kinase (Ras-ERK) and tens in homolog of chromosome 10-serine-threonine protein kinase-Akt (PTEN-Akt) lie upstream of HIF1α and can control HIF-1α accumulation [14,15]. Hematologic mantle cell lymphoma, which harbors activated Ras-ERK and aberrant activation of PI3k/Akt, shows upregulated expression of HIF-1𝛼 through these factors [16].
Recently, it has been reported that inhibition of ATR can also increase tumor cell killing in cancer cells that exhibit high levels of replication stress [17]. However, for ATR inhibition, combined use of an ATR inhibitor and DNA damage agent is necessary, so that the effects of combined administration of ATR inhibitors with DNA damage agents have been investigated [18]. Use of a DNA damage agent alone or in combination with ATR inhibition is limited by the dose-limiting toxicities associated with DNA damage agents on normal proliferating tissues [19,20]. Furthermore, pancreatic cancers [21] and mantle cell lymphoma [22] with cyclin D1 accumulation are insensitive or resistant to DNA damage drugs. They were sensitive to 3EZ, 20Ac-ingenol, irrespective of the mechanism of cyclin D1 accumulation, via decrease in the cellular accumulation of cyclin D1[23,24]. 3EZ, 20Ac-ingenol is a catalytic inhibitor-type topoisomerase I (topo I) with less cytotoxicity [25], and we reported that it promotes activation of the ATR-dependent p53 pathway through DNA damage response (DDR) activation during the S phase and upregulates PTEN expression, which specifically induces apoptosis in cancer cells showing cyclin D1 accumulation [23,24]. 3EE, 20Ac-ingenol, a stereoisomer of 3EZ, 20Ac-ingenol which is also a catalytic inhibitor-type topo 1 [26], showed the same effects of inhibition of cell proliferation and decreased cellular cyclin D1 accumulation in Jeko-1 and Panc-1 cells, and cellular apoptosis induced via ATR-dependent p53 activation [23,24]. In this study, we investigated apoptosis induction in these cell lines under the hypoxic condition through ATR- and HIF-1α-dependent p53 accumulation via cooperative activation of ATR by two DNA replication stresses induced by 3EE, 20Ac-ingenol and hypoxia.
2. Results
2.1. Effects of 3EE, 20Ac-Ingenol on cell proliferative activity of Jeko-1 and Panc-1 cells under normoxic and hypoxic conditions
The effect of 3EE, 20Ac-ingenol on the cell proliferative activity was investigated by MTT assay in Jeko-1 and Panc-1 cells under normoxic (20% O2) and hypoxic (0.5%-1.3% O2) conditions. Treatment for 72 h with different concentrations of 3EE, 20Ac-ingenol reduced the viability of the cancer cells in a dose-dependent manner in the Jeko-1 and Panc-1 cells; the proliferative activity of the Jeko-1 (Figure 1A) and Panc-1 (Figure 1B) cells gradually decreased as the concentration of 3EE, 20Ac-ingenol in the medium increased, with the peak inhibition of approximately 60%-70% reached at 10 μM. The IC50 of 3EE, 20Ac-ingenol for the Jeko-1 cells was about 2 μM, whereas that for the Panc-1 cells was about 1 μM.
In order to investigate the effects of 3EE, 20Ac-ingenol in inducing chemosensitivity in the Jeko-1 and Panc-1 cell lines under the hypoxic condition, we compared the differences in the cell viability in the presence of 3EE, 20Ac-ingenol under hypoxic culture conditions. Under the hypoxic condition, 72-h treatment with different concentrations of 3EE, 20Ac-ingenol (0-10 μM) reduced the viability of the cancer cells, with a higher degree of reduction in the viability of the Panc-1 cells (Figure 1B) as compared with the Jeko-1 cells (Figure 1A). The IC50 value of 3EE, 20Ac-ingenol for the Panc-1 cells was about 1.5 μM, and although the proliferation-inhibitory activity was slightly lower under the hypoxic condition than under the normoxic condition, the cells remained highly sensitive to the effect of 3EE, 20Ac-ingenol. While HIF-1𝛼 is known to be resistant to topo I inhibitor agents under the hypoxic condition [6], 3EE, 20Ac-ingenol inhibited the proliferation of Panc-1 cells without causing resistance to hypoxia (Figure 1B). However, the Jeko-1 cell line showed resistance to 3EE, 20Ac-ingenol treatment under the hypoxic condition (Figure 1A). Thus, the sensitivity of the two cell lines to 3EE, 20Ac-ingenol varied under the hypoxic condition.
2.2. Effects of 3EE, 20Ac-ingenol on ATR activation and p53 accumulation
The DNA damage kinase, ATR, which is activated in the DNA damage response (DDR) that responses to DNA double-stranded breaks or replication fork stalling, contributes to control progression to the S phase of the cell cycle, DNA replication, DNA repair, and apoptosis [27]. Furthermore, ATR is activated by replication stress in regions of single-stranded DNA under severe hypoxia, and contributes to hypoxia-dependent HIF-1𝛼 accumulation [9]. ATR is also activated as part of the enhanced DDR by the genomic instability induced by 3EE, 20Ac-ingenol even in normoxia, contributing to progression to the S phase of the cell cycle in cells showing cyclin D1 accumulation [24], and by double-strand breaks (DSBs) caused by DNA damage agents containing topo poison inhibitors, contributing to DSB repair [28]. In this study, we examined whether ATR might also be activated as part of the DDR induced by 3EE 20Ac-ingenol treatment under the hypoxic condition as under the normoxic condition. In investigating the activation by 3EE, 20Ac-ingenol, we considered that under the hypoxic condition, ATR activation in the Jeko-1 and Panc-1 cells may occur via two DDR pathways, one activated by hypoxia and the other by 3EE, 20Ac-ingenol. We found that under hypoxia, First ATR was activated as part of the DDR elicited by hypoxia (Figure 2B, lane 1). The second ATR activation pathway induced by 3EE, 20Ac-ingenol has already been reported previously [24]; in this study, this ATR activation was observed from 12 h to 48 h as part of the enhanced DDR elicited by 3EE, 20Ac-ingenol treatment (Figure S1A,B). We examined whether, with the progression of hypoxia, 3EE, 20Ac-ingenol can still induce ATR activation in the cells in addition to the first activation induced by hypoxia. After preincubation for 24 h under the hypoxic condition, the first ATR activation continued in the absence of 3EE, 20Ac-ingenol in the control Panc-1 cells, and the second ATR activation was induced by the addition of 3EE, 20Ac-ingenol under progressive hypoxia. The second ATR activation was clearly detected as a more intense protein band as compared with the band observed in the control cells exposed to hypoxia alone (Figure 2B, lane 2 vs. 1), suggesting that under this condition, ATR activation is derived from two different pathways. In the Jeko-1 cells, although slight ATR activation was induced under hypoxia (first pathway) and continued after the addition of 3EE, 20Ac-ingenol (second pathway) (Figure 2A, lanes 1,2), the ATR activation was no longer detected after 48 h, and continued to be undetectable until 72 h (Figure 2A, lanes 3,4); this finding suggests that because no noticeable activation of hypoxia-dependent ATR activation is observed, the activation may not occur in these cells.
Phosphorylation of Ser15 in p53 is principally mediated by ATR and ATM in response to genotoxic stress, and is followed subsequently by phosphorylation of other residues at various sites [29]. Under the hypoxic condition, the p-p53 band associated with the first ATR activation pathway during the pre-and control incubation periods was already weakly detected in the control Panc-1 cells (Figure 2B, lane 1). Following 3EE, 20Ac-ingenol treatment, the strong p-p53 band associated with the two ATR activation pathways described above was observed at 24 h (Figure 2B, lane 2); this band was more intense than that observed in the cells exposed to hypoxia alone (control) (Figure 2B, lane 2 vs. 1). After phosphorylation of the serine 15 residue of p53, a band representing further phosphorylation of p-p53 at various other sites and residues was also observed in the Panc-1 cells. Phospho-p53 (Ser15) antibody (#9284) detects only levels of p53 phosphorylated at serine 15, and does not cross-react with p53 phosphorylated (#9282) at other sites. After the initial increase, the amount of p-p53 decreased at 48 h after treatment. We examined the amount of p53 protein accumulation in the Jeko-1 and Panc-1 cells exposed to hypoxia. Slight p53 accumulation was observed in the Panc-1 control cells after pre- and control incubation, and intense activation was observed after 48 h of 3EE, 20Ac-ingenol treatment and continued up to 72 h (Figures 2B and S2). In the Jeko-1 cells, unlike in the Panc-1 cells, neither p-p53 (dada not shown) nor p53 accumulation (Figures 2A and S2) was observed following exposure to hypoxia.
