
Submitted:
18 May 2023
Posted:
19 May 2023
You are already at the latest version
Abstract
Sepsis is associated with dysregulated cortisol secretion, leading to abnormal levels of cortisol in the blood. In the early stages of sepsis, cortisol levels are typically elevated due to increased secretion from the adrenal glands. However, as the disease progresses, cortisol levels may decline due to impaired adrenal function, leading to relative adrenal insufficiency. The latter is thought to be caused by a combination of factors, including impaired adrenal function, decreased production of corticotropin-releasing hormone (CRH) and adrenocorticotropic hormone (ACTH) by the hypothalamus and pituitary gland, and increased breakdown of cortisol by enzymes such as 11β-hydroxysteroid dehydrogenase type 2 (11β-HSD2). The dysregulation of cortisol secretion in sepsis is thought to contribute to the pathophysiology of the disease by impairing the body's ability to mount an appropriate inflammatory response. The actions of cortisol are mediated through two types of corticosteroid receptors: the mineralocorticoid receptor (MR) and the glucocorticoid receptor (GCR). In sepsis, the expression and activity of GCR is altered, contributing to the pathophysiology of the disease. GCR expression and activity are typically downregulated in sepsis, impairing the body's ability to mount an appropriate inflammatory response. Given the dysregulation of cortisol secretion and corticosteroid receptors in sepsis, there has been considerable interest in the use of steroids as a treatment for sepsis. The rationale for steroid-based treatment is to supplement the body's endogenous cortisol production, restore normal cortisol levels, and improve the body's ability to mount an appropriate inflammatory response. However, the use of steroids in sepsis remains controversial, and clinical trials have yielded mixed results. Some studies have shown that steroid-based treatment can improve shock reversal, reduce the need for vasopressors, and improve overall survival in patients with septic shock, whereas other studies have found no benefit or even harm from steroid-based treatment in sepsis.
Keywords:
CIRCI
; sepsis
; critical illness
; adrenal
; cortisol
; glucocorticoid receptor
1. Introduction—Critical illness
Critical illness refers to a state of poor health where the vital organs are not functioning properly, and immediate care is necessary to prevent the risk of imminent death. This condition has the potential for reversal. Critical care aims to treat the critical condition of the patient rather than the underlying condition. The care of critically ill patients involves a multidisciplinary approach and takes place in an intensive care unit (ICU) with experienced personnel. Examples of patients requiring critical care include those who have undergone major surgery, those with surgical complications, severely injured individuals, those with serious infections, burns, respiratory or circulatory failure [1]. Over time, critical illness management has evolved from organ support and vital sign monitoring to the identification of specific syndromes. Recently, biological heterogeneity within current critical states has been recognized through findings of translational research [2].
1.1. Sepsis and septic shock
Sepsis is a critical condition characterized by dysfunction of the organs due to an uncontrolled response of the body to an infection. Organ dysfunction can be indicated by an increase in the Sequential [Sepsis-related] Organ Failure Assessment (SOFA) score of 2 points or more, which is linked with a mortality rate of over 10% during hospitalization. Sepsis is a significant contributor to both illness and death, and it commonly affects a substantial number of ICU patients either upon admission or during their stay [3].
Septic shock is a specific type of sepsis that is characterized by severe circulatory, cellular, and metabolic dysfunction, leading to a higher risk of mortality compared to sepsis alone. The identification of septic shock in patients can be done through clinical evaluation, where a vasopressor is needed to maintain a mean arterial pressure of 65 mm Hg or higher and a serum lactate level greater than 2 mmol/L in the absence of hypovolemia. The combination of these two factors has been linked to hospital mortality rates exceeding 40%. Septic shock continues to be the primary cause of death in non-coronary ICUs [4].
1.2. HPA axis and critical illness and/or sepsis
Severe stress on the body is evident during critical illness. The stress response is primarily orchestrated by two major components, the sympathetic nervous system and the hypothalamic-pituitary-adrenal (HPA) axis [2]. The amygdala triggers a distress signal, and subsequently, the hypothalamus activates the sympathetic nervous system by transmitting signals through autonomic nerves to the adrenal glands. In response, these glands release epinephrine into the bloodstream, resulting in significant physiological changes that affect almost all organs. When stressors activate the HPA axis, the hypothalamic paraventricular nucleus releases corticotropin-releasing hormone (CRH), which, together with vasopressin, triggers the pituitary corticotropes to release adrenocorticotropic hormone (ACTH) into the systemic circulation. This hormone acts on the adrenal gland's fasciculate layer, which is responsible for the synthesis and secretion of glucocorticoids, with cortisol being the primary glucocorticoid in humans (Figure 1).
Parts of the figure were drawn by using pictures from Servier Medical Art. Servier Medical Art by Servier is licensed under a Creative Commons Attribution 3.0 Unported License (https://creativecommons.org/licenses/by/3.0/).
Cortisol binds to two receptor types, the glucocorticoid receptor (GCR) and the mineralocorticoid receptor (MR), to produce essential cardiovascular, metabolic, and immune regulatory responses necessary for survival. Glucocorticoids play a critical role in maintaining vascular tone, endothelial integrity, and vascular permeability during acute illness. Additionally, they enhance the vasoconstrictor effects of both endogenous and exogenous catecholamines.
1.3. Critical-illness-related corticosteroid insufficiency
Surviving critical illness depends on maintaining adequate adrenocortical function, which is typically reflected in elevated plasma cortisol levels among most critically ill patients. However, the increased systemic cortisol availability observed in critical illness is not solely attributable to a centrally activated HPA axis but rather to peripheral adaptations [5]. These adaptations include the release of circulating cortisol from plasma binding proteins, such as transcortin (CBG) and albumin, resulting in an increase in the free and active form of cortisol. Only the unbound "free" cortisol fraction is lipid soluble and can penetrate the cell membrane to bind with the cytoplasmic GCR. Additionally, cortisol breakdown in the liver and kidney is suppressed, further maintaining high levels of free cortisol in the circulation and target tissues. In addition, an increase in CRH and ACTH, leading to high cortisol production, also plays a role [2].
Some critically ill patients do not exhibit the expected increase in cortisol levels. This state was previously referred to as relative adrenal insufficiency (RAI), which means that cortisol production is inadequate in relation to the body's needs [6].
The Surviving Sepsis Campaign guidelines from 2008 introduced the term critical-illness-related corticosteroid insufficiency (CIRCI) to describe a condition where the cellular activity of corticosteroids is inadequate for the severity of the patient's illness [7]. CIRCI can be caused by a decrease in adrenal steroid production (adrenal insufficiency) or tissue resistance to glucocorticoids, leading to an exaggerated and prolonged proinflammatory response. The mechanisms responsible for HPA axis dysfunction during critical illness are complex and may involve reduced production of CRH, ACTH, and cortisol, as well as dysfunction of their receptors. Damage to the adrenals caused by infarction or hemorrhage has also been suggested as a contributing factor. Although CIRCI may affect a broad range of critically ill patients, most research has focused on patients in the acute phase of septic shock. This condition may present clinically as hypotension and lack of response to catecholamine infusions. The diagnosis of CIRCI is typically based on a delta total serum cortisol of less than 9 µg/dL after administration of 250 µg of i.v. cosyntropin or a random total cortisol of less than 10 µg/dL [8].
The 2017 update of the guidelines for the diagnosis and treatment of CIRCI by the Society of Critical Care Medicine (SCCM) and the European Society of Intensive Care Medicine (ESICM) acknowledged the complexity of the syndrome, and questioned the reliability of a single diagnostic test [9]. While acknowledging that clinicians do not commonly use diagnostic criteria in their routine practice, the task force recommended the use of either random or delta cortisol for diagnosis, and suggested the high-dose (250-μg) ACTH stimulation test rather than the low-dose (1-μg) test. Although free cortisol is the active hormone, the authors proposed the use of plasma total cortisol due to its wider availability compared to free cortisol measurements. The use of salivary unbound cortisol was not recommended as it is not cost-effective or practical [9].
1.4. COVID-19 and Adrenal function
The coronavirus disease 2019 (COVID-19), is caused by the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). It affects predominantly the lungs but also other organs, including the endocrine glands [10]. The virus enters into the cells through the angiotensin-converting enzyme 2 (ACE2) receptor, in the presence of transmembrane serine protease 2 (TMPRSS2) [10]. The endocrine system possesses both the requisite ACE2 receptor, and the TMPRSS2 protein necessary to permit the SARS-CoV-2 virion cellular access [10].
