
Submitted:
22 May 2023
Posted:
23 May 2023
You are already at the latest version
Abstract
Mycobacterium tuberculosis complex causes tuberculosis (TB), a disease that causes pulmonary inflammation but can also affect other tissues. Despite macrophages having a defined role in TB immunopathogenesis, other innate immune cells, such as neutrophils, are involved in this process. These cells have high phagocytic ability and a microbial-killing machine comprised of enzymes, antimicrobial peptides, and reactive oxygen species. In the last two decades, a new neutrophil immune response, the neutrophil extracellular traps (NETs), has been intensely researched. NETs comprise DNA associated with histones, enzymes, and antimicrobial peptides. These structures are related to antimicrobial immune response and some immuno-pathogenesis mechanisms. This mini review highlights the role of NETs in tuberculosis and how they can be helpful as a diagnostic tool and/or therapeutic target.
Keywords:
NETs
; innate immunity
; mycobacteria.
1. Introduction
Tuberculosis (TB) is an infectious-contagious disease mainly affecting the lungs and other tissues [1]. Several factors have limited TB control, including timely diagnosis and treatment, imperfect diagnostic testing, multi-drug resistance, and treatment abandonment [2,3,4,5]. High TB-burden countries use attenuated Mycobacterium bovis strain-based vaccine (Bacille Calmette-Guerin, BCG) to prevent severe TB at birth or among school-age children. However, this vaccine has limited effects on adult pulmonary TB [6]. In 2020, approximately 10 million people had tuberculosis, but 40% of these cases were undiagnosed and reported [7]. This was followed by a rate of 1.5 million death from TB worldwide. WHO’s End TB Strategy proposes targets of a 95% reduction in the absolute number of TB deaths and a 90% reduction in the TB incidence rate by 2035 [8].
The pathogenesis of TB is complex and involves many immune system cells, such as macrophages, lymphocytes, and neutrophils [9,10]. These last cells may have a beneficial and a detrimental role in TB development [11]. Neutrophil functions comprise various antimicrobial peptides besides crosstalk with different immune cells. Additionally, neutrophils can release extracellular traps (i.e., DNA, histones, and various granular and cytoplasmic proteins) that mediate many immune responses [12,13]. This mini review discusses the role of neutrophil extracellular traps (NETs) in tuberculosis and how it could be helpful to understand this disease, bringing insight into developing new treatments/diagnostic tools.
2. Tuberculosis
Tuberculosis (TB) is one of the oldest infectious diseases in the history [14]. It is caused by the Mycobacterium tuberculosis complex, comprising species including M. bovis, BCG strain of M. bovis, M. africanum, M. caprae, M. microti, M. canettii, and M. pinnipedii. Most TB cases are attributed to M. tuberculosis (Mtb), which accounts for 98% of human cases [15,16].
Patients with TB can be classified into two distinct clinical forms: latent TB infection (LTBI) and active TB disease [17]. Those with latent TB infection are asymptomatic and non-transmissible [18]. At the same time, those with active TB disease show general symptoms such as fever, fatigue, lack of appetite, weight loss, back pain, and night sweat. In addition, those with lung disease may have a persistent cough and hemoptysis (coughing up blood) in its advanced form, which includes pulmonary disease that can result in further transmission. Some patients in the active form of the disease may be asymptomatic and are better described as having subclinical tuberculosis. It is estimated that 5 to 10% of people exposed to Mtb develop its active form [16,19,20,21].
Extra-pulmonary TB (EPTB) form is also an important clinical issue, as it can occur in any part of the body following the initial spreading of Mtb to the pulmonary draining lymphatic system [22]. Mtb is taken up by lymphatic vessel non-professional phagocytes such as endothelial cells and fibroblasts, which then spread via lymph to other organ lymphatic systems. Symptomatic EPTB occurs in nearly 10% of infected people. However, pulmonary TB prevails since alveoli and airways are the main gateway of Mtb air-borne infection and its transmission. EPTB can also undergo latency and active disease independent of pulmonary TB. The lesions of TB include the peritoneum, kidneys, bone marrow, spleen, pleural, meningeal, peritoneal membranes, skin, and genito-urinary tract [19,23,24].
At the start of infection, alveolar macrophages internalize Mtb, which provides a cellular environment for the bacteria to replicate intracellularly [25,26]. This safeguards the host from the mycobacteria and permits the infection to establish itself and persist in the latent phase. These bacteria-filled immune cells can be transported through the alveolar barrier to cause widespread dissemination [19]. Before adaptive immune responses are established, the intracellular replication and spread of Mtb to neighboring pulmonary lymph nodes and other extrapulmonary tissues occur through lymphatics and blood circulation. The failure to restrict bacterial replication may lead to the release of Mtb into the extracellular environment; after that, it actively replicates and releases pathogenic proteins and lipids to interact with hosts. In this way, complex host-pathogen interactions determine the course of Mtb infection [19,27].
At the onset of Mtb infection, the secretion of cytokines and chemokines such as interferons, tumor necrosis factor-α (TNF-α), IL-6, IL-12, IL-17, IL-23, CCL2, CCL3, CCL5, CXCL8, and CXCL10, attracts additional phagocytes to the site of infection, which puts immune pressure on the bacteria [28,29]. In addition, they stimulate adaptive immune responses but also cause inflammation and destruction of lung tissue that can be triggered by M1 macrophages responding to Mtb via iNOS-RNS, ROS, TNF-α, and IL-6, associated with TLR 2/4 pathways, including MAPK and Jun activation. The arrival of neutrophils is an initial defense against infection but can also limit immunity against the bacteria and offer a conducive environment for its replication. Innate immunity plays a crucial role in the long-term control of Mtb infection, controlling inflammation and directing adaptive immune responses [15,19,30,31].
Monocytes and neutrophils are attracted to the site of Mtb infection [10,31]. Still, monocyte recruitment may inadvertently promote bacterial dissemination through polarization from M1 to an anti-inflammatory M2 phenotype expressing IL-10, TGF-β1, IL-10, CCL-24, and arginase-1, while inhibiting soluble antibacterial factors with TNF-α, IL-12, IL-6, and NO, favoring bacterial replication. This M2 phenotype is associated with TB, leprosy, and other chronic inflammatory diseases such as asthma and parasitic infections, but also with the resolution of non-infectious inflammatory lesions. Moreover, monocytes can transport Mtb to pulmonary lymph nodes and coordinate with dendritic cells to stimulate CD4+ T cell production after infection. Neutrophil and monocyte recruitment is a hosting strategy to contain bacterial replication, but it is coopted by the bacterium to facilitate its growth and dissemination [9,19,31].
During Mtb infection, dendritic cells (DCs) are essential in priming adaptive immune responses [32]. Professional antigen-presenting cells initiate adaptive immunity by presenting Mtb antigens in the context of major histocompatibility complex (MHC), costimulatory molecules, and cytokines. Depletion of CD11c-expressing cells, a pan-DC marker, after Mtb infection impairs bacterial control and delays the onset of adaptive immunity, highlighting the importance of DCs in mobilizing responses capable of controlling bacterial replication. After infection, DCs migrate to the lung-draining lymph nodes and mature to initiate antigen-specific T cell responses. IL-12 secreted by myeloid cells and essential for IFN-γ induction is required for DC migration during Mtb infection. Antigen-specific T cells can be primed by infected migratory DCs and uninfected resident lymph node DCs. Mtb infection impairs antigen presentation by macrophages and dendritic cells, affecting antigen-specific T cell responses [15,31,32].
