
Submitted:
24 May 2023
Posted:
25 May 2023
You are already at the latest version
Abstract
Despite the numerous advances in target therapy for the treatment of colorectal cancer, aggressive colorectal cancer remains an incurable disease whose negative modulation of the immune system in the tumor microenvironment is still critical for improving the patient's prognosis. Extracellular vesicles (EVs) have received attention for their use as cell-membrane-camouflaged nanoparticles and drug delivery systems in nanomedicine derived by nearly all cell types for intercellular communication and regulation in the tumor microenvironment (TME). In this study, M1 Macrophage EVs (M1EVs) were used as nanocarriers of oxaliplatin (M1EV1) associated with retinoic acid (M1EV2) and Libidibia ferrea (M1EV3), or combined together with retinoic acid and Libidibia ferrea (M1EV4) to evaluate their antiproliferative and immunomodulatory potential on CT-26 and MC-38 colorectal cancer cell lines, as well as their capacity to prevent metastases in mouse models of colorectal cancer such as allographic and peritoneal. Tumors were evaluated by qRT-PCR and immunohistochemistry. The cell death profile and epithelial-mesenchymal transition process (EMT) were analyzed in vitro in colorectal cancer cell lines. Polarization of murine macrophages (RAW264.7 cells) was also carried out. M1EV2 and M1EV3 used alone or especially combined (M1EV4) downregulated the tumor progression by TME immunomodulation, leading to a decrease in primary tumor size and metastases in peritoneum, liver and lungs. STAT3, NF-kB and AKT were the major genes downregulated by systems of M1EVs. Tumor-associated macrophages (TAM) switched M2 phenotype (CD163) towards M1 (CD68) reducing levels of IL-10, TGF-β and CCL22. Furthermore, malignant cells showed overexpression of FADD, APAF-1, caspase-3, and E-cadherin, and decreased expression of MDR-1, survivin, vimentin, CXCR4, and PD-L1 after treatment with systems of M1EVs. The results obtained in this study provided evidence that EVs from M1 antitumor macrophages can transport drugs and increase their immunomodulatory and antitumor activity by stimulating activation or blockage of pathways involved in cell proliferation, migration, cell survival, and drug resistance processes.
Keywords:
EVs
; Macrophages 1
; STAT3
; AKT
; NF-κB
; metastasis
; colorectal cancer.
1. Introduction
Colorectal cancer (CRC) is among the top three cancers with a higher incidence and mortality rate worldwide1–3. This is mainly because the predominant treatment of cancer focuses on inhibiting the growth of cancer, with little emphasis on metastasis4. Limited success has been achieved in terms of treating cancer metastasis5 to face this, target therapy seems to be a new option that has successfully prolonged overall survival for CRC patients6.
Among the biocompatible materials, the one that deserves special attention is EVs. EVs have many of the desirable features of an ideal drug delivery system, such as a long-circulating half-life, the intrinsic ability to target tissues, biocompatibility, biodegradability, and minimal or no inherent toxicity issues7–9. The EVs can deliver conventional chemotherapeutic drugs, increase bioavailability and concentration around tumor tissues, and improve their release profile10. They have also been efficiently used as carriers and tissue-specific drug delivery systems for antitumor agents11,12.
EVs derived from immune cells can activate the immune system by exposing tumor antigens or immune-enhancing molecules and can further be modified to potentiate or redirect its antitumor effects, an interesting concept known as “tumor vaccines”13–15. In addition to their biomedical advantages and immunomodulatory properties, some EVs have also demonstrated intrinsic antitumor activity, due to specific molecules and tumor suppressor agents that they contain12,13,16.
As an important component of innate and adaptive immunity, macrophages may polarize into different functional phenotypes, such as the classically activated M1 and the alternatively activated M2 macrophages (M2-TAM), depending on the external stimulus17. In the context of cancer, M1 macrophages are reportedly associated with tumoricidal activities18–20. Increasing evidence suggests that M2 macrophages could perform immunosuppressive functions and promote tumor progression and metastasis19.
Based on scientific findings, M1 Macrophages are important targets in tumor immunotherapy, especially for potentiating the antitumor response in TME by blocking M2TAM-stimulated STAT3/NF-κB signaling pathways10,21–24. In this context, finding strategies beyond the state-of-the-art in cancer management makes it urgent. There are reports that retinoic acid (RA) generated from vitamin A-derivative retinol acts the innate immunity by binding to intracellular receptors of immune cells25, inhibiting M2-macrophage polarization26,27. Our group has shown that polyphenols, ellagic acid, and gallic acid, present in Libidibia ferrea (LF), a native and endemic species of Brazil, promote antiproliferative activity with induction of intrinsic apoptosis in colorectal cancer cells as well as acting in the decreased of oxygen-reactive species and pro-inflammatory cytokines28,29. Furthermore, ellagic acid reduces the expression of immunosuppressive cytokines by downregulating proficiently NF-κB and STAT330,31 in hepatocarcinoma. In the present work, we analyzed the combination of OXA with EVs of M1 Macrophages loaded with RA and LF to understand their antitumor and antimetastatic properties in colorectal cancer models. For this proposal, the EVs were used as nanocarriers for OXA, RA, and LF to evaluate the antitumor effect in the progression of colorectal cancer.
2. Material and Methods
2.1. Antibodies and reagents
The cytotoxic agent Oxaliplatin used in the experiments (50mg injectable lyophilized powder in a package containing vials) was acquired from LIBBS Pharmaceutical Ltd.a (Sigma-Aldrich, St. Louis, MO, USA). Retinoic acid was purchased from Sigma. The crude extracts of fruits from Libidibia ferrea (LF) using ethanol as solvent at 60% (60T) were obtained from the Therapeutic Innovation laboratory at the Department of Pharmaceutical Sciences-Federal University of Pernambuco (UFPE)28. 1.65g Ellagic acid (EA) and 0.66g gallic acid were quantified from 60T LF by high-performance liquid chromatography (HPLC)28. The EVs were produced and supplied by the Department of Nanobiomaterials and Imaging at Leiden University Medical Center10. The list of antibodies used, and basic information is summarized in table 01S.
2.2. Cell Culture and Cell Lines
Murine colon carcinoma undifferentiated cell lines (CT-26 e MC-38) and macrophage cell lines (RAW 264.7) were obtained from American Type Culture Collection (Rock-Ville, MD, USA). Cell lines were cultivated in Dulbecco’s modified Eagle’s medium (DMEM, Gibco Laboratories, Grand Island, NY, USA) supplemented with 1% antibiotics (penicillin/streptomycin) and 10% fetal bovine serum (FBS). Cells were kept under maintenance until the end of the experiment in a humidified incubator with 5% CO2 at a temperature of 37°C. Cell passage was done 3 times a week by removing the adherent cells with trypsin/EDTA in phosphate-buffered saline (PBS).
2.3. Animals
Male Balb/c mice approximately 7-9 weeks of age weighing 21-28g were purchased at Keizo Asami Immunology Laboratory Facility (FIOCRUZ-PE) and used for the experimental xenographic tumor growth model using undifferentiated carcinoma cell lines from the colon (CT-26). The animals were housed in cages with free access to food and water and treated according to ethical principles for animal experimentation. All surgical and experimental procedures were approved by the ethics committee of the Federal University of Rio Grande do Norte (Nº 222.011/2020 and 265.025/2021).
2.4. Dosage, administration, and group distribution of EVs
The concentrations used in all in vitro experiments corresponded to the quantification of M1EV proteins identified in the nanodrop (µg of protein per mL). For the M1EV protein concentration loaded with RA and L. ferrea without OXA, 20µg/mL was considered. The concentration of OXA in the M1EVs was confirmed by HPLC as described below. For in vivo experiments, the concentration of 2mg/kg of oxaliplatin was standardized.
For the in vivo assays, the respective groups were used: saline group, OXA: 5mg/kg oxaliplatin, 20µg/mL M1EV: M1-EVs, M1EV1: M1EV+OXA, M1EV2: M1EV+OXA+RA, M1EV3: M1EV+OXA+LF, and M1EV4: M1EV+OXA+RA+LF.
For the in vitro assays, cell lines were treated with M1EVs loaded with OXA, RA, and LF (M1EV1; M1EV2 and M1EV3), respectively, or treated with M1EV without OXA but loaded with RA, LF, and RA+LF (M1EVRA: M1EV+RA; M1EVLF: M1EV+LF and M1EVRALF: M1EV+RA+LF), respectively.
2.5. Preparation of EV and OXA loading
To obtain EVs from M1 macrophages, the RAW 264.7 cells were stimulated according to10 to induce their polarization towards an antitumor M1 phenotype 23. Briefly, the RAW cells 264.7 were seeded in medium culture flasks in DMEM and FBS to contribute cell adhesion and growth until 18.4 × 106 cells. The medium was then replaced by DMEM without FBS supplemented with lipopolysaccharides + interferon-gamma (IFN-γ) both at 0.1μg/mL for 48h. Cells were washed and incubated for an additional 48 h with either serum-free media for M1EVs, 100 μg/mL RA, 75 μg/mL L.F, or a combination of both (RA + LF). EVs were collected and purified using a standardized protocol32 of differential centrifugation 33,34 followed by size exclusion separation1 to obtain a monodisperse population enriched in nanosized EVs of around 120nm. The resulting systems (PS-M1-EVs) were characterized by HPLC for analysis of the amount of oxaliplatin incorporated in EVs. The obtained EV fraction was resuspended in PBS (500µl, 1 mg/mL total protein), and characterized for size and polydispersity as described below.
To incorporate OXA, freshly isolated EVs (M1) were incubated with 10 mM solution of OXA (LIBBS Pharmaceutical Ltd.a) in 100% Dimethyl Sulfoxide (DMSO, Sigma Aldrich, cat# D2650-100ML), in a 2:8 ratio, at 4°C for 1 hour. The EVs were centrifuged using size exclusion columns as previously described by our team 35–37and stored at -80°C until used.
2.6. Characterization of EVs
For EVs size distribution and concentration, an aliquot of EVs was diluted in 1mL in sterile PBS and aspirated with a syringe for nanoparticle tracking analysis (NTA) by NanoSight NS300 (Malvern) using an automated syringe pump, camera level of 9 and a detection threshold of 3 for the measurements.
To analyze shape and size, samples were measured by transmission electron microscope (TEM) as previously described 1. Briefly, 20μl EVs were dropped onto a 200-mesh copper grid and incubated at room temperature for 1 min. After fixing with 2% paraformaldehyde for 5 min, EVs were stained with 2% uranyl acetate for 15 min. Rinsed the grids with PBS and dried the grid by contacting the back of the grid with filter paper. Finally, EVs were visualized via a TEM (JEM- 1010, JEOL, Japan). All images were recorded at 18.000× magnification (pixel size 1.2 nm) operating the microscope at 120 kV.
2.7. HPLC
The OXA concentration was then determined using the supernatant after the membrane lysis process on the EVs. The analyzes were performed on a Prominence UFLC-XR chromatographic system (Shimadzu®), consisting of a binary pump (model LC-20AD), online degasser (model DGU-20A3), ALS autosampler (model SIL-20AHT), column oven (model CTO-20A), PDA detector (model SPD-20A) and communication module (model CBM-20A). The data were collected by Shimadzu LC-Solution®software. The oven temperature was set at 30°C with 10μL of injection volume. The wavelength was set at 255 nm. The mobile phase consisted of 10 mM phosphoric acid and acetonitrile (95:5), pumped at 0.5 mL.min-1 in isocratic mode. Due to the high polarity of oxaliplatin, a 10 mM sodium 1-butane sulfonate as a pair was used.
The quantitative analysis was made by a calibration curve (linearity) in a range of 0.3μg/mL-1 to 3.0μg/mL-1. The linearity was determined from three analytical curves in triplicate of the standards. Oxaliplatin concentrations were 0.3, 0.6, 0.9, 1.2, 2.4, and 3.0μg/mL-1. The results were evaluated from the regression coefficient. The limits of detection (LODs) and quantification (LOQs) were calculated with the standard deviation of the slope (σ) and mean value of the slope (S) of the analytical curves.
2.8. Cell uptake
EVs were labeled with a green, fluorescent Vybrant™ Multicolor Cell-Labeling Kit (DiR TermoFisher SCIENTIFIC) by incubation followed by size exclusion separation as previously shown by our group36 . Cells were seeded at 5x103 cells/well in a cover glass and incubated overnight. After the cells adhered to the cover glass, the medium was changed, and the cells were incubated for 24 hours with DIR-labelled EVs. The cell membrane was marked with DIL and the nuclei with DAPI. The cover glasses were mounted on slides with VECTASHIELD Mounting Medium. Samples were examined with a Leica DM5500 B fluorescence microscope and a Leica DFC365 FX digital camera. Digital images were made, analyzed, and stored using Leica Application Suite X (LAS X) software.
2.9. Polarization of RAW 264.7 cells
RAW 264.7 cells were seeded in a 12-well plate at a density of 5 × 105 cells and supplemented with DMEM as described above for 24 h. For stimulation of M2-like polarization, RAW 264.7 cells were cultured with 0.04 µg/mL of IL-4 for 48 h in a serum-free medium as previously described by Araujo et al.23. To evaluate the effect of M1EVs on polarization of the M2-polarized tumor-associated macrophages (M2-TAMs), the RAW 264.7 cells were incubated with M1EVRALF, M1EVLF, M1EVRA, and M1EV after the M2 polarization for 48h. Likewise, M2-polarized TAMs (5 × 104 cells/well) were seeded in 12-well plates (BD Bioscience) in DMEM medium supplemented with 10% FBS. CT-26 cells (1 × 104 cells/insert) were seeded into the upper chamber of the transwell insert with an 8 μm pore size (Corning Inc., Corning, NY, USA) in DMEM with 10% FBS. The following day, the culture inserts with M2-polarized TAMs were treated with M1EVRALF, M1EVLF, M1EVRA and M1EV, and cells were cultured for another 48 h in a humidified incubator with 5% CO2 at a temperature of 37 °C.
The cells were harvested and blocked with 0.5% BSA in PBS for 45 min and then labeled with anti-CD163-PerCP (1:1000) or anti-CD68-FITC (1:1000) antibodies. The cells were protected from light until analyzed by the flow cytometer FACSCalibur (BD Biosciences). All groups were analyzed in duplicates and the experiment was repeated 3 times to confirm the results.
2.10. Enzyme-linked immunosorbent assay for IL-10
RAW 264.7 cells were seeded in a 12-well plate at a density of 5×105 cells and supplemented with DMEM as previously 10 above for 24h. For stimulation of M2-like polarization, RAW 264.7 cells were cultured with 0.04 µg/mL of IL-4 (20 ng·ml−1) for 48 h in a serum-free medium. Afterward, RAW 264.7 cells were treated with M1EV, M1EVRA, M1EVLF, and M1EVRALF for 48h. For M1-like polarization, RAW 264.7 cells were incubated with 0.1 μg/mL lipopolysaccharides (LPS) and 0.1 μg interferon-gamma (IFN-γ) for 48h.
The M2-conditioned medium (CM) from RAW 264.7 cells treated with IL-4, M1EV, M1EVRA, M1EVLF, and M1EVRALF was collected and stored at −80°C to analyze the IL-10 levels. The levels of IL-10 and IL-12 were measured in the CM using a LEGEND MAX™ IL-12 ELISA Kit (BioLegend, Cat#430607, and Cat#430707, respectively) according to the manufacturer's instructions as described previously. Three independent assays with at least three replicates were performed for each experiment.
2.11. Cell Viability assays
CT-26 cells were plated in 96-well flat-bottom plates at a density of 5x103 per 100μL overnight and then treated for 24 and 48hs with tested compounds. Following previously published protocols, to calculate the 50% inhibitory concentrations (IC50) colorectal cells were treated using 5µg/mL, 10µg/mL, and 20µg/mL concentrations of M1EVs. As controls, CT-26 in the DMEM without treatment and CT-26 treated with 50µg/mL, 25µg/mL, and 12.5µg/mL of OXA were analyzed. After incubation, the medium was refreshed for both cell lines and cells were incubated with CellTiter 96 AQueous One Solution (MTS) solution (Promega, Madison, WI, USA) according to the manufacturer’s guidelines. Absorbance was measured at 490 nm using a (SpectraMax ID3 microplate reader, Molecular Devices).
2.12. Detection of Cell Death by Flow Cytometry
The apoptotic profile of CT-26 cells was measured by flow cytometry from Annexin V-FITC Apoptosis Detection Kit (ab 14085). CT-26 was plated in 96 well plates and treated with OXA 25µg/mL, M1EV, M1EVRA, M1EVLF, M1EV1, M1EV2, and M1EV3 for 24 and 48h.
The cells were processed according to the manufacturer's guidelines, double-labeled with Annexin V/FITC and PI, and analyzed by flow cytometer BD FACSCalibur (Biosciences, Franklin Lakes, NJ, USA) for 10.000 events. Data analysis was performed on FlowJo software (BD Biosciences). Annexin V/FITC-only labeled cell populations indicate the early apoptosis stage, while Annexin V/FITC and PI double-labeled cells indicate the advanced apoptosis stage.