There are limited clinical data on HPA axis function during acute COVID-19 infection. The major difficulty is that of performing a detailed evaluation of the HPA axis in patients with COVID-19 who are glucocorticoid dependent, i.e. those with respiratory failure or those treated within an ICU. Tan et al first showed that patients with COVID-19 not receiving steroids mount a marked and appropriate acute cortisol stress response and that this response is significantly higher in COVID-19 compared to non-COVID-19. Furthermore, they found that elevated cortisol was associated with increased mortality and a reduced median survival, suggesting that cortisol probably reflects severity of illness. Moreover, cortisol seemed to be a better independent predictor than were other laboratory markers associated with COVID-19, such as CRP, D-dimer, and neutrophil to leukocyte ratio [11]. Tomo et al recruited 76 RT-PCR-positive COVID-19-positive patients and 79 healthy controls and showed that increased levels of cortisol in cases when compared with controls. In addition, comparing non-survivors with survivors, cortisol levels were significantly elevated in non-survivors [12]. In sharp contrast to the findings of the latter study, another research work found that patients with SARS-CoV-2 who had lower cortisol levels had a greater fatality rate. The authors applied a logistic regression model and found that a rise in cortisol levels by one unit correlated with a 26% lower mortality risk [13]]. A meta-analysis showed that patients with severe COVID-19 had higher cortisol levels than patients with mild-to-moderate COVID-19, however, age and sex may affect this finding [14]. Overall, it seems that there is no consistent association between COVID-19 clinical outcome and the presence of reduced or increased cortisol.
Clinical studies suggest that adrenal insufficiency during COVID-19 may occur, and according to a meta-analysis its prevalence ranges from 3.1% to 64.3% in different studies [15]. Mechanisms leading to HPA axis dysfunction are possibly multifactorial. Firstly, dysregulation of the HPA axis during the course of COVID-19 may be encountered as part of the development of functional CIRCI due to massive cytokines release. Indeed, in a small study, six of the nine COVID-19 critically ill patients had random plasma cortisol concentrations below 10 µg/dl, meeting the criteria for the diagnosis of CIRCI [16]. Iatrogenic causes resulting from prolonged treatment with synthetic glucocorticoids may also lead to HPA axis dysfunction. Second, the expression of two receptors, ACE2 and TMPRSS2, have also been documented in the hypothalamus, pituitary and the adrenals, making them possible direct cytopathic targets of SARS-CoV-2. Adrenal small vessel necrosis, and thrombosis, cortical lipid degeneration, endothellitis, and chronic inflammation have been described [17]. Similarly, in a postmortem study of COVID-19 patients, areas of pituitary necrosis/infarction have been reported [18]. Third, an interesting proposed mechanism, is that antibodies produced by the host to counteract the virus, may hinder the production of ACTH by the host, since there are similarities between certain amino acids of ACTH and those contained by the virus [19].
1.5. COVID-19 and Sepsis/Septic Shock
COVID-19 can lead to sepsis and septic shock. In one study, one out of three COVID-19 patients who were hospitalized experienced sepsis, which was mainly due to SARS-CoV-2, but in 25% of the cases, bacterial infection also played a role [20]. COVID-19 patients with sepsis had a high mortality rate, particularly those with a combination of SARS-CoV-2 and bacterial sepsis. These results confirm the significance of SARS-CoV-2 as a cause of sepsis and emphasize the importance of improving monitoring, detection, prevention, and management of viral and bacterial sepsis in COVID-19 patients who are hospitalized. In an earlier metaanalysis, most patients with Covid19 who required ICU hospitalization exhibited infection-related organ dysfunction and met sepsis criteria [21]. Recent studies have shown that COVID-19 patients who develop sepsis or septic shock have a significantly higher risk of mortality compared to those who do not. In a European study the incidence of sepsis and septic shock was high in COVID-19 patients admitted to the ICU and these patients had higher mortality rates than those without sepsis [22].
2. GCR Expression in Critical Illness and Sepsis
2.1. The GCR
The actions of cortisol are mediated through two types of corticosteroid receptors: the MR and the GCR. The MR is primarily involved in regulating electrolyte balance, while the GCR plays a crucial role in regulating the immune response and inflammation. In sepsis, the expression and activity of both MR and GCR are altered, contributing to the pathophysiology of the disease. Studies have shown that MR expression is upregulated in sepsis, leading to increased sodium retention and fluid accumulation. This can lead to the development of edema, a common complication of sepsis. The GCR mediates the immunological, metabolic, and hemodynamic effects of endogenously produced and exogenously administered glucocorticoids. Alternative splicing of the primary transcript gives rise to two isoforms, GCR-α and GCR-β [23]. GCR-α is the receptor that binds to cortisol, whereas GCR-β has not been well described. Prior to cortisol-binding and translocation to the nucleus, the GCR-α resides in the cytoplasm in a large chaperone complex. The co-chaperone FK506 binding protein 5 (FKBP51) when bound to GCR-α lowers its affinity for cortisol, and negatively regulates the nuclear translocation of GCR-α [24]. After cortisol binding, GCR-α undergoes a conformational change leading to partial dissociation from the chaperone complex exposing its two nuclear localization signals, and the complex moves from the cytosol to the nucleus to regulate gene transcription. This is done by binding to genes containing glucocorticoid-responsive elements (GREs) found in the promoters or intragenic regions of glucocorticoid-target genes [25]. Such genes include the glucocorticoid-inducible gene leucine zipper (GILZ) and serum/glucocorticoid regulated kinase 1 (SGK1), while negatively regulated genes include β-arrestin and osteocalcin (OSC) amongst other inflammatory genes [26]. This transcriptional activation or repression ultimately results in the termination of the inflammatory response [27]. GCR-β has a C-terminal domain that cannot bind to natural or synthetic ligands and is known to suppress GCR-α activity [28-30]. Figure 2 diagrammatically represents cortisol signalling via GCR-α.
Parts of the figure were drawn by using pictures from Servier Medical Art. Servier Medical Art by Servier is licensed under a Creative Commons Attribution 3.0 Unported License (https://creativecommons.org/licenses/by/3.0/)
2.2. GCR Expression and Glucocorticoid Resistance in Critical Illness and Sepsis
Local glucocorticoid availability is determined by the amount of circulating ligand and by tissue-specific expression of 11β-hydroxysteroid dehydrogenase type 1 (11β-HSD1), i.e. the enzyme that converts inert cortisone into metabolically active cortisol. The magnitude of tissue-specific glucocorticoid and GCR action depends on the local cellular availability of cortisol and on the expression and function of GCR. Glucocorticoid resistance refers to the inadequate response of the GCR to regulate the transcription of GCR-responsive genes, despite adequate plasma cortisol concentrations. Glucocorticoid resistance, and hence the extent of cortisol’s effect, may be a consequence of decreased GCR-α mRNA and protein expression, the receptor subtype expressed, reduced GCR affinity for cortisol in a specific target cell and nuclear translocation, and/or DNA binding [31]. Glucocorticoid resistance occurs in sepsis and may be a major contributor of the failure of glucocrticoids to improve septic patients. Evidence for an association between the degree of glucocorticoid unresponsiveness and disease severity and mortality was demonstrated in acute respiratory distress response (ARDS) [32] and septic shock [33].
Most of the data on glucocrticoid resistance in critical illness originates from experimental septic models. Endotoxin and lipopolysaccharide (LPS) injury models have shown decreased ligand affinity and down-regulation of GCR-α expression [34-39]. One group showed that impaired GCR-α dimerization resulted in worse lung barrier function during lipopolysaccharide (LPS)-induced inflammation and glucocorticoid treatment [40]. A study by the same group demonstrated that impairment of GCR-α dimerization aggravated systemic hypotension and worsened lung function during LPS-induced endotoxic shock in mice [41]. Hence, they concluded that the GCR-α dimer is an important mediator of hemodynamic stability and lung function, during LPS-induced systemic inflammation. Other sepsis animals models have demonstrated down-regulation of GCR-α, and/or decreased ligand affinity and up-regulation of GCR-β expression [39,42-45]. In a cecal ligation and puncture (CLP)-induced polymicrobial sepsis model, an intense initial activation of the GCR-α was noted prior to the induction of profound glucocorticoid resistance. Nuclear translocation of GCR and dexamethasone binding were not affected, however DNA binding was. Hence, the authors suggested that the initial augmented GCR-α activity caused the unresponsiveness towards exogenously administered glucocorticoids seen later in the disease, since this initial GCR-α activation could “exhaust” the receptor [46]. Overall, it seems that mRNA expression of GCR-α is downregulated, while mRNA expression of GCR-β is up-regulated in the animal models of sepsis.
