
Submitted:
18 May 2023
Posted:
18 May 2023
You are already at the latest version
Abstract
Protein O-GlcNAcylation has been associated with neurodegenerative diseases such as Alzheimer´s disease (AD). O-GlcNAcylation of Amyloid Precursor Protein (APP) regulates both the trafficking and the processing of the APP through the amyloidogenic pathway, resulting in the release and aggregation of the Aβ1-42 peptide. Microglia clear Aβ aggregates and dead cells to maintain brain homeostasis. Here, using LC-MS/MS we reveal that the Aβ1-42 peptide modifies the microglia O-GlcNAcome. We have identified 55 proteins, focusing our research on Galectin-1 protein, since it is a very versatile protein from a functional point of view. Combining biochemical with genetic approaches we demonstrate that Aβ1-42 peptide specifically targets Galectin-1S8 O-GlcNAcylation via OGT. In addition, this Gal-1-O-GlcNAcylated form, in turn, controls human microglia migration. Given the importance of microglia migration in the progression of AD, this study reports the relationship between Aβ1-42 peptide and Serine 8- O-GlcNAcylation of Galectin to drive microglial migration
Keywords:
Amyloid ß1-42 peptide
; OGT
; O-GlcNAcylation
; Galectin-1
; Galectin-1Serine 8- O-GlcNAcylation
; Gal-1S8A
; microglia
; migration
1. Introduction
Protein O-GlcNAcylation is a dynamic posttranslational modifications (PTM) occurring on thousands of nucleocytoplasmic and mitochondrial proteins that control important cellular processes such as transcription, translation, metabolism or signal transduction [1,2]. O-GlcNAcylation in proteins occurs at serine and threonine residues, which in turn could be also phosphorylated. In an unbalanced situation, one PTM can be favored at the expense of the other [3]. Thus, disruptions in the O-GlcNAcylation/phosphorylation balance have been associated with cancer [4], neurodevelopmental disorders [5] and neurodegeneration such as Alzheimer´s Disease (AD) [6,7,8]. Regarding AD, different studies have demonstrated that in this pathology some proteins are hyper-O-GlcNAcylated while others lose their O-GlcNAcylation [6,7,8,9].
AD is characterized by extracellular amyloid plaques mainly constituted by Aβ1-42 peptide, as well as by intracellular neurofibrillary tangles formed by hyperphosphorylated Tau protein. The accumulation and aggregation of these molecules in the extra- and intracellular spaces results in dystrophic neurites and loss of neurons and synapses, which causes loss of memory, spatiotemporal disorientation and behavioral changes among other symptoms [10]. Recent studies have associated O-GlcNAcylation with this disease, in fact, when the amyloid precursor protein (APP) is O-GlcNAcylated at threonine 576, this PTM promotes the trafficking and the processing of the APP through the amyloidogenic pathway, which results in the generation of Aβ1-42 peptide [9]. However, O-GlcNAcylation in Tau protein may have a neuroprotective role. Its O-GlcNacylation at serine and threonine residues prevents Tau hyperphosphorylation via competition with the phospho sites [11,12]. In addition, O-GlcNAcylation can also play a neuroprotective role by inhibiting necrosis in AD [13]. Therefore, the toxicity or the protective role of O-GlcNAcylation in AD is protein-specific.
The presence of reactive glia in AD around the extracellular amyloid plaques has been well established, with microglia being of those cells [14]. Microglia are responsible for the inflammatory response that provokes neuronal death and it is also recruited to the amyloid plaques in order to phagocytose Aβ1-42 peptides. For this, microglia firstly require a whole mechanism of migration towards the plaques. The process mentioned is especially relevant in initial stages of the disease as it prevents the formation of amyloid plaques [15,16]. However, with aging, microglia lose its phagocytic function and the clearance of Aβ1-42 is insufficient, favoring the pathology of AD [17]. In fact, microglia from old AD-mice express low levels of Aβ-binding receptors, as well as low levels of Aβ-degrading enzymes compared to microglia from young AD-mice [18]. Hence, owing to its functional duality, with a pro-inflammatory function on the one hand and its beneficial phagocytic function on the other, the role of microglia in AD is still controversial.
Here, we have revealed that Aβ1-42 peptide modifies the protein O-GlcNAcylation pattern in human microglia, identifying 55 proteins. One of these is Galectin-1 which is specifically O-GlcNAcylated in the Serine 8. This O-GlcNAcylation, in turn, controls microglia migration mediated by Aβ1-42 peptide. Given the importance of migration in the progression of AD, in this study we have described a possible mechanism to control these cellular response through Gal-1 Ser8 O-GlcNAcylation.
2. Materials and Methods
2.1. Materials
Aβ1-42 peptide was produced and purified in our laboratory as reported in [19]. Wheat germ agglutinin (WGA)-agarose beads, Thiamet G, OSMI-I and collagen type I were from Sigma-Aldrich. Protein A-SepharoseTMCL-4B beads and Nickel-Agarose beads were from GE Healthcare. Dulbecco´s Modified Eagle Medium (DMEM) high glucose, fetal bovine serum (FBS), 2-mercaptoethanol, penicillin and streptomycin were from Gibco.
2.2. Plasmid construct and TAT-OBD-Gal-1 protein purification
Complementary DNA encoding for a 20–amino acid peptide (residues 1-20) of Gal-1 that contains the Serine 8 (called OGT-binding domain or OBD-Gal-1) was amplified by polymerase chain reaction using the following oligonucleotides: Fw: CTCGAGATGGCTTGTGGTC Rv: GGAATTCCCTCGCCTCGC and cloned in vector pTAT-HA. The pTAT-OBD-Gal-1 vector was transformed into Escherichia coli (BL21) and proteins expressed by Isopropyl-β-D-thiogalactopyranoside (IPTG) induction. Recombinant TAT-OBD-Gal-1 protein was purified by sequential chromatography on Nickel-Agarose (GE) and anion exchange chromatography [20].
2.3. Site-Directed mutagenesis
The mutagenesis of Gal-1S8A was performed with the QuickChange II Site-Directed Mutagenesis Kit according to manufacturer´s instructions (Agilent Technologies). Original plasmid mDsRed-Gal-1 was a gift from Michael Davidson (Addgene plasmid #55831; http://n2t.net/addgene/55831; RRID: Addgene_55831). It was amplified with the following oligonucleotides mDsRed-Gal-1S8A Fw (5´-GCTTGTGGTCTGGTCGCCGCCAACCTGAATCTCAAACC-3’) and mDsRed-Gal-1S8A Rv (5’-GGTTTGAGATTCAGGTTGGCGGCGACCAGACCACAAGC-3’).
2.4. Cell culture
Immortalized human microglia (Cat no T0251, abm, USA) were cultured in DMEM high-glucose, supplemented with 10% FBS and 100 U/mL penicillin and 100 μg/mL streptomycin. Cells were seeded in plates that had previously been coated with 0.01% (v/v) collagen type I and incubated at 37 ºC in an atmosphere with 5% CO2. For experiments, cells were washed 3x with PBS and then deprived with 1% FBS for 24 hours. On the day of the experiments, cells were untreated or treated with 5 μM Amyloid-β1-42 (Aβ1-42) peptide as indicated. To examine the effects of OSMI-I and Thiamet G, those inhibitors were added at 25 μM and 10 μM respectively for16 hours before stimulation with Aβ1-42 peptide.
2.5. Microglia transient transfection
Microglia were washed twice with PBS and resuspended in DMEM high glucose without FBS. 4x106 cells/200 μL medium were then transferred to an electroporation cuvette of 0.4 cm (Sigma-Aldrich) containing between 5-10 μg cDNA of interest as indicated in the results section. The electroporation was carried out at 260 V and 950 μF in a Gene Pulser XcellTM electroporator (Bio-Rad). After that, electroporated cells were collected and DMEM with FBS was added. Transfected cells were incubated for 48 hours in the incubator at 37 ºC before the experiments. Microglia was also transfected with Lipofectamine™ 3000 (Invitrogen, Thermo Fisher Scientific) according to the manufacturer´s instructions 48 hours prior to the experiments.
2.6. Wheat Germ Agglutinin-Affinity Precipitation and protein identification by LC-MS/MS
Cells were stimulated or not with 5 µM Aβ1-42 peptide at 37 °C for 30 minutes and washed twice with cold PBS. Next, cells were lysed with RIPA buffer (50 mM Tris-HCl pH 7.4, 1% Igepal® CA-630, 0.25% sodium deoxycholate, 150 mM NaCl, 1 mM EDTA, 1 mM PMSF, 1 mM Na3VO4, 1 mM NaF and protease inhibitor cocktail (P8340 from Sigma-Aldrich)) for 20 min at 4 °C in an orbital shaker. Then, the lysates were centrifuged at 13500 rpm for 10 min at 4 °C and the supernatants were incubated with previously washed WGA-agarose beads for 1 hour at 4 °C in an orbital shaker. After that, the complexes were washed three times with binding buffer at 4000 rpm for 1 min at 4 °C. The O-GlcNAcylated proteins were dissociated from the WGA-agarose beads by the addition of loading buffer (LB) and samples were analyzed by SDS-PAGE.
