
Submitted:
23 May 2023
Posted:
25 May 2023
You are already at the latest version
Abstract
Biotic and abiotic factors influence the formation of fungal-algal pairings in lichen symbiosis. It is poorly known which factors determine the specific associations, especially when distantly related fungi are considered. Here, we investigated the effect of different drivers on the association patterns of taxonomically diverse lichenized fungi and their trebouxioid symbiotic partners. In 200 samples collected across three biomes, we found 41 species of lichenized fungi, associating with 16 species of trebouxioid green algae, of which 62% were previously unreported. Species identity of the fungal and algal partner had the greatest effect on the outcome of the symbiosis compared with abiotic factors such as climatic variables and geographic distance. Partner specificity was found to be higher in tropical regions than in temperate and arctic regions. Co-phylogenetic analyses indicated congruent phylogenies of trebouxioid algae and associated fungi, rejecting their random associations. Evolutionary mechanisms contributing most to the observed phylogenetic patterns were ‘loss’ and ‘failure to diverge’ of the algal partners. This study broadens our knowledge of fungi-algae symbiotic patterns in view of Trebouxia-associated fungi.

Keywords:
Trebouxia
; lichen
; symbiosis
; association
; coevolution
1. Introduction
Lichens are important components of Fungi, accounting for about 20% of known fungal species [1]. Lichenization events have happened independently several times in the evolutionary history of Ascomycota and Basidiomycota [2,3]. The peculiar mutualistic symbiosis of lichens, composed of fungi, green algae, or cyanobacteria [4,5], helps them predominantly exist in various extreme environments such as polar, plateau, and desert regions [6], playing a crucial role in the early formation of terrestrial ecosystems [7].
Up to now, there are nearly 20,000 accepted lichen-forming fungi (LFF) species [1], while lichen-forming algae (LFA) species are no more than 200 [8]. The composition of LFF and LFA in lichen symbiosis is not symmetrical [9], however, the association between LFF and LFA is far more complex than the ratio of species number. Both biotic and abiotic factors including cophylogeny of LFF and LFA, geography, and reproductive strategy of LFF, influence the formation of fungal-algal pairings in the lichen symbiosis.
Various degrees of availability, selectivity, and specificity between LFF and LFA are the most frequent descriptions for driving the lichen associations. Specificity is defined as the narrow taxonomic range of interacting partners, whereas selectivity refers to the frequency of association with distinct partners [10]. Generally, compared with LFA-LFF association with higher selectivity, higher specificity of LFF-LFA association has been discovered at any taxonomic level. For example, at the order level, the LFF Arthoniales, Ostropales, Pyrenulales, and Trypetheliales, mainly distributed in the tropics, are preferentially associated with green algae Trentepohliales, and Lecanorales and Teloschistales are mainly associated with green algae Trebouxiales [11]. Most LFF genera and species choose one green alga or cyanobacterial genus and one specific species as their LFA, respectively [12,13,14], except for some scarce examples such as species in Lobaria, Peltigera, and Pseudocyphellaria of Peltigerales, which can associate with two different LFA Domains like cyanobacteria and green algae [14,15]. Until now, almost all researches on symbiont association patterns were performed using a specific lichen taxon as a model, in which genus level and family level associations are most common [8,12,15,16,17]. It is poorly known the association pattern between LFF and LFA when distantly related fungi are considered.
Different lichen taxa usually show different responses to diverse geography on the LFF-LFA association. Some LFF are not affected by the geographical factors that show strong specificity to the LFA across remarkable global ranges, such as the associations between the genus Oropogon and limited Trebouxia OTUs, Omphalina and Coccomyxa, and Evernia mesomorpha and Trebouxia jamesii s.l. [12,18,19]. While in some lichens, geographic distributions lead to different patterns of symbioses, such as Stereocaulon and Sticta [16,17,20]. The ecological preference of LFA would indirectly affect lichen distribution, different LFF associate with the same LFA generally exhibiting similar ecological properties [21,22]. Asexual reproduction is proceeded by vegetative propagules, such as isidia, soredia, and lobules in lichens, by which lichens colonize new habitats by co-dispersing LFF and LFA (vertical transmission). Sexual reproduction depends on the spores of LFF, and new symbioses form until LFF meets the suitable LFA (horizontal transmission) [23]. Asexual reproduction would be apt to structure stable lichen mutualistic associations but possibly result in low LFA diversity. In contrast, the propagation distance would be farther, and high LFA diversity occurs through sexual reproduction [21]. Nevertheless, these studies are either still not considering distantly related fungi or only limited to a certain geographical zone, it is not clear the association pattern between LFF and LFA when multi types of geographical zones as well as different LFF orders are considered simultaneously.
Among the known lichen symbionts, 50 % –70% are symbiotic with green algae Trebouxiaceae, of which the LFA genus Trebouxia is the most common [8]. There is an ongoing interest in understanding the diversity and evolutionary relationships in Trebouxia-associated lichens [12]. Recently, a total of 109 to 113 candidate species lineages were determined, a four-fold increase from the 29 formally described Trebouxia species, thereby underestimating Trebouxia diversity [5]. Nonetheless, very few among the nearly 1,600 samples in these studies were collected from China, encompassing a cryptic or unknown diversity of Trebouxia considering its extensive ecogeographical range. Therefore, it provides an impetus for intensively studying the diversity of Trebouxia-associated lichens in China to understand their evolutionary pattern. Furthermore, Trebouxia-associated lichens occur in different fungal orders and across different climatic zones.
In this study, we chose Trebouxia-associated lichen symbionts as a model, considering different taxonomic levels (including order) and distinct biomes (mainly from China) at the same time. A total of 200 samples were included here, among which we found 41 species of lichenized fungi, associating with 16 species of trebouxioid green algae, of which 62% previously unreported. We demonstrated that species identity of the fungal and algal partner had the greatest effect on the outcome of the symbiosis. Partner specificity was higher in tropical regions. Co-phylogenetic analyses indicated congruent phylogenies of fungi and algae, in which loss’ and ‘failure to diverge’ of the algal partners were observed to contribute most to the coevolutionary mechanisms. Overall, we demonstrated a biotic factor (partner identity) is the more important driver of symbiosis of Trebouxia spp. and associated fungal partners over a broader taxonomic and ecological context, which broadens our knowledge of fungi-algae symbiotic patterns.
2. Results
2.1. Molecular sequence data and phylogenetic analysis
We generated 1,238 DNA sequences for this study, including 196 ITS, 188 nuLSU, 176 mtSSU, and 136 RPB1 sequences from LFF and 186 ITS, 170 nuSSU, and 186 rbcL sequences from LFA (Table S1). The BLASTn of ITS sequences against the GenBank database indicated the LFF was grouped into 41 species, 29 genera, 14 families, eight orders, and two classes in Ascomycota, of which 40 species belonging to Lecanoromycetes, and one species to Candelariomycetes, by the high identity of ITS sequences with the described species. The species delimitation was confirmed with their phylogenetic topology using the ITS dataset (Figure S1) and the four-locus concatenated phylogeny (Figure S2).
All the LFA belong to Trebouxia based on their ITS sequence analysis using the BLASTn tool. The phylogenetic topology of Trebouxia was constructed based on the combined ITS and rbcL sequences by incorporating sequences from the study by Muggia, Nelsen, Kirika, Barreno, Beck, Lindgren, Lumbsch and Leavitt [5] (Figure S3). Three DNA species delimitation analyses (GMYC, bPTP, and ABGD) were used to confirm the species boundaries (Figure S4). Sixteen species were recovered, and ten were undescribed (Figures S3 and S5).
