
Submitted:
18 May 2023
Posted:
19 May 2023
You are already at the latest version
Abstract
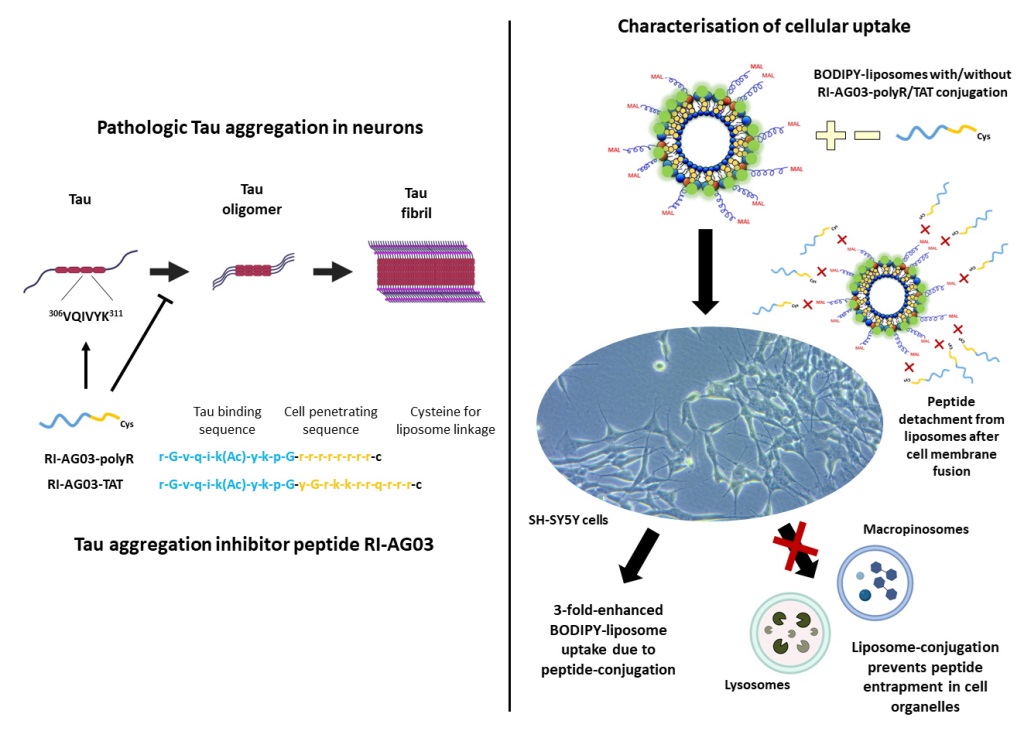
Given the pathological role of hyperphosphorylated Tau aggregation in Alzheimer’s disease (AD) our laboratory previously developed the novel Tau aggregation inhibitor peptide, RI-AG03. As Tau aggregates accumulate intracellularly, it is essential that Tau-targeting peptides efficiently traverse the cell membrane. In addition, these peptides need to avoid sequestration into cell organelles, particularly those involved in degradation, to ensure interaction with Tau in the cytoplasm. Here, we examine the cellular uptake and intracellular trafficking of RI-AG03 when the peptide is unconjugated or linked to nanoliposome carrier particles. The impact of adding the cell penetrating peptide (CPP) sequences, polyarginine (polyR) and transactivator of transcription (TAT), to RI-AG03 was also characterised. Our data show that the conjugation of CPP-containing RI-AG03 to liposomes dramatically increases cellular liposomes uptake. The majority of this uptake occurs via direct cell penetration. However, we find that macropinocytosis is also an important mechanism for unconjugated and RI-AG03-polyR-conjugated, but not RI-AG03-TAT-conjugated, liposome uptake. In agreement with this macropinocytosis-mediated internalisation, liposome lipids localise to lysosomes and macropinosomes following cellular uptake. However, while unconjugated RI-AG03 peptides also localise to lysosomes, liposome conjugation prevents this localisation, with the peptide detaching from the liposomes at the cell surface and evading organelle entrapment. Overall, this study demonstrates that CPP sequences and nanoliposomes carriers enhance the intracellular delivery of RI-AG03, furthering the possibility of the peptide’s use in targeting Tau pathology in AD.
Keywords:
Alzheimer’s Disease
; Tauopathy
; Cell penetrating peptide
; Polyarginine
; TAT
; Liposomes
; Nanoparticles
; Tau pathology
; Tau aggregation inhibitor
; Endocytosis
1. Introduction
Alzheimer’s disease (AD) pathology is characterized by the extracellular accumulation of amyloid plaques consisting of aggregated amyloid beta (Aβ)-peptides and the intracellular accumulation of neurofibrillary tangles (NFTs) consisting of aggregated hyperphosphorylated Tau protein [1]. In recent years, most therapeutic efforts have focused on the reduction of pathologic Aβ species (reviewed in e.g. [2] and [3]) [4,5]. However, the alternative and complementary approach of reducing Tau aggregates has gained considerable traction [6]. To date, Tau-targeted therapies have focused on the inhibition of Tau kinases and Tau aggregation, or immunotherapies that enhance clearance of the protein [6,7].
Under non-pathological conditions, Tau functions as a microtubule-assembling and stabilising protein localised to neuronal axons [8]. Through alternative splicing, six Tau isoforms are produced from the Microtubule-Associated Protein Tau (MAPT) gene on chromosome 17. Depending on the inclusion of exon 10, Tau isoforms contain either 3 or 4 microtubule-binding repeat domains (R) along with either zero, one or two N-terminal inserts (N) [9,10]. Mutations in the MAPT gene or the age-associated build-up of wild-type Tau protein initiate the accumulation, hyperphosphorylation and conformational rearrangement of Tau. This results in microtubular detachment and the sequential aggregation of Tau into toxic soluble oligomers, paired helical filaments (PHFs) and insoluble NFTs within neurons [9,10]. Importantly, Tau oligomers and the proteolysed core of PHFs, containing the microtubule repeat domain region of Tau (R1 - R4), spread between neurons to cross-seed Tau aggregation [11,12,13,14]. This results in the propagation of Tau throughout the central nervous system. Interestingly, the spread of Tau, as assessed by Braak staging in AD [15], is more strongly correlated with cognitive decline and the appearance of clinical symptoms than Aβ pathology [9,10,16], suggesting that the propagation of Tau is a primary driver of the disease.
Based on the aggregation-inducing 16KLVFF20 sequence within Aβ, we previously developed peptides to reduce Aβ-peptide aggregation [17]. The most effective peptides, OR2 and its proteolytically resistant form RI-OR2, prevent the aggregation of recombinant Aβ into oligomers and fibrils in vitro, whilst protecting SH-SY5Y cells from Aβ toxicity [17,18]. Since it has previously been shown that conjugating a transactivator of transcription (TAT) cell-penetrating peptide (CPP) sequence to peptides and nanoparticles improves their blood brain barrier (BBB) translocation [19,20], this CPP was added to our peptide to generate RI-OR2-TAT for in vivo utility [21]. We showed that fluorescein-labelled RI-OR2-TAT crossed the BBB in APP/PSEN1 mice following intraperitoneal injection [21]. Moreover, RI-OR2-TAT decreased plaque load, microgliosis and oxidative stress, while enhancing neurogenesis in the hippocampal dentate gyrus in the APP/PS1 mouse model of AD, confirming the in vivo efficacy of the peptide.
To further enhance delivery of RI-OR2-TAT, multiple copies of the peptide were attached to distearoyl phosphatidylethanolamine (DSPE)-polyethylene glycol (PEG)-maleimide (Mal) liposomes to form peptide inhibitor nanoparticles (PINPs) [22]. This liposome-conjugated form of RI-OR2 achieved 50% inhibition of Aβ1-42 aggregation at a much lower molar ratio (1:2000 drug:Aβ) than the unconjugated RI-OR2 peptide (1:5 drug:Aβ). Furthermore, the PINPs were found to translocate across an in vitro BBB model (hCMEC/D3 cell monolayer), while improving object recognition memory in the Tg2576 (APPSWE) AD mouse model in vivo.
Given recent interest in Tau as a therapeutic target for AD, we decided to apply a similar strategy to develop a novel peptide that targets Tau aggregation. The 306VQIVYK311 in the R3 domain (present in all six Tau isoforms) and 275VQIINK280 sequence in R2 (present in 4R Tau isoforms that contain the translated exon 10) are thought to drive Tau beta-sheet formation and aggregation [23,24]. We tested various peptides based on the 306VQIVYK311 sequence and identified RI-AG03 (see Figure 1A for sequence) as being particularly efficacious in attenuating the aggregation of heparin-seeded recombinant TauΔ1-250 in vitro, with a 94% reduction in Tau aggregation seen at equimolar drug:Tau concentrations [25].
Due to the intraneuronal localisation and propagation of Tau [7], intracellular delivery of Tau-targeting therapeutics is essential. Thus, to enhance both BBB penetration and neuronal uptake, RI-AG03 contains an octaarginine (polyarginine; polyR) CPP sequence [27,28,29,30]. Interestingly, arginine-rich peptides may also show anti-aggregatory effects on amyloids (reviewed in [31]). In agreement with this suggestion, we found that adding three or nine arginine residues to an earlier version of our peptide, AG02, enhanced inhibition of TauΔ1-250 aggregation in vitro by AG02 by 20 % or 30 %, respectively [25].
Cellular uptake of extracellular material, including RI-AG03-polyR or liposomes, occurs in either an energy-independent manner or through ATP-dependent transport across the cell membrane, also known as endocytosis. Various form of endocytosis exists, including clathrin-mediated endocytosis (CME) and clathrin-independent mechanisms, such as caveolae-mediated endocytosis (CavME) and macropinocytosis [32,33,34]. Liposomes, due to their lipophilic properties, can directly fuse with and translocate across the cell membrane [35,36,37]. Alternatively, liposomes can be internalised by endocytosis. Liposome concentration and properties including cargo type, size, shape, surface charge and hydrophobicity determine the preferred cellular uptake pathways involved [33,38]. The surface modification of liposomes can also regulate the cellular uptake pathways involved. Unconjugated CPPs can be taken up by direct membrane translocation, CME, CavME or macropinocytosis, dependent on a range of conditions, such as CPP type and concentration, choice of the employed cell line or cell density (reviewed in [38]). The conjugation of positively charged CPPs to liposomes, is thought to improve liposome apposition and fusion with the negatively charged plasma membrane, resulting in enhanced cellular liposome uptake [35].
While RI-AG03-polyR PINPs have previously been shown to prevent aggregation of recombinant Tau [25], their efficacy in cellular models has not been confirmed and the intracellular uptake mechanisms for RI-AG03-polyR PINPs have not yet been characterised. Therefore, in the current study, we characterise the primary cellular uptake mechanisms and intracellular trafficking of RI-AG03-polyR PINPs in a human neuronal cell line (neuroblastoma SH-SY5Y cells).
RI-AG03 was synthesised with either a polyR or TAT sequence to compare the impact of these CPPs on cell internalisation (Figure 1). RI-AG03-polyR and RI-AG03-TAT were attached to liposomes by click chemistry between a cysteine residue added to the end of the CPP sequence and maleimide groups (Mal) protruding from the liposomes [39]. In order to visualise the subcellular distribution of the peptide and liposomes, respectively, 6-FAM and Cy5-labelled peptides and BODIPY-cholesterol-containing liposomes were used. Endocytosis of unconjugated and peptide-conjugated BODIPY-liposomes was evaluated using pharmacological inhibitors for CME (chlorpromazine), CavME (filipin) and macropinocytosis (EIPA selective for micropinocytosis and cytochalasin D for macropinocytosis and partial CME/CavME inhibition) [40,41].
Our data show that conjugating RI-AG03 containing either the polyR or TAT sequence to liposomes increased cellular liposome uptake three-fold. Unconjugated liposomes and liposome-conjugated RI-AG03, with the polyR or TAT CPP, were mainly internalised via direct membrane penetration. However, cellular uptake of unconjugated and RI-AG03-polyR-linked liposomes was also partially mediated by macropinocytosis. Importantly, following cellular uptake, we show that RI-AG03 dissociates from liposomes. We also found that the peptide does not co-localise with any of the cell organelles we characterised, suggesting that conjugating RI-AG03 to liposomes prevents cell organelle entrapment of the peptide. Collectively, these results give new insight into the cellular uptake mechanisms of our liposome-conjugated Tau aggregation inhibitor peptides, supporting the use of CPPs to enhance cellular uptake and confirming their intracellular availability as Tau-targeted therapeutics.
