
Submitted:
23 May 2023
Posted:
23 May 2023
You are already at the latest version
Preprints on COVID-19 and SARS-CoV-2
Abstract
Recent studies have confirmed that lung microvascular endothelial injury plays a critical role in the pathophysiology of COVID-19. Our group and others have demonstrated the beneficial effects of H2S in several pathological processes and provided a rationale for considering the therapeutic implications of H2S in COVID-19 therapy. Here, we evaluated the effect of the slow-releasing H2S donor, GYY4137, on the barrier function of a lung endothelial cell monolayer in vitro, after challenging the cells with plasma samples from COVID-19 patients or inactivated SARS-CoV-2 virus. We also assessed how the cytokine/chemokine profile of patients’ plasma, endothelial barrier permeability, and disease severity correlated with each other. Alterations of barrier permeability after treatments with patient plasma, inactivated virus, and GYY4137 were monitored and assessed by electrical impedance measurements in real-time. We present evidence that GYY4137 treatment reduced endothelial barrier permeability after plasma challenge and completely reversed the endothelial barrier disruption caused by inactivated SARS-CoV-2 virus. We also showed that disease severity correlated with the cytokine/chemokine profile of the plasma but not with barrier permeability changes in our assay. Overall, these data demonstrate that treatment with H2S-releasing compounds has the potential to ameliorate SARS-CoV-2–associated lung endothelial barrier disruption.
Keywords:
endothelial barrier
; COVID-19
; SARS-CoV-2
; hydrogen sulfide
; cytokine
; TNF-α
1. Introduction
As of May 2023, just after the third anniversary of its officially declared outbreak, Coronavirus Disease 2019 (COVID-19) has caused more than 6.9 million deaths worldwide. The causative agent of this disease, the Severe Acute Respiratory Syndrome Coronavirus-2 (SARS-CoV-2), has infected over 760 million people globally, and this number is still rising rapidly [1]. Despite substantial advances in prevention and treatment strategies, this pandemic still poses global health and economic challenges due to the constantly emerging novel variants of the virus and the long-term consequences of the infection, collectively termed post-COVID conditions or long COVID [2,3,4]. COVID-19 was initially viewed as a respiratory disease, but growing evidence supports the critical role of endothelial dysfunction not only in the pulmonary vasculature but in other organs, both in acute cases and during long COVID [5,6,7,8,9].
Severe manifestations of COVID-19 are characterized by progressive respiratory failure resulting from diffuse alveolar damage with inflammatory infiltrates and alveolar edema, intra-alveolar fibrin deposition and hemorrhage, endothelialitis, as well as pulmonary and systemic coagulopathy that form obstructive microthrombi in the lung and other organs [6,10,11]. Additional pathological findings in the vasculature of COVID-19 lungs include disruption of intercellular junctions, basal membrane contact loss, neutrophilic capillaritis/endothelialitis, pulmonary thromboembolism, pulmonary infarctions, and venous thrombosis [12,13]. The contribution of different factors to COVID-19‒related endotheliopathy is still under debate. A growing body of evidence points to high levels of pro-inflammatory cytokines and chemokines produced in the lung tissue by infected alveolar epithelial cells and alveolar macrophages [14,15,16], platelet activation [17,18], as well as to direct contact of endothelial cells (ECs) with SARS-CoV-2 spike protein [19,20,21,22]. At the same time, recent data indicate that direct viral infection of ECs is less likely to play a major role in these processes [12,22,23,24,25].
In the lung, alveolar epithelial cells are surrounded by an extracellular matrix and adjacent pulmonary microvascular ECs, forming the alveolar-capillary endothelial barrier through inter-endothelial junctions [26,27]. The permeability properties of this barrier are tightly regulated through interactions between ECs, surrounding tissue, and biologically active molecules in the blood [26,28]. Lung microvascular endothelial injury has been linked to the most severe complications of COVID-19, acute respiratory distress syndrome (ARDS), multiorgan failure, and death [8,29]. Recent studies have demonstrated that lung endothelial barrier damage and dysfunction - characterized by increased vascular permeability and loss of barrier integrity leading to the leakage of fluid and plasma proteins into the surrounding tissue - play a critical role in the pathophysiology of the disease and contribute to the development of ARDS [8,16,17]. In the current clinical practice, no specific therapeutic strategies aim to restore the endothelial barrier in COVID-19 patients.
Hydrogen sulfide (H2S) is a gaseous signaling molecule produced in various mammalian cell types, including ECs [30,31]. Among a wide array of physiological functions, it plays a fundamental role in vascular homeostasis, modulates inflammatory responses, and reduces vascular leakage [30,32,33,34,35]. Several studies have demonstrated that H2S improves endothelial barrier function in a variety of experimental conditions. H2S inhalation attenuated pathologically enhanced blood-brain barrier permeability in animal models of cardiac arrest and resuscitation [36,37]. H2S treatment prevented lipopolysaccharide (LPS)-induced hyperpermeability in EC cultures [38] and protected against LPS inhalation-induced acute lung injury by reducing neutrophil transmigration and inhibiting pro-inflammatory signaling in animal models [39,40]. The H2S donor NaHS attenuated increased endothelial permeability and inflammation in murine lung specimens challenged by particulate matter inhalation [41]. In contrast, some reports demonstrated that decreased endogenous H2S production and altered sulfur metabolism reduced vascular permeability [42,43]. These currently available studies indicate that H2S signaling modulates EC barrier function in a context-dependent manner, and its potential disease-specific impact needs to be determined by targeted research [30]. To our knowledge, no such data exists about H2S in the context of SARS-CoV-2–mediated lung endothelial barrier disruption.
Recent findings have provided a rationale for considering the therapeutic implications of H2S donor molecules in COVID-19 therapy [44,45]. Our group and others have shown that H2S significantly attenuates the replication of several respiratory viruses and virus-induced inflammation [46,47]. Moreover, it has been speculated that H2S blocks SARS-CoV-2 entry into host cells by interfering with angiotensin-converting enzyme 2 (ACE2) and transmembrane protease serine 2 (TMPRSS2) expression [45]. In addition, impaired endogenous H2S availability is linked to cardiovascular [48] , metabolic [49], and pulmonary diseases [50], which are all risk factors for developing severe COVID-19. Potential alterations in the endogenous H2S plasma level in COVID-19 patients are still under debate, but data suggest that impaired H2S availability contributes to COVID-19–associated endotheliopathy and a more severe outcome of this disease [51,52,53]. Furthermore, inhalation of the H2S donor, sodium thiosulfate, elicited protective effects in COVID-19 patients by reducing symptoms and accelerating recovery [54]. All these results suggest that H2S may exhibit beneficial effects in the pathomechanism of COVID-19, but the therapeutic potential of slow-releasing H2S compounds in lung endothelial barrier disruption associated with SARS-CoV-2 infection has never been explored.
In this study, we aimed to assess the effect of the slow-releasing H2S donor GYY4137 on the barrier function of a human lung microvascular EC monolayer in vitro, after challenging the cells with plasma samples from COVID-19 patients or inactivated SARS-CoV-2 virus. To our knowledge, these data are the first of their kind. We also evaluated inflammatory cytokine levels in the patients’ plasma and determined their correlation with disease severity and impact on the endothelial barrier function.
2. Materials and Methods
2.1. Human plasma samples
De-identified clinical plasma samples and clinical data of hospitalized COVID-19 patients were received from the University of Texas Medical Branch (UTMB) Biorepository for Severe Emerging Infections (BSEI). Samples were collected from consented patients under the Clinical Characterization Protocol for Severe Emerging Infections (UNMC IRB # 146-20-FB/UTMB-IRB # 20−0066), PI, Dr. David Brett-Major, U. Nebraska Medical Center; UTMB site-PI, Dr. Susan McLellan. Note that multiple samples (longitudinal sampling) were collected from COVID-19 patients, classified as Mild (n = 7, total samples = 11), Moderate (n = 6, total samples = 12), Severe (n = 4, total samples = 12), or Critical (n = 9, total samples = 26) based on their oxygen therapy requirement by the following definitions: Mild - room air, Moderate - nasal cannula, Severe - non-invasive ventilation (continuous positive airway pressure/bilevel positive airway pressure/high-flow nasal cannula/large-reservoir oxygen mask), Critical – invasive ventilation (intubation; mechanical ventilation/extracorporeal membrane oxygenation). Patients typically moved through the various levels of oxygen devices and were classified into a disease severity category based on the highest level of oxygen delivery required. Control plasma samples (n = 20) from healthy subjects (one sample per subject) were collected under IRB protocol in Dr. Cardenas’ lab (HSC-GEN-09-0314). All samples were inactivated on dry ice by gamma irradiation (5 Mrad) prior to usage following institutionally approved Standard Operating Procedures for SARS-CoV-2 work.
2.2. Cell culture
Human Lung Microvascular Endothelial Cells (HLMVECs, #540-05a), Attachment Factor Solution, and microvascular endothelial growth medium were purchased from Cell Applications (San Diego, CA, USA). HLMVECs are primary endothelial cells isolated from the lung capillaries of a 19-year-old Hispanic male. Cells were used up to passage six without losing their morphologic and phenotypic characteristics, per company recommendations. HLMVECs were grown in microvascular endothelial growth medium in a 5% CO2 atmosphere at 37 °C in coated cell culture flasks, dishes, and microplates following supplier’s instructions.
2.3. Virus
SARS-CoV-2 Omicron BA.1 (Lineage B.1.1.529 Strain: EHC_C19_2811C; GISAID: EPI_ISL_7171744) was provided by the World Reference Center for Emerging Viruses and Arboviruses (WRCEVA) at UTMB, propagated on TMPRSS2-expressing Vero E6 cells, sucrose purified and resuspended in HLMVEC basal medium with 1% BSA. The stock was then titrated by plaque assay as previously described [55]. Virus stock was then inactivated on dry ice by gamma irradiation (5 Mrad) before usage. Cultures from mock-infected cells were purified and processed similarly to vehicle control. All infectious work was performed in a biosafety level 3 laboratory (BSL3, Dr. Freiberg) at the Galveston National Laboratory, UTMB. The irradiated virus stock and corresponding vehicle control were transferred to a BSL-2 laboratory (Dr. Modis) for endothelial permeability assays. For this project, an irradiated equivalent dose of 5x104 PFU was used per reaction in 200 µL volume.
2.4. Reagents
Recombinant human TNF-α protein was purchased from R&D systems (Minneapolis, MN, USA). The slow-releasing H2S donor molecule, GYY4137, was purchased from Cayman (Ann Arbor, MI, USA). Bovine Serum Albumin (BSA) was purchased from Millipore Sigma (Burlington, MA, USA).
2.5. Bio-plex multiplex immunoassay
Analysis of inflammatory markers in human plasma samples was performed using a Bio-Plex Pro Human cytokine group 1 panel 27-plex kit (Bio-Rad, Hercules, CA, USA) following the manufacturer’s instructions.
2.6. Endothelial permeability assay
An electrical impedance assay was adapted from the manufacturer’s protocol using the xCELLigence Real-Time Cell Analysis system (Agilent, Santa Clara, CA). HLMVECs were seeded in a precoated 96-well cell culture plate equipped with gold electrodes (E-plate; Agilent, Santa Clara, CA) at a density of 14,000 cells per well, predefined by previous experiments [56,57]. Cells were incubated under growth conditions for 48 hours to form a confluent monolayer (indicated by a plateau of electrical impedance). The growth medium was then replaced by a prewarmed starvation medium containing 1% BSA and no growth supplements for 2 hours before starting the treatment protocol. TNF-α and GYY4137 working solutions were prepared in starvation medium. For vehicle control of TNF-α and GYY4137 treatment, starvation medium was used. As controls for plasma treatment and virus challenge, growth medium and matching virus-free medium (respectively) were used. First, the HLMVEC monolayer was treated with human plasma, TNF-α, inactivated SARS-CoV-2 virus, or vehicle control. Thirty minutes later, GYY4137 or vehicle control was added to the wells, and the E-plates were further incubated in a 5% CO2 atmosphere at 37 °C for 24 hours. All treatments were carried out in starvation medium, and each treatment group had 3-6 replicates on each plate. Plasma samples and inactivated virus stocks were used at 1:10 dilution with matching vehicle controls. TNF-α and GYY4137 were used at 10 ng/mL and 300 µM, respectively, unless otherwise indicated. Electrical impedance for each well was recorded as Cell Index (CI) every 10 min in real-time throughout this process. CI readings were normalized (Normalized Cell Index, NCI) to the last recorded CI value before the first treatment to minimize well-to-well noise resulting from pre-treatment differences (seeding, electrode sensitivity, etc.). During data analysis, NCI values were further normalized (Relative Normalized Cell Index, RNCI) to the negative control (treated with vehicle control only; Control) readings from the same E-plate to account for plate-to-plate variability. Unless otherwise indicated, measurements at 12 hours after treatment were analyzed and plotted.