2.2. Mutualistic association pattern between LFF and LFA
Fifty-one associations were observed between 41 LFF and 16 LFA (Figure 1; Table S1). At the class level, the LFF species in Lecanoromycetes were associated with all 16 LFA species. At the order level, the LFF orders, including samples of more than two species, were associated with multiple LFA species, that is, the species of Lecanorales, Caliciales, Teloschistales, and Pertusariales, were associated with ten, six, three, and two LFA species, respectively. Similarly, at the family level, the LFF species of Parmeliaceae, Megasporaceae, Teloschistaceae, and Physciaceae, including samples of more than two species, were associated with six, four, three, and two LFA species, respectively. At the genus level, Circinaria was the only genus that included samples of more than two species, which was associated with two LFA species. Each LFF species was associated with one to four LFA species at the species level. Diplotomma alboatrum had the most potent selectivity but the poorest specificity to LFA, with the association ratio being 1 vs. 4.
Each Trebouxia species was compatible with one to 11 LFF species, and 11 of 16 Trebouxia species were associated with more than one LFF (Figure 1). Five LFA species, viz. Trebouxia arboricola, T. cretacea, T. impressa, T. jamesii, and Trebouxia sp. 10 were associated with four to ten LFF species across multiple families, genera, and species, among which the first four LFA species even were associated with multiple species distributed into different orders of LFF. Trebouxia cretacea had the most robust selectivity but the poorest specificity to LFF, which accepted 11 LFF species across two classes, seven orders, seven families, and eight genera.
Four pairs of associations between LFF and LFA have unique specificity no matter the source of the samples, i.e., Hypogymnia hypotrypa vs. Trebouxia sp. 5, Menegazzia subsimilis vs. T. sp. 4, Parmotrema tinctorum vs. T. corticola, and Psoroma fruticulosum vs. T. sp. 6. Trebouxia sp. 8 was found to symbiosis with two LFF species, namely Heterodermia japonica and H. speciose; however, 95% samples (21 of 22) were H. japonica. Most of these specific associations happened in tropic/subtropic zones, indicating that the LFF-LFA association tends to be more specific in warmer climates. The mutualistic associations corresponding to the three climate zones are summarized in Table 1. Interaction network analyses further revealed that the tropic and subtropic networks had lower web connectance (0.15), linkage density (2.13), and nestedness (22.92) and were segregated into more compartments (6) compared with the cold and temperate networks.
2.3. Correlation among LFF, LFA, and ecological factors
We performed variation partitioning analysis to assess the relative contributions of climatic variables, geographical distance, LFF or LFA, distribution types, reproductive modes, and host specificity to shape the mutualistic association of lichens (Figure 2). For the LFA, climatic variables, geographical distance, LFF, and the other factors explained 36% of the variation (Figure 2a). The LFF explained the most significant proportion of the variation with 12% independent effect and 14% in combination with other variables. Distribution types, reproduction modes, and host specificity independently explained 3% of the variation, whereas 16% was shared with other variables. Geographical distance and climatic variables independently explained 1% and 2% of the variation, and 5% and 10% shared with other variables, respectively. On the other hand, climatic variables, geographical distance, LFA, and other factors explained 28% of the variation of the LFF (Figure 2b). The LFA explained the most significant proportion of variation with 11% independent effect and 12% in combination with other variables. The climatic variables independently explained 3% of the total variation and 7% shared with other variables, followed by 2% and 9% of variation explained by distribution types, reproduction modes, and host specificity independently and in combination with other variables, respectively. The geographical distance explained 3% when combined with other variables while zero independently.
2.4. Cophylogenetic analyses
The cophylogenetic analyses were performed using the well-supported LFF and LFA phylogenies. The global-fit tests (ParaFit and PACo) supported overall congruence between tree topologies of fungal hosts and Trebouxia species (ParaFit Global = 0.08, P = 0.001; PACo m2 global value = 2.73, P = 0, Figure S6), and rejected the null hypothesis of random association. Sixteen out of the 51 mutualistic associations (31.4%) contributed significantly to Parafit Global based on ParaFit1 values and 18 associations (35.3%) based on ParaFit2 values (P ≤ 0.05).
Most cost scenarios tested in Jane 4.0 supported significant congruence between LFF and LFA phylogenies. All random solutions were worse than the solution reconstructed by the program, except for costs H and Q (Table 2). Among the significant reconstructions, cost regime B yielded the lowest overall cost that suggested one cospeciation, eight duplications, six duplications with host-switches, 90 losses, and 35 failures to diverge events leading to the congruent phylogenies between LFF and LFA (Figure S7). The roles of losses and failure to diverge were vital and all the cost regimes showed the same solution regardless of the penalized values.
3. Discussion
Accurate species delimitation of symbiotic partners is a crucial prerequisite. Otherwise, a biased identity will result in masked interactions [15]. We performed phylogenetic analysis using the dataset in Muggia, Nelsen, Kirika, Barreno, Beck, Lindgren, Lumbsch and Leavitt [5] as the backbone to construct the phylogeny of Trebouxia species. Besides, we used three species delimitation approaches to define the algal species boundary. The results corresponded well with those of Muggia, Nelsen, Kirika, Barreno, Beck, Lindgren, Lumbsch and Leavitt [5] (Figure S3). Sixteen Trebouxia species were delimitated, of which ten (62.5%) are potentially new species to science, which supports the cryptic Trebouxia species diversity has been dramatically underestimated. Some inconsistent delimited results occurred that would make the potential Trebouxia species diversity higher. For example, within the Heterodermia japonica/H. speciosa clade, four or six Trebouxia species were recognized by GMYC (ITS), bPTP (ITS), ABGD (ITS + rbcL), and bPTP (ITS + rbcL), whereas only one species was adopted by GMYC (ITS + rbcL), ABGD (ITS), and Muggia, Nelsen, Kirika, Barreno, Beck, Lindgren, Lumbsch and Leavitt [5] (Figures S2, S4 and S5). We chose the last delimitation considering some samples in this clade growing in the same locality, even on the same bark, such as QJ2-1, QJ2-2, and QJ2-8. In a clade composed of 18 samples representing eight lichen species (Bryoplaca tetraspora, Ochrolechia tartarea, Gondwania regalis, Megaspora verrucosa, Lecanora fuscobrunnea, Physcia caesia, Physcia dubia, and Rhizoplaca chrysoleuca), all the delimitation methods accepted only one Trebouxia species except for GMYC (ITS) supporting two (Figures S4,A5). Hence, only one Trebouxia species was accepted according to the rule of the majority.
Our data showed that 32 (78%) of the 41 LFF species were associated with only one LFA species. In comparison, 11 (69%) of 16 LFA species accepted more than one LFF species (Figure 1). These asymmetric mutualistic association patterns indicated higher selectivity and weaker specificity from the perspective of LFA choosing LFF over the reverse. Previous interaction network studies have hypothesized that the asymmetry may result from the uneven distribution of fungal hosts and algal species, called an abundance-asymmetry hypothesis [24,25]. This hypothesis was under further development that species abundance would determine interspecific interaction frequency and strength and the resulting asymmetry structure if individuals in a community interact randomly [26]. The known species abundance differs about one hundred times between LFF and LFA (Kroken & Taylor, 2000; Lücking et al., 2017), which inevitably impacts the mutualistic association patterns. The reproduction modes also affect the asymmetric mutualistic association patterns. LFF have both sexual and asexual reproduction modes [27]. Macrolichens, like most species included in our studies, generally develop asexual structures like isidia and soredia (Bowler & Rundel, 1975), by which LFF disperse together with their LFA. Asexual reproduction was thought to be the only propagation strategy in Trebouxia-containing lichens [28]. Therefore, LFF tends to act as a specialist in mutualistic association. However, for LFA, Trebouxia species are seldom found in a free-living state and even cannot be alive when lichens die; therefore, they tend to act as generalists in the mutualistic association chosen by LFF, especially in a harsh environment [29,30].