Results
Conjugating RI-AG03-polyR and RI-AG03-TAT to Liposomes Enhances Cellular Liposome Uptake
We initially investigated whether unconjugated liposomes or peptide-conjugated liposomes were taken up by SH-SY5Y neuroblastoma cells over a 4 h time period. Cells were left untreated or treated with either unconjugated BODIPY-liposomes, RI-AG03-polyR BODIPY-liposomes or RI-AG03-TAT BODIPY-liposomes. Median cell fluorescence was significantly influenced by treatment type (F(3, 32) = 32.42, p < 0.001; one-way ANOVA). While treatment of SH-SY5Y cells with unconjugated BODIPY-liposomes led to a 94-fold increase in median cell fluorescence, this was non-significant (Figure 2). Treatment of cells with RI-AG03-polyR-conjugated BODIPY-liposomes and RI-AG03-TAT-conjugated BODIPY-liposomes resulted in a significant 309-fold (p < 0.001, Tukey’s HSD) and 311-fold (p < 0.001, Tukey’s HSD) increase, respectively, in median cell fluorescence relative to untreated cells. Moreover, conjugating RI-AG03-polyR or RI-AG03-TAT to BODIPY-liposomes also significantly increased cellular fluorescence (approximately 3-fold) relative to unconjugated BODIPY liposomes (p < 0.001 in both cases). Therefore, linking RI-AG03 with either a polyR or TAT CPP to liposomes increases their cellular uptake.
Confocal microscopy (Figure 2B) showed that treatment with unconjugated and peptide-conjugated BODIPY-liposomes increases fluorescence in the cell cytoplasm, supporting the cellular uptake of liposome components. Cells treated with RI-AG03-polyR or RI-AG03-TAT-conjugated BODIPY-liposomes exhibited an apparent greater fluorescence in the cytoplasm than that of cells treated with unconjugated BODIPY-liposomes (Figure 2B), supporting more effective internalisation of liposome components when the peptides were conjugated to liposomes.
Unconjugated and RI-AG03-polyR Conjugated Liposomes, but not RI-AG03-TAT-Conjugated Liposomes, Are Partially Internalised by Macropinocytosis
By utilising the endocytosis inhibitors chlorpromazine, filipin, cytochalasin D and EIPA [40,41], we sought to determine the cellular uptake mechanisms involved in the uptake of unconjugated and peptide-conjugated liposomes. Initially, we confirmed that the inhibitor concentrations used were non-toxic (Supplementary Material, Figure S1A and S1B) to ensure that cell death would not affect the measurement of liposome uptake.
Endocytosis inhibitor treatment significantly altered BODIPY-fluorescence levels in SH-SY5Y cells treated with unconjugated liposomes (F(4, 40)= 11.30, p < 0.001; one-way ANOVA; Figure 3A). Both the CME inhibitor chlorpromazine and the CavME inhibitor filipin had no effect on cell BODIPY fluorescence levels when the cells were treated with unconjugated BODIPY-liposomes. However, both the macropinocytosis inhibitor cytochalasin D (p < 0.001) and the macropinocytosis inhibitor EIPA (p = 0.010) significantly reduced cellular BODIPY fluorescence levels by -19 % and -13 %, respectively, relative to no-inhibitor controls, when cells were treated with unconjugated BODIPY-liposomes. These results suggest that unconjugated BODIPY-liposomes are not internalised by CME or CavME, but that they are partially taken up via macropinocytosis. However, since the effects of cytochalasin D and EIPA were modest, most unconjugated BODIPY-liposome uptake by SH-SY5Y cells is likely through energy-independent membrane fusion and translocation, as previously characterised for liposomes [33,42].
For RI-AG03-polyR-conjugated BODIPY-liposomes, chlorpromazine and filipin again had no effect on cell BODIPY fluorescence levels (Figure 3A). In addition, cytochalasin D treatment did not influence cellular uptake of these peptide-liposomes. By contrast, relative to no-inhibitor controls, EIPA significantly decreased (-28%) cell BODIPY fluorescence levels in RI-AG03-polyR-conjugated BODIPY-liposome-treated cells (p < 0.05). These data suggest that uptake of RI-AG03-polyR-conjugated BODIPY-liposomes is independent of CME and CavME, but that macropinocytosis is partly involved. In the case of RI-AG03-TAT-conjugated BODIPY-liposomes, none of the endocytosis inhibitors altered the cellular BODIPY fluorescence levels (Figure 3C). Thus, cellular uptake of RI-AG03-TAT-conjugated BODIPY-liposomes seems to be independent of energy-dependent CME, CavME and macropinocytosis, and may involve uptake via energy-independent mechanisms.
Collectively, these data demonstrate that unconjugated, RI-AG03-polyR-conjugated and RI-AG03-TAT-conjugated BODIPY-liposome components predominantly enter cultured SH-SY5Y cells through direct membrane fusion and translocation. A smaller proportion of the cellular uptake of unconjugated and RI-AG03-polyR-conjugated BODIPY-liposomes, but not RI-AG03-TAT-conjugated BODIPY-liposomes, involves macropinocytosis.
BODIPY Fluorescence from Unconjugated and Peptide-Conjugated BODIPY-Liposomes Localises to Macropinocytosis-Associated Cell Organelles Following Uptake
Next, we investigated how the BODIPY-labelled cholesterol in our liposomes, either unconjugated or linked to RI-AG03-polyR or RI-AG03-TAT, distributed subcellularly following cellular uptake.
Immunocytochemistry revealed that BODIPY from unconjugated BODIPY-liposomes, RI-AG03-polyR-BODIPY-liposomes and RI-AG03-TAT-BODIPY-liposomes, to some extent, partially localised to macropinosomes, as demonstrated by co-localisation with pHrodo™ Red Dextran (Figure 4A). The modest co-localisation of BODIPY fluorescence from unconjugated BODIPY-liposomes and RI-AG03-polyR-conjugated BODIPY-liposomes with macropinosomes parallels our earlier results, which showed that these BODIPY-liposomes were partially internalised by macropinocytosis (Figure 3A&B). While BODIPY from RI-AG03-TAT-conjugated BODIPY-liposomes also showed some co-localisation with pHrodo™ Red Dextran, despite largely endocytosis-independent internalisation (Figure 3C), this co-localisation appeared less frequent in comparison to that from unconjugated and RI-AG03-polyR-conjugated BODIPY-liposomes (Figure 4B). Furthermore, BODIPY from all three liposome types weakly co-localised with lysosomes, as determined by co-localisation with LysoTracker Deep Red (Figure 4B). This suggests that the liposomes and peptide-conjugated liposomes potentially bypass these degradative compartments.
Following Internalisation of RI-AG03 Peptide-Liposomes, the Intracellular Distribution of the Peptide Differs from that of the Liposome Vehicle
Thus far, we have utilised BODIPY-labelled cholesterol as a putative marker for the subcellular distribution of unconjugated and RI-AG03-conjugated liposomes. However, the cellular distribution of BODIPY-cholesterol might not reflect that of the peptides. Therefore, we synthesised RI-AG03 peptides with 6-carboxyfluorescein (6-FAM) fluorescent moieties to characterise the subcellular distribution of both free and liposome-conjugated RI-AG03.
The unconjugated 6-FAM-RI-AG03-polyR and 6-FAM-RI-AG03-TAT peptides showed poor co-localisation with the macropinosome marker pHrodo™ Red Dextran (Figure 5). However, there was no co-localisation of 6-FAM fluorescence with macropinosomes when the peptides were conjugated to liposomes (Figure 5). This contrasts with the observed co-localisation of BODIPY fluorescence to Macropinosomes, suggesting the differential trafficking of the previously conjugated peptide and the lipid liposome components (Figure 4A).
The free forms of both 6-FAM-RI-AG03-polyR and 6-FAM-RI-AG03-TAT also showed partial co-localisation with LysoTracker Deep Red in lysosomes (Figure 6). Interestingly, in contrast to BODIPY fluorescence from RI-AG03-polyR- and RI-AG03-TAT-conjugated BODIPY-liposomes (Figure 4B), there was little evidence that 6-FAM-labelled peptides conjugated to liposomes co-localised with lysosomes (Figure 6). This suggests that, when conjugated to liposomes, RI-AG03-polyR/TAT do not undergo processing in lysosomes and that there is differential subcellular trafficking of the peptide and lipid liposome components.
To exclude the possibility that liposome-conjugated 6-FAM-RI-AG03 peptides are sequestered into the plasma membrane, preventing their entry into the cell and subsequent co-localisation with intracellular organelles, we also characterised co-localisation with the plasma membrane using CellLight™ Plasma Membrane-RFP. BODIPY fluorescence from unconjugated and RI-AG03-polyR/TAT-conjugated BODIPY-liposomes showed partial co-localisation with the plasma membrane after 16 hours of incubation (Figure 7). In addition, unconjugated 6-FAM-RI-AG03-polyR or 6-FAM-RI-AG03-TAT peptides also partially co-localised with the cell membrane (Figure 7). This suggests that, at 16 hours of incubation, a proportion of the unconjugated 6-FAM-peptides and BODIPY-liposome vehicle were integrated, or in transit through, the cell membrane. However, similar to macropinosomes (Figure 5) and lysosomes (Figure 6), 6-FAM-RI-AG03-polyR and 6-FAM-RI-AG03-TAT conjugated to liposomes did not appear to co-localise with the cell membrane at this time point (Figure 7). In addition, both 6-FAM-RI-AG03-polyR and 6-FAM-RI-AG03-TAT conjugated to liposomes did not show co-localisation with fluorescent probes targeting early endosomes, the endoplasmic reticulum or the Golgi at this time point (Supplemental Material, Figures S2-S4). Collectively, these data suggest that the liposome vehicle and the initially conjugated peptides have distinct intracellular trafficking patterns during and after their internalisation by SH-SY5Y cells.
RI-AG03 Dissociates from Its Liposome Vehicle Following Cellular Membrane Fusion
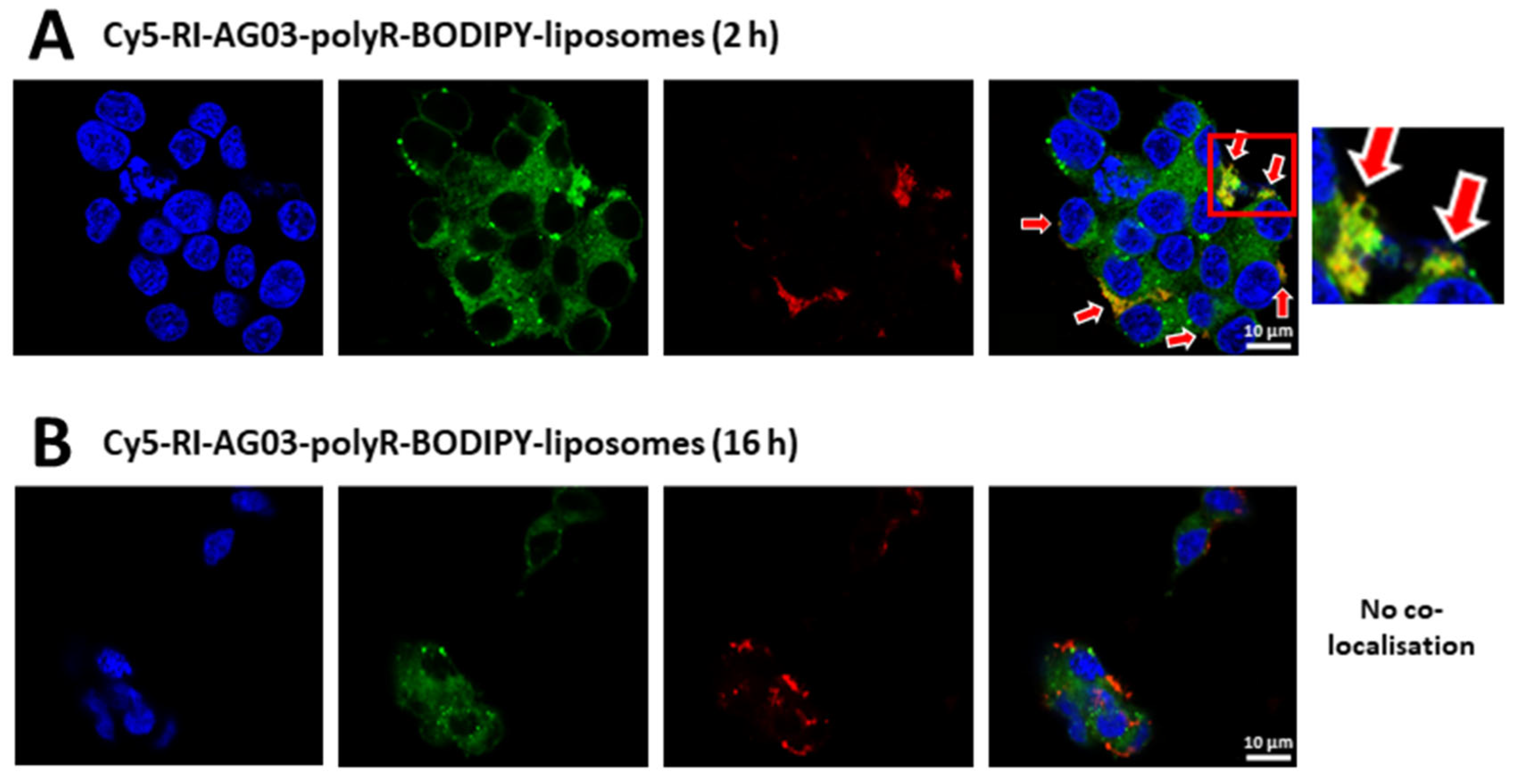
The disparate co-localisation of 6-FAM-peptide-lipsomes and peptide-BODIPY-liposomes with cell organelles led us to investigate whether the peptide detaches from its liposome vehicle before moving to an alternative cellular destination. As such, we used a novel construct that consisted of a Cy5-labelled RI-AG03-polyR peptide that was linked to BODIPY-liposomes in order to simultaneously monitor the subcellular distribution of the peptide and the liposomal carrier in the same cells following 2 and 16 h incubations.
