
Submitted:
25 May 2023
Posted:
26 May 2023
You are already at the latest version
Abstract
Iron-based materials are widely applied in Fenton chemistry and they have promising prospects in the processing of wastewater. The composition complexity and rich chemistry of iron and/or oxides, however, hamper the precise understanding on the active sites and the working mechanism which still remain highly controversial. Herein, iron oxides of four different model systems are designed through a conventional precipitation method plus H2 reduction treatment. These systems feature Fe@Fe3O4 with abundant oxygen vacancy, Fe0 and Fe3O4 particles with interface structures, and Fe3O4-dominated nanoparticles of different sizes. These materials are applied in the decomposition of methyl orange as a model reaction to assess the Fenton chemistry. The Fe@Fe3O4 with core-shell structures exhibited significantly higher decomposition activity than the other Fe3O4-rich nanoparticles. A thin Fe3O4 layer formed by auto-oxidation of iron particles when exposing to air can boost the activity as compared with the Fe0 and Fe3O4 particles with interface structures but poor oxygen vacancy. The unique hetero-structure with the co-existence of both metallic iron and oxygen vacancy displayed excellent redox propensity which might account for the superior Fenton activity. This finding provides a new perspective to understand and design highly efficient iron-based Fenton catalysts.
Keywords:
Advanced oxidation process
; Core-shell structure
; Fenton chemistry
; Fe@Fe3O4 interface
; Methyl orange decomposition
; Oxygen vacancy
1. Introduction
The widespread pollution of wastewater due to the discharge of high-concentration chemical solutions containing diverse organic compounds from the industry has posed a formidable challenge to a sustainable society[1,2,3]. The conventional approaches of wastewater management relying on the biodegradation or physiochemical methods (such as the combination of chlorination and adsorption) are still not optimal to handle the industrial wastewater. In this context, advanced oxidation processes (AOP) are catching great attentions as one of the most promising alternative solutions[4,5,6,7]. Compared with the traditional biodegradation method which might be suspected to biomass poisoning, AOP is a non-selective technology, which depends on the oxidation of organic compounds with the in situ generated radical species with strong oxidizing ability. Therefore, it is extremely flexible to handle the diversity of organic pollutants from the different chemical sectors.
As one of the most cost-effective AOP technologies, the Fenton process has drawn great interest in the wastewater treatment[4,5]. The classic Fenton chemistry is initiated by the formation of hydroxyl radicals (HO*) through the activation of H2O2 by Fe2+ ions. The as-formed HO* and together other with other radicals of strong oxidizing potentials can then convert the organic pollutants into smaller molecules or CO2[5]. To further enhance the oxidation efficiency, similar Fenton-like processes such as photo-Fenton[8,9,10], electro-Fenton[11], and sono-Fenton[12], have also been studies by different researchers. Despite of the difference of various techniques, the development of highly active Fenton catalysts is of optimal importance. The classic Fenton reaction was performed by soluble iron salts as homogeneous catalysts conducted at an optimal pH of 3.0. In addition to the requirement of neutralization before the discharge, the precipitation and subsequent disposal of iron hydroxides are the major problems[7]. To overcome this disadvantage, different heterogeneous Fenton systems have been proposed which can be roughly classified into two categories: (i) the iron-containing catalysts, including iron minerals, clay-based catalysts, and other iron-containing catalysts; and (ii) non-ferrous catalysts based on cerium[13], chromium[14], cobalt[15], copper[16], manganese[17], ruthenium[18], and polyoxometalates[19]. Despite of the extensive explorations, iron-based Fenton systems possess several advantages such as the high reactivity of Fe2+/Fe3+[20], the abundance and low cost of the metal, and low toxicity and environmental compatibility, rendering them highly appealing.
Iron minerals, including magnetite (Fe3O4) [21,22,23], hematite (α-Fe2O3) [24], maghemite (γ-Fe2O3)[25], goethite (α-FeOOH)[26], akaganèite (β-FeOOH)[27,28], lepidocrocite (γ-FeOOH), etc., are among the most extensively studied Fenton systems. In addition, zero-valence iron (ZVI)[29,30,31] and hybridized structures (e.g., Fe@Fe2O3[32,33,34,35], and Fe2+/Fe@Fe2O3[36,37]) are also widely explored. Despite of the different compositions and structures of these catalytic materials, it is generally accepted that Fe2+ species are crucial for the generation of HO*[4,5,6,7]. However, there remains highly controversial questions regarding the nature of the active sites for the metallic iron and oxides. For instance, Yuan et al.[30] employed ZVI in methyl orange decomposition and found activity drop in consecutive runs. Analysis of the recovered catalyst confirmed the formation of oxide layers that was suggested to be responsible for the activity degrading. On the contrary, recent studied showed that by combining ZVI with iron oxides such as Fe@Fe2O3 core-shell structures are able to achieve excellent Fenton activity[32]. It has been proposed that Fe0 can transfer two electrons to O2 to generate H2O2, which further reacts with generated Fe2+ to produce HO* under acidic to neutral pH conditions[38]. The above opposing opinions highlight the necessity of a more in-depth understanding of the Fe0 and iron oxides interfaces. Furthermore, ZVI is known to be susceptible to auto-oxidation when exposed in air. This means the outermost surfaces of the bulk metallic iron is likely covered by iron oxide layers. The nature of these layers and the impact on the Fenton activity are still not well understood.
In this contribution, we synthesized different iron oxides-based model systems through a conventional precipitation method in conjunction with H2 reduction treatment. These systems showed very different structural features: (1) Fe0-rich cores covered with Fe3O4 layers (Fe@Fe3O4), (2) Fe0 and Fe3O4 nanoparticles with interface structures, and (3) Fe3O4-dominated nanoparticles of different sizes. These materials are then applied as the Fenton catalysts in the decomposition of methyl orange that was selected as the model reaction to assess the catalytic activity. Our catalytic results showed that the first two systems with the co-existence of both Fe0 and Fe3O4 phases were significantly more active than the Fe3O4-dominated nanoparticles of different sizes, and the Fe@Fe3O4 with core-shell structures exhibited more than twice higher decomposition activity than the Fe0 and Fe3O4 nanoparticles with interface structures. We also demonstrated that a thin Fe3O4 layer up to tens of nanometers was formed by the auto-oxidation of iron particles when exposing to air, leading to similar core-shell structures of the Fe@Fe3O4 model system and consequently comparable Fenton activity. Furthermore, our results also indicated that the redox propensity might be a more critical activity descriptor than the contents of Fe2+ and oxygen vacancy in the Fenton chemistry, and the co-existence of both zero-valent iron and rich oxygen can boost the Fenton activity.
2. Results and discussion
2.1. Synthesis and characterization of the iron oxides
2.1.1. Synthesis of the iron oxides
Different iron oxides were prepared by the precipitation methods by varying the iron precursors and the surfactants (Figure 1). The resultant precipitates derived from Fe(NH4)2(SO4)2·6H2O, Fe(NO3)3·9H2O, and FeCl3·6H2O were denoted as Fe2O3-S-P, Fe2O3-N-E, and Fe2O3-Cl-P, respectively. These samples were further subject to H2 reduction before the application in MO decomposition. In addition, a commercial sample denoted as Fe3O4-C was purchased as the reference.