The active disease develops in weeks or more extended periods due to Mtb slow replication within macrophages and variation in strain virulence. This triggers macrophage activation and the induction of adaptive immune responses, resulting in the fusion of phagosomes and lysosomes and the secretion of cytokines such as IFN-α, TNF-α, IFN-β, IL-1β, IL-6, and IL-12 [33,34]. While these cytokines aid in the host’s bactericidal response, they may also cause severe lung inflammation and tissue destruction, a sign of active tuberculosis. HIV infection is the main risk factor linked to the progression from latent TB infection to active TB. HIV weakens the immune system by decreasing the numbers of CD4+ T cells and impairing the function of both CD4+ T cells and CD8+ T cells, essential for protection against active TB [9,35,36]. Diabetes Mellitus (DM) patients are more susceptible to TB due to reduced Mtb phagocytosis, altered cytokines, and impaired glucose tolerance. Furthermore, drug–drug interactions between TB and DM treatments can reduce the effectiveness of both treatments and the potential worsening of drug side effects. Consequently, it is essential to establish early diagnosis and treatment for both TB and DM to prevent the increased risk of cardiovascular complications and death [37]. Additional risk factors for reactivation TB include aging, malnutrition, and medical conditions that impede immunity, such as renal failure, cancer, and immunosuppressive therapy [9,35,36].
The development of adaptive immunity against Mtb begins right after the bacteria spreads into the lymph nodes, where granulomas form. This immune response is mediated by T lymphocytes (CD4+ and CD8+) that release cytotoxins and recruit B lymphocytes and plasma cells [38]. CD4+ T cells, especially Th1 cells that produce interferon-γ (IFN-γ), are critical for developing a successful adaptive immunity. MHC class II and I molecules expressed by APCs also play a vital role in the presentation of bacterial antigens to T lymphocytes, allowing them to produce IFN-γ and other cytokines, like interleukin-2 (IL-2) and tumor necrosis factor-alpha (TNF-α). Studies have suggested that reducing CD4+ T cells in HIV-1 infection is connected to a higher bacterial load and a broader spread of tuberculosis during TB-HIV co-infection, emphasizing the importance of T-cell-mediated adaptive immunity in the defense against Mtb [38,39,40].
CD8+ T lymphocytes are critical in the cellular response against mycobacteria, as they can eliminate infected cells or bacilli. Cytotoxic serine proteases, held in cytolytic granules, are a part of the granzyme (Gzms) family and have a significant role in the operations of CD8+ T cells. CD4+ cytotoxic T cells, which secrete GzmB and perforin, have also been identified. Granzyme K, discharged by activated CD8+ T cells, encourages reactive oxygen species (ROS) production in target cells and leads to fast cell death by causing single-stranded nicks of chromosomal DNA and cell membrane damage. In TB patients, CD8+ T cells with GzmK are more abundant and clonally expanded in tuberculous pleural effusion than in peripheral blood. GzmK is associated with inflammation, as it is upregulated during acute and chronic inflammation [41].
One of the hallmarks of TB is granuloma formation [26,42]. Granulomas represent a complex immune response of the host against Mtb infection responsible for tuberculosis. They aim to confine the bacteria to a specific location and prevent dissemination to other body parts. In pulmonary tuberculosis, granulomas comprise macrophages, dendritic cells, neutrophils, fibroblasts, T lymphocytes, and B lymphocytes, which organize into a compact and organized structure. Plasma cells, CD4+, CD8+, and B T lymphocytes are arranged at the periphery, forming a peripheral lymphatic rim in the granuloma that limits mycobacterial replication [9,15,19,31,43]. Adaptive immunity is crucial in Mtb containment in granulomatous latency [19].
3. Neutrophils
Neutrophils comprise a significant portion of the circulating leukocytes in humans, accounting for approximately 50-70%. These cells have a characteristic segmented nucleus, a roughly 7-10 micrometers diameter, and play a critical role, especially in antimicrobial response [44,45,46,47,48,49]. The clinical significance of their abundance lies in predicting severe infections that may follow congenital or acquired conditions causing a decrease in neutrophil count in the bloodstream [50]. These cells were thought to be transcriptionally inactive. However, recent findings led to a paradigm shift, revealing subpopulation heterogeneity and crucial inflammatory and repair functions [51,52].
During development in the bone marrow, myeloid progenitors undergo sequential maturation to generate mature neutrophils that are released to the circulation in a process that gauges the extramedullary pool size. Although the kinetics of circulating neutrophils and their precursors remains somewhat uncertain, there has been increasing evidence about the remarkable plasticity that can be molded by the different tissues where they can be redistributed throughout your lifetime [53,54]. That can be explained by the transcriptional diversity of neutrophils during maturation, known as “neutrotime”, a transcriptional continuum for neutrophils in the bone marrow, blood, and spleen, suggesting a dominant developmental spectrum that underlies neutrophil heterogeneity under normal conditions [55]. Such diversity also includes low-density neutrophils (LDNs), which may manifest immunosuppressive or pro-inflammatory properties. Proinflammatory LDNs are known as low-density granulocytes (LDGs), and immunosuppressive LDNs are known as granulocytic myeloid-derived suppressor cells (G-MDSCs). Additionally, specific tumor-associated neutrophils (TANs) can be further categorized into N1 and N2 subsets, representing functions that are either anti-tumorigenic or pro-tumorigenic, respectively [56,57].
Neutrophil cytoplasm contains granules and secretory vesicles that play a crucial role in their function. As the first line of defense, neutrophils are the initial responders against a wide range of pathogens. These effector functions include phagocytosis with phagolysosome activity, degranulation of antimicrobial molecules and various enzymes involved in inflammation regulation, formation of neutrophil extracellular traps (NETs), and playing a role in tissue repair and wound healing by releasing growth factors [12,58,59].
The development of inflammation can occur in response to both infectious and sterile injuries, with PAMPs and DAMPs triggering similar pro-inflammatory responses through the activation of PRRs [60,61,62]. Neutrophils are recruited to both types of inflammation, but their recruitment and subsequent pro-inflammatory responses can be detrimental in sterile injuries such as obesity-associated inflammation [63]. The ability of neutrophils to distinguish between infectious and sterile inflammation is currently unknown, as both types of inflammation can activate similar receptors and intracellular pathways, leading to similar outcomes. However, different neutrophil types (pro-inflammatory versus pro-resolving) may be recruited depending on the type of inflammation and the specific ligands and cells involved [56].
Neutrophil recruitment to inflamed tissues is a multi-step process involving upregulating adhesion molecules on endothelial cells, allowing for neutrophil tethering, rolling, and eventually firm adhesion [12,64]. The interaction between adhesion molecules and chemokine receptors on neutrophils and their ligands on endothelial cells mediates this process. Neutrophil activation and chemotaxis toward the site of inflammation are also critical steps in this process [12,63]. Once adhered, neutrophils may display probing behavior before crawling toward endothelial cell junctions to transmigrate across the endothelial layer. The transmigration process can occur either paracellularly or transcellularly, with the latter being mediated by specialized structures called transmigratory cups. While the mechanisms of neutrophil migration through the endothelial basement membrane are not fully understood, it is suggested that neutrophils preferentially migrate through regions with low levels of extracellular matrix molecules and gaps between pericyte regions [65,66].
Neutrophils, traditionally viewed as pro-inflammatory cells, have been shown to exhibit anti-inflammatory and healing characteristics [63]. In addition, they aid in removing dead cells and bacteria, perform wound debridement, and express proteases that benefit tissue repair. Neutrophils also facilitate monocyte recruitment and promote their removal from the tissue, which contributes to the resolution of inflammation and promotes healing and tissue repair. Additionally, it is seen that neutrophils release growth factors that promote angiogenesis, which can be exploited by cancer cells to promote their growth and spread [67].