2.13. EMT Analysis by Immunofluorescence
Immunostaining was performed to identify proteins involved in epithelial-mesenchymal-transition (EMT), such as: E-cadherin and Vimentin. On a cover glass, CT-26 were seeded at 2x104 cells/well density and placed in a 24-well plate containing DMEM supplemented with 10% FBS. After complete adhesion, cells were treated with 20 µg/mL of M1EV, M1EVRA, M1EVLF e M1EVRALF and maintained for 48h. After this time, cells were washed with PBS, fixed in 4% buffered paraformaldehyde, permeated with TritonX-100 (0.3%), and then incubated with anti-E-Cadherin and Vimentin antibodies overnight in a humid chamber. Antibodies were diluted (1:200) in Diamond antibody diluent (Cell Marque, Rocklin, CA, USA). The cells were then washed and incubated with Alexa®Fluor 488 at 1:300 in blocking solution for another 60 min and 1:1000 DAPI (Life Technologies) for nuclear staining. Samples were analyzed using a Leica DM5500 B fluorescence microscope (Leica Microsystems, Wetzlar, Germany) using an appropriate filter to detect the excitation of the staining.
2.14. Analysis of gene expression in CT-26 and MC38 cell lines by qRT-PCR
CT-26 and MC-38 cell lines were observed by indirect co-culture tests with M2-polarized macrophage cell lines RAW 264.7. In a 12-well plate, MC38 cells and CT-26 cells were plated at a density of 2x105 cells/well in DMEM supplemented with 10% FBS. At the same time, M2-polarized were plated at a density of (1x104 cells/insert) into the 5 µm pore size transwell insert chamber (Corning Inc., Corning, NY, USA) in DMEM supplemented with 10% of FBS.
After 24h the MC38 cells and CT-26 within the insert were treated with 20µg/mL of M1EV, M1EVRA, M1EVLF, and M1EVRALF and maintained for another 48h. After treatment, MC38 and CT-26 were harvested with TRIzol® reagent (Invitrogen, CA, USA). 48hs later the cells were processed into chloroform and absolute ethanol for complete RNA extraction. Then total RNA was purified using SV Total RNA Isolation System (Promega, WI, USA) according to the manufacturer's instructions. Then, the RNA was immediately converted to cDNA by reverse transcriptase using a High-capacity RNA-to-cDNA™ kit (Applied Biosystems, Waltham, MA, USA).
Were evaluated the expression of genes: STAT3 (F, 5-’GGGCCTGGTGTGAACTACTC-3’ and R, 5-′GGTATTGCTGCAGGTCGTTG-3’ 59.8°C), SURVIVIN (F, 5-’AGAACAAAATTGCAAAGGAGACCA-3’ and R, 5-’GGCATGTCACTCAGGTCCAA-3’,59.8°C), MDR1 (F, 5-’TCAGCAACAGCAGTCTGGAG-3’ and R, 5-’ACTATGAGCACACCAGCACC-3’, 60°C), FAAD (F, 5-’AGAAGAAGAACGCCTCGGTG-3’ and R, 5-’GCTCACAGATTCCTGGGCTT-3’; 55.5°C), APAF-1 (F, 5-’TTCCAGTGGCAAGGACACAG-3’ and R, 5-’CCACTCTCCACAGGGACAAC-3’;60°C), NF-κB (F, 5-’CCGTCTGTCTGCTCTCTCT-3’ and R, 5-’CGTAGGGATCATCGTCTGCC-3’; 59.1°C), Cadherin (F, 5-’TGATGATGCCCCCAACACTC-3’ and R, 5’CCAAGCCCTTGGCTGTTTTC3’; 60°C), and Vimentin (F, 5-’TCCAGAGAGAGGAAGCCGAA-3’ and R, 5-’CTTTCATACTGCTGGCGCAC-3’ 59.93°C).
Real-time quantitative PCR analysis of genes of interest was performed using PowerUp™ SYBR® Green Master Mix (Applied Biosystems). The experiments were performed in triplicate. The standard qRT-PCR conditions and relative expression were calculated using the 2−ΔΔCt as previously described (33).
2.15. In Vivo2.16. Colorectal cancer allographic model and treatment regimens
To assess the potential for inhibiting tumor growth by M1EVs in the animal model (34) of allographic colorectal cancer tumor growth, CT-26 cells (1x106/mice) were inoculated subcutaneously injected into the right flank of male Balb/c mice. When the tumor volume achieved 3-4mm (35), the animals were organized into seven groups: Saline, M1EV, OXA, M1EV1, M1EV2, M1EV3, and M1EV4 with five animals each and treated intratumorally. The treatments were readministered every 5 days (3 treatments in 15 days). The tumor size was measured following the description (36,37). The equation mm3 = (width × length2)×0.52 (38) was used to calculate their volume.
At the end of the experiment the animals were euthanized (80mg/kg, I.P.) 2% thiopental (Cristália, São Paulo, Brazil), and the subcutaneous tumor was removed and collected, half of the tumor was stored at -80°C for qPCR analysis and the other part were placed in 10% paraformaldehyde for histopathological analysis.
All animal protocols have been carried out in accordance with the Guide for the Care and Use of Laboratory Animals as adopted by the U.S. National Institutes of Health and were approved by our institutional Animal Use Ethics Committee CEUA N. 222.011/2020.
2.17. Immunofluorescence
To order to understand tumor immunity, activation of the immune response via immunological control points as well as modulation of the metastatic process, expression of cytokines such as IL-10, PD-L1, and MMP-2 was studied. The expression of these targets was analyzed through immunofluorescence in tumor fragments collected from the mice used in the CRC allographic model. Three random fragments of the tumor tissue sections from each animal (three animals per group) were deparaffinized as previously described (24).
The sections were incubated overnight with mouse anti-MMP2, PDL-1, and IL-10 primary antibodies (1:300 in blocking solution 1% normal goat serum; Abcam, USA and Santa Cruz Biotechnology, USA, respectively) at 4°C, washed three times in PBS/0,2% triton X-100 for 5 min and incubated with Alexa®Fluor 555 goat anti-rabbit or goat anti-mouse secondary antibody (1:400 in BSA 1%). After washing, the glass coverslips were removed and rested on a mounting medium containing DAPI (Abcam) on glass slides for labeling the colors. The cells were analyzed using a fluorescence microscope (ZEISS), and digital images were collected to assess the average fluorescence intensity using the ZEN blue software (ZEISS).
2.18. Immunohistochemistry
Immunohistochemical staining of thin sections of the allographic tumors and peritoneal tumors (3μm) from three animals per group was obtained from each group. The tissue fragments were prepared as previously described (24). Tissue sections were incubated overnight at 4°C with primary antibodies anti-AKT (Santa Cruz Biotechnology, Cat# sc-5298), anti-PI3K (Santa Cruz Biotechnology, Cat# sc-1737), anti-E-cadherin (Cat # MA1-06304) (all 1:400; Cell Marque, Rocklin, CA, USA), anti-AKT (Santa Cruz Biotechnology, Cat# sc-5298), anti-CD163 (ProteinTech, Cat#16646-1-AP), anti-CXCL12 (RD Systems, Cat# COJ0519041) and anti-NF-kB((Santa Cruz Biotechnology, Cat#sc-8008) (all 1:400)). Slices were washed with phosphate buffer and incubated with a streptavidin/HRP-conjugated secondary antibody (VECTASTAIN Universal Quick HRP Kit) for 30 min. Immunoreactivity to the various proteins was visualized with a colorimetric-based detection kit following the protocol provided by the manufacturer. The cell nucleus was differentiated with Mayer's hematoxylin for 5 min.
The immunoreactivity was analyzed based on the scores of Charafe-Jauffret(35). The slides were analyzed under a microscope (Nikon E200 LED, Department of Morphology/UFRN) with objectives (40x and 100x). The intensity of cell immunostaining was scored as follows: 1 = absence of positive cells, 2 = small number of positive cells or isolated cells, 3 = moderate number of positive cells, and 4 = large number of positive cells10,27. Marking intensity was assessed by two examiners previously trained in a double-blind manner.
2.19. Peritoneal colorectal cancer model
The aggressive metastatic disseminated peritoneal colorectal carcinoma model was developed by intraperitoneal injection of 1x106 CT-26 colon cancer cells (per mice) into the peritoneal cavity of male Balb/c mice to generate peritoneal tumors, as previously described (39). After 03 days, mice were divided into four groups: Saline, OXA, M1EV1, and M1EV4 with five animals each and treated intraperitoneally. The treatments were readministered every 4 days (3 treatments in 12 days). Animals were euthanized with (80 mg/kg, I.P.) 2% thiopental (Cristália, São Paulo, Brazil) on the day. According to diameter under 1.0mm or over 3.0 mm by microscopy, the number of peritoneal nodules was counted, and data are shown from representative experiments.
Part of the peritoneal tumor masse was harvested and immediately frozen at −80°C for qPCR analysis. Other tumor fragments, liver, and lungs were immersed in 10% paraformaldehyde for histopathological analysis. The protocol was approved by the Committee on the Ethics of Animal Experiments (CEUA) of the Federal University of Rio Grande do Norte (UFRN) (permit number: 265.025/2021).
2.20. Metastasis Analysis of the liver and lungs from mice with peritoneal colorectal cancer
Livers and lungs were collected from all animals from the Intraperitoneal colorectal cancer model and included in the study for microscopic analysis to evaluate the effect of metastases by the proposed treatment. As previously described24, the liver and lungs were prepared for histological analysis. Sections of 5μm thickness were obtained for H&E staining and examined by light microscopy (Nikon Eclipse 2000 equipped with Nikon DS-Fi2; Nikon Corporation, Tokyo, Japan). For histopathological analysis of the liver and lungs, the number of metastatic niches and lymphocytic infiltrates were considered as previously described by Cavalcante et al.24. The values obtained from this analysis were evaluated in two aspects: proportion of the score vs. intensity of the score24. Likewise, histological analysis of peritoneal tumors was performed with emphasis on 4 criteria: anaplastic cells, lymphocytic infiltrate, blood vessels, and necrosis. Each specimen was scored according to Feng 38.
2.21. Gene expression of resistance to apoptosis and drug, EMT and immunosuppression by qRT-PCR
Gene expression analysis was performed with tumor fragments from both animal models performed. The standard qRT-PCR conditions and relative expression were calculated using the 2−ΔΔCt as previously described27.
For tumor fragments of mice with allographic colorectal cancer, the quantitative real-time PCR analysis in this study evaluated the expression of genes: β-actin (F, 5′-CCACCATGTACCCAGGCATT-3′ and R, 5′-CGGACTCATCGTACTCCTGC-3′, 60°C); FAAD (F, 5′-AGAAGAAGAACGCCTCGGTG-3′, and R, 5′-GCTCACAGATTCCTGGGCTT-3′, 56°C), APAF-1 (F,5′-TTCCAGTGGCAAGGACACAG-3′, and R, 5′-CCACTCTCCACAGGGACAAC-3′, 60°C); NF-κB (F, 5′CCGTCTGTCTGCTCTCTCT-3′, and R, 5′-CGTAGGGATCATCGTCTGCC-3′, 60°C); SURVIVIN (F, 5′-AGAACAAAATTGCAAAGGAGACCA-3′, and R, 5′-GGCATGTCACTCAGGTCCAA-3′, 60°C); STAT-3 (F, 5′-GGGCCTGGTGTGAACTACTC-3′ and R, 5′GGTATTGCTGCAGGTCGTTG-3′ 60°C); CXCR4 (F, 5′-CATGGAACCGATCAGTGTGAG-3′, and R, 5′-TGAAGGCCAGGATGAGAACG-3, 60°C); CD8 (F, 5′-GCTCAGTCATCAGCAACTCG-3′, and R, 5′-ATCACAGGCGAAGTCCAATC-3′, 58°C); CD68 (F, 5′CCCCTACTCCAACGTCCAAC-3′, and R, 5′-CAACTCTCCCTTCTCACCCA-3′, 60°C) and SNAIL (F, 5′-GACTCCTTCCAGCCTTGGTC-3′, and R, 5′-CCAGTAACCACCCTGCTGAG-3′, 60°C).
For tumors collected from animals submitted to the intraperitoneal colorectal cancer model, the quantitative real-time PCR analysis in this study evaluated the expression of genes: β-actin (F, 5′-CCACCATGTACCCAGGCATT-3′ and R, 5′-CGGACTCATCGTACTCCTGC-3′, 60°C); STAT-3 (F, 5′-GGGCCTGGTGTGAACTACTC-3′ and R, 5′-GGTATTGCTGCAGGTCGTTG-3′ 60°C); CXCR4 (F, 5′-CATGGAACCGATCAGTGTGAG-3′, and R, 5′-TGAAGGCCAGGATGAGAACG-3; PD-L1 (F, 5’-CCAGCCACTTCTGAGCATGA-3’ and R, 5’-CAGACAGCAAGAGCCTGTCA-3’, 60°C).
2.22. Statistical Analysis
All the experiments were performed in triplicate. Results were analyzed by two-way ANOVA and Bonferroni post-test. Values of p<0.05 were considered indicative of statistical significance (*p<0.05, **p<0.01, ***p<0.001, and ****p<0.0001). The Kruskal-Wallis and Dunn tests were used to compare medians for non-parametric tests (Graph Pad Prism 5.0 Software, La Jolla, CA, USA).
3. Results
3.1. Validation of EVs
The characterization of EVs isolated from M1-polarized RAW 264.7 was performed to study the size, morphology, and protein concentration (Figure 1A-G). Images obtained from the vesicles analyzed a well-defined round morphology, with a size range of 100–120nm, and were obtained in a concentration of around 0.5 to 1.2×108 particles (Figure 1 E and F). Quantitative analyzes of drug loading by HPLC indicated that the concentration of OXA corresponds to 1-2mg/mL (Figure 1G).
3.2. M1EVs Down-Regulate Macrophage Polarization towards M2 and Levels of IL-10.
To evaluate whether M1EVs can modify the polarization of TAMs, we polarized RAW 264.7 macrophages towards M2-TAMs phenotype in the presence of IL-4 and M1EV without OXA but with RA (M1EVRA), LF (M1EVLF), and RA+LF (M1EVRALF) (Figure 2A-D).
Flow cytometric analysis showed that expression of the M1-related marker CD68 was 58.1% in M2-polarized TAMs (Figure 2 A) and the expression was significantly increased to 81.9%, 73.7%, 84.7%, and 85.6% after addition of M1EV, M1EVRA, M1EVLF, and M1EVRALF after the M2 polarization, respectively (Figure 2A). On the other hand, M2-marker CD163 was significantly higher in control RAW 264.7 cells stimulated with IL-4 (59.5%), demonstrating that IL-4 successfully induced the alteration of classical macrophages to M2-polarized TAMs (Figure 2B). After treatment with M1EV, M1EVRA, M1EVLF, and M1EVRALF after the M2 polarization, the expression significantly decreased to 52,9%, 43,3%, 53,4%, and 48,7%, respectively (Figure 2 B).
To study whether colorectal cancer cells could act in the polarization of RAW 264.7 towards M2-TAM and whether this mechanism could be blocked by M1EV, M1EVRA, M1EVLF, and M1EVRALF, M2-TAMs were indirectly co-cultured with CT-26 cells and treated with M1EV, M1EVRA, M1EVLF, and M1EVRALF in a transwell system. After 24 h, the expression of CD163 and CD68 was analyzed in M2-TAM cells by flow cytometry (Figure 2 C and D). The results showed that the CT-26 cells treated with M1EV (77%), M1EVRA(76.3%), M1EVLF(85.3%), and M1EVRALF(84.6%) up-regulated the expression of CD68 in M2-TAM (66.8%) while the expression of CD163 was down-regulated to 33.1%, 31.4%, 23.1% and 29.7%, respectively, when compared with M2-TAM(62.6%).
The anti-inflammatory effect of the cytokine IL-10 was also assessed 48h after the treatment of M2-TAM with M1EV, M1EVRA, M1EVLF, and M1EVRALF. The supernatants were collected, and the levels of murine IL-10 were determined (Figure 2 E). RAW 264.7 treated with IL-4 (M2-TAM) showed increased levels of IL-10 (p < 0.001) when compared to the RAW group (Figure 2E). This result revealed that the polarization towards M2-TAM was successful. However, only M1EVRALF-treated M2-TAM had a similar level of IL-10 to the RAW 264.7 which meant a down-secretion of IL-10 by M2-TAM.
3.3. M1EV Systems Decreased the Cell Viability and Increased the Cell Death in CT-26 Cell Line.
Cell viability and apoptosis were measured by MTS assay (Figure 1S) and Flow Cytometry (Figure 2S) in 24 and 48 h, respectively. In this viability assay, CT-26 cells were treated with M1EVs loaded with OXA (M1EV1), OXA+RA (M1EV2), and OXA+LF (M1EV3) in the 12,5 µg/mL, 25 µg/mL, and 50 µg/mL to OXA and 5µg/mL, 10µg/mL, and 20µg/mL to M1EV+OXA+RA or + LF or +RALF. The cells treated with 10µg/mL and 20µg/mL M1EV (p<0.001), 5 µg/mL, 10 µg/mL and 20 µg/mL M1EV1 (p<0.0001), 20 µg/mL M1EV2 (p<0.0001) and M1EV3 (0<0.05) decreased the cell viability in 24 hrs. On the other hand, all concentrations tested reduced cell viability (p<0.0001) in 48 hours.