Most human clinical studies have investigated cortisol availability in critical illness, with only a few exploring the role of GCR. The data from these studies suggest the existence of glucocrticoid resistance, especially in sepsis. More specifically, in septic patients glucocorticoid treatment induced expression of miR-124, which in turn down-regulated GCR-α and limited the anti-inflammatory effects of glucocorticoids, prompting the authors to suggest that treatment with steroids might aggravate GC resistance in patients with increased GCR-β mRNA levels [47]. The expression of GCR-β in peripheral mononuclear cells of septic patients, and the effect of serum from septic patients on GCR expression and glucocorticoid sensitivity in cultured immune cells was evaluated in another study [48]. A transient increase in GCR-β mRNA expression was observed in sepsis, while serum from septic patients could induce glucocorticoid resistance in vitro [48]. Reduced GCR-α mRNA expression in peripheral blood mononuclear leucocytes in patients with sepsis or septic shock have been reported [49]. Another study examined GCR isoform abundance in tissues harvested from patients immediately after death from sepsis or non-septic critical illness, and found decreased GCR-α and increased GCR-β receptor numbers in the heart and liver [42]. In septic shock, GCR-α expression was increased in T-lymphocytes, regardless of glucocorticoid treatment, while the GCR binding capacity was reduced in neutrophils of glucocorticoid-treated patients, suggesting the hampered response to exogenous or endogenous glucocorticoids since neutrophils are the predominant circulating leucocyte in septic shock, [50]. More recently, septic non-survivors showed lower GCR-α expression and higher cortisol levels than septic survivors. Moreover, the septic patients exhibited upregulated plasma cortisol levels along with downregulated GCR-α expression in peripheral blood mononuclear cells (PBMCs) compared to controls, whereas GCR-β showed the opposite trend [51]. The reduced GCR-α expression and/or affinity in the blood of critically ill patients suggests that these patients become glucocorticoid-resistant requiring stress-doses of glucocorticoids. On the other hand, one study showed that despite variation, the GCR number and affinity in mononuclear cells from patients during the haemodynamic compensatory phase of sepsis did not differ from control subjects, suggesting that glucocorticoids could be effective in the haemodynamic compensatory phase of sepsis [52]. Increased GCR-α mRNA and protein expression was shown in the acute phase of sepsis compared to systemic inflammatory response syndrome (SIRS) and healthy subjects, implying no need for exogenous steroids at this phase [53]. Only one study, in ventilated critically ill patients within the first 24-hrs post-intubation and within three days of extubation, has demonstrated decreased cytosolic GCR protein levels, and subsequent downregulation of cortisol binding [54]. Finally, our group showed that polymorphonuclear cells (PMNs), isolated from critically ill patients who had not received steroids, displayed a highly variable expression of GCR-α and GCR-β mRNA, with the expression levels of both receptors decreasing during ICU stay [55]. In the follow-up study, and compared to healthy controls, the mRNA expression of both GCR-α and GCR-β was increased, while during the sub-acute phase, the expression of both isoforms was lower compared to controls, as was the expression of FKBP5 and GILZ [56]. A recent report agrees with the results from these two studies. More specifically, Téblick and co-workers quantified the gene expression of key regulators of local glucocorticoid action, including 11β-HSD1, GCR-α, GCR-β, FKBP51, and GILZ in various immune cells and tissues[57]. The expression profiles were compared in relation to duration of critical illness and glucocorticoid availability. In the patients’ neutrophils, GCR-α and GILZ were significantly suppressed throughout ICU stay, while low-to-normal GCR-α and increased GILZ expression was found in the patients’ monocytes. FKBP51 was increased in the monocytes, and transiently increased in the neutrophils, whereas GCR-β was undetectable. In the septic patients, increased systemic glucocorticoid availability in most tissues was associated with suppressed GCR-α, increased FKBP51 and unaltered GILZ. Only in the lung, and the adjacent diaphragm and adipose tissues, an increase in circulating glucocorticoids resulted in higher GCR-α activity [57]. Thus, the authors proposed that these tissue-specific and time-independent adaptations to critical illness facilitated GCR-α action primarily to the lung, protecting against damage in other cells and tissues, including neutrophils. Throughout critical illness, GCR action was inhibited in neutrophils, possibly due to suppression of GCR-α expression, and hence glucocorticoid resistance could not be overcome by further increasing glucocorticoid availability. These findings do not seem to support the notion of a maladaptive generalised glucocorticoid resistance requiring treatment with glucocorticoids [57]. Finally, a novel human GCR variant, G459V, exhibited a hyperactive response when treated with hydrocortisone. More specifically, its activity was more than 30 times greater than the reference GCR-α. Unexpectedly, G459V showed considerably increased activity when treated with the GCR antagonist RU486. Thus, the authors concluded that variants of GCR can potentially alter the response to stress and steroid treatment, which could explain the mitigated clinical response in sepsis [58].
In respiratory syncytial virus (RSV) bronchiolitis-infected infants, the α:β GCR mRNA expression ratio was decreased significantly in severe disease compared to mild and normal controls. Furthermore, the expression of GCR-β positively correlated with the clinical score of severity, and might in part explain the insensitivity to glucocorticoid treatment in RSV-infection [59]. Another study measured the cellular GCR-α activity in PBMCs from critically ill children, and found significantly lower total and cytoplasmic, but not nuclear, GCR-α protein levels in critically ill children compared to healthy controls [60]. The authors suggested that the lower total and cytoplasmic receptor levels in critically ill children limit the GCR-mediated response to exogenous glucocorticoid therapy. The undisturbed translocation indicated that residual receptors retain their functionality and accessibility to therapeutic treatments. In another study in critically ill paediatric patients, those with shock and increased illness severity had lower GCR-α expression in CD4 and CD8 lymphocytes, while GCR-α expression did not correlate with cortisol levels [61]. Finally, in a different paediatric septic shock cohort, GCR-α expression did not differ between SIRS, sepsis, and septic shock. Decreased expression of the GCR-α protein, however, correlated with poor outcomes in septic shock, especially in patients with high cortisol levels [62].
2.3. GCR Εxpression in COVID-19
Data on COVID-19 and GCR are even more limited. Our group demonstrated that critically ill COVID-19 patients had increased expression of GCR-α and GILZ, as well as increased serum cortisol levels, compared to equally severe non-COVID-19 critically ill patients [63]. Our results are indicative of the strong activation of endogenous cortisol response to SARS-CoV-2, providing additional rationale for corticotherapy in critically ill patients with COVID-19 that might, however, not be enough to prevent death [64]. Single-cell RNA sequencing data from bronchoalveolar lavage fluid (BALF) of severe COVID-19 patients receiving corticosteroid therapy revealed that alveolar macrophages, smooth muscle cells, and endothelial cells co-express GCR and IL-6. GCR expression was decreased in severely ill COVID-19 patients compared to mild patients, prompting the authors to suggest that this may be a reflection of the pathologic down-regulation of this endogenous immunomodulatory mechanism, which might be restored with corticosteroid therapy [65]. Very recently it was demonstrated that in moderate-severe COVID-19 patients, GCR gene expression was significantly higher in the patients responding to corticosteroid treatment compared to the non-responders. GCR isoforms and mutations did not seem to correlate with clinical response. Moreover, GILZ expression positively correlated with GCR expression. This study clarified the relationship between GCR expression with therapeutic responses to corticosteroids [66].
3. Glucocorticoids in Septic Shock: Therapeutics, Clinical Trials and Future Research
Currently, the rationale for the use of corticosteroids in septic shock is based on the need to harness the exaggerated and impaired pro-inflammatory response during this condition. Corticosteroids are needed for normal cardiac function and have both inotropic and vascular actions. The inhibition of nitric oxide (NO) synthase by corticosteroids leads to the inhibition of sepsis-induced vasodilation mediated by NO [67].
Before the 1970s, high doses of glucocorticoids were empirically given to patients with septic shock. The perceived aim was to prevent an allergic or hypersensitivity reaction to the causative toxin, without the intention of replacing the adrenal cortex's natural hormone production. At the time a recommended dose was 300-500 mg of hydrocortisone every eight hours for three to five days [68].
The first organized trials investigating the use of glucocorticoids in septic shock were conducted in the 1970s and 1980s. One of the earliest randomized controlled trials (RCTs) investigating the use of glucocorticoids in septic shock was conducted in 1976. In this trial, septic shock patients were randomized either to receive 3 mg/kg dexamethasone or 30 mg/kg methylprednisolone (or the equivalent at multiple doses) or saline as placebo. Two hundred fifty four patients with septic shock received dexamethasone (DEX) or methylprednisolone (MPS), and 246 received saline. In the latter the mortality rate was 41% whereas in the steroid-treated patients, mortality was significantly lower at 13%, with no difference between DEX vs MPS [69]. Another early RCT was conducted by Sprung and colleagues in 1984. The study enrolled 59 patients with septic shock and randomized them to receive either DEX/MPS (n=43) or placebo (n=16). The study found a significant improvement shock reversal with steroids but no improvement in survival versus placebo [70].
However, in the late 1980s and 1990s, it was generally agreed that corticosteroids should not be used to treat sepsis and septic shock due to studies showing no improvement in patient survival rates. Furthermore, the use of steroids in critically ill patients was feared, being linked to adverse effects, especially superinfections and polyneuromyopathy [71].
