Submitted:
24 May 2023
Posted:
25 May 2023
You are already at the latest version
Abstract
People with specific TREM2 gene variants are more prone to develop Alzheimer's disease (AD). The TREM2 receptor regulates the number of myeloid cells, phagocytosis, and the inflammatory response via interacting with apolipoproteins and amyloid. Higher TREM2 expression has been found to protect against AD. When TREM2 activity increases, the activity of genes involved in the activation of microglia cells decreases. This can improve the efficiency of phagocytosis. When TREM2 is highly expressed and the inflammasome is activated, the results are not always congruent. Therefore, this study aimed to discover how sTREM2 levels in CSF and plasma samples relate to other indices of AD pathology. We examined 98 AD plasma samples, 35 plasma samples of subjects with mild cognitive impairment (MCI), 11 healthy controls (HC) plasma samples, as well as 155 AD CSF samples, 90 MCI CSF samples, and 50 HC CSF samples. CSF sTREM2 levels were higher in the AD group than in the MCI and HC groups, in contrast to plasma sTREM2. This shows that CSF sTREM2 levels could be used to distinguish between healthy and AD patients. CSF sTREM2 levels were significantly correlated with neurofibrillary changes, cognitive decline, and inflammasome activity in AD patients. While our findings are consistent with previous research, future studies will need to include more patients and employ standardized methodological approaches to add CSF sTREM2 to the list of biomarkers for AD.
Keywords:
Alzheimer’s disease
; cerebrospinal fluid
; inflammasome
; microglia
; mild cognitive impairment
; plasma
; tau protein
; TREM2
1. Introduction
Carriers of certain TREM2 gene variants (R47H, D87N, L211P, H157Y, R62H, and T96K) have an increased risk of Alzheimer’s disease (AD). Therefore, the TREM2 receptor is commonly investigated in the context of AD [1,2]. After APOE gene variants [3], the R47H variant of TREM2 is the second most common risk factor for AD, and it can increase the risk of developing AD by two- to fourfold [2]. ApoE is one of the TREM2 receptor ligands, and its interaction, if altered, can have a significant effect on the onset of AD symptoms [4]. The TREM2 receptor is indispensable for interacting with apolipoproteins, amyloid β (Aβ), and regulating the number of myeloid cells, phagocytosis, and inflammatory response [1,2]. TREM2 is also essential for synaptic pruning and regulates synaptic clearance via phagocytosis, whereas the absence of TREM2 results in aberrant synaptic clearance [4]. Microglial cholesterol metabolism also depends on TREM2 signaling, as TREM2 deficiency is associated with impaired intracellular cholesterol storage and efflux [5]. Intriguingly, TREM2 indirectly regulates the myelinating process by influencing cholesterol metabolism and clearing myelin debris [5], which is severely disrupted by the TREM2 p.Q33X mutation in microglia, which interferes with lysosomal function and contributes to Nasu-Hakola disease [6,7]. The downstream signaling of TREM2 induces transcriptional changes that promote the transition from homeostatic to disease-associated microglia phenotype (DAM) [8]. The TREM2 receptor also mediates autophagy, one of the mechanisms for degrading cellular proteins, particularly abnormally aggregated, which is impaired in AD [9]. Humans with TREM2 risk variants and TREM2-deficient mice accumulate vesicles resembling autophagy [9].
The protective effect of increased TREM2 expression against AD symptoms has previously been reported [10]. The TREM2 receptor interacts with the Aβ to initiate its phagocytosis [11]. Higher levels of cerebrospinal fluid (CSF) sTREM2 are associated with delayed Aβ accumulation in the brain, as measured by amyloid-PET scan [12]. In the 5XFAD model of AD, protracted TREM2 stimulation via injection of human TREM2 (hTREM2) agonistic mAb (AL002c) attenuated pathological changes of Aβ and neurite injury [13]. However, a recent study suggests that chronic TREM2 antibody administration does not influence Aβ plaque burden [14]. In addition, chronic TREM2 activation increased the dissemination of tau neurofibrillary degeneration and loss of the synapses surrounding plaques in the region with the highest pathological tau changes and neuritic dystrophy [14]. In contrast, the soluble form of the TREM2 receptor (sTREM2) promoted Aβ plaque degradation [15], indicating that two receptor isoforms may play distinct functions in relation to pathological changes and disease stages. In accordance with that statement, a recent study on APP/PS1 mice demonstrated that increased TREM2 activation indirectly inhibits tau protein phosphorylation and neuronal loss by inhibiting glycogen synthase kinase-3β, the major actor of tau hyperphosphorylation [16]. In addition, AD-associated microglial activation stage 2 markers in non-demented individuals are predictive of a delayed accumulation of tau [17]. The increased activity of TREM2 in the transgenic mouse model is associated with decreased expression of genes essential for the pro-inflammatory response of microglia [18] and can increase its phagocytotic activity [12,13]. TREM2 expression and inflammasome activation produce contradictory outcomes [19,20,21,22,23]. Microglia cells with the R47H variant have a diminished rate of inflammasome activation [19], whereas macrophages with higher TREM2 expression inhibit inflammasome activation [20]. In contrast, their pyroptosis is increased in the absence of TREM2 [21]. A similar effect was observed in C57/BL6 mice where overexpression of the TREM2 attenuated postoperative neuroinflammation [24]. TREM2 overexpression also ameliorates LPS-induced oxidative stress and inflammation in the BV2 cell line [25]. In contrast, one study discovered that the AD mouse model TREM2 knock-out (APPPS1; Trem2-/-) has decreased expression of the pro-inflammatory cytokines IL-1β and IL-6 and increased expression of anti-inflammatory markers associated with reduced Aβ and tau protein pathological alterations [22]. Microglia activated by high glucose levels increase TREM2 expression and pro-inflammatory cytokine release, whereas downregulation of TREM2 reduces inflammation caused by high glucose levels [23]. Soluble TREM2 (sTREM2) is a form of TREM2 receptor that is released after its extracellular domain is cleaved by ADAM10 and ADAM17 metalloproteases [11], and its function remains poorly understood. Another process can be involved in generating sTREM2. Namely, the soluble form of the receptor can be produced by transcription of the alternative transcript lacking a transmembrane domain [26], and it appears that approximately 25% of the sTREM2 may be due to the expression of this isoform. As discussed, TREM2 shedding from the membrane may function as a negative regulator of TREM2 signaling [27]. It is still debatable whether the soluble form of TREM2 has an agonistic effect or whether it competes with the presumably protective full-length membrane TREM2 [11,27].
It has been hypothesized that sTREM2 initiates the survival of microglia and the secretion of pro-inflammatory cytokines [28]. In the 5xFAD transgenic mouse model, injection of sTREM2 into the hippocampus increases microglial clustering around amyloid plaques and reduces amyloid plaque load by uptake and degradation of Aβ, thus rescuing deficits of spatial memory and long-term potentiation [15]. However, the depletion of microglia abolishes the neuroprotective effect of sTREM2 [15]. Recently, a rare p.H157Y variant of TREM2 was identified at the extracellular domain cleavage site; this variant is associated with increased production of sTREM2 and increased risk of AD [29]. In vivo experiments with Trem2 H157Y knock-in mice revealed that increased levels of sTREM2 are advantageous for the animals, resulting in improved synapse function and amelioration of amyloid pathological alterations [29]. However, the study didn’t include other pathological factors, such as tau protein changes, which could be one explanation for why this mutation has a different effect in humans [29]. In a separate study examining the effect of the R47H variant on tau pathology in the PS19 mouse model of tauopathy expressing either human TREM2 common or R47H variant, it was discovered that impaired TREM2 signaling (in the case of the R47H variant) significantly attenuated neurodegenerative changes caused by tau pathology. It is hypothesized that altered TREM2 receptor signaling influences microglial activation and, as a result, ameliorates microglia-mediated degeneration [30]. It appears that tau neurofibrillary degeneration associated with TREM2 signaling is highly dependent on ApoE variants [31]. TREM2 knock-out P301S tau mice with expressed ApoE4 variation have significantly higher neurodegenerative and tau protein alterations than mice with the same ApoE variant and an expressed TREM2 gene [31]. In the THY-Tau22 transgenic murine model of tauopathy, TREM2 deficiency decreased microglial activation, which exacerbated pathological changes in later stages [32]. The TREM2 signaling pathways are extraordinarily complex, as evidenced by their varying effects on diverse pathological alterations, differences in signaling between soluble and membrane isoform signaling, and discordance between animal models and human AD patients. In amyloid mouse models of AD, for instance, brain expression or injection of wild-type sTREM2 decreases amyloid pathological changes, indicating that wild-type sTREM2 is protective against amyloid pathology [33].