The treatment with 20 µg/mL M1EV3 increased in 41,2% the total apoptosis in 24hrs when compared with the control cells without treatment. Likewise, cells treated with M1EV3 showed a consistent result when analyzed after 48h, being 56,32% of the total cell apoptosis induction. Based on the results shown by cell viability and flow cytometer assays, we decided to add the M1EV4 group (M1EV+OXA+RA+LF) for the next experiments.
3.4. M1EV Systems Modulated EMT by Up-regulating E-cadherin and Down-Regulating Vimentin.
To analyze EMT in the CT-26 cells after treatment with M1EV systems, the expression of E-cadherin and Vimentin was analyzed as shown in figure 3A-L. For this end, M1EVs without OXA but with RA, LA, and RA+LF were used. All treated cells were compared to the CT-26 control cells without treatment. For E-Cadherin, there was a statistically significant increase in expression for the cell groups of treatment: M1EVRA and M1RALF (p<0.0001) when compared to CT-26 cells without treatment (Figure 3A-E and L). On the other hand, cells treated with M1EVLF (p<0.01) and M1EVRALF (p<0.001) reduced the expression of vimentin (Figure 3 F-K).
3.5. Gene Expression Analysis of Resistance to Apoptosis and Drug, EMT and STAT3 and NF-κB Transcription Factors in CT26 and MC38 Cell Lines in response to the Treatment with M1EV Systems.
To understand the anti-tumor effects of the M1EV systems without OXA but with RA, LF, and RA+LF on CT26 and MC38 colorectal cells under the direct influence of M2-TAM in a co-culture assay, we performed the analysis of gene expression of NF-κB, FAAD, APAF-1, MDR1, Survivin, STAT3, Vimentin, and E-Cadherin by rtPCR (Figure 4B-H). For this assay, we compared the RAW 264.7+IL-4-contacted cancer cells treated with RAW 264.7+IL-4-contacted cancer cells without treatment.
For extrinsic and intrinsic apoptosis markers, FAAD and Apaf-1 respectively, the results indicated up-regulation in both cell lines for the treatments with M1EVLF when compared to RAW 264.7+IL-4 -co-cultured CT-26 cells, pointing a cell apoptosis stimulation without the presence of oxaliplatin (Figure 4B-C). However, FAAD expression was higher in MC38 cells when they were treated with M1EVRA, M1EVLF, and M1EVRALF (p<0.0001, p<0.00,1, and p<0.0001, respectively). On the other hand, CT-26 cells showed a higher APAF-1 expression than the MC38 cell line when treated with M1EVRA, M1EVLF, and M1EVRALF (all, p<0.0001).
The gene expression of Survivin, a member of the inhibitors of the apoptosis family, and MDR1 expression, a target responsible for the process of drug resistance (MDR1) were analyzed (Figure 4D-E). MC38 cells showed lower expression of Survivin when treated with M1EVRALF (p<0.05) while MDR1 expression was down-regulated in treatments with M1EVRA and M1EVLF (both, p<0.0001) in CT-26 cells.
Gene expression of EMT by the analysis of E-Cadherin and vimentin was also observed in our study (Figure 4G-H). In both cell lines, M1EVRA, M1EVLF, and M1EVRALF were able to decrease the expression of Vimentin (all, p<0.0001). Likewise, the expression of E-Cadherin was significantly increased in both cell lines when treated with M1EVRA, M1EVLF, and M1EVRALF (CT-26 cells: p<0.05, p<0.001and p<0.0001 and MC38 cells: p<0.0001 and p<0.001, respectively).
Finally, as important orchestrators of tumor progression, the genetic expression of STAT3 and NF-κB signaling pathways were also studied (Fig. A and F). NF-κB expression was down-regulated in both CT-26 and MC38 cell lines after the treatment with M1EVRA, M1EVLF, and M1EVRALF (all, p<0.0001). On the other hand, only CT-26 cells had reduced STAT3 expression after treatment with M1EVRA, M1EVLF, and M1EVRALF (all, p<0.001).
3.6. In vivo Antitumor Effect of M1EV loaded with OXA, RA, and LF.
3.6.1. Allographic Colorectal Cancer
To evaluate the anti-tumor and immunomodulatory effects of the M1EV with OXA, RA, LF, and RA+LF in mice, allographic tumors were analyzed. Photomicrographs of the tumors showed that the tumor growth curve in all the groups was affected by the treatments. The groups treated with M1EV (p<0.05), M1EV1 (p<0.0001), and M1EV4 (p<0.0001) had a higher tumor growth inhibition when compared with the saline group (Figure 5 A1-G1 and B). Likewise, tumor weight showed a similar result to the tumor growth curve related to M1EV (p<0.05), M1EV1 (p<0.0001), and M1EV4 (p<0.0001) groups (Figure 1 C).
3.6.2. M1EV loaded with OXA and Combined With RA, LF, and/or RA+LF Modulated the Tumor Progression in TME of Primary Tumors.
3.6.2.1. Protein Expression Analysis of Tumor Progression
The tumor immunosuppression and invasion were assessed and analyzed through protein expression of IL-10, PD-L1, and MMP2 by immunofluorescence (Figure 6 A-X).
For immunosuppression analysis, the expression of PD-L1 was overexpression in the group treated with OXA. However, M1EV systems holding OXA (M1EV1, p<0.01), RA (M1EV2, p<0.05) and RA + LF (M1EV4, p<0.0001) reduced the PD-L1 expression when compared with saline group. On the other hand, the IL-10 was up-regulated in the OXA group (p<0.01) and M1EV (P<0.05) and down-regulated in the M1EV1, M1EV2, M1EV3, and M1EV4 groups, even without statistical significance. Regarding the tumor invasion, the treatment with M1EV1, M1EV2, and M1EV4 (all, p<0.0001) was capable of down-regulating the MMP2 expression.
Analysis of cell cycle regulation by PI3K/AKT signaling pathway and EMT in TME was performed. The results showed that in the groups treated with M1EV1, M1EV2, M1EV3, and M1EV4, there was a significant decrease in the expression of AKT (Figure 7 A and E) and PI3K (Figure 7 B and F) when compared to the saline group (both, p<0.0001). Interestingly, the group treated with M1EV4 (p<0.0001) showed a significant increase in the expression of E-Cadherin (Figure 8A3-G3 and H.3).
3.6.2.2. Gene Expression Analysis of Primary Tumors by qRT-PCR.
The expression of genes coding for transcription factors (NF-κB and STAT3) as well as EMT (SNAIL), apoptosis (FAAD and APAF-1), and Immunocompetence (CD8 and CD68) in the TME were assessed by analysis of gene expression in tumor fragments (Figure 8A-G).
Up-regulation of FAAD expression was observed in the groups treated with OXA(p<0.01), M1EV1, M1EV2, M1EV3 and M1EV4 (all, p<0.001) when compared with saline group. Likewise, the APAF-1 expression increased in groups treated with OXA (p<0.05), M1EV1, M1EV2, M1EV3 and M1EV4 (all, p<0.0001). More interesting, M1EV1, M1EV2, M1EV3 and M1EV4 (all, p<0.001) increased significantly the expression of FAAD and APF-1 when compared to OXA (Figure 8 A and B).
NF-κB gene expression was reduced in all treated groups (p<0.0001) (Figure 8C). On the other hand, STAT3 gene expression was down-regulated in all treatments (p<0.0001), except in M1EV1 and M1EV3 (Figure 8 D).
The efficacy of the anti-tumor immune response was studied by analysis of the CD8 gene expression, and the results indicated an up-regulation of this gene after the treatment with M1EV and M1EV4 (p<0.0001), M1EV2 (p<0.05) and M1EV3 (p<0.01) (Figure 8E).
For the M1-type macrophage marker gene, the gene expression of CD68 was significantly increased for the group treated with M1EV (p<0.0001), and M1EV, M1EV2, and M1EV3 (p<0.001) (Figure 8 F).
The SNAIL gene expression, a transcription factor involved in the EMT, was downregulated when treated with M1EV1 (p<0.01) (Figure 8G).
3.7. Peritoneal Colorectal Cancer
3.7.1. Antitumor Effect of M1EV4 in Colorectal Cancer Peritoneal Metastasis
The results shown previously pointed out that M1EV4 presented a higher anti-tumor effect than M1EV1, M1EV2, and M1EV3 in allographic tumors. Therefore, M1EV4 was tested to study its antitumor in a metastatic colorectal cancer model.
Figure 9 shows how the peritoneal tumors were affected by the treatments with OXA, M1EV1, and M1EV4 (Figure 9 A-D). The peritoneal tumors decreased the weight after the treatment with OXA, M1EV1, and M1EV4 (all, p<0.0001) when compared with the saline group as seen in Figure 9 B. Likewise, these treatments were able to reduce the number of tumors in mice (Figure 9 C). However, the efficiency of M1EV4 in reducing the tumor weight (p<0.001) and number of tumors (p<0.001) was higher than OXA (Figure 9 B and C). Furthermore, histopathological criteria were evaluated to study the antitumor benefit of our treatment. Together, the results pointed out that M1EV4 had a higher antitumor effect by decreasing the anaplastic cells (p<0.01), as well as, increasing the Lymphocytic infiltrated (p<0.001) in metastatic TME.
3.7.2. Protein and gene expression profile of immunosuppression in peritoneal tumors
To order to understand the tumor immunosuppressive profile via AKT/NF-κB/STAT3 in colon cancer peritoneal metastasis, protein and gene expressions of AKT, NF-κB, STAT3, CXCL12, CXCR4, CD163, and PD-L1 were analyzed in peritoneal tumors after treatment with OXA, M1EV1, and M1EV4 (Figure 10 A-W). Interestingly, the protein expression of AKT and NF-κB decreased in all the treated groups, especially in M1EV4 (p<0.0001) when compared with the saline group (Figure 10 A-H). Likewise, the protein expression of CXCL12 and CD163 had a higher reduction in M1EV4 (both, p<0.0001) (Figure 10 I-P). On the hand, the gene expression of STAT3, CXCR4, and PD-L1 did not show a statistically significant difference when the saline group was compared with the M1EV4 group. However, they had a considerable expression reduction. Intriguingly, the OXA group pointed a higher expression of STAT3 , CXCR4, and PD-L1 than the saline group (p<0.05, p<0.0001 and p<0.001, respectively), M1EV1 (p<0.05, p<0.01 and p<0.05, respectively) and M1EV4 (p<0.05, p<0.0001 and p<0.001, respectively).
The histopathological analysis of the liver showed the presence of tumor cells infiltrated throughout the organ, in all groups tested (arrowheads; Figure 11 A-D). However, the number and diameter of metastatic niches appear to be smaller when treated with M1EV1 (p<0.001) and M1EV4 (p<0.0001), than the saline (Figure 11 A-D and I). Massive metastatic niches were also found in the lungs of saline-treated animals (Figure 11- E). On the other hand, M1EV1 and M1EV4 (both, p<0.01) discreetly reduced metastases in the lungs (Figure 11 E-H and J).
4. Discussion
Important signaling pathway crosstalk, especially between STAT3 and NF-κB, has been implicated in the cancer hallmarks, such as resistance to apoptosis and drug, EMT, and immunosuppression (23,24,27,39. In this work, OXA-coated extracellular vesicles from macrophages 1 (M1EVs) combined with retinoic acid and Libidia Ferrea were demonstrated to modulate the tumor progression of colorectal cancer. Gene and protein profiles correlated to the tumor progression were significantly altered. Our results showed that NF-κB, STAT3, and AKT were the most importantly downregulated signaling pathways due to their crucial role in the crosstalk between malignant cells and tumor-associated immune cells.
In this line, the interaction between the stroma and cancer cells in the primary TME is a fundamental key point to understanding the factors involved in cancer metastasis 40. In TME, M2-TAMs are alternatively activated cells by IL-4 and IL-13, which eventually activate the JAK/STAT-3 pathway to induce the production of immunosuppressor cytokines like IL-10 and PD-L1, favoring resistance to apoptosis and drug as well as invasion 41–44. Furthermore, the NF-kB pathway inhibited by PI3K-AKT signaling in M2-TAMs causes activation of an immunosuppressive transcriptional sequence in the inflammatory course of tumor progression 45. There are several reports that the transcription factor Foxo1, which is a key gene in M1 macrophages is inhibited by Akt signaling 46,47.
In this context, TAMs polarization course towards M2 disrupted by NP-mediated NF-κB, STAT3, and AKT downregulation seems to have triggered the reduced expression of PD-L1 and IL-10, chemokines released by M2-polarized TAM. M2-TAMs PD-L1 and IL-10 expression upregulates the crosstalk of AKT/NF-kB pathways in tumor cells resulting in epithelial-mesenchymal transition 27,48,49, as well as down-regulates CD8+ T cells by upregulating the STAT3 expression in TME 27,50.
In this study, to order to activate the immune system and down-regulate the tumor progression of colorectal cancer, we used extracellular vesicles (EV) from M1-like macrophages as drug carriers. Similar studies demonstrated that EV-mediated endocytosis can promote the delivery of different drugs such as doxorubicin10,51,52, paclitaxel53,54, and curcumin55,56 and are known as EVs or exosome-like vesicles carriers of drugs (41,42). EVs from different cells, especially immune and cancer cells, can regulate the TME by exposing cell antigens or immune-enhancing receptors, promoting effects as "tumor vaccines" 49,50,57. It has been reported that EVs are internalized into cells by way of fusion and/or receptor-mediated endocytosis (41,42). Our results showed that CT-26 cells internalized EVs very efficiently within 24h after the exposure to them corroborating with other reports of EV uptake in literature (10,43,44). This result strengthens the hypothesis that EVs is an important vehicle for releasing drugs inside the cells due to their biocompatibility (9), representing the beyond of “state-of-art” in cancer treatment due to their endogenous origin, which minimizes their immunogenicity and toxicity58.
Our in vitro and in vivo results suggested that different systems of M1EVs had a strong apoptotic effect, corroborating with other studies that used EVs as drug carriers 10,51,59. However, the apoptotic effect in both signaling pathways, intrinsic and extrinsic ways, was higher when M1EVs were loaded with OXA, RA, and LF (M1EV4). Interestingly, when CT-26 and MC38 cell lines were co-cultured with M2-TAM and treated with M1EVs without OXA but loaded with RA and LF, the gene expression of P-glycoprotein (MDR1) and Survivin was down-regulated significantly. MDR1 is localized on the plasma membrane of resistant cancer cells to several antitumor drugs, such as vinblastine, vincristine, doxorubicin, daunorubicin, oxaliplatin, teniposide and paclitaxel 60,61 while Survivin is a gene that inhibits the intrinsic apoptosis pathway by accumulating in the mitochondria, enhancing cell resistance to apoptosis 62,63. Together, these results showed that M1EVs with or without OXA but loaded with RA and LF had a significant effect in down-regulating the resistance to the drug and apoptosis.
In primary TME of colorectal cancer allographic tumors in mice, a modulatory loop of immunosuppressive cytokines is fed by the interaction between stromal cells, specifically M2-TAM, and cancer cells resulting in the immune escape and EMT 24,27 In this line, we investigated the expression of different markers of immunosuppression (PD-L1 and IL-10), Immunocompetence (CD8 and CD68), and EMT (E-cadherin, SNAIL, and MMP-2). Different systems of M1EVs loaded with OXA, RA, and LF showed a high efficiency in reducing immunosuppression and EMT. On the other hand, different systems of M1EV upregulated the immunocompetence by increasing the expression of CD8 lymphocytes and CD68 M1 macrophages in TME. When we analyzed our results deeply, we concluded that M1EV4 which loaded all compounds had a higher antitumor effect when compared with other systems of M1EVs. More importantly, in vitro, assays demonstrated that the protein and gene expression of E-cadherin increased, and Vimentin reduced significantly in cancer cell lines after treatment with M1EVs without OXA but loaded with RA and LF, especially when RA and LF were combined.
According to the “Seed and Soil” hypothesis, metastatic capacity is determined in two ways, such as an internal oncogenic driving force and a pro-tumor environment for tumor cells64. Based on the results of primary tumors described previously, we moved forward for investigating the expression of different hallmarks of metastasis in mice peritoneal cancer, and more importantly, we tested the antitumor effect of M1EV4 in these hallmarks. Several studies have reported that tumor progression and metastasis are regulated by several factors related to M2-TAM, immunosuppressive cytokines, metastatic regulators, and dysregulated transcription factors in TME (24,27,64. In this context, upregulation of STAT3, NF-kB, and AKT signaling pathways seemed to stimulate the CXCL12/CXCR4 axis that promotes tumor progression and metastasis as well as regulates M2-TAM recruitment and immunosuppression24,27,64,65. The expression of genes coding for transcription factor (STAT3) as well as metastasis regulators (CXCR4) and immunosuppression (PD-L1) in the TME was markedly downregulated by M1EV4 loaded with OXA, RA, and LF in metastatic peritoneal tumors. Likewise, protein expression of transcription factors (AKT and NF-kB), metastatic factors (CXCL12), and M2-TAM immunosuppression (CD163) also were reduced after treatment with M1EV4. Reinforcing these molecular findings, clinically M1EV4 was able to reduce metastatic niches in peritoneum, liver and lungs in mice treated for two weeks.