2.1.2. Compositional and structural analyses of the iron oxides
The compositions of the as-prepared iron oxides were first checked by powder X-ray diffraction patterns (PXRD, Figure 2). The representative diffraction lines corresponding to α-Fe2O3 (PDF # 33-0664) can be found for all the samples, confirming the successful synthesis of the targeted materials. In addition, these materials showed clearly differences in the diffraction intensities with the order of Fe2O3-Cl-P ~ Fe2O3-N-E >> Fe2O3-S-P. The crystallite sizes of Fe2O3 were then estimated by using the Scherrer equation and the (104) facet. The sizes of Fe2O3-Cl-P, Fe2O3-N-E, and Fe2O3-S-P were determined to be 44, 33, and 15 nm, respectively. The morphologies of as-derived Fe2O3 were studied by combined electron microscopic techniques (Figure 3). Scanning electron microscopy (SEM) revealed the formation of condensed aggregates for Fe2O3-S-P and Fe2O3-N-E, and the more homogeneous nanospheres for Fe2O3-Cl-P, which were likely resulting from the combined use of different iron precursors and surfactants. In line with the SEM observation, transmission electron microscopy (TEM) also confirmed the relatively smaller sizes of the irregular nanoparticles for Fe2O3-S-P and Fe2O3-N-E.
2.1.3. Compositional and structural analyses of the reduced iron oxides
The PXRD patterns of the Fe2O3 and the commercial Fe3O4-C after H2 reduction at 350 oC were presented in Figure 4. The results can be grouped into two classes. Both Fe2O3-N-E and Fe2O3-S-P showed the predominant diffraction lines corresponding to the (110) facet of metallic iron (Fe0 PDF # 06-0696) with the coexistence of relatively weak diffractions related to the (311) and (220) facets of Fe3O4 (PDF # 19-0629). On the contrary, all the typical diffraction patterns of Fe3O4 were found for Fe2O3-Cl-P and Fe3O4-C, but with quite different intensities. The above results suggested the different redox ability of these samples which will be studied in detail in the next sections. Furthermore, it means Fe3O4 was probably the predominant phase with the coexisted Fe0 in Fe2O3-S-P and Fe2O3-N-E, while Fe3O4 was the main phase in Fe2O3-Cl-P and Fe3O4-C. Estimation of the crystallite sizes of Fe0 based on the (110) facet revealed comparable values between Fe2O3-S-P and Fe2O3-N-E (57 vs. 52 nm), while the sizes of Fe3O4 based on the (220) facet were determined to be 24, 53, 36, and 56 nm, respectively, for Fe2O3-S-P, Fe2O3-N-E, Fe2O3-Cl-P and Fe3O4-C.
The morphologies of the reduced samples were assessed by TEM (Figure 5). Fe2O3-S-P and Fe2O3-N-E showed irregular-shaped particles while Fe2O3-Cl-P can still preserve the sphere-like morphology. As compared with un-reduced iron oxides, the particle sizes became much larger, hinting the severe aggregation during the reduction treatments. Besides, Fe3O4-C also showed big particles of irregular shapes. To further assess the structural differences, high-resolution TEM (HRTEM) analyses of the reduced samples were conducted (inset in Figure 5 and Figure S1). Among all these materials, a clear core-shell structure was evidenced only in Fe2O3-S-P. Detailed analyses of this material revealed the continuous lattice fringes of Fe(111) with a distance of 2.0268 Å, and in the periphery the lattice fringes of Fe3O4(111) with a distance of 2.5320 Å were found (Figure 6). These observations therefore ambiguously pointed to the formation of Fe0 as the core covered with a thin shell of Fe3O4. Like in the case of Fe2O3-S-P, the lattice fringes of both Fe(110) and Fe3O4(220) facets were observed in Fe2O3-N-E. These two facets were also in close proximity, suggesting the presence of Fe- Fe3O4 interfaces. In contrast, lattice fringe analyses of Fe2O3-Cl-P and Fe3O4-C only found the presence of Fe3O4 phase. In general, the TEM observations were highly consistent with the PXRD results.
2.1.4. Redox properties of the iron oxides
The redox properties of a catalytic material play an important role in the Fenton chemistry. Therefore, different techniques were applied to assess the redox properties of the as-derived iron oxides. The phase transitions of different iron compositions in flowing H2 from room temperature to 700 oC were followed by in situ PXRD (Figure 7). Both Fe2O3-S-P and Fe2O3-N-E showed very similar phase transition patterns. Namely, the Fe3O4 phase in these two samples vanished at much lower temperatures as compared with Fe2O3-Cl-P (658-673 vs. 746 oC). Meanwhile, the appearance of Fe0 diffractions emerged at significantly lower temperatures (493-523 vs. 627 oC). Differing from the three as-prepared Fe2O3 materials which showed the reduction to different degrees, the in situ PXRD patterns of Fe3O4-C indicated that this material was extremely difficult to reduce. The Fe3O4 phase predominated with only very fainted diffractions related to Fe0. Overall, these results suggested the very different reduction propensities of the materials with the order of Fe2O3-S-P ~ Fe2O3-N-E > Fe2O3-Cl-P >> Fe3O4-C.
2.1.5. Surface iron species and oxygen vacancy
The different surface iron and oxygen species of the reduced iron oxides were studied by X-ray photoelectron spectroscopy (XPS, Figure 8), as they were suggested to have important influences on the redox and Fenton chemistry. The Fe 2p spectra of all the samples showed a doublet at 736-706 eV with a satellite around 718 eV. The spectra were analyzed by peak fitting with Fe2+ and Fe3+ species with known peak positions from the literature results. It was determined that the shares of Fe2+ were comparable for Fe2O3-S-P, Fe2O3-Cl-P and Fe3O4-C (44-46%), and was much higher than that of Fe2O3-N-E (35%). The O 1s of these reduced samples were also analyzed by fitting the spectra. It was revealed that Fe2O3-S-P, Fe2O3-Cl-P and Fe3O4-C possessed similar shares of oxygen vacancy (Ov, 30-36%), while it was the lowest for Fe2O3-N-E (14%).