More recently, there has been an increased recognition of relative adrenal insufficiency in critically ill patients. The administration of glucocorticoids may counter the state of adrenal insufficiency induced by critical illness, which contributes to shock. Studies from the late 1990s and early 2000s have shown that lower doses of steroids administered over an extended period of time can provide hemodynamic benefits. Since then, numerous RCTs and meta-analyses have investigated the efficacy of glucocorticoids in septic shock, with varying results. The first major trial was the Corticosteroid Therapy of Septic Shock (CORTICUS) trial, which was conducted in 2008. This trial included 499 patients with septic shock (of whom 251 received hydrocortisone initially at 50 mg IV four times daily for 5 days and tapering for 6 days) and found no significant difference in mortality rates between those who received hydrocortisone and those who received a placebo[72-75]. Since the CORTICUS trial, several other trials have been conducted, including the Adjunctive Glucocorticoid Therapy in Patients with Septic Shock (ADRENAL) trial and the Activated Protein C and Corticosteroids for Human Septic Shock (APROCCHSS) trial [76-79].
In the ADRENAL trial hydrocortisone was given at 200mg/day for seven days (or until discharge from the ICU) while in the APROCCHSS trial hydrocortisone 50 mg was imtravenously administered four times daily (plus fludrocortisone 50 μg once daily) for seven days. The ADRENAL trial included 3658 patients and found no significant difference in mortality rates between those who received hydrocortisone (n=511/1832; 28%) and those who received a placebo (n=526/1826; 29%). In contrast to these results, the APROCCHSS trial, which included 898 patients found a significant reduction in mortality after 180 days in the trial with hydrocortisone treatment (47%) versus placebo (52%).
While these studies suggest that glucocorticoids may be beneficial - to a degree - in septic shock, their use remains controversial and future research is warranted. One area of research is the use of biomarkers to identify patients who are most likely to benefit from glucocorticoids.
Earlier studies found that patients with septic shock who had high levels of cortisol and low levels of ACTH at baseline were more likely to benefit from glucocorticoids [80]. Other research showed that patients with septic shock who had higher levels of interleukin-6 (IL-6) at baseline were more likely to benefit from early goal-directed therapy [81]. These findings suggest that some biomarkers may be useful in identifying patients who are most likely to benefit from glucocorticoids.
Another area of research is the time of initiation, the dose and the duration of glucocorticoid treatment. The CORTICUS, ADRENAL or APROCCHSS studies used different hydrocortisone administration regimens. Future studies should investigate further the optimal dose and duration of glucocorticoid treatment in septic shock[67] .
The concommitant administration of fludrocortisone with hydrocortisone is still being explored. A recent multicenter observational cohort study involving 88,275 patients with septic shock who were administered norepinephrine and initiated hydrocortisone treatment, found that the inclusion of fludrocortisone alongside hydrocortisone was linked to a reduction of 3.7% in the difference between mortality or discharge rates compared to using hydrocortisone alone [82].
The role of CBG in regulating the action of glucocorticoids is essential. It regulates the availability of glucocorticoids by sequestering them in the bloodstream and CBG affects the biological activity of glucocorticoids by modulating their binding to their intracellular receptors. The binding of glucocorticoids to CBG alters their conformation, which modulates their affinity for GCRs and, consequently, their ability to regulate gene expression [83]. By delving deeper into the role of CBG in inflammatory diseases in general and in septic shock in particular, novel therapeutic approaches based on the concept of efficient delivery of cortisol to tissues could be discovered.
Gender vis-à-vis hydrocortisone administered in septic shock is another domain to be explored. The ADRENAL trial revealed that there were gender-based differences in the impact of hydrocortisone on sepsis. Specifically, hydrocortisone was found to increase the likelihood of shock recurrence in females, with an odds ratio of 1.48 [1.03-2.14; p=0.03] [84].
4. Current Guidelines for Glucocorticoids in Septic Shock
In 2008, the Surviving Sepsis Campaign guidelines recommended the use of hydrocortisone in patients with septic shock who were unresponsive to fluid resuscitation and vasopressor therapy. The guidelines suggested a dose of 200 mg/day of hydrocortisone for seven days. The use of other glucocorticoids, such as MPS and DEX, was not recommended due to the lack of evidence [7]. The 2021 Surviving Sepsis Campaign guidelines recommend the use of hydrocortisone in patients with septic shock who are unresponsive to fluid resuscitation and vasopressor therapy and have a baseline cortisol level of less than 10 μg/dL [85]. The guidelines suggest a lower dose of hydrocortisone (200 mg/day) for up to three days. The guidelines also suggest considering the use of DEX in patients with septic shock who are unresponsive to fluid resuscitation and vasopressor therapy and have a baseline cortisol level of less than 10 μg/dL. The recommended dose of DEX is 6 mg/day for up to 10 days.
5. COVID-19 and treatment with Steroids
The current state of medical knowledge and practice regarding the administration of DEX in ICU/critically ill patients with COVID-19 is that it can be an effective treatment to reduce mortality in certain patients. The use of DEX was first studied in the RECOVERY trial, a large randomized controlled trial in the UK that investigated several potential treatments for COVID-19 [86]. The trial found that DEX reduced mortality by about one-third in ventilated patients and by about one-fifth in patients receiving oxygen without mechanical ventilation. However, DEX was not found to be beneficial for patients who did not require respiratory support. Since the publication of the RECOVERY trial results, DEX has been recommended by several international guidelines for the treatment of COVID-19 in hospitalized patients, including those in the ICU. For example, the National Institutes of Health (NIH) COVID-19 Treatment Guidelines currently recommend the use of DEX in hospitalized patients who require supplemental oxygen or mechanical ventilation at a dose of 6mg/day [87]. It is important to note that while DEX has shown promise in reducing mortality in certain patients, it is not a cure for COVID-19 and should be used judiciously.
6. Conclusion
In conclusion, glucocorticoids remain controversial in the management of septic shock; they may be beneficial but their use should be individualized, based on patient characteristics and biomarkers. Future research in this area should focus on identifying the patients who are most likely to benefit from glucocorticoids, and determining the optimal dose and duration of treatment.
Author Contributions
Conceptualization, I.I., A.G.V., I.D.; Investigation, I.I., A.G.V., C.K., C.S.V., I.D.; Resources, I.I., A.G.V., I.D., X.X.; Writing – Original Draft Preparation, I.I., A.G.V., C.K., C.S.V., S.O., A.K., I.D.; Writing – Review & Editing, I.I., A.G.V., C.K., C.S.V., S.O., A.K., I.D.; Visualization, I.I., A.G.V.; Supervision, I.I., I.D.; Project Administration, I.I., I.D.
Conflicts of Interest
The authors report that they have no conflict of interest.