The results show that, after a 2 h treatment with Cy5-RI-AG03-polyR-BODIPY-liposomes, co-localisation of the peptide and liposome carrier was visible (Figure 8A). However, after 16 h, no co-localisation between both was evident anymore (Figure 8B). Therefore, following the membrane fusion and cellular uptake of peptide-liposomes, the peptide appears to dissociate from the liposome carrier.
Discussion
This study demonstrates that unconjugated BODIPY-liposomes and, to a greater extend, RI-AG03-polyR- or RI-AG03-TAT-conjugated BODIPY-liposomes are readily taken up by SH-SY5Y cells (Figure 2). Because our liposomes contain a high proportion of cholesterol (47.5 %) (Figure 1B), this likely facilitated their internalisation, as incorporating cholesterol into liposomes has previously been shown to increase uptake by SH-SY5Y cells, BBB-associated brain microvascular endothelial cells and glia-like Schwann cells [43]. The same study also showed that cholesterol incorporation did not heighten liposome uptake by skeletal muscle-like NIH-3T3 fibroblasts. This suggests that the composition of our liposomes might favour BBB translocation and fusion with both Tau-containing neurons and oligodendrocytes (the CNS counterpart of peripheral Schwann cells) [44,45,46,47], with lesser uptake by skeletal muscle cells. The surface charge of the liposomes, which can be modified by the conjugation of CPPs, also impacts cellular uptake [33]. Positively charged (cationic) nanoparticles are electrostatically attracted to negatively charged (anionic) bilipid membranes [48]. Although we have not characterised the zeta potential of liposomes in this study, earlier studies and the charge characteristics and relative composition of our liposomal components (47.5% cholesterol: negative, 47.5% SM: positive and 5% DSPE-PEG(2000)-Mal: negative) suggest that unconjugated liposomes will exhibit a negative surface charge [49,50,51,52]. In contrast, polyR and TAT are positively charged CPPs (net charge +8) [53], and analysis of the amino acid compositions of RI-AG03-polyR and RI-AG03-TAT support a total positive charge of +10.9 for each peptide. It has previously been shown that anionic liposome formulations acquire positive zeta charge following polyR conjugation [54]. Thus, linkage of RI-AG03-polyR or RI-AG03-TAT to our liposomes likely imparts a positive surface charge that promotes peptide-liposome fusion with the negatively charged cellular plasma membrane [35]. This charge effect might contribute to the 3-fold enhanced uptake of RI-AG03-polyR- or RI-AG03-TAT-conjugated BODIPY-liposomes as compared to unconjugated BODIPY-liposomes (Figure 2A). In agreement with this, cationic gold nanospheres (+ 20 mV) are internalised more readily than neutral (- 4 mV) and anionic (- 10 mV) gold nanospheres [55]. Moreover, similar to our RI-AG03-polyR-liposomes, the surface conjugation of polyR to PEG(2000)-containing liposomes resulted in greater transfection of H4II-E cells, as compared to non-polyR-coated liposomes [56]. Comparable to RI-AG03-TAT-liposomes, coating polyethylenimine/PEG-liposomes with TAT peptides also increases the transfection efficiency of SH-SY5Y cells [57]. Collectively, the results of these experiments and our studies indicate that the cellular uptake of liposomes is enhanced when cationic peptides containing polyR or TAT CPPs are conjugated to the liposome surface, which may in part result from changes to the surface charge of the liposomes.
Using various endocytosis inhibitors and immunocytochemistry, we revealed that SH-SHY5Y cells partially internalised unconjugated liposomes and RI-AG03-polyR-conjugated liposomes through macropinocytosis (Figure 3A-C). Macropinocytosis involves the formation of cell membrane protrusions (lamellipodia) that engulf large volumes of extracellular fluid. This nonselective mechanism enables cellular uptake of molecules that are too large for other endocytosis pathways. The internalised macropinosomes range from 0.5 - 10 μm in diameter and are, in part, delivered to lysosomes [33,58]. The co-localisation of BODIPY fluorescence from unconjugated and RI-AG03-polyR-conjugated liposomes with macropinosomes and lysosomes supports a key role for this cellular uptake and processing mechanism in SH-SY5Y cells (Figure 4). Although the internalisation of RI-AG03-TAT-liposomes appeared to be mostly independent of macropinocytosis in SH-SY5Y cells (Figure 3C), they were also found to weakly co-localised with macropinosome and lysosome markers (Figure 4A,B). It is likely that random membrane fusion events, as typical for liposomes [33,42], contributes in part to liposome internalisation by macropinocytosis and trafficking into macropinocytosis-associated cell organelles (macropinosomes and lysosomes). In support of this suggestion, unconjugated and RI-AG03-polyR-conjugated BODIPY-liposomes, whose uptake was partially dependent on macropinocytosis (Figure 3A,B), showed greater co-localisation with macropinosomes than RI-AG03-TAT-BODIPY-conjugated liposomes (Figure 4A,B). Given that neurons are likely to be bioenergetically impaired in AD, as indicated by impaired glucose metabolism and insulin resistance [59,60], the cellular uptake of tau targeting peptides, such as RI-AG03-TAT, by ATP-independent mechanisms may be therapeutically advantageous.
The size of liposomes affects their endocytic uptake in cells [33]. Nanoparticles ranging from a few to several hundred nanometers in size and those having a positive surface charge are commonly internalised by macropinocytosis [33]. By contrast, negatively charged particles ranging from 120 - 150 nm, with a maximum diameter of 200 nm, are preferentially taken up via CME or CavME. While our liposomes are extruded with 100 nm pores, the actual size may be larger due to the incorporated PEG groups and peptide-conjugation. For example, we showed that incorporating different ratios of a PEG-linked curcumin derivate (Y) into control liposomes, originally sized ~53 nm, increased the liposome diameter approximately three-fold (> 180 nm) [52]. Similarly, our unpublished in-house measurements indicate that the RI-AG03-conjugated liposomes used in this study are likely to be around ~280 nm. Thus, the size of unconjugated and peptide-conjugated liposomes is likely too large for CME- and CavME-mediated uptake, that is selective for cargo sizes between 120 - 150 nm, with an upper limit of ~200 nm [33]. This is in agreement with the fact that we found no evidence for CME- and CavME-mediated liposome uptake and that macropinocytosis partially mediated the internalisation of both unconjugated and RI-AG03-polyR-conjugated BODIPY-liposomes (Figure 3A,B).
Liposome composition also plays a key role in influencing cellular uptake. Extraction of cholesterol from the plasma membrane prevents the recruitment of RAS-related C3 botulinum toxin substrate 1 (Rac1) to the plasma membrane and inhibits macropinocytosis [61]. Given the high cholesterol content (47.5%) of the liposomes used in this study, they could act as a micropinocytosis initiating signal through Rac1 recruitment, promoting liposome uptake by this mechanism. Further studies are needed to confirm this suggestion.
The surface charge of liposomes also affects endocytosis, with the presumable positive surface charge on peptide-conjugated liposomes likely to promote macropinocytosis [33,35]. This is consistent with the observation that the macropinocytosis inhibitor EIPA reduced the cellular internalisation of RI-AG03-polyR-conjugated liposomes to a greater extent that unconjugated BODIPY-liposomes (Figure 3A,B). This indicates that the conjugation of RI-AG03-polyR to liposomes increased the relative proportion of macropinocytosis-mediated liposome uptake in SH-SY5Y cells.
The CPP present also affects liposomal cellular uptake mechanisms, and we found evidence for this with both the polyR and TAT CPPs. Nona-arginine peptides induce cell membrane multi-lamellarity, increasing energy-independent uptake [62]. The surface conjugation of cationic polyR to liposomes also enhances the apposition and fusion of liposomes with lipid bilayers, promoting uptake [35,36]. In addition, high concentrations (40µM) of nona-arganine and TAT promote uptake through direct membrane penetration, CavME and micropinocytosis rather than through CME [63]. This suggests that coating liposomes with polyR or TAT may enhance liposomal cell membrane translocation. Such an increase in direct membrane translocation was observed for RI-AG03-TAT-conjugated liposomes, whose internalisation was increased 3-fold when compared to unconjugated liposomes (Figure 2A). This occurred in an endocytosis-independent manner (Figure 3C). RI-AG03-polyR-conjugated liposomes also showed greater uptake, being 3-fold greater than unconjugated liposomes (Figure 2A). However, in this case increased uptake was partially mediated by increased macropinocytosis (Figure 3A, B). Thus, the presence of CPP-containing peptides can enhance cellular liposome uptake by both energy-independent and endocytosis-mediated uptake, dependent upon the CPP present.
One important limitation to our studies is that we are unable to investigate several less well characterised endocytosis mechanisms, such as CLIC/GEEC-driven endocytosis, flotillin-mediated endocytosis and circular dorsal ruffles [32], as specific inhibitors for these endocytic pathways are currently lacking [40,41]. Thus, we cannot ruled out liposome uptake by these alternative endocytosis mechanisms in SH-SY5Y cells.
In addition, treatment of SH-SY5Y cells with chlorpromazine concentrations that completely inhibit CME was not possible. Typically, chlorpromazine is used at 50 - 100 µM to inhibit CME in cells [41], with an IC50 of 17.4 µM reported in U2OS cells [64]. We were only able to employ 10 µM chlorpromazine, as 12.5 µM caused SH-SY5Y cell detachment and concentrations ≥ 15 µM were highly toxic (Supplementary material, Figure S1). This is consistent with published data showing chlorpromazine toxicity in undifferentiated SH-SY5Y cells (LC50 = 5 µM) [65]. Therefore, CME was only partially blocked in our experiments, with complete CME inhibition likely being toxic to SH-SY5Y cells. Since BODIPY fluorescence from unconjugated, RI-AG03-polyR- and RI-AG03-TAT-conjugated liposomes partially co-localised with early endosomes (Supplementary material, Figure S2), which are CME-associated cell organelles [66], it is likely that CME partially contributes to liposome cellular uptake.
A major challenge for drug-conjugated nanocarriers is to avoid entrapment in cell degradative compartments, such as endosomes and lysosomes [36]. It has been proposed that the linkage of polyR, TAT and other CPPs to liposomes facilitates endolysosome membrane fusion, leading to the ejection of liposome-encapsulated cargo into the cytoplasm [35]. For example, Ruan et al., (2007) demonstrated that TAT-conjugated quantum dots were internalised by macropinocytosis, becoming trapped in the inner macropinosome membrane [67]. Thus, linking a CPP to a nanocarrier does not necessarily improve cell organelle escape, and the utility of additional endosomal escape strategies might be necessary [35]. In the current study we found that unconjugated, RI-AG03-polyR-conjugated and RI-AG03-TAT-conjugated BODIPY-liposomes co-localised with macropinosomes (Figure 4A), lysosomes (Figure 4B) and the cell membrane (Figure 7) in SH-SY5Y cells. However, when 6-FAM-labelled RI-AG03-polyR and RI-AG03-TAT were conjugated to non-fluorescent liposomes, there was no co-localisation of the fluorescent peptide with these organelles (Figure 5, 6 and 7), early endosomes, the ER or Golgi (Supplementary material, Figure S2-4). Moreover, we also found that liposome conjugated 6-FAM-RI-AG03-polyR detached from BODIPIY-labelled liposomes after fusing with the SH-SY5Y cell membrane (Figure 8). Therefore, this shows that following cellular uptake, the conjugated peptide dissociates from the liposome vehicle and escapes entrapment in degrative cell organelles, possibly trafficking into the cytoplasm. Given that unconjugated 6-FAM-RI-AG03-polyR/TAT peptides partially co-localised with these degradative cell organelles, but 6-FAM-RI-AG03-polyR/TAT-conjugated liposomes did not (Figure 5, 6, 7 and S2), peptide-conjugation to liposomes might alter peptide trafficking in favour of enhanced cytoplasmic delivery. Moreover, the lack of 6-FAM-RI-AG03-polyR/TAT peptide co-localisation with the plasma membrane when conjugated to liposomes (Figure 7) suggests that the peptide does not remain stuck in the cellular membrane after dissociation from its liposome vehicle. However, further studies are necessary to confirm cytoplasmic delivery of the peptide, and its interaction with Tau in the cell environment.