2.3. Effects of 3EE, 20Ac-ingenol treatment on HIF-1𝛼 accumulation and Caspase-3 activation
Cellular adaptation to DNA replication stress under hypoxia is known to be controlled by ATR through HIF-1𝛼 accumulation [9,10]. We examined whether DNA replication stress induced by 3EE, 20Ac-ingenol might also induce HIF-1𝛼 accumulation through ATR activation using whole cell lysates. Under normoxia, no HIF-1𝛼 expression was observed in either the control or 3EE, 20Ac-ingenol-treated Jeko-1 cells (Figure 3A, lanes 1,2). Under hypoxia, slight accumulation of HIF-1𝛼 was observed in the absence of 3EE, 20Ac-ingenol (control) (Figure 3A, lane 3); this HIF-1𝛼 expression was considered as an adaptive response to hypoxia, and the HIF-1𝛼 accumulation was still low; also, the amount of accumulation scarcely changed after 3EE 20Ac-ingenol treatment of these cells. In the case of the Panc-1 cells, under the normoxia, as in the Jeko-1 cells, no HIF-1𝛼 accumulation was detected in either the control state or after 3EE, 20Ac-ingenol treatment (Figure 3B, lanes 1,2). However, under the hypoxic condition, HIF-1𝛼 accumulation was observed both in the absence and presence of 3EE, 20Ac-ingenol, with intense accumulation of HIF-1𝛼 noted in the control cells and lower amounts noted in the 3EE, 20Ac-ingenol-treated cells after 24 h (Figure 3B, lanes 3,4). HIF-1𝛼 expression is controlled by ATR as an adaptive response to hypoxia [10] and translocates from the cytoplasm to the nuclei [30]. Then, we compared the changes in the intracellular localization of HIF-1𝛼 in the Panc-1 cells after 3EE, 20Ac-ingenol treatment under the hypoxic condition and investigated the time-course of HIF-1𝛼 accumulation (Figure 3C,D). All the samples were preincubated for 24 h; subsequently, HIF-1𝛼 accumulation was observed both in the absence (control) and presence of 3EE, 20Ac-ingenol. HIF-1𝛼 accumulation in the control cells (first ATR activation pathway) exposed to hypoxia started earlier than the HIF-1𝛼 accumulation derived from 3EE, 20Ac-ingenol treatment (second ATR activation pathway) by only the amount of preincubation time under hypoxia. We considered that the adaptive response to hypoxia leading to HIF-1𝛼 accumulation and the response to 3EE, 20Ac-ingenol treatment merge, and the summation of HIF-1𝛼 accumulation as well as the first ATR activation pathway may be observed following exposure to hypoxia. First HIF-1𝛼 protein in the pre- and control (absence of 3EE, 20Ac-ingnol) conditions was translated under hypoxia, but while a small amount of HIF-1𝛼 protein remained in the cytoplasm after 24 h of control incubation (48 h from pre-incubation) (Figure 3C, lane 1), most of it was translocated to the nuclei (Figure 3D, lane 1). The initially translated HIF-1𝛼 in the cytoplasm was first observed in the nuclei of the control cells at 48 h after preincubation. The protein band of the subsequently translated HIF-1𝛼 after 3EE, 20Ac-ingenol treatment under the hypoxic condition was observed in the cytoplasm of the treated Panc-1 cells at 24 h (Figure 3C, lane 2). By this time, the initially translated HIF-1𝛼 protein via the first ATR activation pathway that had translocated to the nuclei had been eliminated (Figure 3D, lane 2) in the 3EE, 20Ac-ingenol-treated cells. At 48 h after 3EE, 20Ac-ingenol treatment, the subsequently translated HIF-1𝛼 protein was also no longer detected in the cytoplasm (Figure 3C, lane 3), while an increase of the amount of this protein was observed in the nuclei (Figure 3D, lane 3). However, this accumulation of the translocated HIF-1𝛼 in the nuclei was also only transient, with the levels decreasing early (Figure 3D, lane 4). These results may suggest that the translated HIF-1𝛼 in the cells following 3EE, 20Ac-ingenol treatment under hypoxia follows exactly the same path as the initially translated HIF-1𝛼 after exposure to hypoxia alone, but after a lag of 48 h.
HIF-1𝛼 is the major transcription factor in cells under the hypoxic condition and is a key regulator of the adaptive responses to hypoxia. Although HIF-1𝛼 usually promotes tumor cell survival under hypoxia, under severe hypoxia, it has been reported to trigger apoptosis [11,31]. Our results showed activation of caspase-3 in the Panc-1 cells after 24 h exposure to mild hypoxia, which continued to increase until 72 h after 3EE, 20Ac-ingenol treatment (Figure 3E). Although slight accumulation of HIF-1𝛼 was observed in the Jeko-1 cells (Figure 3A, lane 3), it occurred in the absence of ATR activation. Therefore, HIF-dependent p53 accumulation might not be induced in these cells. Furthermore, almost no caspase-3 activation was detected either in the Jeko-1 cells (data not shown).
2.4. Effects of 3EE, 20Ac-ingenol treatment on the PD-L1 and PTEN expressions
PD-L1 expression has been shown to be activated by DNA replication-induced stress caused by DNA damage agents under the normoxic condition [28]. Under the normoxic condition, slight expression of PD-L1 was seen in the control Jeko-1 cells; the expression level began to increase at 12 h after 3EE, 20Ac-ingenol treatment of the cells, decreasing slightly thereafter from 24 h to 48 h (Figure S1C). PD-L1 protein was also detected in the control Panc-1 cells under the normoxic condition, with the level increasing at 12 h after 3EE, 20Ac-ingenol treatment. In the Panc-1 cells also, similar to the case in the Jeko-1 cells, reduction of the PD-L1 expression levels was observed starting from 24 h after 3EE, 20Ac-ingenol treatment, with the reduced levels persisting until 48 h after the addition of 3EE, 20Ac-ingenol (Figure S1D).
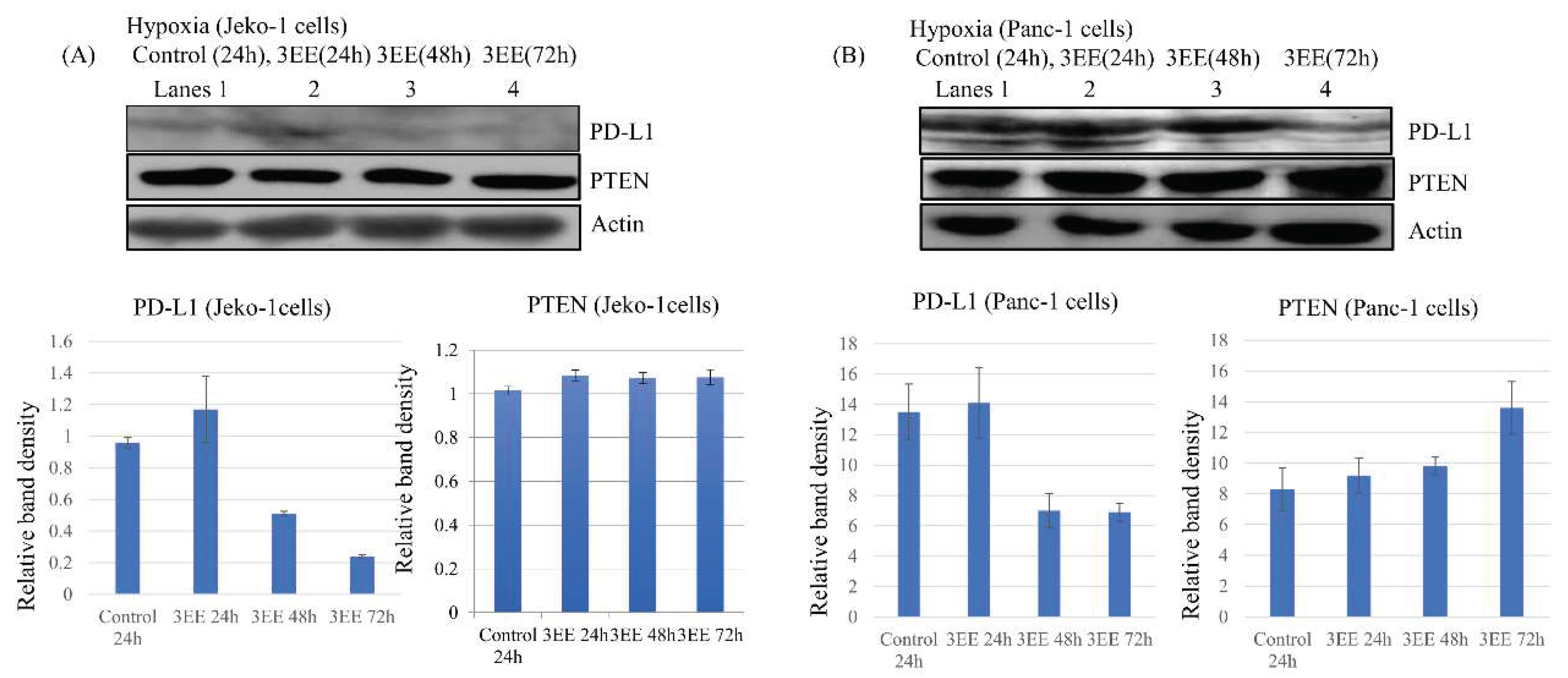
PD-L1 expression has been shown to be activated by hypoxia-dependent HIF-1𝛼 accumulation [3,5,32]. Therefore, we investigated the effects of 3EE 20Ac-ingenol treatment on the PD-L1 expression in the Jeko-1 (Figure 4A) and Panc-1 (Figure 4B) whole cells under the hypoxic condition. The Jeko-1 cells showed a slight increase of PD-L1 expression until 24 h after 3EE, 20Ac-ingenol in addition to low expression of it under the hypoxic condition (Figure 4A, lanes 1,2), but the levels decreased from 48 h to 72 h after 3EE, 20Ac-ingenol treatment (Figure 4A, lanes 3,4). This slight increase of PD-L1 expression in the Jeko-1 cells might be related to the activation induced by 3EE, 20Ac-ingenol treatment, as observed under normoxia (Figure S1C). Stronger PD-L1 expression was observed in the control Panc-1 cells under the hypoxic condition (Figure 4B, lane 1). The elevated PD-L1 expression level was maintained until 24 h after the addition of 3EE, 20Ac-ingenol (Figure 4B, lane 2), but decreased by 48 h (Figure 4B, lane 3). The PD-L1 expression decreased further by 72 h after the addition of 3EE, 20Ac-ingenol (Figure 4B, lane 4).