References
- Maslove, D.M.; Tang, B.; Shankar-Hari, M.; Lawler, P.R.; Angus, D.C.; Baillie, J.K.; Baron, R.M.; Bauer, M.; Buchman, T.G.; Calfee, C.S.; et al. Redefining critical illness. Nature Medicine 2022, 28, 1141–1148. [Google Scholar] [CrossRef] [PubMed]
- Van den Berghe, G.; Téblick, A.; Langouche, L.; Gunst, J. The hypothalamus-pituitary-adrenal axis in sepsis- and hyperinflammation-induced critical illness: Gaps in current knowledge and future translational research directions. EBioMedicine 2022, 84, 104284. [Google Scholar] [CrossRef]
- Vincent, J.-L. Sepsis and infection: Two words that should not be confused. Frontiers in Medicine 2023, 10. [Google Scholar] [CrossRef] [PubMed]
- Singer, M.; Deutschman, C.S.; Seymour, C.W.; Shankar-Hari, M.; Annane, D.; Bauer, M.; Bellomo, R.; Bernard, G.R.; Chiche, J.D.; Coopersmith, C.M.; et al. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA 2016, 315, 801–810. [Google Scholar] [CrossRef]
- Téblick, A.; Gunst, J.; Van den Berghe, G. Critical Illness–induced Corticosteroid Insufficiency: What It Is Not and What It Could Be. The Journal of Clinical Endocrinology & Metabolism 2022, 107, 2057–2064. [Google Scholar] [CrossRef]
- Marik, P.E.; Zaloga, G.P. Adrenal insufficiency in the critically ill: A new look at an old problem. Chest 2002, 122, 1784–1796. [Google Scholar] [CrossRef] [PubMed]
- Dellinger, R.P.; Levy, M.M.; Carlet, J.M.; Bion, J.; Parker, M.M.; Jaeschke, R.; Reinhart, K.; Angus, D.C.; Brun-Buisson, C.; Beale, R.; et al. Surviving Sepsis Campaign: International guidelines for management of severe sepsis and septic shock: 2008. Crit Care Med 2008, 36, 296–327. [Google Scholar] [CrossRef] [PubMed]
- Marik, P.E.; Pastores, S.M.; Annane, D.; Meduri, G.U.; Sprung, C.L.; Arlt, W.; Keh, D.; Briegel, J.; Beishuizen, A.; Dimopoulou, I.; et al. Recommendations for the diagnosis and management of corticosteroid insufficiency in critically ill adult patients: Consensus statements from an international task force by the American College of Critical Care Medicine. Crit Care Med 2008, 36, 1937–1949. [Google Scholar] [CrossRef] [PubMed]
- Annane, D.; Pastores, S.M.; Rochwerg, B.; Arlt, W.; Balk, R.A.; Beishuizen, A.; Briegel, J.; Carcillo, J.; Christ-Crain, M.; Cooper, M.S.; et al. Guidelines for the Diagnosis and Management of Critical Illness-Related Corticosteroid Insufficiency (CIRCI) in Critically Ill Patients (Part I): Society of Critical Care Medicine (SCCM) and European Society of Intensive Care Medicine (ESICM) 2017. Crit Care Med 2017, 45, 2078–2088. [Google Scholar] [CrossRef]
- Lazartigues, E.; Qadir, M.M.F.; Mauvais-Jarvis, F. Endocrine Significance of SARS-CoV-2's Reliance on ACE2. Endocrinology 2020, 161. [Google Scholar] [CrossRef]
- Tan, T.; Khoo, B.; Mills, E.G.; Phylactou, M.; Patel, B.; Eng, P.C.; Thurston, L.; Muzi, B.; Meeran, K.; Prevost, A.T.; et al. Association between high serum total cortisol concentrations and mortality from COVID-19. Lancet Diabetes Endocrinol 2020, 8, 659–660. [Google Scholar] [CrossRef] [PubMed]
- Tomo, S.; Banerjee, M.; Karli, S.; Purohit, P.; Mitra, P.; Sharma, P.; Garg, M.K.; Kumar, B. Assessment of DHEAS, cortisol, and DHEAS/cortisol ratio in patients with COVID-19: A pilot study. Hormones 2022, 21, 515–518. [Google Scholar] [CrossRef] [PubMed]
- Ahmadi, I.; Estabraghnia Babaki, H.; Maleki, M.; Jarineshin, H.; Kaffashian, M.R.; Hassaniazad, M.; Kenarkoohi, A.; Ghanbarnejad, A.; Falahi, S.; Kazemi Jahromi, M.; et al. Changes in Physiological Levels of Cortisol and Adrenocorticotropic Hormone upon Hospitalization Can Predict SARS-CoV-2 Mortality: A Cohort Study. Int J Endocrinol 2022, 2022, 4280691. [Google Scholar] [CrossRef] [PubMed]
- Amiri-Dashatan, N.; Koushki, M.; Parsamanesh, N.; Chiti, H. Serum cortisol concentration and COVID-19 severity: A systematic review and meta-analysis. J Investig Med 2022, 70, 766–772. [Google Scholar] [CrossRef] [PubMed]
- Vakhshoori, M.; Heidarpour, M.; Bondariyan, N.; Sadeghpour, N.; Mousavi, Z. Adrenal Insufficiency in Coronavirus Disease 2019 (COVID-19)-Infected Patients without Preexisting Adrenal Diseases: A Systematic Literature Review. Int J Endocrinol 2021, 2021, 2271514. [Google Scholar] [CrossRef] [PubMed]
- Mao, Y.; Xu, B.; Guan, W.; Xu, D.; Li, F.; Ren, R.; Zhu, X.; Gao, Y.; Jiang, L. The Adrenal Cortex, an Underestimated Site of SARS-CoV-2 Infection. Front Endocrinol 2020, 11, 593179. [Google Scholar] [CrossRef] [PubMed]
- Kanczkowski, W.; Gaba, W.H.; Krone, N.; Varga, Z.; Beuschlein, F.; Hantel, C.; Andoniadou, C.; Bornstein, S.R. Adrenal Gland Function and Dysfunction During COVID-19. Horm Metab Res 2022, 54, 532–539. [Google Scholar] [CrossRef]
- Bryce, C.; Grimes, Z.; Pujadas, E.; Ahuja, S.; Beasley, M.B.; Albrecht, R.; Hernandez, T.; Stock, A.; Zhao, Z.; AlRasheed, M.R.; et al. Pathophysiology of SARS-CoV-2: The Mount Sinai COVID-19 autopsy experience. Mod Pathol 2021, 34, 1456–1467. [Google Scholar] [CrossRef]
- Wheatland, R. Molecular mimicry of ACTH in SARS - implications for corticosteroid treatment and prophylaxis. Med Hypotheses 2004, 63, 855–862. [Google Scholar] [CrossRef]
- Shappell, C.N.; Klompas, M.; Kanjilal, S.; Chan, C.; Rhee, C. Prevalence, Clinical Characteristics, and Outcomes of Sepsis Caused by Severe Acute Respiratory Syndrome Coronavirus 2 Versus Other Pathogens in Hospitalized Patients With COVID-19. Crit Care Explor 2022, 4, e0703. [Google Scholar] [CrossRef]
- Karakike, E.; Giamarellos-Bourboulis, E.J.; Kyprianou, M.; Fleischmann-Struzek, C.; Pletz, M.W.; Netea, M.G.; Reinhart, K.; Kyriazopoulou, E. Coronavirus Disease 2019 as Cause of Viral Sepsis: A Systematic Review and Meta-Analysis. Crit Care Med 2021, 49, 2042–2057. [Google Scholar] [CrossRef] [PubMed]
- Karagiannidis, C.; Mostert, C.; Hentschker, C.; Voshaar, T.; Malzahn, J.; Schillinger, G.; Klauber, J.; Janssens, U.; Marx, G.; Weber-Carstens, S.; et al. Case characteristics, resource use, and outcomes of 10 021 patients with COVID-19 admitted to 920 German hospitals: An observational study. The Lancet Respiratory Medicine 2020, 8, 853–862. [Google Scholar] [CrossRef] [PubMed]
- Hollenberg, S.M.; Weinberger, C.; Ong, E.S.; Cerelli, G.; Oro, A.; Lebo, R.; Thompson, E.B.; Rosenfeld, M.G.; Evans, R.M. Primary structure and expression of a functional human glucocorticoid receptor cDNA. Nature 1985, 318, 635–641. [Google Scholar] [CrossRef] [PubMed]
- Binder, E.B. The role of FKBP5, a co-chaperone of the glucocorticoid receptor in the pathogenesis and therapy of affective and anxiety disorders. Psychoneuroendocrinology 2009, 34 (Suppl. S1), S186–S195. [Google Scholar] [CrossRef] [PubMed]
- Kadmiel, M.; Cidlowski, J.A. Glucocorticoid receptor signaling in health and disease. Trends Pharmacol Sci 2013, 34, 518–530. [Google Scholar] [CrossRef] [PubMed]
- Smoak, K.A.; Cidlowski, J.A. Mechanisms of glucocorticoid receptor signaling during inflammation. Mech Ageing Dev 2004, 125, 697–706. [Google Scholar] [CrossRef] [PubMed]
- Gottlicher, M.; Heck, S.; Herrlich, P. Transcriptional cross-talk, the second mode of steroid hormone receptor action. J Mol Med 1998, 76, 480–489. [Google Scholar] [CrossRef]
- Bamberger, C.M.; Bamberger, A.M.; de Castro, M.; Chrousos, G.P. Glucocorticoid receptor beta, a potential endogenous inhibitor of glucocorticoid action in humans. J Clin Invest 1995, 95, 2435–2441. [Google Scholar] [CrossRef]
- Kino, T.; Su, Y.A.; Chrousos, G.P. Human glucocorticoid receptor isoform beta: Recent understanding of its potential implications in physiology and pathophysiology. Cell Mol Life Sci 2009, 66, 3435–3448. [Google Scholar] [CrossRef]