Material & Methods
Peptide Synthesis
RI-AG03-polyR (NH2-r-G-v-q-i-k(Ac)-y-k-p-G-r-r-r-r-r-r-r-r-c), RI-AG03-TAT (NH2-r-G-v-q-i-k(Ac)-y-k-p-G-y-G-r-k-k-r-r-q-r-r-r-c), 6-carboxyfluorescein (6-FAM)-RI-AG03-polyR and 6-FAM-RI-AG03-TAT were synthesised by Severn Biotech Ltd (Kidderminster, UK). Cyanine-5 (Cy5)-RI-AG03-polyR was synthesised by Cambridge Peptides Ltd (Cambridge, UK). To allow liposome linkage, all peptides contained an additional cysteine [39]. The peptides contained D-amino acids (denoted by lower cases) to prevent proteolytic cleavage [18,25], except for glycine, because this amino acid does not possess a D-enantiomer.
Preparation of Liposomes and Click Chemistry Attachment of RI-AGO3
Liposomes were made by dissolving relative molar proportions of the following (all Avanti Polar Lipids Inc., Alabaster, US) in chloroform: 47.5 % sphingomyelin (SM; egg-derived), 47.5 % cholesterol (plant-derived) and 5 % maleimide, 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[maleimide(polyethylene glycol)-2000] (DSPE-PEG(2000)-Mal). For BODIPY-liposome uptake and localisation fluorescence studies, 2 % of the cholesterol was replaced with TopFluor® (BODIPY) cholesterol (Avanti Polar Lipids Inc., Alabaster, US). The lipid mixture was dried under liquid nitrogen and the film resuspended in 1x phosphate-buffered saline (1x PBS; 137 mmol/L NaCl, 2.7 mmol/L KCl, 20 mmol/L Na2HPO4 and 1.8 mmol/L KH2PO4) in a water bath sonicator at 37 °C for 15 min, until fully dissolved. The mixture was then subjected to five freeze-thaw cycles in liquid nitrogen and extruded eleven times using a Mini-Extruder (Avanti Polar Lipids Inc., Alabaster, US) and Hamilton 1000μl Syringes (Avanti Polar Lipids Inc., Alabaster, US), through a 0.1 μm Nuclepore™ Polycarbonate Track-Etched Membrane (Whatman, Maidstone, England) in accordance with the manufacturer’s instructions.
Because DSPE-PEG(2000)-Mal is randomly incorporated into liposomes, with the maleimide group facing either inwards or outwards, only half of the DSPE-PEG(2000)-Mal present (2.5% liposome lipid content) is available for peptide conjugation. To attach the cysteine residue of RI-AG03 to the available DSPE-PEG(2000)-Mal chains via click chemistry, extruded liposomes were incubated with an excess molar proportion of the peptide (= molar concentration of DSPE-PEG(2000)-Mal (2.5 %) x 1.2) for 2 h at 37 °C. The mixture was vortexed once after 1 h and rocked on a plate shaker at room temperature overnight. Unbound peptide was removed by ultracentrifugation for 1 h at 172,000 x g (4 °C) and the liposome pellet was resuspended in PBS in a water bath sonicator at 37 °C, in three 15 min sonication cycles with vortexing between cycles. To remove any liposome clumps following resuspension, the liposomes were centrifuged at max speed (17 x g) in a benchtop centrifuge for 4 min, through 0.22 μm Corning® Costar® Spin-X® centrifuge tube cellulose acetate filters (Corning Inc., Corning, US). Liposome concentrations were quantified using the LabAssay™ Phospholipid kit (FUJIFILM Wako Shibayagi Corporation, Osaka, Japan).
Cell Culture
Human neuroblastoma, SH-SY5Y, cells (ATCC®; CRL-2266; Manassas, USA)) were maintained in Dulbecco's Modified Eagle Medium (DMEM)-F12 (Gibco, Brigg, UK) containing 10 % (v/v) heat-inactivated fetal bovine serum (FBS; Sigma-Aldrich/Merck Life Science UK Limited, Dorset, UK) and 1 % (v/v) antibiotic-antimycotic solution (Sigma-Aldrich/Merck Life Science UK Limited, Dorset, UK) at 37 °C and 5 % CO2.
Flow Cytometry
SH-SY5Y cells were seeded at a density of 350,000 cells in 12 well plates and allowed to settle overnight. Cells were then treated with a final concentration of 75 μM unconjugated, RI-AG03-polyR-conjugated or RI-AG03-TAT-conjugated BODIPY-liposomes for 4 h. To investigate endocytosis pathways, cells were treated with vehicle solution (dimethyl sulfoxide; DMSO) or a final concentration of 10 µM chlorpromazine hydrochloride, 7.5 µM cytochalasin D, 5 µg/mL filipin III from streptomyces filipinensis or 50 µM 5-(N-ethyl-N-isopropyl)amiloride (EIPA, all from Merck Life Science UK Limited, Dorset, UK) dissolved in DMSO for 30 min prior to further co-incubated with the liposomes for 4 h at 37 °C. Cells were then trypsinised, pelleted and resuspended in 1 mL 1x PBS. An equal volume of 4 % (w/v) paraformaldehyde (PFA) in 1x PBS was added, for a final concentration of 2 % PFA, and the cells were fixed for 30 min at 4 °C in the dark. Median cellular fluorescence was quantified by flow cytometry (CytoFLEX; Beckman Coulter, High Wycombe, UK) with a minimum cell count of 10,000 cells per sample. The experiments were performed in triplicates (three wells per group, treated with the same liposome stock) and repeated over three independent experiments with fresh liposome stocks (total samples per group: n = 9).
Immunocytochemistry
SH-SY5Y cells were seeded onto poly-L-lysine-coated coverslips at a density of 150,000 cells per well in 24-well plates and allowed to adhere overnight. Cells were then treated with either 6-FAM-RI-AG03-polyR/TAT peptide, unconjugated BODIPY-liposomes, RI-AG03-polyR/TAT-conjugated to BODIPY-liposomes, 6-FAM-RI-AG03-polyR/TAT-conjugated to BODIPY-liposomes or Cy5-RI-AG03-polyR-liposomes, and incubated for 2 h or 16 h to investigate cell organelle trafficking. To co-detect cell organelles, the following live cells stains (all Invitrogen, Massachusetts, US) were used according to the manufacturer’s instructions: LysoTracker Deep Red (for lysosomes), pHrodo™ Red Dextran, 10,000 MW ( for macropinosomes), CellLight™ Plasma Membrane-RFP (for cell membrane), CellLight™ Early Endosomes-RFP (for early endosomes), CellLight™ ER-RFP (for endoplasmic reticulum) and CellLight™ Golgi-RFP (for Golgi). The cells were flushed in 1x PBS, fixed in 4 % (w/v) PFA in 1x PBS (4oC for 30 min). The coverslips were then mounted with ProLong™ Diamond Antifade Mountant containing DAPI (Invitrogen, Massachusetts, US), sealed and stored at 4 °C in the dark. At least 3 images per treatment group from duplicate coverslips were taken at 63x magnification using a ZEISS LSM880 confocal microscope.
Viability Assays
SH-SY5Y cells were seeded in 96 well plates, allowed to settle overnight and, at a density of ~60 - 80 % confluency, treated with a final concentration of 75 µM unconjugated liposomes or peptide-liposomes or endocytosis inhibitors, including chlorpromazine, filipin, cytochalasin D and EIPA, for 4.5 h (n = 6; Supplementary material, Figure S1). Cell viability was assessed using Cell Counting Kit - 8 (Sigma-Aldrich/Merck Life Science UK Limited, Dorset, UK) according to the manufacturer’s instructions.
Statistical Analysis
Statistical analysis was performed with JASP (University of Amsterdam, Amsterdam, The Netherlands). To compare the cellular uptake of unconjugated BODIPY-liposomes or peptide-BODIPY-liposomes in the absence or presence of endocytosis inhibitors, one-way and two-way ANOVA followed by Tukey’s post-hoc test were applied. One-way ANOVA and Tukey’s post-hoc correction were used for the viability assays. Significance was set at p < 0.05.
Conclusions
Conjugating RI-AG03 to liposomes, independent of the CPP sequence (TAT or polyR) included in the peptide, enhances cellular liposome uptake by SH-SY5Y cells. Whilst RI-AG03-polyR-conjugated liposomes are moderately internalised by macropinocytosis, cellular uptake of RI-AG03-TAT-conjugated liposomes is independent of ATP-dependent endocytosis mechanisms. This potentially makes RI-AG03-TAT peptide the preferential choice for RI-AG03 delivery, given the energy-impairment of neurons in AD and Tauopathies. Finally, we provide evidence that RI-AG03 detaches from its liposome vehicle following cellular uptake, and that it avoids peptide entrapment in degrading cell organelles. This suggests that liposome conjugation may enhance the cellular availability of RI-AG03, which may increase its utility in preventing cellular tau aggregation. Thus, RI-AG03 conjugation to liposomes (PINPs) may be useful for the treatment of Tau pathology in neurodegenerative diseases.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
Conceptualization, David Allsop; Data curation, Niklas Reich; Investigation, Niklas Reich; Methodology, Niklas Reich; Supervision, David Allsop, Edward Parkin and Neil Dawson; Visualization, David Allsop; Writing – original draft, Niklas Reich and Neil Dawson; Writing – review & editing, Niklas Reich, Edward Parkin and Neil Dawson.
Funding
The research was supported by Lancaster University’s Defying Dementia charity (https://www.lancaster.ac.uk/giving/defying-dementia/).
Acknowledgments
BioRender.com was used to create the graphical abstract.
Conflicts of interest
Niklas Reich, Edward Parkin and Neil Dawson have no conflict of interest. David Allsop has a patent for the Tau aggregation inhibitor peptide RI-AG03
References
- Breijyeh, Z., and R. Karaman. "Comprehensive Review on Alzheimer's Disease: Causes and Treatment." Molecules 25, no. 24 (2020). [CrossRef]
- Pinheiro, L., and C. Faustino. "Therapeutic Strategies Targeting Amyloid-Beta in Alzheimer's Disease." Current Alzheimer Research 16, no. 5 (2019): 418-52.
- Panza, F., M. Lozupone, G. Logroscino, and B. P. Imbimbo. "A Critical Appraisal of Amyloid-Beta-Targeting Therapies for Alzheimer Disease." Nat Rev Neurol 15, no. 2 (2019): 73-88.
- Tampi, R. R., B. P. Forester, and M. Agronin. "Aducanumab: Evidence from Clinical Trial Data and Controversies." Drugs Context 10 (2021). [CrossRef]
- Vaz, M., V. Silva, C. Monteiro, and S. Silvestre. "Role of Aducanumab in the Treatment of Alzheimer's Disease: Challenges and Opportunities." Clinical Interventions in Aging 17 (2022): 797-810. [CrossRef]
- Congdon, E. E., and E. M. Sigurdsson. "Tau-Targeting Therapies for Alzheimer Disease." Nat Rev Neurol 14, no. 7 (2018): 399-415. [CrossRef]
- Sandusky-Beltran, L. A., and E. M. Sigurdsson. "Tau Immunotherapies: Lessons Learned, Current Status and Future Considerations." Neuropharmacology 175 (2020): 108104. [CrossRef]
- Guo, T., W. Noble, and D. P. Hanger. "Roles of Tau Protein in Health and Disease." Acta Neuropathologica 133, no. 5 (2017): 665-704. [CrossRef]
- Kametani, F., and M. Hasegawa. "Reconsideration of Amyloid Hypothesis and Tau Hypothesis in Alzheimer's Disease." Front Neurosci 12 (2018): 25.
- Gulisano, W., D. Maugeri, M. A. Baltrons, M. Fa, A. Amato, A. Palmeri, L. D'Adamio, C. Grassi, D. P. Devanand, L. S. Honig, D. Puzzo, and O. Arancio. "Role of Amyloid-Beta and Tau Proteins in Alzheimer's Disease: Confuting the Amyloid Cascade." Journal of Alzheimers Disease 64, no. s1 (2018): S611-S31.
- Simic, G., M. Babic Leko, S. Wray, C. Harrington, I. Delalle, N. Jovanov-Milosevic, D. Bazadona, L. Buee, R. de Silva, G. Di Giovanni, C. Wischik, and P. R. Hof. "Tau Protein Hyperphosphorylation and Aggregation in Alzheimer's Disease and Other Tauopathies, and Possible Neuroprotective Strategies." Biomolecules 6, no. 1 (2016): 6.
- Wang, Y., V. Balaji, S. Kaniyappan, L. Kruger, S. Irsen, K. Tepper, R. Chandupatla, W. Maetzler, A. Schneider, E. Mandelkow, and E. M. Mandelkow. "The Release and Trans-Synaptic Transmission of Tau Via Exosomes." Molecular Neurodegeneration 12, no. 1 (2017): 5. [CrossRef]
- Saman, S., W. Kim, M. Raya, Y. Visnick, S. Miro, S. Saman, B. Jackson, A. C. McKee, V. E. Alvarez, N. C. Lee, and G. F. Hall. "Exosome-Associated Tau Is Secreted in Tauopathy Models and Is Selectively Phosphorylated in Cerebrospinal Fluid in Early Alzheimer Disease." Journal of Biological Chemistry 287, no. 6 (2012): 3842-9. [CrossRef]
- Mirbaha, H., B. B. Holmes, D. W. Sanders, J. Bieschke, and M. I. Diamond. "Tau Trimers Are the Minimal Propagation Unit Spontaneously Internalized to Seed Intracellular Aggregation." Journal of Biological Chemistry 290, no. 24 (2015): 14893-903. [CrossRef]
- Braak, H., and E. Braak. "Neuropathological Stageing of Alzheimer-Related Changes." Acta Neuropathologica 82, no. 4 (1991): 239-59. [CrossRef]
- Wischik, C. M., C. R. Harrington, and J. M. Storey. "Tau-Aggregation Inhibitor Therapy for Alzheimer's Disease." Biochemical Pharmacology 88, no. 4 (2014): 529-39.