It has been reported that the increase in PD-L1 expression is associated with the loss of PTEN [33], and HIF-1𝛼 expression is also inhibited by activation of PTEN or inhibition of PI3K/Akt [15,34]. To determine whether PTEN is recruited in the regulation of DDR elicited by 3EZ 20Ac-ingenol treatment under the hypoxic condition, we measured the PTEN protein level in the cells by western blotting (Figure 4A,B). In the Jeko-1cells, no increase in PTEN was observed from 24 h to 72 h after addition of 3EE, 20Ac-ingenol under hypoxia (Figure 4A). On the other hand, in the Panc-1 cells, increase of PTEN was observed at 24 h following the addition of 3EE, 20Ac-ingenol under the hypoxic condition, and the amount of PTEN continued to increase until 72 h after addition of the agent. (Figures 4B and S3B).
3. Discussion
Although, 3EE, 20Ac-ingenol effectively inhibited cell proliferation in both Jeko-1 and Panc-1 cells under the normoxic condition, the sensitivity of the cells to hypoxia varied between the two cell lines (Figures 1A,B). Under the hypoxic condition, HIF-1𝛼 accumulation is reported as being capable of inducing resistance to various anticancer agents [6,7,8], but treatment of the cells with 3EE, 20Ac-ingenol under this condition restored the sensitivity of the cells to anticancer agents, attenuated the cellular resistance to hypoxia (Figure 1B), and induced apoptosis through ATR-dependent and HIF-1𝛼-dependent p53 accumulation in the Panc-1 cells overexpressing of cyclin D1 (Figures 3C,D,E). However, no such attenuation of the resistance to cell proliferation by restoration of the sensitivity to hypoxia (Figure 1A) or ATR activation and p53 accumulation (Figure 2A) as an adaptive response to hypoxia was observed following 3EE, 20Ac-ingenol treatment in the hematologic cancer cell line, Jeko-1. The mechanisms of cancer cell survival against hypoxia differ among hematological cancers, Jeko-1cells [14,15,16], and solid cancers, Panc-1 cells [1,2]. In the Jeko-1 cells [16], slight HIF-1𝛼 accumulation was observed in the absence of ATR activation under the hypoxic condition, and the mechanism of HIF-1𝛼 expression was different from that occurring via ATR activation as an adaptive response to hypoxia in the Panc-1 cells (Figure 2A) [9,10]. These findings could be explained by the differences in the mechanisms of cell survival against hypoxia.
Under severe hypoxia, HIF-1𝛼 promotes stabilization of p53 through control of ATR activation, although accumulation of p53 also downregulates HIF-1𝛼 expression [9,10]. With prolonged exposure to severe hypoxia (< 0.1% O2 or anoxia), the amount of p53 in the cells increases, resulting in the degradation of HIF-1𝛼. Progressive decrease of HIF-1𝛼 and accumulation of p53 with time may contribute to cell death by progressing ones [11,12,13,31]. In Panc-1 cells overexpressing cyclin D1, under the mild hypoxic condition, the cells showed HIF-1𝛼 accumulation (Figure 3D) and p53 phosphorylation and p53 stabilization (Figure 2B) through ATR activation. p53 serine 15 is phosphorylated by both ATR and ATM activations in response to hypoxia in an ATR-dependent manner, leading to p53 accumulation [35]. HIF-1𝛼 promotes p53 phosphorylation at serine 15 in response to hypoxia, inducing HIF-1𝛼 and p53-dependent apoptosis [36]. The p53 activated (p-p53) by 3EE, 20Ac-ingenol through the various responses (Figure 2B, lane 2) decreased the amount of HIF-1𝛼 translated initially (control; Figure 3D, lane 1) at 24 h after addition of 3EE, 20Ac-ingenol (Figure 3D, lane 2). After 48 h, the subsequently translated HIF-1𝛼 expressed in the Panc-1 cells after 3EE, 20Ac-ingenol treatment and translocated to the nuclei (Figure 3D, lane 3) also began to decrease in the same way as the initially translated HIF-1𝛼 (Figure 3D, lane 4), with further increase of p53 accumulation (Figure 2B, lanes 3,4 and Figure S2). High p-p53 and p53 accumulation was observed in the Panc-1 cells, which show cyclin D1 overexpression, through the high HIF-1𝛼 translation following 3EE, 20Ac-ingenol treatment under the hypoxic condition, which resulted in HIF-1𝛼 degradation and cellular apoptosis through caspase-3 activation (Figure 3E, lanes 3,4).
PD-L1 expression is upregulated through HIF-1𝛼 activation in response to replication-associated stress upon exposure to hypoxia and/or DNA damage (DNA-double strand breaks) by chemotherapeutic agents [5,28]; We consider that the DNA replication stress induced by hypoxia and 3EE, 20Ac-ingenol induces PD-L1 expression in the Panc-1 cells (Figure 4B, lanes 1, 2), but only transiently induces PD-L1 expression following only 3EE, 20Ac-ingenol treatment in the Jeko-1 cells (Figure 4A, lane 2). PD-L1 not only serves as an immune checkpoint inhibitor, but also promotes chemoresistance and cell growth, and exerts anti-apoptotic effects on the cancer cells [3,4,37,38]. However, knockdown of HIF-1𝛼 inhibits hypoxia-induced PD-L1 expression [32], and suppression of PD-L1 is known to block cell proliferation [39] and induce apoptosis in cancer cells [31,32,40]. Although the DNA replication stress induced in Panc-1 cells by 3EE, 20Ac-ingenol treatment may lead to the HIF-1𝛼 dependent (Figure 3C, lane 2) PD-L1 expression, similar to that following exposure of the cells to hypoxia (Figure 3D lane 1), the amount of activated PD-L1 decreased (Figure 4B, lane 4) with the early disappearance of HIF-1𝛼 through p53 accumulation (Figure 3D, lane 4), resulting in inhibition of cell growth (Figure 1B) and induction of apoptosis (Figure 3E) in the Panc-1 cells. HIF-1𝛼 activated through ATR activation in the Jeko-1 cells was not observed under hypoxia, and the increase of PD-L1 expression might be activated by 3EE, 20Ac-ingenol treatment instead of the HIF-1𝛼. The activated PD-L1 expression decreased over time as normoxia (Figure S1C).
ATR which is activated by replication stress induced by exposure of the cells to severe hypoxia controls efficient adaptation of the cells to hypoxia through regulating HIF-1𝛼 expression and p53 accumulation [9,10,11]. In addition to the initial ATR activation elicited by hypoxia, more intense ATR activation was detected in the Panc-1 cells that show cyclin D1 accumulation as part of the enhanced DDR induced by 3EE, 20Ac-ingenol treatment under the hypoxic condition (Figure 2B, lane 1 vs. 2-4). The first translation peak of HIF-1𝛼 associated with hypoxic stress (first ATR activation pathway) was observed in the nuclei of control Panc-1 cells as an adaptive response to mild hypoxia (0.5%-1.3% O2) (Figure 3D, lane 1). Under the hypoxic condition, the subsequently translated HIF-1𝛼 associated with the DNA replication stress induced by 3EE, 20Ac-ingenol (Figure 3C, lane 2) combines with the DNA replication stress induced by hypoxia, and the summation of HIF-1𝛼 is translocated in to nuclei, which forms the second peak (Figure 3D, lane 3). The characteristic of HIF-1𝛼 expression by 3EE, 20Ac-ingenol under hypoxia could show to be shared in the nuclei by the two peaks derived from hypoxia alone and by 3EE, 20Ac-ingenol plus hypoxia by examining its changes of the localization (Figure 3D, lanes 1,3). The HIF-1𝛼 accumulated by the two pathways contributed to p53 stabilization under mild hypoxia. In experiments using transfection of a p53-expressing plasmid, hypoxic translation of HIF-1𝛼 was attenuated by p53, and high p53 expression degraded the HIF-1𝛼 protein [13]. Transient expression of HIF-1𝛼 is observed with the overexpression of p53 under anoxia, followed by a rapid decrease in the amount of HIF-1𝛼 protein. Although HIF-1𝛼 disappears more rapidly under anoxiaas compared with that under normoxia, under both normoxia and anoxia, HIF-1𝛼 completely disappeared by 24 h with plasmid-induced p53 overexpression [13]. The cooperative effect of the ATR activations occurring in response to similar replication stresses induced by 3EE, 20Ac-ingenol and hypoxia (Figure 2B, lane 1 vs. 2-4) resulted in accumulation of p53 phosphorylated at serine 15 (Figure 2B, lanes 1,2) and p53 stabilization (Figure 2B, lanes 3,4) through HIF-1𝛼 accumulation in the Panc-1 cells (Figure 3D, lanes 1,3). Transient expression and early disappearance of HIF-1𝛼 in the Panc-1 cells was promoted by higher activation of p53 (Figure 3D, lane 1 vs. 2, lane 3 vs. 4). The p-p53 and p53 accumulation associated with ATR activation was observed at 24 h and 72 h. Many topo 1 catalytic inhibitors that induce a decatenation checkpoint causing G2 phase arrest are less toxic and, therefore, cannot induce apoptosis [25]. 3EZ, 20Ac-ingenol was also shown, in a previous study, to induce G2/M phase arrest in DT40 cells [26]. How does the catalytic-type topoisomerase inhibitor 3EE, 20Ac-ingenol activate the ATR/p53 pathway through S phase arrest under the mild hypoxic condition in cells overexpressing cyclin D1? It has been reported that stabilization of Cdt1 produced by inhibition of proteasome in cells showing nuclear cyclin D1 accumulation continually primes DNA re-replication during the S phase and compromises the intra S phase checkpoint [41]. As one function, cyclin D1 led to activation of the downstream biochemical events, including the expressions of cyclin A and proliferating cell nuclear antigen, and cyclin E- and cyclin A-associated kinase activation, which initiates DNA replication [42]. In the Panc-1 cells that progressed to the S phase through cyclin D1 overexpression, replication forks become stalled due to decatenation inhibition of the topo 1 catalytic activity caused by 3EE, 20Ac-ingenol, which may induce the intra S phase checkpoint. We consider that the 3EE, 20Ac-ingenol causes the DNA replication stress to activate ATR, which results in phosphorylation of p53 and strong HIF-1𝛼 dependent p53 accumulation under mild hypoxia. However, the detailed mechanisms remain unknown, and further studies are needed to examine how DNA replication stresses activate ATR at the S phase checkpoint.