2.2. Performance in MO decomposition
The catalytic performance of the derived iron oxides was evaluated in the decomposition of methyl orange that was chosen as a model reaction to assess the Fenton chemistry. All the materials were reduced in H2 at 350 oC prior to the tests. At first, the activity was evaluated at a lower MO concentration of 100 μg/mL (Figure 9a). The conversion steadily increased for Fe2O3-Cl-P and Fe3O4-C. While full conversion was already achieved at 60 min for Fe2O3-C, the conversion only slowly increased and stayed at about 22% at 120 min for Fe2O3-Cl-P. In stark contrast, full conversion was readily achieved upon the addition of H2O2 for both Fe2O3-S-P and Fe2O3-N-E. To further discriminate the activity difference, the decomposition activity was further evaluated at a higher MO concentration of 500 μg/mL (Figure 9b). Under such conditions, full conversion was still readily reached at 30 min for Fe2O3-S-P. In contrast, the conversions at 120 min were only 80% and 35%, respectively, for Fe2O3-N-E and Fe3O4-C. To quantitatively compare the Fenton activity of the four samples, the initial reaction rates were calculated by considering the linear parts of the conversion-time profiles (Figure 9c). Fe2O3-S-P exhibited the highest rate of 370 mgMO gcat.-1 min-1, which is two times higher than that of Fe2O3-N-E and significantly outperforms Fe2O3-Cl-P and Fe3O4-C by 1-2 orders of magnitude. In order to confirm that the reaction proceeded in a heterogeneous manner, we conducted another test by replacing the iron oxides with trace Fe(NO3)3 so as to mimicking the leaching of iron species into the solution. It turned out that the MO decomposition occurred rough slowly in the first 30 min and a low conversion of ca. 26% was reached for 120 min (Figure S2). The extremely slow kinetics as compared with the highly active Fe2O3-S-P and Fe2O3-N-E thus suggested the heterogeneous nature of the decomposition reaction on the latter.
2.3. Origins of the divergent catalytic performance
2.3.1. Structure-performance relationships
The Fenton activity of iron-based materials can be influenced by many factors, which makes it difficult to discriminate the key activity descriptors. Among the suspected properties, the Fe2+ species, the oxygen vacancy, and redox properties might play important roles in influencing the activity. Therefore, tentative correlations were made between the initial reaction rates with the shares of Fe2+ and Ov derived from the XPS fitting, and the TFe (the temperature corresponding to the appearance of Fe0 phase from in situ PXRD, Figure 10). Noted that TFe was adopted to roughly reflect the redox properties of the iron oxides. The results demonstrated that there was no clear dependence of the initial activity on either the shares of Fe2+ or Ov, whereas increasing activity as a function of decreasing TFe was evidenced. Therefore, it seems that the redox propensity might be a reasonable activity descriptor in the Fenton chemistry.
2.3.2. Importance of the Fe@Fe3O4 interfaces
The detailed characterization of the iron oxides revealed that both Fe0 and Fe3O4 were formed on Fe2O3-S-P and Fe2O3-N-E, while only Fe3O4 existed on Fe2O3-Cl-P and Fe3O4-C. Thus, it is reasonable to speculate that the presence of Fe0 was crucial to the high Fenton activity. To further discriminate the MO decomposition performance of the different compositions of iron species, Fe2O3-S-P reduced in H2 at varying temperatures from 300-450 oC was conducted. The PXRD results confirmed that Fe2O3 was the major phase after 300 oC reduction (Figure 11). After reduction at 350 oC, Fe0 was the major phase with the coexistence of Fe3O4. After further increasing the temperature to 450 oC, Fe0 was the only phase.
Having deriving different compositions of the iron-based materials from Fe2O3-S-P, their activity in MO decomposition was evaluated (Figure 12). The results showed that the samples reduced at 350 and 450 oC displayed basically the same activity, whereas no appreciable conversion can be observed for the unreduced sample and after 300 oC reduction. Combining with the activity evaluation, it can be concluded that Fe0 or Fe0/Fe3O4 were responsible for the catalytic activity, but both Fe3O4 or Fe2O3, when used alone, were completely inactive in Fenton chemistry.
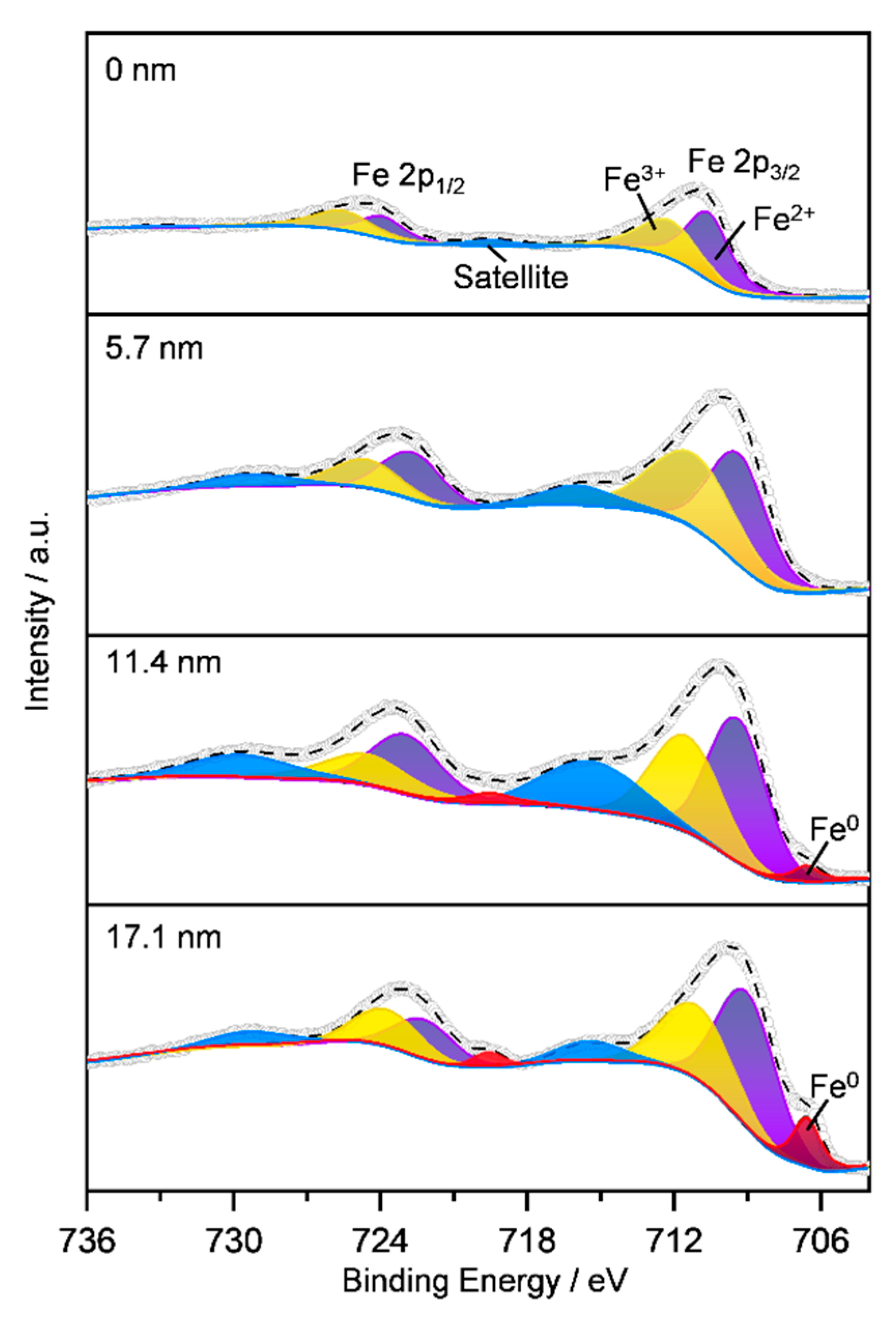
Although the iron oxide in Fe2O3-S-P was completely converted into Fe0 after 450 oC reduction based on PXRD, the exposure to air should easily lead to the formation of a thin oxide layer. It means for this sample, both Fe0 and Fe3O4 might co-exist, like in the case of the sample reduced at 350 oC. To proof the speculation, XPS with depth profile analysis were performed on Fe2O3-S-P after 450 oC and exposure to air (Figure 13). The fitting of the Fe 2p XPS spectra evidently revealed the exclusive presence of both Fe3+ and Fe2+ species at the outer surfaces, while the contribution of Fe0 emerged up to 11.4 nm depth and the share became larger subject to further sputtering. The results thus confirmed the formation of an iron oxide layer of ca. 11-17 nm thickness owing to the auto-oxidation of metallic iron. In addition, the previous TEM and PXRD results of Fe2O3-S-P after 350 oC reduction also indicated the presence of the Fe@Fe3O4 core-shell structure and a thickness of Fe3O4 layer of <24 nm. Therefore, the similar activity of Fe2O3-S-P subject to 350-400 oC reduction can be attributed to the presence of similar Fe@Fe3O4 core-shell structures. On the other hand, Fe2O3-N-E with both Fe0 and Fe3O4 as the major phases without the apparent core-shell structures exhibited the relatively lower activity as compared to Fe2O3-S-P. Albeit an interface between both phases was confirmed by HRTEM, no core-shell structures can be observed, plus the Fe3O4 particle sizes (ca. 54 nm) were much larger as compared with the layer thickness in Fe2O3-S-P. These factors might lead to reduced number of interface structures and thus the lower activity.