In TB immunopathogenesis, neutrophils have a dual role [11,68,69]. High levels of these cells can be found in patients with active TB in the bloodstream and bronchoalveolar fluid [70,71,72]. Additionally, these cells are involved in central nervous system tuberculosis development [73]. Furthermore, neutrophils can phagocyte mycobacteria, and their antimicrobial machinery can display some antimycobacterial activities [44]. Further, it has been demonstrated that alveolar mucosa increases the capacity of the neutrophil to recognize and kill Mtb [74]. However, the dysregulated granulocytic influx is related to disease progression [75], and massive neutrophilic infiltration has been found in mouse models of TB [76]. Thus, neutrophil is also implicated in the exacerbation of lung tissue damage. In the following section, we describe the role of neutrophil extracellular traps in tuberculosis.
4. Neutrophil extracellular trap in tuberculosis
Neutrophil extracellular traps (NETs) are web-like structures composed of chromatin and antimicrobial proteins released by neutrophils. The primary function of NETs is to capture and kill microorganisms [77]. The DNA strands within NETs act as a physical barrier, immobilizing bacteria and fungi. At the same time, the antimicrobial proteins associated with the NETs, such as histones, neutrophil elastase, and myeloperoxidase, possess potent microbicidal properties that can directly kill pathogens [78]. Moreover, NETs also play a role in modulating immune responses. They can activate other immune cells, such as macrophages and dendritic cells, to enhance their antimicrobial activities. Additionally, NETs can interact with immune system components, including antibodies and complement proteins, facilitating the recognition and clearance of pathogens [79]. While NETs are crucial for host defense, their dysregulation has been associated with various inflammatory and autoimmune diseases. Excessive or prolonged NET formation can damage tissue, as NETs contain toxic components that can harm healthy cells and contribute to chronic inflammation [78,79,80,81].
Although NETs have infection control actions [82], the role of these neutrophil traps is still controversial and being investigated in diseases caused by mycobacteria [83]. Initially, it was only known that NETs could capture microorganisms. However, it is now recognized that such structures exert a direct microbicidal action against infectious agents [84]. Studies have already demonstrated the microbicidal activity of NETs against various pathogens, including bacteria, fungi, protozoa, and viruses [77,85,86,87,88]. However, other studies reveal that these extracellular structures only can capture but not kill Mtb, something already observed in vitro and in vivo, which may be related to the complex structure of its cell wall and its multiple detoxification mechanisms [89,90,91,92,93].
A current and relevant question regarding the role of NETs in tuberculosis is whether their release can be more beneficial or harmful to the host. This is because Mtb and BCG can mediate the release of NETs stimulation, and excessive formation of these structures can cause tissue damage [89,92,94]. Different mechanisms of ‘NETosis’ induced by Mtb and associated tissue damage have already been reported. Dang et al. [83] observed that the extracellular sphingomyelinase Rv0888 of Mtb in recombinant M. smegmatis could act as a virulence factor, inducing the release of NETs with associated myeloperoxidase (MPO), which leads to the aggravation of lung lesions through apoptosis via caspase-3. Additionally, extracellular traps induced by Mtb stimulate macrophages to release IL-6, TNF-α, and IL-1β, inflammatory cytokines that can recruit more neutrophils to the site of infection, exacerbating the inflammatory response and lung injury [95]. In addition, NETs associated with the NF-kB-dependent matrix metalloproteinase-8 (MMP-8) enzyme leading to collagen degradation in the lung tissue of humans with TB, and increased levels of NETs in patients’ sputum have been observed [96]. Teixeira et al. [97] also observed that NETosis could be induced by type 1 IFN through the blocking of GM-CSF, which leads to the exacerbated formation of NETs and consequent growth of mycobacteria and worsening of pulmonary pathology, reinforcing the evidence that neutrophil traps when not controlled, contribute to the maintenance of pulmonary immunopathology in tuberculosis. Moreover, Su et al. [98] demonstrated that the induction of low-density granulocyte generation occurs during mycobacterium tuberculosis infection, facilitated by the promotion of neutrophil extracellular trap formation through the ROS pathway.
Another mechanism capable of inducing NETosis is the release of ESAT-6 by the mycobacterial ESX-1 system [99]. Francis et al. [99] demonstrated that the ESAT-6 protein acts as a leukocidin, causing neutrophil necrosis through a high influx of Ca2+ that leads to NET release, correlating the ESX-1 system as one of the virulence factors of Mtb. Moreover, the release of these NETs not only removes the threat of neutrophils and creates a necrotic environment rich in nutrients and conducive to extracellular bacillary growth. Therefore, in addition to aggravating tissue damage, NETs, when released excessively, also contribute in other ways to the progression of the pathology, and Mtb seems to know how to exploit this well.
On the other hand, one of the characteristics of active pulmonary tuberculosis is the presence of necrotic cores containing leukocytes and extracellular bacteria, which can undergo cavitation. Therefore, these lesions are associated with the potential for airborne transmission of the disease [99,100]. In addition, these granulomas are hypoxic, leading to decreased formation of NETs, probably caused by the decrease of reactive oxygen species (ROS) [101]. However, the consequences of this decrease are still unclear.
Furthermore, studying markers that help indicate tuberculosis’s stages is paramount for its treatment. In this regard, Schechter et al. [102] observed that the release of NETs in TB could be identified through blood plasma by detecting extracellular DNA, human neutrophil elastase (HNE), and MPO. In addition, treatment with antibiotics decreased plasma levels of NETs. Similarly, De Melo et al. [103] reported higher levels of citrullinated histone H3, a marker of NETs, in the peripheral blood of patients with pulmonary tuberculosis and lung tissue damage. Additionally, Meier et al. [104] also observed increased levels of NETs in the total blood of tuberculosis progressors up to 6 months before the diagnosis of the disease, as well as their additional activation in tuberculosis patients. Such evidence suggests the detection of NETs could be an essential biomarkers in tuberculosis [72]. Furthermore, it again brings to light the formation of NETs associated with the immunopathology of the disease.
As previously mentioned, Mtb can induce the release of NETs but is not killed by these traps, which is interesting considering that NETs can lead to the death of other microorganisms. However, the mechanisms by which the mycobacterium evades extracellular traps have yet to be fully elucidated. Nevertheless, it is known that many bacterial species have developed ways to escape death by NETs. For example, Streptococcus pyogenes, Staphylococcus aureus, and S. pneumoniae produce DNases, which are seen as virulence factors for impairing NETosis [105,106,107]. Additionally, S. pneumoniae has a polysaccharide capsule that reduces the binding of NETs and modifies the lipoteichoic acids of its cell wall, altering its charge and disrupting its affinity with antimicrobial factors[107]. With the same goal, Group A Streptococcus (GAS) produces the M1 protein, inhibiting the action of the broad-spectrum antibacterial cathelicidin LL-37. Thus, inadequate LL-37 activity helps the survival of GAS within NETs [108]. Furthermore, it was demonstrated that LL37 complexed with DNA are internalized by macrophages after neutrophils release and then inhibit mycobacteria growth intracellularly [94]. Thus, a evade this mechanism seems a plausible way to mycobacteria escape NETs antimicrobial activity.