To determine whether sTREM2 protects against AD and AD models and at what stage of the disease, it is crucial to conduct additional longitudinal studies on the AD population. Analysis of the TREM2 levels in the cerebrospinal fluid (CSF) generally revealed elevated levels in AD patients; however, other studies did not confirm these findings [34], and some reported variable TREM2 levels dependent on the disease stage, with the highest levels typically associated with earlier symptomatic stages (for review, see [33]). Intriguingly, an increase in TREM2 CSF levels correlates positively with total and phosphorylated tau CSF levels, whereas an increase in Aβ correlates with a decrease in TREM2 [1,34,35,36,37]. Lower Aβ-PET signal was associated with higher CSF TREM2 levels, indicating that microglial activity may be protective against the formation of Aβ plaques [12]. Therefore, sTREM2 may be a useful biomarker of the pathological state of AD; however, future research should include additional studies with a greater number of patients and more uniform analysis procedures. This study aimed to determine the relationship between sTREM2 levels in CSF and plasma samples and other markers of AD pathology.
2. Materials and Methods
2.1. Cerebrospinal fluid and blood collection
This study included patients admitted to the University Hospital Center “Zagreb” in Zagreb, Croatia. AD diagnosis was based on NIA-AA (National Institute on Aging-Alzheimer's Association guidelines for the neuropathologic assessment of Alzheimer's disease) [38,39], NINCDS-ADRDA (National Institute of Neurological and Communicative Disorders and Stroke and Alzheimer's Disease and Related Disorders Association), and DSM-IV-TR (Diagnostic and Statistical Manual of Mental Disorders) criteria. The diagnosis of MCI (Mild Cognitive Impairment) was determined by criteria described by Petersen et al. [40], and Albert et. al. [41]. CSF samples were obtained by lumbar puncture (performed at intervertebral spaces L3/L4 or L4/L5). Following 10 min of centrifugation at 2000 g, CSF samples were aliquoted and stored at −80 °C in polypropylene tubes. Venous blood samples were collected in the morning on an empty stomach. Samples were collected using plastic syringes containing 1 mL of acid citrate dextrose as an anticoagulant. Thrombocyte-free plasma was extracted through centrifugation, first at 1100 g for 3 min and then at 5087 g for 15 min. Plasma samples were stored at −20 °C. All procedures were approved by the Ethical Committee of the Clinical Hospital Center “Zagreb” (protocol no. 02/21 AG, class 8.1–18/82-2 from 24 April 2018, and protocol no. 02/21 AG, class 8.1–19/201-2 from 23 September 2019) and the Central Ethical Committee of the University of Zagreb School of Medicine (protocol no. 380-59-10106-18-111/126, class 641-01/18-02/01 from 20 June 2018, and protocol no. 380-59-10106-19-111/251, class 641-01/19-02/01 from October 2019). We analyzed 98 AD, 35 MCI, and 11 HC plasma samples, as well as 155 AD CSF samples, 90 MCI CSF samples, and 50 HC CSF samples.
2.2. ELISA procedure
Levels of the biomarkers (sTREM2, ASC, p-tau181, t-tau, and Aβ1-42) in CSF and sTREM2 in plasma samples were determined with Enzyme-Linked Immunosorbent Assay (ELISA) by manufacturer protocols for:
- TREM2 (Human TREM2 ELISA Kit, Abcam, Cambridge, United Kingdom),
- Aβ1-42 (Innotest β-amyloid1-42, Fujirebio, Gent, Belgium),
- total tau (Innotest hTau Ag, Fujirebio, Gent, Belgium), p-tau181 (Innotest Phospho-Tau(181P), Fujirebio, Gent, Belgium), and
- ASC (Human PYCARD/ASC/TMS1 Sandwich ELISA, LSBio, Seattle, Washington, United States).
Aβ1-42, t-tau, p-tau181, and ASC ELISA measurements were performed on 96-well plates coated with capture antibodies (indirect capture method). Samples and standards were added and incubated in the plates. The ASC ELISA samples were diluted 1:2. After incubation, the detection antibody was added to the washed wells. For the indirect detection of Aβ1-42, t-tau, p-tau181, and ASC biotin-labeled detection antibodies were used in conjunction with the streptavidin-HRP complex.
The sTREM2 ELISA kit consisted of a 96-well plate coated with an anti-tag antibody that immobilizes the complex of capture antibody-analyte-detector antibody. After adding samples (1:50 dilution) and standards to the wells, a capture/detector antibody mixture was added. HRP chromogenic substrate was applied following incubation and washing. All analytes’ absorbances were measured at 450 nm at Glomax Explorer (Promega, Madison, WI, USA), and protein concentrations were calculated using a 4-parameter algorithm in GraphPad Prism 8.0 (GraphPad Software Inc., San Diego, CA, USA).
3. Results
In Table 1, the basic characteristics of the MCI, AD, and HC groups are presented.
3.1. sTREM2 levels in CSF and plasma samples
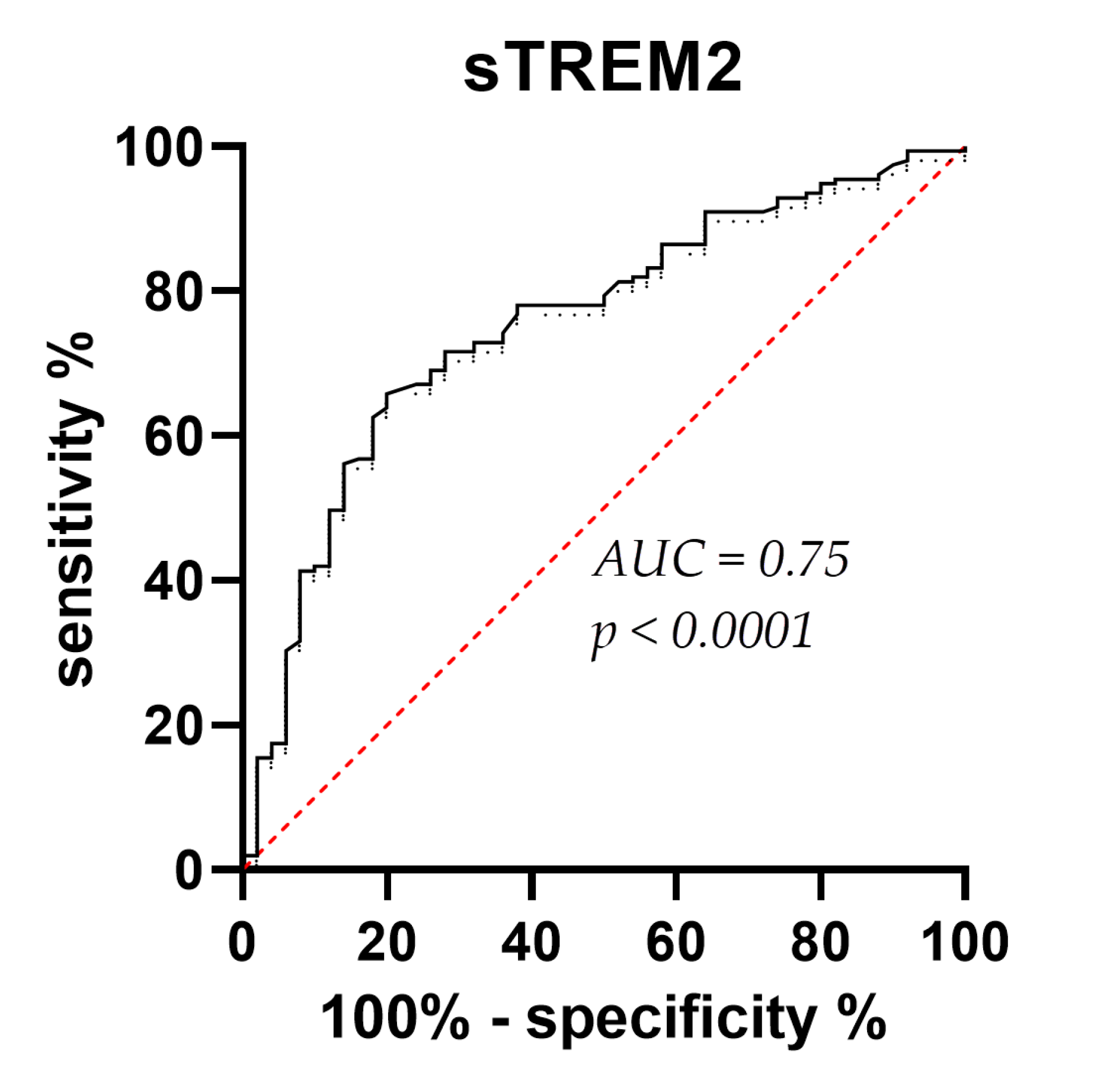
CSF sTREM2 concentrations were significantly higher in AD than in MCI (p = 0.01) and HC (p < 0.0001), and in MCI than in the HC group (p = 0.01) (Figure 1). Plasma sTREM2 concentrations were comparable between groups. The correlation between sTREM2 levels and age, sex, Mini-Mental State Examination (MMSE) score, ASC protein concentrations, and primary CSF biomarkers for AD (total and phosphorylated tau protein and Aβ concentrations) was analyzed. CSF sTREM2 levels correlated significantly positively with patient age (rs = 0.26, p < 0.0001; Figure 2a), plasma sTREM2 concentrations (rs = 0.2, p = 0.02; Figure 2b), CSF p-tau181 (rs = 0.28, p < 0.0001; Figure 2c), CSF ASC protein levels (rs = 0.19, p = 0.003; Figure 2d), positively and highly significantly with t-tau protein levels (rs = 0.31, p < 0.0001; Figure 2e), and negatively and significantly with MMSE score (rs = - 0.19, p = 0.002; Figure 2f). There was no relationship between the levels of sTREM2 and Aβ1-42 in CSF (rs = 0.08, p = 0.18). Age was also positively correlated with plasma sTREM2 concentrations (rs = 0.26, p = 0.002). As shown in Figure 3, the sensitivity and specificity of sTREM2 in CSF as a biomarker for AD were 65.81% and 80.00%, respectively, at a cut-off value of 23950 pg/ml (AUC = 0.75; p < 0.001).