- Austen, B. M., K. E. Paleologou, S. A. Ali, M. M. Qureshi, D. Allsop, and O. M. El-Agnaf. "Designing Peptide Inhibitors for Oligomerization and Toxicity of Alzheimer's Beta-Amyloid Peptide." Biochemistry 47, no. 7 (2008): 1984-92.
- Taylor, M., S. Moore, J. Mayes, E. Parkin, M. Beeg, M. Canovi, M. Gobbi, D. M. Mann, and D. Allsop. "Development of a Proteolytically Stable Retro-Inverso Peptide Inhibitor of Beta-Amyloid Oligomerization as a Potential Novel Treatment for Alzheimer's Disease." Biochemistry 49, no. 15 (2010): 3261-72.
- Tian, X. H., F. Wei, T. X. Wang, D. Wang, J. Wang, X. N. Lin, P. Wang, and L. Ren. "Blood-Brain Barrier Transport of Tat Peptide and Polyethylene Glycol Decorated Gelatin-Siloxane Nanoparticle." Materials Letters 68 (2012): 94-96. [CrossRef]
- Al Humaidan, E.L., S.L. Pedersen, A. Burkhart, C.L.M. Rasmussen, T. Moos, P. Fuchs, E.F.A. Fernandes, B. Ozgür, K. Strømgaard, A. Bach, B. Brodin, and M. Kristensen. "The Cell-Penetrating Peptide Tat Facilitates Effective Internalization of Psd-95 Inhibitors into Blood–Brain Barrier Endothelial Cells but Less Efficient Permeation across the Blood–Brain Barrier in Vitro and in Vivo." Front. Drug Deliv. 2, no. 854703 (2022). [CrossRef]
- Parthsarathy, V., P. L. McClean, C. Holscher, M. Taylor, C. Tinker, G. Jones, O. Kolosov, E. Salvati, M. Gregori, M. Masserini, and D. Allsop. "A Novel Retro-Inverso Peptide Inhibitor Reduces Amyloid Deposition, Oxidation and Inflammation and Stimulates Neurogenesis in the Appswe/Ps1deltae9 Mouse Model of Alzheimer's Disease." Plos One 8, no. 1 (2013): e54769.
- Gregori, M., M. Taylor, E. Salvati, F. Re, S. Mancini, C. Balducci, G. Forloni, V. Zambelli, S. Sesana, M. Michael, C. Michail, C. Tinker-Mill, O. Kolosov, M. Sherer, S. Harris, N. J. Fullwood, M. Masserini, and D. Allsop. "Retro-Inverso Peptide Inhibitor Nanoparticles as Potent Inhibitors of Aggregation of the Alzheimer's Abeta Peptide." Nanomedicine 13, no. 2 (2017): 723-32.
- von Bergen, M., S. Barghorn, L. Li, A. Marx, J. Biernat, E. M. Mandelkow, and E. Mandelkow. "Mutations of Tau Protein in Frontotemporal Dementia Promote Aggregation of Paired Helical Filaments by Enhancing Local Beta-Structure." Journal of Biological Chemistry 276, no. 51 (2001): 48165-74.
- von Bergen, M., P. Friedhoff, J. Biernat, J. Heberle, E. M. Mandelkow, and E. Mandelkow. "Assembly of Tau Protein into Alzheimer Paired Helical Filaments Depends on a Local Sequence Motif ((306)Vqivyk(311)) Forming Beta Structure." Proceedings of the National Academy of Sciences of the United States of America 97, no. 10 (2000): 5129-34.
- Aggidis A., Chatterjee S., Townsend D., Fullwood N.J., Ortega E. R., Tarutani A., Hasegawa M., Lucas H., Mudher A., and Allsop D. "Peptide-Based Inhibitors of Tau Aggregation as a Potential Therapeutic for Alzheimer’s Disease and Other Tauopathies." bioRxiv, (2021). [CrossRef]
- Chandrasekaran, S., M. J. McGuire, and M. R. King. "Sweeping Lymph Node Micrometastases Off Their Feet: An Engineered Model to Evaluate Natural Killer Cell Mediated Therapeutic Intervention of Circulating Tumor Cells That Disseminate to the Lymph Nodes." Lab Chip 14, no. 1 (2014): 118-27. [CrossRef]
- Futaki, S., W. Ohashi, T. Suzuki, M. Niwa, S. Tanaka, K. Ueda, H. Harashima, and Y. Sugiura. "Stearylated Arginine-Rich Peptides: A New Class of Transfection Systems." Bioconjug Chem 12, no. 6 (2001): 1005-11. [CrossRef]
- Mitchell, D. J., D. T. Kim, L. Steinman, C. G. Fathman, and J. B. Rothbard. "Polyarginine Enters Cells More Efficiently Than Other Polycationic Homopolymers." J Pept Res 56, no. 5 (2000): 318-25. [CrossRef]
- Gotanda, Y., F. Y. Wei, H. Harada, K. Ohta, K. I. Nakamura, K. Tomizawa, and K. Ushijima. "Efficient Transduction of 11 Poly-Arginine Peptide in an Ischemic Lesion of Mouse Brain." J Stroke Cerebrovasc Dis 23, no. 8 (2014): 2023-30. [CrossRef]
- Pham, W., B. Q. Zhao, E. H. Lo, Z. Medarova, B. Rosen, and A. Moore. "Crossing the Blood-Brain Barrier: A Potential Application of Myristoylated Polyarginine for in Vivo Neuroimaging." Neuroimage 28, no. 1 (2005): 287-92. [CrossRef]
- Mamsa, S. S. A., and B. P. Meloni. "Arginine and Arginine-Rich Peptides as Modulators of Protein Aggregation and Cytotoxicity Associated with Alzheimer's Disease." Frontiers in Molecular Neuroscience 14 (2021): 759729. [CrossRef]
- Doherty, G. J., and H. T. McMahon. "Mechanisms of Endocytosis." Annu Rev Biochem 78 (2009): 857-902.
- Foroozandeh, P., and A. A. Aziz. "Insight into Cellular Uptake and Intracellular Trafficking of Nanoparticles." Nanoscale Res Lett 13, no. 1 (2018): 339. [CrossRef]
- Vercauteren, D., R. E. Vandenbroucke, A. T. Jones, J. Rejman, J. Demeester, S. C. De Smedt, N. N. Sanders, and K. Braeckmans. "The Use of Inhibitors to Study Endocytic Pathways of Gene Carriers: Optimization and Pitfalls." Molecular Therapy 18, no. 3 (2010): 561-9. [CrossRef]
- El-Sayed, A., S. Futaki, and H. Harashima. "Delivery of Macromolecules Using Arginine-Rich Cell-Penetrating Peptides: Ways to Overcome Endosomal Entrapment." AAPS J 11, no. 1 (2009): 13-22. [CrossRef]
- Pei, D., and M. Buyanova. "Overcoming Endosomal Entrapment in Drug Delivery." Bioconjug Chem 30, no. 2 (2019): 273-83. [CrossRef]
- Ross, C., M. Taylor, N. Fullwood, and D. Allsop. "Liposome Delivery Systems for the Treatment of Alzheimer's Disease." Int J Nanomedicine 13 (2018): 8507-22. [CrossRef]
- Shi, N. Q., X. R. Qi, B. Xiang, and Y. Zhang. "A Survey on "Trojan Horse" Peptides: Opportunities, Issues and Controlled Entry to "Troy"." J Control Release 194 (2014): 53-70.
- Mason, A. F., and P. Thordarson. "Synthesis of Protein Bioconjugates Via Cysteine-Maleimide Chemistry." J Vis Exp, no. 113 (2016).
- Dutta, D., and J. G. Donaldson. "Search for Inhibitors of Endocytosis: Intended Specificity and Unintended Consequences." Cell Logist 2, no. 4 (2012): 203-08.
- Ivanov, A. I. "Pharmacological Inhibition of Endocytic Pathways: Is It Specific Enough to Be Useful?" Methods Mol Biol 440 (2008): 15-33.
- Curtis, E. M., A. H. Bahrami, T. R. Weikl, and C. K. Hall. "Modeling Nanoparticle Wrapping or Translocation in Bilayer Membranes." Nanoscale 7, no. 34 (2015): 14505-14. [CrossRef]
- Lee, S., A. T. Ashizawa, K. S. Kim, D. J. Falk, and L. Notterpek. "Liposomes to Target Peripheral Neurons and Schwann Cells." Plos One 8, no. 11 (2013): e78724. [CrossRef]
- Vogel, J. W., Y. Iturria-Medina, O. T. Strandberg, R. Smith, E. Levitis, A. C. Evans, O. Hansson, Initiative Alzheimer's Disease Neuroimaging, and Study Swedish BioFinder. "Author Correction: Spread of Pathological Tau Proteins through Communicating Neurons in Human Alzheimer's Disease." Nature Communications 12, no. 1 (2021): 4862. [CrossRef]
- Narasimhan, S., L. Changolkar, D. M. Riddle, A. Kats, A. Stieber, S. A. Weitzman, B. Zhang, Z. Li, E. D. Roberson, J. Q. Trojanowski, and V. M. Y. Lee. "Human Tau Pathology Transmits Glial Tau Aggregates in the Absence of Neuronal Tau." J Exp Med 217, no. 2 (2020). [CrossRef]
- Zareba-Paslawska, J., K. Patra, L. Kluzer, T. Revesz, and P. Svenningsson. "Tau Isoform-Driven Cbd Pathology Transmission in Oligodendrocytes in Humanized Tau Mice." Frontiers in Neurology 11 (2020): 589471. [CrossRef]
- Higuchi, M., B. Zhang, M. S. Forman, Y. Yoshiyama, J. Q. Trojanowski, and V. M. Lee. "Axonal Degeneration Induced by Targeted Expression of Mutant Human Tau in Oligodendrocytes of Transgenic Mice That Model Glial Tauopathies." Journal of Neuroscience 25, no. 41 (2005): 9434-43. [CrossRef]
- Li, S., and N. Malmstadt. "Deformation and Poration of Lipid Bilayer Membranes by Cationic Nanoparticles." Soft Matter 9, no. 20 (2013): 4969-76. [CrossRef]
- Magarkar, A., V. Dhawan, P. Kallinteri, T. Viitala, M. Elmowafy, T. Rog, and A. Bunker. "Cholesterol Level Affects Surface Charge of Lipid Membranes in Saline Solution." Sci Rep 4 (2014): 5005. [CrossRef]
- Rye, K. A., N. J. Hime, and P. J. Barter. "The Influence of Sphingomyelin on the Structure and Function of Reconstituted High Density Lipoproteins." Journal of Biological Chemistry 271, no. 8 (1996): 4243-50. [CrossRef]
- Oswald, M., S. Geissler, and A. Goepferich. "Determination of the Activity of Maleimide-Functionalized Phospholipids During Preparation of Liposomes." Int J Pharm 514, no. 1 (2016): 93-102. [CrossRef]
- Taylor, M., S. Moore, S. Mourtas, A. Niarakis, F. Re, C. Zona, B. La Ferla, F. Nicotra, M. Masserini, S. G. Antimisiaris, M. Gregori, and D. Allsop. "Effect of Curcumin-Associated and Lipid Ligand-Functionalized Nanoliposomes on Aggregation of the Alzheimer's Abeta Peptide." Nanomedicine 7, no. 5 (2011): 541-50.
- Juliano, R., M. R. Alam, V. Dixit, and H. Kang. "Mechanisms and Strategies for Effective Delivery of Antisense and Sirna Oligonucleotides." Nucleic Acids Res 36, no. 12 (2008): 4158-71. [CrossRef]
- Opanasopit, P., J. Tragulpakseerojn, A. Apirakaramwong, T. Ngawhirunpat, T. Rojanarata, and U. Ruktanonchai. "The Development of Poly-L-Arginine-Coated Liposomes for Gene Delivery." Int J Nanomedicine 6 (2011): 2245-52. [CrossRef]
- Cho, E. C., J. Xie, P. A. Wurm, and Y. Xia. "Understanding the Role of Surface Charges in Cellular Adsorption Versus Internalization by Selectively Removing Gold Nanoparticles on the Cell Surface with a I2/Ki Etchant." Nano Lett 9, no. 3 (2009): 1080-4.