Furthermore, bacteria can escape from NETs through other mechanisms, such as releasing peptidases, forming biofilm, and inhibiting ROS production [106]. Concerning Mtb, to what extent this mycobacterium exploits these escape mechanisms has yet to be fully clear. Still, it is known that mycobacteria have a complex cell wall structure with a high lipid content, which hinders the penetration of antibacterial agents [109]. Additionally, Mtb is also sensitive to the action of LL-37, and here, the inhibition of this peptide by the pathogen is also something that can be investigated. Likewise, its detoxifying mechanisms should be better studied to bring to light, in greater completeness, the factors that contribute to the survival of Mtb in NETs, which would aid in discovering new therapeutic targets for tuberculosis.
5. Conclusions
The role of neutrophil extracellular traps (NETs) in diseases caused by mycobacteria, particularly tuberculosis, is still controversial and under investigation. While studies have shown the microbicidal activity of NETs against various pathogens, including bacteria, fungi, protozoa, and viruses, some research suggests that NETs can capture but not kill M. tuberculosis, possibly due to the complex structure of its cell wall and evasion mechanisms. Also, excessive formation of NETs in TB can lead to tissue damage and exacerbate the inflammatory response and lung injury (Figure 1). However, NETs levels in the serum plasm of TB patients could be explored more to find a new way to detect the disease progression. Understanding the interaction between M. tuberculosis and NETs could lead to identifying new therapeutic targets for TB.
Author Contributions
Conceptualization, L.H.A.C.-S.; writing—original draft preparation, L.H.A.C.-S., F.S.A., A.G.A., F.C.C. and S.E.R.V.; writing—review and editing, T.S.L.K.; and supervision, T.S.L.K. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by fellowships “Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES) (Number: 88887.704195/2022-00; 88887.717944/2022-00)” and “Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) (Number: 440939/2020-8)”.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Garrido-Cardenas, J.A.; de Lamo-Sevilla, C.; Cabezas-Fernández, M.T.; Manzano-Agugliaro, F.; Martínez-Lirola, M. Global tuberculosis research and its future prospects. Tuberculosis (Edinb). 2020, 121. [Google Scholar] [CrossRef] [PubMed]
- Nogueira, B.M.F.; Krishnan, S.; Barreto-Duarte, B.; Araújo-Pereira, M.; Queiroz, A.T.L.; Ellner, J.J.; Salgame, P.; Scriba, T.J.; Sterling, T.R.; Gupta, A.; et al. Diagnostic biomarkers for active tuberculosis: progress and challenges. EMBO Mol. Med. 2022, 14. [Google Scholar] [CrossRef] [PubMed]
- Dicks, K. V.; Stout, J.E. Molecular Diagnostics for Mycobacterium tuberculosis Infection. Annu. Rev. Med. 2019, 70, 77–90. [Google Scholar] [CrossRef] [PubMed]
- Lucena, L.A. de; Dantas, G.B. da S.; Carneiro, T.V.; Lacerda, H.G. Factors Associated with the Abandonment of Tuberculosis Treatment in Brazil: A Systematic Review. Rev. Soc. Bras. Med. Trop. 2023, 56. [Google Scholar] [CrossRef] [PubMed]
- Yadav, P. Challenges & Solutions for Recent Advancements in Multi-Drugs Resistance Tuberculosis: A Review. Microbiol. insights 2023, 16, 117863612311524. [Google Scholar] [CrossRef]
- Singh, S.; Saavedra-Avila, N.A.; Tiwari, S.; Porcelli, S.A. A century of BCG vaccination: Immune mechanisms, animal models, non-traditional routes and implications for COVID-19. Front. Immunol. 2022, 13. [Google Scholar] [CrossRef]
- Pai, M.; Kasaeva, T.; Swaminathan, S. Covid-19’s Devastating Effect on Tuberculosis Care - A Path to Recovery. N. Engl. J. Med. 2022, 386, 1490–1493. [Google Scholar] [CrossRef]
- World Health Organization (WHO) Global Tuberculosis Report 2022. Geneva World Heal. Organ. 2022.
- Kanabalan, R.D.; Lee, L.J.; Lee, T.Y.; Chong, P.P.; Hassan, L.; Ismail, R.; Chin, V.K. Human tuberculosis and Mycobacterium tuberculosis complex: A review on genetic diversity, pathogenesis and omics approaches in host biomarkers discovery. Microbiol. Res. 2021, 246. [Google Scholar] [CrossRef]
- Scriba, T.J.; Coussens, A.K.; Fletcher, H.A. Human Immunology of Tuberculosis. Microbiol. Spectr. 2016, 4. [Google Scholar] [CrossRef]
- Borkute, R.R.; Woelke, S.; Pei, G.; Dorhoi, A. Neutrophils in Tuberculosis: Cell Biology, Cellular Networking and Multitasking in Host Defense. Int. J. Mol. Sci. 2021, 22. [Google Scholar] [CrossRef]
- Liew, P.X.; Kubes, P. The Neutrophil’s Role During Health and Disease. Physiol. Rev. 2019, 99, 1223–1248. [Google Scholar] [CrossRef] [PubMed]
- Burn, G.L.; Foti, A.; Marsman, G.; Patel, D.F.; Zychlinsky, A. The Neutrophil. Immunity 2021, 54, 1377–1391. [Google Scholar] [CrossRef] [PubMed]
- Loddenkemper, R.; Murray, J.F. History of Tuberculosis. In Essential Tuberculosis; Springer, Cham: Cham, 2021; pp. 3–9. [Google Scholar]
- Guerra-De-Blas, P.D.C.; Torres-González, P.; Bobadilla-Del-Valle, M.; Sada-Ovalle, I.; Ponce-De-León-Garduño, A.; Sifuentes-Osornio, J. Potential Effect of Statins on Mycobacterium tuberculosis Infection. J. Immunol. Res. 2018, 2018. [Google Scholar] [CrossRef] [PubMed]
- Pai, M.; Behr, M.A.; Dowdy, D.; Dheda, K.; Divangahi, M.; Boehme, C.C.; Ginsberg, A.; Swaminathan, S.; Spigelman, M.; Getahun, H.; et al. Tuberculosis. Nat. Rev. Dis. Prim. 2016, 2. [Google Scholar] [CrossRef] [PubMed]
- Dowdy, D.W.; Raviglione, M.C. Basic and Descriptive Epidemiology of Tuberculosis. In Essential Tuberculosis; Springer, Cham, 2021; pp. 29–36.