This study proposes a sophisticated engineering of extracellular vesicles as drug carriers to control primary tumors and metastatic disease. In this study, we provided evidence that EVs from M1 Macrophages increase (I) anti-tumor activity in TME by (I) immunomodulatory molecule carrier (II) potentialize and increase the bioavailability of oxaliplatin in lower dose and/or (II) stimulating the activation or blocking of signaling pathways involved in cell proliferation, migration, and survival processes.
5. Conclusions
The understanding of cancer and metastatic processes and the signaling pathways involved can provide several potential targets for drug intervention. However, the focus should be on the simultaneous inhibition of cancer growth and metastasis in which would produce clinically effective therapeutic results. Finally, clinical trials should be encouraged to incorporate correlative endpoints in their studies to identify accurate biomarkers of response to retinoid therapy. The central idea of this project was to combine the anti-cancer properties of L. ferrea with the immunomodulation capacity of retinoic acid in low doses of oxaliplatin encapsulated in extracellular vesicles whose membrane comes from antitumor M1 Macrophages.
Author Contributions
RFAJ designed and supervised the experimental study; PLA and CJ developed and characterized the EVs loaded with drugs from M1; LALS fractioned Libidia Ferrea and quantified ellagic acid and gallic acid. TGC, CFSA, and ASO quantified oxaliplatin incorporated in EVs by HPLC. TGC, VBG, ILM, SVPS, CJ, PLA, and SOPS performed the experiments and formatted figures and tables; RFAJ, TGC, PMMG, ASO, and CFSA analyzed the data; RFAJ and TGS wrote the manuscript; RFAJ edited the final version of the manuscript. All authors have approved the final version of the manuscript and are accountable for all aspects of this study.
Funding
Thais Gomes de Carvalho received financial support No 968572355 of ERASMUS (UE-LUMC). We acknowledge the support of a postdoctoral fellowship from Raimundo Fernandes de Araujo Junior by CNPq/ 200989/2022-7 and CNPq (Productivity in Research, Grant No 3018770/2019). This project has received funding from the innovation program under the Marie Skłodowska-Curie grant agreement No 777682 (CANCER), 734684 (CHARMED) as well as KWF Kankerbestrijding (Dutch Cancer Society), grant number 13167.
Conflicts of Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Ismail, N.I.; Othman, I.; Abas, F.; Lajis, N.H.; Naidu, R. Mechanism of Apoptosis Induced by Curcumin in Colorectal Cancer. Int. J. Mol. Sci. 2019, 20, 2454. [Google Scholar] [CrossRef] [PubMed]
- Arnold, M.; Sierra, M.S.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global patterns and trends in colorectal cancer incidence and mortality. Gut 2017, 66, 683–691. [Google Scholar] [CrossRef] [PubMed]
- Mármol, I.; Sánchez-De-Diego, C.; Pradilla Dieste, A.; Cerrada, E.; Rodriguez Yoldi, M. Colorectal Carcinoma: A General Overview and Future Perspectives in Colorectal Cancer. Int. J. Mol. Sci. 2017, 18, 197. [Google Scholar] [CrossRef] [PubMed]
- Guan, X. Cancer metastases: challenges and opportunities. Acta Pharm. Sin. B 2015, 5, 402–418. [Google Scholar] [CrossRef] [PubMed]
- Weber, GF. Why does cancer therapy lack effective anti-metastasis drugs? Cancer Letters. 2013. [CrossRef]
- Xie, Y.-H.; Chen, Y.-X.; Fang, J.-Y. Comprehensive review of targeted therapy for colorectal cancer. Signal Transduct. Target. Ther. 2020, 5, 1–30. [Google Scholar] [CrossRef]
- Turturici, G.; Tinnirello, R.; Sconzo, G.; Geraci, F. Extracellular membrane vesicles as a mechanism of cell-to-cell communication: advantages and disadvantages. Am. J. Physiol. Physiol. 2014, 306, C621–C633. [Google Scholar] [CrossRef]
- Aryani A, Denecke B. Exosomes as a Nanodelivery System: a Key to the Future of Neuromedicine? Mol Neurobiol. 2016, 53, 818–34. [Google Scholar] [CrossRef]
- Ha, D.; Yang, N.; Nadithe, V. Exosomes as therapeutic drug carriers and delivery vehicles across biological membranes: current perspectives and future challenges. Acta Pharm. Sin. B 2016, 6, 287–296. [Google Scholar] [CrossRef]
- Jorquera-Cordero, C.; Lara, P.; Cruz, L.J.; Schomann, T.; van Hofslot, A.; de Carvalho, T.G.; Guedes, P.M.D.M.; Creemers, L.; Koning, R.I.; Chan, A.B.; et al. Extracellular Vesicles from M1-Polarized Macrophages Combined with Hyaluronic Acid and a β-Blocker Potentiate Doxorubicin’s Antitumor Activity by Downregulating Tumor-Associated Macrophages in Breast Cancer. Pharmaceutics 2022, 14, 1068. [Google Scholar] [CrossRef]
- Raimondo, S.; Giavaresi, G.; Lorico, A.; Alessandro, R. Extracellular Vesicles as Biological Shuttles for Targeted Therapies. Int. J. Mol. Sci. 2019, 20, 1848. [Google Scholar] [CrossRef] [PubMed]
- Chulpanova, D.S.; Kitaeva, K.V.; James, V.; Rizvanov, A.A.; Solovyeva, V.V. Therapeutic Prospects of Extracellular Vesicles in Cancer Treatment. Front. Immunol. 2018, 9, 1534. [Google Scholar] [CrossRef]
- Lara, P.; Chan, A.B.; Cruz, L.J.; Quest, A.F.G.; Kogan, M.J. Exploiting the Natural Properties of Extracellular Vesicles in Targeted Delivery towards Specific Cells and Tissues. Pharmaceutics 2020, 12, 1022. [Google Scholar] [CrossRef] [PubMed]
- Taghikhani, A.; Farzaneh, F.; Sharifzad, F.; Mardpour, S.; Ebrahimi, M.; Hassan, Z.M. Engineered Tumor-Derived Extracellular Vesicles: Potentials in Cancer Immunotherapy. Front. Immunol. 2020, 11, 221. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Yin, Y.; Lai, R.C.; Lim, S.K. Immunotherapeutic Potential of Extracellular Vesicles. Front. Immunol. 2014, 5, 518–518. [Google Scholar] [CrossRef]
- Goughnour PC, Park MC, Kim SB, Jun S, Yang WS, Chae S, et al. Extracellular vesicles derived from macrophages display glycyl-tRNA synthetase 1 and exhibit anti-cancer activity. J Extracell Vesicles. 2020, 10, e12029. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Liang, H.; Zen, K. Molecular Mechanisms That Influence the Macrophage M1–M2 Polarization Balance. Front. Immunol. 2014, 5, 614. [Google Scholar] [CrossRef]
- Liu X, Wang G, You Z, Qian P, Chen H, Dou Y, et al. Inhibition of hypoxia-induced proliferation of pulmonary arterial smooth muscle cells by a mTOR siRNA-loaded cyclodextrin nanovector. Biomaterials 2014, 35, 4401–4416. [CrossRef]
- Martinez, F.O.; Gordon, S. The M1 and M2 paradigm of macrophage activation: Time for reassessment. F1000Prime Rep. 2014, 6, 13. [Google Scholar] [CrossRef]
- Wang, Y.; Smith, W.; Hao, D.; He, B.; Kong, L. M1 and M2 macrophage polarization and potentially therapeutic naturally occurring compounds. Int. Immunopharmacol. 2019, 70, 459–466. [Google Scholar] [CrossRef]
- Ruffell, B.; Coussens, L.M. Macrophages and Therapeutic Resistance in Cancer. Cancer Cell 2015, 27, 462–472. [Google Scholar] [CrossRef] [PubMed]
- Xue, Y.; Tong, L.; Liu, F.L.; Liu, A.; Zeng, S.; Xiong, Q.; Yang, Z.; He, X.; Sun, Y.; Xu, C. Tumor-infiltrating M2 macrophages driven by specific genomic alterations are associated with prognosis in bladder cancer. Oncol. Rep. 2019, 42, 581–594. [Google Scholar] [CrossRef] [PubMed]
- Junior, R.F.d.A.; Eich, C.; Jorquera, C.; Schomann, T.; Baldazzi, F.; Chan, A.B.; Cruz, L.J. Ceramide and palmitic acid inhibit macrophage-mediated epithelial–mesenchymal transition in colorectal cancer. Mol. Cell. Biochem. 2020, 468, 153–168. [Google Scholar] [CrossRef] [PubMed]
- Cavalcante RS, Ishikawa U, Silva ES, Silva-Júnior AA, Araújo AA, Cruz LJ, et al. STAT3/NF-κB signalling disruption in M2 tumour-associated macrophages is a major target of PLGA nanocarriers/PD-L1 antibody immunomodulatory therapy in breast cancer. Br J Pharmacol. 2021, 178, 2284–304. [CrossRef]
- Hall, J.A.; Grainger, J.R.; Spencer, S.P.; Belkaid, Y. The Role of Retinoic Acid in Tolerance and Immunity. Immunity 2011, 35, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Shao, X.-J.; Xiang, S.-F.; Chen, Y.-Q.; Zhang, N.; Cao, J.; Zhu, H.; Yang, B.; Zhou, Q.; Ying, M.-D.; He, Q.-J. Inhibition of M2-like macrophages by all-trans retinoic acid prevents cancer initiation and stemness in osteosarcoma cells. Acta Pharmacol. Sin. 2019, 40, 1343–1350. [Google Scholar] [CrossRef]
- Júnior, R.F.d.A.; A Lira, G.; Schomann, T.; Cavalcante, R.S.; Vilar, N.F.; de Paula, R.C.M.; Gomes, R.F.; Chung, C.K.; Jorquera-Cordero, C.; Vepris, O.; et al. Retinoic acid-loaded PLGA nanocarriers targeting cell cholesterol potentialize the antitumour effect of PD-L1 antibody by preventing epithelial-mesenchymal transition mediated by M2-TAM in colorectal cancer. Transl. Oncol. 2023, 31, 101647. [Google Scholar] [CrossRef]
- Guerra ACV de A, Soares LAL, Ferreira MRA, Araújo AA de, Rocha HA de O, Medeiros JS de, et al. Libidibia ferrea presents antiproliferative, apoptotic and antioxidant effects in a colorectal cancer cell line. Biomedicine and Pharmacotherapy. 2017.
- Falcão, T.R.; Rodrigues, C.A.O.; de Araújo, A.A.; de Medeiros, C.A.C.X.; Soares, L.A.L.; Ferreira, M.R.A.; Vasconcelos, R.C.; Júnior, R.F.d.A.; Lopes, M.L.D.d.S.; Guerra, G.C.B. Crude extract from Libidibia ferrea (Mart. ex. Tul.) L.P. Queiroz leaves decreased intra articular inflammation induced by zymosan in rats. BMC Complement. Altern. Med. 2019, 19, 1–10. [Google Scholar] [CrossRef]
- Zaazaa, A.M.; Lokman, M.S.; Shalby, A.B.; Ahmed, H.H.; A El-Toumy, S. Ellagic Acid Holds Promise Against Hepatocellular Carcinoma in an Experimental Model: Mechanisms of Action. Asian Pacific Journal of Cancer Prevention 2018, 19, 387–393. [Google Scholar] [CrossRef]
- Chao, P.-C.; Hsu, C.-C.; Yin, M.-C. Anti-inflammatory and anti-coagulatory activities of caffeic acid and ellagic acid in cardiac tissue of diabetic mice. Nutr. Metab. 2009, 6, 33–33. [Google Scholar] [CrossRef]
- Thery, C.; Clayton, A.; Amigorena, S.; Raposo, G. Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Curr. Protoc. Cell Biol. 2006, 3. [Google Scholar] [CrossRef] [PubMed]
- Saari, H.; Lázaro-Ibáñez, E.; Viitala, T.; Vuorimaa-Laukkanen, E.; Siljander, P.; Yliperttula, M. Microvesicle- and exosome-mediated drug delivery enhances the cytotoxicity of Paclitaxel in autologous prostate cancer cells. J. Control. Release 2015, 220, 727–737. [Google Scholar] [CrossRef] [PubMed]
- Rao, Q.; Zuo, B.; Lu, Z.; Gao, X.; You, A.; Wu, C.; Du, Z.; Yin, H. Tumor-derived exosomes elicit tumor suppression in murine hepatocellular carcinoma models and humans in vitro. Hepatology 2016, 64, 456–472. [Google Scholar] [CrossRef] [PubMed]
- Xie, F.; Huang, Y.; Zhan, Y.; Bao, L. Exosomes as drug delivery system in gastrointestinal cancer. Front. Oncol. 2023, 12, 1101823. [Google Scholar] [CrossRef]
- Lara, P.; Palma-Florez, S.; Salas-Huenuleo, E.; Polakovicova, I.; Guerrero, S.; Lobos-Gonzalez, L.; Campos, A.; Muñoz, L.; Jorquera-Cordero, C.; Varas-Godoy, M.; et al. Gold nanoparticle based double-labeling of melanoma extracellular vesicles to determine the specificity of uptake by cells and preferential accumulation in small metastatic lung tumors. J. Nanobiotechnol. 2020, 18, 20. [Google Scholar] [CrossRef]
- Lara P, Huis In ‘T Veld R V. , Jorquera-Cordero C, Chan AB, Ossendorp F, Cruz LJ. Zinc-phthalocyanine-loaded extracellular vesicles increase efficacy and selectivity of photodynamic therapy in co-culture and preclinical models of colon cancer. Pharmaceutics 2021, 13. [Google Scholar]
- Feng, M.; Feng, J.; Chen, W.; Wang, W.; Wu, X.; Zhang, J.; Lai, M. Lipocalin2 suppresses metastasis of colorectal cancer by attenuating NF-κB-dependent activation of snail and epithelial mesenchymal transition. Mol. Cancer 2016, 15, 1–18. [Google Scholar] [CrossRef]
- He, Y.; Júnior, R.F.d.A.; Cavalcante, R.S.; Yu, Z.; Schomann, T.; Gu, Z.; Eich, C.; Cruz, L.J. Effective breast cancer therapy based on palmitic acid-loaded PLGA nanoparticles. Mater. Sci. Eng. C 2023, 145, 213270. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Zhang, H.; Jiang, X.; Qian, C.; Liu, Z.; Luo, D. Factors involved in cancer metastasis: a better understanding to “seed and soil” hypothesis. Mol. Cancer 2017, 16, 176. [Google Scholar] [CrossRef]
- Su, S.; Liu, Q.; Chen, J.; Chen, J.; Chen, F.; He, C.; Huang, D.; Wu, W.; Lin, L.; Huang, W.; et al. A Positive Feedback Loop between Mesenchymal-like Cancer Cells and Macrophages Is Essential to Breast Cancer Metastasis. Cancer Cell 2014, 25, 605–620. [Google Scholar] [CrossRef]
- Ruffell, B.; Coussens, L.M. Macrophages and Therapeutic Resistance in Cancer. Cancer Cell 2015, 27, 462–472. [Google Scholar] [CrossRef] [PubMed]
- Sica, A.; Mantovani, A. Macrophage plasticity and polarization: in vivo veritas. J. Clin. Investig. 2012, 122, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Horlad, H.; Ma, C.; Yano, H.; Pan, C.; Ohnishi, K.; Fujiwara, Y.; Endo, S.; Kikukawa, Y.; Okuno, Y.; Matsuoka, M.; et al. An IL-27/Stat3 axis induces expression of programmed cell death 1 ligands (PD-L1/2) on infiltrating macrophages in lymphoma. Cancer Sci. 2016, 107, 1696–1704. [Google Scholar] [CrossRef] [PubMed]
- Kaneda, M.M.; Messer, K.S.; Ralainirina, N.; Li, H.; Leem, C.J.; Gorjestani, S.; Woo, G.; Nguyen, A.V.; Figueiredo, C.C.; Foubert, P.; et al. PI3Kγ is a molecular switch that controls immune suppression. Nature 2016, 539, 437–442. [Google Scholar] [CrossRef] [PubMed]
- Fukao, T.; Koyasu, S. PI3K and negative regulation of TLR signaling. Trends Immunol. 2003, 24, 358–363. [Google Scholar] [CrossRef]
- Covarrubias, A.J.; Aksoylar, H.I.; Horng, T. Control of macrophage metabolism and activation by mTOR and Akt signaling. Semin. Immunol. 2015, 27, 286–296. [Google Scholar] [CrossRef] [PubMed]
- Hussain, S.M.; Kansal, R.G.; Alvarez, M.A.; Hollingsworth, T.J.; Elahi, A.; Miranda-Carboni, G.; Hendrick, L.E.; Pingili, A.K.; Albritton, L.M.; Dickson, P.V.; et al. Role of TGF-β in pancreatic ductal adenocarcinoma progression and PD-L1 expression. Cell. Oncol. 2021, 44, 673–687. [Google Scholar] [CrossRef]
- Bonde, A.K.; Tischler, V.; Kumar, S.; Soltermann, A.; Schwendener, R.A. Intratumoral macrophages contribute to epithelial-mesenchymal transition in solid tumors. BMC Cancer 2012, 12, 35. [Google Scholar] [CrossRef] [PubMed]
- Sun Z, Fourcade J, Pagliano O, Chauvin JM, Sander C, Kirkwood JM, et al. IL10 and PD-1 cooperate to limit the activity of tumor-specific CD8+ T cells. Cancer Res. 2015, 75, 1635–44. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Li, S.; Song, J.; Ji, T.; Zhu, M.; Anderson, G.J.; Wei, J.; Nie, G. A doxorubicin delivery platform using engineered natural membrane vesicle exosomes for targeted tumor therapy. Biomaterials 2014, 35, 2383–2390. [Google Scholar] [CrossRef]
- Hadla, M.; Palazzolo, S.; Corona, G.; Caligiuri, I.; Canzonieri, V.; Toffoli, G.; Rizzolio, F.; Tsai, L.-C.; Hsieh, H.-Y.; Lu, K.-Y.; et al. Exosomes increase the therapeutic index of doxorubicin in breast and ovarian cancer mouse models. Nanomedicine 2016, 11, 2431–2441. [Google Scholar] [CrossRef]
- Yang, T.; Martin, P.; Fogarty, B.; Brown, A.; Schurman, K.; Phipps, R.; Yin, V.P.; Lockman, P.; Bai, S. Exosome Delivered Anticancer Drugs Across the Blood-Brain Barrier for Brain Cancer Therapy in Danio Rerio. Pharm. Res. 2015, 32, 2003–2014. [Google Scholar] [CrossRef]
- Kim, M.S.; Haney, M.J.; Zhao, Y.; Mahajan, V.; Deygen, I.; Klyachko, N.L.; Inskoe, E.; Piroyan, A.; Sokolsky, M.; Okolie, O.; et al. Development of exosome-encapsulated paclitaxel to overcome mdr in cancer cells. Nanomed. Nanotechnol. Biol. Med. 2016, 12, 655–664. [Google Scholar] [CrossRef]
- Zhuang, X.; Xiang, X.; Grizzle, W.; Sun, D.; Zhang, S.; Axtell, R.C.; Ju, S.; Mu, J.; Zhang, L.; Steinman, L.; et al. Treatment of Brain Inflammatory Diseases by Delivering Exosome Encapsulated Anti-inflammatory Drugs From the Nasal Region to the Brain. Mol. Ther. 2011, 19, 1769–1779. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.; Zhuang, X.; Xiang, X.; Liu, Y.; Zhang, S.; Liu, C.; Barnes, S.; Grizzle, W.; Miller, D.; Zhang, H.-G. A Novel Nanoparticle Drug Delivery System: The Anti-inflammatory Activity of Curcumin Is Enhanced When Encapsulated in Exosomes. Mol. Ther. 2010, 18, 1606–1614. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Yin, Y.; Lai, R.C.; Lim, S.K. Immunotherapeutic Potential of Extracellular Vesicles. Front. Immunol. 2014, 5, 518–518. [Google Scholar] [CrossRef] [PubMed]
- Rashed, M.H.; Bayraktar, E.; Helal, G.K.; Abd-Ellah, M.F.; Amero, P.; Chavez-Reyes, A.; Rodriguez-Aguayo, C. Exosomes: From Garbage Bins to Promising Therapeutic Targets. Int. J. Mol. Sci. 2017, 18, 538. [Google Scholar] [CrossRef]
- Tian, Y.; Li, S.; Song, J.; Ji, T.; Zhu, M.; Anderson, G.J.; Wei, J.; Nie, G. A doxorubicin delivery platform using engineered natural membrane vesicle exosomes for targeted tumor therapy. Biomaterials 2014, 35, 2383–2390. [Google Scholar] [CrossRef]
- Kong FB, Deng QM, Deng HQ, Dong CC, Li L, He CG, et al. Siva-1 regulates multidrug resistance of gastric cancer by targeting MDR1 and MRP1 via the NF-KB pathway. Mol Med Rep. 2020, 22, 1558–66. [Google Scholar] [CrossRef]
- Zhang, Z.-Z.; Yu, W.-X.; Zheng, M.; Liao, X.-H.; Wang, J.-C.; Yang, D.-Y.; Lu, W.-X.; Wang, L.; Zhang, S.; Liu, H.-K.; et al. PIN1 Inhibition Sensitizes Chemotherapy in Gastric Cancer Cells by Targeting Stem Cell–like Traits and Multiple Biomarkers. Mol. Cancer Ther. 2020, 19, 906–919. [Google Scholar] [CrossRef]
- Li, D.; Hu, C.; Li, H. Survivin as a novel target protein for reducing the proliferation of cancer cells (Review). Biomed. Rep. 2018, 8, 399–406. [Google Scholar] [CrossRef] [PubMed]
- Dohi, T.; Beltrami, E.; Wall, N.R.; Plescia, J.; Altieri, D.C. Mitochondrial survivin inhibits apoptosis and promotes tumorigenesis. J. Clin. Investig. 2004, 114, 1117–1127. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Yung, M.M.H.; Ngan, H.Y.S.; Chan, K.K.L.; Chan, D.W. The Impact of the Tumor Microenvironment on Macrophage Polarization in Cancer Metastatic Progression. Int. J. Mol. Sci. 2021, 22, 6560. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.-B.; Gu, Z.-Q.; Jian, K.; Qi, J. CXCR4-mediated Stat3 activation is essential for CXCL12-induced cell invasion in bladder cancer. Tumor Biol. 2013, 34, 1839–1845. [Google Scholar] [CrossRef]
Figure 1.