3.2. Figures, Tables, and Schemes
Figure 1.
Significantly higher levels of CSF sTREM2 in AD compared to MCI (*p = 0.01), and HC (****p < 0.0001), as well as in MCI versus HC group (*p = 0.01).
Figure 1.
Significantly higher levels of CSF sTREM2 in AD compared to MCI (*p = 0.01), and HC (****p < 0.0001), as well as in MCI versus HC group (*p = 0.01).
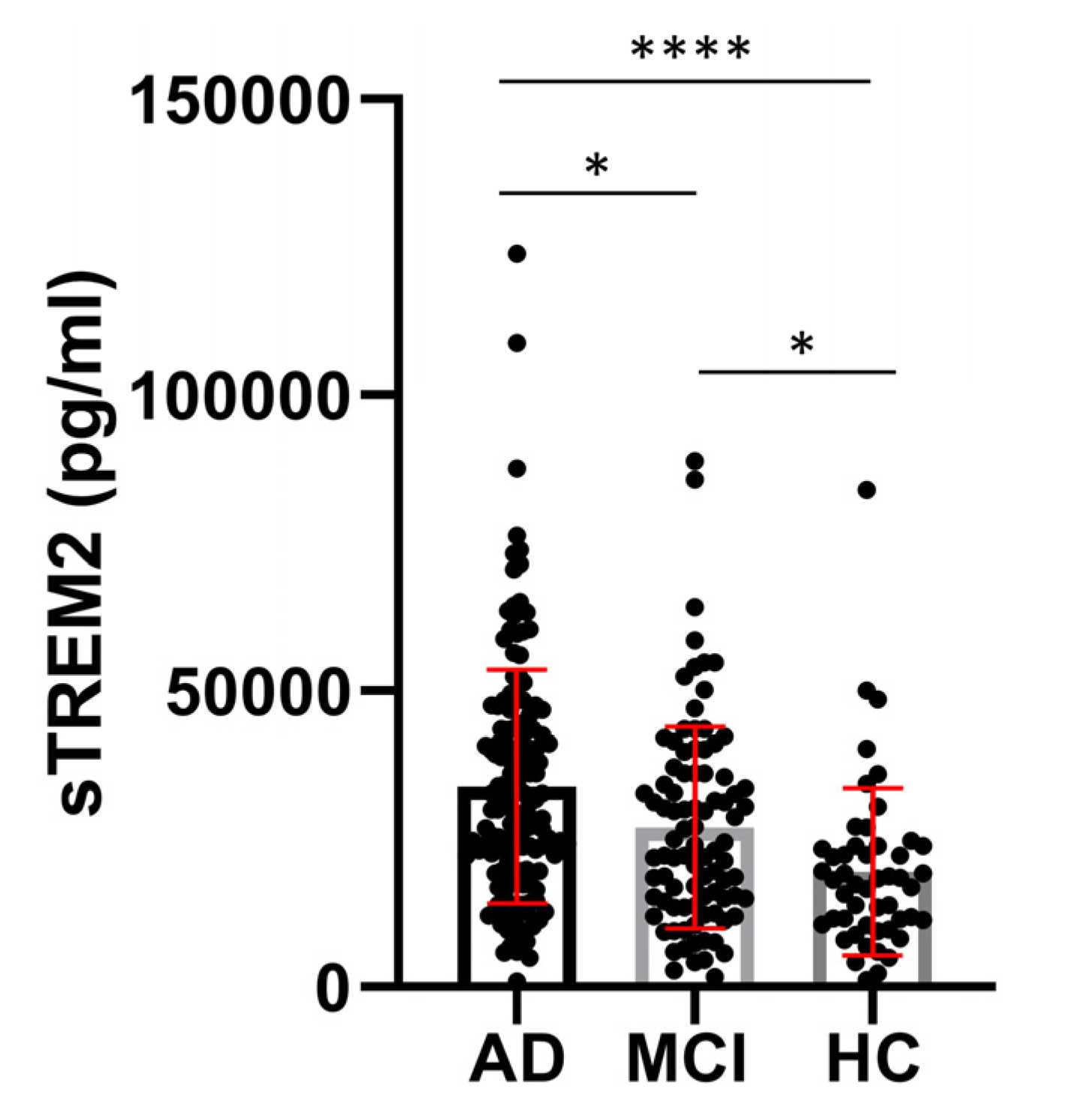
Figure 2.
CSF sTREM2 levels correlate positively and highly significantly with age (rs = 0.26, p < 0.0001) (a), positively and significantly with plasma sTREM2 levels (rs = 0.2, p = 0.02) (b), positively and highly significantly with p-tau181 (rs = 0.28, p < 0.0001) (c), positively and significantly with ASC levels (rs = 0.19, p = 0.003) (d), positively and highly significantly with t-tau protein concentrations (rs = 0.31, p < 0.0001) (e), and negatively and significantly with MMSE score (rs = - 0.19, p = 0.002) (f).
Figure 2.
CSF sTREM2 levels correlate positively and highly significantly with age (rs = 0.26, p < 0.0001) (a), positively and significantly with plasma sTREM2 levels (rs = 0.2, p = 0.02) (b), positively and highly significantly with p-tau181 (rs = 0.28, p < 0.0001) (c), positively and significantly with ASC levels (rs = 0.19, p = 0.003) (d), positively and highly significantly with t-tau protein concentrations (rs = 0.31, p < 0.0001) (e), and negatively and significantly with MMSE score (rs = - 0.19, p = 0.002) (f).
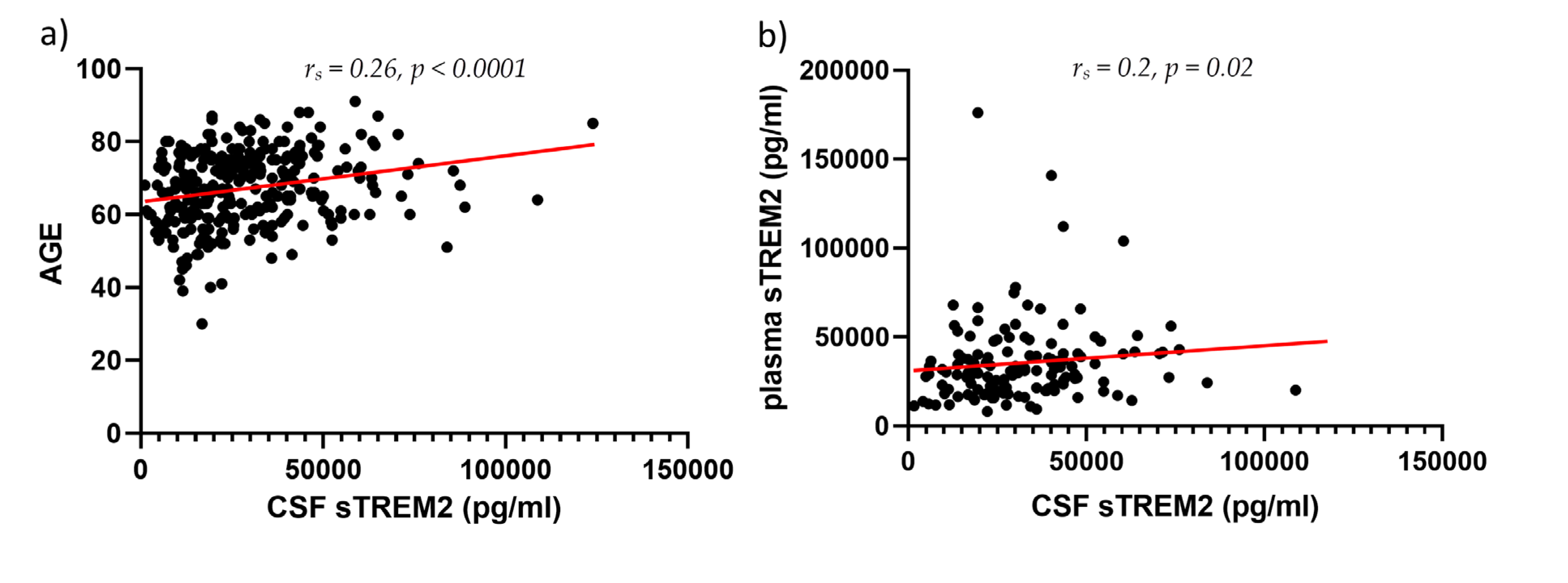
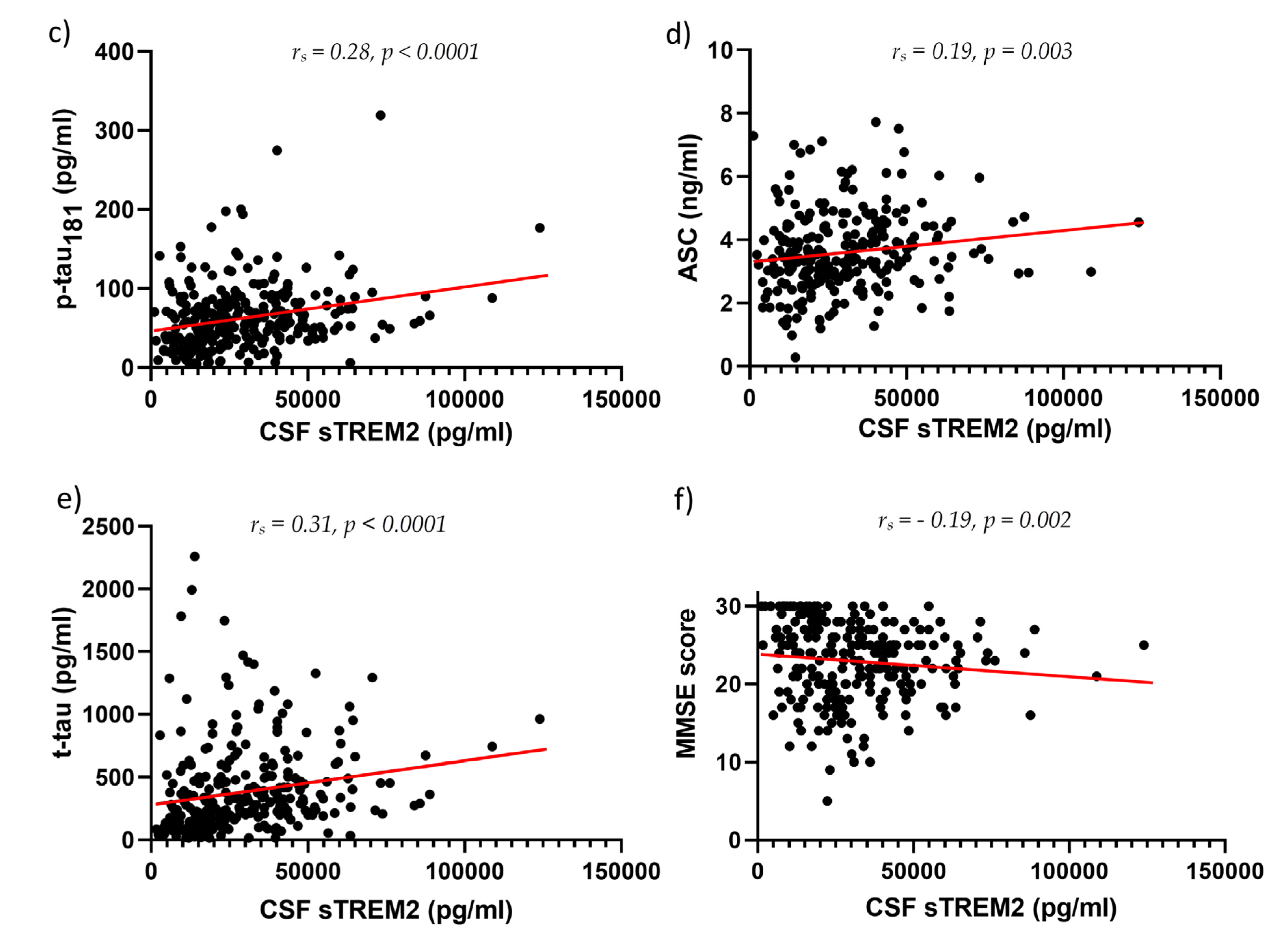
Figure 3.
Receiver Operating Characteristic (ROC) curve for CSF sTREM2 levels. AUC (area under the curve) = 0.75; p < 0.0001.
Figure 3.
Receiver Operating Characteristic (ROC) curve for CSF sTREM2 levels. AUC (area under the curve) = 0.75; p < 0.0001.
4. Discussion
Our findings demonstrate convincingly that CSF levels of sTREM2 are significantly greater in the AD group than in the MCI and HC groups. Apart from rare exceptions [34,42], the vast majority of previous studies [43,44] have, like the present study, found elevated concentrations of sTREM2 in the CSF to be strongly associated with AD. Importantly, we found that concentrations of sTREM2 in CSF are substantially higher in the MCI group compared to the HC group, confirming previous suggestions that sTREM2 could be used as the biomarker to detect conversion from MCI to AD [44] and can indicate earlier changes in MCI stage. Our findings also demonstrate that, statistically, plasma levels of sTREM2 do not differ between the three groups analyzed. This once again confirms that prospective AD biomarkers in plasma are poor indicators of what is occurring in the brain, and as a result, these biomarkers have unsatisfactory outcomes. There are numerous reasons, but some of the most important are as follows: 1) probably only a small fraction of sTREM2 enters the peripheral biofluid system, 2) like other proteins, sTREM2 can be degraded by proteases or form complexes with various blood proteins, and 3) it can also be cleared in the liver, kidney, and by macrophages in peripheral organs and tissues [45]. The third reason suggests that the optimal blood-collection site should be changed from the cubital vein to the internal jugular vein, as this would reduce blood dilution, degradation, and organ clearance effects [45]. Regardless of the aforementioned explanations, our results indicate that plasma sTREM2 concentrations do not adequately differentiate microglial activity between the MCI, AD, and HC subject groups. On the other hand, differences in CSF sTREM2 levels suggest that the innate immune response in the brains of AD patients is much stronger and that microglia are more active in the MCI stage than during healthy aging. Using transgenic mouse models of amyloid deposition, it has been demonstrated that genetic variability in the microglial response to amyloid deposition may be a significant risk factor for AD [46]. In the same study that confirmed the involvement of four mouse orthologs of GWAS-established risk genes for AD, including TREM2, at least four new putative risk genes were identified with a high degree of statistical rigor, as increased expression of these genes in microglia was observed only in the presence of amyloid [46].