2.7. Data and statistical analysis
Data are shown as mean ± SD. Statistical analyses included Rout outlier test (Q=0.1%), D'Agostino-Pearson and Shapiro-Wilk normality tests, one-way, or two-way ANOVA followed by Tukey’s multiple comparisons test, Wilcoxon matched-paired signed rank t-test, Kruskal-Wallis non-parametric one-way ANOVA test followed by Dunn's multiple comparisons, and simple linear regression as indicated in figure legends. All statistical analysis was performed using GraphPad Prism 9 analysis software (GraphPad Software Inc., La Jolla, CA). The experiments were repeated at least three times independently, with at least three replicates of each assay group or condition. A value of P ≤ 0.05 was considered statistically significant. In the Figures, “ns” indicates non-significant differences between two groups where needed for emphasis.
3. Results
3.1. Patient characteristics
In this study, we used de-identified clinical plasma samples and clinical data of hospitalized COVID-19 patients received from the UTMB Biorepository for Severe Emerging Infections. For control, we used plasma from healthy subjects. COVID-19 patients were classified as Mild, Moderate, Severe, or Critical based on the highest level of their oxygen therapy requirement by the following definitions: Mild (n=7) - room air, Moderate (n=6) - nasal cannula (NC), Severe (n=4) - non-invasive ventilation, Critical (n=9) - invasive ventilation. Demographic data, clinical characteristics, and initial laboratory findings for COVID-19 patients included in the study are summarized by disease severity groups in Table 1. Note that longitudinal data of clinical laboratory markers acquired during the patients’ hospitalization are not included in this table. Although the means of several clinical measurements show marked differences between patient groups at the time of admission, none of these differences reached statistical significance.
3.2. Plasma cytokine/chemokine levels show a strong correlation with disease severity
Elevated plasma levels of inflammatory markers [including tumor necrosis factor-alpha (TNF-α), macrophage inflammatory protein-1 alpha (MIP-1α), interleukin 8 (IL-8), interferon-gamma-induced protein 10 (IP-10), and others] have been reliably associated with more severe disease and serious symptoms including alveolar-capillary barrier disruption and intra-alveolar hemorrhage [58,59,60,61]. Based on these reports, we first sought to correlate disease severity with plasma biomolecules using the Bio-Plex Pro Human Cytokine 27-plex Assay (Bio-Rad). Interestingly, the plasma levels of ten signaling molecules [TNF-α, MIP-1α, interleukin-1 receptor antagonist (IL-1ra), granulocyte colony-stimulating factor (G-CSF), IL-8, IP-10, interferon-gamma (IFN-γ), monocyte chemoattractant protein-1 (MCP-1), platelet-derived growth factor (PDGF), and vascular endothelial growth factor (VEGF)] were found significantly elevated in patient samples compared to healthy controls, but only six (TNF-α, MIP-1α, IL-1ra, G-CSF, IL-8, and IP-10) showed a gradually rising trend of mean values in correlation with disease severity (Fig. 1A-F). Remarkably, none of them were found to be significantly elevated in the mild plasma group compared to samples of healthy individuals, and four of them (TNF-α, MIP-1α, IL-1ra, and G-CSF) only showed a significant increase in severe and critical samples (Fig. 1A-D). IL-8 and IP-10 demonstrated the most consistent correlation with disease severity, presenting significantly increased levels in moderate, severe, as well as critical samples (Fig. 1E-F). Additionally, elevated expression levels of MCP-1, PDGF, VEGF, and IFN-γ exhibited only a partial correlation with disease severity (data not shown).
3.3. Clinical laboratory markers are elevated in COVID-19 patients’ blood
Serum levels of lactate dehydrogenase (LDH), C-reactive protein (CRP), and D-dimer are routinely tested in hospitalized COVID-19 patients to assess acute inflammation, blood clotting tendencies, and increased tissue damage, respectively. We plotted all available longitudinal records of these clinical markers for each patient group (Fig. 1G-I) to evaluate their correlation with disease severity and to compare them to the cytokine/chemokine profiles established earlier. Note that these measurements for the healthy control group were not available, however, comparing these results to the normal reference range (LDH <280 U/L; CRP <1 mg/dL; D-dimer <0.5 µg/mL) [62,63,64] revealed that all three markers in all disease groups were elevated. In addition, LDH levels in the critical group were significantly increased even compared to the mild and moderate groups (Fig. 1G). None of the groups presented distinct and significantly different CRP readings, although the critical group had a markedly higher mean value than the others. Elevated D-dimer levels correlated better with the critical categorization as values for that group were significantly higher relative to severe, moderate, and mild groups. Overall, the three clinical markers together clearly distinguished the critical patient group from the others, but the close correlation of gradually increasing levels with disease severity found with inflammatory cytokines and chemokines was not observed in these tests.
3.4. GYY4137 treatment improves endothelial barrier function
In our next set of experiments, we assessed the effects of biomolecules and patient plasma treatment on the barrier function of a confluent HLMVEC monolayer modeling the lung endothelial barrier in vitro, using the xCELLigence Real-Time Cell Analysis system (Agilent). This method uses electrochemical impedance measurements (Cell Index, CI) to determine the permeability of the endothelial monolayer with CI values, as well as Normalized and Relative Normalized Cell Index values (NCI and RNCI, respectively, as described in the Materials and Methods section). Higher CI, NCI, and RNCI values represent increased barrier function and a more intact endothelial monolayer. Reduced CI values show compromised endothelial barrier and increased endothelial permeability. Since this study aimed to evaluate the potential beneficial effects of exogenous H2S on the lung microvascular endothelial barrier in COVID-19 patients, first, we tested the impact of H2S alone on our model. Using different concentrations (10-1,000 µM) of the slow-releasing H2S donor, GYY4137, we established the real-time NCI curves of these treatments (Fig. 2A) and determined their effects on the endothelial barrier at the 12-hour time point after treatment (Fig. 2B). We found that the real-time NCI curves for all tested GYY4137 concentrations consistently ran above the mock-treated control curve throughout the monitored 50-hour-long period after treatment (Fig. 2A). Considering that under the starvation conditions of the assay the NCI of the untreated control (Fig. 2A, B; green line and bar, respectively) started to fall 24 hours after treatment, we limited the use of the assay to 24 hours in all following experiments. We also determined that treatment with all tested GYY4137 concentrations significantly raised the RNCI compared to control (Fig. 2B) without any changes in cell viability and cytotoxicity (data not shown). Based on these results and earlier studies [65], we selected a 300 µM concentration of GYY4137 to test its barrier-protective effect in combination with treatments with other agents. To verify the functionality and biological relevance of our model, we next attempted mimicking the loss of endothelial barrier integrity using TNF-α as control since it has repeatedly been shown to interfere with ECs tightness and cause increased barrier permeability [66,67,68]. We treated the HLMVEC monolayer with increasing concentrations of TNF-α (1 pg/mL-100 ng/mL) followed by 300 µM GYY4137 or mock treatment 30 minutes later. We found that 1-100 pg/mL TNF-α did not alter the RNCI, while 1-100 ng/mL TNF-α treatment resulted in decreased RNCI values in a dose-dependent manner at 12 hours (Fig. 2C) without increasing cellular toxicity (data not shown). We measured very similar RNCI values after 24 hours as well (data not shown). Importantly, treating the cells with 300 µM GYY4137 30 minutes after TNF-α treatment completely restored RNCI levels suggesting the reestablishment of intercellular junctions. Altogether, these data verified that our human lung endothelial barrier model using the xCELLigence system is adequate to study changes in endothelial barrier permeability when subjected to various challenges.
3.5. Human plasma treatment alters endothelial barrier function, and GYY4137 modifies these effects
After establishing the impact of GYY4137 and TNF-α treatments, we evaluated the effects of patient plasma treatment on the HLMVEC monolayer. Plasma treatments resulted in significant alterations in NCI levels (Fig. 2D), and each patient plasma sample produced a unique and robust EC barrier pattern recuperated very closely by replications (n=3-6, data not shown). Based on our previous results, we used 10 ng/mL TNF-α treatment as positive control. We observed that some of the plasma treatments resulted in a comparable level of endothelial barrier disruption to TNF-α (continuous blue line), especially up to 12 hours after treatment (continuous red and black lines corresponding to a severe and a critical sample, respectively). Interestingly, 300 µM GYY4137 treatment (dotted lines of the corresponding color) significantly alleviated the endothelial barrier disruption caused by plasma treatments at 12 hours, and most of this beneficial effect was diminished at 24 hours. None of the treatments caused detectable cytotoxicity measured by LDH-assay (data not shown). Most importantly, adding GYY4137 after treating the HLMVECs with human plasma significantly increased EC barrier function, suggesting the barrier-protective impact of H2S donation.
3.6. Endothelial barrier disruption caused by plasma from COVID-19 patients does not correlate with disease severity or plasma cytokine/chemokine levels
Recent literature demonstrated various levels of correlation between endothelial barrier disruption and the plasma cytokine levels and/or disease severity of patients with different pathological conditions, including COVID-19 [59,69,70]. Surprisingly, when we compared the effect of plasma from different disease severity groups on the RNCI of the HLMVEC monolayer, we found no statistical differences at 12 (Fig. 3A) or 24 hours (data not shown) suggesting that our model was equally responding to samples from healthy individuals and COVID-19 patients. Similarly, when we calculated the barrier-altering effect of GYY4137 treatment after plasma treatment, we only detected a slightly increasing trend towards the more severe disease groups, but this trend did not reach the level of statistical difference (Fig. 3B). Moreover, none of the biomolecules that showed a strong correlation with disease severity in experiments described earlier (Fig. 1A-F) presented correlation with the RNCI after plasma treatment (Fig. 3C). In fact, when we matched the barrier disrupting effect of each plasma sample with its cytokine/chemokine profile, none of the inflammatory signaling molecules assessed in this study correlated with the corresponding RNCI values (data not shown). Altogether our results suggest that the change in endothelial barrier permeability in our model is neither linked to COVID-19 disease severity nor the individual plasma concentration of any of the inflammatory markers analyzed.
3.7. GYY4137 increases endothelial barrier function in a disruption-dependent manner
Next, we evaluated the effect of GYY4137 by plotting the RNCI values of plasma- and GYY4137-treated samples against the corresponding RNCI results of samples treated with plasma only. We found a significant overall RNCI increase at 12 hours as a result of GYY4137 treatment (Fig. 3D) and, to a much lesser extent, at 24 hours as well (data not shown). Importantly, this analysis demonstrated that adding GYY4137 after treating the HLMVECs with human plasma significantly elevated EC barrier function, suggesting that GYY4137 combated plasma-induced barrier disruption. We then further explored the potential factors upon which the effect of GYY4137 treatment may depend. We found that greater initial barrier damage was met with a relatively greater healing effect by the administration of GYY4137 (Fig. 3E). As a result, when we grouped our plasma samples based on their initial barrier-damaging effect (regardless of which disease severity group the patient belonged to), the more damaged groups of samples benefited more from GYY4137 treatment. Interestingly, instead of a gradual increase in restoring endothelial barrier integrity, we observed a relatively sharp increase at approximately 25% barrier disruption and a milder gradual rising trend both below and after this threshold.
3.8. Inactivated SARS-CoV-2 Omicron BA.1 increases endothelial barrier permeability, which can be prevented by GYY4137 treatment
A direct contribution of the SARS-CoV-2 virus to the endothelial barrier disruption observed in the lungs of COVID-19 patients has been debated since the beginning of the pandemic. Most recent studies have suggested that contact with viral proteins (most notably the spike protein and its S1 subunit) rather than viral infection of the endothelial cells may play a role in endothelial activation and barrier disruption [19,20]. Based on these studies, we assessed the impact of inactivated SARS-CoV-2 Omicron BA.1 (Omicron) in our in vitro model. The endothelial monolayer was treated with an equivalent of 5x104 PFU per well or similarly prepared mock (control). We found that compared to vehicle control, virus treatment caused a significant decrease in NCI and RNCI values at both 12 and 24 hours after treatment (Fig. 4A, B). Specifically, the NCI curve of virus treatment ran parallel to the 10 ng/mL TNF-α control curve up to the 12-hour time point causing approximately 60% as much decrease in barrier function (Fig. 4A). Throughout the next 12 hours, the virus-treated curve remained steady. Overall, inactivated virus treatment caused approximately 20-25% increase in endothelial barrier permeability both at 12 and 24 hours compared to control suggesting a direct contribution of SARS-CoV-2 Omicron BA.1 proteins to lung endothelium damage. Similar to our results with plasma from COVID-19 patients, treating our lung endothelial barrier model initially damaged by inactivated virus challenge with GYY4137 (300 µM) significantly improved endothelial barrier function (Fig. 4A, B). However, unlike with the plasma samples, this restorative effect by the H2S-donor did not diminish over time and remained at virtually identical levels to mock-treated control up until the 24-hour time point (Fig. 4A). None of the treatments caused detectable cytotoxicity measured by LDH-assay (data not shown). Most importantly, GYY4137 treatment prevented disruption of endothelial barrier integrity caused by virus challenge (Fig. 4B) altogether suggesting that GYY4137 exerts a therapeutic effect in lung endotheliopathy seen in COVID-19 patients.