- Kim, H. K., E. Davaa, C. S. Myung, and J. S. Park. "Enhanced Sirna Delivery Using Cationic Liposomes with New Polyarginine-Conjugated Peg-Lipid." Int J Pharm 392, no. 1-2 (2010): 141-7. [CrossRef]
- Suk, J. S., J. Suh, K. Choy, S. K. Lai, J. Fu, and J. Hanes. "Gene Delivery to Differentiated Neurotypic Cells with Rgd and Hiv Tat Peptide Functionalized Polymeric Nanoparticles." Biomaterials 27, no. 29 (2006): 5143-50. doi:10.1016/j.biomaterials.2006.05.013.
- Lim, J. P., and P. A. Gleeson. "Macropinocytosis: An Endocytic Pathway for Internalising Large Gulps." Immunol Cell Biol 89, no. 8 (2011): 836-43. [CrossRef]
- Mosconi, L., A. Pupi, and M. J. De Leon. "Brain Glucose Hypometabolism and Oxidative Stress in Preclinical Alzheimer's Disease." Ann N Y Acad Sci 1147 (2008): 180-95.
- Neth, B. J., and S. Craft. "Insulin Resistance and Alzheimer's Disease: Bioenergetic Linkages." Frontiers in Aging Neuroscience 9 (2017): 345. doi:10.3389/fnagi.2017.00345.
- Grimmer, S., B. van Deurs, and K. Sandvig. "Membrane Ruffling and Macropinocytosis in A431 Cells Require Cholesterol." Journal of Cell Science 115, no. Pt 14 (2002): 2953-62. [CrossRef]
- Allolio, C., A. Magarkar, P. Jurkiewicz, K. Baxova, M. Javanainen, P. E. Mason, R. Sachl, M. Cebecauer, M. Hof, D. Horinek, V. Heinz, R. Rachel, C. M. Ziegler, A. Schrofel, and P. Jungwirth. "Arginine-Rich Cell-Penetrating Peptides Induce Membrane Multilamellarity and Subsequently Enter Via Formation of a Fusion Pore." Proc Natl Acad Sci U S A 115, no. 47 (2018): 11923-28. [CrossRef]
- Duchardt, F., M. Fotin-Mleczek, H. Schwarz, R. Fischer, and R. Brock. "A Comprehensive Model for the Cellular Uptake of Cationic Cell-Penetrating Peptides." Traffic 8, no. 7 (2007): 848-66. [CrossRef]
- Daniel, J. A., N. Chau, M. K. Abdel-Hamid, L. Hu, L. von Kleist, A. Whiting, S. Krishnan, P. Maamary, S. R. Joseph, F. Simpson, V. Haucke, A. McCluskey, and P. J. Robinson. "Phenothiazine-Derived Antipsychotic Drugs Inhibit Dynamin and Clathrin-Mediated Endocytosis." Traffic 16, no. 6 (2015): 635-54. [CrossRef]
- Hakeem, I.J., N.J. Hodges, and F. Michelangeli. "Cytotoxicity of Sh-Sy5y Neuroblastoma Cells to the Antipsychotic Drugs, Chlorpromazine and Trifluoperazine, Is Via a Ca2+-Mediated Apoptosis Process and Differentiation of These Cells with Retinoic Acid Makes Them More Resistant to Cell Death." Clin Oncol Res (2020).
- McMahon, H. T., and E. Boucrot. "Molecular Mechanism and Physiological Functions of Clathrin-Mediated Endocytosis." Nat Rev Mol Cell Biol 12, no. 8 (2011): 517-33. [CrossRef]
- Ruan, G., A. Agrawal, A. I. Marcus, and S. Nie. "Imaging and Tracking of Tat Peptide-Conjugated Quantum Dots in Living Cells: New Insights into Nanoparticle Uptake, Intracellular Transport, and Vesicle Shedding." J Am Chem Soc 129, no. 47 (2007): 14759-66.
Figure 1.
Design of RI-AG03 derivatives employed in the current study. A. Sequence of the Tau aggregation inhibitor peptide RI-AG03. The peptide consists of a Tau binding sequence (blue) that interacts with the pro-aggregatory ‘VQIVYK’ sequence in microtubule-binding repeat domain 3 (dark blue box) that is present in all 6 isoforms of Tau. The positively charged cell penetrating sequences polyR (orange) or TAT (red) were attached to the peptide to facilitate cellular uptake. A cysteine was added at the end of the CPP to enable linkage of the peptide to liposomes via click chemistry. B. Unconjugated liposomes and BODIPY-liposomes to which peptides were attached through click chemistry between the cysteine residue of the peptide and protruding maleimide groups (Mal) of the liposomes. For the generation of fluorescent BODIPY-liposomes, 2 % cholesterol was replaced with BODIPY-cholesterol. Adapted from [26].
Figure 1.
Design of RI-AG03 derivatives employed in the current study. A. Sequence of the Tau aggregation inhibitor peptide RI-AG03. The peptide consists of a Tau binding sequence (blue) that interacts with the pro-aggregatory ‘VQIVYK’ sequence in microtubule-binding repeat domain 3 (dark blue box) that is present in all 6 isoforms of Tau. The positively charged cell penetrating sequences polyR (orange) or TAT (red) were attached to the peptide to facilitate cellular uptake. A cysteine was added at the end of the CPP to enable linkage of the peptide to liposomes via click chemistry. B. Unconjugated liposomes and BODIPY-liposomes to which peptides were attached through click chemistry between the cysteine residue of the peptide and protruding maleimide groups (Mal) of the liposomes. For the generation of fluorescent BODIPY-liposomes, 2 % cholesterol was replaced with BODIPY-cholesterol. Adapted from [26].
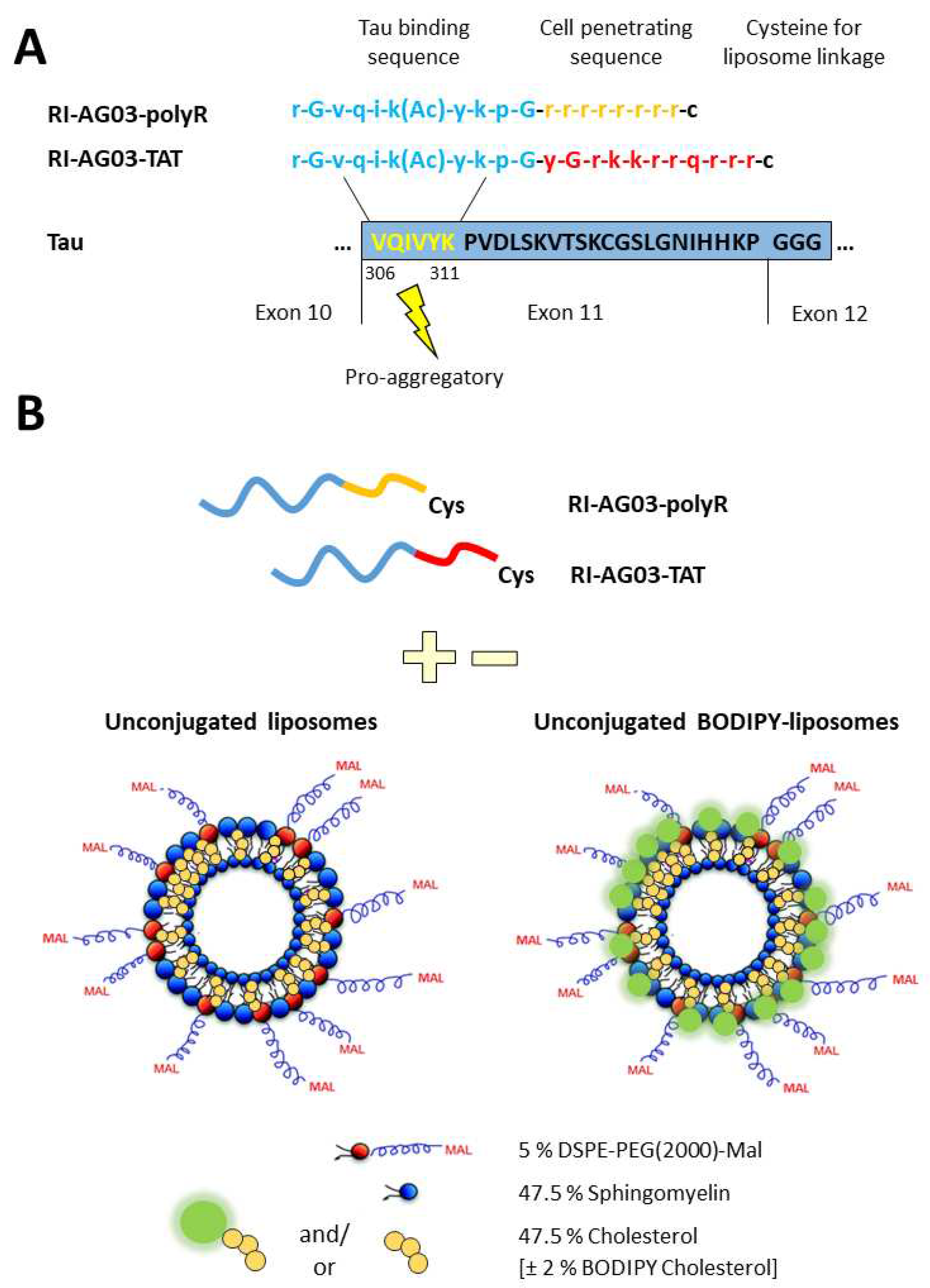
Figure 2.
Internalisation of BODIPY fluorescence from unconjugated BODIPY-liposomes and RI-AG03 peptide conjugated BODIPY-liposomes by SH-SY5Y cells. A. Cells were treated with a final concentration of 75 µM unconjugated, RI-AG03-polyR-conjugated or RI-AG03-TAT-conjugated BODIPY-liposomes for 4 h and fluorescence quantified via flow cytometry. The graph shows the Mean ± SEM cell fluorescence values derived from three independent experiments with n = 3 technical replicates and freshly prepared liposome stocks in each run. *** p < 0.001, Tukey’s HSD. B. Representative confocal images of untreated, unconjugated BODIPY-liposome and peptide-conjugated BODIPY-liposome treated SH-SY5Y cells. Images were taken with consistent settings for the pinhole size, laser strength and gain.
Figure 2.
Internalisation of BODIPY fluorescence from unconjugated BODIPY-liposomes and RI-AG03 peptide conjugated BODIPY-liposomes by SH-SY5Y cells. A. Cells were treated with a final concentration of 75 µM unconjugated, RI-AG03-polyR-conjugated or RI-AG03-TAT-conjugated BODIPY-liposomes for 4 h and fluorescence quantified via flow cytometry. The graph shows the Mean ± SEM cell fluorescence values derived from three independent experiments with n = 3 technical replicates and freshly prepared liposome stocks in each run. *** p < 0.001, Tukey’s HSD. B. Representative confocal images of untreated, unconjugated BODIPY-liposome and peptide-conjugated BODIPY-liposome treated SH-SY5Y cells. Images were taken with consistent settings for the pinhole size, laser strength and gain.
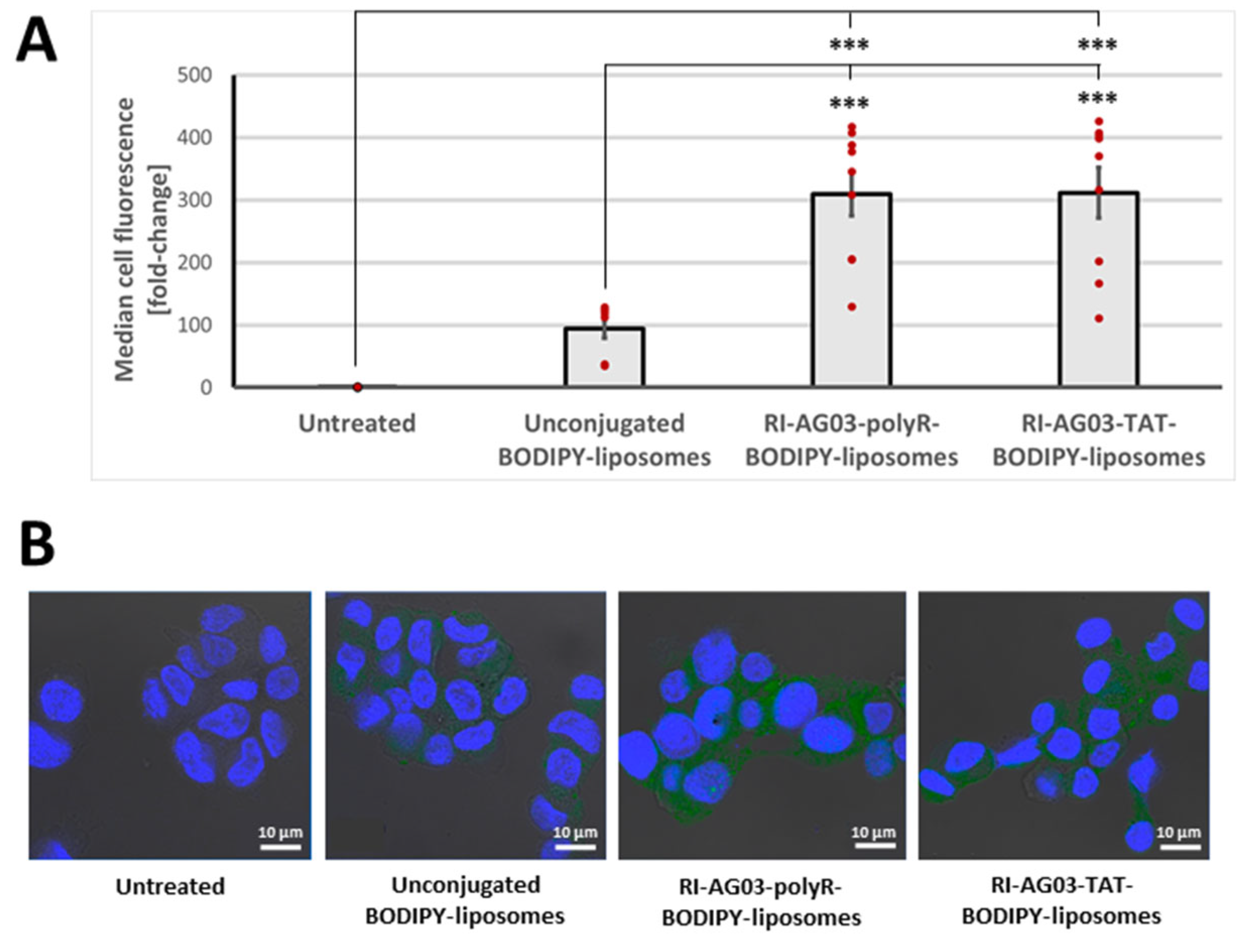
Figure 3.