For the O-GlcNAcylated proteomic analysis, 6.4 x 107 cells and WGA-agarose beads were used. After purification, the O-GlcNAcylated proteins were eluted from the complexes by the addition of 20 mM GlcNAc in binding buffer for 10 minutes at 4 °C in an orbital shaker. The samples were then centrifuged at 4000 rpm for 1 min at 4 °C and the supernatants were collected and analyzed by LC-MS/MS in the Proteomics General Service (SGIker) of the University of the Basque Country (UPV/EHU). The Q Exactive mass spectrometer (Thermo Scientific) coupled to Easy-nLC 1000 nanoUPLC System (Thermo Scientific) chromatograph was used. The resulting spectra were processed with Proteome Discoverer 1.4 (Thermo Scientific). Swiss-Prot human database was used for peptide identification.
WGA purification was also used for the specific analysis of Gal-1 O-GlcNAcylation. Microglia were transfected with 10 µg of either a plasmid encoding for mDsRed-Gal-1 or mDsRed-Gal-1S8A by electroporation. Cells were serum-deprived for 24 hours and they were stimulated or not with 5 µM Aβ1-42 peptide for 30 minutes. Lysates were obtained in RIPA buffer containing the OGA inhibitor Thiamet G and O-GlcNAcylated proteins were purified with WGA-agarose beads at 4 °C. The affinity complexes were then washed three times with RIPA + Thiamet G and proteins were eluted. The eluted proteins and the total input fractions were analyzed by SDS-PAGE and the presence of Gal-1 was determined by a Western blot using specific anti-DsRed antibody.
2.7. in vitro OGT assay
Microglia were transfected with pDest-N-Myc-OGT by electroporation. 24 hours later, transfected cells were serum-deprived for an additional 24 hours. On the day of the experiment, cells were stimulated or not with 5 µM Aβ1-42 peptide for 30 minutes and they were lysed with RIPA buffer containing OGA inhibitor (10 µM Thiamet G). 500 µg protein per condition were incubated for 2 hours at 4 °C with anti-Myc antibody bound to protein A-Sepharose beads. Immunocomplexes were washed and the transferase reaction was made by adding 100 μL of 1 mM UDP-GlcNAc and 500 ng GST-Gal 1 as substrates. The reaction was incubated at 37 °C for 3 hours and it was stopped by the addition of loading buffer. The proteins were separated by SDS-PAGE and OGT activity was measured by analyzing GST-Gal-1 O-GlcNAcylation by SDS-PAGE and Western blot with specific antibodies.
2.8. Scratch wound healing assay
Cells were seeded 24 hours prior to experiment serum-deprived with 1% FBS and the experiment was done in this serum-free medium to ensure that there was no proliferation during the migration assay. A scratch was done on the cell monolayer and cells were stimulated or not with 5 µM Aβ1-42 peptide. In experiments with inhibitors, these compounds were added 16 h prior to stimulation as indicated in the results section. Photographs of the wound were taken in four random fields with the 4x objective in an EVOS™ Digital Color Fluorescence Microscope (Invitrogen) at the beginning of the experiment and after 24 hours of incubation at 37 °C. The percentage of migration was calculated in Fiji-ImageJ by measuring the width of the wound before (0h) and after (24h) cells had migrated.
2.9. Transwell Migration Assay
Transwell assay was done based on Xu et al., 2016 by using Costar Transwell System with a 8-µm pore size polycarbonate membrane [21]. 250,000 human microglia cells that had been serum-deprived with 1% FBS for 24 hours were seeded in the upper chamber in 200 µL of serum-free medium and in the presence or in the absence of 5 µM Aβ1-42 peptide. 600 µL of serum-free medium were placed in the lower chamber. Cells were incubated for 24 hours at 37 °C. Microglia that had not migrated were removed from the upper side of the membrane with a cotton swab and cells that had migrated to the lower side were fixed with 4% PFA for 10 minutes. Migrated cells were then stained with Mayer´s Hematoxylin for 20 minutes. Four randomly chosen areas were photographed with the 10x objective in an EVOS™ Digital Color Fluorescence Microscope (Invitrogen) and cells were counted in Fiji-ImageJ in order to calculate the percentage of migrating cells.
2.10. Statistical analysis.
All data were expressed as the mean ± S.E.M. Statistical analyses were performed using GraphPad Prism statistical software (versión 5.0; GraphPad software (SanDiego, CA, USA)). Comparisons between experimental groups were made using Mann-Whitney statistical analysis. To determine the significance between data means, (* p < 0.05, ** p < 0.01, *** p < 0.001).
3. Results
3.1. Aβ1-42 peptide Increases Protein O-GlcNAcylation in Human Microglia
Firstly, we investigated if the protein O-GlcNAcylation pattern was modified in the presence of Aβ1-42 peptide in human microglia. For that, 1 x 107 cells were treated or not with 5 μM Aβ1-42 peptide for 30 minutes, washed and lysed. Then O-GlcNAcylated proteins were purified with Wheat germ agglutinin (WGA) agarose beads and analyzed by SDS-PAGE followed by Western blotting. WGA-purified proteins from Aβ1-42 peptide -stimulated microglia showed an increased O-GlcNAcylation pattern (Figure 1A, lane 2 compared to lane 1). Total lysates (input) were used as internal control (Figure 1A, lanes 3, 4). The quantification of three independent experiments demonstrated that the stimulation resulted in a 1.17 ± 0.03 fold increase in microglial protein O-GlcNAcylation, compared to the untreated control (Figure 1 A, Histogram).
These results prompted us to identify the specific proteins that were differentially O-GlcNAcylated in Aβ1-42 peptide -stimulated microglia. With this purpose, cells were stimulated or not with 5 µM Aβ1-42 peptide for 30 minutes and O-GlcNAcylated proteins were purified and analyzed by LC-MS/MS as described in the Materials and Methods section. As shown in the Venn diagram of Figure 1B, the results from the LC-MS/MS identified 44 proteins in untreated cells, 182 proteins in both conditions and 55 proteins were exclusively present in the Aβ1-42 peptide-stimulated lysates (Figure 1B and Table S1).
Of these 55 proteins, Galectin-1 (Gal-1) was chosen among several others owing to its functional versatility. In fact, Gal-1 controls many signaling cascades both in an intra- and extracellular manner with potential implications in the control of microglial migration. Analyzing the specific peptides of Gal-1 obtained by LC-MS/MS, we identified the ACGLVASNLNLKPGECLR peptide. This peptide is located at the N-terminal from aa 2 to aa 19 and contained a serine, Ser8, which could be O-GlcNAcylated. This result suggested that the O-GlcNAcylation in this specific Ser could play an important role in the function of the protein.
Next, we first examined whether Aβ1-42 peptide induced Gal-1 O-GlcNAcylation in human microglia and secondly verified if this PTM occurred on Ser8. Therefore, microglia were transfected with 10 µg of either a plasmid encoding for mDsRed-Gal-1 or mDsRed-Gal-1S8A by electroporation and Gal-1 O-GlcNAcylation was analyzed as indicated in the Materials and Methods section. As shown in Figure 1C, Aβ1-42 peptide stimulation resulted in an increase in WGA-purified Gal-1, indicating increased Gal-1 O-GlcNAcylation stoichiometry (Figure 1C, upper panel, lane 2). From a quantitative point of view, the stimulation resulted in a 2.01 ± 0.02-fold increase of O-GlcNAcylated Gal-1 (Figure 1C, column 2), compared to control (Figure 1C, column 1). The addition of free GlcNAc demonstrated that WGA-agarose beads were specifically purifying O-GlcNAcylated proteins (Figure 1C, upper panel, lane and column 3).
Regarding the mutated Gal-1, our results indicated that the exchange of the Ser8 by Ala resulted in a strong decrease in the Gal-1 O-GlcNAcylation. Without Aβ1-42 peptide stimulation, the O-GlcNAcylation levels were similar to the Gal-1WT in non-stimulated cells (1.22 ± 0.09) and in the presence of Aβ1-42 peptide the levels were 1.04 ± 0.03. Thus, Aβ1-42 peptide was not able to induce an increase in O-GlcNAcylation in Gal-1S8A. Altogether, our findings suggested that Aβ1-42 peptide mediate the Ser8 O-GlcNAcylation of Gal-1 in human microglia.
Given that Aβ1-42 peptide resulted in a rise in microglial protein O-GlcNAcylation, Gal-1 being among those proteins, our next goal was to investigate whether Aβ1-42 peptide produced an increase in OGT enzymatic activity towards Gal-1. This was measured by an in vitro OGT activity assay.
As shown in Figure 1D, Aβ1-42 peptide provoked a rise in OGT activity towards Gal-1, measured as an increase in the O-GlcNAcylation of its substrate Gal-1. For the quantification, O-GlcNAcylated Gal-1 (Figure 1D, upper panel) was normalized with total Gal-1 (Figure 1D, middle panel). Western blot with anti-Myc antibody confirmed the immunoprecipitation of the enzyme OGT (Figure 1D, lower panel). Quantitatively, in the presence of Aβ1-42 peptide, OGT was 2-fold as active as in non-stimulated conditions. The quantification of the Western blot demonstrated a fold change of 2.08 ± 0.33 compared to control (Figure 1D).