Furthermore, in our study, different symbiotic partners showed different association patterns across different climatic zones and ecoregions. The network analyses supported that boreal/arctic and temperate zones had higher web connectance, linkage density, and nestedness than the tropic/subtropic zones, indicating higher reciprocal specificity between the symbiotic partners in the tropics, which is consistent with the finding of Singh, et al. [31]. More than 56% of mutualistic associations are specifically one-to-one in the tropics compared to 10%–12% in boreal/arctic and temperate zones. There is a higher selectivity of LFA in arctic/temperate regions than tropic/subtropic regions as it associates with eight to ten LFF in the former regions but at most five LFF in the latter regions, suggesting frequent mycobiont switches happened especially in harsh environments such as Antarctic [10,32]. On the contrary, the patterns of LFF specificities toward LFA remain unchanged across different ecoregions and climatic zones, in which most LFF maintain relationships with one LFA except for a very few cases, such as Circinaria tortuosa, Diplotomma alboatrum, Gondwania regalis, Lobothallia semisterilis, Ochrolechia tartarea, Parmotrema clavuliferum, and Ramalina calicaris associating two or three LFA species. Among all the mutualistic associations, the most apparent modularity can be well shown by Hypogymnia hypotrypa because both LFF and LFA acted as one-to-one specialists. This high reciprocal specificity is most possibly related to their vertical transmissions via soredia and the limited niche of LFA, which was also found in Evernia mesomorpha, belonging to the same family as Hypogymnia hypotrypa [13,19].
The symbiotic partners have the most significant impact that drives mutualism, with 11%–12% independent effects (12%–14% in combination with other variables). The contribution of LFF and LFA to their corresponding partners is based on the genetic distance of their phylogenies. Therefore, the effects of the symbiotic partners could be understood as their cophylogenetic trend; that is, the effect value here is roughly equivalent to the degree of cophylogenetic evolution. Variation partitioning analyses showed that the geographical location (GPS) and 19 climate factors related to the temperature and precipitation contributed no more than 3%. Similar results were found in many other symbiosis analyses, in which the symbiotic partners mainly drove the mutualism, whereas ecological mechanisms and geographical distributions such as extreme environments or cooler climate zones partly explained the variation [12,16,23].
There are still 64% and 72% unknown interpretations that need further exploration. One of these unknown factors is historical climatological and geological processes that have turned out to affect the current species distribution patterns [33]. Whether or to what extent this factor brings the effect is not known in this study. Besides, the research model in this study is Trebouxia-associated lichens that distribute across different genera, families, and orders, which behave in much more complex interactions than the studies focusing on species or genus level. A much higher explanation of the variation within one fungal genus could be easily obtained [16,31].
Cophylogeny evaluates the dependency of two associated groups of organisms to illuminate how ecological and evolutionary processes affect species diversification [34]. Our study rejected the null hypothesis of a random association between LFF and LFA, as previous studies reported [5,17,35,36], indicating the significant congruence between Trebouxia algae and its associated LFF. Loss and failure of LFA to diverge played a much more critical role than cospeciation, duplication, and host switch driving the coevolution in lichen mutualism (Figure S7). It has been verified that cospeciation between symbionts seldom happened except for those with strictly vertical transmission instead of the reciprocal selection by mutualistic partners such as lichens [37]. Host-switches, failure of the LFA to diverge, and losses are commonly found to be the prevalent events shaping lichen symbionts, such as Protoparmelia-Trebouxia, Sticta-Symbiochloris, and Cladonia-Trebouxia [17,31,38]. Failure of the LFA to diverge with its fungal hosts is known to be responsible for the occurrence of the generalist algal species [17,39], as shown by one Trebouxia species associated with multiple fungal species across the different taxonomic levels in our study. Losses of LFA are a consequence of extinction or incomplete lineage sorting that has been reported as another significant event during the coevolutionary process of lichens [31]. It is usually caused by the inability of the algae to parasite the fungal hosts when the host speciates incipiently with small population size [37,39]. Furthermore, this study's evolutionary events leading to Trebouxia-associated symbionts do not correspond to the climate zones. In contrast, macroclimate may influence unique lichen association patterns, such as Protoparmelia-Trebouxia symbionts driven by different evolutionary mechanisms in different climatic regions [31].
Our study highlights the LFF-LFA association in Trebouxia-related lichens. The sampling scope is not restricted to species, genus, family or order but across a broader taxonomic scale covering a comprehensive framework of lichen phylogeny. In addition, our study involves different climatic zones and ecological types, including local, regional to global scales other than a specific geographical scale. Although the sampling size of this study is not very large that cannot present the whole picture of the Trebouxia-related lichens, this study gave us an overview of the LFF-LFA mutualistic associations on larger taxonomic and ecological scales. More climate zones, ecological types, taxa, and sample sizes should be considered in the future exploration of mechanisms underlying the coevolution of mutualistic associations.
4. Materials and Methods
4.1. Taxon sampling
Two hundred Trebouxia lichen samples were collected from a wide range of geographical scales from the mainland (forest and sand desert) and Taiwan (forest) of China, Japan (forest), Israel (sand desert), and Antarctic (cold desert) across three climate zones (temperate, tropic/subtropic, and cold) in 2014–2018 (Figure 3, Table S1). The samples represented a wide range of taxonomical scales across different orders, families, genera, and species, mostly belonging to the class Lecanoromycetes.
4.2. DNA extraction, amplification, and sequencing
Total genomic DNA was extracted from lichen thalli using a modified CTAB method [40]. For LFA, we amplified the nuclear internal transcribed spacer region (ITS), the nuclear small subunit ribosomal RNA gene (SSU), and the chloroplast ribulose-bisphosphate carboxylase-RuBisCO gene (rbcL). For LFF, four nuclear markers were amplified, i.e., ITS, the large nuclear subunit ribosomal RNA gene (nuLSU), the largest subunit of RNA polymerase II (RPB1), and the small mitochondrial subunit ribosomal RNA gene (mtSSU). PCR amplification was performed in 25 μl reaction volume consisting of 12.5 μl of 2 × Taq Mix DNA polymerase (CWBIO, China), 9.5 μl of ddH2O, 1 μl each (10 μM) of the primer set (forward and reverse), and 1 μl DNA template. Primers and PCR conditions are listed in Table S2. PCR products were examined on 0.8% agarose gels, purified using a gel purification kit (Shanghai Huashun Bioengineering Corporation, China) following the manufacturer’s instructions, and sequenced using ABI 3730 XL Sequencer by Shanghai BioSune Corporation of China.
4.3. DNA alignment and phylogenetic analysis
DNA sequence contigs were assembled and manually edited using the SeqMan module of the DNASTAR Lasergene Packages 7 (Madison, WI, USA). Sequences of each locus were aligned separately with MAFFT v 7.471 [41] using the G-INS-i algorithm. Gaps were treated as missing data, and ambiguously aligned regions were excluded using Gblocks v0.91b [42]. These sequence data have been submitted to the GenBank databases under accession number in Table S1. Sequence alignments were deposited at TreeBase (http://purl.org/phylo/treebase/phylows/study/TB2:S30069?x-access-code=9b532bf68bf8b1b507058d77e93697e4&format=html)
Individual gene topologies of LFA and LFF were reconstructed using the RAxML v.8 [43] program implemented on the CIPRES Science Gateway v.3.3 [44] with a GTRGAMMA model and 1,000 bootstrap (BS) replicates. Before concatenating the data sets, the individual gene topologies were checked for congruence. A conflict was significant when two data partitions supported conflicting monophyletic groups with ML bootstrap values of ≥70% in both trees [45]. Independent gene data sets were combined to achieve maximum phylogenetic resolution without evidence of conflicting nodes.
Bayesian inference was performed on the LFA and LFF concatenated data sets in MrBayes v.3.2.7. [46] on the CIPRES Science Gateway v.3.3. The best-fitting models of nucleotide substitutions were selected using the corrected Akaike information criterion (AICc) as suggested by jModelTest [47]. Two parallel Markov chain Monte Carlo (MCMC) runs were performed, each using four chains and 10,000,000 generations, sampling trees every 10,000th generation. A 50% majority-rule consensus tree was generated from the combined sample of both runs after discarding the initial 25% as burn-in.
The phylogenetic trees were visualized using FigTree v.1.4.0 [48]. All clades with BS of ≥70% and posterior probability (PP) of ≥0.95 were considered significantly supported.