4. Discussion
One of the novel findings of the present study is that treatment with GYY4137, a well-characterized, slow-releasing H2S donor, ameliorates endothelial barrier disruption caused by plasma samples from COVID-19 patients in vitro, regardless of disease severity. In our real-time in vitro assay model, patient plasma altered endothelial barrier permeability in a highly sample-specific manner, causing barrier damage comparable to TNF-α control, a cytokine known to disrupt endothelial junctions [66,67,68]. Surprisingly, plasma-induced barrier disruption did not correlate with disease status based on the patient’s oxygen requirement: Plasma from some patients with mild disease caused as much or even more damage as some critical patients’ plasma, while plasma from some other individuals in each disease severity group did not elicit any increase in endothelial barrier permeability or even triggered a decrease. Additionally, we determined the cytokine/chemokine profile of each plasma sample and found a high correlation between disease severity and the concentration of several biomolecules, most notably IL-8 and IP-10. In fact, their plasma levels mirrored increasing disease severity much closer than any of the three regularly used clinical markers, LDH, CRP, and D-dimer [71,72,73]. On the other hand, none of the cytokines and chemokines we assessed demonstrated correlation between plasma levels and the corresponding in vitro barrier function assay results. Finally, inactivated SARS-CoV-2 Omicron BA.1 virus particles elicited a very robust endothelial barrier disruption in our assay that was completely reversed by adding GYY4137. Taken together, these data characterize the effects of patient plasma and inactivated virus particles on the lung microvascular endothelial barrier and provide the basis for further efforts to develop novel treatment modalities that specifically target H2S signaling in COVID-19 patients.
The COVID-19 pandemic has presented an enormous challenge to countries and health systems worldwide, unprecedented in recent history. This challenge is far from being over due to the constantly evolving nature of the causative agent, SARS-CoV-2, and the debilitating long-term effects of the infection [2,3]. The two complementary approaches, prevention and treatment, in the global fight against this pandemic have been increasingly successful but require tremendous amounts of money and effort: As of May 2023, more than three years after the first reported cases, the results still leave room for improvement with about 100,000 new cases and 500-1000 deaths recorded daily worldwide [1]. Vaccine development and distribution, together with other preventive measures, are used as the first line of defense with limited success around the world [74,75]. Serving as the second line, the efficacy of therapeutic approaches in reducing morbidity and mortality of the disease has been gradually increasing, but there is still a great need for novel effective and inexpensive drugs to fill in the gaps of currently available treatment modalities, especially outside the highest-income countries [76,77]. In light of recent discoveries about its anti-inflammatory, vasculoprotective, and antiviral effects, H2S has been proposed as a potential defense against COVID-19 [44,45]. In this study, we aimed to assess the effect of an H2S-donor, GYY4137, on the barrier function of lung endothelial cells, after challenging them with plasma samples from COVID-19 patients, including non-survivors or inactivated SARS-CoV-2 virus. In parallel, we also sought to demonstrate any correlation between inflammatory markers in patient plasma and disease severity.
Damaged endothelial barrier and increased microvascular permeability are hallmarks of severe COVID-19 pathology, greatly contributing to disease severity and mortality [6,8,11]. H2S has been shown to modulate vascular permeability in several reports, and its effects on the endothelial barrier function have been suggested to be potentially disease- and organ-specific [30,36,38]. In this regard, we established a lung microvasculature-specific assay and tested the effects of the H2S-donor, GYY4137, in the context of COVID-19–associated biological samples (patient’s plasma and inactivated virus particles). We found a significant increase of barrier function (termed CI, NCI, and RNCI as described in the Materials and Methods section) 12 hours after GYY4137 treatment regardless of the existence and nature of pre-treatments. Interestingly, this effect was maintained, diminished or even reversed at later time points depending on the initial treatment. These results are in line with recent reports showing that H2S inhalation or H2S-donor treatment reduced pathologically increased vascular permeability in the brains of rats after cardiac arrest [37] or in the lungs of mice after particulate matter inhalation [41], respectively. Remarkably, larger initial barrier disruption by COVID-19 patients’ plasma evoked a relatively greater barrier function increase by GYY4137 treatment. Beyond the obvious base-effect (from a lower base, the same nominal increase constitutes a higher percent), we found an unexplained phenomenon that a 25% or larger RNCI decrease caused by plasma treatment was followed by a significantly greater increase evoked by GYY4137 than when the initial barrier damage was smaller. This could be caused by some of the tight or adherens junctions between ECs that are preferentially restored first or faster by GYY4137 and confer different levels of connecting strength between cells [78,79], but to fully understand the reason for this bi-phasic effect will require further investigations. Taken together, our data support the notion that an increased level of H2S in the lung microenvironment, either by increased endogenous production or by pharmaceutical intervention may be beneficial in severe COVID-19.
Another question we addressed was how the cytokine/chemokine profile of patients’ plasma, the endothelial barrier disruption caused by this plasma, and disease severity correlate with each other. As expected, we found higher levels of inflammatory cytokines and chemokines in the plasma of COVID-19 patients with more severe disease, confirming previously reported data [15,59,61]. Moreover, six (TNF-α, MIP-1α, IL-1ra, G-CSF, IL-8, IP-10) of the assessed signaling molecules demonstrated gradually increasing plasma levels consistent with disease severity, much more so than routine clinical markers, LDH, CRP, and D-dimer [71,72,73]. In fact, we found that while these markers (especially LDH and D-dimer) clearly distinguished the critical group from the others, they did not separate the other groups. While there are obvious advantages to recognizing the critical phase in the course of COVID-19 using blood tests designed to detect these molecules, a panel of the six cytokines/chemokines listed above could provide a better resolution to monitor disease progression. Surprisingly, some biomolecules that have been reported to be potential markers for COVID-19 disease severity, including VEGF [80,81], MCP-1 [60], IFN-γ [82], PDGF [81] and others, either did not or only partially correlate with disease severity, maybe due to the relatively small number (10-12) of samples in some of the groups. Nevertheless, our data support previous findings that monitoring blood cytokine/chemokine levels, especially for IL-8 [58,61] and IP-10 [60,83], could be used as additional biomarkers to help identify and manage COVID-19 patients with different disease severity. On the other hand, we found no correlation between plasma cytokine/chemokine levels and endothelial barrier disruption in our in vitro assay. In light of this finding, it is not surprising that the measured in vitro barrier disruption does not correlate with disease severity either. This counterintuitive result is, in fact, in line with recent literature [84] dissecting the factors in COVID-19 patients’ plasma potentially causing endothelial barrier disruption. Kovacs-Kasa et al. verified that the factor(s) in patients’ plasma disrupting microvascular integrity were heat-labile, but no single or set of cytokine(s) could be accounted for enhanced vascular permeability. They also disproved the potential role of ACE2-binding and complement factors C3a and C5a in the phenomenon. Recent studies implicated several molecular mechanisms, including altered expression and function of adhesion and junction proteins (ICAM-1 and 2, VCAM-1, E- and P-selectin, claudins, occludins, VE-cadherin, Connexin-43, and others) [78,79,85], and/or pathologically modified signaling by integrins, TGF-β, complement, the glycocalyx, mitochondria and (most recently) microRNAs [12,19,86,87], to contribute to lung endothelial barrier disruption, but the causative agents in the plasma initiating these processes are still widely debated. Since the plasma levels of the cytokines/chemokines assessed in this study are several magnitudes lower than necessary to significantly lower CI values in our assay (e.g., for TNF-α, 1-100 pg vs. 1-100 ng respectively), direct effect from these molecules in vitro could not be expected. However, lung tissue levels of these mediators are estimated to be potentially over 1,000-fold higher than in plasma during severe inflammation [88,89], reaching the necessary levels for endothelial activation and barrier disruption in vivo. It is also worth noting that disease severity groups were solely based on the patients’ oxygen requirements without consideration of any other clinical characteristics. A more complex classification system including several clinical markers and symptoms could yield different results. Therefore, further in vitro and in vivo studies will be needed to resolve this debate.
Circulating virus particles and/or viral spike protein in the patient’s blood have also been implicated in inducing increased microvascular permeability. The potential role and significance of the spike protein (or other viral proteins) in endothelial barrier disruption are still highly controversial [20,69,78,86,90] and not always assessed when using these assays. For example, endothelial damage has been reported in the lung after using (1) the spike protein that induced degradation of junction proteins [78] as well as altered integrin and transforming growth factor beta signaling [19], (2) the nucleoprotein that induced EC activation via Toll-like receptor 2 and mitogen-activated protein kinase signal pathways [13] and (3) non-infectious pseudovirus expressing the spike protein that compromised mitochondria and impeded endothelial NO synthase activity [86]. To this end, we tested the effects of inactivated SARS-CoV-2 Omicron BA.1 virus in our in vitro assay and demonstrated that challenging the HLMVEC monolayer by 5x104 PFU/well infective dose equivalent inactivated virus results in a drop of CI values comparable to the effect of 10 ng/mL TNF-α. We chose to work with a B.1.1.529 variant as this lineage was circulating in the population at the time of experimentation. Most importantly, inactivated virus-induced endothelial barrier disruption was completely reversed by 300 µM GYY4137. We speculate that the barrier-disruptive potential of the virus particle and/or the spike protein could be lineage- and even sublineage-dependent, explaining the inconsistent data about endothelial barrier disruption available from similar studies using proteins derived from Wuhan or WA1/2020 isolates [69]. For example, it has been well-demonstrated that Omicron linages feature increased ACE2-affinity and immune evasion capabilities due to several mutations, most of which alter the antigenicity of the spike protein and at the same time modify its structure and function as well [91,92,93]. As a result of their unique virological features in comparison to other SARS-CoV-2 strains, Omicron variants exhibit less efficient TMPRSS2 usage, less spike cleavage, lower fusogenicity, and an altered entry mechanism [94,95]. Similarly, the use of different endothelial cells could also be the source of inconsistent findings because of the different genetic backgrounds of the original donors [69]. Further studies addressing virus-host cell interactions with respect to the spectrum of genetic variations of both could be warranted to better assess the clinical relevance of this pathomechanism.
Our study has certain limitations. First, the number of plasma samples per group was somewhat uneven; there were more critical and healthy samples available than samples belonging to the other three disease severity groups. This weakness of the study design may have introduced a bias towards more significant differences between the two larger sample groups than the others, but we do not believe that it fundamentally altered any of our findings. A follow-up study with larger sample numbers could verify our data. Second, we focused on only one aspect of COVID-19–associated endotheliopathies, the alterations of barrier function using ECs only. While a more complex study could put the results in more context, our simplified monoculture-based approach had the advantage of providing clear answers to some of the basic questions: 1. Are there factors in patient plasma capable of altering endothelial barrier function alone? 2. What correlations exist among the cytokine/chemokine profile of plasma samples, the endothelial barrier disruption caused by them and disease severity? 3. Can inactivated virus alone, as a surrogate of using viral proteins, cause endothelial barrier disruption? And most importantly: 4. Does treatment with an H2S-donor provide beneficial effects against SARS-CoV-2–associated lung microvascular barrier disruption? Third, we only tested one virus variant and primary lung ECs from one donor as proof of concept. As discussed above, these choices have introduced a genetic bias for the virus-host cell interactions, and we believe that our results justify more comprehensive follow-up studies. Finally, we only tested the effects of a one-time treatment with a single H2S donor molecule, GYY4137. Other H2S-releasing agents may have more sustained pharmacological effects as recently reviewed by Szabo and Papapetropoulos [96]. Further studies will be necessary to clarify the effects of repeated treatments using several different H2S-releasing compounds to verify whether potential clinical trials could be warranted for pharmacological increase/stabilization of the endothelial barrier as a third pillar for the treatment of COVID-19 in addition to immunomodulators and anti-virals.
5. Conclusions
Overall, our data demonstrate that treatment with H2S-releasing compounds has the potential to ameliorate SARS-CoV-2–associated lung endothelial barrier disruption. Although much work remains to be done to fully understand and dissect the molecular mechanisms involved as well as the therapeutic implications of this approach in treating COVID-19, this work provides the basis for future investigations.
6. Patents
O.E and A.N.F reported a patent for treating viral infections using hydrogen sulfide donors (US-9504701-B2).
Author Contributions
Conceptualization, O.E., P. S., J.C.C., A. N. F., and K.M.; methodology, O.E., P. S., E.L., J.C.C., and K.M.; formal analysis, O.E., P.S., and K.M.; investigation, O.E., P.S., G.T., C.L.V., B.J.L., E.L., T.L.J., and K.M.; resources, C.B.L., S.M., J.C.C., A.N.F., and K.M.; data curation, O.E, P.S., G.T., C.L.V., B.J.S., T.L.J., and C.B.L.; writing—original draft preparation, O.E, P.S., and K.M.; writing—review and editing, O.E, P.S., E.L., C.B.L., S.M., J.C.C., A.N.F., and K.M.; funding acquisition, K.M. All authors have read and agreed to the published version of the manuscript.”