2.3.3. Discussions on the origin of superior activity
The radicals formation such as OH* and HO2* was generally acknowledged to be responsible for the MO decomposition in the Fenton chemistry. Indeed, the reaction was immediately jeopardized upon the addition of a radical trapper such as dimethyl sulfoxide (Figure S3), demonstrating the radical-induced nature of this reaction. Both Fe2O3-Cl-P-and Fe3O4-C showed comparable rich surface Fe2+ species and Ov but without the presence of ZVI. In contrast, Fe2O3-N-E containing ZVI but with a lower number of Ov also displayed inferior activity. These results hinted that the superior Fenton activity of Fe2O3-S-P with the Fe@Fe3O4 core-shell structures might be attributed to the abundance of both Fe0 and Ov. The rate-limiting step in the heterogeneous Fenton reaction is viewed as the formation of Fe2+ species that are responsible to generate highly active OH* through equation 1. The previous studies have proposed that Fe2+ species might be produced by the oxidation of ZVI by H2O2 or molecular oxygen in acidic conditions[39,40]. In addition, the studies on the Fe@Fe2O3 systems also suggested the excellent reducing properties of metallic iron to achieve a high Fe2+/Fe3+ redox cycling rate, wherein ZVI can directly inject two electrons to activate molecular oxygen[41,42]. On the other hand, the rich number of Ov has been demonstrated to be beneficial to the generation of OH*[43,44,45,46]. Li et al. [45] confirmed that H2O2 adsorbed on the electron-rich location readily underwent dissociation induced by Ov (equation 2). Chen et al.[43] reported the accelerated electron transfer process and threefold yield of OH* for the Ov-rich micro ZVI interface. The synergistic cooperation catalysis between ZVI and iron oxides was proposed in equation 3. Following this equation, the surface bonded Fe2+ species were generated between the iron oxides and Ov. The surface bonded Fe2+ species were suggested to be more reactive then ferrous ions in activating H2O2, thus avoiding the rate-limiting step in the classic Fenton reaction.
H2O2 + Fe2+ → Fe3+ + OH* + OH-
H2O2 + Ov → OH* + OH-
FexOy + Ov → e- + Fe2+(sur)
3. Materials and Methods
3.1. Synthesis of the iron oxides
All the reagents were obtained from commercial suppliers and used without further purification. Fe(NH4)2(SO4)2·6H2O(99.5%), Fe(NO3)3·9H2O and FeCl3·6H2O were purchased from Aladdin. Polyethylene glycol 400 (PEG-400), ethylene glycol (EG, ≥99.5%) and sodium carbonate (Na2CO3, ≥99.8%) were purchased from Sinopharm. Ammonium hydroxide solution (NH3·H2O,25%-28%) and Fe3O4-C (99.5%) were purchased from Macklin.
The other three iron oxides were prepared by a precipitation method. A solution containing Fe(NH4)2(SO4)2·6H2O (0.02 mol/L) and polyethylene glycol 400 (200 mL) was gradually heated to 120 °C under mechanical stirring and maintained at 120 °C for 1 h. Then a Na2CO3 aqueous solution (200 mL, 0.2 mol/L) was added gradually dropwise through a syringe pump at a rate of 5.5 mL/min. The mixture was aged at 120 °C for 1 h. The precipitate was washed with water and ethanol, dried at 120 °C for 12 h and calcined at 500 °C for 5 h. The solid obtained was denoted as Fe2O3-S-P. Fe2O3-N-E and Fe2O3-Cl-P were obtained following the similar precipitation methods by using Fe(NO3)3·9H2O and FeCl3·6H2O as the precursors, EG and PEG-400 as the solvents, and sodium carbonate concentrations of 0.02 and 0.2 M, respectively.
3.2. Material characterizations
The composition and crystalline structures of the catalysts was characterized by powder X-ray diffraction (PXRD) with an x'Pert3 Panalytical X-ray diffractometer (PANalytical, Almelo, The Netherlands) using Cu Kα radiation (λ = 0.154178 nm). The tube voltage and current were 40 kV and 40 mA, respectively. In situ PXRD patterns were recorded with Rigaku SmartLab-9 kW D/teX Ultra250 diffractometer using a Cu Kα radiation source operated at 40 kV and 200 mA. The powder samples were spread into a high-temperature chamber and the diffraction patterns were recorded at 2θ of 30–90o with a scanning rate of 10o min-1 at room temperature. Then, the sample was heated to 823 K successively at a rate of 10 oC min-1 under 10% H2/He flow of 30 mL min-1.
Scanning electron microscopy (SEM) was conducted on a Quanta 200F instrument operating at 10 kV and 50 pA. The powder sample was dispersed in dry form onto fresh carbon paint deposited on an aluminum holder.
Transmission electron microscopy (TEM) images were taken on a FEI Tecnai G2F 20 microscopy operating at 120 kV. The specimen was prepared by ultrasonically dispersing the powder sample in ethanol, depositing droplets of the suspensions onto carbon-coated copper or gold grids, and drying in air.
X-ray photoelectron spectroscopy (XPS) analysis of the materials was performed on a Thermo ESCALAB 250Xi spectrometer using a 15 kV Al Kα X-ray source as a radiation source. The binding energy was calibrated using the C 1s peak (284.6 eV) as the reference. For the depth profile analysis, the samples were sputtered with Ar ions.