- Oakley, R.H.; Sar, M.; Cidlowski, J.A. The human glucocorticoid receptor beta isoform. Expression, biochemical properties, and putative function. J Biol Chem 1996, 271, 9550–9559. [Google Scholar] [CrossRef]
- Bamberger, C.M.; Schulte, H.M.; Chrousos, G.P. Molecular determinants of glucocorticoid receptor function and tissue sensitivity to glucocorticoids. Endocr Rev 1996, 17, 245–261. [Google Scholar] [CrossRef] [PubMed]
- Meduri, G.U.; Muthiah, M.P.; Carratu, P.; Eltorky, M.; Chrousos, G.P. Nuclear factor-kappaB- and glucocorticoid receptor alpha- mediated mechanisms in the regulation of systemic and pulmonary inflammation during sepsis and acute respiratory distress syndrome. Evidence for inflammation-induced target tissue resistance to glucocorticoids. Neuroimmunomodulation 2005, 12, 321–338. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.; Pretorius, C.J.; Ungerer, J.P.; Cardinal, J.; Blumenthal, A.; Presneill, J.; Gatica-Andrades, M.; Jarrett, P.; Lassig-Smith, M.; Stuart, J.; et al. Glucocorticoid Sensitivity Is Highly Variable in Critically Ill Patients With Septic Shock and Is Associated With Disease Severity. Crit Care Med 2016, 44, 1034–1041. [Google Scholar] [CrossRef] [PubMed]
- Da, J.; Chen, L.; Hedenstierna, G. Nitric oxide up-regulates the glucocorticoid receptor and blunts the inflammatory reaction in porcine endotoxin sepsis. Crit Care Med 2007, 35, 26–32. [Google Scholar] [CrossRef]
- Koulouras, V.P.; Li, R.; Chen, L.; Hedenstierna, G.G. Effects of inhaled carbon monoxide and glucocorticoids in porcine endotoxin sepsis. Int J Clin Exp Med 2011, 4, 53–66. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Xu, R.B. Changes in canine leukocyte glucocorticoid receptors during endotoxin shock. Circ Shock 1988, 26, 99–105. [Google Scholar]
- Reichardt, H.M.; Umland, T.; Bauer, A.; Kretz, O.; Schutz, G. Mice with an increased glucocorticoid receptor gene dosage show enhanced resistance to stress and endotoxic shock. Mol Cell Biol 2000, 20, 9009–9017. [Google Scholar] [CrossRef]
- Stith, R.D.; McCallum, R.E. Down regulation of hepatic glucocorticoid receptors after endotoxin treatment. Infect Immun 1983, 40, 613–621. [Google Scholar] [CrossRef]
- Bergquist, M.; Nurkkala, M.; Rylander, C.; Kristiansson, E.; Hedenstierna, G.; Lindholm, C. Expression of the glucocorticoid receptor is decreased in experimental Staphylococcus aureus sepsis. J Infect 2013, 67, 574–583. [Google Scholar] [CrossRef]
- Vettorazzi, S.; Bode, C.; Dejager, L.; Frappart, L.; Shelest, E.; Klaßen, C.; Tasdogan, A.; Reichardt, H.M.; Libert, C.; Schneider, M.; et al. Glucocorticoids limit acute lung inflammation in concert with inflammatory stimuli by induction of SphK1. Nat Commun 2015, 6, 7796. [Google Scholar] [CrossRef]
- Wepler, M.; Preuss, J.M.; Merz, T.; Hartmann, C.; Wachter, U.; McCook, O.; Vogt, J.; Kress, S.; Gröger, M.; Fink, M.; et al. Impaired Glucocorticoid Receptor Dimerization Aggravates LPS-Induced Circulatory and Pulmonary Dysfunction. Front Immunol 2019, 10, 3152. [Google Scholar] [CrossRef] [PubMed]
- Abraham, M.N.; Jimenez, D.M.; Fernandes, T.D.; Deutschman, C.S. Cecal Ligation and Puncture Alters Glucocorticoid Receptor Expression. Crit Care Med 2018, 46, e797–e804. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, J.E.; Feng, Y.; Velazquez, H.; Sessa, W.C. Endothelial glucocorticoid receptor is required for protection against sepsis. Proc Natl Acad Sci U S A 2013, 110, 306–311. [Google Scholar] [CrossRef] [PubMed]
- Kamiyama, K.; Matsuda, N.; Yamamoto, S.; Takano, K.; Takano, Y.; Yamazaki, H.; Kageyama, S.; Yokoo, H.; Nagata, T.; Hatakeyama, N.; et al. Modulation of glucocorticoid receptor expression, inflammation, and cell apoptosis in septic guinea pig lungs using methylprednisolone. Am J Physiol Lung Cell Mol Physiol 2008, 295, L998–L1006. [Google Scholar] [CrossRef] [PubMed]
- Dekelbab, B.H.; Witchel, S.F.; DeFranco, D.B. TNF-alpha and glucocorticoid receptor interaction in L6 muscle cells: A cooperative downregulation of myosin heavy chain. Steroids 2007, 72, 705–712. [Google Scholar] [CrossRef] [PubMed]
- Vandewalle, J.; Timmermans, S.; Paakinaho, V.; Vancraeynest, L.; Dewyse, L.; Vanderhaeghen, T.; Wallaeys, C.; Van Wyngene, L.; Van Looveren, K.; Nuyttens, L.; et al. Combined glucocorticoid resistance and hyperlactatemia contributes to lethal shock in sepsis. Cell Metab 2021, 33, 1763–1776. [Google Scholar] [CrossRef] [PubMed]
- Ledderose, C.; Mohnle, P.; Limbeck, E.; Schutz, S.; Weis, F.; Rink, J.; Briegel, J.; Kreth, S. Corticosteroid resistance in sepsis is influenced by microRNA-124--induced downregulation of glucocorticoid receptor-alpha. Crit Care Med 2012, 40, 2745–2753. [Google Scholar] [CrossRef]
- Guerrero, J.; Gatica, H.A.; Rodriguez, M.; Estay, R.; Goecke, I.A. Septic serum induces glucocorticoid resistance and modifies the expression of glucocorticoid isoforms receptors: A prospective cohort study and in vitro experimental assay. Crit Care 2013, 17, R107. [Google Scholar] [CrossRef]
- Molijn, G.J.; Koper, J.W.; van Uffelen, C.J.; de Jong, F.H.; Brinkmann, A.O.; Bruining, H.A.; Lamberts, S.W. Temperature-induced down-regulation of the glucocorticoid receptor in peripheral blood mononuclear leucocyte in patients with sepsis or septic shock. Clin Endocrinol 1995, 43, 197–203. [Google Scholar] [CrossRef]
- Bergquist, M.; Lindholm, C.; Strinnholm, M.; Hedenstierna, G.; Rylander, C. Impairment of neutrophilic glucocorticoid receptor function in patients treated with steroids for septic shock. Intensive Care Med Exp 2015, 3, 59. [Google Scholar] [CrossRef]
- Li, J.; Xie, M.; Yu, Y.; Tang, Z.; Hang, C.; Li, C. Leukocyte glucocorticoid receptor expression and related transcriptomic gene signatures during early sepsis. Clin Immunol 2021, 223, 108660. [Google Scholar] [CrossRef] [PubMed]
- Sigal, G.A.; Maria, D.A.; Katayama, M.L.; Wajchenberg, B.L.; Brentani, M.M. Glucocorticoid receptors in mononuclear cells of patients with sepsis. Scand J Infect Dis 1993, 25, 245–248. [Google Scholar] [CrossRef] [PubMed]
- Vardas, K.; Ilia, S.; Sertedaki, A.; Charmandari, E.; Briassouli, E.; Goukos, D.; Apostolou, K.; Psarra, K.; Botoula, E.; Tsagarakis, S.; et al. Increased glucocorticoid receptor expression in sepsis is related to heat shock proteins, cytokines, and cortisol and is associated with increased mortality. Intensive Care Med Exp 2017, 5, 10. [Google Scholar] [CrossRef] [PubMed]
- Siebig, S.; Meinel, A.; Rogler, G.; Klebl, E.; Wrede, C.E.; Gelbmann, C.; Froh, S.; Rockmann, F.; Bruennler, T.; Schoelmerich, J.; et al. Decreased cytosolic glucocorticoid receptor levels in critically ill patients. Anaesth Intensive Care 2010, 38, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Vassiliou, A.G.; Floros, G.; Jahaj, E.; Stamogiannos, G.; Gennimata, S.; Vassiliadi, D.A.; Tsagarakis, S.; Tzanela, M.; Ilias, I.; Orfanos, S.E.; et al. Decreased glucocorticoid receptor expression during critical illness. Eur J Clin Invest 2019, 49, e13073. [Google Scholar] [CrossRef]
- Vassiliou, A.G.; Stamogiannos, G.; Jahaj, E.; Botoula, E.; Floros, G.; Vassiliadi, D.A.; Ilias, I.; Tsagarakis, S.; Tzanela, M.; Orfanos, S.E.; et al. Longitudinal evaluation of glucocorticoid receptor alpha/beta expression and signalling, adrenocortical function and cytokines in critically ill steroid-free patients. Mol Cell Endocrinol 2020, 501, 110656. [Google Scholar] [CrossRef]
- Téblick, A.; Van Dyck, L.; Van Aerde, N.; Van der Perre, S.; Pauwels, L.; Derese, I.; Debaveye, Y.; Wouters, P.J.; Vanhorebeek, I.; Langouche, L.; et al. Impact of duration of critical illness and level of systemic glucocorticoid availability on tissue-specific glucocorticoid receptor expression and actions: A prospective, observational, cross-sectional human and two translational mouse studies. EBioMedicine 2022, 80, 104057. [Google Scholar] [CrossRef]