3.2. O-GlcNAc homeostasis controls Aβ1-42-induced microglial migration
Taking into account, that OGT mediated the O-GlcNAcylation of Gal-1, a protein implicated in cellular migration, we investigated whether O-GlcNAcylation regulates microglia migration. This was analyzed by a scratch wound healing assay using the OGT inhibitor OSMI-I [22] and the OGA inhibitor Thiamet G [23]. Briefly, 8x105 cells were seeded in a 6-well plate per well 48 hours prior to the experiment and they were then serum-starved for 24 hours. The day of the experiment, 25 μM OSMI-I, 10 μM Thiamet G or DMSO (as vehicle) were added overnight. Next, a scratch was done in the cell monolayer and 5 μM Aβ1-42 peptide was added in the corresponding wells. As expected, 24 h in the presence of Aβ1-42 peptide produced a strong increase in microglia migration (Figure 2D) compared to time 0 (Figure 2B), which resulted in a 440.85 ± 29.15% increase in migration compared to control (Figure 2M). The vehicle (DMSO) did not alter migration. Regarding the effect of OSMI-I, this compound reduced basal microglia migration (Figure 2G) and OSMI-I abolished the Ab1-42-dependent increase in microglial migration (Figure 2H compared to Figure 2D). The migration rate was 40.93 ± 9.09% in the presence of OSMI-I and in the absence of the Aβ1-42 peptide (Figure 2M), and addition of amyloid beta did not affect migration of OSMI-treated cells (40.92 ± 9.72% , Figure 2M). On the contrary, the OGA inhibitor Thiamet G provoked a substantial increase in microglia migration. In fact, when cells were not stimulated with Aβ1-42 peptide, migration reached 539.24 ± 53.28% (Figures K, M). This was similar when the Aβ1-42 peptide was added to microglia, which resulted in a 549.94 ± 66.27% of migration (Figures 2L, M).
In order to verify these results, the effect of O-GlcNAcylation in microglia migration was analyzed by a transwell assay. 250,000 cells were seeded in the upper well of the transwell chamber, they were treated with OSMI-I, Thiamet G or DMSO for 1 hour followed by 5 μM Aβ1-42 oligomers and 24 hours microglia migration was examined. Aβ1-42 peptide increased microglia migration (5516.97 ± 44.89%) (Figure 3B) compared to control cells, (Figure 3A). Again, OSMI-I completely blocked microglial migration induced by Aβ1-42 peptide. From a quantitative point of view, migration rate was 108.12 ± 13.06% (Figure 3D,G) and 87.79 ± 11.41% (Figure 3C,G) with and without Aβ1-42 oligomers stimulation respectively. As expected, Thiamet G provoked an uncontrolled migration, even in the absence of Aβ1-42 peptide, where the migration rate was 5736.69 ± 64.68% (Figure 3E,F), and the presence of Aβ1-42 peptide did not modify this rate 5665.27 ± 164.93% (Figure 3F,M). These findings suggested that protein O-GlcNAcylation could control microglia migration.
3.3. O-GlcNAc homeostasis controls Aβ1-42-induced microglial migration through galectin-1 Ser8 O-GlcNAcylation
The OGT inhibitor, OSMI-I, reduces global O-GlcNAcylation in cells and affects cell viability (24). Taking into account that we had identified by LC/MS the Gal-1 ACGLVASNLNLKPGECLR peptide susceptible to be O-GlcNAcylated in Serine 8, we generated and purified the TAT-OGT binding domain-Gal-1 (TAT-OBD-Gal-1) peptide. This permeant peptide would interfere in the natural interaction between the OGT and Gal-1, blocking the Gal-1 O-GlcNAcylation and downstream signaling pathway.
Briefly, immortalized human microglia (800,000 cells/well) were seeded in 6-well plates 48 hours prior to the experiment. Cells were serum-starved for 24 hours. Then, they were treated with 25 µg of either TAT-EGFP control peptide or 25 µg TAT-OBD-Gal-1 for one hour. A scratch was done in the cell monolayer, 5 µM Aβ1-42 peptide was added and images were taken at 0 h and 24 h of migration in an Evos microscope. As shown in Figure 4, TAT-EGFP-treated cells responded to Aβ1-42 peptide by increasing their migration to 1225.45 ± 16% (Figure 4D, I) comparing to control, expressed as 100% (Figure 4C and I). On the contrary, TAT-OBD-Gal-1 treatment completely blocked microglia migration. In fact, the migration rate was 138.45 ± 15.51% without Aβ1-42 peptide stimulation (Figure 4G, I), which was similarly maintained at 127.25 ± 40.59% with Aβ1-42 peptide (Figure 4H, I).
Next, a transwell assay was performed with TAT-EGFP and TAT-OBD-Gal-1 peptides. In this regard, serum-deprived immortalized human microglia were seeded in the upper well of the transwell chamber (250,000 cells per condition), they were treated with 25 µg either TAT-EGFP control peptide or 25 µg TAT-OBD-Gal-1 for one hour followed by 5 µM Aβ1-42 peptide. Migration was analyzed 24 hours later. As shown in Figure 5, TAT-EGFP-treated cells responded to Aβ1-42 peptide by increasing significantly their migration to 731.73 ± 25.45% (Figure 5B, E) comparing to non-stimulated cells (Figure 5A, E). On the contrary, TAT-OBD-Gal-1 completely blocked microglia migration. These microglia migrated 84.53 ± 14.46% without Aβ1-42 peptide stimulation (Figure 5C, E) which was similarly maintained at 95.38 ± 8.14% with Aβ1-42 peptide (Figure 5D, E). Altogether, these results suggested that Aβ1-42 peptide would involve the tandem OGT/Gal-1 in microglia migration.
In order to examine the role of the Gal-1S8 O-GlcNAcylation in the control of microglia migration, human microglia were transfected with 10 μg of plasmids encoding for either mDsRed-Gal-1wt or mDsRed-Gal-1S8A (non-O-GlcNAc Gal-1) and 24 hours later cells were serum-starved for 24 hours. Then, a scratch was done in the cell monolayer, microglia were treated or not with 5 μM Aβ1-42 peptide and migration was analyzed by taking images at 0 h and 24 h. As shown in Figure 6, microglia that had been transfected with Gal-1wt responded to Aβ1-42 peptide (159.82 ± 3.62%) (Figure 6D,I) compared to control (Figure 6C,I). Gal-1S8A-transfected cells, however, resulted in an uncontrolled migration both in the absence (193.82 ± 5.08%, Figure 6G, I) and in the presence of Aβ1-42 peptide (192.96 ± 1.45%, Figure 6H, I). When we performed the microglia migration experiment in a transwell assay, the migration pattern was similar like wound healing assay. Microglia that had been transfected with Gal-1WT responded to Aβ1-42 peptide by migrating 1177.4 ± 136.16% (Figure 7B and E) compared to control (Figure 7A and E). On the other hand, Gal-1S8A-transfected cells presented an uncontrolled migration both in the absence (1936.20 ± 155.72%, Figure 7C and E) and in the presence of Aβ1-42 peptide (2274.29 ± 155.86%, Figure 78D and E).
Hence, our findings suggested that the Gal-1 Ser8O-GlcNAcylation fine-tunes microglial migration, and abolishing Gal-1 Ser8 O-GlcNAcylation resulted in a hyper-migration phenotype in microglia, indicative of uncontrolled microglial migration. Overall, our data suggest that Aβ1-42 peptide regulates microglial migration through modulation of Gal-1 Ser8 O-GlcNAcylation.
4. Discussion
Protein O-GlcNAcylation pattern is altered in the brains of AD patients, affecting myriad proteins, including synaptic, cytoskeleton and memory-associated proteins, all of them relevant in the context of AD [6,8]. Accordingly, our study on microglia has revealed that Aβ1-42 peptide provoked an increase in protein O-GlcNAcylation, identifying 55 proteins among which we found Gal-1. In addition, LC/MS analysis suggested that the Gal-1 O-GlcNAcylation could occur on serine 8, verifying it both by biochemical and genetic approaches. The fact that Gal-1 is modified by the GlcNAc group points out that it could be an important PTM for cell biology [24,25,26,27]. In fact, we have demonstrated that this O-GlcNAcylation is relevant for migration in human microglia. Furthermore, we have identified that the residue that is being modified is the Ser8 from Gal-1. As a matter of fact, it is interesting to highlight that the Gal-1 is reported as phosphorylated at that same Ser 8 residue [28,29]. Taking into account, that phosphorylation and O-GlcNAcylation could compete for the same residues, a disequilibrium between both PTMs could provoke an aberrant function of Gal-1 in microglia, in our case in the context of AD. Aβ1-42 peptide could favor the balance towards the O-GlcNAcylation as a detriment to phosphorylation. Hence, it would be interesting to investigate if AD patients present reduced phosphorylation, together with increased O-GlcNAcylation, in Gal-1. It would also be important to study whether this imbalance depends on the stage of the disease. We postulate that this imbalance towards Gal-1 O-GlcNAcylation could be happening at initial stages, where microglia migration is increased. Thus, restoring the O-GlcNAcylation/phosphorylation equilibrium could be a therapeutic strategy in pathologies where this equilibrium is altered. In this regard, some investigations have done attempts with OGA inhibitors in order to increase tau protein O-GlcNAcylation and decrease phosphorylation [30,31]. Nevertheless, we believe that this approach should be specific for a particular protein. In fact, not all proteins present an equilibrium displaced towards O-GlcNAcylations in AD, as some of them are displaced towards phosphorylation [32,33,34].
Examining the functionality of O-GlcNAcylated Gal-1 induced by Aβ1-42 peptide in the control of microglia migration, our findings suggested that O-GlcNAcylated Gal-1 maintained this response in a controlled manner. On the contrary, when we mutated Serine 8 to Alanine, which prevented O-GlcNAcylation at this site, although we expected to block microglia migration induced by Aβ1-42 peptide, this mutation led to boost the migration in an uncontrolled manner. This Gal-1 mutant behaved as whether it was a constitutively active form. This increased migration could seem contradictory if we compare it with the inhibition of migration observed with OGT inhibitor OSMI-I or the TAT-OBD-Gal-1 interfering peptide. The fact that OGT has a wide variety of substrates means that inhibiting its activity or using an interfering peptide could block the O-GlcNAcylation of many proteins rather than only Gal-1. Therefore, the response is different than when we specifically target the O-GlcNAcylation of Gal-1. Thus, in this paper we have also described a peptide that could be blocking OGT activity.