3.3. Activity evaluation of the iron-based materials in decomposition of methyl orange
The activity evaluation of the developed iron-based materials in the decomposition of methyl orange (MO) was performed in a 250 mL three-necked round flask, equipped with a magnetic stirrer and a temperature thermometer. The temperature was controlled by a pre-heated water bath. For a typical reaction, 100 mL of aqueous MO solutions of 100 or 500 μg/mL were charged into the flask reactor. The pH of the solution was then adjusted to 3.0 by diluted HCl solution. After that, a certain amount of the solid catalysts was added and the suspension was stirred for 30 min to reach the adsorption-desorption equilibrium. Finally, the reaction was initiated upon the addition of a diluted H2O2 solution, and the concentration of MO was continuously monitored by taking a small portion of the liquid samples for analysis on a UV-vis spectrometer (TU-1810, Beijing Puxi General Motors Instrument Co.) at λmax = 464 nm (see Figure S4 for more details).
4. Conclusions
We have developed iron oxides of four different model systems through the conventional precipitation method by combining with subsequent H2 reduction treatment. These materials possess very different compositions and structural features, including the Fe@Fe3O4 core-shell structures with rich oxygen vacancy (Fe2O3-S-P), the mixtures of Fe0 and Fe3O4 particles with interface structures but low oxygen vacancy (Fe2O3-N-E), and Fe3O4-dominated nanoparticles of different sizes and rich in oxygen vacancy (Fe2O3-Cl-P and Fe3O4-C). These materials when used as Fenton catalysts in the methyl orange decomposition displayed significantly different activity with the order of Fe2O3-S-P > Fe2O3-N-E > Fe3O4-C > Fe2O3-Cl-P. The passivate layer of metallic iron of up to ca. 24 nm can lead to the similar Fe@Fe3O4 core-shell structures with excellent activity. This study further demonstrated that the numbers of Fe2+ or oxygen vacancy alone might not be the only activity descriptors for the iron-based materials, while the formation of metallic iron coupled with the rich oxygen vacancy can significantly boost the Fenton activity, thus highlighting the necessity of more in-depth analyses of the role of the hybrid Fe@FexOy.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Figure S1: title; Table S1: title; Video S1: title.
Author Contributions
Conceptualization, X. Mou and W. Kan; investigation, R. Zheng and R. Tan; XRD and XPS, Y. Lv; TEM, R. Lin; writing, review and editing, X. Mou, P. Fang and W. Kan. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Acknowledgments
The authors would like to thank Deiyou Chen from Shiyanjia Lab (www.shiyanjia.com) for TEM measurements.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Tran, N.H.; Reinhard, M.; Gin, K.Y.-H. Occurrence and fate of emerging contaminants in municipal wastewater treatment plants from different geographical regions-a review. Wat. Res. 2018, 133, 182–207. [Google Scholar] [CrossRef] [PubMed]
- Hodges, B.C.; Cates, E.L.; Kim, J.H. Challenges and prospects of advanced oxidation water treatment processes using catalytic nanomaterials. Nat. Nanotechnol. 2018, 13, 642–650. [Google Scholar] [CrossRef] [PubMed]
- Boczkaj, G.; Fernandes, A. Wastewater treatment by means of advanced oxidation processes at basic pH conditions: A review. Chem. Eng. J. 2017, 320, 608–633. [Google Scholar] [CrossRef]
- Bokare, A.D.; Choi, W. Review of iron-free Fenton-like systems for activating H2O2 in advanced oxidation processes. J. Hazard. Mater. 2014, 275, 121–135. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Zhu, R.; Xi, Y.; Zhu, J.; Zhu, G.; He, H. Strategies for enhancing the heterogeneous Fenton catalytic reactivity: A review. Appl. Catal. B 2019, 255. [Google Scholar] [CrossRef]
- He, J.; Yang, X.; Men, B.; Wang, D. Interfacial mechanisms of heterogeneous Fenton reactions catalyzed by iron-based materials: A review. J. Environ. Sci. 2016, 39, 97–109. [Google Scholar] [CrossRef] [PubMed]
- Rahim Pouran, S.; Abdul Raman, A.A.; Wan Daud, W.M.A. Review on the application of modified iron oxides as heterogeneous catalysts in Fenton reactions. J. Clean. Prod. 2014, 64, 24–35. [Google Scholar] [CrossRef]
- Zepp, R.G.; Faust, B.C.; Hoigne, J. Hydroxyl radical formation in aqueous reactions (pH 3-8) of iron (II) with hydrogen peroxide: the photo-Fenton reaction. Environ. Sci. Technol. 1992, 26, 313–319. [Google Scholar] [CrossRef]
- Oller, I.; Malato, S.; Sánchez-Pérez, J.; Gernjak, W.; Maldonado, M.; Pérez-Estrada, L.; Pulgarín, C. A combined solar photocatalytic-biological field system for the mineralization of an industrial pollutant at pilot scale. Catal. Today 2007, 122, 150–159. [Google Scholar] [CrossRef]
- Malato, S.; Fernández-Ibáñez, P.; Maldonado, M.I.; Blanco, J.; Gernjak, W. Decontamination and disinfection of water by solar photocatalysis: recent overview and trends. Catal. Today 2009, 147, 1–59. [Google Scholar] [CrossRef]
- Brillas, E.; Sirés, I.; Oturan, M.A. Electro-Fenton process and related electrochemical technologies based on Fenton’s reaction chemistry. Chem. Rev. 2009, 109, 6570–6631. [Google Scholar] [CrossRef]
- Adewuyi, Y.G. Sonochemistry in environmental remediation. 1. Combinative and hybrid sonophotochemical oxidation processes for the treatment of pollutants in water. Environ. Sci. Technol. 2005, 39, 3409–3420. [Google Scholar] [CrossRef]
- Cai, W.; Chen, F.; Shen, X.; Chen, L.; Zhang, J. Enhanced catalytic degradation of AO7 in the CeO2–H2O2 system with Fe3+ doping. Appl. Catal. B 2010, 101, 160–168. [Google Scholar] [CrossRef]
- Bokare, A.D.; Choi, W. Advanced oxidation process based on the Cr (III)/Cr (VI) redox cycle. Environ. Sci. Technol. 2011, 45, 9332–9338. [Google Scholar] [CrossRef]
- Gabriel, J.; Baldrian, P.; Verma, P.; Cajthaml, T.; Merhautová, V.; Eichlerová, I.; Stoytchev, I.; Trnka, T.; Stopka, P.; Nerud, F. Degradation of BTEX and PAHs by Co (II) and Cu (II)-based radical-generating systems. Appl. Catal. B 2004, 51, 159–164. [Google Scholar] [CrossRef]
- Gabriel, J.; Shah, V.; Nesměrák, K.; Baldrian, P.; Nerud, F. Degradation of polycyclic aromatic hydrocarbons by the copper (II)-hydrogen peroxide system. Folia Microbiol. 2000, 45, 573–575. [Google Scholar] [CrossRef]
- Han, Y.-F.; Chen, F.; Zhong, Z.; Ramesh, K.; Chen, L.; Jian, D.; Ling, W.W. Complete oxidation of low concentration ethanol in aqueous solution with H2O2 on nanosized Mn3O4/SBA-15 catalyst. Chem. Eng. J. 2007, 134, 276–281. [Google Scholar] [CrossRef]