4.4. Species delimitation
The LFF identification was mainly based on DNA sequences combined with the phenotypic examination. The phylogenetic relationship of LFF species was first assessed with ITS trees using sequences generated newly for this study and downloaded from GenBank with ML and Bayesian methods and re-evaluated by the four-locus phylogeny. To determine the identity of LFA and their phylogenetic positions, we incorporated ITS and rbcL sequences from this study in the Trebouxia dataset from Muggia, Nelsen, Kirika, Barreno, Beck, Lindgren, Lumbsch and Leavitt [5] for phylogenetic analysis. The identity of Trebouxia species was then tested using three species delimitation approaches, i.e., Automatic Barcoding Gap Discovery (ABGD) [49], a Bayesian implementation of the PTP approach (bPTP) [50], and the coalescent-based General Mixed Yule Coalescent (GMYC) approach [51].
ABGD infers a model-based confidence limit for intraspecific divergence and detects the barcode gap as the first significant gap beyond this limit to partition the data. The primary data partitions are then recursively split to get finer partitions until no further partitions can be detected [49]. The ABGD analyses were performed at the web interface (https://bioinfo.mnhn.fr/abi/public/abgd/abgdweb.html) for each data set of ITS, SSU, and rbcL separately. Genetic distances were calculated using the JC69 model, with a prior P ranging from 0.001 to 0.1 and a relative gap width ranging from 1.0 to 1.5. The bPTP analyses were conducted on the bPTP Web Server (http://species.h-its.org/ptp/) based on unrooted phylogenetic trees inferred by Maximum Likelihood. The analyses were run for 200,000 generations, using 0.1 burn-in and 100 thinning. Both ML and Bayesian solutions were examined. For the GMYC analyses, ultrametric trees were estimated from the combined sequence dataset using the BEAST v2.6.3 program [52]. We ran MCMC chains for 15 million generations under the coalescent model with constant population size and a constant clock as the tree prior. Default values were used for the remaining priors. The chain mixture and convergence were evaluated in TRACER v.1.7.2 [53] after removing 10% of the samples as burn-in. The effective sample size (ESS) values greater than 200 were considered a good indicator. The consensus tree was generated using TreeAnnotator 1.8.2 [52] after discarding the first 3,000 trees. The GMYC analysis was executed under the single-threshold model using the SPLITS package [51] available for R 4.0.5 [54].
4.5. Interaction network analyses
The mutualistic association network of Trebouxia species and their associated fungal symbionts (Table S3) was represented in R using the function plotweb in the bipartite package based on the species trees [55]. Bipartite network analyses were conducted to measure the complexity of the interactions with five commonly used qualitative indexes, namely connectance, linkage density, links per species, number of compartments, and nestedness, using the functions networklevel in the R package bipartite [55,56].
4.6. Variation partitioning analysis
The relative effects of climatic variables, geographical distance, the symbiotic partner, and other factors, such as distribution types, reproduction modes, and host specificity (Table S4) on the variance in LFA and LFF diversity were analyzed by variation partitioning analysis based on redundancy analysis, using the varpart function in the vegan package [57]. The phylogenetic distances of LFA or LFF were calculated using the Patristic software with their phylogeny as the input [58], and the first ten PcoA axes were used as a response variable. The 19 climatic variables for the locations of 200 samples (Table S4) were obtained from the Worldclim database (www.worldclim.org) with a grid cell resolution of 2.5 min, using the software DIVA-GIS 7.5 [59]. These 19 environmental variables were implemented forward selection using the function RDA in vegan with the distribution of LFA and LFF, respectively. Geographical distance values (latitude and longitude) were transformed to the principal coordinates of neighbor matrices (PCNM) vectors representing the geographical distances at various spatial scales. PCNM vectors were calculated based on the pairwise geographical distances obtained by the function pcnm in the vegan package. All four PCNMs were used for the analysis. The presence/absence matrix of 30 variables, including distribution types, reproduction modes, and host specificity, was combined as a mixed factor. Similarly, forward selection with the distribution of LFA and LFF was applied, respectively.
4.7. Cophylogenetic analyses
Two distance-based methods, including PataFit (Legendre et al., 2002) and Procrustean Approach to Cophylogeny (PACo) [60], and an event-based method, i.e., Jane 4.0 [61], were used to assess the cophylogenetic patterns between LFF and LFA. One sample corresponding to each LFF-LFA association was chosen for the cophylogenetic analysis to assess potential cophylogenetic patterns.
Distance-based analysis was performed to assess the overall congruence between host and parasite phylogenies accompanying quantification of the relative contribution of individual host-parasite associations to the overall congruence. ParaFit was implemented in R package APE [62]. The ParaFitGlobal fit between parasite and host phylogenies was computed, and statistical significance was tested by 999 permutations to assess whether parasites are randomly associated with their hosts. If the observed ParaFitGlobal statistic is larger than the randomized statistic in more than 95% of cases, the null hypothesis of independent evolution can be rejected, and congruence can be assumed. The contribution of individual host-parasite associations is conducted using the ParaFitLink1 and ParaFitLink2 tests. A significant link suggests that a particular host-parasite association contributes to the global congruence between the host and parasite trees. PACo analysis was performed in R package APE and VEGAN [57], in which the dependence of algal phylogeny on the fungal host phylogeny was tested through 100,000 permutations. The contribution of each host-parasite association to the global fit is measured using jackknife estimation of their respective squared residuals and confidence intervals associated with each host-parasite association.
The event-based analyses were performed using Jane 4.0, which supports polytomies and accommodates parasites with multiple hosts. The topology-based program assigns costs to five coevolutionary events: cospeciation, duplication, host switch, loss/extinction, and failure to diverge [61]. It is difficult to estimate the relative cost of events; instead, a default event cost scheme (cospeciation = 0, duplication = 1, host switch = 2, loss/extinction = 1, failure to diverge = 1), as well as other 16 cost regimes derived from the default regime were selected to test the overall costs of reconstructions in Jane 4.0. All regimes were analyzed with a population size of 23, and the number of generations has been set to 45, as Conow et al. (2010) recommended. The statistical significance of reconstructions was evaluated with 100 random tips mapping permutations.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Figure S1: Phylogeny of 41 species of lichen-forming fungi based on Randomized Axelerated Maximum Likelihood (RAxML) analysis using the internal transcribed spacer region (ITS) sequences. The species names and accession numbers of type strains were marked in red. The number in each node represents bootstrap support (BS) and posterior probability (PP). BS values of ≥70 and PP values of ≥0.95 were plotted on the branches. Scale in 0.02 substitution per site; Figure S2: Phylogeny of lichen-forming fungi based on the Randomized Axelerated Maximum Likelihood analysis of the concatenated four-locus dataset including internal transcribed spacer region (ITS), the large nuclear subunit ribosomal RNA gene (nuLSU), the small mitochondrial subunit ribosomal RNA gene (mtSSU), and the largest subunit of RNA polymerase II (RPB1) sequences. The number in each node represents bootstrap support (BS) and posterior probability (PP). BS values of ≥70 and PP values of ≥0.95 were plotted on the branches. Scale in 0.02 substitution per site; Figure S3: Phylogeny of lichen-forming algae Trebouxia species based on the Randomized Axelerated Maximum Likelihood (RAxML) analysis of a concatenated two-locus dataset including internal transcribed spacer region (ITS) and chloroplast ribulose-bisphosphate carboxylase-RuBisCO (rbcL) gene sequences with reference sequences from the study of Muggia et al. (2020) marked in black. The number in each node represents bootstrap support (BS) and posterior probability (PP). BS values of ≥70 and PP values of ≥0.95 were plotted on the branches. Scale in 0.09 substitution per site; Figure S4: Comparison of different Trebouxia species delimitations resulting from three species delimitation approaches, i.e., Automatic Barcoding Gap Discovery (ABGD), a Bayesian implementation of the PTP (bPTP) [50], and the coalescent-based General Mixed Yule Coalescent (GMYC), along with the species scenarios proposed by Muggia et al. (2020) and. The phylogeny was constructed by Randomized Axelerated Maximum Likelihood (RAxML) analysis based on the combined internal transcribed spacer region (ITS) and chloroplast ribulose-bisphosphate carboxylase-RuBisCO gene (rbcL) gene sequences. Scale in 0.02 substitution per site. The numbers in the seven columns represent the species numbers defined by different methods; Figure S5: Phylogeny of lichen-forming algae Trebouxia species based on the Randomized Axelerated Maximum Likelihood (RAxML) analysis of a concatenated three-locus dataset including internal transcribed spacer region (ITS), the large nuclear subunit ribosomal RNA gene (nuSSU), and chloroplast ribulose-bisphosphate carboxylase-RuBisCO (rbcL) sequences. The number in each node represents bootstrap support (BS) and posterior probability (PP). BS values of ≥70 and PP values of ≥0.95 were plotted on the branches. Scale in 0.01 substitution per site; Figure S6: Boxplot of the jackknifed squared residuals with upper 95% confidence intervals associated with each host-symbiont link from Procrustean Approach to Cophylogeny (PACo) analysis. Asterisks on the top of the bars indicate significant congruence, as supported by ParaFit; Figure S7: The least costly cophylogenetic scenario between lichen-forming algae (LFA) and their lichen-forming fungi (LFF) hosts, reconstructed using Jane 4.0. The cost regime settings were as follows: cospeciation = 0, duplication = 1, duplication with host switch = 2, losses = 1, and failures to diverge = −1, corresponding to cost regime E (Table 2). Black branches represent the LFF host phylogeny, and blue branches the LFA parasite phylogeny. The LFF names are in bold. Yellow and red solid circles represent duplications dashed lines with purple circles represent losses, dented lines with black asterisks represent failures to diverge. Habitat information is provided with the LFF species. AR is abbreviated for arctic/alpine or boreal zone, TM is abbreviated for temperate zone, STR is abbreviated for subtropic zone, and TR is abbreviated for tropic zone; Table S1: Lichen samples used in this study, including voucher information and GenBank accession numbers; Table S2: Molecular markers and primer sequences used in this study; Table S3: The climate type of lichen distributions; Table S4: The information on geographical distance, bioclimatic variables, and other factors of lichen-forming fungi and algae.