Gal-1 is a protein that can function as a monomer or as a homodimer [24]. Its dimerization is essential to activate signaling cascades such as Ras/MAPK in the intracellular space [35] or CD45 phosphatase in the extracellular space [36]. We speculate that the presence of the GlcNAc group in Gal-1 favors its monomeric form over the dimeric form, while the S8A mutant would facilitate Gal-1 homodimerization. Many functions of the Gal-1 as a homodimer are related to its ability to function as a lectin by binding to glycoproteins of the cell membrane and components of the extracellular matrix [24], controlling cell responses such as proliferation, survival, inflammation in cancer or cell migration [24,37]. In the context of the central nervous system (CNS), it is reported that astrocytes secrete Gal-1 which binds to CD45 glycoprotein in microglia to prevent the development of experimental autoimmune encephalomyelitis [36]. Nevertheless, these authors did not investigate if the microglia itself was able to secrete Gal-1 protein and regulate its activation state through an autocrine mechanism. Future research will be needed to clarify whether O-GlcNAcylated Gal-1 is secreted, and secondly, whether it produces an autocrine/paracrine regulation in microglia and other cell types in the CNS.
Conclusion
This study has revealed that in human microglia, Aβ1-42 peptide induced an increase in the protein O-GlcNAcylation and OGT activity towards Gal-1. Moreover, we have identified that one of those proteins is Gal-1. The O-GlcNAcylation of Gal-1 in Ser8 is key for Aβ1-42 peptide-induced microglia migration. In addition, we have generated a Gal-1 (S8A) that behaves as a constitutively active form of Gal-1, absolutely dysregulating microglia migration. Given the importance of microglial migration in AD we do not discard that Gal-1 could play an important role during the progression of AD, especially at initial stages where microglia are recruited to the amyloid plaques.
Supplementary Materials
Table S1: O-GlcNAcylated proteins exclusively identified in the Aβ1-42 peptide-stimulated microglia.
Author Contributions
Conceptualization, F.L. and J.L.Z.; methodology, A.S.A., M.L.M.; investigation, AS.A., F.L., J.L.Z.; formal analysis, A.S.A., F.L.; writing— original draft preparation, A.S.A., F.L. and J.L.Z.; writing—review and editing, F.L. and J.L.Z. All authors have read and agreed to the published version of the manuscript.
Funding
A.A.S. was predoctoral fellowship (PRE_2017_1_0016) from the Basque Government. M.L.M. was a fellowship from Foundation “Jesús de Gangoiti y Barrera”. J.L.Z. was supported by the Instituto de Salud Carlos III (PI18/00207), Basque Government (PIBA_2020_1_0048) and University of Basque Country Grant (US19/04).
Data availability statement
Not applicable.
Acknowledgments
We thank Dr El Bachir Affar (Département de Biochimie et Medicine Moléculaire, Faculté de Médicine, Université de Montréal, Canada), for the generous gift of the plasmid (pDest-N-Myc-OGT) encoding for OGT.
Conflicts of interest
The authors declare that they have no conflicts of interest with the contents of this article. All authors qualify for authorship, approved the final version of the manuscript, and agree to be accountable for all aspects of the research in ensuring that questions related to the accuracy or integrity of any part of the study are appropriately investigated and resolved.
References
- Torres, C.R.; Hart, G.W. Topography and polypeptide distribution of terminal N acetylglucosamine residues on the surfaces of intact lymphocytes. Evidence for O-linked GlcNAc. J Biol Chem 1984, 259, 3308–17. [Google Scholar] [CrossRef]
- Wenzel, D.M.; Olivier-Van Stichelen, S. The O-GlcNAc cycling in neurodevelopment and associated diseases. Biochem Soc Trans 2022, 50, 1693–1702. [Google Scholar] [CrossRef]
- Bond, M.R.; Hanover, J.A. A little sugar goes a long way: the cell biology of O -GlcNAc. J Cell Biol 2015, 208, 869–80. [Google Scholar] [CrossRef] [PubMed]
- Slawson, C.; Hart, G.W. O-GlcNAc signalling: implications for cancer cell biology. Nat Rev Cancer 2011, 11, 678–84. [Google Scholar] [CrossRef] [PubMed]
- Pravata, V.M.; Omelkova, M.; Stavridis, M.P.; Desbiens, C.M.; Stephen, H.M.; et al. An intellectual disability syndrome with single-nucleotide variants in O-GlcNAc transferase. Eur J Hum Genet 2020, 28, 706–14. [Google Scholar] [CrossRef] [PubMed]
- Yuzwa, S.A.; Vocadlo, D.J. O-GlcNAc and neurodegeneration: biochemical mechanisms and potential roles in Alzheimer's disease and beyond. Chem Soc Rev 2014, 43, 6839–58. [Google Scholar] [CrossRef]
- Wang, S.; Yang, F.; Petyuk, V.A.; Shukla, A.K.; Monroe, M.E.; et al. Quantitative proteomics identifies altered O-GlcNAcylation of structural, synaptic and memory associated proteins in Alzheimer's disease. J Pathol 2017, 243, 78–88. [Google Scholar] [CrossRef] [PubMed]
- Forster, S.; Welleford, A.S.; Triplett, J.C.; Sultana, R.; Schmitz, B.; Butterfield, D.A. Increased O-GlcNAc levels correlate with decreased O-GlcNAcase levels in Alzheimer disease brain. Biochim Biophys Acta 2014, 1842, 1333–9. [Google Scholar] [CrossRef]
- Chun, Y.S.; Kwon, O.H.; Chung, S. O-GlcNAcylation of amyloid-beta precursor protein at threonine 576 residue regulates trafficking and processing. Biochem Biophys Res Commun 2017, 490, 486–91. [Google Scholar] [CrossRef]
- Soria Lopez, J.A.; Gonzalez, H.M.; Leger, G.C. Alzheimer's disease. Handb Clin Neurol 2019, 167, 231–55. [Google Scholar]
- Liu, F.; Iqbal, K.; Grundke-Iqbal, I.; Hart, G.W.; Gong. C. X. O-GlcNAcylation regulates phosphorylation of tau: a mechanism involved in Alzheimer's disease. Proc Natl Acad Sci U S A 2004, 101, 10804–9. [Google Scholar] [CrossRef] [PubMed]
- Gong, C.X.; Iqbal, K. Hyperphosphorylation of microtubule-associated protein tau: a promising therapeutic target for Alzheimer disease. Curr Med Chem 2008, 15, 2321–8. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Ha, H.J.; Chung, E.S.; Baek, S.H.; Cho, Y.; et al. O-GlcNAcylation ameliorates the pathological manifestations of Alzheimer's disease by inhibiting necroptosis. Sci Adv 2021, 7. [Google Scholar] [CrossRef] [PubMed]
- Verkhratsky, A.; Zorec, R.; Rodriguez, J.J.; Parpura, V. Astroglia dynamics in ageing and Alzheimer's disease. Curr Opin Pharmacol 2016, 26, 74–9. [Google Scholar] [CrossRef]
- Lai, A.Y. ; McLaurin, JClearance of amyloid-beta peptides by microglia and macrophages: the issue of what, when and where. Future Neurol 2012, 7, 165–76. [Google Scholar] [CrossRef] [PubMed]
- Feng, W.; Zhang, Y.; Wang, Z.; Xu, H.; Wu, T.; et al. Microglia prevent beta-amyloid plaque formation in the early stage of an Alzheimer's disease mouse model with suppression of glymphatic clearance. Alzheimers Res Ther 2020, 12, 125. [Google Scholar] [CrossRef] [PubMed]
- Streit, W.J.; Sammons, N.W.; Kuhns, A.J.; Sparks, D.L. Dystrophic microglia in the aging human brain. Glia 2004, 45, 208–12. [Google Scholar] [CrossRef]
- Hickman, S.E.; Allison, E.K.; El Khoury, J. Microglial dysfunction and defective beta-amyloid clearance pathways in aging Alzheimer's disease mice. J Neurosci 2008, 28, 835460. [Google Scholar] [CrossRef]
- Walsh, D.M.; Thulin, E.; Minogue, A.M.; Gustavsson, N.; Pang, E.; et al. A facile method for expression and purification of the Alzheimer's disease-associated amyloid beta-peptide. FEBS J 2009, 276, 1266–81. [Google Scholar] [CrossRef]
- Haddad, E.; Zugaza, J.L.; Louache, F.; Debili, N.; Crouin, C.; et al. The interaction between Cdc42 and WASP is required for SDF-1-induced T-lymphocyte chemotaxis. Blood 2001, 97, 33–8. [Google Scholar] [CrossRef]
- Xu, F.; Xu, Y.; Zhu, L.; Rao, P.; Wen, J.; et al. 'Fasudil inhibits LPS- induced migration of retinal microglial cells via regulating p38-MAPK signaling pathway'. Mol Vis 2016, 22, 836–46. [Google Scholar] [PubMed]
- Ortiz-Meoz, R.F.; Jiang, J.; Lazarus, M.B.; Orman, M.; Janetzko, J.; et al. A small molecule that inhibits OGT activity in cells. ACS Chem Biol 2015, 10, 1392–7. [Google Scholar] [CrossRef]
- Yuzwa, S.A.; Macauley, M.S.; Heinonen, J.E.; Shan, X.; Dennis, R.J.; et al. A potent mechanism-inspired O-GlcNAcase inhibitor that blocks phosphorylation of tau in vivo. Nat Chem Biol 2008, 4, 483–90. [Google Scholar] [CrossRef] [PubMed]
- Camby, I.; Le Mercier, M.; Lefranc, F.; Kiss, R. Galectin-1: a small protein with major functions. Glycobiology 2006, 16, 137R–157R. [Google Scholar] [CrossRef]