Funding
This research was funded by an institutional research award to K.M. from the Department of Surgery at the University of Texas Medical Branch.
Institutional Review Board Statement
The specimens utilized for this study were collected using the Clinical Characterization Protocol for Severe Emerging Infections and conducted in accordance with the Declaration of Helsinki. The protocol was approved by the Institutional Review Board of the University of Nebraska Medical Center (Dr. David Brett-Major, protocol # 0146-20-FB, approved 02/22/2020) with local context review by the Institutional Review Board of the University of Texas Medical Branch (Dr. Susan McLellan, protocol # 20-0066, approved 06/09/2020). Control plasma was collected from healthy subjects under a protocol approved by the University of Texas Health Science Center Institutional Review Board at Houston (Dr. Jessica C. Cardenas, protocol # HSC-GEN-09-0314 approved 8/13/2018).
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
Raw data generated in the current project are available, upon request, from the author of the correspondence (K.M.).
Acknowledgments
The authors are grateful to the patients who have made this study possible by donating samples to the Biorepository for Severe Emerging Infections at the University of Texas Medical Branch, funded in part by a grant from the Sealy & Smith Foundation.
Conflicts of Interest
The authors declare no conflict of interest.
References
- World Health Organization (WHO) Coronavirus (COVID-19) Dashboard. Accessed on 18 May 2023, https://covid19.who.int.
- 2. Gebo KA, Heath SL, Fukuta Y, Zhu X, Baksh S, Abraham AG, Habtehyimer F, Shade D, Ruff J, Ram M, Laeyendecker O, Fernandez RE, Patel EU, Baker OR, Shoham S, Cachay ER, Currier JS, Gerber JM, Meisenberg B, Forthal DN, Hammitt LL, Huaman MA, Levine A, Mosnaim GS, Patel B, Paxton JH, Raval JS, Sutcliffe CG, Anjan S, Gniadek T, Kassaye S, Blair JE, Lane K, McBee NA, Gawad AL, Das P, Klein SL, Pekosz A, Casadevall A, Bloch EM, Hanley D, Tobian AAR, Sullivan DJ. Early Treatment, Inflammation and Post-COVID Conditions. medRxiv. 2023.
- Phillips S, Williams MA. Confronting Our Next National Health Disaster - Long-Haul Covid. N Engl J Med. 2021, 385, 577–579. [Google Scholar] [CrossRef] [PubMed]
- Center for Disease Control and Prevention (CDC), Long COVID or Post-COVID Conditions. Accessed on 18 May 2023, https://www.cdc.gov/coronavirus/2019-ncov/long-term-effects/index.html.
- 5. Ahamed J, Laurence J. Long COVID endotheliopathy: Hypothesized mechanisms and potential therapeutic approaches. J Clin Invest. 2022; 132.
- Chen W, Pan JY. Anatomical and Pathological Observation and Analysis of SARS and COVID-19: Microthrombosis Is the Main Cause of Death. Biol Proced Online. 2021, 23, 4. [Google Scholar]
- Halawa S, Pullamsetti SS, Bangham CRM, Stenmark KR, Dorfmuller P, Frid MG, Butrous G, Morrell NW, de Jesus Perez VA, Stuart DI, O'Gallagher K, Shah AM, Aguib Y, Yacoub MH. Potential long-term effects of SARS-CoV-2 infection on the pulmonary vasculature: A global perspective. Nat Rev Cardiol. 2022, 19, 314–331. [Google Scholar] [CrossRef]
- Libby P, Luscher T. COVID-19 is, in the end, an endothelial disease. Eur Heart J. 2020, 41, 3038–3044. [Google Scholar] [CrossRef]
- Nalbandian A, Sehgal K, Gupta A, Madhavan MV, McGroder C, Stevens JS, Cook JR, Nordvig AS, Shalev D, Sehrawat TS, Ahluwalia N, Bikdeli B, Dietz D, Der-Nigoghossian C, Liyanage-Don N, Rosner GF, Bernstein EJ, Mohan S, Beckley AA, Seres DS, Choueiri TK, Uriel N, Ausiello JC, Accili D, Freedberg DE, Baldwin M, Schwartz A, Brodie D, Garcia CK, Elkind MSV, Connors JM, Bilezikian JP, Landry DW, Wan EY. Post-acute COVID-19 syndrome. Nat Med. 2021, 27, 601–615. [Google Scholar] [CrossRef] [PubMed]
- Bourgonje AR, Abdulle AE, Timens W, Hillebrands JL, Navis GJ, Gordijn SJ, Bolling MC, Dijkstra G, Voors AA, Osterhaus AD, van der Voort PH, Mulder DJ, van Goor H. Angiotensin-converting enzyme 2 (ACE2), SARS-CoV-2 and the pathophysiology of coronavirus disease 2019 (COVID-19). J Pathol. 2020, 251, 228–248. [Google Scholar] [CrossRef]
- Deshmukh V, Motwani R, Kumar A, Kumari C, Raza K. Histopathological observations in COVID-19: A systematic review. J Clin Pathol. 2021, 74, 76–83. [Google Scholar] [CrossRef]
- Nicosia RF, Ligresti G, Caporarello N, Akilesh S, Ribatti D. COVID-19 Vasculopathy: Mounting Evidence for an Indirect Mechanism of Endothelial Injury. Am J Pathol. 2021, 191, 1374–1384. [Google Scholar] [CrossRef]
- Qian Y, Lei T, Patel PS, Lee CH, Monaghan-Nichols P, Xin HB, Qiu J, Fu M. Direct Activation of Endothelial Cells by SARS-CoV-2 Nucleocapsid Protein Is Blocked by Simvastatin. J Virol. 2021, 95, e0139621. [Google Scholar] [CrossRef]
- 14. Barilli A, Visigalli R, Ferrari F, Bianchi MG, Dall'Asta V, Rotoli BM. Immune-Mediated Inflammatory Responses of Alveolar Epithelial Cells: Implications for COVID-19 Lung Pathology. Biomedicines. 2022; 10.
- Fajgenbaum DC, June CH. Cytokine Storm. N Engl J Med. 2020, 383, 2255–2273. [Google Scholar] [CrossRef]
- Rauch A, Dupont A, Goutay J, Caplan M, Staessens S, Moussa M, Jeanpierre E, Corseaux D, Lefevre G, Lassalle F, Faure K, Lambert M, Duhamel A, Labreuche J, Garrigue D, De Meyer SF, Staels B, Van Belle E, Vincent F, Kipnis E, Lenting PJ, Poissy J, Susen S, Lille CRN, Members of the LSC. Endotheliopathy Is Induced by Plasma From Critically Ill Patients and Associated With Organ Failure in Severe COVID-19. Circulation. 2020, 142, 1881–1884. [Google Scholar] [CrossRef]
- Bonaventura A, Vecchie A, Dagna L, Martinod K, Dixon DL, Van Tassell BW, Dentali F, Montecucco F, Massberg S, Levi M, Abbate A. Endothelial dysfunction and immunothrombosis as key pathogenic mechanisms in COVID-19. Nat Rev Immunol. 2021, 21, 319–329. [Google Scholar] [CrossRef] [PubMed]
- Canzano P, Brambilla M, Porro B, Cosentino N, Tortorici E, Vicini S, Poggio P, Cascella A, Pengo MF, Veglia F, Fiorelli S, Bonomi A, Cavalca V, Trabattoni D, Andreini D, Omodeo Sale E, Parati G, Tremoli E, Camera M. Platelet and Endothelial Activation as Potential Mechanisms Behind the Thrombotic Complications of COVID-19 Patients. JACC Basic Transl Sci. 2021, 6, 202–218. [Google Scholar] [CrossRef]
- Biering SB, Gomes de Sousa FT, Tjang LV, Pahmeier F, Zhu C, Ruan R, Blanc SF, Patel TS, Worthington CM, Glasner DR, Castillo-Rojas B, Servellita V, Lo NTN, Wong MP, Warnes CM, Sandoval DR, Clausen TM, Santos YA, Fox DM, Ortega V, Naar AM, Baric RS, Stanley SA, Aguilar HC, Esko JD, Chiu CY, Pak JE, Beatty PR, Harris E. SARS-CoV-2 Spike triggers barrier dysfunction and vascular leak via integrins and TGF-beta signaling. Nat Commun. 2022, 13, 7630. [Google Scholar] [CrossRef] [PubMed]
- Colunga Biancatelli RML, Solopov PA, Sharlow ER, Lazo JS, Marik PE, Catravas JD. The SARS-CoV-2 spike protein subunit S1 induces COVID-19-like acute lung injury in Kappa18-hACE2 transgenic mice and barrier dysfunction in human endothelial cells. Am J Physiol Lung Cell Mol Physiol. 2021, 321, L477–L484. [Google Scholar] [CrossRef] [PubMed]
- 21. Rotoli BM, Barilli A, Visigalli R, Ferrari F, Dall'Asta V. Endothelial Cell Activation by SARS-CoV-2 Spike S1 Protein: A Crosstalk between Endothelium and Innate Immune Cells. Biomedicines. 2021; 9.
- Wagner JUG, Bojkova D, Shumliakivska M, Luxan G, Nicin L, Aslan GS, Milting H, Kandler JD, Dendorfer A, Heumueller AW, Fleming I, Bibli SI, Jakobi T, Dieterich C, Zeiher AM, Ciesek S, Cinatl J, Dimmeler S. Increased susceptibility of human endothelial cells to infections by SARS-CoV-2 variants. Basic Res Cardiol. 2021, 116, 42. [Google Scholar] [CrossRef] [PubMed]
- 23. Bordoni V, Mariotti D, Matusali G, Colavita F, Cimini E, Ippolito G, Agrati C. SARS-CoV-2 Infection of Airway Epithelium Triggers Pulmonary Endothelial Cell Activation and Senescence Associated with Type I IFN Production. Cells. 2022; 11.
- Muhl L, He L, Sun Y, Andaloussi Mae M, Pietila R, Liu J, Genove G, Zhang L, Xie Y, Leptidis S, Mocci G, Stritt S, Osman A, Anisimov A, Hemanthakumar KA, Rasanen M, Hansson EM, Bjorkegren J, Vanlandewijck M, Blomgren K, Makinen T, Peng XR, Hu Y, Ernfors P, Arnold TD, Alitalo K, Lendahl U, Betsholtz C. The SARS-CoV-2 receptor ACE2 is expressed in mouse pericytes but not endothelial cells: Implications for COVID-19 vascular research. Stem Cell Reports. 2022, 17, 1089–1104. [Google Scholar] [CrossRef] [PubMed]
- Schimmel L, Chew KY, Stocks CJ, Yordanov TE, Essebier P, Kulasinghe A, Monkman J, Dos Santos Miggiolaro AFR, Cooper C, de Noronha L, Schroder K, Lagendijk AK, Labzin LI, Short KR, Gordon EJ. Endothelial cells are not productively infected by SARS-CoV-2. Clin Transl Immunology. 2021, 10, e1350. [Google Scholar] [CrossRef]
- Lucas R, Verin AD, Black SM, Catravas JD. Regulators of endothelial and epithelial barrier integrity and function in acute lung injury. Biochem Pharmacol. 2009, 77, 1763–1772. [Google Scholar] [CrossRef]
- Vandenbroucke E, Mehta D, Minshall R, Malik AB. Regulation of endothelial junctional permeability. Ann N Y Acad Sci. 2008, 1123, 134–145. [Google Scholar]
- Lum H, Malik AB. Regulation of vascular endothelial barrier function. Am J Physiol. 1994, 267, L223–L241. [Google Scholar]
- Ackermann M, Verleden SE, Kuehnel M, Haverich A, Welte T, Laenger F, Vanstapel A, Werlein C, Stark H, Tzankov A, Li WW, Li VW, Mentzer SJ, Jonigk D. Pulmonary Vascular Endothelialitis, Thrombosis, and Angiogenesis in Covid-19. N Engl J Med. 2020, 383, 120–128. [Google Scholar] [CrossRef] [PubMed]
- Cirino G, Szabo C, Papapetropoulos A. Physiological roles of hydrogen sulfide in mammalian cells, tissues, and organs. Physiol Rev. 2023, 103, 31–276. [Google Scholar] [CrossRef] [PubMed]
- Olson KR, DeLeon ER, Liu F. Controversies and conundrums in hydrogen sulfide biology. Nitric Oxide. 2014, 41, 11–26. [Google Scholar] [CrossRef]
- Abdollahi Govar A, Toro G, Szaniszlo P, Pavlidou A, Bibli SI, Thanki K, Resto VA, Chao C, Hellmich MR, Szabo C, Papapetropoulos A, Modis K. 3-Mercaptopyruvate sulfurtransferase supports endothelial cell angiogenesis and bioenergetics. Br J Pharmacol. 2020, 177, 866–883. [Google Scholar] [CrossRef] [PubMed]
- Kanagy NL, Szabo C, Papapetropoulos A. Vascular biology of hydrogen sulfide. Am J Physiol Cell Physiol. 2017, 312, C537–C549. [Google Scholar] [CrossRef]
- Mendiola PJ, Naik JS, Gonzalez Bosc LV, Gardiner AS, Birg A, Kanagy NL. Hydrogen Sulfide Actions in the Vasculature. Compr Physiol. 2021, 11, 2467–2488. [Google Scholar]
- Pan LL, Liu XH, Gong QH, Wu D, Zhu YZ. Hydrogen sulfide attenuated tumor necrosis factor-alpha-induced inflammatory signaling and dysfunction in vascular endothelial cells. PLoS ONE. 2011, 6, e19766. [Google Scholar]
- Geng Y, Li E, Mu Q, Zhang Y, Wei X, Li H, Cheng L, Zhang B. Hydrogen sulfide inhalation decreases early blood-brain barrier permeability and brain edema induced by cardiac arrest and resuscitation. J Cereb Blood Flow Metab. 2015, 35, 494–500. [Google Scholar] [CrossRef]
- Li H, Zhu L, Feng J, Hu X, Li C, Zhang B. Hydrogen Sulfide Decreases Blood-Brain Barrier Damage via Regulating Protein Kinase C and Tight Junction After Cardiac Arrest in Rats. Cell Physiol Biochem. 2018, 47, 994–1006. [Google Scholar] [CrossRef]
- 38. Bourque C, Zhang Y, Fu M, Racine M, Greasley A, Pei Y, Wu L, Wang R, Yang G. H2S protects lipopolysaccharide-induced inflammation by blocking NFkappaB transactivation in endothelial cells. Toxicol Appl Pharmacol. 2018; 338, 20–29.