Characterization of EVs derived from M1-polarized RAW 264.7. Representative photomicrographs of the uptake analysis in CT-26 cells over 24 hours (A-D). In blue the nucleus marked with DAPI (A), in red the cytoplasmic marking of CT-26 with DIL (B), in white, the EVs marked inside the cells (C), and in figure (D) merged cells stained with all dyes. Analysis of the EVs morphology by transmission electron microscopy (TEM) (E). Analysis of the size of the EVs obtained after completing the process of obtaining and standardizing the EVs (F). This image represents the quantitative analysis of the concentration of oxaliplatin detected by HPLC (G).
Figure 1.
Characterization of EVs derived from M1-polarized RAW 264.7. Representative photomicrographs of the uptake analysis in CT-26 cells over 24 hours (A-D). In blue the nucleus marked with DAPI (A), in red the cytoplasmic marking of CT-26 with DIL (B), in white, the EVs marked inside the cells (C), and in figure (D) merged cells stained with all dyes. Analysis of the EVs morphology by transmission electron microscopy (TEM) (E). Analysis of the size of the EVs obtained after completing the process of obtaining and standardizing the EVs (F). This image represents the quantitative analysis of the concentration of oxaliplatin detected by HPLC (G).
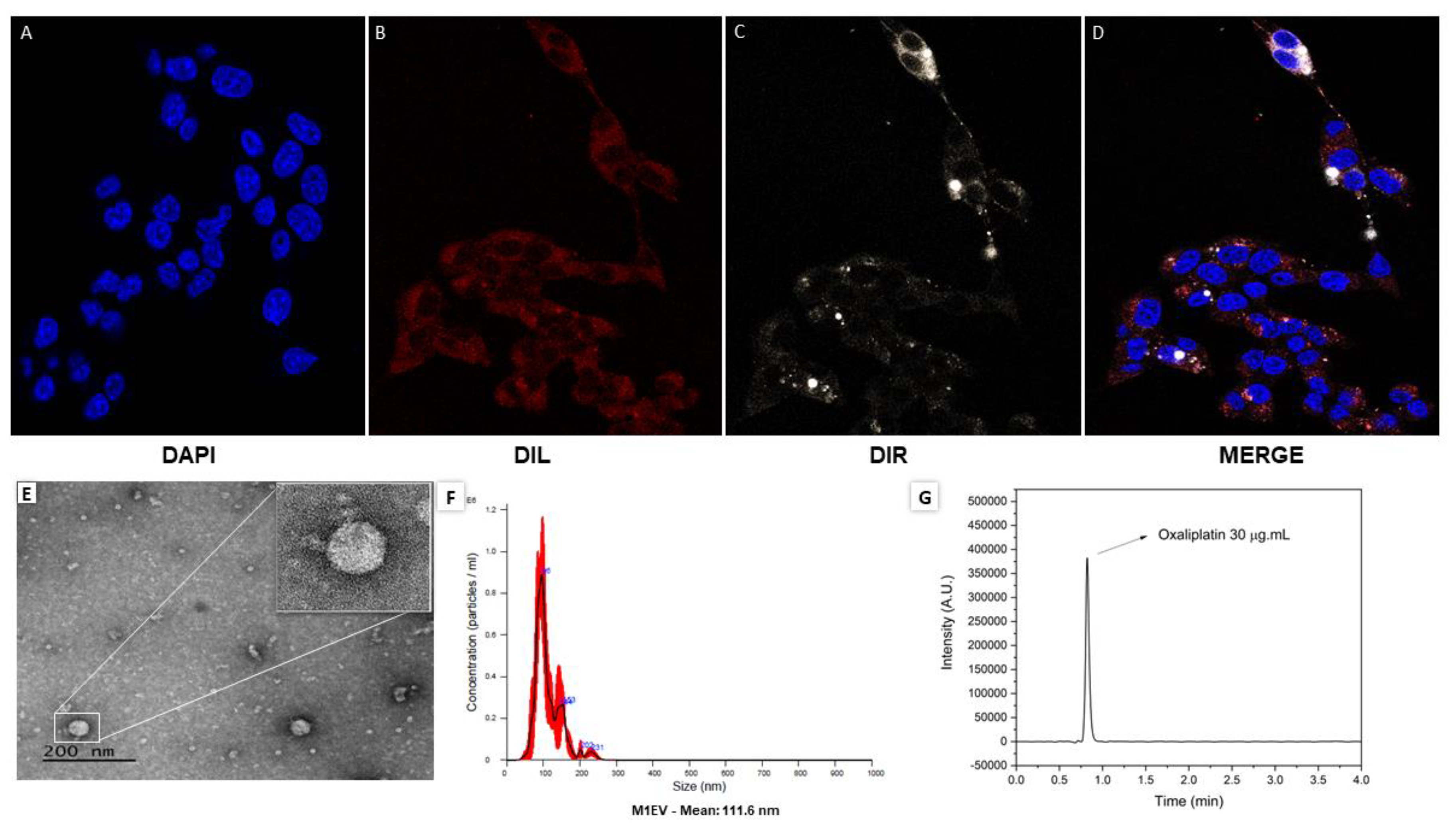
Figure 2.
M1EV, M1EVRA, M1EVLF, and M1EVRALF decreased M2-TAM polarization. (A-B) Representative flow cytometry profile of CD68 and CD163 expression on RAW 264.7 control cells and upon treatment with IL-4, or IL-4 and M1EV, M1EVRA, M1EVLF, and M1EVRALF. (C-D) Expression of CD68 and CD163 in Raw 264.7 cells treated with IL-4, or IL-4 and M1EV, M1EVRA, M1EVLF, and M1EVRALF in co-culture with CT-26 cells. (E) Level of IL-10 from cell culture supernatants of IL-4-polarized RAW 264.7 cells, in the presence and absence of M1EV, M1EVRA, M1EVLF, and M1EVRALF. Unstained (CT-26 control cells), RAW: RAW 264.7 cells (control cells), RAW+IL4: RAW 264.7 cells with IL-4, M1EV: M1-EVs 20µg/mL, M1EVRA: M1-EVs with retinoic acid, M1EVLF: M1-EVs with L. ferrea, and M1EVRALF: M1-EVs with retinoic acid and L. ferrea. For analyses of the cytokines, all treatment groups were compared to the RAW + IL-4 group (**p<0.01, ***p<0.001, and ****p<0.0001).
Figure 2.
M1EV, M1EVRA, M1EVLF, and M1EVRALF decreased M2-TAM polarization. (A-B) Representative flow cytometry profile of CD68 and CD163 expression on RAW 264.7 control cells and upon treatment with IL-4, or IL-4 and M1EV, M1EVRA, M1EVLF, and M1EVRALF. (C-D) Expression of CD68 and CD163 in Raw 264.7 cells treated with IL-4, or IL-4 and M1EV, M1EVRA, M1EVLF, and M1EVRALF in co-culture with CT-26 cells. (E) Level of IL-10 from cell culture supernatants of IL-4-polarized RAW 264.7 cells, in the presence and absence of M1EV, M1EVRA, M1EVLF, and M1EVRALF. Unstained (CT-26 control cells), RAW: RAW 264.7 cells (control cells), RAW+IL4: RAW 264.7 cells with IL-4, M1EV: M1-EVs 20µg/mL, M1EVRA: M1-EVs with retinoic acid, M1EVLF: M1-EVs with L. ferrea, and M1EVRALF: M1-EVs with retinoic acid and L. ferrea. For analyses of the cytokines, all treatment groups were compared to the RAW + IL-4 group (**p<0.01, ***p<0.001, and ****p<0.0001).
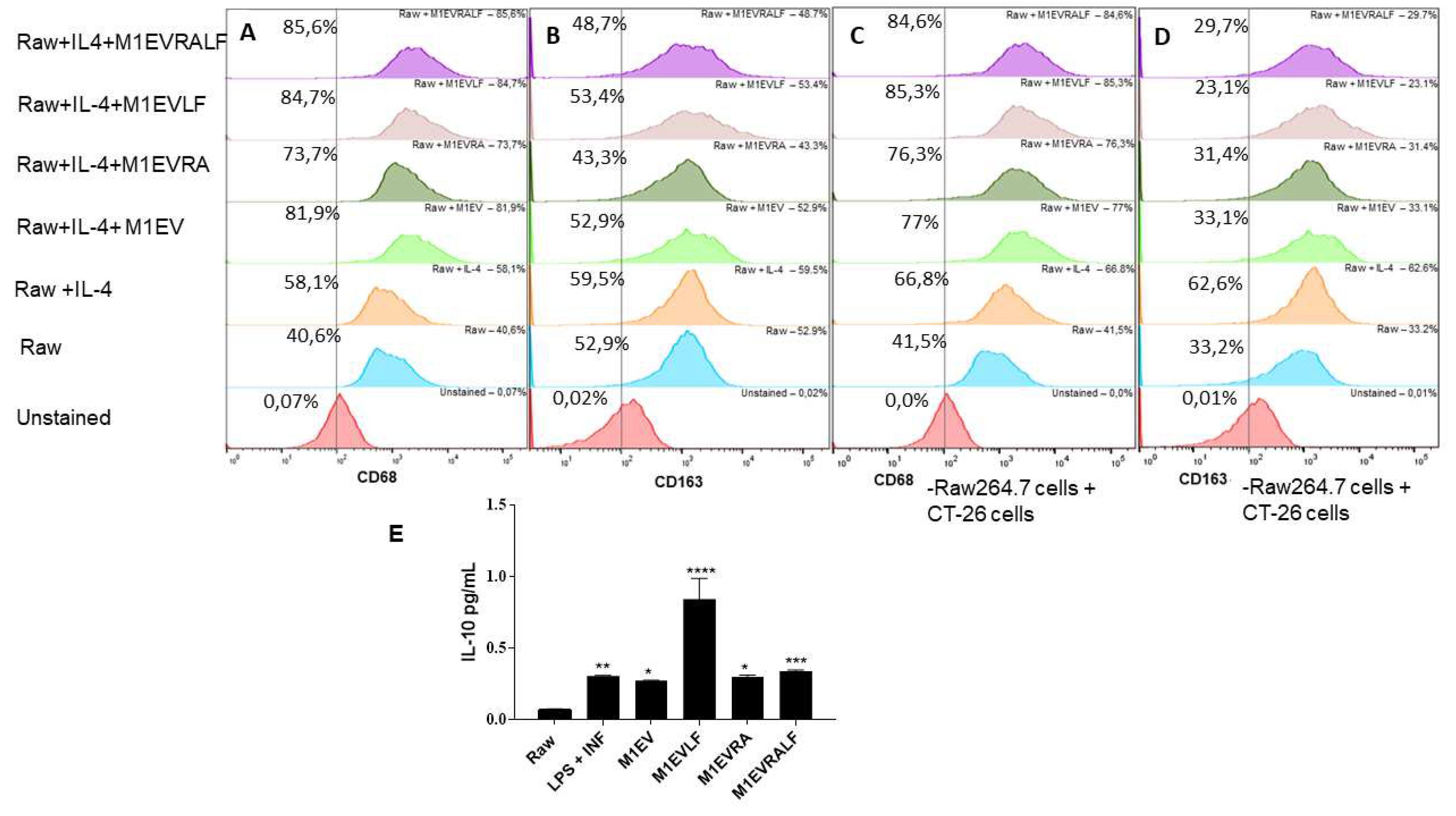
Figure 3.
M1EVs, M1EVRA, M1EVLF and M1EVRALF up-regulated the expression of E-cadherin and down-regulated the expression of Vimentin in CT-26 cells. Immunofluorescence of expression of E-Cadherin and Vimentin in response to the M1EV systems without OXA but loaded with RA, LF and RA+LF (A-J). Graphical representation of the mean Fluorescence Intensity (AU) for the anti-E-Cadherin (K) and anti-Vimentin (L) immunoreactivity in CT-26 cells from each group (green) with DAPI nuclear stained (blue). CT-26 (control cells), M1EVRA: M1-EVs with retinoic acid, M1EV: M1-EVs, M1EVLF: M1-EVs with L. ferrea, and M1EVRALF: M1-EVs with retinoic acid and L. ferrea. Results are presented expressed by mean ± SEM. All treated cells were compared to the CT-26 control group (**p<0.01, ***p<0.001, and ****p<0.0001). Magnification 400x (Scale Bars are 20µM).