Previously, variations in sTREM2 levels were determined at distinct disease stages. The MCI group had the greatest levels of sTREM2, which may be indicative of microglia’s response to early neuronal death [35] and efferocytosis [47]. Efferocytosis enables microglia to efficiently remove apoptotic and necrotic cells as well as cellular debris. In addition, it stimulates the production of anti-inflammatory cytokines, such as TGF-β and IL-10, while suppressing the release of pro-inflammatory cytokines. Thus, a deficiency in efferocytosis leads to the accumulation of apoptotic cells and cholesterol-rich brain tissue cellular detritus, resulting in chronic inflammation and autoimmunity [47]. Other authors [36,37] have discovered that sTREM2 concentrations are exceedingly sensitive to the onset of pathological changes during the progression of the disease [36,37,44]. In early non-symptomatic stages with low Aβ and normal tau levels, sTREM2 levels are lower, and sTREM2 concentrations increase when tau protein changes manifest as elevated CSF levels of p-tau and t-tau [36,37]. The TREM2 receptor is known to interact with Aβ and initiates its phagocytosis [11]. Ewers and colleagues [12] have demonstrated that elevated sTREM2 CSF levels prevent Aβ accumulation in the brain. In the 5xFAD model of AD, protracted TREM2 stimulation by injection of human TREM2 (hTREM2) agonistic mAb (AL002c) was protective against Aβ pathological changes and neurite damage [13]. Even though the role of the TREM2 receptor in the pathogenesis of AD is not completely understood, it is reasonable to assume that in the early stages of the disease, it may play a protective role, but as the disease progresses and adaptive immune response gets more involved, it becomes harmful [11,48]. In addition, TREM2 may only be protective against a subset of the pathological alterations in AD, specifically the previously mentioned Aβ load, but not tau pathology. Recent research [48] found that the number of T cells, particularly cytotoxic T cells, was significantly elevated in regions of pathological tau changes in the brains of mice with tauopathy and AD patients.
The cross-sectional design of the present investigation is a limitation; a longitudinal study would be preferable for tracking TREM2 level changes across disease stages. Notwithstanding, correlation analysis reveals a robust relationship between microglial activity and pathological alterations of tau proteins. In our study, sTREM2 CSF levels were positively correlated with both t-tau and p-tau181 CSF levels - findings in good agreement with previous studies in which tau-related neurodegeneration was reported to be associated with an increase in CSF sTREM2 (for review, see [11,27]), but not with Aβ1-42 CSF levels, similar to a recent report by Suarez-Cálvet et al. [36]. Based on the current investigation and the study of Suarez-Cálvet et al., it is logical to conclude that the pathology of the longest form of amyloid (Aβ1-42) in the absence of downstream tau-related neurodegeneration is associated with a decrease in CSF sTREM2. Intriguingly, a recent study found a positive association between sTREM2 in CSF and the shorter variants of amyloid – Aβx-40 [49], which are not typically associated with AD [50,51]. These findings may be explained by the possibility that AD patients have a higher removal rate of soluble Aβ1-42 because of increased cellular Aβ1-42 uptake [52].