Author Contributions
Conceptualization, X.-L.W and X.-Z.L; validation, X.-L.W and X.-Z.L; formal analysis, Y.-B.Z, X.-Z.L, D.-Y.H, Y.-Y.W, Q.-X.Y and Q.R.; investigation, Y.-B.Z,, X.-L.W ; writing—original draft preparation, X.-L.W; writing—review and editing, X.-Z.L and Y.-B.Z.;. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the National Natural Science Foundation of China (32070096), the Space Application System of China Manned Space Program (KJZ-YY-WSM05) and Beijing Natural Science Foundation (5232020), and Youth Innovation Promotion Association of Chinese Academy of Sciences (2012076, 2017125).
Data Availability Statement
The DNA sequence alignments were deposited at TreeBase (http://purl.org/phylo/treebase/phylows/study/TB2:S30069?x-access-code=9b532bf68bf8b1b507058d77e93697e4&format=html).
Acknowledgments
We are grateful to the Chinese Arctic and Antarctic Administration for the help in carrying out the project in the Great Wall Station during the 34th Chinese National Antarctic Expedition, and Enago (https://www.enago.cn/) for its linguistic assistance during the preparation of this manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Lücking, R.; Hodkinson, B.P.; Leavitt, S.D. The 2016 classification of lichenized fungi in the Ascomycota and Basidiomycota–Approaching one thousand genera. The Bryologist 2017, 119, 361–416. [Google Scholar] [CrossRef]
- Gargas, A.; DePriest, P.T.; Grube, M.; Tehler, A. Multiple origins of lichen symbioses in fungi suggested by SSU rDNA phylogeny. Science 1995, 268, 1492–1495. [Google Scholar] [CrossRef] [PubMed]
- Lutzoni, F.; Pagel, M.; Reeb, V. Major fungal lineages are derived from lichen symbiotic ancestors. Nature 2001, 411, 937–940. [Google Scholar] [CrossRef]
- Lücking, R.; Nelsen, M.P. Ediacarans, protolichens, and lichen-derived Penicillium: A critical reassessment of the evolution of lichenization in fungi. Transformative Paleobotany 2018, 551–590. [Google Scholar] [CrossRef]
- Muggia, L.; Nelsen, M.P.; Kirika, P.M.; Barreno, E.; Beck, A.; Lindgren, H.; Lumbsch, H.T.; Leavitt, S.D. Formally described species woefully underrepresent phylogenetic diversity in the common lichen photobiont genus Trebouxia (Trebouxiophyceae, Chlorophyta): an impetus for developing an integrated taxonomy. Molecular Phylogenetics and Evolution 2020, 149, 106821. [Google Scholar] [CrossRef] [PubMed]
- Lumbsch, H.T.; Rikkinen, J. Evolution of lichens. In The fungal community: its organization and role in the ecosystem., Dighton, J., White, J.F., Eds.; CRC Press: Boca Raton, Florida, 2017; pp. 53–62. [Google Scholar]
- Taylor, T.; Hass, H.; Kerp, H. The oldest fossil ascomycetes. Nature 1999, 399, 648–648. [Google Scholar] [CrossRef]
- Kroken, S.; Taylor, J.W. Phylogenetic species, reproductive mode, and specificity of the green alga Trebouxia forming lichens with the fungal genus Letharia. Bryologist 2000, 645–660. [Google Scholar] [CrossRef]
- Hill, D.J. Asymmetric co-evolution in the lichen symbiosis caused by a limited capacity for adaptation in the photobiont. The Botanical Review 2009, 75, 326–338. [Google Scholar] [CrossRef]
- Yahr, R.; Vilgalys, R.; Depriest, P.T. Strong fungal specificity and selectivity for algal symbionts in Florida scrub Cladonia lichens. Molecular Ecology 2004, 13, 3367–3378. [Google Scholar] [CrossRef]
- Thüs, H.; Muggia, L.; Pérez-Ortega, S.; Favero-Longo, S.E.; Joneson, S.; O’Brien, H.; Nelsen, M.P.; Duque-Thüs, R.; Grube, M.; Friedl, T.; et al. Revisiting photobiont diversity in the lichen family Verrucariaceae (Ascomycota). European Journal of Phycology 2011, 46, 399–415. [Google Scholar] [CrossRef]
- Leavitt, S.D.; Kraichak, E.; Nelsen, M.P.; Altermann, S.; Divakar, P.K.; Alors, D.; Esslinger, T.L.; Crespo, A.; Lumbsch, T. Fungal specificity and selectivity for algae play a major role in determining lichen partnerships across diverse ecogeographic regions in the lichen-forming family Parmeliaceae (Ascomycota). Molecular Ecology 2015, 24, 3779–3797. [Google Scholar] [CrossRef]
- Chagnon, P.L.; Magain, N.; Miadlikowska, J.; Lutzoni, F. Strong specificity and network modularity at a very fine phylogenetic scale in the lichen genus Peltigera. Oecologia 2018, 187, 767–782. [Google Scholar] [CrossRef]
- Rikkinen, J. Cyanolichens: An Evolutionary Overview. In Cyanobacteria in Symbiosis, Rai, A.N., Bergman, B., Rasmussen, U., Eds.; Springer Netherlands: Dordrecht, 2002; pp. 31–72. [Google Scholar]
- Pardo-De la Hoz, C.J.; Magain, N.; Lutzoni, F.; Goward, T.; Restrepo, S.; Miadlikowska, J. Contrasting symbiotic patterns in two closely related lineages of trimembered lichens of the genus Peltigera. Frontiers in Microbiology 2018, 9, 2770–2770. [Google Scholar] [CrossRef] [PubMed]
- Vančurová, L.; Muggia, L.; Peksa, O.; Řídká, T.; Škaloud, P. The complexity of symbiotic interactions influences the ecological amplitude of the host: a case study in Stereocaulon (lichenized Ascomycota). Molecular Ecology 2018, 27, 3016–3033. [Google Scholar] [CrossRef]
- Lindgren, H.; Moncada, B.; Lücking, R.; Magain, N.; Simon, A.; Goffinet, B.; Sérusiaux, E.; Nelsen, M.P.; Mercado-Díaz, J.A.; Widhelm, T.J.; et al. Cophylogenetic patterns in algal symbionts correlate with repeated symbiont switches during diversification and geographic expansion of lichen-forming fungi in the genus Sticta (Ascomycota, Peltigeraceae). Molecular Phylogenetics and Evolution 2020, 150, 106860. [Google Scholar] [CrossRef] [PubMed]
- Zoller, S.; Lutzoni, F. Slow algae, fast fungi: exceptionally high nucleotide substitution rate differences between lichenized fungi Omphalina and their symbiotic green algae Coccomyxa. Molecular Phylogenetics and Evolution 2003, 29, 629–640. [Google Scholar] [CrossRef] [PubMed]
- Piercey-Normore, M.D. Vegetatively reproducing fungi in three genera of the Parmeliaceae share divergent algal partners. The Bryologist 2009, 112, 773–785. [Google Scholar] [CrossRef]