- Campaniço, A.; Harjivan, S.G.; Warner, D.F.; Moreira, R.; Lopes, F. Addressing Latent Tuberculosis: New Advances in Mimicking the Disease, Discovering Key Targets, and Designing Hit Compounds. Int. J. Mol. Sci. 2020, Vol. 21, Page 8854 2020, 21, 8854. [Google Scholar] [CrossRef]
- Ferluga, J.; Yasmin, H.; Al-Ahdal, M.N.; Bhakta, S.; Kishore, U. Natural and trained innate immunity against Mycobacterium tuberculosis. Immunobiology 2020, 225. [Google Scholar] [CrossRef] [PubMed]
- Simmons, J.D.; Stein, C.M.; Seshadri, C.; Campo, M.; Alter, G.; Fortune, S.; Schurr, E.; Wallis, R.S.; Churchyard, G.; Mayanja-Kizza, H.; et al. Immunological mechanisms of human resistance to persistent Mycobacterium tuberculosis infection. Nat. Rev. Immunol. 2018, 18, 575–589. [Google Scholar] [CrossRef]
- Warner, D.F.; Koch, A.; Mizrahi, V. Diversity and disease pathogenesis in Mycobacterium tuberculosis. Trends Microbiol. 2015, 23, 14–21. [Google Scholar] [CrossRef]
- Bruchfeld, J.; Forsman, L.D.; Fröberg, G.; Niward, K. Extrapulmonary Tuberculosis. In Essential Tuberculosis; Springer, Cham: Cham, 2021; pp. 259–266. [Google Scholar]
- Rodriguez-Takeuchi, S.Y.; Renjifo, M.E.; Medina, F.J. Extrapulmonary Tuberculosis: Pathophysiology and Imaging Findings. Radiographics 2019, 39, 2023–2037. [Google Scholar] [CrossRef]
- Wilkinson, R.J.; Rohlwink, U.; Misra, U.K.; Van Crevel, R.; Mai, N.T.H.; Dooley, K.E.; Caws, M.; Figaji, A.; Savic, R.; Solomons, R.; et al. Tuberculous meningitis. Nat. Rev. Neurol. 2017 1310 2017, 13, 581–598. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, S. Pathogenesis, immunology, and diagnosis of latent Mycobacterium tuberculosis infection. Clin. Dev. Immunol. 2011, 2011. [Google Scholar] [CrossRef] [PubMed]
- Jellinger, K.A.; Bustin, S.; Alsayed, S.S.R.; Gunosewoyo, H. Tuberculosis: Pathogenesis, Current Treatment Regimens and New Drug Targets. Int. J. Mol. Sci. 2023, Vol. 24, Page 5202 2023, 24, 5202. [Google Scholar] [CrossRef]
- Palucci, I.; Delogu, G. Host Directed Therapies for Tuberculosis: Futures Strategies for an Ancient Disease. Chemotherapy 2018, 63, 172–180. [Google Scholar] [CrossRef] [PubMed]
- Domingo-Gonzalez, R.; Prince, O.; Cooper, A.; Khader, S.A. Cytokines and Chemokines in Mycobacterium tuberculosis Infection. Microbiol. Spectr. 2016. [Google Scholar] [CrossRef] [PubMed]
- Mundra, A.; Yegiazaryan, A.; Karsian, H.; Alsaigh, D.; Bonavida, V.; Frame, M.; May, N.; Gargaloyan, A.; Abnousian, A.; Venketaraman, V. Pathogenicity of Type I Interferons in Mycobacterium tuberculosis. Int. J. Mol. Sci. 2023, 24. [Google Scholar] [CrossRef] [PubMed]
- Ernst, J.D. Mechanisms of M. tuberculosis Immune Evasion as Challenges to TB Vaccine Design. Cell Host Microbe 2018, 24, 34–42. [Google Scholar] [CrossRef]
- Sia, J.K.; Rengarajan, J. Immunology of Mycobacterium tuberculosis Infections. Microbiol. Spectr. 2019, 7. [Google Scholar] [CrossRef]
- Kim, H.; Shin, S.J. Pathological and protective roles of dendritic cells in Mycobacterium tuberculosis infection: Interaction between host immune responses and pathogen evasion. Front. Cell. Infect. Microbiol. 2022, 12, 891878. [Google Scholar] [CrossRef]
- Zumla, A.; Rao, M.; Parida, S.K.; Keshavjee, S.; Cassell, G.; Wallis, R.; Axelsson-Robertsson, R.; Doherty, M.; Andersson, J.; Maeurer, M.J. Inflammation and tuberculosis: host-directed therapies. J. Intern. Med. 2015, 277, 373–387. [Google Scholar] [CrossRef]
- Domingo-Gonzalez, R.; Prince, O.; Cooper, A.; Khader, S.A. Cytokines and Chemokines in Mycobacterium tuberculosis Infection. Microbiol. Spectr. 2016, 4. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P. Adult pulmonary tuberculosis as a pathological manifestation of hyperactive antimycobacterial immune response. Clin. Transl. Med. 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Bell, L.C.K.; Noursadeghi, M. Pathogenesis of HIV-1 and Mycobacterium tuberculosis co-infection. Nat. Rev. Microbiol. 2018, 16, 80–90. [Google Scholar] [CrossRef]
- Ssekamatte, P.; Sande, O.J.; van Crevel, R.; Biraro, I.A. Immunologic, metabolic and genetic impact of diabetes on tuberculosis susceptibility. Front. Immunol. 2023, 14. [Google Scholar] [CrossRef] [PubMed]
- Jasenosky, L.D.; Scriba, T.J.; Hanekom, W.A.; Goldfeld, A.E. T cells and adaptive immunity to Mycobacterium tuberculosis in humans. Immunol. Rev. 2015, 264, 74–87. [Google Scholar] [CrossRef] [PubMed]
- Chai, Q.; Lu, Z.; Liu, C.H. Host defense mechanisms against Mycobacterium tuberculosis. Cell. Mol. Life Sci. 2020, 77, 1859–1878. [Google Scholar] [CrossRef]
- Urdahl, K.B. Understanding and overcoming the barriers to T cell-mediated immunity against tuberculosis. Semin. Immunol. 2014, 26, 578–587. [Google Scholar] [CrossRef]
- Jiang, J.; Cao, Z.; Xiao, L.; Su, J.; Wang, J.; Liang, J.; Yang, B.; Liu, Y.; Zhai, F.; Wang, R.; et al. Single-cell profiling identifies T cell subsets associated with control of tuberculosis dissemination. Clin. Immunol. 2023, 248. [Google Scholar] [CrossRef]
- Ehlers, S.; Schaible, U.E. The granuloma in tuberculosis: Dynamics of a host-pathogen collusion. Front. Immunol. 2012, 3, 411. [Google Scholar] [CrossRef]
- Gideon, H.P.; Hughes, T.K.; Tzouanas, C.N.; Wadsworth, M.H.; Tu, A.A.; Gierahn, T.M.; Peters, J.M.; Hopkins, F.F.; Wei, J.R.; Kummerlowe, C.; et al. Multimodal profiling of lung granulomas in macaques reveals cellular correlates of tuberculosis control. Immunity 2022, 55, 827–846. [Google Scholar] [CrossRef]