- Nio-Kobayashi, J.; Itabashi, T. Galectins and Their Ligand Glycoconjugates in the Central Nervous System Under Physiological and Pathological Conditions. Front Neuroanat 2021, 15, 767330. [Google Scholar] [CrossRef] [PubMed]
- Mathew, M.P.; Abramowitz, L.K.; Donaldson, J.G.; Hanover, J.A. Nutrient-responsive O-GlcNAcylation dynamically modulates the secretion of glycan-binding protein galectin 3. J Biol Chem 2012, 298, 101743. [Google Scholar] [CrossRef]
- Cedeno-Laurent, F.; Dimitroff, C.J. Galectin-1 research in T cell immunity: past, present and future. Clin Immunol 2021, 142, 107–16. [Google Scholar] [CrossRef] [PubMed]
- Mertins, P.; Yang, F.; Liu, T.; Mani, D.R.; Petyuk, V.A.; et al. Ischemia in tumors induces early and sustained phosphorylation changes in stress kinase pathways but does not affect global protein levels. Mol Cell Proteomics 2014, 13, 1690–704. [Google Scholar] [CrossRef]
- Mertins, P.; Mani, D.R.; Ruggles, K.V.; Gillette, M.A.; Clauser, K.R.; et al. Proteogenomics connects somatic mutations to signalling in breast cancer. Nature 2016, 534, 55–62. [Google Scholar] [CrossRef]
- Selnick, H.G.; Hess, J.F.; Tang, C.; Liu, K.; Schachter, J.B.; et al. Discovery of MK-8719, a Potent O-GlcNAcase Inhibitor as a Potential Treatment for Tauopathies. J Med Chem 2019, 62, 10062–97. [Google Scholar] [CrossRef]
- Yuzwa, S.A.; Macauley, M.S.; Heinonen, J.E.; Shan, X.; Dennis, R.J.; et al. A potent Mechanism inspired O-GlcNAcase inhibitor that blocks phosphorylation of tau in vivo'. Nat Chem Biol 2008, 4, 483–90. [Google Scholar] [CrossRef] [PubMed]
- Robertson, L.A.; Moya, K.L.; Breen, K.C. The potential role of tau protein O glycosylation in Alzheimer's disease'. J Alzheimers Dis 2004, 6, 489–95. [Google Scholar] [CrossRef] [PubMed]
- Forster, S.; Welleford, A.S.; Triplett, J.C.; Sultana, R.; Schmitz, B.; et al. Increased O GlcNAc levels correlate with decreased O-GlcNAcase levels in Alzheimer disease brain. Biochim Biophys Acta 2014, 1842, 1333–9. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Yang, F.; Petyuk, V.A.; Shukla, A.K.; Monroe, M.E.; et al. Quantitative proteomics identifies altered O-GlcNAcylation of structural, synaptic and memory-associated proteins in Alzheimer's disease'. J Pathol 2017, 243, 78–88. [Google Scholar] [CrossRef]
- Blazevits, O., Y.G. Mideksa, M. Solman, A. Ligabue, N. Ariotti, H. et al. Galectin-1 dimers can scaffold Raf-effectors to increase H-ras nanoclustering. Sci Rep 2016, 6, 24165. [Google Scholar] [CrossRef]
- Starossom, S.C.; Mascanfroni, I.D.; Imitola, J.; Cao, L.; Raddassi, K.; et al. Galectin-1 deactivates classically activated microglia and protects from inflammation-induced neurodegeneration. Immunity 2012, 37, 249–63. [Google Scholar] [CrossRef]
- Elola, M.T.; Ferragut, F.; Mendez-Huergo, S.P.; Croci, D.O.; Bracalente, C.; Rabinovich, G.A. Galectins: Multitask signaling molecules linking fibroblast, endothelial and immune cell programs in the tumor microenvironment. Cell Immunol 2018, 333, 34–45. [Google Scholar] [CrossRef]
Figure 1.
Aβ1-42 peptide-stimulated posttranslational modifications by O-GlcNAcylation in microglia. A) Microglia cells were stimulated with 5 μM Aβ1-42 peptide for 30 minutes, lysed in RIPA buffer and O-GlcNAcylated proteins were purified by WGA-agarose beads. Purified and total input fractions were analyzed by SDS-PAGE and Western blot with α-O-GlcNAc. The blot is representative of three independent experiments and histogram represents the mean±S.E.M. of three independent experiments (N=3) compared to control as fold change. Mann-Whitney statistical analysis shows a significant difference. *** p<0.001. B) Microglia was stimulated or not with 5 µM Aβ1-42 peptide for 30 minutes O-GlcNAcylated proteins were purified with WGA-agarose beads. Proteins present in each group were identified by LC-MS/MS. Numbers inside the Venn diagram indicate the amount of proteins that were identified exclusively in each group and the number of proteins that were common in both groups. The Venn diagram is representative of three independent experiments, with the difference of proteins identified between them being ±5. C) Either mDsRed-Gal1-9 or mDsRed-Gal1-9S8A were transfected in microglia, they were stimulated with 5 µM Aβ1-42 peptide and O-GlcNAcylated proteins were purified with WGA-agarose beads. Free GlcNAc was added as a control of specificity of the purification. Purified and total input fractions were analyzed by SDS-PAGE and Western blot with α-DsRed. The blot is representative of three independent experiments (N=3) and histogram represents the mean ± S.E.M. of each WGA-purified condition, normalized by their corresponding total input, and compared to the non-stimulated control. Mann-Whitney statistical analysis shows a significant difference. ***p<0.001; ###p<0.001. D) Myc-OGT was transfected in microglia, they were stimulated with 5 µM Aβ1-42 peptides and an immunoprecipitation was performed with α-myc. Substrates UDP-GlcNAc and GST-Gal-1 were added to the complexes and O-GlcNAcylation reaction was incubated. OGT activity was visualized by SDS-PAGE and Western blot with α-O-GlcNAc for Gal-1. The blot is representative of three independent experiments (N=3). α-O-GlcNAc was used for analyzing OGT activity; α-GST was used in order to check total substrate; and α-myc in order to confirm the immunoprecipitation efficiency. O-GlcNAcylated Gal-1 was normalized by its corresponding total substrate (α-GST) in each condition. These normalized values were compared to the non-stimulated control (fold change). Histogram represents the fold change means ± S.E.M. of each condition. Mann-Whitney statistical analysis showed a significant difference. *p<0.05.
Figure 1.
Aβ1-42 peptide-stimulated posttranslational modifications by O-GlcNAcylation in microglia. A) Microglia cells were stimulated with 5 μM Aβ1-42 peptide for 30 minutes, lysed in RIPA buffer and O-GlcNAcylated proteins were purified by WGA-agarose beads. Purified and total input fractions were analyzed by SDS-PAGE and Western blot with α-O-GlcNAc. The blot is representative of three independent experiments and histogram represents the mean±S.E.M. of three independent experiments (N=3) compared to control as fold change. Mann-Whitney statistical analysis shows a significant difference. *** p<0.001. B) Microglia was stimulated or not with 5 µM Aβ1-42 peptide for 30 minutes O-GlcNAcylated proteins were purified with WGA-agarose beads. Proteins present in each group were identified by LC-MS/MS. Numbers inside the Venn diagram indicate the amount of proteins that were identified exclusively in each group and the number of proteins that were common in both groups. The Venn diagram is representative of three independent experiments, with the difference of proteins identified between them being ±5. C) Either mDsRed-Gal1-9 or mDsRed-Gal1-9S8A were transfected in microglia, they were stimulated with 5 µM Aβ1-42 peptide and O-GlcNAcylated proteins were purified with WGA-agarose beads. Free GlcNAc was added as a control of specificity of the purification. Purified and total input fractions were analyzed by SDS-PAGE and Western blot with α-DsRed. The blot is representative of three independent experiments (N=3) and histogram represents the mean ± S.E.M. of each WGA-purified condition, normalized by their corresponding total input, and compared to the non-stimulated control. Mann-Whitney statistical analysis shows a significant difference. ***p<0.001; ###p<0.001. D) Myc-OGT was transfected in microglia, they were stimulated with 5 µM Aβ1-42 peptides and an immunoprecipitation was performed with α-myc. Substrates UDP-GlcNAc and GST-Gal-1 were added to the complexes and O-GlcNAcylation reaction was incubated. OGT activity was visualized by SDS-PAGE and Western blot with α-O-GlcNAc for Gal-1. The blot is representative of three independent experiments (N=3). α-O-GlcNAc was used for analyzing OGT activity; α-GST was used in order to check total substrate; and α-myc in order to confirm the immunoprecipitation efficiency. O-GlcNAcylated Gal-1 was normalized by its corresponding total substrate (α-GST) in each condition. These normalized values were compared to the non-stimulated control (fold change). Histogram represents the fold change means ± S.E.M. of each condition. Mann-Whitney statistical analysis showed a significant difference. *p<0.05.