- Hu, Z.; Leung, C.-F.; Tsang, Y.-K.; Du, H.; Liang, H.; Qiu, Y.; Lau, T.-C. A recyclable polymer-supported ruthenium catalyst for the oxidative degradation of bisphenol A in water using hydrogen peroxide. New J. Chem. 2011, 35, 149–155. [Google Scholar] [CrossRef]
- Mizuno, N.; Yamaguchi, K.; Kamata, K. Epoxidation of olefins with hydrogen peroxide catalyzed by polyoxometalates. Coord. Chem. Rev. 2005, 249, 1944–1956. [Google Scholar] [CrossRef]
- Huang, J.; Jones, A.; Waite, T.D.; Chen, Y.; Huang, X.; Rosso, K.M.; Kappler, A.; Mansor, M.; Tratnyek, P.G.; Zhang, H. Fe(II) Redox Chemistry in the Environment. Chem. Rev. 2021, 121, 8161–8233. [Google Scholar] [CrossRef]
- Danielsen, K.M.; Hayes, K.F. pH Dependence of carbon tetrachloride reductive dechlorination by magnetite. Environ. Sci. Technol. 2004, 38, 4745–4752. [Google Scholar] [CrossRef]
- Gorski, C.A.; Nurmi, J.T.; Tratnyek, P.G.; Hofstetter, T.B.; Scherer, M.M. Redox Behavior of Magnetite: Implications for contaminant reduction. Environ. Sci. Technol. 2010, 44, 55–60. [Google Scholar] [CrossRef]
- Zhong, Y.; Yu, L.; Chen, Z.F.; He, H.; Ye, F.; Cheng, G.; Zhang, Q. Microwave-Assisted Synthesis of Fe3O4 Nanocrystals with Predominantly Exposed Facets and Their Heterogeneous UVA/Fenton Catalytic Activity. ACS Appl. Mater. Interface. 2017, 9, 29203–29212. [Google Scholar] [CrossRef]
- Jaramillo-Páez, C.; Navío, J.A.; Hidalgo, M.; Bouziani, A.; El Azzouzi, M. Mixed α-Fe2O3/Bi2WO6 oxides for photoassisted hetero-Fenton degradation of methyl orange and phenol. J. Photochem. Photobiol. A 2017, 332, 521–533. [Google Scholar] [CrossRef]
- Ma, Y.; Wang, B.; Wang, Q.; Xing, S. Facile synthesis of α-FeOOH/γ-Fe2O3 by a pH gradient method and the role of γ-Fe2O3 in H2O2 activation under visible light irradiation. Chem. Eng. J. 2018, 354, 75–84. [Google Scholar] [CrossRef]
- Krumina, L.; Lyngsie, G.; Tunlid, A.; Persson, P. Oxidation of a dimethoxyhydroquinone by ferrihydrite and goethite nanoparticles: iron reduction versus surface catalysis. Environ. Sci. Technol. 2017, 51, 9053–9061. [Google Scholar] [CrossRef]
- Su, S.; Liu, Y.; Liu, X.; Jin, W.; Zhao, Y. Transformation pathway and degradation mechanism of methylene blue through β-FeOOH@ GO catalyzed photo-Fenton-like system. Chemosphere 2019, 218, 83–92. [Google Scholar] [CrossRef]
- He, D.; Chen, Y.; Situ, Y.; Zhong, L.; Huang, H. Synthesis of ternary g-C3N4/Ag/γ-FeOOH photocatalyst: an integrated heterogeneous Fenton-like system for effectively degradation of azo dye methyl orange under visible light. Appl. Surf. Sci. 2017, 425, 862–872. [Google Scholar] [CrossRef]
- Chen, Z.X.; Jin, X.Y.; Chen, Z.; Megharaj, M.; Naidu, R. Removal of methyl orange from aqueous solution using bentonite-supported nanoscale zero-valent iron. J. Colloid. Interface. Sci. 2011, 363, 601–607. [Google Scholar] [CrossRef]
- Xie, S.; Huang, P.; Kruzic, J.J.; Zeng, X.; Qian, H. A highly efficient degradation mechanism of methyl orange using Fe-based metallic glass powders. Sci. Rep. 2016, 6, 21947. [Google Scholar] [CrossRef]
- Yuan, N.; Zhang, G.; Guo, S.; Wan, Z. Enhanced ultrasound-assisted degradation of methyl orange and metronidazole by rectorite-supported nanoscale zero-valent iron. Ultrason. Sonochem. 2016, 28, 62–68. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Ai, Z.; Ho, W.; Zhang, L. Core–shell Fe–Fe2O3 nanostructures as effective persulfate activator for degradation of methyl orange. Sep. Purif. Technol. 2013, 108, 159–165. [Google Scholar] [CrossRef]
- Huang, Q.; Cao, M.; Ai, Z.; Zhang, L. Reactive oxygen species dependent degradation pathway of 4-chlorophenol with Fe@ Fe2O3 core–shell nanowires. Appl. Catal. B 2015, 162, 319–326. [Google Scholar] [CrossRef]
- Shen, W.; Lin, F.; Jiang, X.; Li, H.; Ai, Z.; Zhang, L. Efficient removal of bromate with core-shell Fe@ Fe2O3 nanowires. Chem. Eng. J. 2017, 308, 880–888. [Google Scholar] [CrossRef]
- Shi, J.; Ai, Z.; Zhang, L. Fe@ Fe2O3 core-shell nanowires enhanced Fenton oxidation by accelerating the Fe (III)/Fe (II) cycles. Wat. Res. 2014, 59, 145–153. [Google Scholar] [CrossRef]
- Liu, W.; Ai, Z.; Cao, M.; Zhang, L. Ferrous ions promoted aerobic simazine degradation with Fe@Fe2O3 core–shell nanowires. Appl. Catal. B 2014, 150, 1–11. [Google Scholar] [CrossRef]
- Ai, Z.; Gao, Z.; Zhang, L.; He, W.; Yin, J.J. Core–shell structure dependent reactivity of Fe@Fe2O3 nanowires on aerobic degradation of 4-chlorophenol. Environ. Sci. Technol. 2013, 47, 5344–5352. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Ai, Z.; Zhang, L. Fe@Fe2O3 core-shell nanowires enhanced Fenton oxidation by accelerating the Fe(III)/Fe(II) cycles. Wat. Res. 2014, 59, 145–153. [Google Scholar] [CrossRef]
- Costa, R.C.; Moura, F.C.; Ardisson, J.; Fabris, J.; Lago, R. Highly active heterogeneous Fenton-like systems based on Fe0/Fe3O4 composites prepared by controlled reduction of iron oxides. Appl. Catal. B 2008, 83, 131–139. [Google Scholar] [CrossRef]
- Leupin, O.X.; Hug, S.J. Oxidation and removal of arsenic (III) from aerated groundwater by filtration through sand and zero-valent iron. Wat. Res. 2005, 39, 1729–1740. [Google Scholar] [CrossRef]
- Wu, H.; Ai, Z.; Zhang, L. Anoxic and oxic removal of humic acids with Fe@Fe2O3 core–shell nanowires: A comparative study. Wat. Res. 2014, 52, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Cao, M.; Ai, Z.; Zhang, L. Dramatically enhanced aerobic atrazine degradation with Fe@ Fe2O3 core–shell nanowires by tetrapolyphosphate. Environ. Sci. Technol. 2014, 48, 3354–3362. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Su, J.; Meng, Y.; Yu, M.; Zheng, M.; Sun, Y.; Xi, B. Oxygen vacancy promoted heterogeneous Fenton-like degradation of sulfamethazine by chlorine-incorporated micro zero-valent iron. Chem. Eng. J. 2023, 463. [Google Scholar] [CrossRef]
- Deng, Y.; Tian, X.; Shen, G.; Gao, Y.; Lin, C.; Ling, L.; Cheng, F.; Liao, S.; Zhang, S. Coupling hollow Fe3O4 nanoparticles with oxygen vacancy on mesoporous carbon as a high-efficiency ORR electrocatalyst for Zn-air battery. J. Colloid. Interface. Sci. 2020, 567, 410–418. [Google Scholar] [CrossRef]
- Li, H.; Shang, J.; Yang, Z.; Shen, W.; Ai, Z.; Zhang, L. Oxygen vacancy associated surface Fenton chemistry: surface structure dependent hydroxyl radicals generation and substrate dependent reactivity. Environ. Sci. Technol. 2017, 51, 5685–5694. [Google Scholar] [CrossRef]
- Ruiz Puigdollers, A.; Schlexer, P.; Tosoni, S.; Pacchioni, G. Increasing oxide reducibility: the role of metal/oxide interfaces in the formation of oxygen vacancies. ACS Catal. 2017, 7, 6493–6513. [Google Scholar] [CrossRef]
Figure 1.