- Parker, H.A.; Forrester, L.; Kaldor, C.D.; Dickerhof, N.; Hampton, M.B. Antimicrobial Activity of Neutrophils Against Mycobacteria. Front. Immunol. 2021, 12. [Google Scholar] [CrossRef] [PubMed]
- Naumenko, V.; Turk, M.; Jenne, C.N.; Kim, S.J. Neutrophils in viral infection. Cell Tissue Res. 2018, 371, 505–516. [Google Scholar] [CrossRef] [PubMed]
- Cavalcante-Silva, L.H.A.; Carvalho, D.C.M.; Lima, É. de A.; Galvão, J.G.F.M.; da Silva, J.S. d. F.; Sales-Neto, J.M. de; Rodrigues-Mascarenhas, S. Neutrophils and COVID-19: The road so far. Int. Immunopharmacol. 2021, 90. [Google Scholar] [CrossRef]
- Rosales, C. Neutrophil: A Cell with Many Roles in Inflammation or Several Cell Types? Front. Physiol. 2018, 9, 113. [Google Scholar] [CrossRef]
- Galani, I.E.; Andreakos, E. Neutrophils in viral infections: Current concepts and caveats. J. Leukoc. Biol. 2015, 98, 557–564. [Google Scholar] [CrossRef] [PubMed]
- Urban, C.F.; Backman, E. Eradicating, retaining, balancing, swarming, shuttling and dumping: a myriad of tasks for neutrophils during fungal infection. Curr. Opin. Microbiol. 2020, 58, 106–115. [Google Scholar] [CrossRef] [PubMed]
- Newburger, P.E. Autoimmune and other acquired neutropenias. Hematol. Am. Soc. Hematol. Educ. Progr. 2016, 2016, 38–42. [Google Scholar] [CrossRef]
- Kumar, S.; Dikshit, M. Metabolic Insight of Neutrophils in Health and Disease. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef]
- Shim, H.B.; Deniset, J.F.; Kubes, P. Neutrophils in homeostasis and tissue repair. Int. Immunol. 2022, 34, 399–407. [Google Scholar] [CrossRef]
- Palomino-Segura, M.; Sicilia, J.; Ballesteros, I.; Hidalgo, A. Strategies of neutrophil diversification. Nat. Immunol. 2023, 24, 575–584. [Google Scholar] [CrossRef]
- Hidalgo, A.; Chilvers, E.R.; Summers, C.; Koenderman, L. The Neutrophil Life Cycle. Trends Immunol. 2019, 40, 584–597. [Google Scholar] [CrossRef]
- Grieshaber-Bouyer, R.; Radtke, F.A.; Cunin, P.; Stifano, G.; Levescot, A.; Vijaykumar, B.; Nelson-Maney, N.; Blaustein, R.B.; Monach, P.A.; Nigrovic, P.A.; et al. The neutrotime transcriptional signature defines a single continuum of neutrophils across biological compartments. Nat. Commun. 2021, 12. [Google Scholar] [CrossRef] [PubMed]
- Ley, K.; Hoffman, H.M.; Kubes, P.; Cassatella, M.A.; Zychlinsky, A.; Hedrick, C.C.; Catz, S.D. Neutrophils: New insights and open questions. Sci. Immunol. 2018, 3, eaat4579. [Google Scholar] [CrossRef] [PubMed]
- Deniset, J.F.; Kubes, P. Neutrophil heterogeneity: Bona fide subsets or polarization states? J. Leukoc. Biol. 2018, 103, 829–838. [Google Scholar] [CrossRef]
- Cassatella, M.A.; Östberg, N.K.; Tamassia, N.; Soehnlein, O. Biological Roles of Neutrophil-Derived Granule Proteins and Cytokines. Trends Immunol. 2019, 40, 648–664. [Google Scholar] [CrossRef] [PubMed]
- Phillipson, M.; Kubes, P. The Healing Power of Neutrophils. Trends Immunol. 2019, 40, 635–647. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Akira, S. The role of pattern-recognition receptors in innate immunity: Update on toll-like receptors. Nat. Immunol. 2010, 11, 373–384. [Google Scholar] [CrossRef]
- Netea, M.G.; Balkwill, F.; Chonchol, M.; Cominelli, F.; Donath, M.Y.; Giamarellos-Bourboulis, E.J.; Golenbock, D.; Gresnigt, M.S.; Heneka, M.T.; Hoffman, H.M.; et al. A guiding map for inflammation. Nat. Immunol. 2017, 18, 826–831. [Google Scholar] [CrossRef]
- Medzhitov, R. Inflammation 2010: New Adventures of an Old Flame. Cell 2010, 140, 771–776. [Google Scholar] [CrossRef]
- Kolaczkowska, E.; Kubes, P. Neutrophil recruitment and function in health and inflammation. Nat. Rev. Immunol. 2013, 13, 159–175. [Google Scholar] [CrossRef]
- Chatfield, S.M.; Thieblemont, N.; Witko-Sarsat, V. Expanding Neutrophil Horizons: New Concepts in Inflammation. J. Innate Immun. 2018, 10, 422–431. [Google Scholar] [CrossRef] [PubMed]
- Muller, W.A. Mechanisms of Leukocyte Transendothelial Migration. Annu. Rev. Pathol. Mech. Dis. 2011, 6, 323–344. [Google Scholar] [CrossRef] [PubMed]
- Hyun, Y.; Hong, C. Deep insight into neutrophil trafficking in various organs. J. Leukoc. Biol. 2017, 102, 617–629. [Google Scholar] [CrossRef] [PubMed]
- Powell, D.R.; Huttenlocher, A. Neutrophils in the Tumor Microenvironment. Trends Immunol. 2016, 37, 41–52. [Google Scholar] [CrossRef]
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.M.; Wang, W.; Song, Z.G.; Hu, Y.; Tao, Z.W.; Tian, J.H.; Pei, Y.Y.; et al. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef]
- Hilda, J.N.; Das, S.; Tripathy, S.P.; Hanna, L.E. Role of neutrophils in tuberculosis: A bird’s eye view. Innate Immun. 2020, 26, 240–247. [Google Scholar] [CrossRef]
- Panteleev, A. V.; Nikitina, I.Y.; Burmistrova, I.A.; Kosmiadi, G.A.; Radaeva, T. V.; Amansahedov, R.B.; Sadikov, P. V.; Serdyuk, Y. V.; Larionova, E.E.; Bagdasarian, T.R.; et al. severe tuberculosis in humans correlates best with neutrophil abundance and lymphocyte deficiency and does not correlate with antigen-specific CD4 T-cell response. Front. Immunol. 2017, 8, 963. [Google Scholar] [CrossRef]