The accumulated p53 also activates PTEN expression [43], which inhibits PI3K/Akt [44] and ERK [45]. Under normoxia, PD-L1 expression is regulated by inhibition of ERK [46]. Higher levels of PD-L1 protein are seen in cells with genetic deletion of PTEN than in cells with wild-type PTEN under normoxia, which can be attenuated by inhibition of PI3K/Akt; thus, PD-L1 expression is inhibited by PTEN activation [33,47,48]. 3EZ, 20Ac-ingenol induces downregulation of p-Akt through upregulation of PTEN [23]. PD-L-1 was highly expressed (27.8%) in pancreatic ductal adenocarcinoma under normoxia, and the cell-intrinsic PD-L1 facilitates in tumor growth through Hippo signaling pathway by independence of the immune system [49]. In this study, under normoxia, 3EE, 20Ac-ingenol upregulated the expression of PTEN in the Panc-1 cells (Figure S3A,B) and also inhibited p-ERK (Figure S4). Under the hypoxic condition, upregulation of PD-L1 expression depends on increased expression of HIF-1𝛼. [5]. Hypoxia-induced PD-L1 expression is inhibited by knockdown of HIF-1𝛼 [32]. Furthermore, HIF-1𝛼 is inhibited by activation of the PTEN/Akt pathway [15,34]. Under the hypoxic condition also, 3EE, 20Ac-ingenol upregulated PTEN expression in the Panc-1 cells (Figures 4B and S3B), and under this condition, the PTEN/Akt pathway activated by 3EE, 20Ac-ingenol not only decreases the accumulation of HIF-1𝛼, but may also decrease PD-L1 expression. PD-L1 expression in Panc-1 cells that occurred under both the normoxic (Figure S1D, lane 4) and hypoxic condition (Figure 4B, lane 4) was clearly decreased by 3EE, 20Ac-ingenol treatment. Furthermore, PD-L1 is upregulated through INF-𝛾 activation, which serves as an immune inhibitor [3,5,50]. Ingenol-type diterpenoil compaounds isolated from Euphorbia kausui exert a wide range of pharmacological activities, including tumor inhibitory activities, immune regulatory activities, and modulatory effects on INF-γ [51,52]. It has been reported previously that 3EZ, 20Ac-ingenol more specifically inhibited the proliferation of cancer cells overexpressing cyclin D1 which showed resistance to irinotecan as compared with that of cancer cells that did not show cyclin D1 accumulation [22,53]. In this study, we showed that 3EE, 20Ac-ingenol treatment effectively inhibited both HIF-1𝛼 and PD-L1 upregulations that occurred under the hypoxic condition to induce apoptosis and may also offer promise for boosting antitumor immunity.
4. Materials and Methods
4.1. Cell lines and cellular proliferation
Jeko-1 cell was obtained from the American Type Culture Collections. Panc-1 cells was provided by the RIKEN BRC through the National Bio-Resource Project of the MEXT, Japan. The diterpene compound, 3-O-(2′E,4′E-decadienoyl)-20-O- acetylingenol (3EE, 20Ac-ingenol) was purified from the roots of Euphorbia kausui [54] and dissolved in dimethyl sulfoxide. The cancer cells were incubated in RPMI 1640 supplemented with 10% fetal calf serum at 37 °C. For hypoxia, after the cells were preincubated during 24 h under hypoxic conditions (0.5-1.3% O2) using the nBIONIX hypoxic culture kit (Sugiyamagen), the Jeko-1 cell line, a mantle cell lymphoma cell and the Panc-1 cell line, a pancreatic cancer cell were furthermore incubated for 72 h with 3EE, 20 Ac-ingenol and without it (control) under the same condition. Cell growth was determined by an MTT assay using the Cell Proliferation Kit I (Roche Applied Science) as described previously [26].
4.2. Immunoblotting
Jeko-1 and Panc-1 cells were cultured for various time points in the presence of 2μM and 3 μM 3EZ, 20Ac-ingenol, respectively and washed with PBS. The cells were fractionated into a nuclear and cytoplasmic fraction using Nuclear Cytoplasmic Extraction Reagents (Thermo Scientific). The protein concentrations were determined using the Bradford reagent for protein assays (Bio-Rad Laboratories). A total of 20 μg protein of the cell lysates was resolved on 8%, 10%, or 15% SDS-polyacrylamide gels and transferred onto a polyvinylidene difluoride membrane. The blots were made using anti-PTEN, anti-p53 (#9282), anti-p-p53 (Ser15)(#9284), anti PD L1 (Cell Signaling Technology), anti-ATR (Santa Cruz, Cell Signaling Technology and Abcam), anti-active caspase-3 (R&D systems), anti HIF-1α(Gene Tex), anti Lamin A/C (Proteintech Group), and anti-actin (Sigma) antibodies followed by detection, using an enhanced chemiluminescence system.
5. Conclusions
Under the hypoxic condition, 3EE, 20Ac-ingenol activates phosphorylation of p53 and HIF-1𝛼-dependent p53 accumulation and facilitates HIF-1𝛼 degradation in the solid cancer cell line, Panc-1 cells showing cyclin D1 overexpression, which results in caspase 3 activation and apoptosis of the cells. However, under the hypoxic condition, no such specific effect of cyclin D1 overexpression was observed in the hematological cancer cell line, Jeko-1 cells. The characteristics of the specific apoptosis induction by 3EE, 20Ac-ingenol in cancer cells showing cyclin D1 overexpression is observed in both hematological and solid cancer cell lines under normoxia but in solid cancer cell line under hypoxia.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Acknowledgements
We acknowledge the support for the research in part from Nihon University to S. Miyata and S. Kitanaka.
Declaration of Competing Interest
The authors declare no conflict of interest.