This interdependence between sTREM2 levels and biomarkers of tau protein alterations has been reported previously, confirming the significance of the TREM2 receptor in tauopathy pathogenesis [11]. However, the precise roles of the TREM2 receptor in tauopathies remain undetermined. Its primary functions appear to include promoting phagocytosis, moderating the response to neuronal injury, and modulating neuroinflammation [7]. Recent research examining the effect of the R47H variant on tau pathology in the PS19 mouse model of tauopathy revealed that this variant improves microglia response and causes neurodegenerative alterations related to pathological changes of tau proteins [30]. In the THY-Tau22 mouse model of tauopathy, another study revealed that TREM2 deficiency worsens tau pathological changes in later stages owing to the lower microglial activation rate [32]. Depending on the APOE genotype, TREM2 signaling has distinct effects on pathological changes of the tau protein. Tau pathological changes and neurodegeneration are significantly more severe in mice with a silenced TREM2 gene and ApoE4 than in mice with an active TREM2 gene [31]. This is further evidence of the complexity of TREM2 signaling and a reminder of the distinctions between animal models of AD and humans. A positive correlation between sTREM2 concentrations and the age of the subjects suggests that microglial activation increases with age, whereas a negative correlation between sTREM2 concentrations and MMSE score suggests that sTREM2 could be a good indicator of cognitive deficits and that neurodegenerative changes and microglial activation are highly interrelated.
In addition, sTREM2 CSF levels were positively correlated with CSF ASC protein levels, indicating a potential link between the release of the soluble TREM2 form and the activation of the inflammasomes. Multiple studies [19,20,21,23] have demonstrated the relationship between inflammasomes and TREM2 receptors. Microglia cells with the R47H TREM2 gene variant, which increases the risk of AD onset, have a decreased activation of the NLRP3 inflammasome activation upon ligand binding to the TREM2 receptor [19]. Overexpression of TREM2 in macrophages inhibits activation of NLRP3 inflammasome [20], whereas silencing the TREM2 gene substantially increases macrophage pyroptosis [21]. These results suggest that TREM2 receptor expression performs a protective and anti-inflammatory role (for review, see [53]). In the context of AD, this property of TREM2 would imply greater control over inflammatory response and protection from over-activation and cross-activation of the inflammasomes, including the neuronal NLRP1 inflammasome [54], but this does not imply that silencing the inflammatory response is always the best course of action. For a healthy immune response, the timing and sequence of events are of the utmost importance, as AD pathological changes may affect the interaction between two receptors differently. In interpreting the results, it is important to remember that a previous study [55] has already demonstrated a correlation between elevated sTREM2 levels and pathological changes of the white matter, specifically small vessel disease and amyloid angiopathy, regardless of the severity of other pathological changes. This study demonstrates that CSF sTREM2 concentrations can differentiate between healthy and AD patients. With a cutoff value of 23950 pg/ml, CSF sTREM2 had a sensitivity of 66.81% and a high specificity of 80%.
AD is also characterized by a dysregulation of glucose metabolism [56,57,58,59], and a recent study found that inflammation initiated by elevated glucose levels in the BV2 microglial cell line results in a higher rate of NLRP3 inflammasome activation [23]. Additional experiments have shown that the TREM2 receptor can modulate the inflammatory response initiated by high glucose levels and the NLRP3 inflammasome [23]. Choi and colleagues found that 5xFAD mouse microglia consume significantly more glucose than wild-type microglia and that as the disease progresses, microglia express a greater number of glucose transporters (GLUT1 and GLUT2), have insufficient glycolysis, and a higher level of oxidative phosphorylation [28]. In addition, they discovered a positive correlation between the levels of CSF sTREM2 and glucose intake in the human hippocampus [28], which, given the activation of inflammasomes in glia in response to high glucose concentrations, could be a link between sTREM2 levels and the activation of inflammasomes. Despite the apparent connection between TREM2 signaling and the inflammasome activation pathway, the role and interaction of sTREM2 and inflammasome activation remain undefined. Even though other monocytes can also express the TREM2 receptor and release its soluble form into the blood [60], our results indicate a positive correlation between CSF and plasma sTREM2, which suggests that the state of the periphery indicates alterations in the brain to some extent. Several studies have failed to find a correlation between CSF sTREM2 levels and peripheral blood sTREM2 levels [61,62], and one study reported a negative correlation between CSF and plasma sTREM2 levels [63]. Despite obvious differences in CSF sTREM2 levels, a later statement can explain the lack of significant differences in plasma sTREM2 levels between the analyzed groups due to the reasons already discussed (for review, see [45]). Lastly, plasma sample groups were substantially smaller than CSF sample groups, which may explain why the differences were not statistically significant.
5. Conclusions
In contrast to plasma sTREM2, CSF sTREM2 levels were significantly higher in the AD group than in the MCI and HC groups, indicating a high diagnostic potential for distinguishing diseased from healthy individuals. The significant difference in sTREM2 levels between patients with MCI and HC subjects highlights sTREM2’s potential even further. AD patients with elevated CSF sTREM2 levels reliably predict neurofibrillary degeneration, cognitive decline, and inflammasome activation. To confirm the addition of CSF sTREM2 to the list of mandatory biomarkers for AD, future research should include more patients and follow the same pre-analytical, analytical, and post-analytical procedures. Considering the complexity of the TREM2 signaling in relation to various pathological aspects of AD, future longitudinal studies on AD patients are necessary.
Author Contributions
Conceptualization, G. Š.; methodology, E.Š.P., M.B.L., L.L.H., G.Š.; software, E.Š.P., M.B.L., G.Š.; validation, F.B., G.Š.; formal analysis, E.Š.P., M.B.L., G.Š.; investigation, E.Š.P., M.B.L., L.L.H., K.B., Ž.V., M.B., N.K., F.B., G.Š.; resources, G.Š.; data curation, E.Š.P., G.Š.; writing—original draft preparation, E.Š.P., G.Š.; writing—review and editing, E.Š.P., M.B.L., L.L.H., K.B., Ž.V., M.B., N.K., F.B., G.Š.; visualization, E.Š.P., M.B.L., G.Š.; supervision, G.Š.; project administration, E.Š.P., L.L.H., G.Š.; funding acquisition, G.Š. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Hrvatska zaklada za znanost (The Croatian Science Foundation), grant number IP-2019-04-3584 (“Role of blood-brain barrier, innate immunity, and tau protein oligomerization in the pathogenesis of Alzheimer’s disease”), and the Scientific Centre of Excellence for Basic, Clinical, and Translational Neuroscience CoRE-NEURO grant number KK01.1.1.01.0007 (“Experimental and clinical research of hypoxic-ischemic damage in perinatal and adult brain”) funded by the European Union through the European Regional Development Fund.
Institutional Review Board Statement
The study was approved by the Ethical Committee of the Clinical Hospital Center “Zagreb” (protocol no. 02/21 AG, class 8.1–18/82-2 from 24 April 2018, and protocol no. 02/21 AG, class 8.1–19/201-2 from 23 September 2019) and the Central Ethical Committee of the University of Zagreb School of Medicine (protocol no. 380-59-10106-18-111/126, class 641-01/18-02/01 from 20 June 2018, and protocol no. 380-59-10106-19-111/251, class 641-01/19-02/01 from October 2019).
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study, including the consent to publish the results of this paper. Written informed consent has been obtained from the patients to publish this paper.