- Faller S, Hausler F, Goeft A, von Itter MA, Gyllenram V, Hoetzel A, Spassov SG. Hydrogen sulfide limits neutrophil transmigration, inflammation, and oxidative burst in lipopolysaccharide-induced acute lung injury. Sci Rep. 2018, 8, 14676. [Google Scholar] [CrossRef] [PubMed]
- 40. Jiang L, Jiang Q, Yang S, Huang S, Han X, Duan J, Pan S, Zhao M, Guo S. GYY4137 attenuates LPS-induced acute lung injury via heme oxygenase-1 modulation. Pulm Pharmacol Ther. 2019; 54, 77–86.
- Wang T, Wang L, Zaidi SR, Sammani S, Siegler J, Moreno-Vinasco L, Mathew B, Natarajan V, Garcia JG. Hydrogen sulfide attenuates particulate matter-induced human lung endothelial barrier disruption via combined reactive oxygen species scavenging and Akt activation. Am J Respir Cell Mol Biol. 2012, 47, 491–496. [Google Scholar] [CrossRef] [PubMed]
- 42. Santos BM, Garattini EG, Branco LGS, Leite-Panissi CRA, Nascimento GC. The therapeutic potential of cystathionine gamma-lyase in temporomandibular inflammation-induced orofacial hypernociception. Physiol Behav. 2018; 188, 128–133.
- Yuan S, Pardue S, Shen X, Alexander JS, Orr AW, Kevil CG. Hydrogen sulfide metabolism regulates endothelial solute barrier function. Redox Biol. 2016, 9, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Citi V, Martelli A, Brancaleone V, Brogi S, Gojon G, Montanaro R, Morales G, Testai L, Calderone V. Anti-inflammatory and antiviral roles of hydrogen sulfide: Rationale for considering H(2) S donors in COVID-19 therapy. Br J Pharmacol. 2020, 177, 4931–4941. [Google Scholar] [CrossRef] [PubMed]
- Yang, G. H2S as a potential defense against COVID-19? Am J Physiol Cell Physiol. 2020, 319, C244–C249. [Google Scholar] [CrossRef] [PubMed]
- Bazhanov N, Escaffre O, Freiberg AN, Garofalo RP, Casola A. Broad-Range Antiviral Activity of Hydrogen Sulfide Against Highly Pathogenic RNA Viruses. Sci Rep. 2017, 7, 41029. [Google Scholar] [CrossRef] [PubMed]
- Li H, Ma Y, Escaffre O, Ivanciuc T, Komaravelli N, Kelley JP, Coletta C, Szabo C, Rockx B, Garofalo RP, Casola A. Role of hydrogen sulfide in paramyxovirus infections. J Virol. 2015, 89, 5557–5568. [Google Scholar] [CrossRef]
- Mani S, Li H, Untereiner A, Wu L, Yang G, Austin RC, Dickhout JG, Lhotak S, Meng QH, Wang R. Decreased endogenous production of hydrogen sulfide accelerates atherosclerosis. Circulation. 2013, 127, 2523–2534. [Google Scholar] [CrossRef]
- Jain SK, Bull R, Rains JL, Bass PF, Levine SN, Reddy S, McVie R, Bocchini JA. Low levels of hydrogen sulfide in the blood of diabetes patients and streptozotocin-treated rats causes vascular inflammation? Antioxid Redox Signal. 2010, 12, 1333–1337. [Google Scholar] [CrossRef]
- Wang P, Wu L, Ju Y, Fu M, Shuang T, Qian Z, Wang R. Age-Dependent Allergic Asthma Development and Cystathionine Gamma-Lyase Deficiency. Antioxid Redox Signal. 2017, 27, 931–944. [Google Scholar] [CrossRef]
- Dominic P, Ahmad J, Bhandari R, Pardue S, Solorzano J, Jaisingh K, Watts M, Bailey SR, Orr AW, Kevil CG, Kolluru GK. Decreased availability of nitric oxide and hydrogen sulfide is a hallmark of COVID-19. Redox Biol. 2021, 43, 101982. [Google Scholar]
- 52. Oza PP, Kashfi K. Utility of NO and H2S donating platforms in managing COVID-19: Rationale and promise. Nitric Oxide. 2022; 128, 72–102.
- Renieris G, Katrini K, Damoulari C, Akinosoglou K, Psarrakis C, Kyriakopoulou M, Dimopoulos G, Lada M, Koufargyris P, Giamarellos-Bourboulis EJ. Serum Hydrogen Sulfide and Outcome Association in Pneumonia by the SARS-CoV-2 Coronavirus. Shock. 2020, 54, 633–637. [Google Scholar] [CrossRef] [PubMed]
- 54. Onikienko S, Vinokurov M, Yurinskaya M, Zemlyanoi A, Abkin S, Shaykhutdinova E, Palikov V, Ivanov A, Smirnova O, Fedyakina I, Bychkova N, Zatsepina O, Garbuz D, Evgen'ev M. The Effects of H(2)S and Recombinant Human Hsp70 on Inflammation Induced by SARS and Other Agents In Vitro and In Vivo. Biomedicines. 2022; 10.
- Escaffre O, Freiberg AN. Polyphenylene carboxymethylene (PPCM) microbicide repurposed as antiviral against SARS-CoV-2. Proof of concept in primary human undifferentiated epithelial cells. Antiviral Res. 2021, 194, 105162. [Google Scholar] [CrossRef]
- Lopez E, Peng Z, Kozar RA, Cao Y, Ko TC, Wade CE, Cardenas JC. Antithrombin III Contributes to the Protective Effects of Fresh Frozen Plasma Following Hemorrhagic Shock by Preventing Syndecan-1 Shedding and Endothelial Barrier Disruption. Shock. 2020, 53, 156–163. [Google Scholar] [CrossRef] [PubMed]
- Lopez E, Fukuda S, Modis K, Fujiwara O, Enkhtaivan B, Trujillo-Abarca R, Ihara K, Lima-Lopez F, Perez-Bello D, Szabo C, Prough DS, Enkhbaatar P. Arginine vasopressin receptor 2 activation promotes microvascular permeability in sepsis. Pharmacol Res. 2021, 163, 105272. [Google Scholar] [CrossRef]
- 58. Tufa A, Gebremariam TH, Manyazewal T, Getinet T, Webb DL, Hellstrom PM, Genet S. Inflammatory mediators profile in patients hospitalized with COVID-19: A comparative study. Front Immunol. 2022; 13, 964179.
- Chen G, Wu D, Guo W, Cao Y, Huang D, Wang H, Wang T, Zhang X, Chen H, Yu H, Zhang X, Zhang M, Wu S, Song J, Chen T, Han M, Li S, Luo X, Zhao J, Ning Q. Clinical and immunological features of severe and moderate coronavirus disease 2019. J Clin Invest. 2020, 130, 2620–2629. [Google Scholar] [CrossRef] [PubMed]
- Chen Y, Wang J, Liu C, Su L, Zhang D, Fan J, Yang Y, Xiao M, Xie J, Xu Y, Li Y, Zhang S. IP-10 and MCP-1 as biomarkers associated with disease severity of COVID-19. Mol Med. 2020, 26, 97. [Google Scholar] [CrossRef] [PubMed]
- Li L, Li J, Gao M, Fan H, Wang Y, Xu X, Chen C, Liu J, Kim J, Aliyari R, Zhang J, Jin Y, Li X, Ma F, Shi M, Cheng G, Yang H. Interleukin-8 as a Biomarker for Disease Prognosis of Coronavirus Disease-2019 Patients. Front Immunol. 2020, 11, 602395. [Google Scholar]
- Farhana A, Lappin SL: Biochemistry, Lactate Dehydrogenase. In: StatPearls. edn. Treasure Island (FL); 2023.
- Nehring SM, Goyal A, Patel BC: C Reactive Protein. In: StatPearls. edn. Treasure Island (FL); 2023.
- Bounds EJ, Kok SJ: D Dimer. In: StatPearls. edn. Treasure Island (FL); 2023.
- 65. Untereiner AA, Olah G, Modis K, Hellmich MR, Szabo C. H2S-induced S-sulfhydration of lactate dehydrogenase a (LDHA) stimulates cellular bioenergetics in HCT116 colon cancer cells. Biochem Pharmacol. 2017; 136, 86–98.