References
- Talks, K.L.; Turley, H.; Gatter, K.C.; Maxwell, P.H.; Pugh, C.W.; Ratcliffe, P.J.; Harris, A.L. The expression and distribution of the hypoxia-inducible factors HIF-1alpha and HIF-2alpha in normal human tissues, cancers, and tumor-associated macrophages. Am J Pathol. 2000, 157, 411–421. [Google Scholar] [CrossRef] [PubMed]
- Majmundar, A.J.; Wong, W.J.; Simon, M.C. Hypoxia-inducible factors and the response to hypoxic stress. Mol Cell. 2010, 40, 294–309. [Google Scholar] [CrossRef] [PubMed]
- Barsoum, I.B.; Smallwood, C.A.; Siemens, D.R.; Graham, C.H. A mechanism of hypoxia-mediated escape from adaptive immunity in cancer cells. Cancer Res. 2014, 743, 665–674. [Google Scholar] [CrossRef] [PubMed]
- Harrington, B.K.; Wheeler, E.; Hornbuckle, K.; Shana'ah, A.Y.; Youssef, Y.; Smith, L.; Hassan, Q., 2nd; Klamer, B.; Zhang, X.; Long, M.; Baiocchi, R.A.; Maddocks, K.; Johnson, A.J.; Byrd, J.C. Modulation of immune checkpoint molecule expression in mantle cell lymphoma. Leuk Lymphoma. 2019, 60, 2498–2507. [Google Scholar] [CrossRef]
- Noman, M.Z.; Desantis, G.; Janji, B.; Hasmim, M.; Karray, S.; Dessen, P.; Bronte, V.; Chouaib, S. PD-L1 is a novel direct target of HIF-1α, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J Exp Med. 2014, 211, 781–790. [Google Scholar] [CrossRef] [PubMed]
- Cai, T.Y.; Liu, X.W.; Zhu, H.; Cao, J.; Zhang, J.; Ding, L.; Lou, J.S.; He, Q.J.; Yang, B. Tirapazamine sensitizes hepatocellular carcinoma cells to topoisomerase I inhibitors via cooperative modulation of hypoxia-inducible factor-1α. Mol Cancer Ther. 2014, 13, 630–642. [Google Scholar] [CrossRef] [PubMed]
- Yokoi, K.; Fidler, I.J. Hypoxia increases resistance of human pancreatic cancer cells to apoptosis induced by gemcitabine. Clin Cancer Res. 2004, 10, 2299–2306. [Google Scholar] [CrossRef]
- Koch, S.; Mayer, F.; Honecker, F.; Schittenhelm, M.; Bokemeyer, C. Efficacy of cytotoxicagents used in the treatment of testicular germ cell tumours under normoxic and hypoxic conditions in vitro. Br J Cancer. 2003, 89, 2133–2139. [Google Scholar] [CrossRef]
- Hammond, E.M.; Denko, N.C; Dorie, M.J.; Abraham, R.T.; Giaccia, A.J. Hypoxia links ATR and p53 through replication arrest. Mol Cell Biol. 2002, 22, 1834–1843. [Google Scholar] [CrossRef]
- Fallone, F.; Britton, S.; Nieto, L.; Salles, B.; Muller, C. ATR controls cellular adaptation to hypoxia through positive regulation of hypoxia-inducible factor 1 (HIF-1) expression. Oncogene. 2013, 32, 4387–4396. [Google Scholar] [CrossRef]
- Carmeliet, P.; Dor, Y.; Herbert, J.M.; Fukumura, D.; Brusselmans, K.; Dewerchin, M.; Neeman, M.; Bono, F.; Abramovitch, R.; Maxwell, P.; Koch, C.J.; Ratcliffe, P.; Moons, L.; Jain, R.K.; Collen, D.; Keshert, E. Role of HIF-1alpha in hypoxia-mediated apoptosis, cell proliferation and tumour angiogenesis. Nature. 1998, 394, 485–490. [Google Scholar] [CrossRef]
- An, W.G.; Kanekal, M.; Simon, M.C.; Maltepe, E.; Blagosklonny, M.V.; Neckers, L.M. Stabilization of wild-type p53 by hypoxia-inducible factor 1alpha. Nature. 1998, 392, 405–408. [Google Scholar] [CrossRef] [PubMed]
- Schmid, T.; Zhou, J.; Köhl, R.; Brüne, B. p300 relieves p53-evoked transcriptional repression of hypoxia-inducible factor-1 (HIF-1). Biochem J. 2004, 380(Pt 1), 289–295. [Google Scholar] [CrossRef]
- Liu, L.; Zhang, H.; Sun, L.; Gao, Y.; Jin, H.; Liang, S.; Wang, Y.; Dong, M.; Shi, Y.; Li, Z.; Fan, D. ERK/MAPK activation involves hypoxia-induced MGr1-Ag/37LRP expression and contributes to apoptosis resistance in gastric cancer. Int J Cancer. 2010, 127, 820–829. [Google Scholar] [CrossRef] [PubMed]
- Jiang, B.H.; Jiang, G.; Zheng, J.Z.; Lu, Z.; Hunter, T.; Vogt, P.K. Phosphatidylinositol 3-kinase signaling controls levels of hypoxia-inducible factor 1. Cell Growth Differ. 2001 l, 12, 363–369. [Google Scholar]
- Argyriou, P.; Papageorgiou, S.G.; Panteleon. V.; Psyrri, A.; Bakou, V.; Pappa, V.; Spathis, A.; Economopoulou, P.; Papageorgiou, E.; Economopoulos, T.; Rontogianni, D. Hypoxia-inducible factors in mantle cell lymphoma: implication for an activated mTORC1→HIF-1α pathway. Ann Hematol. 2011, 90, 315–322. [Google Scholar] [CrossRef] [PubMed]
- Fokas, E.; Prevo, R.; Hammond, E.M.; Brunner, T.B.; McKenna, W.G.; Muschel, RJ. Targeting ATR in DNA damage response and cancer therapeutics. Cancer Treat Rev. 2014, 40, 109–117. [Google Scholar] [CrossRef]
- Toledo, L.I.; Murga, M.; Zur, R.; Soria, R.; Rodriguez, A.; Martinez, S.; Oyarzabal, J.; Pastor, J.; Bischoff, J.R; Fernandez-Capetillo, O. A cell-based screen identifies ATR inhibitors with synthetic lethal properties for cancer-associated mutations. Nat Struct Mol Biol. 2011, 18, 721–727. [Google Scholar] [CrossRef]
- Thomas, A.; Pommier, Y. Targeting Topoisomerase I in the Era of Precision Medicine. Clin. Cancer Res. 2019, 25, 6581–6589. [Google Scholar] [CrossRef]
- Jo, U.; Senatorov, I.S.; Zimmermann, A.; Saha, L.K.; Murai, Y.; Kim, S.H.; Rajapakse, V.N.; Elloumi, F.; Takahashi, N.; Schultz, C.W.; Thomas, A.; Zenke, F.T.; Pommier, Y. Novel and Highly Potent ATR Inhibitor M4344 Kills Cancer Cells With Replication Stress, and Enhances the Chemotherapeutic Activity of Widely Used DNA Damaging Agents. Mol Cancer Ther. 2021, 20, 1431–1441. [Google Scholar] [CrossRef]
- Giuliani, J.; Bonetti, A. FOLFIRINOX is a cost-effective combination chemotherapy in first-line for advanced pancreatic Cancer. Pancreatology. 2019, 19, 325–330. [Google Scholar] [CrossRef]
- Mohanty, S.; Mohanty, A.; Sandoval, N.; Tran, T.; Bedell, V.; Wu, J.; Scuto, A.; Murata-Collins, J.; Weisenburger, D.D.; Ngo, V.N. Cyclin D1 depletion induces DNA damage in mantle cell lymphoma lines. Leuk Lymphoma. 2017, 58, 676–688. [Google Scholar] [CrossRef] [PubMed]
- Miyata, S.; Wang, L.Y.; Kitanaka, S. 3EZ, 20Ac-ingenol induces cell-specific apoptosis in cyclin D1 over-expression through the activation of ATR and downregulation of p-Akt. Leuk Res. 2018, 64, 46–51. [Google Scholar] [CrossRef]
- Miyata, S.; Nakamura, T.; Kitanaka, S. 3EZ, 20Ac-ingenol-induced Apoptosis in Chemoresistant Cancers With Cyclin D1 Accumulation. Anticancer Res. 2020, 40, 6237–6246. [Google Scholar] [CrossRef] [PubMed]
- Wu, N.; Wu, X.W.; Agama, K.; Pommier, Y.; Du, J.; Li, D.; Gu, L.Q.; Huang, Z.S.; An, L.K. A novel DNA topoisomerase I inhibitor with different mechanism from camptothecin induces G2/M phase cell cycle arrest to K562 cells. Biochemistry. 2010, 49, 10131–10136. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, Y.; Kanbe,M. ; Watanabe, M.; Dan, K.; Matsuzaki, K.; Kitanaka, S.; Miyata S. 3EZ,20Ac-ingenol, a catalytic inhibitor of topoisomerases, downregulates p-Akt and induces DSBs and apoptosis of DT40 cells. Arch Pharm Res. 2013, 36, 1029–1038. [Google Scholar] [CrossRef]
- Cimprich, K.A.; Cortez, D. ATR: an essential regulator of genome integrity. Nat Rev Mol Cell Biol. 2008, 9, 616–627. [Google Scholar] [CrossRef] [PubMed]
- Sato, H.; Niimi, A.; Yasuhara, T.; Permata, T.B.M.; Hagiwara, Y.; Isono, M.; Nuryadi, E.; Sekine, R.; Oike, T.; Kakoti, S.; Yoshimoto, Y.; Held, K.D.; Suzuki, Y.; Kono, K.; Miyagawa, K.; Nakano, T.; Shibata, A. DNA double-strand break repair pathway regulates PD-L1 expression in cancer cells. Nat Commun. 2017, 8, 1751. [Google Scholar] [CrossRef]
- Meek, D.W.; Anderson, C.W. Posttranslational modification of p53: cooperative integrators of function. Cold Spring Harb Perspect Biol. 2009, 1, a000950. [Google Scholar] [CrossRef]
- Moritz, W.; Meier, F.; Stroka, D.M.; Giuliani, M.; Kugelmeier, P.; Nett, P.C.; Lehmann, R.; Candinas, D.; Gassmann, M.; Weber, M. Apoptosis in hypoxic human pancreatic islets correlates with HIF-1alpha expression. FASEB J. 2002, 16, 745–747. [Google Scholar] [CrossRef]