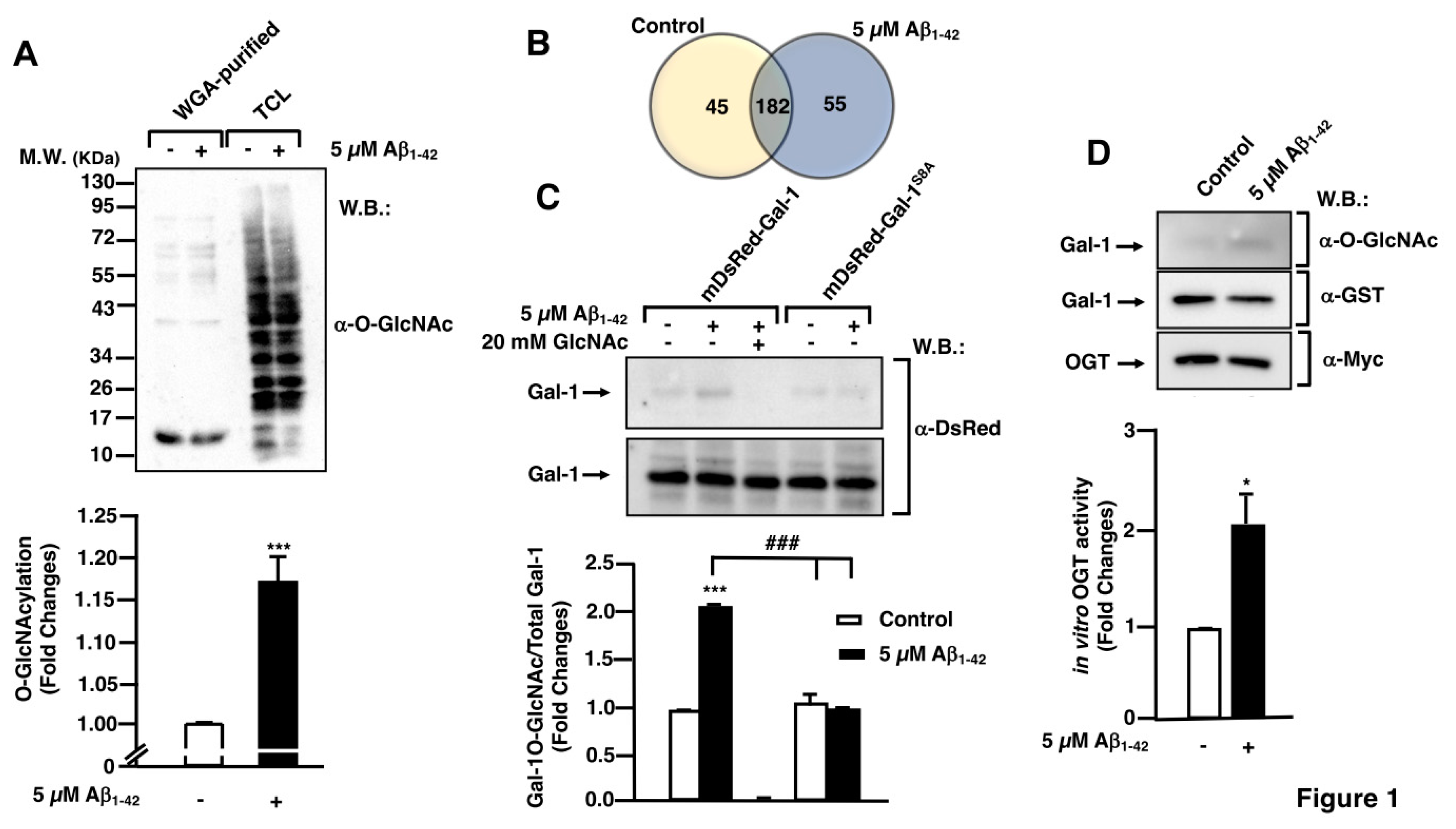
Figure 2.
Protein O-GlcNAcylation mediated by Aβ1-42 peptide control microglia migration: analysis of cell migration by in vitro wound healing assay. Microglia were treated with either vehicle (DMSO), OSMI or Thiamet G and they were stimulated or not with 5 µM Aβ1-42 peptide. Cell migration was analyzed after 24 hours of incubation. A-L. Representative photographs of a randomly chosen field of one independent experiment out of three. Scale bar in the lower part of each photograph is 1000 μm. M. Histogram represents the quantification of the migration. Values indicate the mean ± S.E.M. of the migrated distance (expressed as the % of control) in four randomly chosen fields of three independent experiments (N=3. Mann-Whitney statistical analysis shows a significant difference. ##p<0.01; **p<0.01; *** p<0.001.
Figure 2.
Protein O-GlcNAcylation mediated by Aβ1-42 peptide control microglia migration: analysis of cell migration by in vitro wound healing assay. Microglia were treated with either vehicle (DMSO), OSMI or Thiamet G and they were stimulated or not with 5 µM Aβ1-42 peptide. Cell migration was analyzed after 24 hours of incubation. A-L. Representative photographs of a randomly chosen field of one independent experiment out of three. Scale bar in the lower part of each photograph is 1000 μm. M. Histogram represents the quantification of the migration. Values indicate the mean ± S.E.M. of the migrated distance (expressed as the % of control) in four randomly chosen fields of three independent experiments (N=3. Mann-Whitney statistical analysis shows a significant difference. ##p<0.01; **p<0.01; *** p<0.001.
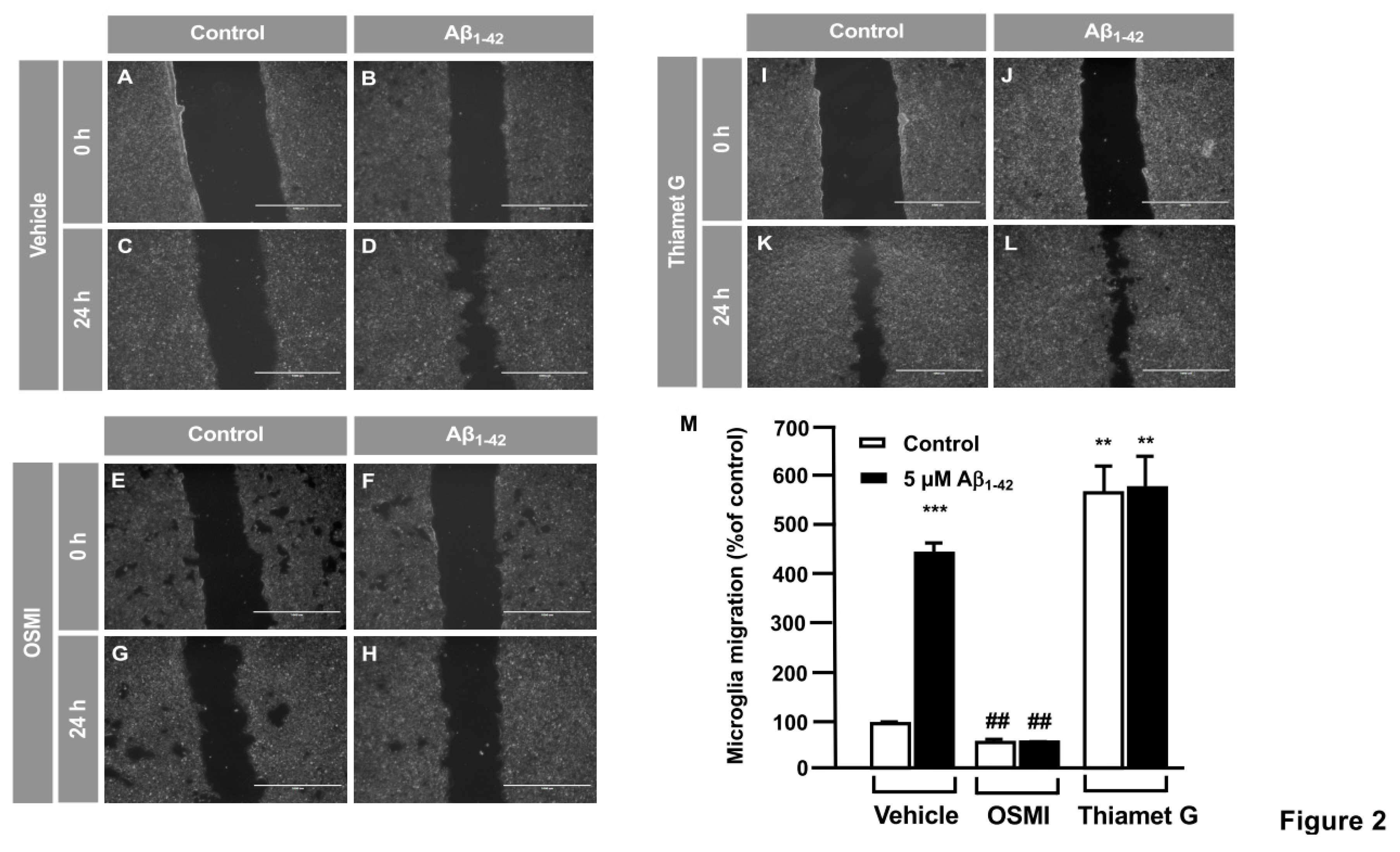
Figure 3.