- Liu P, Bian Y, Fan Y, Zhong J, Liu Z. Protective Effect of Naringin on In Vitro Gut-Vascular Barrier Disruption of Intestinal Microvascular Endothelial Cells Induced by TNF-alpha. J Agric Food Chem. 2020, 68, 168–175. [Google Scholar] [CrossRef]
- Rochfort KD, Collins LE, McLoughlin A, Cummins PM. Tumour necrosis factor-alpha-mediated disruption of cerebrovascular endothelial barrier integrity in vitro involves the production of proinflammatory interleukin-6. J Neurochem. 2016, 136, 564–572. [Google Scholar] [CrossRef]
- Yu J, Ma Z, Shetty S, Ma M, Fu J. Selective HDAC6 inhibition prevents TNF-alpha-induced lung endothelial cell barrier disruption and endotoxin-induced pulmonary edema. Am J Physiol Lung Cell Mol Physiol. 2016, 311, L39–L47. [Google Scholar] [CrossRef] [PubMed]
- Joffre J, Rodriguez L, Matthay ZA, Lloyd E, Fields AT, Bainton RJ, Kurien P, Sil A, Calfee CS, Woodruff PG, Erle DJ, Hendrickson C, Krummel MF, Langelier CR, Matthay MA, Kornblith LZ, Hellman J, Consortium C-M-PfET, Covid-19 Associated Coagulopathy I, Thrombosis Study G. COVID-19-associated Lung Microvascular Endotheliopathy: A "From the Bench" Perspective. Am J Respir Crit Care Med. 2022, 206, 961–972. [Google Scholar] [CrossRef] [PubMed]
- Otifi HM, Adiga BK. Endothelial Dysfunction in Covid-19 Infection. Am J Med Sci. 2022, 363, 281–287. [Google Scholar] [CrossRef]
- Ali, N. Elevated level of C-reactive protein may be an early marker to predict risk for severity of COVID-19. J Med Virol. 2020, 92, 2409–2411. [Google Scholar] [CrossRef]
- Henry BM, Aggarwal G, Wong J, Benoit S, Vikse J, Plebani M, Lippi G. Lactate dehydrogenase levels predict coronavirus disease 2019 (COVID-19) severity and mortality: A pooled analysis. Am J Emerg Med. 2020, 38, 1722–1726. [Google Scholar] [CrossRef]
- Lippi G, Favaloro EJ. D-dimer is Associated with Severity of Coronavirus Disease 2019: A Pooled Analysis. Thromb Haemost. 2020, 120, 876–878. [Google Scholar] [CrossRef] [PubMed]
- Moore S, Hill EM, Dyson L, Tildesley MJ, Keeling MJ. Retrospectively modeling the effects of increased global vaccine sharing on the COVID-19 pandemic. Nat Med. 2022, 28, 2416–2423. [Google Scholar] [CrossRef]
- Watson OJ, Barnsley G, Toor J, Hogan AB, Winskill P, Ghani AC. Global impact of the first year of COVID-19 vaccination: A mathematical modelling study. Lancet Infect Dis. 2022, 22, 1293–1302. [Google Scholar] [CrossRef]
- Oliver JC, Silva EN, Soares LM, Scodeler GC, Santos AS, Corsetti PP, Prudencio CR, de Almeida LA. Different drug approaches to COVID-19 treatment worldwide: An update of new drugs and drugs repositioning to fight against the novel coronavirus. Ther Adv Vaccines Immunother. 2022, 10, 25151355221144845. [Google Scholar]
- Usher, AD. The global COVID-19 treatment divide. Lancet. 2022, 399, 779–782. [Google Scholar] [CrossRef]
- Raghavan S, Kenchappa DB, Leo MD. SARS-CoV-2 Spike Protein Induces Degradation of Junctional Proteins That Maintain Endothelial Barrier Integrity. Front Cardiovasc Med. 2021, 8, 687783. [Google Scholar] [CrossRef]
- Wu X, Xiang M, Jing H, Wang C, Novakovic VA, Shi J. Damage to endothelial barriers and its contribution to long COVID. Angiogenesis. 2023, 1–18. [Google Scholar]
- Kong Y, Han J, Wu X, Zeng H, Liu J, Zhang H. VEGF-D: A novel biomarker for detection of COVID-19 progression. Crit Care. 2020, 24, 373. [Google Scholar] [CrossRef] [PubMed]
- Pine AB, Meizlish ML, Goshua G, Chang CH, Zhang H, Bishai J, Bahel P, Patel A, Gbyli R, Kwan JM, Won CH, Price C, Dela Cruz CS, Halene S, van Dijk D, Hwa J, Lee AI, Chun HJ. Circulating markers of angiogenesis and endotheliopathy in COVID-19. Pulm Circ. 2020, 10, 2045894020966547. [Google Scholar]
- Gadotti AC, de Castro Deus M, Telles JP, Wind R, Goes M, Garcia Charello Ossoski R, de Padua AM, de Noronha L, Moreno-Amaral A, Baena CP, Tuon FF. IFN-gamma is an independent risk factor associated with mortality in patients with moderate and severe COVID-19 infection. Virus Res. 2020, 289, 198171. [Google Scholar] [CrossRef] [PubMed]
- Lev S, Gottesman T, Sahaf Levin G, Lederfein D, Berkov E, Diker D, Zaidman A, Nutman A, Ilan Ber T, Angel A, Kellerman L, Barash E, Navon R, Boico O, Israeli Y, Rosenberg M, Gelman A, Kalfon R, Simon E, Avni N, Hainrichson M, Zarchin O, Gottlieb TM, Oved K, Eden E, Tadmor B. Observational cohort study of IP-10's potential as a biomarker to aid in inflammation regulation within a clinical decision support protocol for patients with severe COVID-19. PLoS ONE. 2021, 16, e0245296. [Google Scholar]
- Kovacs-Kasa A, Zaied AA, Leanhart S, Koseoglu M, Sridhar S, Lucas R, Fulton DJ, Vazquez JA, Annex BH. Elevated Cytokine Levels in Plasma of Patients with SARS-CoV-2 Do Not Contribute to Pulmonary Microvascular Endothelial Permeability. Microbiol Spectr. 2022, 10, e0167121. [Google Scholar]
- Jin Y, Ji W, Yang H, Chen S, Zhang W, Duan G. Endothelial activation and dysfunction in COVID-19: From basic mechanisms to potential therapeutic approaches. Signal Transduct Target Ther. 2020, 5, 293. [Google Scholar] [CrossRef]
- Lei Y, Zhang J, Schiavon CR, He M, Chen L, Shen H, Zhang Y, Yin Q, Cho Y, Andrade L, Shadel GS, Hepokoski M, Lei T, Wang H, Zhang J, Yuan JX, Malhotra A, Manor U, Wang S, Yuan ZY, Shyy JY. SARS-CoV-2 Spike Protein Impairs Endothelial Function via Downregulation of ACE 2. Circ Res. 2021, 128, 1323–1326. [Google Scholar] [CrossRef]
- Papadopoulos KI, Papadopoulou A, Aw TC. Beauty and the beast: Host microRNA-155 versus SARS-CoV-2. Hum Cell. 2023, 36, 908–922. [Google Scholar] [CrossRef]
- Bouros D, Alexandrakis MG, Antoniou KM, Agouridakis P, Pneumatikos I, Anevlavis S, Pataka A, Patlakas G, Karkavitsas N, Kyriakou D. The clinical significance of serum and bronchoalveolar lavage inflammatory cytokines in patients at risk for Acute Respiratory Distress Syndrome. BMC Pulm Med. 2004, 4, 6. [Google Scholar]
- Szabo PA, Dogra P, Gray JI, Wells SB, Connors TJ, Weisberg SP, Krupska I, Matsumoto R, Poon MML, Idzikowski E, Morris SE, Pasin C, Yates AJ, Ku A, Chait M, Davis-Porada J, Guo XV, Zhou J, Steinle M, Mackay S, Saqi A, Baldwin MR, Sims PA, Farber DL. Longitudinal profiling of respiratory and systemic immune responses reveals myeloid cell-driven lung inflammation in severe COVID-19. Immunity. 2021, 54, 797–814 e796. [Google Scholar] [CrossRef]
- 90. Rauti R, Shahoha M, Leichtmann-Bardoogo Y, Nasser R, Paz E, Tamir R, Miller V, Babich T, Shaked K, Ehrlich A, Ioannidis K, Nahmias Y, Sharan R, Ashery U, Maoz BM. Effect of SARS-CoV-2 proteins on vascular permeability. Elife. 2021; 10.
- Behrens GMN, Cossmann A, Hoffmann M. Omicron spike protein: A clue for viral entry and immune evasion. Signal Transduct Target Ther. 2022, 7, 339. [Google Scholar] [CrossRef] [PubMed]
- Ou J, Lan W, Wu X, Zhao T, Duan B, Yang P, Ren Y, Quan L, Zhao W, Seto D, Chodosh J, Luo Z, Wu J, Zhang Q. Tracking SARS-CoV-2 Omicron diverse spike gene mutations identifies multiple inter-variant recombination events. Signal Transduct Target Ther. 2022, 7, 138. [Google Scholar] [CrossRef] [PubMed]
- Syed AM, Ciling A, Taha TY, Chen IP, Khalid MM, Sreekumar B, Chen PY, Kumar GR, Suryawanshi R, Silva I, Milbes B, Kojima N, Hess V, Shacreaw M, Lopez L, Brobeck M, Turner F, Spraggon L, Tabata T, Ott M, Doudna JA. Omicron mutations enhance infectivity and reduce antibody neutralization of SARS-CoV-2 virus-like particles. Proc Natl Acad Sci U S A. 2022, 119, e2200592119. [Google Scholar] [CrossRef]
- Hu B, Chan JF, Liu H, Liu Y, Chai Y, Shi J, Shuai H, Hou Y, Huang X, Yuen TT, Yoon C, Zhu T, Zhang J, Li W, Zhang AJ, Zhou J, Yuan S, Zhang BZ, Yuen KY, Chu H. Spike mutations contributing to the altered entry preference of SARS-CoV-2 omicron BA.1 and BA.2. Emerg Microbes Infect. 2022, 11, 2275–2287. [Google Scholar] [CrossRef]
- Meng B, Abdullahi A, Ferreira I, Goonawardane N, Saito A, Kimura I, Yamasoba D, Gerber PP, Fatihi S, Rathore S, Zepeda SK, Papa G, Kemp SA, Ikeda T, Toyoda M, Tan TS, Kuramochi J, Mitsunaga S, Ueno T, Shirakawa K, Takaori-Kondo A, Brevini T, Mallery DL, Charles OJ, Collaboration C-NBC-, Genotype to Phenotype Japan C, Ecuador CC, Bowen JE, Joshi A, Walls AC, Jackson L, Martin D, Smith KGC, Bradley J, Briggs JAG, Choi J, Madissoon E, Meyer KB, Mlcochova P, Ceron-Gutierrez L, Doffinger R, Teichmann SA, Fisher AJ, Pizzuto MS, de Marco A, Corti D, Hosmillo M, Lee JH, James LC, Thukral L, Veesler D, Sigal A, Sampaziotis F, Goodfellow IG, Matheson NJ, Sato K, Gupta RK. Altered TMPRSS2 usage by SARS-CoV-2 Omicron impacts infectivity and fusogenicity. Nature. 2022, 603, 706–714. [Google Scholar] [CrossRef]
- Szabo C, Papapetropoulos A. International Union of Basic and Clinical Pharmacology. CII: Pharmacological Modulation of H2S Levels: H2S Donors and H2S Biosynthesis Inhibitors. Pharmacol Rev. 2017, 69, 497–564. [Google Scholar] [CrossRef]
Figure 1.
Disease severity positively correlates with plasma cytokine profiles and routine laboratory data. (A-F) Cytokine levels in plasma samples from COVID-19 patients and healthy volunteers were measured using Bio-Plex Pro Human Cytokine 27-plex Assay (Bio-Rad) and plotted grouped by disease severity. Only the statistical differences compared to the healthy control group are highlighted in these panels; (G-I) Clinical laboratory marker measurements (all available longitudinal data) of the same patient cohort were plotted grouped by disease severity. All statistical differences found between patient groups are labeled. All results in this figure are presented as dot plots of individual mean values for each sample. Bars represent group means and standard deviations. The statistical significance was assessed by Kruskal-Wallis non-parametric one-way ANOVA test followed by Dunn's multiple comparisons. *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001.
Figure 1.
Disease severity positively correlates with plasma cytokine profiles and routine laboratory data. (A-F) Cytokine levels in plasma samples from COVID-19 patients and healthy volunteers were measured using Bio-Plex Pro Human Cytokine 27-plex Assay (Bio-Rad) and plotted grouped by disease severity. Only the statistical differences compared to the healthy control group are highlighted in these panels; (G-I) Clinical laboratory marker measurements (all available longitudinal data) of the same patient cohort were plotted grouped by disease severity. All statistical differences found between patient groups are labeled. All results in this figure are presented as dot plots of individual mean values for each sample. Bars represent group means and standard deviations. The statistical significance was assessed by Kruskal-Wallis non-parametric one-way ANOVA test followed by Dunn's multiple comparisons. *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001.
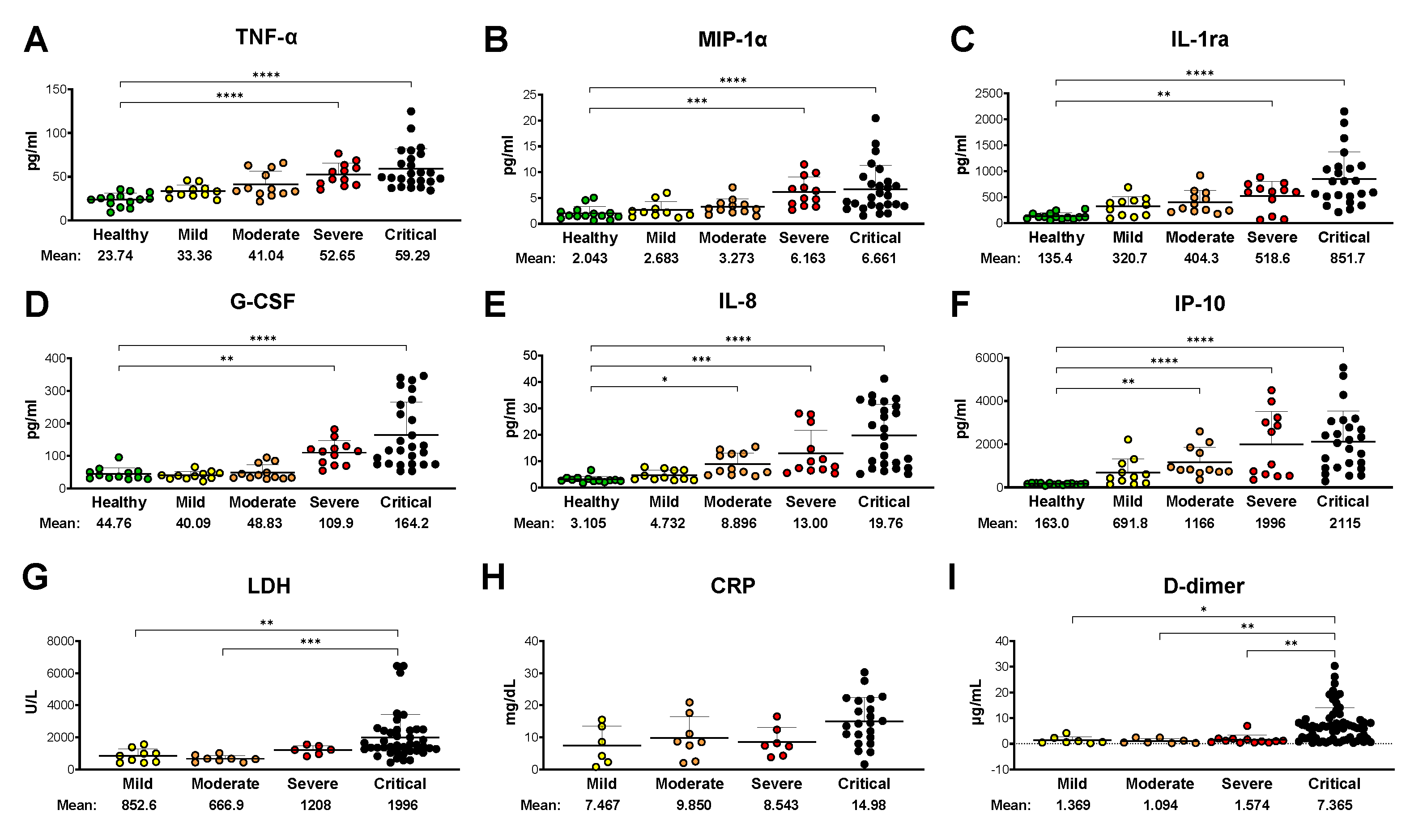
Figure 2.