Data Availability Statement
All the data reported are available on request from the corresponding author.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- Zhong:, L.; Chen, X.F. The emerging roles and therapeutic potential of soluble TREM2 in Alzheimer's disease. Front Aging Neurosci. 2019, 11, 328. [Google Scholar] [CrossRef] [PubMed]
- Jay, T.R.; von Saucken, V.E.; Landreth, G.E. TREM2 in neurodegenerative diseases. Mol Neurodegener. 2017, 12, 56. [Google Scholar] [CrossRef] [PubMed]
- Corder, E.H.; Saunders, A.M.; Strittmatter, W.J.; Schmechel, D.E.; Gaskell, P.C.; Small, G.W.; Roses, A.D.; Haines, J.L.; Pericak-Vance, M.A. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer's disease in late onset families. Science. 1993, 261, 921–923. [Google Scholar] [CrossRef] [PubMed]
- Qin, Q.; Wang, M.; Yin, Y.; Tang, Y. The specific mechanism of TREM2 regulation of synaptic clearance in Alzheimer's disease. Front Immunol. 2022, 13, 845897. [Google Scholar] [CrossRef] [PubMed]
- Li, R.Y.; Qin, Q.; Yang, H.C.; Wang, Y.Y.; Mi, Y.X.; Yin, Y.S.; Wang, M.; Yu, C.J.; Tang, Y. TREM2 in the pathogenesis of AD: a lipid metabolism regulator and potential metabolic therapeutic target. Mol Neurodegener. 2022, 17, 40. [Google Scholar] [CrossRef]
- Filipello, F.; You, S.F.; Mirfakhar, F.S.; Mahali, S.; Bollman, B.; Acquarone, M.; Korvatska, O.; Marsh, J.A.; Sivaraman, A.; Martinez, R.; Cantoni, C.; De Feo, L.; Ghezzi, L.; Minaya, M.A.; Renganathan, A.; Cashikar, A.G.; Satoh, J.I.; Beatty, W.; Iyer, A.K.; Cella, M.; Raskind, W.H.; Piccio, L.; Karch, C.M. Defects in lysosomal function and lipid metabolism in human microglia harboring a TREM2 loss of function mutation. Acta Neuropathol. 2023, 145, 749–772. [Google Scholar] [CrossRef]
- Šimić, G.; Španić, E.; Langer Horvat, L.; Hof, P.R. Blood-brain barrier and innate immunity in the pathogenesis of Alzheimer's disease. Prog Mol Biol Transl Sci. 2019, 168, 99–145. [Google Scholar] [CrossRef]
- Keren-Shaul, H.; Spinrad, A.; Weiner, A.; Matcovitch-Natan, O.; Dvir-Szternfeld, R.; Ulland, T.K.; David, E.; Baruch, K.; Lara-Astaiso, D.; Toth, B.; Itzkovitz, S.; Colonna, M.; Schwartz, M.; Amit, I. A unique microglia type associated with restricting development of Alzheimer's disease. Cell. 2017, 169, 1276–1290.e17. [Google Scholar] [CrossRef]
- Ulland, T.K.; Song, W.M.; Huang, S.C.; Ulrich, J.D.; Sergushichev, A.; Beatty, W.L.; Loboda, A.A.; Zhou, Y.; Cairns, N.J.; Kambal, A.; Loginicheva, E.; Gilfillan, S.; Cella, M.; Virgin, H.W.; Unanue, E.R.; Wang, Y.; Artyomov, M.N.; Holtzman, D.M.; Colonna, M. TREM2 maintains microglial metabolic fitness in Alzheimer's disease. Cell. 2017, 170, 649–663.e13. [Google Scholar] [CrossRef]
- Wang, Y.; Ulland, T.K.; Ulrich, J.D.; Song, W.; Tzaferis, J.A.; Hole, J.T.; Yuan, P.; Mahan, T.E.; Shi, Y.; Gilfillan, S.; Cella, M.; Grutzendler, J.; DeMattos, R.B.; Cirrito, J.R.; Holtzman, D.M.; Colonna, M. TREM2-mediated early microglial response limits diffusion and toxicity of amyloid plaques. J Exp Med. 2016, 213, 667–675. [Google Scholar] [CrossRef]
- Yang, J.; Fu, Z.; Zhang, X.; Xiong, M.; Meng, L.; Zhang, Z. TREM2 ectodomain and its soluble form in Alzheimer's disease. J Neuroinflammation. 2020, 17, 204. [Google Scholar] [CrossRef] [PubMed]
- Ewers, M.; Biechele, G.; Suárez-Calvet, M.; Sacher, C.; Blume, T.; Morenas-Rodriguez, E.; Deming, Y.; Piccio, L.; Cruchaga, C.; Kleinberger, G.; Shaw, L.; Trojanowski, J.Q.; Herms, J.; Dichgans, M.; Brendel, M.; Haass, C.; Franzmeier, N. Higher CSF sTREM2 and microglia activation are associated with slower rates of β-amyloid accumulation. EMBO Mol Med. 2020, 12, e12308. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Mustafa, M.; Yuede, C.M.; Salazar, S.V.; Kong, P.; Long, H.; Ward, M.; Siddiqui, O.; Paul, R.; Gilfillan, S.; Ibrahim, A.; Rhinn, H.; Ilaria Tassi, I.; Rosenthal, A.; Schwabe, T.; Colonna, M. Anti-human TREM2 induces microglia proliferation and reduces pathology in an Alzheimer's disease model. J Exp Med. 2020, 217, e20200785. [Google Scholar] [CrossRef]
- Jain, N.; Lewis, C.A.; Ulrich, J.D.; Holtzman, D.M. Chronic TREM2 activation exacerbates Aβ-associated tau seeding and spreading. J Exp Med. 2023, 220, e20220654. [Google Scholar] [CrossRef] [PubMed]
- Zhong, L.; Xu, Y.; Zhuo, R.; Wang, T.; Wang, K.; Huang, R.; Wang, D.; Yue Gao, Y.; Zhu, Y.; Sheng, X.; Chen, K.; Wang, N.; Zhu, L.; Can, D.; Marten, Y.; Shinohara, M.; Liu, C.C.; Du, D.; Sun, H.; Wen, L.; Xu, H.; Bu, G.; Chen, X.F. Soluble TREM2 ameliorates pathological phenotypes by modulating microglial functions in an Alzheimer's disease model. Nat Commun. 2019, 10, 1365. [Google Scholar] [CrossRef] [PubMed]
- Peng, X.; Guo, H.; Zhang, X.; Yang, Z.; Ruganzu, J.B.; Yang, Z.; Wu, X.; Bi, W.; Ji, S.; Yang, W. TREM2 inhibits tau hyperphosphorylation and neuronal apoptosis via the PI3K/Akt/GSK-3β signaling pathway in vivo and in vitro. Mol Neurobiol. 2023, 60, 2470–2485. [Google Scholar] [CrossRef]
- Pereira, J.B.; Janelidze, S.; Strandberg, O.; Whelan, C.D.; Zetterberg, H.; Blennow, K.; Palmqvist, S.; Stomrud, E.; Mattsson-Carlgren, N.; Hansson, O. Microglial activation protects against accumulation of tau aggregates in nondemented individuals with underlying Alzheimer's disease pathology. Nat Aging. 2022, 2, 1138–1144. [Google Scholar] [CrossRef]
- Lee, C.Y.D.; Daggett, A.; Gu, X.; Jiang, L.L.; Langfelder, P.; Li, X.; Wang, N.; Zhao, Y.; Park, C.S.; Cooper, Y.; Ferando, I.; Mody, I.; Coppola, G.; Xu, H.; Yang, X.W. Elevated TREM2 gene dosage reprograms microglia responsivity and ameliorates pathological phenotypes in Alzheimer's disease models. Neuron. 2018, 97, 1032–1048.e5. [Google Scholar] [CrossRef]
- Cosker, K.; Mallach, A.; Limaye, J.; Piers, T.M.; Staddon, J.; Neame, S.J.; Hardy, J.; Pocock, J.M. Microglial signalling pathway deficits associated with the patient-derived R47H TREM2 variants linked to AD indicate inability to activate inflammasome. Sci Rep. 2021, 11, 13316. [Google Scholar] [CrossRef]
- Yang, S.; Yang, Y.; Wang, F.; Luo, Q.Y.; Zhang, Y.; Zheng, F.; Shu, Q.; Chen, Q.; Fang, X. TREM2 dictates antibacterial defense and viability of bone marrow-derived macrophages during bacterial infection. Am J Respir Cell Mol Biol. 2021, 65, 176–188. [Google Scholar] [CrossRef]
- Wang, Y.; Cao, C.; Zhu, Y.; Fan, H.; Liu, Q.; Liu, Y.; Chen, K.; Wu, Y.; Liang, S.; Li, M.; Li, L.; Liu, X.; Zhang, Y.; Wu, C.; Lu, G.; Wu, M. TREM2/β-catenin attenuates NLRP3 inflammasome-mediated macrophage pyroptosis to promote bacterial clearance of pyogenic bacteria. Cell Death Dis. 2022, 13, 771. [Google Scholar] [CrossRef] [PubMed]