Figure 3.
M1EVs, M1EVRA, M1EVLF and M1EVRALF up-regulated the expression of E-cadherin and down-regulated the expression of Vimentin in CT-26 cells. Immunofluorescence of expression of E-Cadherin and Vimentin in response to the M1EV systems without OXA but loaded with RA, LF and RA+LF (A-J). Graphical representation of the mean Fluorescence Intensity (AU) for the anti-E-Cadherin (K) and anti-Vimentin (L) immunoreactivity in CT-26 cells from each group (green) with DAPI nuclear stained (blue). CT-26 (control cells), M1EVRA: M1-EVs with retinoic acid, M1EV: M1-EVs, M1EVLF: M1-EVs with L. ferrea, and M1EVRALF: M1-EVs with retinoic acid and L. ferrea. Results are presented expressed by mean ± SEM. All treated cells were compared to the CT-26 control group (**p<0.01, ***p<0.001, and ****p<0.0001). Magnification 400x (Scale Bars are 20µM).
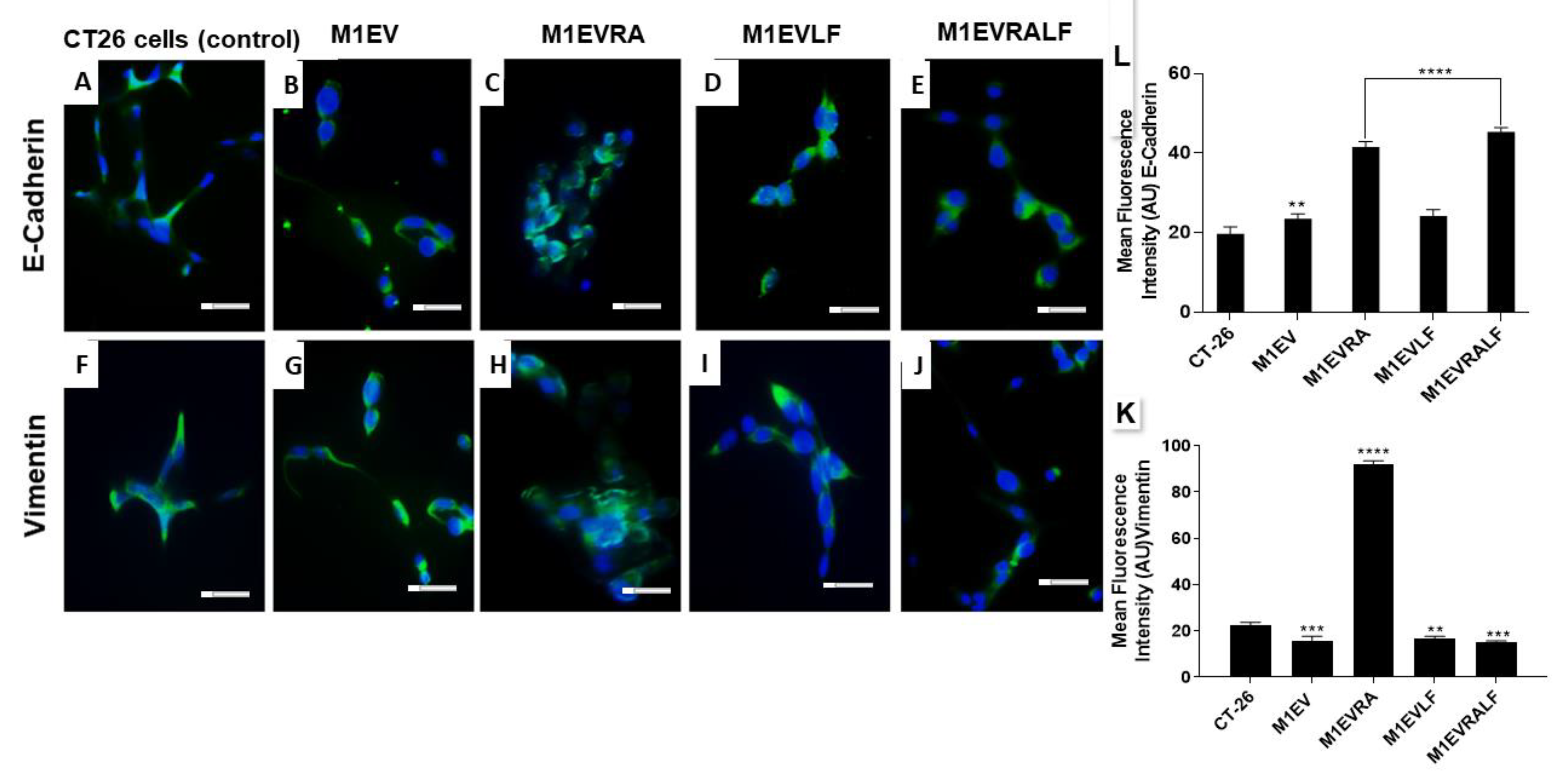
Figure 4.
Relative mRNA expression from NF-κB (A), FAAD (B), Apaf-1 (C), MDR1 (D), SURVIVIN (E), STAT3 (F), Vimentin (G), and E-Cadherin (H) genes in CT-26 cell line co-cultured with Raw 264.7 cells and treated with M1EV systems without OXA but loaded with RA, LF and RA+LF. CT-26 (control cells), MC38 (control cells), RAW: RAW 264.7 cells (control cells), RAW+IL4: RAW 264.7 cells with IL-4, M1EV: M1-EVs 20µg/mL, M1EVRA: M1-EVs with retinoic acid, M1EVLF: M1-EVs with L. ferrea, and M1EVRALF: M1-EVs with retinoic acid and L. ferrea. Results are presented as a fold-change of the media values, normalized to Beta-actin (β-actin), and expressed by mean ± SEM. All treated cells were compared to the RAW+IL-4 group (ns: non-significant, *p<0.05, **p<0.01, ***p<0.001, and ****p<0.0001).
Figure 4.
Relative mRNA expression from NF-κB (A), FAAD (B), Apaf-1 (C), MDR1 (D), SURVIVIN (E), STAT3 (F), Vimentin (G), and E-Cadherin (H) genes in CT-26 cell line co-cultured with Raw 264.7 cells and treated with M1EV systems without OXA but loaded with RA, LF and RA+LF. CT-26 (control cells), MC38 (control cells), RAW: RAW 264.7 cells (control cells), RAW+IL4: RAW 264.7 cells with IL-4, M1EV: M1-EVs 20µg/mL, M1EVRA: M1-EVs with retinoic acid, M1EVLF: M1-EVs with L. ferrea, and M1EVRALF: M1-EVs with retinoic acid and L. ferrea. Results are presented as a fold-change of the media values, normalized to Beta-actin (β-actin), and expressed by mean ± SEM. All treated cells were compared to the RAW+IL-4 group (ns: non-significant, *p<0.05, **p<0.01, ***p<0.001, and ****p<0.0001).
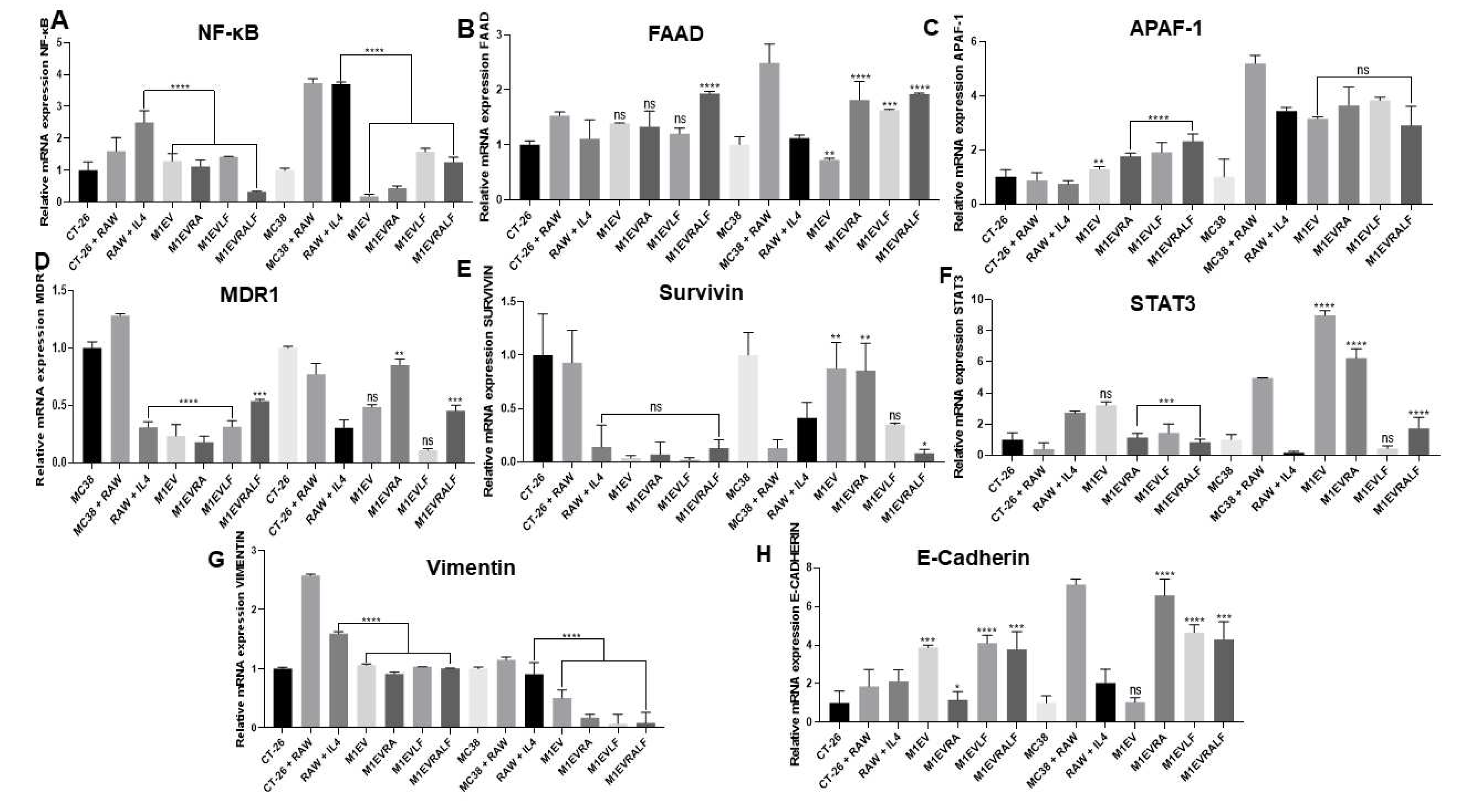
Figure 5.
Inhibition of tumor growth. The excised tumors were presented on a panel for comparative analysis of tumor size. Saline group (A.1), OXA (B.1), M1EV (C.1), M1EV1 (D.1), M1EV2 (E.1), M1EV3 (F.1), and M1EV4 (G.1). The growth curve of allographic colorectal tumors in BALB/c mice shows a reduction in tumor volumes after treatment with M1EV, M1EV1, and M1EV4 (B). Arrows indicate therapeutic interventions via peritumor administration. Graphical representation of the analysis of the collected tumor mass (C). The arrows indicate the days on which the animals received the treatments. OXA: 5mg/kg oxaliplatin, M1EV: 20µg/Kg M1-EVs, M1EV1: M1-EVs with 2mg/Kg oxaliplatin, M1EV2: M1-EVs with 2mg/Kg oxaliplatin and retinoic acid, M1EV3: M1-EVs with 2mg/Kg oxaliplatin and L. ferrea, and M1EV4: M1-EVs with 2mg/Kg oxaliplatin, retinoic acid, and L. ferrea. Results are presented expressed by mean ± SEM. All treated groups were compared to the saline control group (*p<0.05, **p<0.01, ***p<0.001, and ****p<0.0001).
Figure 5.
Inhibition of tumor growth. The excised tumors were presented on a panel for comparative analysis of tumor size. Saline group (A.1), OXA (B.1), M1EV (C.1), M1EV1 (D.1), M1EV2 (E.1), M1EV3 (F.1), and M1EV4 (G.1). The growth curve of allographic colorectal tumors in BALB/c mice shows a reduction in tumor volumes after treatment with M1EV, M1EV1, and M1EV4 (B). Arrows indicate therapeutic interventions via peritumor administration. Graphical representation of the analysis of the collected tumor mass (C). The arrows indicate the days on which the animals received the treatments. OXA: 5mg/kg oxaliplatin, M1EV: 20µg/Kg M1-EVs, M1EV1: M1-EVs with 2mg/Kg oxaliplatin, M1EV2: M1-EVs with 2mg/Kg oxaliplatin and retinoic acid, M1EV3: M1-EVs with 2mg/Kg oxaliplatin and L. ferrea, and M1EV4: M1-EVs with 2mg/Kg oxaliplatin, retinoic acid, and L. ferrea. Results are presented expressed by mean ± SEM. All treated groups were compared to the saline control group (*p<0.05, **p<0.01, ***p<0.001, and ****p<0.0001).
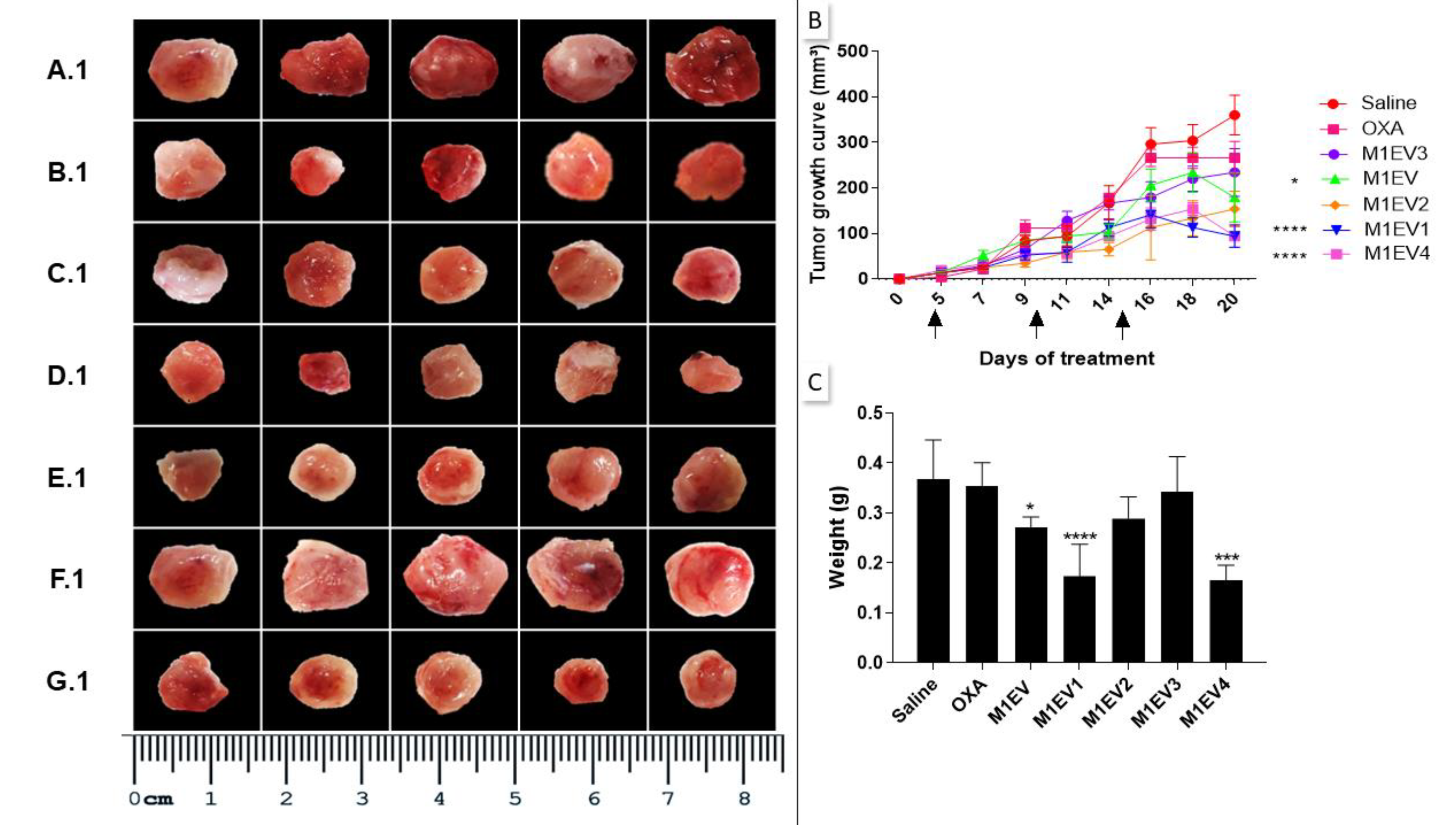
Figure 6.