Effects of endocytosis inhibitors on cellular BODIPY fluorescence levels for SHSY5Y cells treated with (A) unconjugated, (B) RI-AG03-polyR-conjugated and (C) RI-AG03-TAT-conjugated BODIPY-liposomes. Data shown as the Mean ± SEM median cell fluorescence values from three independent repeats (3 samples per run) normalised to no inhibitor controls. * p < 0.05 and *** p < 0.001 relative to no-inhibitor control (post-hoc Tukey’s HSD).
Figure 3.
Effects of endocytosis inhibitors on cellular BODIPY fluorescence levels for SHSY5Y cells treated with (A) unconjugated, (B) RI-AG03-polyR-conjugated and (C) RI-AG03-TAT-conjugated BODIPY-liposomes. Data shown as the Mean ± SEM median cell fluorescence values from three independent repeats (3 samples per run) normalised to no inhibitor controls. * p < 0.05 and *** p < 0.001 relative to no-inhibitor control (post-hoc Tukey’s HSD).
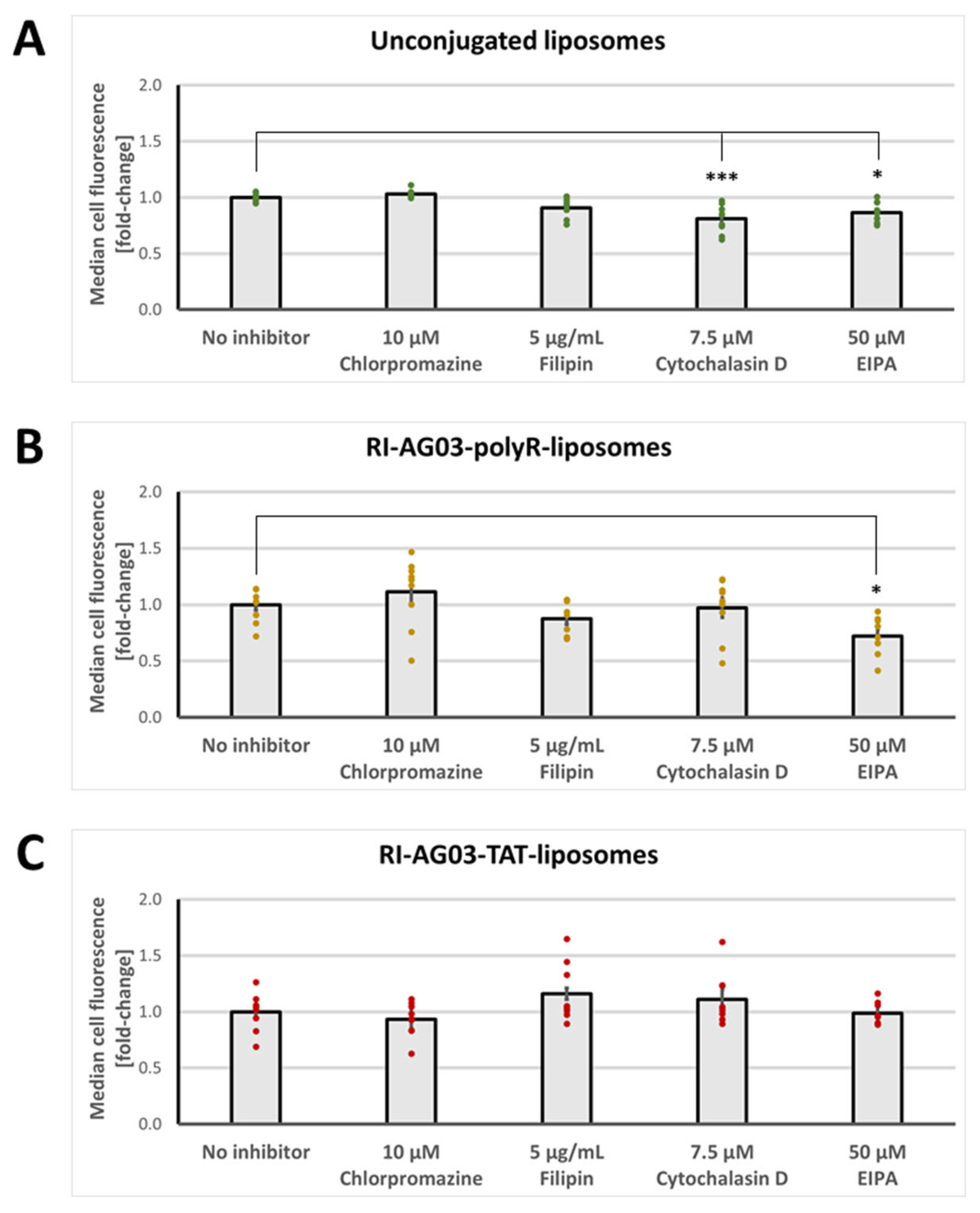
Figure 4.
Co-localisation of BIDOPY fluorescence from unconjugated and peptide-conjugated BODIPY-liposomes with macropinosomes and lysosomes and in SH-SY5Y cells. Cells were co-incubated with BODIPY-liposomes (green) and pHrodo™ Red Dextran (macropinosomes) or LysoTracker Deep Red (lysosomes; both in red) for 16 h. Arrows indicate observed co-localisation.
Figure 4.
Co-localisation of BIDOPY fluorescence from unconjugated and peptide-conjugated BODIPY-liposomes with macropinosomes and lysosomes and in SH-SY5Y cells. Cells were co-incubated with BODIPY-liposomes (green) and pHrodo™ Red Dextran (macropinosomes) or LysoTracker Deep Red (lysosomes; both in red) for 16 h. Arrows indicate observed co-localisation.
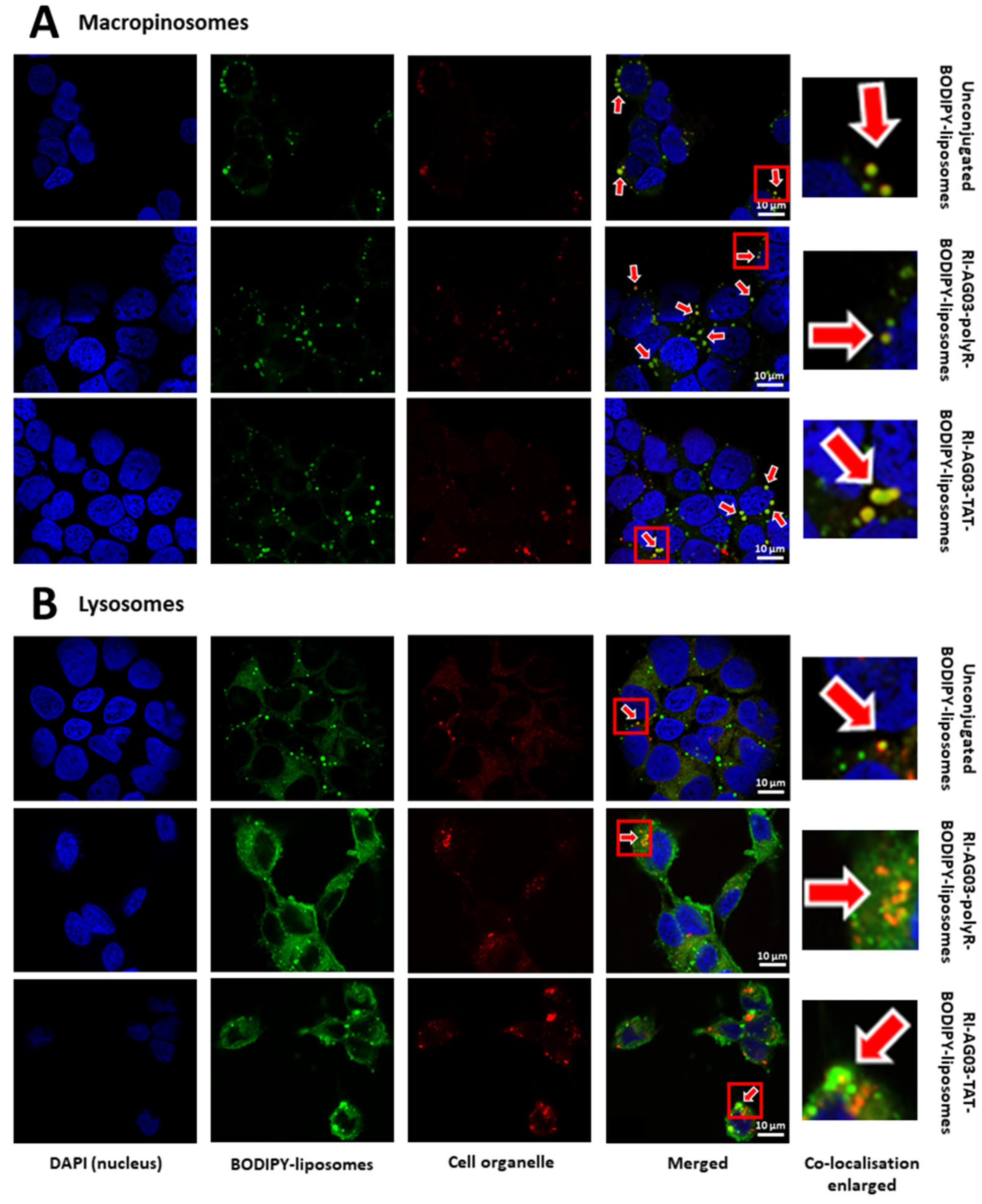
Figure 5.
Co-localisation of 6-FAM-RI-AG03-polyR/TAT peptides and 6-FAM-peptide-conjugated liposomes with macropinosomes. SH-SY5Y cells were co-treated with unconjugated 6-FAM-labelled RI-AG03-polyR/TAT peptides or peptide-conjugated liposomes and the lysosomal live marker LysoTracker Deep Red for 16 h. Arrows show observed co-localisation.
Figure 5.
Co-localisation of 6-FAM-RI-AG03-polyR/TAT peptides and 6-FAM-peptide-conjugated liposomes with macropinosomes. SH-SY5Y cells were co-treated with unconjugated 6-FAM-labelled RI-AG03-polyR/TAT peptides or peptide-conjugated liposomes and the lysosomal live marker LysoTracker Deep Red for 16 h. Arrows show observed co-localisation.