- Kilic, M.; Kasperczyk, H.; Fulda, S.; Debatin, K.M. Role of hypoxia inducible factor-1 alpha in modulation of apoptosis resistance. Oncogene. 2007, 26, 2027–2038. [Google Scholar] [CrossRef]
- Zhou, L.; Cha, G.; Chen, L.; Yang, C.; Xu, D.; Ge, M. HIF1α/PD-L1 axis mediates hypoxia-induced cell apoptosis and tumor progression in follicular thyroid carcinoma. Onco Targets Ther. 2019, 12, 6461–6470. [Google Scholar] [CrossRef]
- Parsa, A.T.; Waldron, J.S.; Panner, A.; Crane, C.A.; Parney, I.F.; Barry. J.J.; Cachola, K.E.; Murray, J.C.; Tihan, T.; Jensen, M.C.; Mischel, P.S.; Stokoe, D.; Pieper, R.O. Loss of tumor suppressor PTEN function increases B7-H1 expression and immunoresistance in glioma. Nat Med. 2007, 13, 84–88. [Google Scholar] [CrossRef]
- Joshi, S.; Singh, A.R.; Durden, D.L. MDM2 regulates hypoxic hypoxia-inducible factor 1α stability in an E3 ligase, proteasome, and PTEN-phosphatidylinositol 3-kinase-AKT-dependent manner. J Biol Chem. 2014, 289, 22785–22797. [Google Scholar] [CrossRef] [PubMed]
- Hammond, E.M.; Dorie, M.J.; Giaccia, A.J. ATR/ATM targets are phosphorylated by ATR in response to hypoxia and ATM in response to reoxygenation. J Biol Chem. 2003, 278, 12207–12213. [Google Scholar] [CrossRef]
- Moeller, B.J.; Dreher, M.R.; Rabbani, Z.N.; Schroeder, T.; Cao, Y.; Li, C.Y.; Dewhirst, M.W. Pleiotropic effects of HIF-1 blockade on tumor radiosensitivity. Cancer Cell. 2005, 8, 99–110. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.; Strome, S.E.; Salomao, D.R.; Tamura, H.; Hirano, F.; Flies, D.B.; Roche, P.C.; Lu, J.; Zhu, G.; Tamada, K.; Lennon, V.A.; Celis, E.; Chen, L. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med. 2002, 8, 793–800. [Google Scholar] [CrossRef] [PubMed]
- Black, M.; Barsoum, I.B.; Truesdell, P.; Cotechini, T.; Macdonald-Goodfellow, S.K.; Petroff, M.; Siemens, D.R.; Koti, M.; Craig, A.W.; Graham, C.H. Activation of the PD-1/PD-L1 immune checkpoint confers tumor cell chemoresistance associated with increased metastasis. Oncotarget. 2016, 7, 10557–10567. [Google Scholar] [CrossRef]
- Clark, C.A.; Gupta, H.B.; Sareddy, G.; Pandeswara, S.; Lao, S.; Yuan, B.; Drerup, J.M.; Padron, A.; Conejo-Garcia, J.; Murthy, K.; Liu, Y.; Turk, M.J.; Thedieck, K.; Hurez, V.; Li, R.; Vadlamudi, R.; Curiel, T.J. Tumor-Intrinsic PD-L1 Signals Regulate Cell Growth, Pathogenesis, and Autophagy in Ovarian Cancer and Melanoma. Cancer Res. 2016, 76, 6964–6974. [Google Scholar] [CrossRef]
- Azuma, T.; Yao, S.; Zhu, G.; Flies, A.S.; Flies, S.J.; Chen, L. B7-H1 is a ubiquitous antiapoptotic receptor on cancer cells. Blood. 2008, 111, 3635–3643. [Google Scholar] [CrossRef]
- Kim, J.K.; Diehl, J.A. Nuclear cyclin D1: an oncogenic driver in human cancer. J Cell Physiol. 2009, 220, 292–296. [Google Scholar] [CrossRef] [PubMed]
- Albrecht, J.H.; Hansen, L.K. Cyclin D1 promotes mitogen-independent cell cycle progression in hepatocytes. Cell Growth Differ. 1999, 10, 397–404. [Google Scholar]
- Stambolic, V.; MacPherson, D.; Sas, D.; Lin, Y.; Snow, B.; Jang, Y.; Benchimol, S.; Mak, T.W. Regulation of PTEN transcription by p53. Mol Cell. 2001, 8, 317–325. [Google Scholar] [CrossRef] [PubMed]
- Stambolic, V.; Suzuki, A.; de la Pompa, J.L.; Brothers, G.M.; Mirtsos, C.; Sasaki, T.; Ruland, J.; Penninger, J.M.; Siderovski, D.P.; Mak, T.W. Negative regulation of PKB/Akt-dependent cell survival by the tumor suppressor PTEN. Cell. 1998, 95, 29–39. [Google Scholar] [CrossRef]
- Gu, J.; Tamura, M.; Yamada, K.M. Tumor suppressor PTEN inhibits integrin- and growth factor-mediated mitogen-activated protein (MAP) kinase signaling pathways. J Cell Biol. 1998, 143, 1375–1383. [Google Scholar] [CrossRef]
- Liu, J.; Hamrouni, A.; Wolowiec, D.; Coiteux, V.; Kuliczkowski, K.; Hetuin, D.; Saudemont, A.; Quesnel, B. Plasma cells from multiple myeloma patients express B7-H1 (PD-L1) and increase expression after stimulation with IFN-{gamma} and TLR ligands via a MyD88-, TRAF6-, and MEK-dependent pathway. Blood. 2007, 110, 296–304. [Google Scholar] [CrossRef]
- Crane, C.A.; Panner, A.; Murray, J.C.; Wilson, S.P.; Xu, H.; Chen, L.; Simko, J.P.; Waldman, F.M.; Pieper, R.O.; Parsa, A.T. PI(3) kinase is associated with a mechanism of immunoresistance in breast and prostate cancer. Oncogene. 2009, 28, 306–312. [Google Scholar] [CrossRef]
- Jiang, X.; Zhou, J.; Giobbie-Hurder, A.; Wargo, J.; Hodi, F.S. The activation of MAPK in melanoma cells resistant to BRAF inhibition promotes PD-L1 expression that is reversible by MEK and PI3K inhibition. Clin Cancer Res. 2013, 19, 598–609. [Google Scholar] [CrossRef] [PubMed]
- Pu, N.; Gao, S.; Yin, H.; Li, J.A.; Wu, W.; Fang, Y.; Zhang, L; Rong, Y. ; Xu, X; Wang, D.; Kuang, T.; Jin, D,; Yu, J.; Lou, W. Cell-intrinsic PD-1 promotes proliferation in pancreatic cancer by targeting CYR61/CTGF via the hippo pathway. Cancer Lett. 2019, 460, 42–53. [Google Scholar] [CrossRef]
- Shurin, M.R.; Umansky, V. Cross-talk between HIF and PD-1/PD-L1 pathways in carcinogenesis and therapy. J Clin Invest. 2022, 132, e159473. [Google Scholar] [CrossRef]
- Yan, X.; Zhang, L.; Cao, Y.; Yao, W.; Tang, Y.; Ding, A. An Ingenol Derived from Euphorbia kansui Induces Hepatocyte Cytotoxicity by Triggering G0/G1 Cell Cycle Arrest and Regulating the Mitochondrial Apoptosis Pathway in Vitro. Molecules. 2016, 21, 813. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Gao, L.; Li, Z.; Yan, X.; Yang, Y.; Tang, Y.; Cao, Y.; Ding, A. Bio-guided isolation of the cytotoxic terpenoids from the roots of Euphorbia kansui against human normal cell lines L-O2 and GES-1. Int J Mol Sci. 2012, 13, 11247–11259. [Google Scholar] [CrossRef] [PubMed]
- Miyata, S.; Fukuda, Y.; Tojima, H.; Matsuzaki, K.; Kitanaka, S.; Sawada, H. Mechanism of the inhibition of leukemia cell growth and induction of apoptosis through the activation of ATR and PTEN by the topoisomerase inhibitor 3EZ, 20Ac-ingenol. Leuk Res. 2015, 39, 927–932. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.Y.; Wang, N.L.; Yao, X.S.; Miyata, S.; Kitanaka, S. Diterpenes from the roots of kansui and their in vitro effects on the cell division of Xenopus. J Nat Prod. 2002, 65, 1246–1251. [Google Scholar] [CrossRef]
Figure 1.
Effects of 3EE, 20Ac-ingenol on the proliferative activities of Jeko-1 and Panc-1 cells under normoxia and hypoxia. The cells were seeded on to 96-well plates (1.0 × 104 Jeko-1 cells or 3×103 Panc-1 cells per well in 100 μL) and treated with 0 (control), 0.1, 0.5, 1, 5, or 10 μM 3EE, 20Ac-ingenol at 37 °C for 72h under normaxia and hypoxia. The relative cell growth was determined via an MTT assay. The growth of untreated Jeko-1and Panc-1 cells was set as 100%, and the growth of treated Jeko-1and Panc-1 cells was expressed relative to the growth of the untreated cells. All experiments were performed in triplicate, and the data are presented as the mean ± standard deviation. (A) Jeko-1 cells, (B) Panc-1 cells. ★p<0.001.
Figure 1.
Effects of 3EE, 20Ac-ingenol on the proliferative activities of Jeko-1 and Panc-1 cells under normoxia and hypoxia. The cells were seeded on to 96-well plates (1.0 × 104 Jeko-1 cells or 3×103 Panc-1 cells per well in 100 μL) and treated with 0 (control), 0.1, 0.5, 1, 5, or 10 μM 3EE, 20Ac-ingenol at 37 °C for 72h under normaxia and hypoxia. The relative cell growth was determined via an MTT assay. The growth of untreated Jeko-1and Panc-1 cells was set as 100%, and the growth of treated Jeko-1and Panc-1 cells was expressed relative to the growth of the untreated cells. All experiments were performed in triplicate, and the data are presented as the mean ± standard deviation. (A) Jeko-1 cells, (B) Panc-1 cells. ★p<0.001.