- Lowe, D.M.; Bandara, A.K.; Packe, G.E.; Barker, R.D.; Wilkinson, R.J.; Griffiths, C.J.; Martineau, A.R. Neutrophilia independently predicts death in tuberculosis. Eur. Respir. J. 2013, 42, 1752–1757. [Google Scholar] [CrossRef]
- van der Meer, A.J.; Zeerleder, S.; Blok, D.C.; Kager, L.M.; Lede, I.O.; Rahman, W.; Afroz, R.; Ghose, A.; Visser, C.E.; Zahed, A.S.M.; et al. Neutrophil extracellular traps in patients with pulmonary tuberculosis. Respir. Res. 2017, 18. [Google Scholar] [CrossRef]
- Poh, X.Y.; Loh, F.K.; Friedland, J.S.; Ong, C.W.M. Neutrophil-Mediated Immunopathology and Matrix Metalloproteinases in Central Nervous System - Tuberculosis. Front. Immunol. 2022, 12. [Google Scholar] [CrossRef]
- Arcos, J.; Diangelo, L.E.; Scordo, J.M.; Sasindran, S.J.; Moliva, J.I.; Turner, J.; Torrelles, J.B. Lung Mucosa Lining Fluid Modification of Mycobacterium tuberculosis to Reprogram Human Neutrophil Killing Mechanisms. J. Infect. Dis. 2015, 212, 948–958. [Google Scholar] [CrossRef] [PubMed]
- Gopal, R.; Monin, L.; Torres, D.; Slight, S.; Mehra, S.; McKenna, K.C.; Junecko, B.A.F.; Reinhart, T.A.; Kolls, J.; Báez-Saldańa, R.; et al. S100A8/A9 proteins mediate neutrophilic inflammation and lung pathology during tuberculosis. Am. J. Respir. Crit. Care Med. 2013, 188, 1137–1146. [Google Scholar] [CrossRef] [PubMed]
- Lovewell, R.R.; Baer, C.E.; Mishra, B.B.; Smith, C.M.; Sassetti, C.M. Granulocytes act as a niche for Mycobacterium tuberculosis growth. Mucosal Immunol. 2020 141 2020, 14, 229–241. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil Extracellular Traps Kill Bacteria. Science (80-.). 2004, 303, 1532–1535. [Google Scholar] [CrossRef] [PubMed]
- Papayannopoulos, V. Neutrophil extracellular traps in immunity and disease. Nat. Rev. Immunol. 2018, 18, 134–147. [Google Scholar] [CrossRef]
- Brinkmann, V. Neutrophil Extracellular Traps in the Second Decade. J. Innate Immun. 2018, 10, 414–421. [Google Scholar] [CrossRef]
- Narasaraju, T.; Tang, B.M.; Herrmann, M.; Muller, S.; Chow, V.T.K.; Radic, M. Neutrophilia and NETopathy as Key Pathologic Drivers of Progressive Lung Impairment in Patients With COVID-19. Front. Pharmacol. 2020, 11. [Google Scholar] [CrossRef]
- Hahn, J.; Knopf, J.; Maueröder, C.; Kienhöfer, D.; Leppkes, M.; Herrmann, M. Neutrophils and neutrophil extracellular traps orchestrate initiation and resolution of inflammation. Clin. Exp. Rheumatol. 2016, 34, 6–8. [Google Scholar]
- Porto, B.N.; Stein, R.T. Neutrophil Extracellular Traps in Pulmonary Diseases: Too Much of a Good Thing? Front. Immunol. 2016, 7. [Google Scholar] [CrossRef]
- Dang, G.; Cui, Y.; Wang, L.; Li, T.; Cui, Z.; Song, N.; Chen, L.; Pang, H.; Liu, S. Extracellular Sphingomyelinase Rv0888 of Mycobacterium tuberculosis Contributes to Pathological Lung Injury of Mycobacterium smegmatis in Mice via Inducing Formation of Neutrophil Extracellular Traps. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef]
- Yipp, B.G.; Kubes, P. NETosis: how vital is it? Blood 2013, 122, 2784–2794. [Google Scholar] [CrossRef] [PubMed]
- Delgado-Rizo, V.; Martínez-Guzmán, M.A.; Iñiguez-Gutierrez, L.; García-Orozco, A.; Alvarado-Navarro, A.; Fafutis-Morris, M. Neutrophil Extracellular Traps and Its Implications in Inflammation: An Overview. Front. Immunol. 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Hiroki, C.H.; Toller-Kawahisa, J.E.; Fumagalli, M.J.; Colon, D.F.; Figueiredo, L.T.M.; Fonseca, B.A.L.D.; Franca, R.F.O.; Cunha, F.Q. Neutrophil Extracellular Traps Effectively Control Acute Chikungunya Virus Infection. Front. Immunol. 2020, 10. [Google Scholar] [CrossRef] [PubMed]
- Urban, C.F.; Nett, J.E. Neutrophil extracellular traps in fungal infection. Semin. Cell Dev. Biol. 2019, 89, 47–57. [Google Scholar] [CrossRef] [PubMed]
- Guimarães-Costa, A.B.; Nascimento, M.T.C.; Froment, G.S.; Soares, R.P.P.; Morgado, F.N.; Conceição-Silva, F.; Saraiva, E.M. Leishmania amazonensis promastigotes induce and are killed by neutrophil extracellular traps. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 6748–6753. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Kichik, V.; Mondragón-Flores, R.; Mondragón-Castelán, M.; Gonzalez-Pozos, S.; Muñiz-Hernandez, S.; Rojas-Espinosa, O.; Chacón-Salinas, R.; Estrada-Parra, S.; Estrada-García, I. Neutrophil extracellular traps are induced by Mycobacterium tuberculosis. Tuberculosis (Edinb). 2009, 89, 29–37. [Google Scholar] [CrossRef]
- Filio-Rodríguez, G.; Estrada-García, I.; Arce-Paredes, P.; Moreno-Altamirano, M.M.; Islas-Trujillo, S.; Ponce-Regalado, M.D.; Rojas-Espinosa, O. In vivo induction of neutrophil extracellular traps by Mycobacterium tuberculosis in a guinea pig model. Innate Immun. 2017, 23, 625–637. [Google Scholar] [CrossRef]
- Hahn, S.; Giaglis, S.; Chowdury, C.S.; Hösli, I.; Hasler, P. Modulation of neutrophil NETosis: interplay between infectious agents and underlying host physiology. Semin. Immunopathol. 2013, 35, 439–453. [Google Scholar] [CrossRef]
- Nakamura, K.; Nakayama, H.; Sasaki, S.; Takahashi, K.; Iwabuchi, K. Mycobacterium avium-intracellulare complex promote release of pro-inflammatory enzymes matrix metalloproteinases by inducing neutrophil extracellular trap formation. Sci. Rep. 2022, 12. [Google Scholar] [CrossRef]
- Mendoza-Aguilar, M.D.; Arce-Paredes, P.; Aquino-Vega, M.; Rodríguez-Martínez, S.; Rojas-Espinosa, O. Fate of Mycobacterium tuberculosis in peroxidase-loaded resting murine macrophages. Int. J. mycobacteriology 2013, 2, 3–13. [Google Scholar] [CrossRef]