- Singh, G.; Kukwa, M.; Dal Grande, F.; Łubek, A.; Otte, J.; Schmitt, I. A glimpse into genetic diversity and symbiont interaction patterns in lichen communities from areas with different disturbance histories in Białowieża forest, Poland. Microorganisms 2019, 7, 335. [Google Scholar] [CrossRef]
- Beck, A.; Kasalicky, T.; Rambold, G. Myco-photobiontal selection in a Mediterranean cryptogam community with Fulgensia fulgida. New Phytologist 2002, 153, 317–326. [Google Scholar] [CrossRef]
- Peksa, O.; Škaloud, P. Do photobionts influence the ecology of lichens? A case study of environmental preferences in symbiotic green alga Asterochloris (Trebouxiophyceae). Molecular Ecology 2011, 20, 3936–3948. [Google Scholar] [CrossRef]
- Dal Grande, F.; Rolshausen, G.; Divakar, P.K.; Crespo, A.; Otte, J.; Schleuning, M.; Schmitt, I. Environment and host identity structure communities of green algal symbionts in lichens. New Phytologist 2018, 217, 277–289. [Google Scholar] [CrossRef] [PubMed]
- Vázquez, D.P.; Aizen, M.A. Asymmetric specialization: a pervasive feature of plant-pollinator interactions. Ecology 2004, 85, 1251–1257. [Google Scholar] [CrossRef]
- Vázquez, D.P.; Poulin, R.; Krasnov, B.R.; Shenbrot, G.I. Species abundance and the distribution of specialization in host-parasite interaction networks. Journal of Animal Ecology 2005, 946–955. [Google Scholar] [CrossRef]
- Vázquez, D.P.; Melián, C.J.; Williams, N.M.; Blüthgen, N.; Krasnov, B.R.; Poulin, R. Species abundance and asymmetric interaction strength in ecological networks. Oikos 2007, 116, 1120–1127. [Google Scholar] [CrossRef]
- Bowler, P.; Rundel, P. Reproductive strategies in lichens. Botanical Journal of the Linnean Society 1975, 70, 325–340. [Google Scholar] [CrossRef]
- Degelius, G. Biological studies of the epiphytic vegetation on twigs on Fraxinus excelsior. Acta Horti Gotoburgensis Medd Goteborgs Botany Tradgard 1964, 27, 11–55. [Google Scholar]
- Fernández-Mendoz, F.; Domaschke, S.; García, M.A.; Jordan, P.; Martín, M.P.; Printzen, C. Population structure of mycobionts and photobionts of the widespread lichen Cetraria aculeata. Molecular Ecology 2011, 20, 1208–1232. [Google Scholar] [CrossRef]
- Ertz, D.; Guzow-Krzemińska, B.; Thor, G.; Łubek, A.; Kukwa, M. Photobiont switching causes changes in the reproduction strategy and phenotypic dimorphism in the Arthoniomycetes. Scientific Reports 2018, 8, 4952. [Google Scholar] [CrossRef]
- Singh, G.; Dal Grande, F.; Divakar, P.K.; Otte, J.; Crespo, A.; Schmitt, I. Fungal–algal association patterns in lichen symbiosis linked to macroclimate. New Phytologist 2017, 214, 317–329. [Google Scholar] [CrossRef]
- Wirtz, N.; Lumbsch, H.T.; Green, T.G.A.; Türk, R.; Pintado, A.; Sancho, L.; Schroeter, B. Lichen fungi have low cyanobiont selectivity in maritime Antarctica. New Phytologist 2003, 160, 177–183. [Google Scholar] [CrossRef]
- Wiens, J.J.; Donoghue, M.J. Historical biogeography, ecology and species richness. Trends in Ecology & Evolution 2004, 19, 639–644. [Google Scholar] [CrossRef]
- Blasco-Costa, I.; Hayward, A.; Poulin, R.; Balbuena, J.A. Next-generation cophylogeny: unravelling eco-evolutionary processes. Trends in Ecology & Evolution 2021, 36, 907–918. [Google Scholar] [CrossRef]
- Buckley, H.L.; Rafat, A.; Ridden, J.D.; Cruickshank, R.H.; Ridgway, H.J.; Paterson, A.M. Phylogenetic congruence of lichenised fungi and algae is affected by spatial scale and taxonomic diversity. PeerJ 2014, 2, e573. [Google Scholar] [CrossRef] [PubMed]
- Piercey-Normore, M.D. The lichen-forming ascomycete Evernia mesomorpha associates with multiple genotypes of Trebouxia jamesii. New Phytologist 2006, 169, 331–344. [Google Scholar] [CrossRef] [PubMed]
- De Vienne, D.M.; Refrégier, G.; López-Villavicencio, M.; Tellier, A.; Hood, M.E.; Giraud, T. Cospeciation vs host-shift speciation: methods for testing, evidence from natural associations and relation to coevolution. New Phytologist 2013, 198, 347–385. [Google Scholar] [CrossRef]
- Beiggi, S.; Piercey-Normore, M.D. Evolution of ITS ribosomal RNA secondary structures in fungal and algal symbionts of selected species of Cladonia sect. Cladonia (Cladoniaceae, Ascomycotina). Journal of Molecular Evolution 2007, 64, 528–542. [Google Scholar] [CrossRef]
- Millanes, A.M.; Truong, C.; Westberg, M.; Diederich, P.; Wedin, M. Host switching promotes diversity in host-specialized mycoparasitic fungi: uncoupled evolution in the Biatoropsis usnea system. Evolution; international journal of organic evolution 2014, 68, 1576–1593. [Google Scholar] [CrossRef]
- Rogers, S.O.; Bendich, A.J. Extraction of DNA from plant tissues. In Plant Molecular Biology Manual; Springer, 1989; pp. 73–83. [Google Scholar]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Molecular Biology and Evolution 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Castresana, J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Molecular Biology and Evolution 2000, 17, 540–552. [Google Scholar] [CrossRef]
- Stamatakis, A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Miller, M.A.; Pfeiffer, W.; Schwartz, T. Creating the CIPRES Science Gateway for inference of large phylogenetic trees; IEEE, 2010. [Google Scholar]
- Mason-Gamer, R.J.; Kellogg, E.A. Testing for phylogenetic conflict among molecular data sets in the tribe Triticeae (Gramineae). Systematic Biology 1996, 45, 524–545. [Google Scholar] [CrossRef]
- Ronquist, F.; Teslenko, M.; Van Der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Systematic Biology 2012, 61, 539–542. [Google Scholar] [CrossRef] [PubMed]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. jModelTest 2: more models, new heuristics and parallel computing. Nature Methods 2012, 9, 772. [Google Scholar] [CrossRef] [PubMed]
- Rambaut, A. FigTree. Tree figure drawing tool [WWW document] URL http://tree.bio.ed.ac.uk/software/figtree/, 2009.