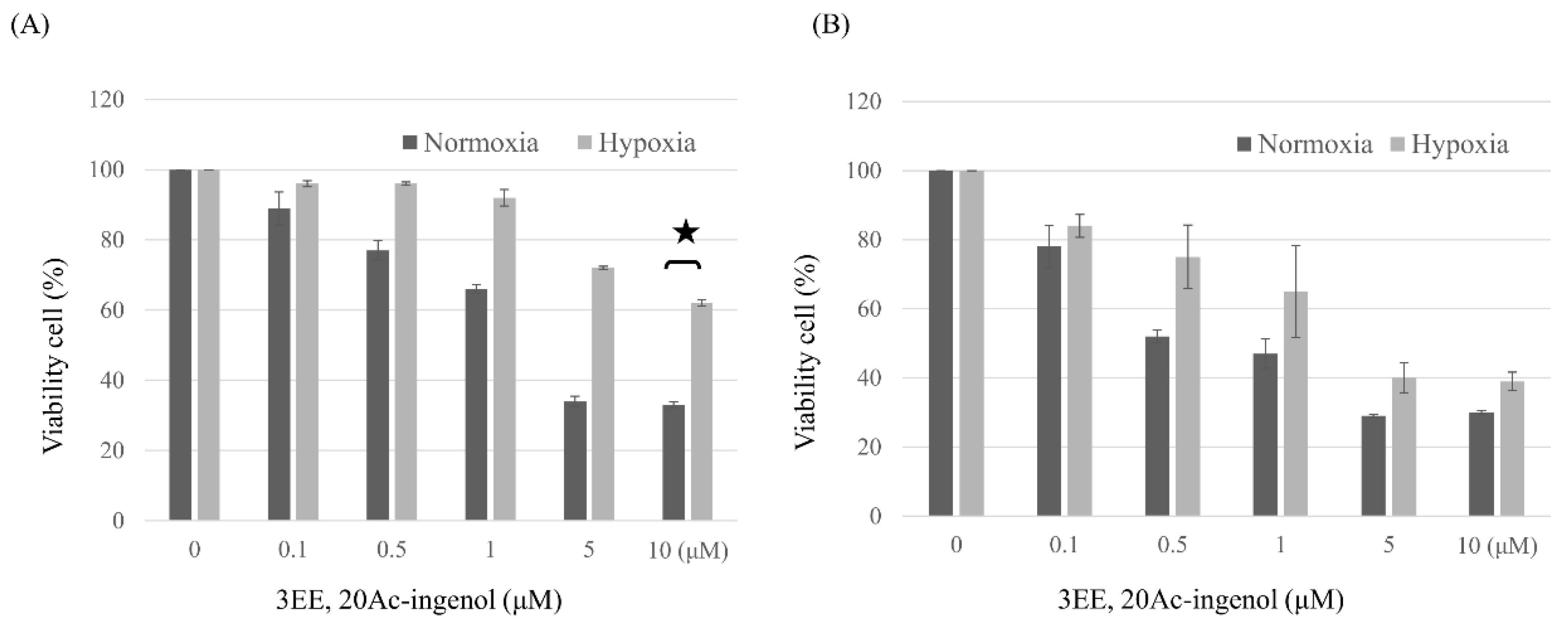
Figure 2.
Effects of 3EE, 20Ac-ingenol on ATR activation and p-p53 and p53 accumulation. For obtaining the effects of hypoxia, the cells were preincubated for 24 h under hypoxic conditions. (A) Jeko-1 cells were treated in the absence (control) for 24h or the presence of 2 μM 3EE, 20Ac-ingenol (3EE) for 24, 48 or 72h under hypoxia. (B) Panc-1 cells were treated by the same procedure except for the presence of 3μM 3EE as Jeko-1 cells. ATR, p-p53 and p53 analyzed by western blot.
Figure 2.
Effects of 3EE, 20Ac-ingenol on ATR activation and p-p53 and p53 accumulation. For obtaining the effects of hypoxia, the cells were preincubated for 24 h under hypoxic conditions. (A) Jeko-1 cells were treated in the absence (control) for 24h or the presence of 2 μM 3EE, 20Ac-ingenol (3EE) for 24, 48 or 72h under hypoxia. (B) Panc-1 cells were treated by the same procedure except for the presence of 3μM 3EE as Jeko-1 cells. ATR, p-p53 and p53 analyzed by western blot.
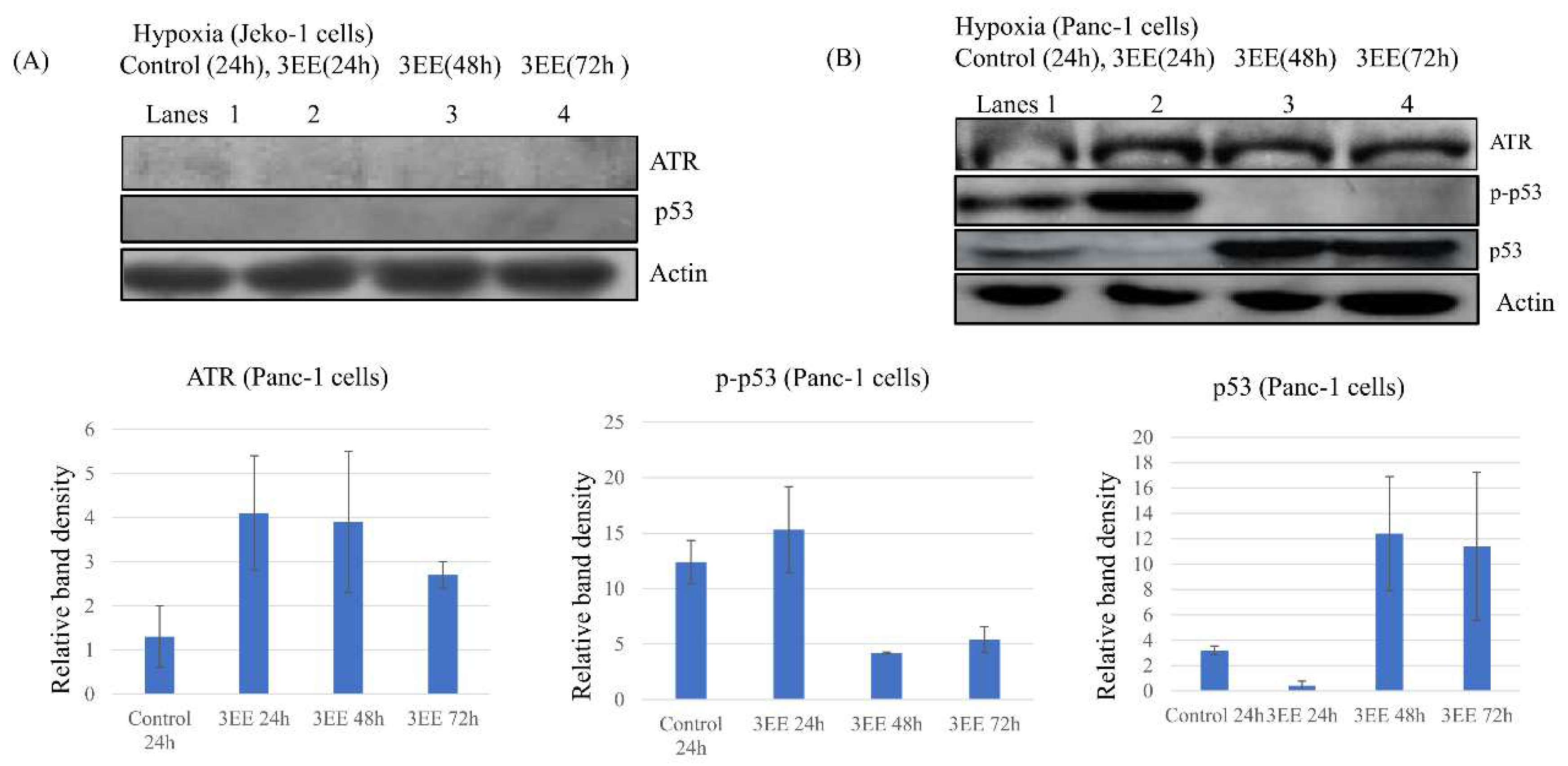
Figure 3.
Effects of 3EE, 20Ac-ingenol on HIF-1𝛼 expression and Caspase-3 activation. (A) Jeko-1 cells were incubated in the absence (control) or presence of 2 μM 3EE, 20Ac-ingenol (3EE) for 24h under normoxia and hypoxia. (B) Panc-1 cells were incubated in the absence (control) or presence of 3 μM 3EE for 24h under normoxia and hypoxia. (C, D, E) Panc-1 cells were treated in the absence (control) for 24h or presence of 3 μM 3EE for 24, 48 or 72h under hypoxia. (C, D) Panc-1 cells were fractionated into nuclear and cytoplasmic fractions with cytoplasmic extraction reagents. (A, B) whole cell. (C) cytoplasm, (D) nucleus, (E) whole cells. HIF-1𝛼 (A,B,C,D) and Caspase 3 (E) analyzed by western blotting.