Protein O-GlcNAcylation mediated by Aβ1-42 peptide control microglia migration: Quantitative and qualitative analysis of microglia migration assessed by in vitro transwell assay. Microglia were treated with either vehicle (DMSO), OSMI or Thiamet G and they were stimulated or not with 5 µM Aβ1-42 peptide. Cell migration was analyzed after 24 hours of incubation. A-F. Representative photographs of a randomly chosen field of one independent experiment out of three. Purple areas correspond to all the cells that migrated, stained with Mayer´s hematoxylin. Scale bar in the lower part of each photograph is 1000 μm. G. Histogram represents the quantification of the migration. Values indicate the mean ± S.E.M. of the stained area (corresponding to the migrated cells) expressed as the % of control in four randomly chosen fields of three independent experiments (N=3). Mann-Whitney statistical analysis shows a significant difference. *** p<0.001.
Figure 3.
Protein O-GlcNAcylation mediated by Aβ1-42 peptide control microglia migration: Quantitative and qualitative analysis of microglia migration assessed by in vitro transwell assay. Microglia were treated with either vehicle (DMSO), OSMI or Thiamet G and they were stimulated or not with 5 µM Aβ1-42 peptide. Cell migration was analyzed after 24 hours of incubation. A-F. Representative photographs of a randomly chosen field of one independent experiment out of three. Purple areas correspond to all the cells that migrated, stained with Mayer´s hematoxylin. Scale bar in the lower part of each photograph is 1000 μm. G. Histogram represents the quantification of the migration. Values indicate the mean ± S.E.M. of the stained area (corresponding to the migrated cells) expressed as the % of control in four randomly chosen fields of three independent experiments (N=3). Mann-Whitney statistical analysis shows a significant difference. *** p<0.001.
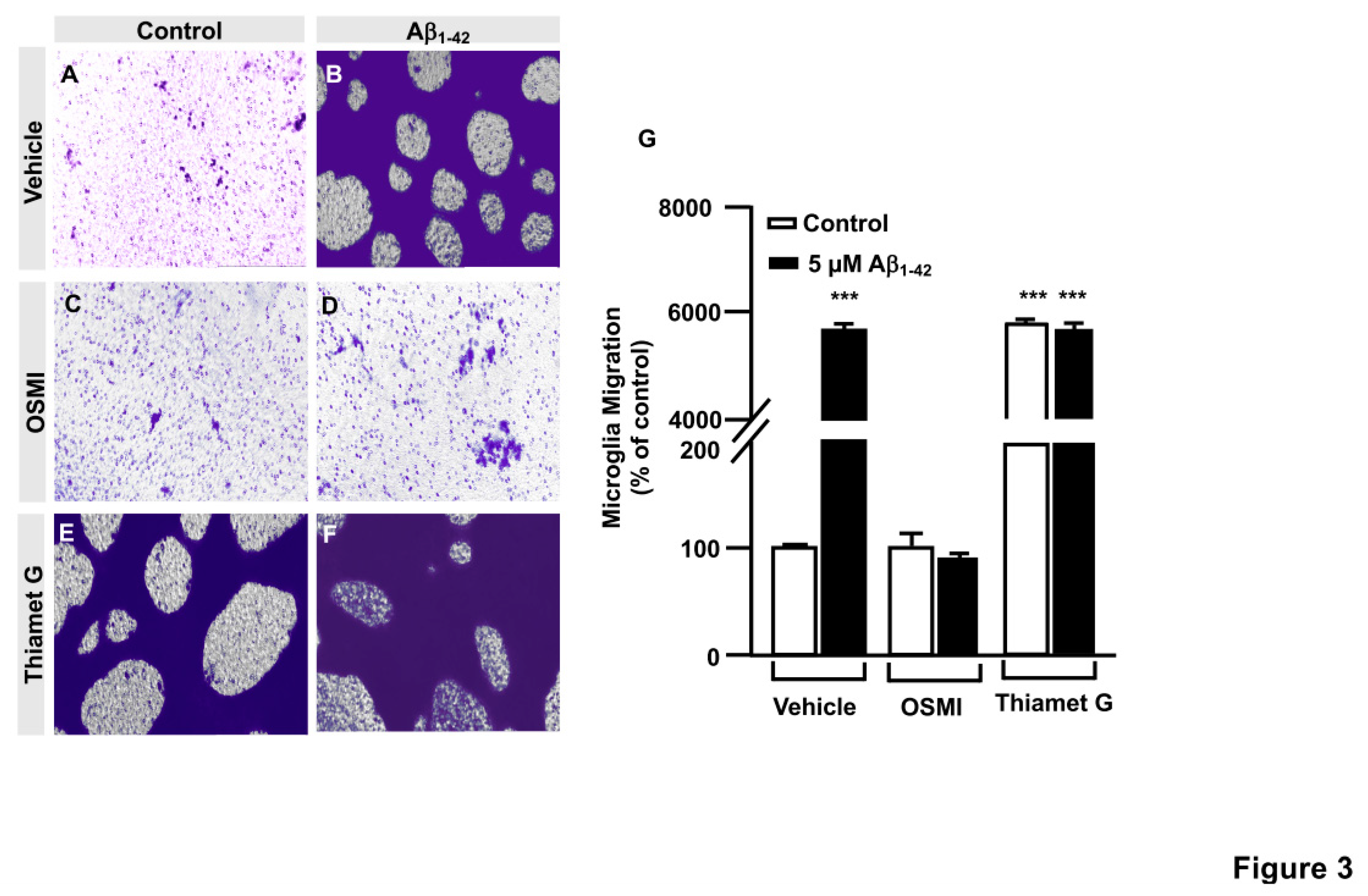
Figure 4.
TAT-OBD-Gal-1 peptide blocks Aβ1-42 oligomers-induced microglia migration: analysis of cell migration by in vitro wound healing assay. Microglia were treated with either TAT-EGFP or TAT-OBD-Gal-1 and they were stimulated or not with 5 µM Aβ1-42 peptide. Cell migration was analyzed after 24 hours of incubation. A-H. Representative photographs of a randomly chosen field of one independent experiment out of three. Scale bar in the lower part of each photograph is 1000 μm. I. Histogram represents the quantification of the migration. Values indicate the mean ± S.E.M. of the migrated distance (expressed as the % of control) in four randomly chosen fields of three independent experiments (N=3). Mann-Whitney statistical analysis showed a significant difference. *** p<0.001.
Figure 4.
TAT-OBD-Gal-1 peptide blocks Aβ1-42 oligomers-induced microglia migration: analysis of cell migration by in vitro wound healing assay. Microglia were treated with either TAT-EGFP or TAT-OBD-Gal-1 and they were stimulated or not with 5 µM Aβ1-42 peptide. Cell migration was analyzed after 24 hours of incubation. A-H. Representative photographs of a randomly chosen field of one independent experiment out of three. Scale bar in the lower part of each photograph is 1000 μm. I. Histogram represents the quantification of the migration. Values indicate the mean ± S.E.M. of the migrated distance (expressed as the % of control) in four randomly chosen fields of three independent experiments (N=3). Mann-Whitney statistical analysis showed a significant difference. *** p<0.001.
Figure 5.
TAT-OBD-Gal-1 peptide blocks Aβ1-42 oligomers-induced microglia migration: Quantitative and qualitative analysis of microglia migration assessed by in vitro transwell assay. microglia were treated with either TAT-EGFP or TAT-OBD-Gal1 and they were stimulated or not with 5 µM Aβ1-42 peptide. Cell migration was analyzed after 24 hours of incubation. A-D. Representative photographs of a randomly chosen field of one independent experiment out of three. H. Histogram represents the quantification of the migration. Values indicate the mean ± S.E.M. of the stained area (expressed as the % of control) in four randomly chosen fields of three independent experiments (N=3). Mann-Whitney statistical analysis showed a significant difference. *** p<0.001.
Figure 5.
TAT-OBD-Gal-1 peptide blocks Aβ1-42 oligomers-induced microglia migration: Quantitative and qualitative analysis of microglia migration assessed by in vitro transwell assay. microglia were treated with either TAT-EGFP or TAT-OBD-Gal1 and they were stimulated or not with 5 µM Aβ1-42 peptide. Cell migration was analyzed after 24 hours of incubation. A-D. Representative photographs of a randomly chosen field of one independent experiment out of three. H. Histogram represents the quantification of the migration. Values indicate the mean ± S.E.M. of the stained area (expressed as the % of control) in four randomly chosen fields of three independent experiments (N=3). Mann-Whitney statistical analysis showed a significant difference. *** p<0.001.