- Grigsby, M.J.; Green, T.L.; Lim, D.; Cho, K.; Greenhalgh, D.G. A Novel Human Glucocorticoid Receptor Variant, G459V, is Hyperactive in Response to Steroids. Shock 2021, 56, 318–324. [Google Scholar] [CrossRef]
- Diaz, P.V.; Pinto, R.A.; Mamani, R.; Uasapud, P.A.; Bono, M.R.; Gaggero, A.A.; Guerrero, J.; Goecke, A. Increased expression of the glucocorticoid receptor beta in infants with RSV bronchiolitis. Pediatrics 2012, 130, e804–e811. [Google Scholar] [CrossRef]
- Indyk, J.A.; Candido-Vitto, C.; Wolf, I.M.; Venkataraman, S.; Munoz, R.; Saladino, R.A.; Witchel, S.F.; Defranco, D.B. Reduced glucocorticoid receptor protein expression in children with critical illness. Horm Res Paediatr 2013, 79, 169–178. [Google Scholar] [CrossRef]
- Shibata, A.R.; Troster, E.J.; Wong, H.R. Glucocorticoid Receptor Expression in Peripheral WBCs of Critically Ill Children. Pediatr Crit Care Med 2015, 16, e132–e140. [Google Scholar] [CrossRef] [PubMed]
- Alder, M.N.; Opoka, A.M.; Wong, H.R. The glucocorticoid receptor and cortisol levels in pediatric septic shock. Crit Care 2018, 22, 244. [Google Scholar] [CrossRef] [PubMed]
- Vassiliou, A.G.; Athanasiou, N.; Keskinidou, C.; Jahaj, E.; Tsipilis, S.; Zacharis, A.; Botoula, E.; Diamantopoulos, A.; Ilias, I.; Vassiliadi, D.A.; et al. Increased Glucocorticoid Receptor Alpha Expression and Signaling in Critically Ill Coronavirus Disease 2019 Patients. Crit Care Med 2021, 49, 2131–2136. [Google Scholar] [CrossRef] [PubMed]
- Annane, D.; Godot, V. Glucocorticoid-Glucocorticoid Receptor Response to Severe Acute Respiratory Syndrome Coronavirus 2. Crit Care Med 2021, 49, 2157–2160. [Google Scholar] [CrossRef] [PubMed]
- Awasthi, S.; Wagner, T.; Venkatakrishnan, A.J.; Puranik, A.; Hurchik, M.; Agarwal, V.; Conrad, I.; Kirkup, C.; Arunachalam, R.; O'Horo, J.; et al. Plasma IL-6 levels following corticosteroid therapy as an indicator of ICU length of stay in critically ill COVID-19 patients. Cell Death Discov 2021, 7, 55. [Google Scholar] [CrossRef] [PubMed]
- Aliska, G.; Nafrialdi, N.; Lie, K.C.; Setiabudy, R.; Putra, A.E.; Widyahening, I.S.; Harahap, A.R. The role of the glucocorticoid receptor and its impact on steroid response in moderate-severe COVID-19 patients. Eur J Pharmacol 2023, 943, 175555. [Google Scholar] [CrossRef]
- Ammar, M.A.; Ammar, A.A.; Wieruszewski, P.M.; Bissell, B.D.; M, T.L.; Albert, L.; Khanna, A.K.; Sacha, G.L. Timing of vasoactive agents and corticosteroid initiation in septic shock. Ann Intensive Care 2022, 12, 47. [Google Scholar] [CrossRef]
- Lansing, A.M. Septic Shock. Can Med Assoc J 1963, 89, 583–588. [Google Scholar]
- Schumer, W. Steroids in the treatment of clinical septic shock. Ann Surg 1976, 184, 333–341. [Google Scholar] [CrossRef]
- Sprung, C.L.; Caralis, P.V.; Marcial, E.H.; Pierce, M.; Gelbard, M.A.; Long, W.M.; Duncan, R.C.; Tendler, M.D.; Karpf, M. The effects of high-dose corticosteroids in patients with septic shock. A prospective, controlled study. N Engl J Med 1984, 311, 1137–1143. [Google Scholar] [CrossRef]
- Sprung, C.L.; Goodman, S.; Weiss, Y.G. Corticosteroid Treatment of Patients in Septic Shock. In Yearbook of Intensive Care and Emegency Medicine 2009, Vincent, J.L., Ed. Springer: Berlin-Heidelberg, 2009; pp. 753–762.
- Bauer, W.; Ball, J.; Grounds, M. Unanswered questions from Corticus and pragmatic suggestions. Crit Care 2008, 12, 426. [Google Scholar] [CrossRef] [PubMed]
- Moreno, R.; Sprung, C.L.; Annane, D.; Chevret, S.; Briegel, J.; Keh, D.; Singer, M.; Weiss, Y.G.; Payen, D.; Cuthbertson, B.H.; et al. Time course of organ failure in patients with septic shock treated with hydrocortisone: Results of the Corticus study. Intensive Care Med 2011, 37, 1765–1772. [Google Scholar] [CrossRef] [PubMed]
- Sprung, C.L.; Annane, D.; Keh, D.; Moreno, R.; Singer, M.; Freivogel, K.; Weiss, Y.G.; Benbenishty, J.; Kalenka, A.; Forst, H.; et al. Hydrocortisone therapy for patients with septic shock. N Engl J Med 2008, 358, 111–124. [Google Scholar] [CrossRef] [PubMed]
- Toma, A.; Stone, A.; Green, R.S.; Gray, S. Steroids for patients in septic shock: The results of the CORTICUS trial. CJEM 2011, 13, 273–276. [Google Scholar] [CrossRef] [PubMed]
- Venkatesh, B.; Myburgh, J.; Finfer, S.; Webb, S.A.; Cohen, J.; Bellomo, R.; McArthur, C.; Joyce, C.J.; Rajbhandari, D.; Glass, P.; et al. The ADRENAL study protocol: Adjunctive corticosteroid treatment in critically ill patients with septic shock. Crit Care Resusc 2013, 15, 83–88. [Google Scholar] [CrossRef] [PubMed]
- Venkatesh, B.; Finfer, S.; Cohen, J.; Rajbhandari, D.; Arabi, Y.; Bellomo, R.; Billot, L.; Glass, P.; Joyce, C.; Li, Q.; et al. Hydrocortisone Compared with Placebo in Patients with Septic Shock Satisfying the Sepsis-3 Diagnostic Criteria and APROCCHSS Study Inclusion Criteria: A Post Hoc Analysis of the ADRENAL Trial. Anesthesiology 2019, 131, 1292–1300. [Google Scholar] [CrossRef] [PubMed]
- Annane, D.; Timsit, J.F.; Megarbane, B.; Martin, C.; Misset, B.; Mourvillier, B.; Siami, S.; Chagnon, J.L.; Constantin, J.M.; Petitpas, F.; et al. Recombinant human activated protein C for adults with septic shock: A randomized controlled trial. Am J Respir Crit Care Med 2013, 187, 1091–1097. [Google Scholar] [CrossRef]
- Annane, D.; Renault, A.; Brun-Buisson, C.; Megarbane, B.; Quenot, J.P.; Siami, S.; Cariou, A.; Forceville, X.; Schwebel, C.; Martin, C.; et al. Hydrocortisone plus Fludrocortisone for Adults with Septic Shock. N Engl J Med 2018, 378, 809–818. [Google Scholar] [CrossRef]
- Russell, J.A. When and how to use predictive biomarkers for corticosteroid treatment of septic shock. Crit Care 2018, 22, 318. [Google Scholar] [CrossRef]
- Bentzer, P.; Fjell, C.; Walley, K.R.; Boyd, J.; Russell, J.A. Plasma cytokine levels predict response to corticosteroids in septic shock. Intensive Care Med 2016, 42, 1970–1979. [Google Scholar] [CrossRef]
- Bosch, N.A.; Teja, B.; Law, A.C.; Pang, B.; Jafarzadeh, S.R.; Walkey, A.J. Comparative Effectiveness of Fludrocortisone and Hydrocortisone vs Hydrocortisone Alone Among Patients With Septic Shock. JAMA Intern Med 2023. [Google Scholar] [CrossRef] [PubMed]
- Meyer, E.J.; Nenke, M.A.; Lewis, J.G.; Torpy, D.J. Corticosteroid-binding globulin: Acute and chronic inflammation. Expert Rev Endocrinol Metab 2017, 12, 241–251. [Google Scholar] [CrossRef] [PubMed]
- Thompson, K.; Venkatesh, B.; Hammond, N.; Taylor, C.; Finfer, S. Sex differences in response to adjunctive corticosteroid treatment for patients with septic shock. Intensive Care Med 2021, 47, 246–248. [Google Scholar] [CrossRef] [PubMed]
- Evans, L.; Rhodes, A.; Alhazzani, W.; Antonelli, M.; Coopersmith, C.M.; French, C.; Machado, F.R.; McIntyre, L.; Ostermann, M.; Prescott, H.C.; et al. Surviving Sepsis Campaign: International Guidelines for Management of Sepsis and Septic Shock 2021. Crit Care Med 2021, 49, e1063–e1143. [Google Scholar] [CrossRef]
- Horby, P.; Lim, W.S.; Emberson, J.R.; Mafham, M.; Bell, J.L.; Linsell, L.; Staplin, N.; Brightling, C.; Ustianowski, A.; Elmahi, E.; et al. Dexamethasone in Hospitalized Patients with Covid-19. N Engl J Med 2021, 384, 693–704. [Google Scholar] [CrossRef]
- Anonymous. Corticosteroids. Coronavirus Disease 2019 (COVID-19) Treatment Guidelines. National Institutes of Health. Available at: https://www.covid19treatmentguidelines.nih.gov/therapies/immunomodulators/corticosteroids/. National Institutes of Health: Bethesda, MD, 2023.
Figure 1.
Schematic outline of the stress response to critical illness; F: cortisol; FMet: F metabolism, including inactivation by 11β-Hydroxysteroid dehydrogenase type 2 to cortisone.