Evaluation of immunosuppression and invasion in the TME. Representative photomicrographs of Immunofluorescence analysis of tumors collected from colorectal cancer allographic model (A-U), 400x, Bar scale: 20µM. Graphical representation of the mean fluorescence intensity (AU) for anti-PD-L1 (V), anti-IL-10 (W), and anti-MMP2 (X) immunoreactivity in tumor fragments from each group (green) with DAPI nuclear counterstained (blue). OXA: 5mg/kg oxaliplatin, M1EV: 20µg/Kg M1-EVs, M1EV1: M1-EVs with 2mg/Kg oxaliplatin, M1EV2: M1-EVs with 2mg/Kg oxaliplatin and retinoic acid, M1EV3: M1-EVs with 2mg/Kg oxaliplatin and L. ferrea, and M1EV4: M1-EVs with 2mg/Kg oxaliplatin, retinoic acid, and L. ferrea. Results are presented expressed by mean ± SEM. All treated groups were compared to the saline control group (*p<0.05, **p<0.01, ***p<0.001, and ****p<0.0001).
Figure 6.
Evaluation of immunosuppression and invasion in the TME. Representative photomicrographs of Immunofluorescence analysis of tumors collected from colorectal cancer allographic model (A-U), 400x, Bar scale: 20µM. Graphical representation of the mean fluorescence intensity (AU) for anti-PD-L1 (V), anti-IL-10 (W), and anti-MMP2 (X) immunoreactivity in tumor fragments from each group (green) with DAPI nuclear counterstained (blue). OXA: 5mg/kg oxaliplatin, M1EV: 20µg/Kg M1-EVs, M1EV1: M1-EVs with 2mg/Kg oxaliplatin, M1EV2: M1-EVs with 2mg/Kg oxaliplatin and retinoic acid, M1EV3: M1-EVs with 2mg/Kg oxaliplatin and L. ferrea, and M1EV4: M1-EVs with 2mg/Kg oxaliplatin, retinoic acid, and L. ferrea. Results are presented expressed by mean ± SEM. All treated groups were compared to the saline control group (*p<0.05, **p<0.01, ***p<0.001, and ****p<0.0001).
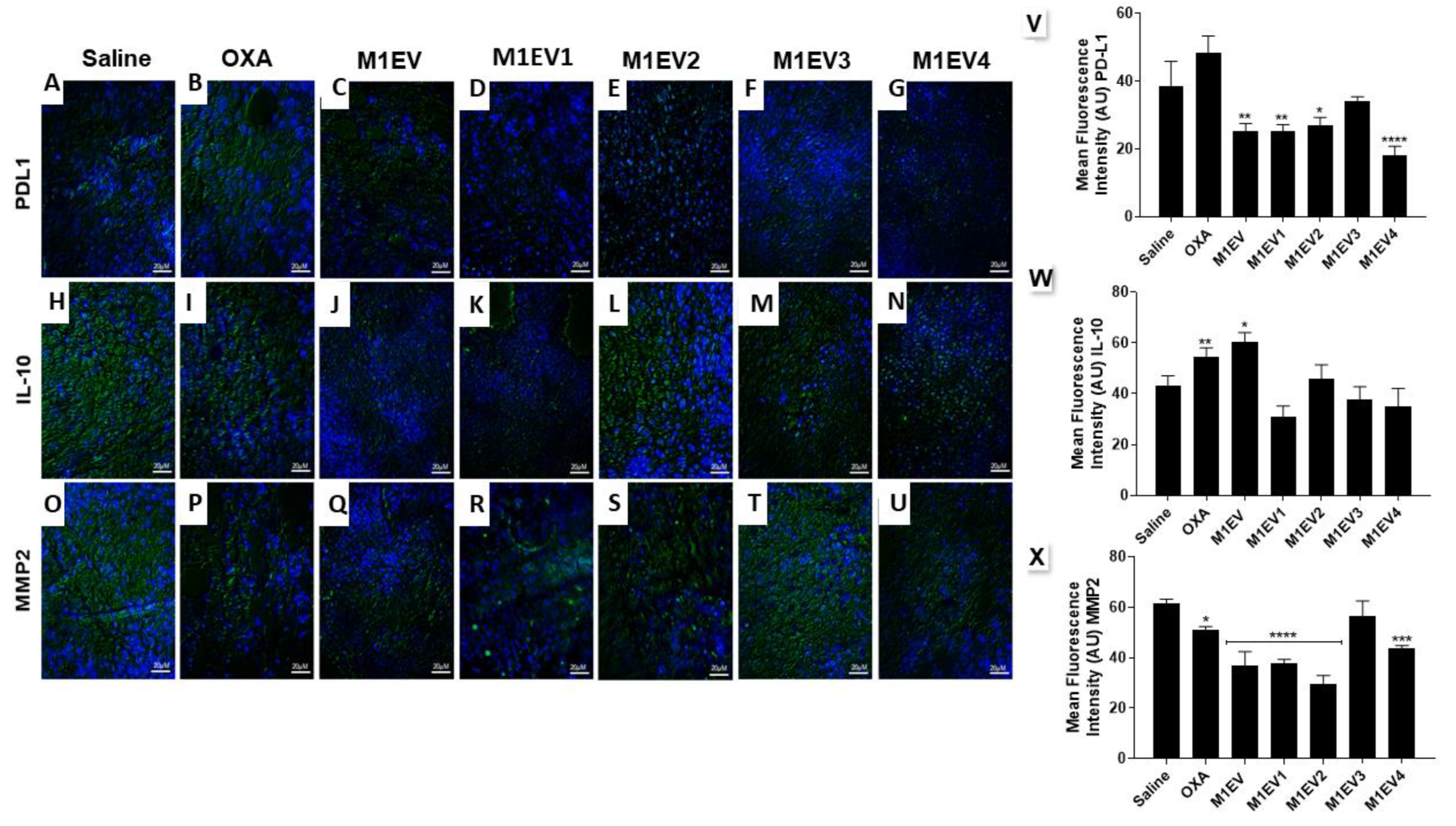
Figure 7.
Influence of RA and LF on in vivo invasion properties of M1-derived EVs loaded with OXA in allographic CT-26 colorectal cancer-bearing mice. The groups were stained for evaluation of AKT-1 (A), PI3K (B), and E-Cadherin (C). Graphical representation of the scores for immunoreactivity in tumor fragments from each group stained for AKT-1 (D), PI3K (E) and E-Cadherin (F). Magnification 400x, Bar scale- 200 µM. OXA: 5mg/kg oxaliplatin, M1EV: 20µg/Kg M1-EVs, M1EV1: M1-EVs with 2mg/Kg oxaliplatin, M1EV2: M1-EV with 2mg/Kg oxaliplatin and retinoic acid, M1EV3: M1-EV with 2mg/Kg oxaliplatin and L. ferrea, and M1EV4: M1-EV with 2mg/Kg oxaliplatin, retinoic acid, and L. ferrea. Results are presented expressed by mean ± SEM. All treatment groups were compared to the saline control group (*p<0.05, **p<0.01, ***p<0.001, and ****p<0.0001).
Figure 7.
Influence of RA and LF on in vivo invasion properties of M1-derived EVs loaded with OXA in allographic CT-26 colorectal cancer-bearing mice. The groups were stained for evaluation of AKT-1 (A), PI3K (B), and E-Cadherin (C). Graphical representation of the scores for immunoreactivity in tumor fragments from each group stained for AKT-1 (D), PI3K (E) and E-Cadherin (F). Magnification 400x, Bar scale- 200 µM. OXA: 5mg/kg oxaliplatin, M1EV: 20µg/Kg M1-EVs, M1EV1: M1-EVs with 2mg/Kg oxaliplatin, M1EV2: M1-EV with 2mg/Kg oxaliplatin and retinoic acid, M1EV3: M1-EV with 2mg/Kg oxaliplatin and L. ferrea, and M1EV4: M1-EV with 2mg/Kg oxaliplatin, retinoic acid, and L. ferrea. Results are presented expressed by mean ± SEM. All treatment groups were compared to the saline control group (*p<0.05, **p<0.01, ***p<0.001, and ****p<0.0001).
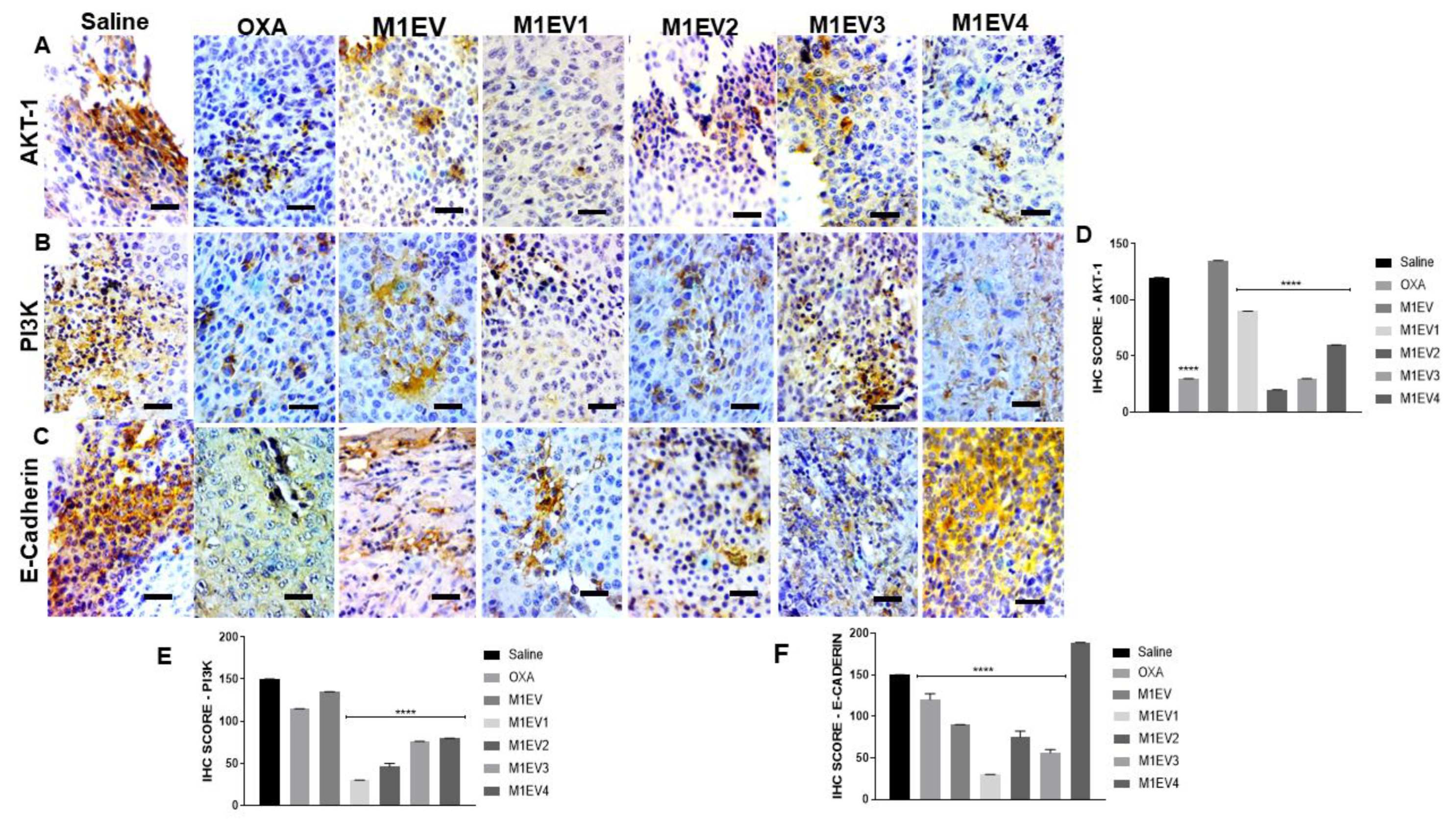
Figure 8.
Relative mRNA expression from analyses of gene expression in tumor fragments from allographic tumor model. FAAD (A), APAF-1 (B), NF-κB (C), STAT3 (D), CD8 (E), CD68 (F), and SNAIL (G) genes. OXA: oxaliplatin 5mg/kg, M1EV: M1-EVs 20µg/mL, M1EV1: M1-EVs with oxaliplatin 2mg/Kg, M1EV2: M1-EVs with oxaliplatin 2mg/Kg and retinoic acid 4mg/mL, M1EV3: M1-EVs with oxaliplatin 2mg/Kg and L. ferrea 2.5mg/mL, and M1EV4: M1-EVs with oxaliplatin 2mg/Kg, retinoic acid 4mg/mL, and L. ferrea 2.5mg/mL. Results are presented as fold-change of the media values, normalized to Beta-actin (β-actin), and expressed by mean ± SEM (*p<0.5, **p<0.01, ***p<0.001, and ****p<0.0001). All treatment groups were compared to the OXA group and the saline control group.
Figure 8.
Relative mRNA expression from analyses of gene expression in tumor fragments from allographic tumor model. FAAD (A), APAF-1 (B), NF-κB (C), STAT3 (D), CD8 (E), CD68 (F), and SNAIL (G) genes. OXA: oxaliplatin 5mg/kg, M1EV: M1-EVs 20µg/mL, M1EV1: M1-EVs with oxaliplatin 2mg/Kg, M1EV2: M1-EVs with oxaliplatin 2mg/Kg and retinoic acid 4mg/mL, M1EV3: M1-EVs with oxaliplatin 2mg/Kg and L. ferrea 2.5mg/mL, and M1EV4: M1-EVs with oxaliplatin 2mg/Kg, retinoic acid 4mg/mL, and L. ferrea 2.5mg/mL. Results are presented as fold-change of the media values, normalized to Beta-actin (β-actin), and expressed by mean ± SEM (*p<0.5, **p<0.01, ***p<0.001, and ****p<0.0001). All treatment groups were compared to the OXA group and the saline control group.
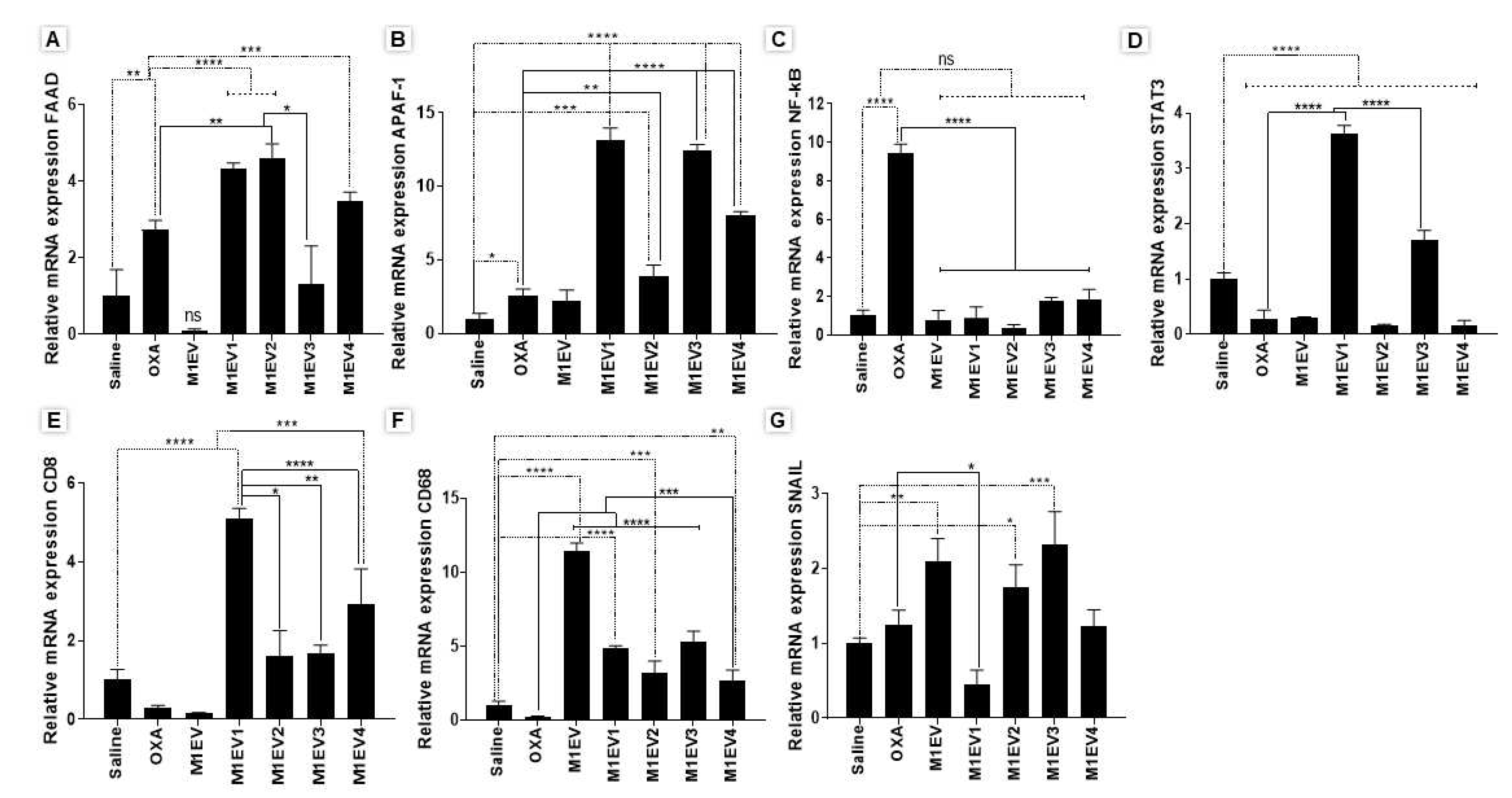
Figure 9.