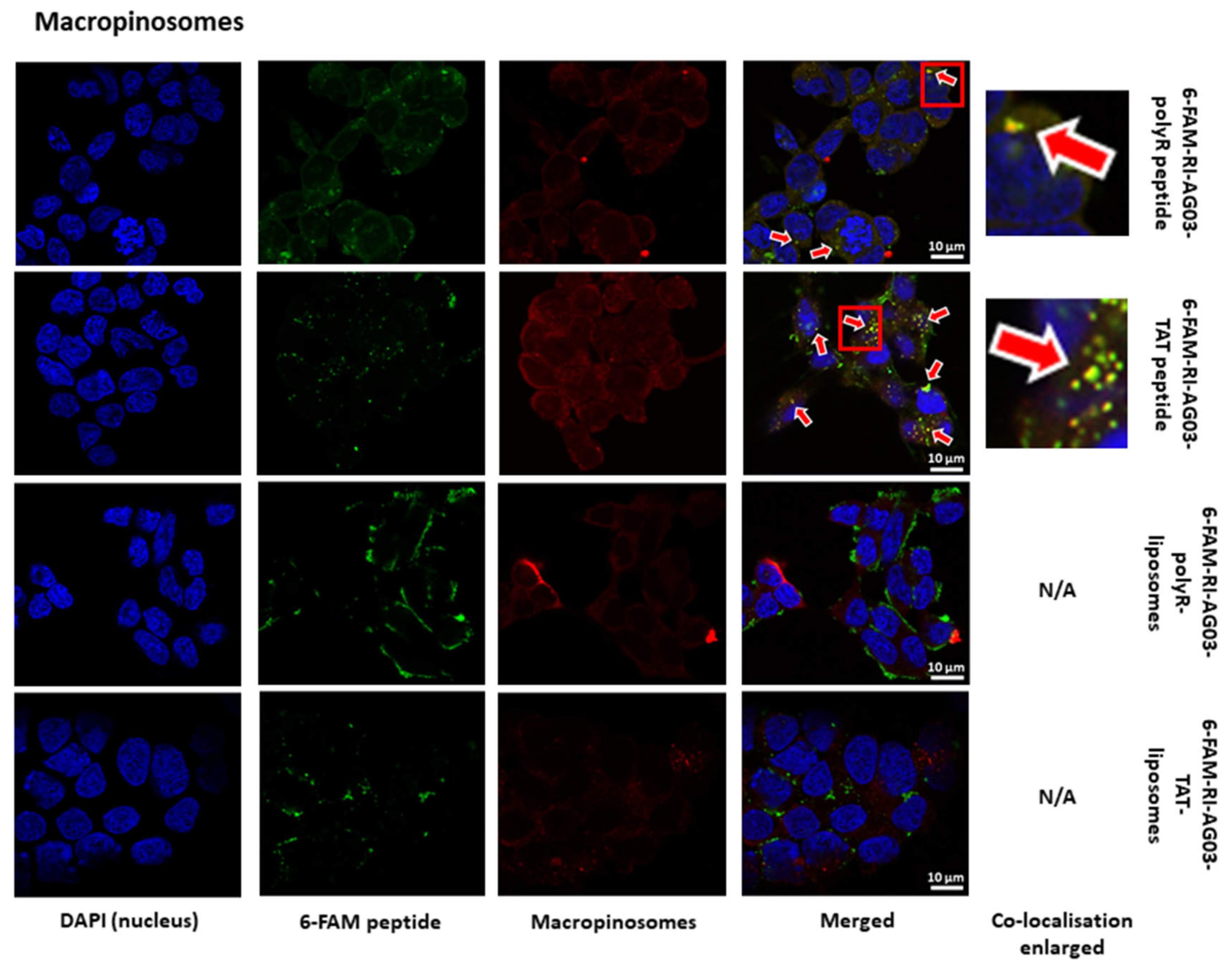
Figure 6.
Subcellular trafficking of 6-FAM-RI-AG03 peptides and 6-FAM-RI-AG03 peptides conjugated to liposomes into lysosomes. SH-SY5Y cells were co-incubated with unconjugated fluorescent 6-FAM-RI-AG03-polyR/TAT peptides or peptide-conjugated liposomes and LysoTracker Deep Red for 16 h. Arrows indicate observed co-localisation.
Figure 6.
Subcellular trafficking of 6-FAM-RI-AG03 peptides and 6-FAM-RI-AG03 peptides conjugated to liposomes into lysosomes. SH-SY5Y cells were co-incubated with unconjugated fluorescent 6-FAM-RI-AG03-polyR/TAT peptides or peptide-conjugated liposomes and LysoTracker Deep Red for 16 h. Arrows indicate observed co-localisation.
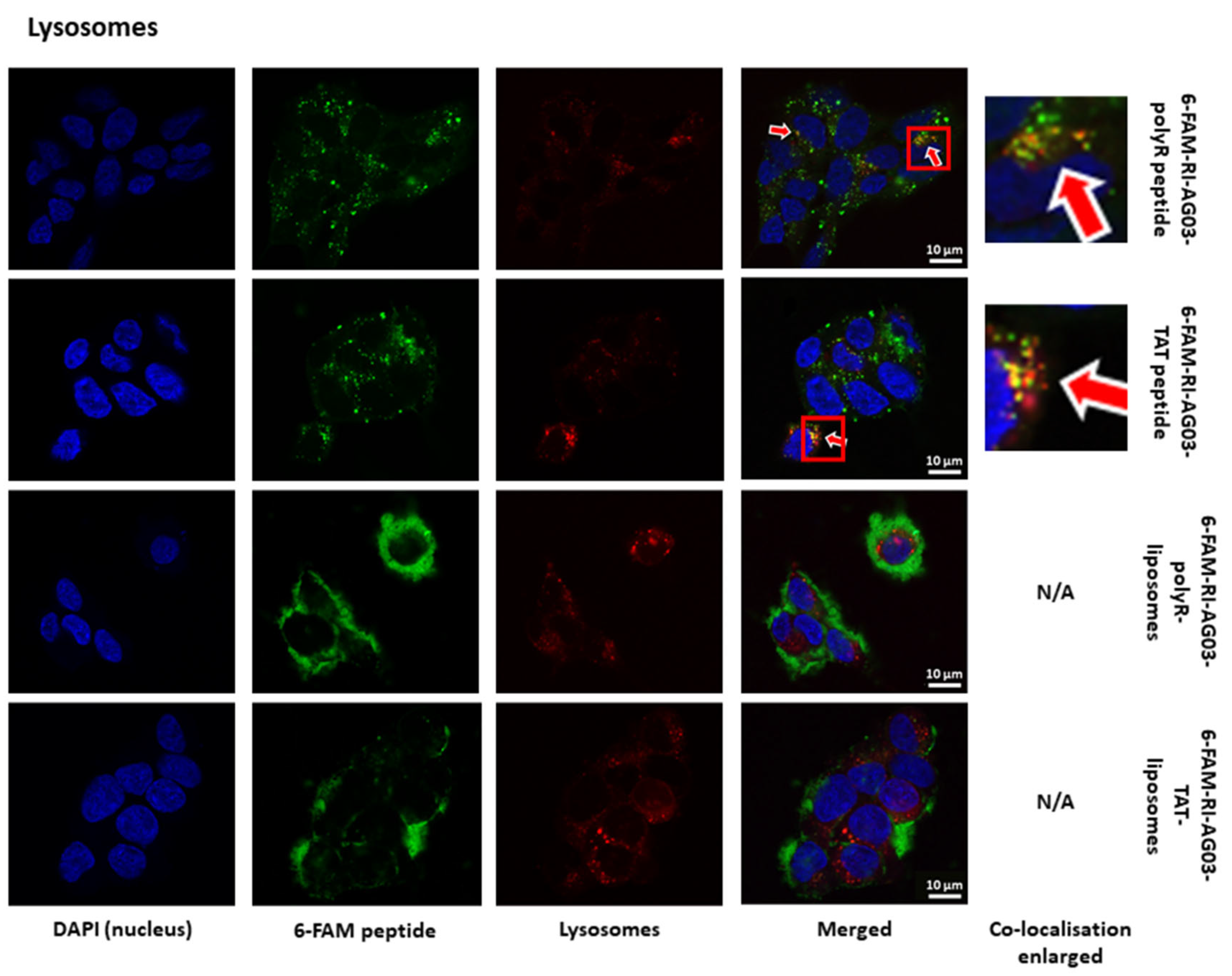
Figure 7.
Co-localisation of BODIPY and 6-FAM fluorescence from unconjugated and RI-AG03-conjugated liposomes with the cell membrane. SH-SY5Y cells were co-treated with unconjugated or RI-AG03-polyR/TAT-conjugated BODIPY-liposomes (green fluorescent liposomes) or unconjugated 6-FAM-RI-AG03-polyR/TAT peptide and 6-FAM-RI-AG03/TAT-conjugated liposomes (green fluorescent peptide) and CellLight™ Plasma Membrane-RFP (red) for 16 h. Arrows show observed co-localisation.
Figure 7.
Co-localisation of BODIPY and 6-FAM fluorescence from unconjugated and RI-AG03-conjugated liposomes with the cell membrane. SH-SY5Y cells were co-treated with unconjugated or RI-AG03-polyR/TAT-conjugated BODIPY-liposomes (green fluorescent liposomes) or unconjugated 6-FAM-RI-AG03-polyR/TAT peptide and 6-FAM-RI-AG03/TAT-conjugated liposomes (green fluorescent peptide) and CellLight™ Plasma Membrane-RFP (red) for 16 h. Arrows show observed co-localisation.
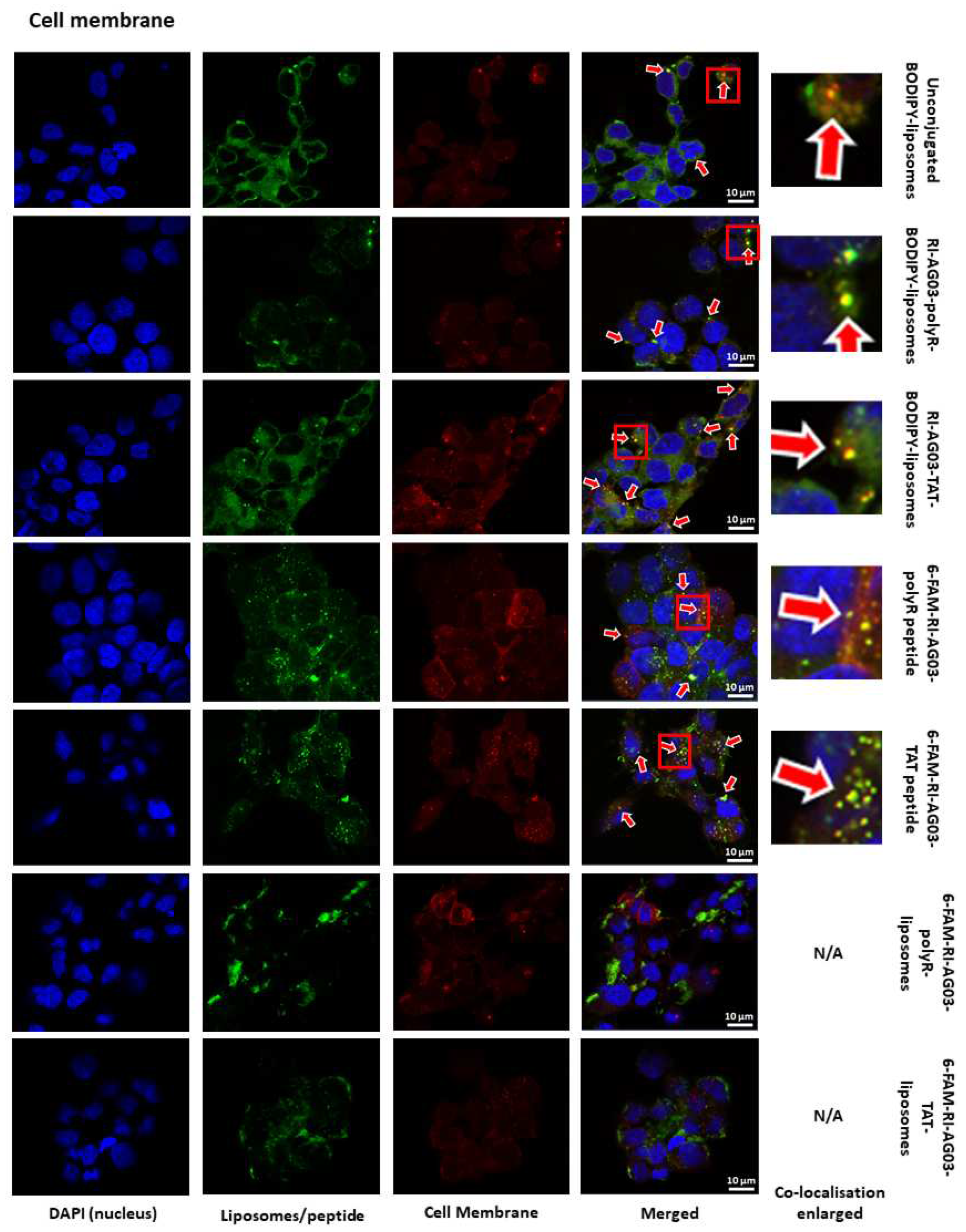
Figure 8.
Dissociation of RI-AG03-polyR from its liposome vehicle following fusion with cells. SH-SY5Y cells were treated with Cy5-RI-AG03-polyR-BODIPY-liposomes for 2 h or 16 h, then images were acquired. Red indicates the Cy5 peptide and green indicates BODIPY fluorescence. Arrows indicate observed co-localisation.
Figure 8.
Dissociation of RI-AG03-polyR from its liposome vehicle following fusion with cells. SH-SY5Y cells were treated with Cy5-RI-AG03-polyR-BODIPY-liposomes for 2 h or 16 h, then images were acquired. Red indicates the Cy5 peptide and green indicates BODIPY fluorescence. Arrows indicate observed co-localisation.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