Figure 3.
Effects of 3EE, 20Ac-ingenol on HIF-1𝛼 expression and Caspase-3 activation. (A) Jeko-1 cells were incubated in the absence (control) or presence of 2 μM 3EE, 20Ac-ingenol (3EE) for 24h under normoxia and hypoxia. (B) Panc-1 cells were incubated in the absence (control) or presence of 3 μM 3EE for 24h under normoxia and hypoxia. (C, D, E) Panc-1 cells were treated in the absence (control) for 24h or presence of 3 μM 3EE for 24, 48 or 72h under hypoxia. (C, D) Panc-1 cells were fractionated into nuclear and cytoplasmic fractions with cytoplasmic extraction reagents. (A, B) whole cell. (C) cytoplasm, (D) nucleus, (E) whole cells. HIF-1𝛼 (A,B,C,D) and Caspase 3 (E) analyzed by western blotting.
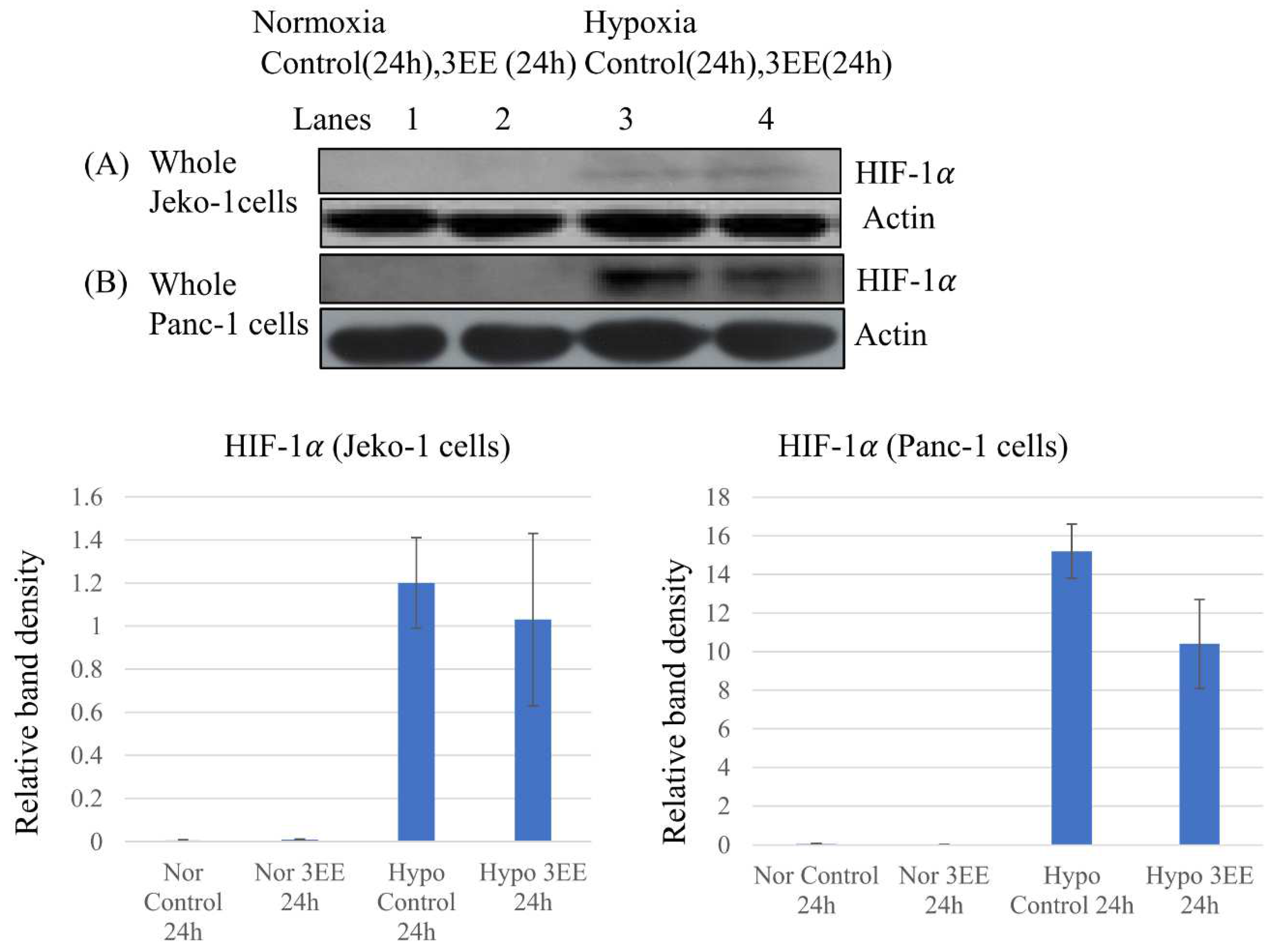
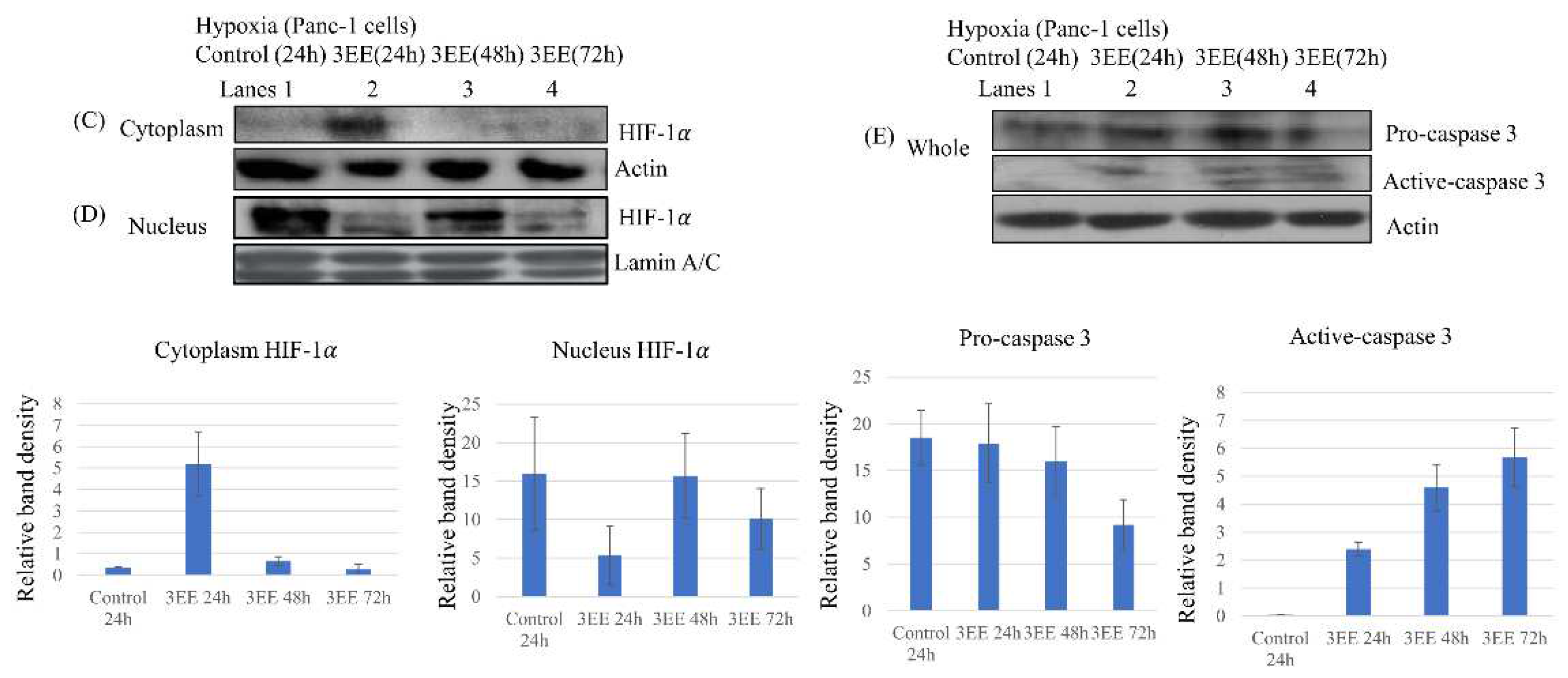
Figure 4.
Effects of 3EE, 20 Ac-ingenol on PD-L1 and PTEN expression. (A) Jeko-1 cells were treated in the absence (control) for 24h or presence of 2 μM 3EE, 20Ac-ingenol (3EE) for 24, 48 or 72h under hypoxia. (B) Panc-1 cells were treated by the same procedure except for the presence of 3μM 3EE as Jeko-1 cells. PD-L1 and PTEN analyzed by western blotting.
Figure 4.
Effects of 3EE, 20 Ac-ingenol on PD-L1 and PTEN expression. (A) Jeko-1 cells were treated in the absence (control) for 24h or presence of 2 μM 3EE, 20Ac-ingenol (3EE) for 24, 48 or 72h under hypoxia. (B) Panc-1 cells were treated by the same procedure except for the presence of 3μM 3EE as Jeko-1 cells. PD-L1 and PTEN analyzed by western blotting.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