M1EV holding OXA, RA, and LF may inhibit nodule formation of colon cancer peritoneal metastasis in mice. (A) Visible tumor nodules were present in the abdominal cavity in representative photos of mice sacrificed 2 weeks after IP injection with CT-26 cells and treatment with 5mg/Kg OXA. (B) Graphical representation of the weight in mg of the tumors collected. (C) Graphical representation from the quantification of tumors per mouse. (D) Histopathological scores of peritoneal tumors. 400x, Bar scale: 200µm OXA: 5mg/kg oxaliplatin, M1EV1: M1-EVs with 2mg/Kg oxaliplatin, M1EV4: M1-EVs with 2mg/Kg oxaliplatin, retinoic acid, and L. ferrea. Results are presented expressed by mean ± SD. All treatment groups were compared to the saline control group. In B and C, treated groups were compared to the OXA group (*p<0.05, **p<0.01, ***p<0.001, and ****p<0.0001).
Figure 9.
M1EV holding OXA, RA, and LF may inhibit nodule formation of colon cancer peritoneal metastasis in mice. (A) Visible tumor nodules were present in the abdominal cavity in representative photos of mice sacrificed 2 weeks after IP injection with CT-26 cells and treatment with 5mg/Kg OXA. (B) Graphical representation of the weight in mg of the tumors collected. (C) Graphical representation from the quantification of tumors per mouse. (D) Histopathological scores of peritoneal tumors. 400x, Bar scale: 200µm OXA: 5mg/kg oxaliplatin, M1EV1: M1-EVs with 2mg/Kg oxaliplatin, M1EV4: M1-EVs with 2mg/Kg oxaliplatin, retinoic acid, and L. ferrea. Results are presented expressed by mean ± SD. All treatment groups were compared to the saline control group. In B and C, treated groups were compared to the OXA group (*p<0.05, **p<0.01, ***p<0.001, and ****p<0.0001).
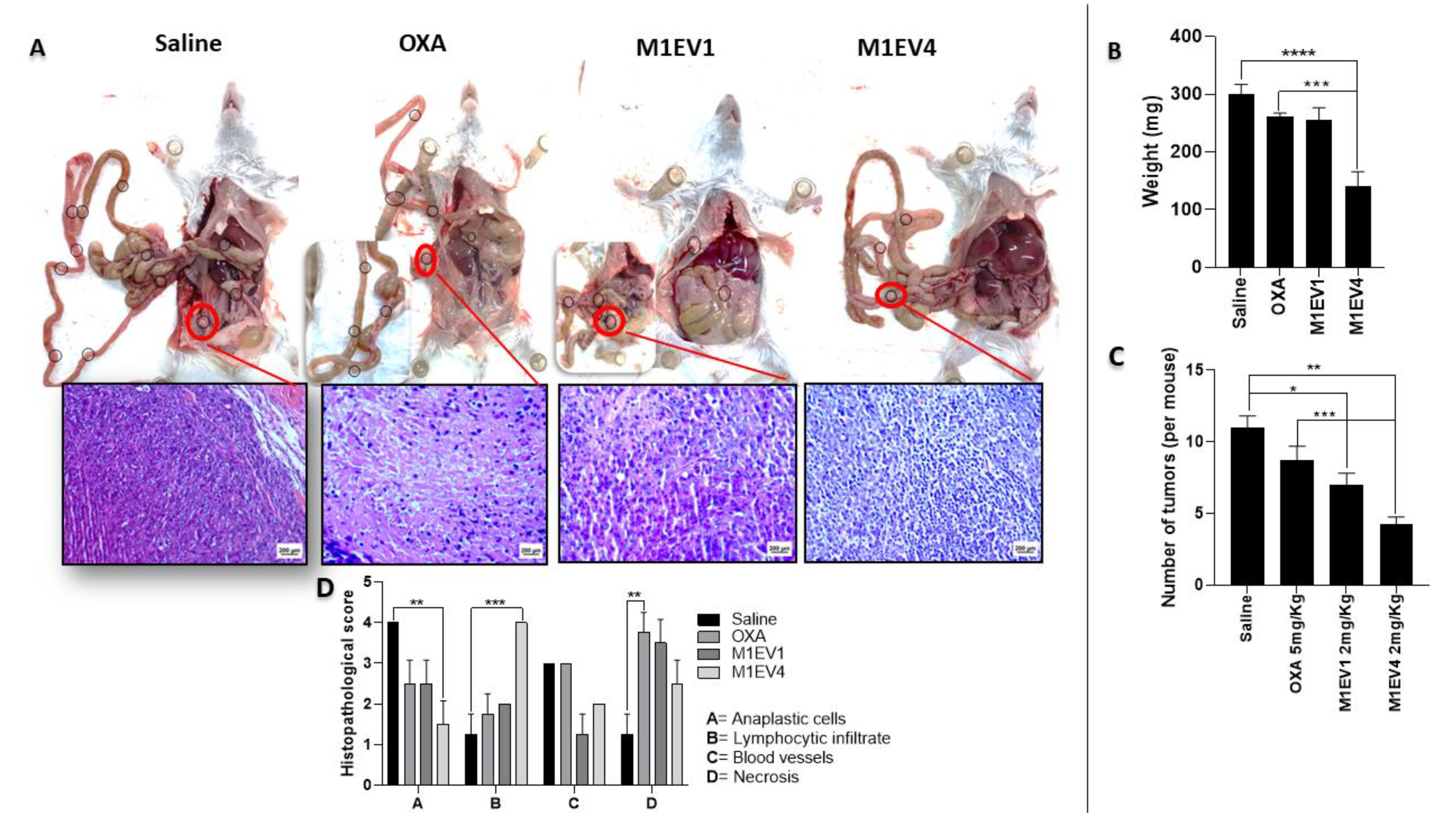
Figure 10.
Evaluation of immunosuppression in colon cancer peritoneal metastasis. The tumors excised from the previously treated BALB/c mice were analyzed for immunostaining by immunohistochemistry (A–T) and relative gene expression (U-X). Images with 100× magnification; 400× magnification is shown in the lower right corner of each image (Bar scale: 200µM). Scores were applied to each image to represent them graphically. The saline (A, E, I, and M), OXA 5mg/kg (B, F, J, and N), M1EV1 (C, G, K, and O), and M1EV4 (D, H, L, and P) groups were stained for evaluation of AKT (A-D), NF-κB (E-H), CXCL12 (I-L), and CD163 (M-P). OXA: 5mg/kg oxaliplatin, M1EV1: M1-EV with 2mg/Kg oxaliplatin, M1EV4: M1-EV with 2mg/Kg oxaliplatin, retinoic acid, and L. ferrea. Magnification 400x. Results are presented expressed by mean ± SD. All treatment groups were compared to the saline control group (*p<0.05, **p<0.01, ***p<0.001, and ****p<0.0001).
Figure 10.
Evaluation of immunosuppression in colon cancer peritoneal metastasis. The tumors excised from the previously treated BALB/c mice were analyzed for immunostaining by immunohistochemistry (A–T) and relative gene expression (U-X). Images with 100× magnification; 400× magnification is shown in the lower right corner of each image (Bar scale: 200µM). Scores were applied to each image to represent them graphically. The saline (A, E, I, and M), OXA 5mg/kg (B, F, J, and N), M1EV1 (C, G, K, and O), and M1EV4 (D, H, L, and P) groups were stained for evaluation of AKT (A-D), NF-κB (E-H), CXCL12 (I-L), and CD163 (M-P). OXA: 5mg/kg oxaliplatin, M1EV1: M1-EV with 2mg/Kg oxaliplatin, M1EV4: M1-EV with 2mg/Kg oxaliplatin, retinoic acid, and L. ferrea. Magnification 400x. Results are presented expressed by mean ± SD. All treatment groups were compared to the saline control group (*p<0.05, **p<0.01, ***p<0.001, and ****p<0.0001).
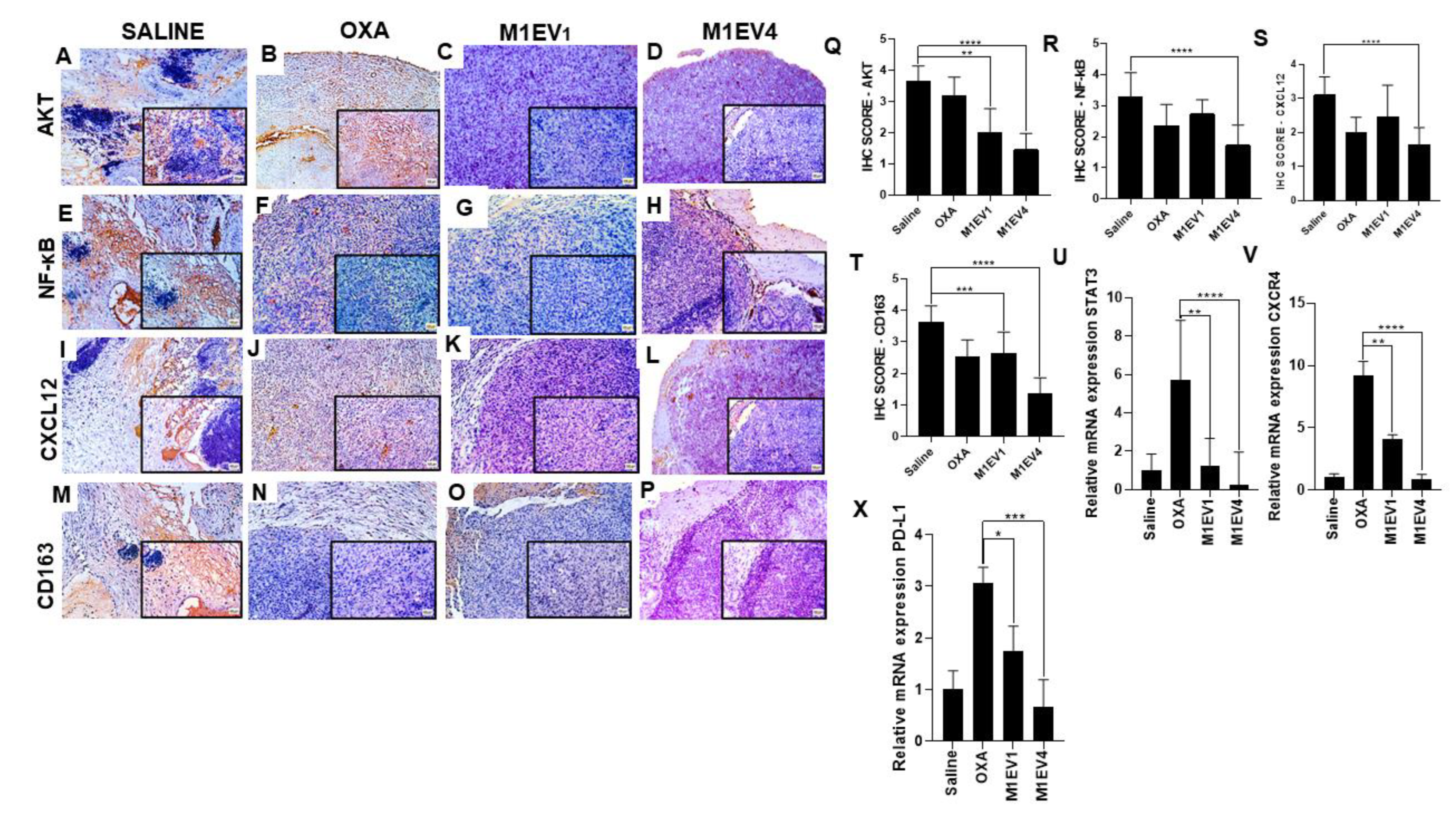
Figure 11.
Evaluation of metastatic niches in liver and Lungs. The evaluation of tumor cell migration to secondary sites was observed in the liver (A-D, 100× and 400×) and lung (E-H, 100× and 400×) from previously treated BALB/c mice. The arrowheads indicate the presence of metastatic niches (A-H), which are best seen in greater magnification (400x). Metastatic niches in the liver (I) and lung (J) were assessed semi-quantitatively by applying scores representing the tumor cells percentage in the tissue parenchyma. OXA: 5mg/kg oxaliplatin, M1EV1: M1-EV with 2mg/Kg oxaliplatin, M1EV4: M1-EV with 2mg/Kg oxaliplatin, retinoic acid, and L. ferrea. Results are presented expressed by mean ± SD. All treatment groups were compared to the saline control group (*p<0.05, **p<0.01, ***p<0.001, and ****p<0.0001). Bar scale: 200µM.
Figure 11.
Evaluation of metastatic niches in liver and Lungs. The evaluation of tumor cell migration to secondary sites was observed in the liver (A-D, 100× and 400×) and lung (E-H, 100× and 400×) from previously treated BALB/c mice. The arrowheads indicate the presence of metastatic niches (A-H), which are best seen in greater magnification (400x). Metastatic niches in the liver (I) and lung (J) were assessed semi-quantitatively by applying scores representing the tumor cells percentage in the tissue parenchyma. OXA: 5mg/kg oxaliplatin, M1EV1: M1-EV with 2mg/Kg oxaliplatin, M1EV4: M1-EV with 2mg/Kg oxaliplatin, retinoic acid, and L. ferrea. Results are presented expressed by mean ± SD. All treatment groups were compared to the saline control group (*p<0.05, **p<0.01, ***p<0.001, and ****p<0.0001). Bar scale: 200µM.
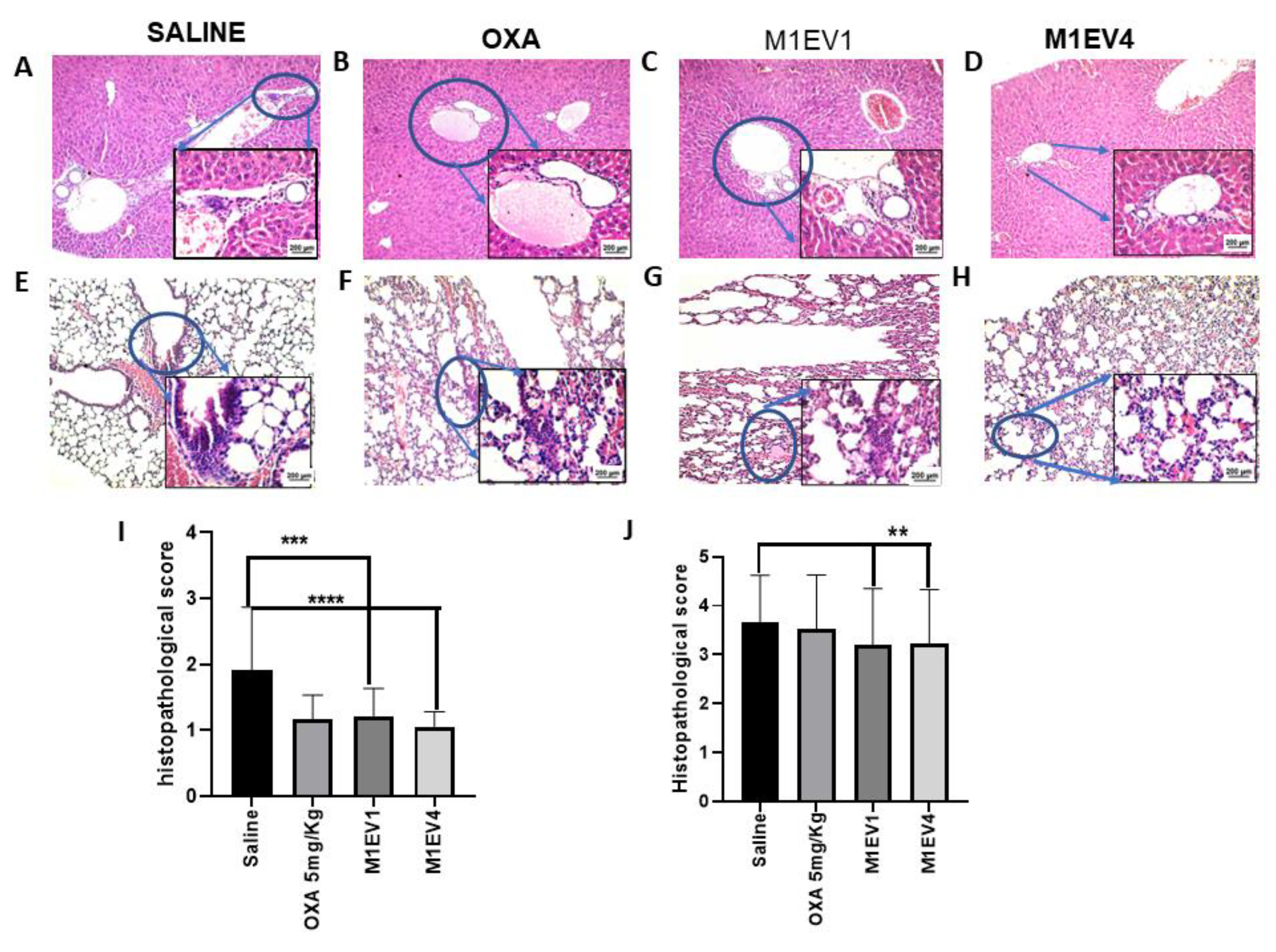
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