- Jay, T.R.; Miller, C.M.; Cheng, P.J.; Graham, L.C.; Bemiller, S.; Broihier, M.L.; Xu, G.; Margevicius, D.; Karlo, J.C.; Sousa, G.L; Cotleur, A.C.; Butovsky, O.; Bekris, L.; Staugaitis, S.M; Leverenz, J.B.; Pimplikar, S.W.; Landreth, G.E.; Howell, G.R.; Ransohoff, R.M.; Lamb, B.T. TREM2 deficiency eliminates TREM2+ inflammatory macrophages and ameliorates pathology in Alzheimer's disease mouse models. J Exp Med. 2015, 212, 287–295. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Long, W.; Gao, M.; Jiao, F.; Chen, Z.; Liu, M.; Yu, L. TREM2 regulates high glucose-induced microglial inflammation via the NLRP3 signaling pathway. Brain Sci. 2021, 11, 896. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Liu, F.; Li, H.; Wang, K.; Cao, X.; Xu, X.; Zhou, Y.; Zou, J.; Zhang, X.; Cui, X. TREM2 ameliorates anesthesia and surgery-induced cognitive impairment by regulating mitophagy and NLRP3 inflammasome in aged C57/BL6 mice. Neurotoxicology. 2022, 90, 216–227. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Liu, F.; Jiang, W.; Wang, K.; Cao, X.; Zou, J.; Zhou, Y.; Li, Z.; Liu, S.; Cui, X.; Zhang, X. TREM2 ameliorates lipopolysaccharide-induced oxidative stress response and neuroinflammation by promoting sirtuin3 in BV2 cells. Neurotox Res. 2022, 40, 56–65. [Google Scholar] [CrossRef] [PubMed]
- Del-Aguila, J.L.; Benitez, B.A.; Li, Z.; Dube, U.; Mihindukulasuriya, K.A.; Budde, J.P.; Farias, F.H.G.; Fernández, M.V.; Ibanez, L.; Jiang, S.; Perrin, R.J.; Cairns, N.J.; Morris, J.C.; Harari, O.; Cruchaga, C. TREM2 brain transcript-specific studies in AD and TREM2 mutation carriers. Mol Neurodegener. 2019, 14, 18. [Google Scholar] [CrossRef] [PubMed]
- Filipello, F.; Goldsbury, C.; You, S.F.; Locca, A.; Karch, C.M.; Piccio, L. Soluble TREM2: Innocent bystander or active player in neurological diseases? Neurobiol Dis. 2022, 165, 105630. [Google Scholar] [CrossRef]
- Zhong, L.; Chen, X.F.; Wang, T.; Wang, Z.; Liao, C.; Wang, Z.; Huang, R.; Wang, D.; Li, X.; Wu, L.; Jia, L.; Zheng, H.; Painter, M.; Atagi, Y.; Liu, C.C.; Zhang, Y.W.; Fryer, J.D.; Xu, H.; Bu, G. Soluble TREM2 induces inflammatory responses and enhances microglial survival. J Exp Med. 2017, 214, 597–607. [Google Scholar] [CrossRef]
- Qiao, W.; Chen, Y.; Zhong, J.; Madden, B.J.; Charlesworth, C.M.; Martens, Y.A.; Liu, C.C.; Knight, J.; Ikezu, T.C.; Kurti, A.; Zhu, Y.; Meneses, A.; Rosenberg, C.L.; Kuchenbecker, L.A.; Vanmaele, L.K.; Li, F.; Chen, K. , Shue, F.; Dacquel, M.V.; Fryer, J.; Pandey, A.; Zhao, N.; Bu, G. Trem2 H157Y increases soluble TREM2 production and reduces amyloid pathology. Mol Neurodegener. 2023, 18, 8. [Google Scholar] [CrossRef]
- Gratuze, M.; Leyns, C.E.; Sauerbeck, A.D.; St-Pierre, M.K.; Xiong, M.; Kim, N.; Serrano, J.R.; Tremblay, M.È.; Kummer, T.T.; Colonna, M.; Ulrich, J.D.; Holtzman, D.M. Impact of TREM2R47H variant on tau pathology-induced gliosis and neurodegeneration. J Clin Invest. 2020, 130, 4954–4968. [Google Scholar] [CrossRef]
- Albanus, R.; Jain, N.; Novotny, B.; Brase, L.; Rodriguez, L.; Mansel, C.; Kipnis, M.; O'Brien, S.; Pasillas, M.P.; Lee, C.; Manis, M.; Colonna, M.; Harari, O.; Glass, C.K.; Ulrich, J.D.; Holtzman, D.M. TREM2-independent microgliosis promotes tau-mediated neurodegeneration in the presence of ApoE4. Neuron. 2023, 111, 202–219.e7. [Google Scholar] [CrossRef]
- Vautheny, A.; Duwat, C.; Aurégan, G.; Joséphine, C.; Hérard, A.S.; Jan, C.; Mitja, J.; Gipchtein, P.; Gaillard, M.C.; Buée, L.; Blum, D.; Hantraye, P.; Bonvento, G.; Brouillet, E.; Cambon, K.; Bemelmans, A.P. THY-Tau22 mouse model accumulates more tauopathy at late stage of the disease in response to microglia deactivation through TREM2 deficiency. Neurobiol Dis. 2021, 155, 105398. [Google Scholar] [CrossRef]
- Brown, G.C.; St George-Hyslop, P. Does soluble TREM2 protect against Alzheimer's disease? Front Aging Neurosci. 2022, 13, 834697. [Google Scholar] [CrossRef] [PubMed]
- Ulrich, J.D.; Ulland, T.K.; Colonna, M.; Holtzman, D.M. Elucidating the role of TREM2 in Alzheimer's disease. Neuron. 2017, 94, 237–248. [Google Scholar] [CrossRef]
- Suárez-Calvet, M.; Kleinberger, G.; Araque Caballero, M.Á.; Brendel, M.; Rominger, A.; Alcolea, D.; Fortea, J.; Lleó, A.; Blesa, R.; Gispert, J.D.; Sánchez-Valle, R.; Antonell, A.; Rami, L.; Molinuevo, J.L.; Brosseron, F.; Traschütz, A.; Heneka, M.T.; Struyfs, H.; Engelborghs, S.; Sleegers, K.; Van Broeckhoven, C.; Zetterberg, H.; Nellgård, B.; Blennow, K.; Crispin, A.; Michael Ewers, M.; Haass, C. sTREM2 cerebrospinal fluid levels are a potential biomarker for microglia activity in early-stage Alzheimer's disease and associate with neuronal injury markers. EMBO Mol Med. 2016, 8, 466–476. [Google Scholar] [CrossRef]
- Suárez-Calvet, M.; Morenas-Rodríguez, E.; Kleinberger, G.; Schlepckow, K.; Araque Caballero, M.A.; Franzmeier, N.; Capell, A.; Fellerer, K.; Nuscher, B.; Eren, E.; Levin, J.; Deming, Y.; Piccio, L.; Karch, C.M.; Cruchaga, C.; Shaw, L.M.; Trojanowski, J.Q.; Weiner, M.; Ewers, M.; Haass, C. Early increase of CSF sTREM2 in Alzheimer's disease is associated with tau related-neurodegeneration but not with amyloid-β pathology. Mol Neurodegener. 2019, 14, 1. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.Z.; Tan, L.; Bi, Y.L.; Shen, X.N.; Xu, W.; Ma, Y.H.; Li, H.Q.; Dong, Q.; Yu, J.T. Dynamic changes of CSF sTREM2 in preclinical Alzheimer's disease: the CABLE study. Mol Neurodegener. 2020, 15, 25. [Google Scholar] [CrossRef]
- Hyman, B.T.; Phelps, C.H.; Beach, T.G.; Bigio, E.H.; Cairns, N.J.; Carrillo, M.C.; Dickson, D.W.; Duyckaerts, C.; Frosch, M.P.; Masliah, E.; Mirra, S.S.; Nelson, P.T.; Schneider, J.A.; Thal, D.R.; Thies, B.; Trojanowski, J.Q.; Vinters, H.V.; Montine, T.J. National Institute on Aging-Alzheimer’s association guidelines for the neuropathologic assessment of Alzheimer’s disease. Alzheimer’s Dement. 2012, 8, 1–13. [Google Scholar] [CrossRef]
- Montine, T.J.; Phelps, C.H.; Beach, T.G.; Bigio, E.H.; Cairns, N.J.; Dickson, D.W.; Duyckaerts, C.; Frosch, M.P.; Masliah, E.; Mirra, S.S.; Nelson, P.T.; Schneider, J.A.; Thal, D.R.; Trojanowski, J.Q.; Vinters, H.V.; Hyman, B.T. National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease: A practical approach. Acta Neuropathol 2012, 123, 1–11. [Google Scholar] [CrossRef]
- Petersen, R.C.; Smith, G.E.; Waring, S.C.; Ivnik, R.J.; Tangalos, E.G.; Kokmen, E. Mild cognitive impairment: Clinical characterization and outcome. Arch Neurol 1999, 56, 303–308. [Google Scholar] [CrossRef]
- Albert, M.S.; DeKosky, S.T.; Dickson, D.; Dubois, B.; Feldman, H.H.; Fox, N.C.; Gamst, A.; Holtzman, D.M.; Jagust, W.J.; Petersen, R.C.; Snyder, P.J.; Carrillo, M.C.; Thies, B.; Phelps, C.H. The diagnosis of mild cognitive impairment due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s association workgroups on. Alzheimer’s Dement 2011, 7, 270–279. [Google Scholar] [CrossRef] [PubMed]
- Heslegrave, A.; Heywood, W.; Paterson, R.; Magdalinou, N.; Svensson, J.; Johansson, P.; Öhrfelt, A.; Blennow, K.; Hardy, J.; Schott, J.; Mills, K.; Zetterberg, H. Increased cerebrospinal fluid soluble TREM2 concentration in Alzheimer's disease. Mol Neurodegener. 2016, 11, 3. [Google Scholar] [CrossRef] [PubMed]