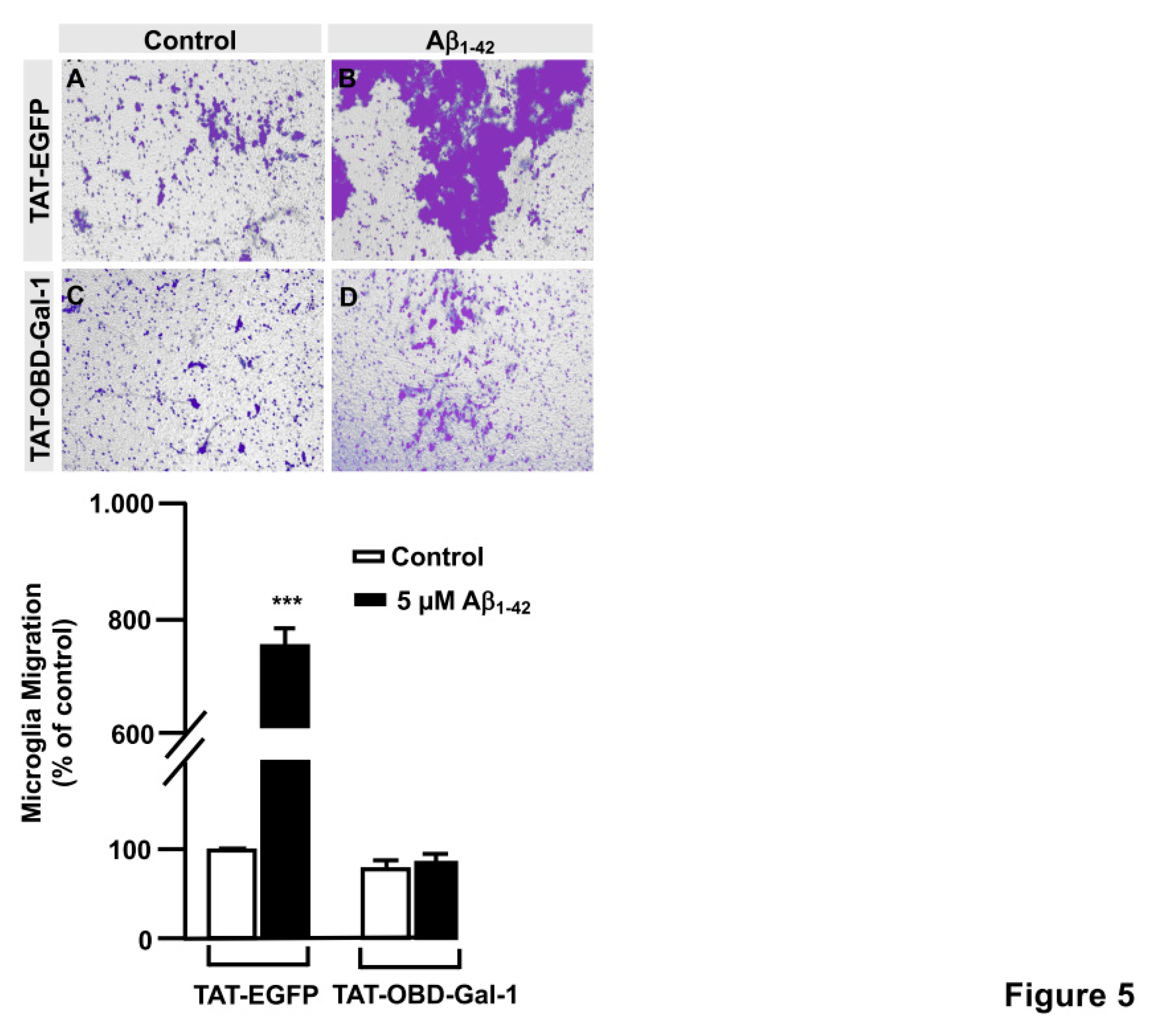
Figure 6.
Galectin-1 O-GlcNAcylation regulates microglia migration in a controlled manner: analysis of cell migration by in vitro wound healing assay. Microglia was transfected with 10 µg of either mDsRed-Gal-1WT or mDsRed-Gal-1S8A and they were stimulated or not with 5 µM Aβ1-42 peptide. Cell migration was analyzed after 24 hours of incubation. A-H. Representative photographs of a randomly chosen field of one independent experiment out of three. Scale bar in the lower part of each photograph is 1000 μm. G. Histogram represents the quantification of the migration. Values indicate the mean ± S.E.M. of the migrated distance (expressed as the % of control) in four randomly chosen fields of three independent experiments (N=3). Mann-Whitney statistical analysis shows a significant difference. *** p<0.001.
Figure 6.
Galectin-1 O-GlcNAcylation regulates microglia migration in a controlled manner: analysis of cell migration by in vitro wound healing assay. Microglia was transfected with 10 µg of either mDsRed-Gal-1WT or mDsRed-Gal-1S8A and they were stimulated or not with 5 µM Aβ1-42 peptide. Cell migration was analyzed after 24 hours of incubation. A-H. Representative photographs of a randomly chosen field of one independent experiment out of three. Scale bar in the lower part of each photograph is 1000 μm. G. Histogram represents the quantification of the migration. Values indicate the mean ± S.E.M. of the migrated distance (expressed as the % of control) in four randomly chosen fields of three independent experiments (N=3). Mann-Whitney statistical analysis shows a significant difference. *** p<0.001.
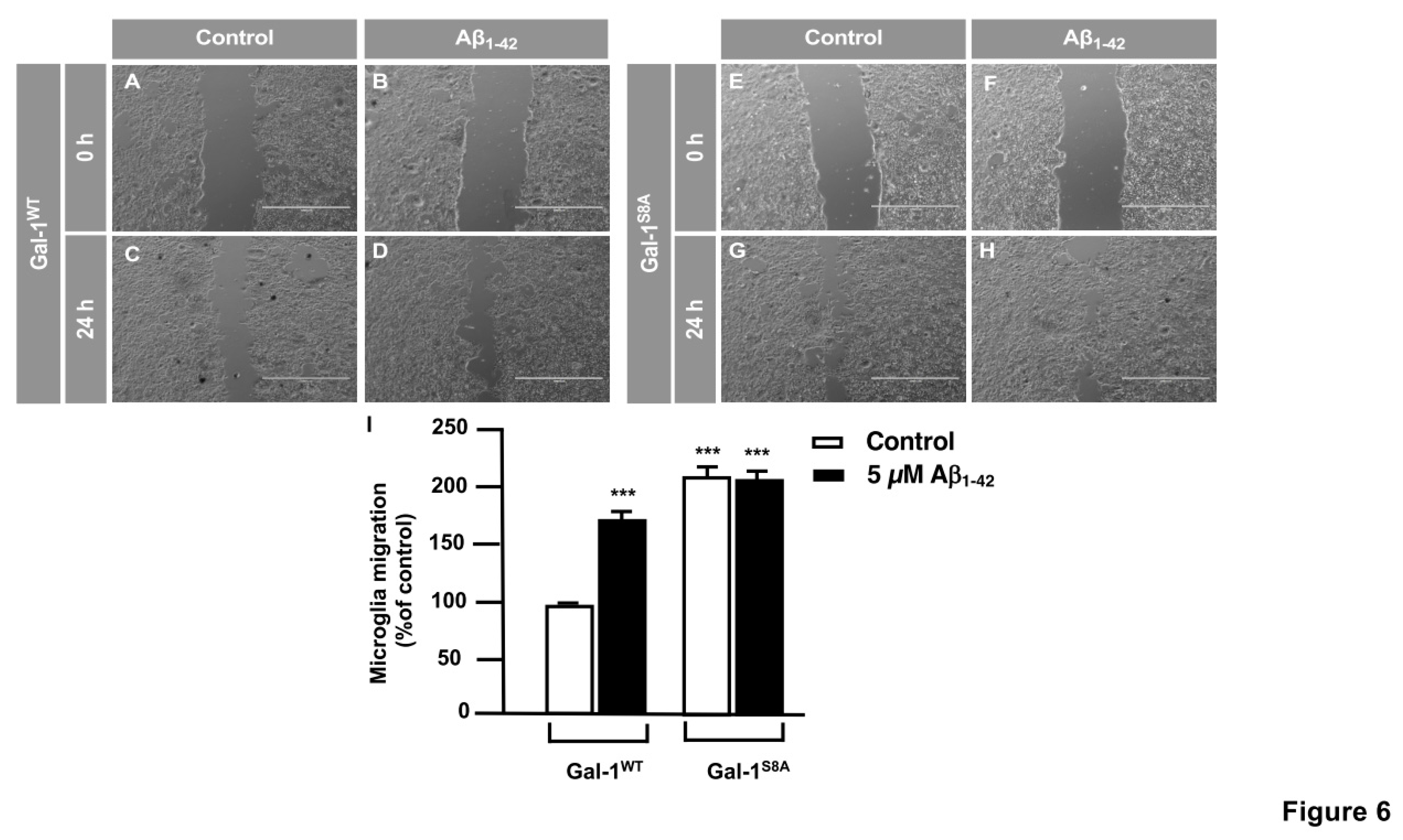
Figure 7.
Galectin-1 O-GlcNAcylation regulates microglia migration in a controlled manner: Quantitative and qualitative analysis of melanoma cell migration assessed by in vitro transwell assay. Microglia cells were transfected with 10 µg of either mDsRed-Gal-1-9WT or mDsRed-Gal-1-9S8A and they were stimulated or not with 5 µM Aβ1-42 peptide. Cell migration was analyzed after 24 hours of incubation. A-D. Representative photographs of a randomly chosen field of one independent experiment out of three. E. Hisotgram represents the quantification of the migration. Values indicate the mean ± S.E.M. of the stained area (expressed as the % of control) in four randomly chosen fields of three independent experiments (N=3). Mann-Whitney statistical analysis shows a significant difference. **p<0.01; *** p<0.001.
Figure 7.
Galectin-1 O-GlcNAcylation regulates microglia migration in a controlled manner: Quantitative and qualitative analysis of melanoma cell migration assessed by in vitro transwell assay. Microglia cells were transfected with 10 µg of either mDsRed-Gal-1-9WT or mDsRed-Gal-1-9S8A and they were stimulated or not with 5 µM Aβ1-42 peptide. Cell migration was analyzed after 24 hours of incubation. A-D. Representative photographs of a randomly chosen field of one independent experiment out of three. E. Hisotgram represents the quantification of the migration. Values indicate the mean ± S.E.M. of the stained area (expressed as the % of control) in four randomly chosen fields of three independent experiments (N=3). Mann-Whitney statistical analysis shows a significant difference. **p<0.01; *** p<0.001.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