Exogenous H2S released by GYY4137 increases endothelial barrier function. HLMVECs were seeded on E-plates and incubated in growth medium for 48 hours to form a confluent monolayer, then starved for 2 hours before treatment. The effects of biomolecules and patient plasma treatment on the barrier function were monitored by electrical impedance measurements (Cell Index) using the xCELLigence Real-Time Cell Analysis system. Higher Cell Index, as well as Normalized and Relative Normalized Cell Index (described in Materials and Methods) values, represent increased barrier function. (A, B) GYY4137 treatment alone raises barrier function in all tested concentrations. Representative ribbon plots (mean of 3-6 replicates of each condition) of different GYY4137 concentrations (A) and histogram (mean ± SD, n=3-9) of normalized data 12 hours after treatment (B) are presented. Statistical differences for each GYY4137 concentration compared to control at 12 hours are labeled. (C) GYY4137 treatment attenuates TNF-α–induced endothelial barrier disruption. Data are shown as mean ± SD of 3-6 measurements 12 hours after treatment. Statistical differences compared to corresponding zero control are labeled. (D) GYY4137 enhances endothelial barrier function at 12 hours after treatment with plasma samples. Ribbon plots show all measurements (as mean of 3-6 replicates of a single sample of each group) for 24 hours after the first treatment. Thirty-minute, 12-hour, and 24-hour time points are marked by blue, dashed vertical lines. The statistical significance was assessed by one-way (B) or two-way (C) ANOVA, followed by Tukey’s multiple comparisons test. ****, p < 0.0001; GYY, GYY4137.
Figure 2.
Exogenous H2S released by GYY4137 increases endothelial barrier function. HLMVECs were seeded on E-plates and incubated in growth medium for 48 hours to form a confluent monolayer, then starved for 2 hours before treatment. The effects of biomolecules and patient plasma treatment on the barrier function were monitored by electrical impedance measurements (Cell Index) using the xCELLigence Real-Time Cell Analysis system. Higher Cell Index, as well as Normalized and Relative Normalized Cell Index (described in Materials and Methods) values, represent increased barrier function. (A, B) GYY4137 treatment alone raises barrier function in all tested concentrations. Representative ribbon plots (mean of 3-6 replicates of each condition) of different GYY4137 concentrations (A) and histogram (mean ± SD, n=3-9) of normalized data 12 hours after treatment (B) are presented. Statistical differences for each GYY4137 concentration compared to control at 12 hours are labeled. (C) GYY4137 treatment attenuates TNF-α–induced endothelial barrier disruption. Data are shown as mean ± SD of 3-6 measurements 12 hours after treatment. Statistical differences compared to corresponding zero control are labeled. (D) GYY4137 enhances endothelial barrier function at 12 hours after treatment with plasma samples. Ribbon plots show all measurements (as mean of 3-6 replicates of a single sample of each group) for 24 hours after the first treatment. Thirty-minute, 12-hour, and 24-hour time points are marked by blue, dashed vertical lines. The statistical significance was assessed by one-way (B) or two-way (C) ANOVA, followed by Tukey’s multiple comparisons test. ****, p < 0.0001; GYY, GYY4137.
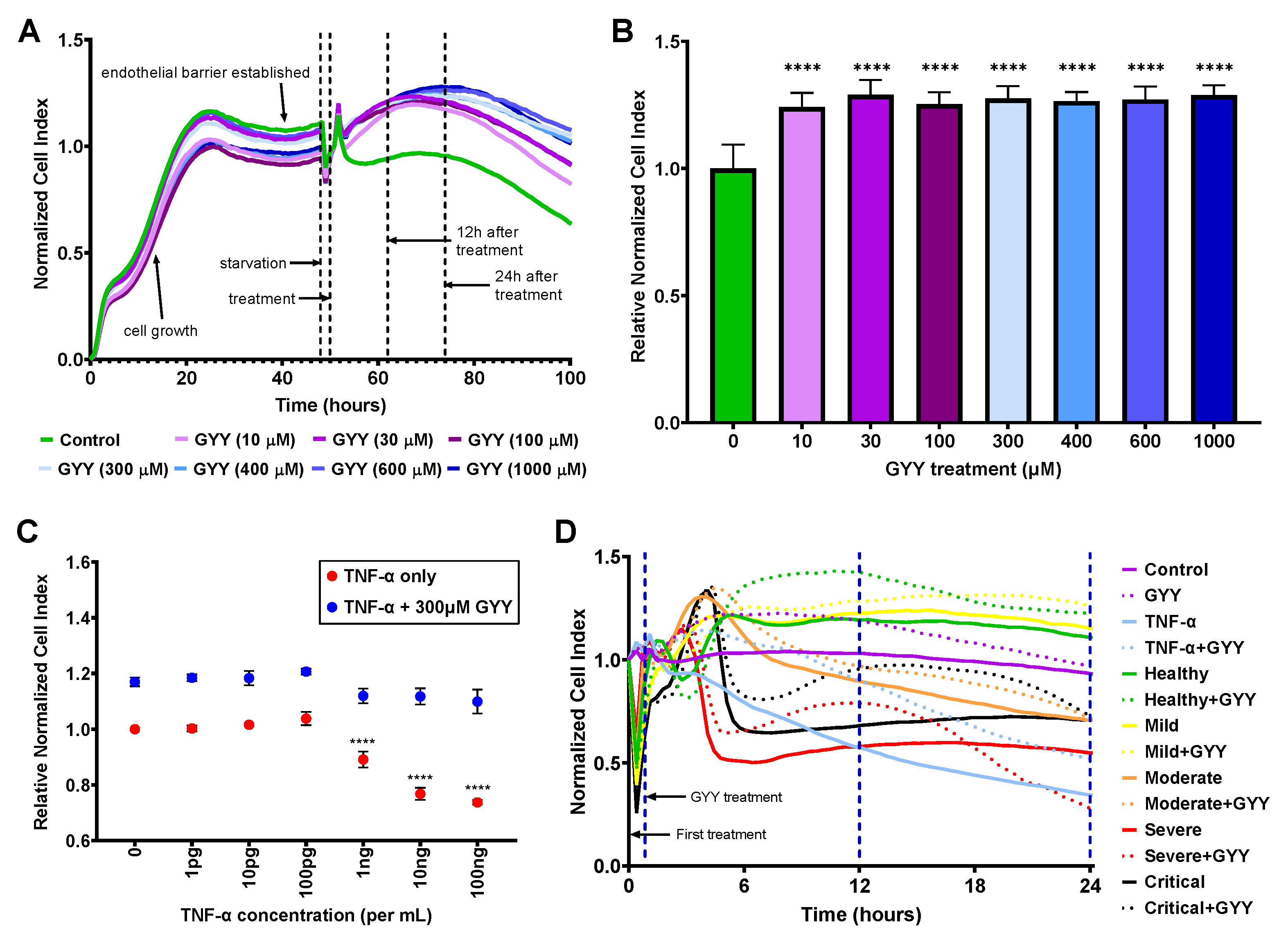
Figure 3.
GYY4137 treatment ameliorates endothelial barrier disruption regardless of disease status. (A, B) Barrier function of an HLMVEC monolayer after treatment with COVID-19 or healthy plasma samples and a second treatment using 300 µM GYY4137 was monitored by electrical impedance measurements using the xCELLigence Real-Time Cell Analysis system. Results are expressed as Relative Normalized Cell Index (RNCI, described in Materials and Methods). Data were collected from at least three independent experiments for each sample (n = 3-6). RNCI values of HLMVEC monolayer after 12h incubation with healthy or COVID-19 plasma samples (A) and the effect of GYY4137 on the RNCI of the plasma-treated HLMVEC monolayer at 12 hours (B) were plotted by disease severity. Data are presented as dot plots of mean values from 3-6 repeated measurements of each sample. Bars represent group means and standard deviations. No statistical differences among disease severity groups were found. (C) Cytokine levels in the plasma samples used for barrier function experiments were measured using Bio-Plex Pro Human Cytokine 27-plex Assay (Bio-Rad) and plotted against the corresponding RNCI values at 12 hours in scatterplots. Slope P-values were calculated to determine statistically significant levels of correlation. None were found. (D) Treatment efficacy plot showing RNCI of HLMVEC monolayer after plasma treatment with or without the addition of GYY4137. Dots represent means of 3-6 measurements for each treatment obtained at the 12-hour timepoint. Statistical difference between RNCI values of untreated and GYY-treated samples is shown. (E) The effect of GYY4137 treatment for each plasma sample was plotted grouped by the level of plasma-induced barrier disruption. Bars represent group means + SD of all measurements (3-6 measurements per sample) 12 hours after treatment. Selected statistical differences among groups are labeled. The statistical significance was assessed by Kruskal-Wallis non-parametric one-way ANOVA test followed by Dunn's multiple comparisons (A, B), simple linear regression (C), 2-tailed, non-parametric, Wilcoxon matched-pairs signed rank t-test (D), and one-way ANOVA followed by Tukey’s multiple comparisons test (E). ****, p < 0.0001; ns, p > 0.05; GYY, GYY4137.
Figure 3.
GYY4137 treatment ameliorates endothelial barrier disruption regardless of disease status. (A, B) Barrier function of an HLMVEC monolayer after treatment with COVID-19 or healthy plasma samples and a second treatment using 300 µM GYY4137 was monitored by electrical impedance measurements using the xCELLigence Real-Time Cell Analysis system. Results are expressed as Relative Normalized Cell Index (RNCI, described in Materials and Methods). Data were collected from at least three independent experiments for each sample (n = 3-6). RNCI values of HLMVEC monolayer after 12h incubation with healthy or COVID-19 plasma samples (A) and the effect of GYY4137 on the RNCI of the plasma-treated HLMVEC monolayer at 12 hours (B) were plotted by disease severity. Data are presented as dot plots of mean values from 3-6 repeated measurements of each sample. Bars represent group means and standard deviations. No statistical differences among disease severity groups were found. (C) Cytokine levels in the plasma samples used for barrier function experiments were measured using Bio-Plex Pro Human Cytokine 27-plex Assay (Bio-Rad) and plotted against the corresponding RNCI values at 12 hours in scatterplots. Slope P-values were calculated to determine statistically significant levels of correlation. None were found. (D) Treatment efficacy plot showing RNCI of HLMVEC monolayer after plasma treatment with or without the addition of GYY4137. Dots represent means of 3-6 measurements for each treatment obtained at the 12-hour timepoint. Statistical difference between RNCI values of untreated and GYY-treated samples is shown. (E) The effect of GYY4137 treatment for each plasma sample was plotted grouped by the level of plasma-induced barrier disruption. Bars represent group means + SD of all measurements (3-6 measurements per sample) 12 hours after treatment. Selected statistical differences among groups are labeled. The statistical significance was assessed by Kruskal-Wallis non-parametric one-way ANOVA test followed by Dunn's multiple comparisons (A, B), simple linear regression (C), 2-tailed, non-parametric, Wilcoxon matched-pairs signed rank t-test (D), and one-way ANOVA followed by Tukey’s multiple comparisons test (E). ****, p < 0.0001; ns, p > 0.05; GYY, GYY4137.