- Puillandre, N.; Lambert, A.; Brouillet, S.; Achaz, G. ABGD, automatic barcode gap discovery for primary species delimitation. Molecular Ecology 2012, 21, 1864–1877. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Kapli, P.; Pavlidis, P.; Stamatakis, A. A general species delimitation method with applications to phylogenetic placements. Bioinformatics 2013, 29, 2869–2876. [Google Scholar] [CrossRef]
- Monaghan, M.T.; Wild, R.; Elliot, M.; Fujisawa, T.; Balke, M.; Inward, D.J.; Lees, D.C.; Ranaivosolo, R.; Eggleton, P.; Barraclough, T.G.; et al. Accelerated species inventory on Madagascar using coalescent-based models of species delineation. Systematic Biology 2009, 58, 298–311. [Google Scholar] [CrossRef]
- Bouckaert, R.; Vaughan, T.G.; Barido-Sottani, J.; Duchêne, S.; Fourment, M.; Gavryushkina, A.; Heled, J.; Jones, G.; Kühnert, D.; Maio, N.D.; et al. BEAST 2.5: An advanced software platform for Bayesian evolutionary analysis. PLoS Computational Biology 2019, 15, e1006650. [Google Scholar] [CrossRef]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior summarization in Bayesian phylogenetics using Tracer 1.7. Systematic Biology 2018, 67, 901–904. [Google Scholar] [CrossRef]
- R Development Core Team. R: A language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing; 2017. [Google Scholar]
- Dormann, C.; Gruber, B.; Fründ, J. Introducing the bipartite package: analysing ecological networks. R News 8: 8–11; 2008. [Google Scholar]
- Delmas, E.; Besson, M.; Brice, M.H.; Burkle, L.A.; Dalla Riva, G.V.; Fortin, M.J.; Gravel, D.; Guimarães Jr, P.R.; Hembry, D.H.; Newman, E.A. Analysing ecological networks of species interactions. Biological Reviews 2019, 94, 16–36. [Google Scholar] [CrossRef]
- Oksanen, J.; Blanchet, F.; Friendly, M.; Kindt, R.; Legendre, P.; McGlinn, D.; Wagner, H. Vegan: Community Ecology Package. R package v.2.3-5 [WWW document] URL https://cran.r-project.org/web/packages/vegan/index.html. 2016.
- Fourment, M.; Gibbs, M.J. PATRISTIC: a program for calculating patristic distances and graphically comparing the components of genetic change. BMC Evolutionary Biology 2006, 6, 1–1. [Google Scholar] [CrossRef]
- Hijmans, R.J.; Guarino, L.; Mathur, P. DIVA-Gis version 7.5.Manual. [WWW document] URL http://www.diva-gis.org. [Accessed 7 August 2021.]. 2012.
- Balbuena, J.A.; Míguez-Lozano, R.; Blasco-Costa, I. PACo: a novel procrustes application to cophylogenetic analysis. PLoS One 2013, 8, e61048. [Google Scholar] [CrossRef] [PubMed]
- Conow, C.; Fielder, D.; Ovadia, Y.; Libeskind-Hadas, R. Jane: a new tool for the cophylogeny reconstruction problem. Algorithms for Molecular Biology 2010, 5, 16. [Google Scholar] [CrossRef] [PubMed]
- Paradis, E.; Claude, J.; Strimmer, K. APE: analyses of phylogenetics and evolution in R language. Bioinformatics 2004, 20, 289–290. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Mutualistic association pattern between lichen-forming fungi (LFF) and lichen-forming algae (LFA). The left and right columns represented the names of LFF and LFA, respectively. The number of lines connecting the LFF and LFA indicated the mutualistic associations. The width of the lines indicated the lichen sample numbers composed of LFF and LFA.
Figure 1.
Mutualistic association pattern between lichen-forming fungi (LFF) and lichen-forming algae (LFA). The left and right columns represented the names of LFF and LFA, respectively. The number of lines connecting the LFF and LFA indicated the mutualistic associations. The width of the lines indicated the lichen sample numbers composed of LFF and LFA.
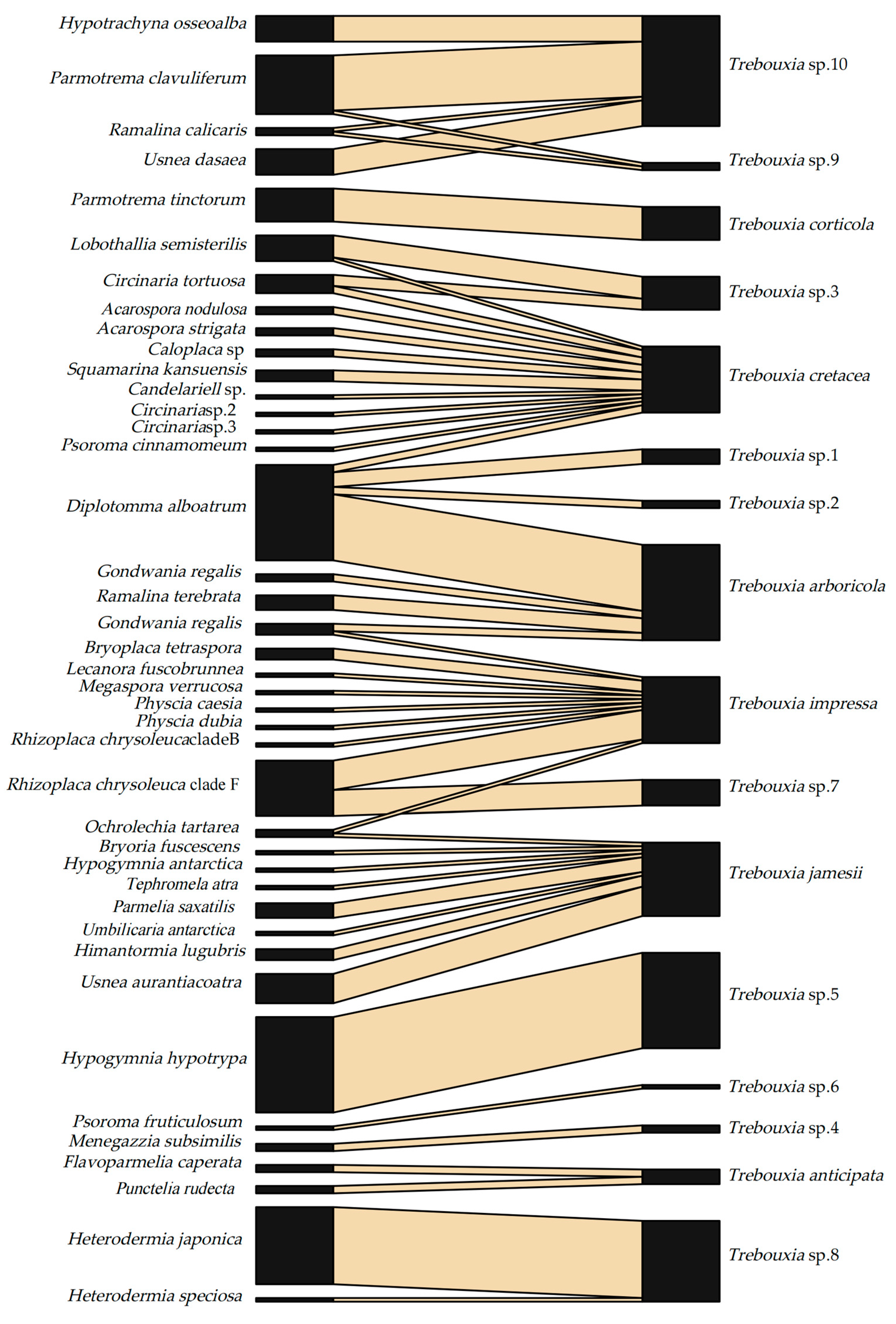
Figure 2.