Schematic illustration of the synthesis procedures of the iron oxides, accompanied with the sample codes and structural characteristics.
Figure 1.
Schematic illustration of the synthesis procedures of the iron oxides, accompanied with the sample codes and structural characteristics.
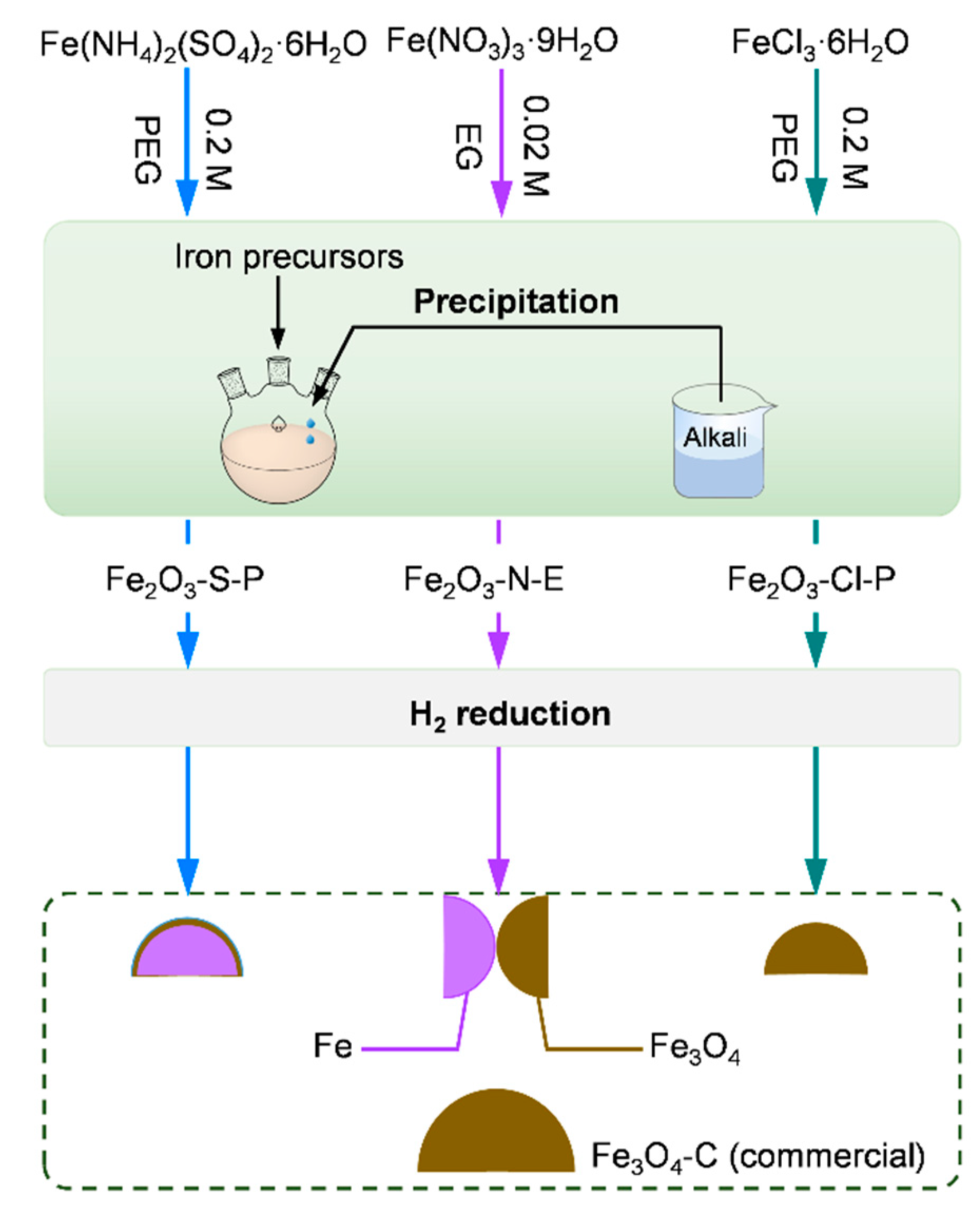
Figure 2.
The PXRD patterns of the as-prepared iron oxides. Vertical lines are the reference standard.
Figure 2.
The PXRD patterns of the as-prepared iron oxides. Vertical lines are the reference standard.