Figure 1.
Schematic outline of the stress response to critical illness; F: cortisol; FMet: F metabolism, including inactivation by 11β-Hydroxysteroid dehydrogenase type 2 to cortisone.
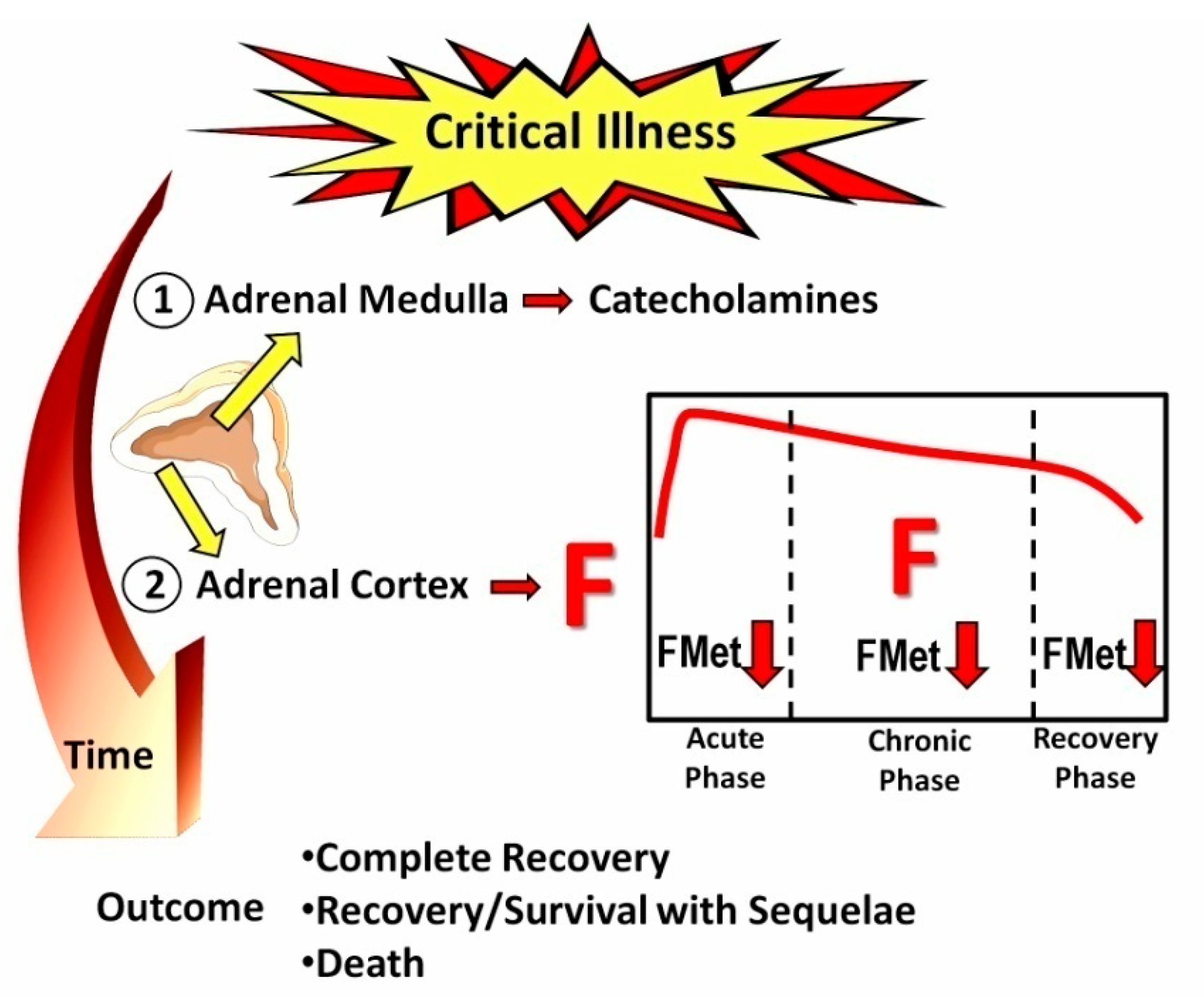
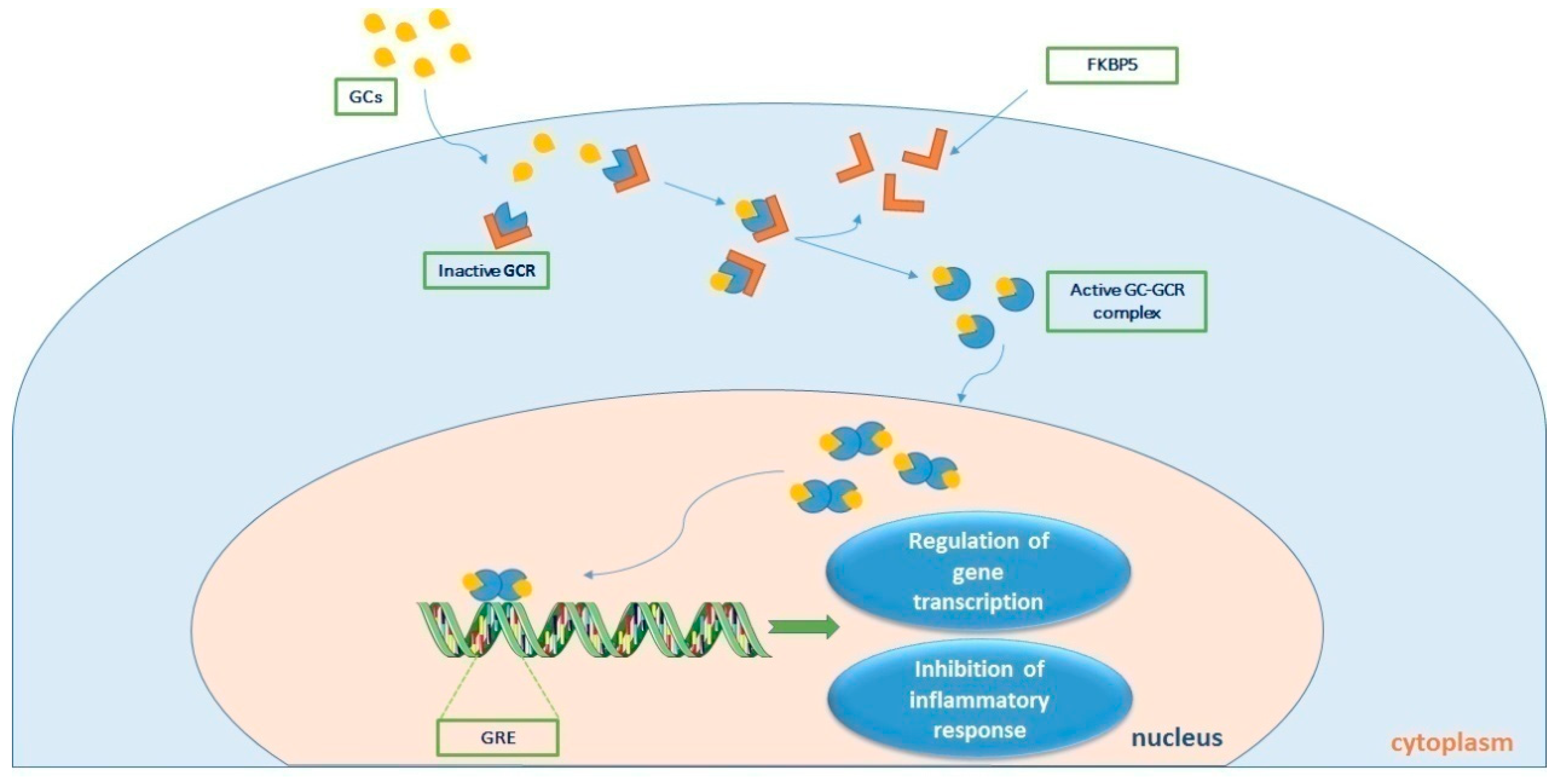
Figure 2.
Τhe process of cortisol signaling via the glucocorticoid receptor (GCR). Upon binding with cortisol, the GCR-cortisol complex is transported from the cytosol to the nucleus. Once in the nucleus, the complex influences transcriptional activation or repression by directly binding to genes containing glucocorticoid (GC) responsive elements (GREs), ultimately leading to the suppression of the inflammatory response. GC-GCR: cortisol-glucocorticoid receptor complex; FKBP5 is a co-chaperone of the GCR.
Figure 2.
Τhe process of cortisol signaling via the glucocorticoid receptor (GCR). Upon binding with cortisol, the GCR-cortisol complex is transported from the cytosol to the nucleus. Once in the nucleus, the complex influences transcriptional activation or repression by directly binding to genes containing glucocorticoid (GC) responsive elements (GREs), ultimately leading to the suppression of the inflammatory response. GC-GCR: cortisol-glucocorticoid receptor complex; FKBP5 is a co-chaperone of the GCR.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