Venn’s diagram showing the results from variation partitioning analysis (VPA). (a). Partitioning variance of lichen-forming fungi (LFF), geographical distance (Geo), 19 bioclimatic variables (Bioclim), and other factors including lichen distribution types, reproduction modes, and host specificity (Multi_fac) onto photobiont diversity and distribution; (b). Partitioning variance of lichen-forming algae (LFA), geographical distance (Geo), 19 bioclimatic variables (Bioclim), and other factors including lichen distribution types, reproduction modes, and host specificity (Multi_fac) onto mycobiont diversity and distribution.
Figure 2.
Venn’s diagram showing the results from variation partitioning analysis (VPA). (a). Partitioning variance of lichen-forming fungi (LFF), geographical distance (Geo), 19 bioclimatic variables (Bioclim), and other factors including lichen distribution types, reproduction modes, and host specificity (Multi_fac) onto photobiont diversity and distribution; (b). Partitioning variance of lichen-forming algae (LFA), geographical distance (Geo), 19 bioclimatic variables (Bioclim), and other factors including lichen distribution types, reproduction modes, and host specificity (Multi_fac) onto mycobiont diversity and distribution.
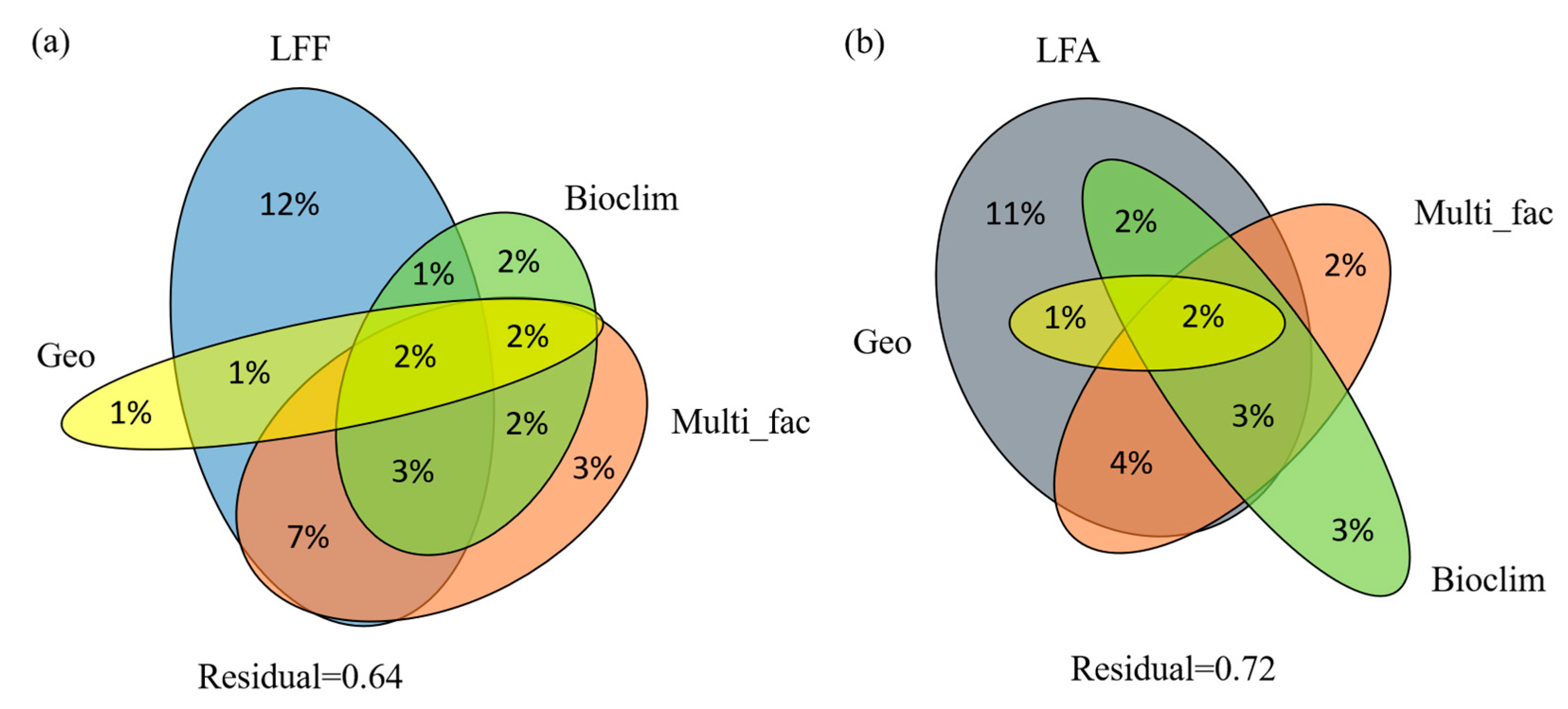
Figure 3.
Collection sites of the lichen materials used in this study were marked by solid blue circles. The map was drawn using the software ArcGIS 10.7.
Figure 3.
Collection sites of the lichen materials used in this study were marked by solid blue circles. The map was drawn using the software ArcGIS 10.7.

Table 1.
The interaction network level parameters.
| Temperate zone | Arctic zone | Tropic/subtropic zone | |
|---|---|---|---|
| Connectance | 0.25 | 0.22 | 0.15 |
| Links per species | 0.89 | 0.87 | 0.76 |
| Linkage density | 4.13 | 3.70 | 2.13 |
| Nestedness | 30.75 | 37.39 | 22.92 |
| Number of compartments | 3 | 3 | 6 |
Table 2.
Cophylogenetic analyses result with event-based approach using Jane 4.0 with different cost regimes.
Table 2.
Cophylogenetic analyses result with event-based approach using Jane 4.0 with different cost regimes.
| Cost regime | C-D-D&S-L-FDa | C | D | D&S | L | FD | Total cost |
|---|---|---|---|---|---|---|---|
| A | 0,1,2,1,1 | 1 | 8 | 6 | 91 | 35 | 146 |
| B | 0,1,2,1,-1 | 1 | 8 | 6 | 90 | 35 | 75 |
| C | 0,2,2,1,1 | 1 | 8 | 6 | 89 | 35 | 152 |
| D | 0,0,2,1,1 | 1 | 9 | 5 | 91 | 35 | 136 |
| E | 0,1,3,1,1 | 1 | 9 | 5 | 91 | 35 | 150 |
| F | 0,1,1,1,1 | 0 | 8 | 7 | 88 | 35 | 138 |
| G | 0,1,2,2,1 | 0 | 8 | 7 | 90 | 35 | 237 |
| H | 0,1,2,0,1* | 2 | 13 | 0 | 113 | 35 | 48 |
| I | 0,1,2,1,2 | 1 | 8 | 6 | 89 | 35 | 179 |
| J | 0,1,2,1,0 | 1 | 8 | 6 | 90 | 35 | 110 |
| K | 1,1,1,1,1 | 0 | 8 | 7 | 88 | 35 | 138 |
| L | 1,1,2,1,1 | 0 | 8 | 7 | 88 | 35 | 145 |
| M | 1,0,0,1,1 | 0 | 7 | 8 | 90 | 35 | 125 |
| N | -1,1,2,1,1 | 1 | 8 | 6 | 89 | 35 | 143 |
| O | 2,1,1,1,1 | 0 | 8 | 7 | 88 | 35 | 138 |
| P | 2,1,1,1,0 | 0 | 8 | 7 | 88 | 35 | 103 |
| Q | 2,1,1,0,0* | 0 | 8 | 7 | 108 | 35 | 15 |
Notes: a Order of cost regimes from left to right is C (cospeciation); D (duplication); D&S (duplication and host switch); L (loss/sorting); and FD (failure to diverge). *This cost regime is not significantly lower than randomly reconstruction.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