- Stephan, A.; Batinica, M.; Steiger, J.; Hartmann, P.; Zaucke, F.; Bloch, W.; Fabri, M. LL37:DNA complexes provide antimicrobial activity against intracellular bacteria in human macrophages. Immunology 2016, 148, 420–432. [Google Scholar] [CrossRef] [PubMed]
- Braian, C.; Hogea, V.; Stendahl, O. Mycobacterium tuberculosis- induced neutrophil extracellular traps activate human macrophages. J. Innate Immun. 2013, 5, 591–602. [Google Scholar] [CrossRef] [PubMed]
- Schechter, M.C.; Buac, K.; Adekambi, T.; Cagle, S.; Celli, J.; Ray, S.M.; Mehta, C.C.; Rada, B.; Rengarajan, J.; Delgado-Rizo, V.; et al. Neutrophil-Derived MMP-8 Drives AMPK-Dependent Matrix Destruction in Human Pulmonary Tuberculosis. Front. Immunol. 2013, 7, 1–13. [Google Scholar] [CrossRef]
- Moreira-Teixeira, L.; Stimpson, P.J.; Stavropoulos, E.; Hadebe, S.; Chakravarty, P.; Ioannou, M.; Aramburu, I.V.; Herbert, E.; Priestnall, S.L.; Suarez-Bonnet, A.; et al. Type I IFN exacerbates disease in tuberculosis-susceptible mice by inducing neutrophil-mediated lung inflammation and NETosis. Nat. Commun. 2020, 11. [Google Scholar] [CrossRef]
- Su, R.; Peng, Y.P.; Deng, Z.; Deng, Y.T.; Ye, J.Q.; Guo, Y.; Huang, Z.K.; Luo, Q.; Jiang, H.; Li, J.M. Mycobacterium tuberculosis Infection Induces Low-Density Granulocyte Generation by Promoting Neutrophil Extracellular Trap Formation via ROS Pathway. Front. Microbiol. 2019, 10. [Google Scholar] [CrossRef]
- Francis, R.J.; Butler, R.E.; Stewart, G.R. Mycobacterium tuberculosis ESAT-6 is a leukocidin causing Ca2+ influx, necrosis and neutrophil extracellular trap formation. Cell Death Dis. 2014, 5. [Google Scholar] [CrossRef]
- Hunter, R.L. The Pathogenesis of Tuberculosis-The Koch Phenomenon Reinstated. Pathog. (Basel, Switzerland) 2020, 9, 1–25. [Google Scholar] [CrossRef]
- Ong, C.W.M.; Fox, K.; Ettorre, A.; Elkington, P.T.; Friedland, J.S. Hypoxia increases neutrophil-driven matrix destruction after exposure to Mycobacterium tuberculosis. Sci. Rep. 2018, 8. [Google Scholar] [CrossRef]
- Schechter, M.C.; Buac, K.; Adekambi, T.; Cagle, S.; Celli, J.; Ray, S.M.; Mehta, C.C.; Rada, B.; Rengarajan, J. Neutrophil extracellular trap (NET) levels in human plasma are associated with active TB. PLoS One 2017, 12. [Google Scholar] [CrossRef]
- De Melo, M.G.M.; Mesquita, E.D.D.; Oliveira, M.M.; Da Silva-Monteiro, C.; Silveira, A.K.A.; Malaquias, T.S.; Dutra, T.C.P.; Galliez, R.M.; Kritski, A.L.; Silva, E.C. Imbalance of NET and Alpha-1-Antitrypsin in Tuberculosis Patients Is Related With Hyper Inflammation and Severe Lung Tissue Damage. Front. Immunol. 2019, 9. [Google Scholar] [CrossRef]
- Meier, S.; Seddon, J.A.; Maasdorp, E.; Kleynhans, L.; du Plessis, N.; Loxton, A.G.; Malherbe, S.T.; Zak, D.E.; Thompson, E.; Duffy, F.J.; et al. Neutrophil degranulation, NETosis and platelet degranulation pathway genes are co-induced in whole blood up to six months before tuberculosis diagnosis. PLoS One 2022, 17. [Google Scholar] [CrossRef] [PubMed]
- Ríos-López, A.L.; González, G.M.; Hernández-Bello, R.; Sánchez-González, A. Avoiding the trap: Mechanisms developed by pathogens to escape neutrophil extracellular traps. Microbiol. Res. 2021, 243, 126644. [Google Scholar] [CrossRef] [PubMed]
- Arazna, M.; Pruchniak, M.P.; Demkow, U. Neutrophil extracellular traps in bacterial infections: strategies for escaping from killing. Respir. Physiol. Neurobiol. 2013, 187, 74–77. [Google Scholar] [CrossRef] [PubMed]
- Manda, A.; Pruchniak, M.P.; Araźna, M.; Demkow, U.A. Neutrophil extracellular traps in physiology and pathology. Cent. J. Immunol. 2014, 39, 116–121. [Google Scholar] [CrossRef]
- Lauth, X.; Von Köckritz-Blickwede, M.; McNamara, C.W.; Myskowski, S.; Zinkernagel, A.S.; Beall, B.; Ghosh, P.; Gallo, R.L.; Nizet, V. M1 Protein Allows Group A Streptococcal Survival in Phagocyte Extracellular Traps through Cathelicidin Inhibition. J. Innate Immun. 2009, 1, 202. [Google Scholar] [CrossRef]
- Brennan, P.J. Structure, function, and biogenesis of the cell wall of Mycobacterium tuberculosis. Tuberculosis 2003, 83, 91–97. [Google Scholar] [CrossRef]
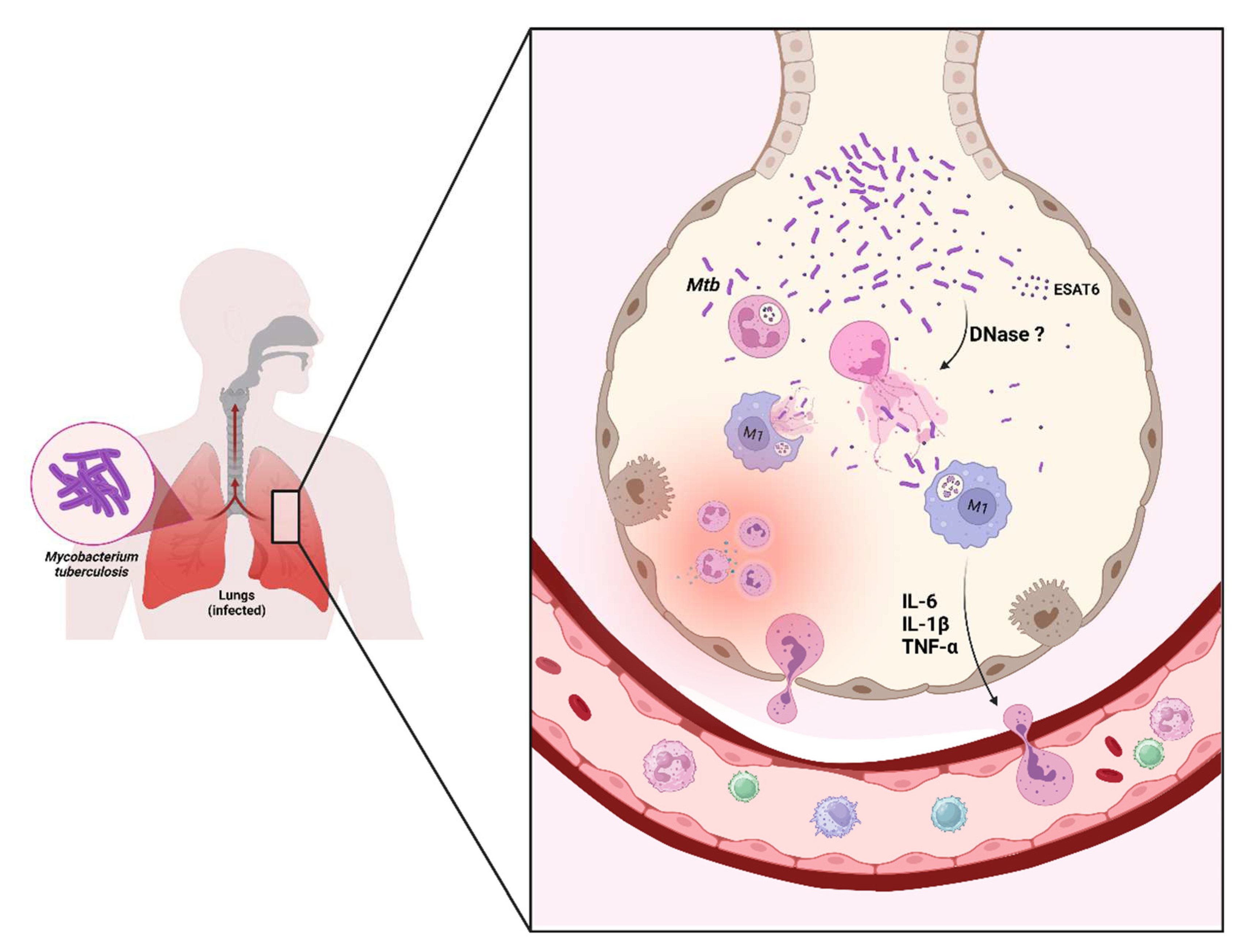
Figure 1.
Mycobacterium tuberculosis and its antigens (e.g., ESAT-6) induce neutrophil extracellular traps release (NETs). This mechanism can capture but not kill M. tuberculosis. Some resistance mechanisms may be associated (e.g., perhaps DNase release). The NETs can induce macrophages to release cytokines (e.g., IL-6, IL-1β, and TNF-α) that favor neutrophil influx. Macrophages can also internalize NETs; in macrophage lysosomes, NETs peptides could kill M. tuberculosis. Created with Biorender.com.
Figure 1.
Mycobacterium tuberculosis and its antigens (e.g., ESAT-6) induce neutrophil extracellular traps release (NETs). This mechanism can capture but not kill M. tuberculosis. Some resistance mechanisms may be associated (e.g., perhaps DNase release). The NETs can induce macrophages to release cytokines (e.g., IL-6, IL-1β, and TNF-α) that favor neutrophil influx. Macrophages can also internalize NETs; in macrophage lysosomes, NETs peptides could kill M. tuberculosis. Created with Biorender.com.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