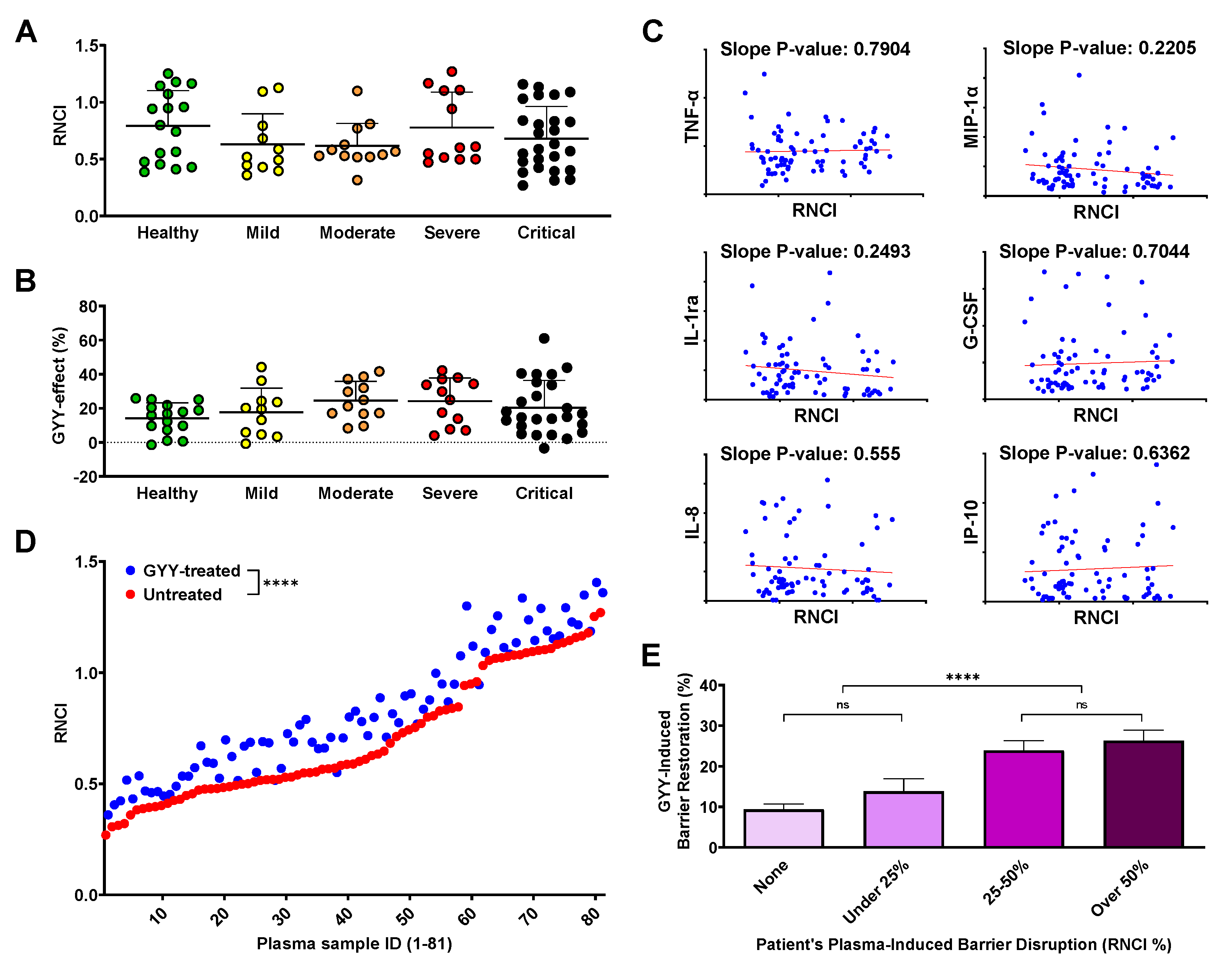
Figure 4.
GYY4137 treatment restores endothelial barrier integrity disrupted by inactivated SARS-CoV-2 Omicron BA.1. Barrier function of an HLMVEC monolayer after treatment with inactivated SARS-CoV-2 or 10 ng/mL TNF-α and a second treatment with 300 µM GYY4137 was monitored by electrical impedance measurements using the xCELLigence Real-Time Cell Analysis system and expressed as Normalized Cell Index and Relative Normalized Cell Index (NCI and RNCI, respectively, described in Materials and Methods). Representative plots of three independent experiments are shown. (A) Ribbon plots show all NCI measurements (as mean of 6-12 replicates) for 24 hours after the first treatment. 12-hour time point is marked by dashed vertical line. (B) Box and whiskers plots represent RNCI data 12 hours after treatment (6-12 replicates per treatment). The box extends from the 25th to 75th percentiles of each sample set, the whiskers go down to the smallest value and up to the largest. The line in the middle of the box is plotted at the median. Selected statistical differences among treatments are labeled. The statistical significance was assessed by one-way ANOVA followed by Tukey’s multiple comparisons test using Graph Pad Prism 9. ****, p < 0.0001; ns, p > 0.05; GYY, GYY4137; Omicron, SARS-CoV-2 Omicron B.1.1.529.
Figure 4.
GYY4137 treatment restores endothelial barrier integrity disrupted by inactivated SARS-CoV-2 Omicron BA.1. Barrier function of an HLMVEC monolayer after treatment with inactivated SARS-CoV-2 or 10 ng/mL TNF-α and a second treatment with 300 µM GYY4137 was monitored by electrical impedance measurements using the xCELLigence Real-Time Cell Analysis system and expressed as Normalized Cell Index and Relative Normalized Cell Index (NCI and RNCI, respectively, described in Materials and Methods). Representative plots of three independent experiments are shown. (A) Ribbon plots show all NCI measurements (as mean of 6-12 replicates) for 24 hours after the first treatment. 12-hour time point is marked by dashed vertical line. (B) Box and whiskers plots represent RNCI data 12 hours after treatment (6-12 replicates per treatment). The box extends from the 25th to 75th percentiles of each sample set, the whiskers go down to the smallest value and up to the largest. The line in the middle of the box is plotted at the median. Selected statistical differences among treatments are labeled. The statistical significance was assessed by one-way ANOVA followed by Tukey’s multiple comparisons test using Graph Pad Prism 9. ****, p < 0.0001; ns, p > 0.05; GYY, GYY4137; Omicron, SARS-CoV-2 Omicron B.1.1.529.
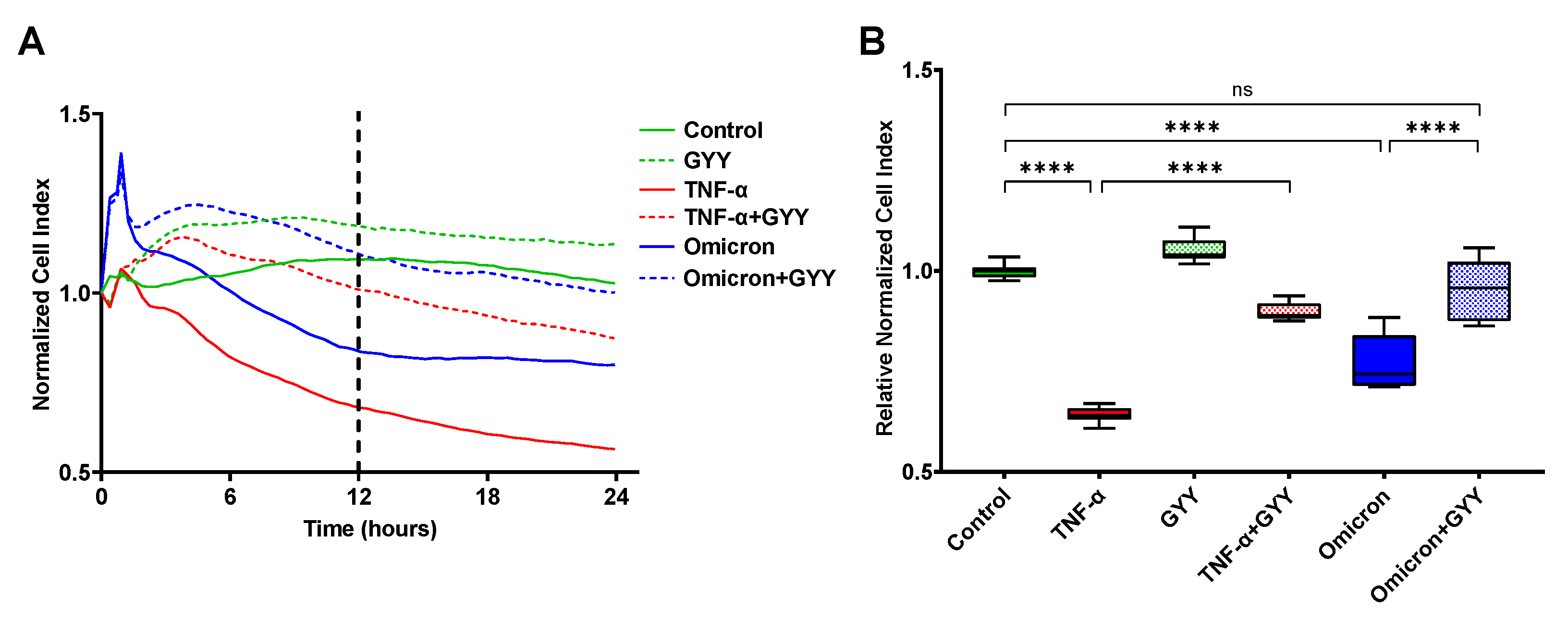

Table 1.
Demographic data, clinical characteristics and laboratory findings of COVID-19 patients included in the study. Data sets of clinical measurements at the time of admission (BMI, temperature, oxygen saturation, respiration rate, LDH, CRP, D-dimer) were analyzed for statistical differences between disease severity groups. None were found. The statistical significance was assessed by Kruskal-Wallis non-parametric one-way ANOVA test followed by Dunn's multiple comparisons and one-way ANOVA followed by Tukey’s multiple comparisons test. a highest level of oxygen delivery required; BMI, body mass index; LDH, lactate dehydrogenase; CRP, C-reactive protein.
Table 1.
Demographic data, clinical characteristics and laboratory findings of COVID-19 patients included in the study. Data sets of clinical measurements at the time of admission (BMI, temperature, oxygen saturation, respiration rate, LDH, CRP, D-dimer) were analyzed for statistical differences between disease severity groups. None were found. The statistical significance was assessed by Kruskal-Wallis non-parametric one-way ANOVA test followed by Dunn's multiple comparisons and one-way ANOVA followed by Tukey’s multiple comparisons test. a highest level of oxygen delivery required; BMI, body mass index; LDH, lactate dehydrogenase; CRP, C-reactive protein.
| Disease Severity | Mild (n=7) | Moderate (n=6) | Severe (n=4) | Critical (n=9) |
|---|---|---|---|---|
| Room aira (%) | 100.0% | 0.0% | 0.0% | 0.0% |
| Nasal cannulaa (%) | 0.0% | 100.0% | 0.0% | 0.0% |
| Non-invasive ventilationa (%) | 0.0% | 0.0% | 100.0% | 0.0% |
| Invasive ventilationa (%) | 0.0% | 0.0% | 0.0% | 100.0% |
| Male (%) | 57.1% | 33.3% | 75.0% | 66.7% |
| Age at Admission, median (years) | 55 | 59 | 54 | 55 |
| Hispanic ethnicity (%) | 28.6% | 33.3% | 0.0% | 33.3% |
| White race (%) | 57.1% | 83.3% | 75.0% | 55.6% |
| Black race (%) | 42.9% | 16.7% | 25.0% | 44.4% |
| Days admitted, median (days) | 5 | 7 | 21 | 36 |
| On dexamethasone (%) | 14.3% | 50.0% | 75.0% | 100.0% |
| Taking remdesivir (%) | 57.1% | 83.3% | 100.0% | 100.0% |
| No antiviral used (%) | 42.9% | 16.7% | 0.0% | 0.0% |
| COVID vaccinated (%) | 0.0% | 0.0% | 0.0% | 0.0% |
| Discharged alive (%) | 100.0% | 100.0% | 75.0% | 11.1% |
| Transferred to another facility (%) | 0.0% | 0.0% | 25.0% | 33.3% |
| Death (%) | 0.0% | 0.0% | 0.0% | 55.6% |
| Clinical characteristics at admission (mean ± SD) | ||||
| BMI | 34.80 ± 6.41 | 31.34 ± 4.50 | 39.53 ± 15.30 | 37.8 ± 9.24 |
| Body weight (kg) | 99.07 ± 19.04 | 85.98 ± 9.45 | 112.71 ± 22.38 | 113.42 ± 32.73 |
| Temperature (Degrees, ᵒC) | 37.10 ± 0.43 | 37.46 ± 1.13 | 38.00 ± 0.69 | 37.35 ± 1.00 |
| Oxygen saturation (%) | 96.14 ± 2.96 | 95.16 ± 2.22 | 90.75 ± 14.08 | 89.33 ± 4.35 |
| Respiration rate (breaths/minute) | 21.85 ± 5.04 | 21.50 ± 3.56 | 27.75 ± 17.63 | 26.77 ± 8.65 |
| LDH (U/L) | 750.45 ± 466.14 | 785.25 ± 204.77 | 1167.33 ± 322.76 | 1065.5 ± 389.10 |
| CRP (mg/dL) | 8.50 ± 6.14 | 13.67 ± 6.59 | 10.36 ± 5.31 | 14.49 ± 5.77 |
| D-dimer (µg/mL) | 1.31 ± 1.48 | 0.57 ± 0.52 | 0.78 ± 0.36 | 1.63 ± 1.77 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