- Bonomi, C.G.; Assogna, M.; Di Donna, M.G.; Bernocchi, F.; De Lucia, V.; Nuccetelli, M.; Fiorelli, D.; Loizzo, S.; Mercuri, N.B.; Koch, G.; Martorana, A.; Motta, C. Cerebrospinal fluid sTREM-2, GFAP, and β-S100 in symptomatic sporadic Alzheimer's disease: Microglial, astrocytic, and APOE contributions along the Alzheimer's disease continuum. J Alzheimers Dis. 2023, 92, 1385–1397. [Google Scholar] [CrossRef]
- Zhao, A.; Jiao, Y.; Ye, G.; Kang, W.; Tan, L.; Li, Y.; Deng, Y.; Liu, J. Alzheimer’s disease neuroimaging initiative (ADNI). Soluble TREM2 levels associate with conversion from mild cognitive impairment to Alzheimer's disease. J Clin Invest. 2022, 132, e158708. [Google Scholar] [CrossRef]
- Huang, S.; Wang, Y.J.; Guo, J. Biofluid biomarkers of Alzheimer's disease: Progress, problems, and perspectives. Neurosci Bull. 2022, 38, 677–691. [Google Scholar] [CrossRef] [PubMed]
- Salih, D.A.; Bayram, S.; Guelfi, S.; Reynolds, R.H.; Shoai, M.; Ryten, M.; Brenton, J.W.; Zhang, D.; Matarin, M.; Botia, J.A.; Shah, R.; Brookes, K.J.; Guetta-Baranes, T.; Morgan, K.; Bellou, E.; Cummings, D.M. , Escott-Price, V.; Hardy, J. Genetic variability in response to amyloid beta deposition influences Alzheimer's disease risk. Brain Commun. 2019; 1, fcz022. [Google Scholar] [CrossRef]
- Andrews, S.J.; Renton, A.E.; Fulton-Howard, B.; Podlesny-Drabiniok, A.; Marcora, E.; Goate, A.M. The complex genetic architecture of Alzheimer's disease: novel insights and future directions. EBioMedicine. 2023, 90, 104511. [Google Scholar] [CrossRef]
- Chen, X.; Firulyova, M.; Manis, M.; Herz, J.; Smirnov, I.; Aladyeva, E.; Wang, C.; Bao, X.; Finn, M.B.; Hu, H.; Shchukina, I.; Kim, M.W.; Yuede, C.M.; Kipnis, J.; Artyomov, M.N.; Ulrich, J.D.; Holtzman, D.M. Microglia-mediated T cell infiltration drives neurodegeneration in tauopathy. Nature. 2023, 615, 668–677. [Google Scholar] [CrossRef]
- Winfree, R.L.; Dumitrescu, L.; Blennow, K.; Zetterberg, H.; Gifford, K.A.; Pechman, K.R.; Jefferson, A.L.; Hohman, T.J. Alzheimer's Disease Neuroimaging Initiative. Biological correlates of elevated soluble TREM2 in cerebrospinal fluid. Neurobiol Aging. 2022, 118, 88–98. [Google Scholar] [CrossRef]
- Attems, J.; Lintner, F.; Jellinger, K.A. Amyloid beta peptide 1-42 highly correlates with capillary cerebral amyloid angiopathy and Alzheimer disease pathology. Acta Neuropathol. 2004, 107, 283–291. [Google Scholar] [CrossRef]
- Attems, J.; Jellinger, K.A. Only cerebral capillary amyloid angiopathy correlates with Alzheimer pathology--a pilot study. Acta Neuropathol. 2004, 107, 83–90. [Google Scholar] [CrossRef]
- Zaretsky, D.V.; Zaretskaia, M.V.; Molkov, Y.I. Alzheimer’s Disease Neuroimaging Initiative. Patients with Alzheimer's disease have an increased removal rate of soluble beta-amyloid-42. PLoS One. 2022, 17, e0276933. [Google Scholar] [CrossRef] [PubMed]
- Španić, E.; Langer Horvat, L.; Hof. P.R.; Šimić, G. Role of microglial cells in Alzheimer's disease tau propagation. Front Aging Neurosci. 2019, 11, 271. [Google Scholar] [CrossRef]
- Španić, E.; Langer Horvat, L.; Ilić, K.; Hof, P.R.; Šimić, G. NLRP1 Inflammasome activation in the hippocampal formation in Alzheimer's disease: Correlation with neuropathological changes and unbiasedly estimated neuronal loss. Cells. 2022, 11, 2223. [Google Scholar] [CrossRef] [PubMed]
- Tsai, H.H.; Chen, Y.F.; Yen, R.F.; Lo, Y.L.; Yang, K.C.; Jeng, J.S.; Tsai, L.K.; Chang, C.F. Plasma soluble TREM2 is associated with white matter lesions independent of amyloid and tau. Brain. 2021, 144, 3371–3380. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Zhong, C. Decoding Alzheimer's disease from perturbed cerebral glucose metabolism: implications for diagnostic and therapeutic strategies. Prog Neurobiol. 2013, 108, 21–43. [Google Scholar] [CrossRef]
- An, Y.; Varma, V.R.; Varma, S.; Casanova, R.; Dammer, E.; Pletnikova, O.; Chia, C.W.; Egan, J.M.; Ferrucci, L.; Troncoso, J.; Levey, A.I.; Lah, J.; Seyfried, N.T.; Legido-Quigley, C.; O'Brien, R.; Thambisetty, M. Evidence for brain glucose dysregulation in Alzheimer's disease. Alzheimers Dement. 2018, 14, 318–329. [Google Scholar] [CrossRef]
- Cho, S.; Lee, H.; Seo, J. Impact of genetic risk factors for Alzheimer's disease on brain glucose metabolism. Mol Neurobiol. 2021, 58, 2608–2619. [Google Scholar] [CrossRef]
- Knezović, A.; Osmanović-Barilar, J.; Ćurlin, M.; Hof, P.R.; Šimić, G.; Riederer, P.; Šalković-Petrišić, M. Staging of cognitive deficits and neuropathological and ultrastructural changes in streptozotocin-induced rat model of Alzheimer's disease. J Neural Transm. 2015, 122, 577–592. [Google Scholar] [CrossRef]
- Choi, H.; Choi, Y.; Lee, E.J.; Kim, H.; Lee, Y.; Kwon, S.; Hwang, D.W.; Lee, D.S. Hippocampal glucose uptake as a surrogate of metabolic change of microglia in Alzheimer's disease. J Neuroinflammation. 2021, 18, 190. [Google Scholar] [CrossRef]
- Piccio, L.; Buonsanti, C.; Cella, M.; Tassi, I.; Schmidt, R.E.; Fenoglio, C.; Rinker, J. 2nd.; Naismith, R.T.; Panina-Bordignon, P.; Passini, N.; Galimberti, D.; Scarpini, E.; Colonna, M.; Cross, A.H. Identification of soluble TREM-2 in the cerebrospinal fluid and its association with multiple sclerosis and CNS inflammation. Brain. 2008, 131, 3081–3091. [Google Scholar] [CrossRef]
- Piccio, L.; Deming, Y.; Del-Águila, J.L.; Ghezzi, L.; Holtzman, D.M.; Fagan, A.M.; Fenoglio, C.; Galimberti, D.; Borroni, B.; Cruchaga, C. Cerebrospinal fluid soluble TREM2 is higher in Alzheimer disease and associated with mutation status. Acta Neuropathol. 2016, 131, 925–933. [Google Scholar] [CrossRef] [PubMed]
- Park, S.H.; Lee, E.H.; Kim, H.J.; Jo, S.; Lee, S.; Seo, S.W.; Park, H.H.; Koh, S.H.; Lee, J.H. The relationship of soluble TREM2 to other biomarkers of sporadic Alzheimer's disease. Sci Rep. 2021, 11, 13050. [Google Scholar] [CrossRef] [PubMed]

Table 1.
Age, number of MMSE points, and CSF levels of sTREM2, t-tau, p-tau181, A1-42, and ASC for the AD, MCI, and HC groups, where (n) represents the sample size analyzed.
Table 1.
Age, number of MMSE points, and CSF levels of sTREM2, t-tau, p-tau181, A1-42, and ASC for the AD, MCI, and HC groups, where (n) represents the sample size analyzed.
| Group | Age (years) mean ± SD (n) |
MMSE (points) mean ± SD (n) |
sTREM2 (pg/ml) mean ± SD (n) |
ASC (ng/ml) mean ± SD (n) |
Aβ1-42 (pg/ml) mean ± SD (n) |
p-tau181 (pg/ml) mean ± SD (n) |
t-tau (pg/ml) mean ± SD (n) |
|---|---|---|---|---|---|---|---|
| AD | 70.58 ± 8.670 (154) | 20.30 ± 4.395 (142) | 33854 ± 19631 (155) | 3.817± 1.348 (126) | 549.5 ± 303.9 (152) | 77.06 ± 45.25 (147) |
522.5 ± 400.9 (146) |
| MCI | 66.42 ± 8.832 (90) | 25.64 ± 2.903 (74) | 26940 ± 17043 (90) | 3.432 ± 1.121 (78) | 665.9 ± 374.8 (86) |
53.32 ± 29,90 (87) |
257.5 ± 177,1 (86) |
| HC | 58.80 ± 10.68 (49) | 29.06 ± 2.199 (32) | 18527 ± 13566 (50) | 3.369 ± 1.272 (39) | 552.8 ± 521.7 (49) |
35.18 ± 22.70 (49) |
222.7 ± 284,0 (48) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