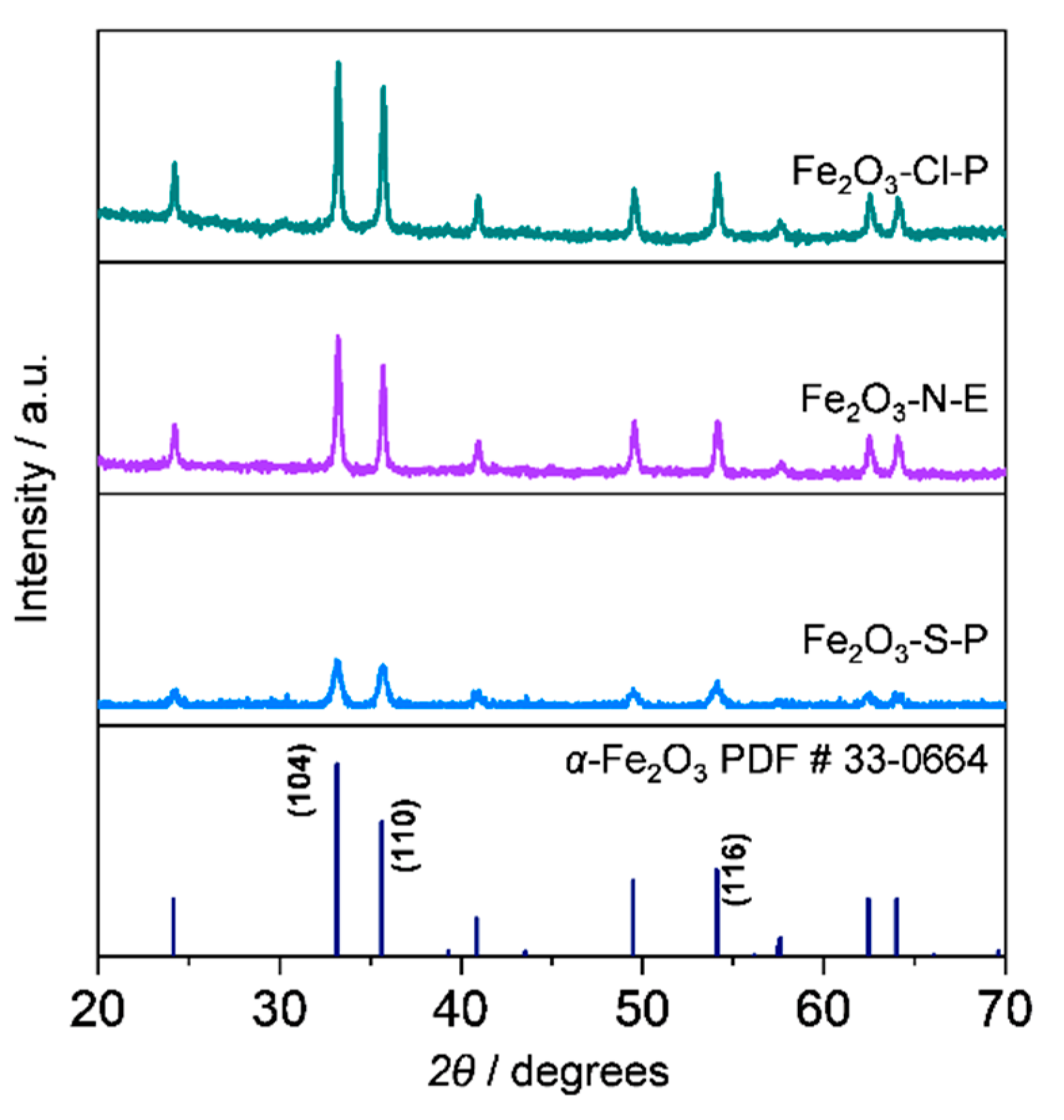
Figure 3.
The SEM (top) and TEM (bottom) images of the as-prepared iron oxides.

Figure 4.
The PXRD patterns of the iron oxides after 350 oC reduction. Vertical lines are the reference standards.
Figure 4.
The PXRD patterns of the iron oxides after 350 oC reduction. Vertical lines are the reference standards.
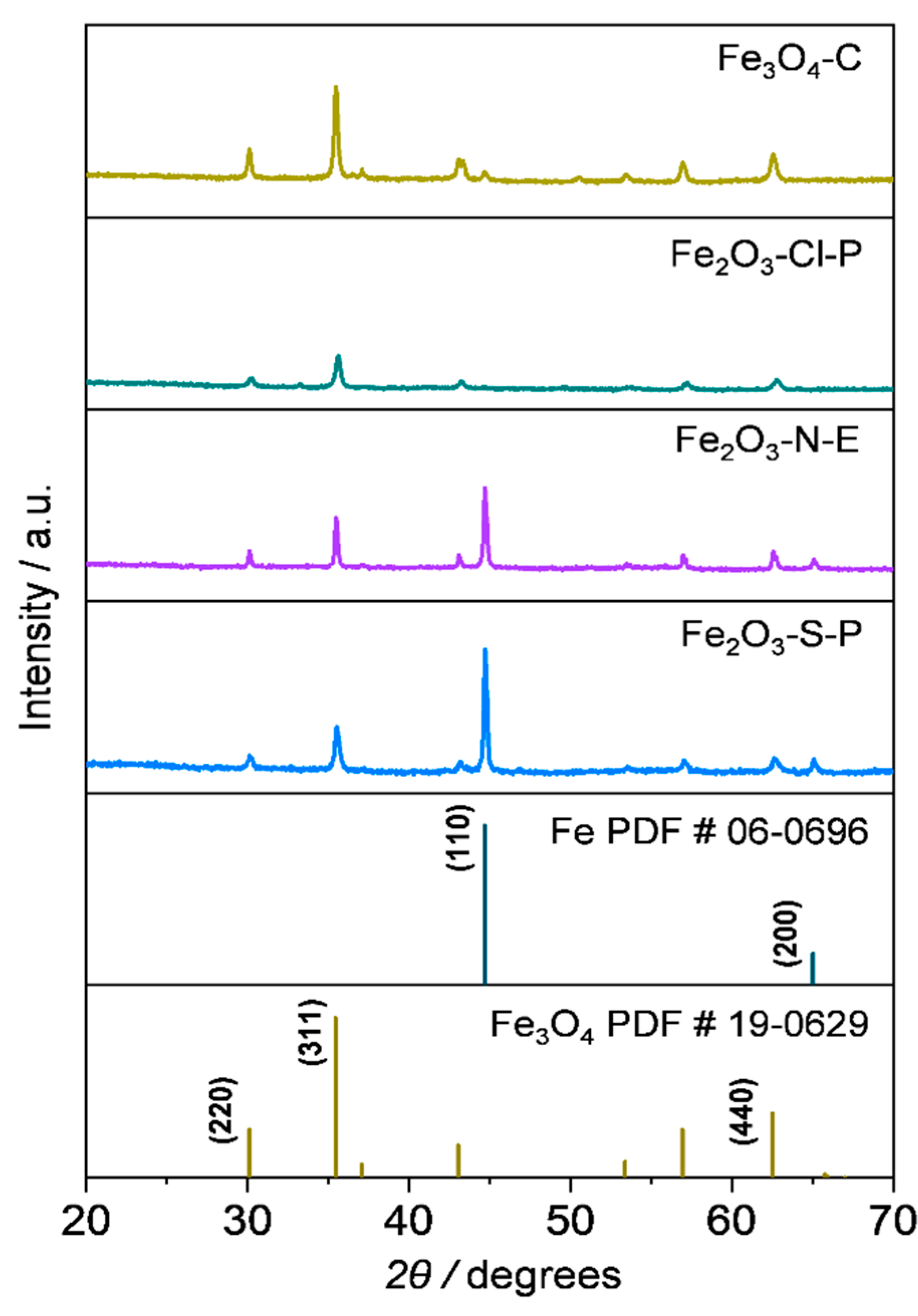
Figure 5.
The TEM images of the iron oxides after 350 oC reduction. The inset in top-left shows the core-shell structures in Fe2O3-S-P.
Figure 5.
The TEM images of the iron oxides after 350 oC reduction. The inset in top-left shows the core-shell structures in Fe2O3-S-P.

Figure 6.
The HRTEM images of the iron oxides after 350 oC reduction.
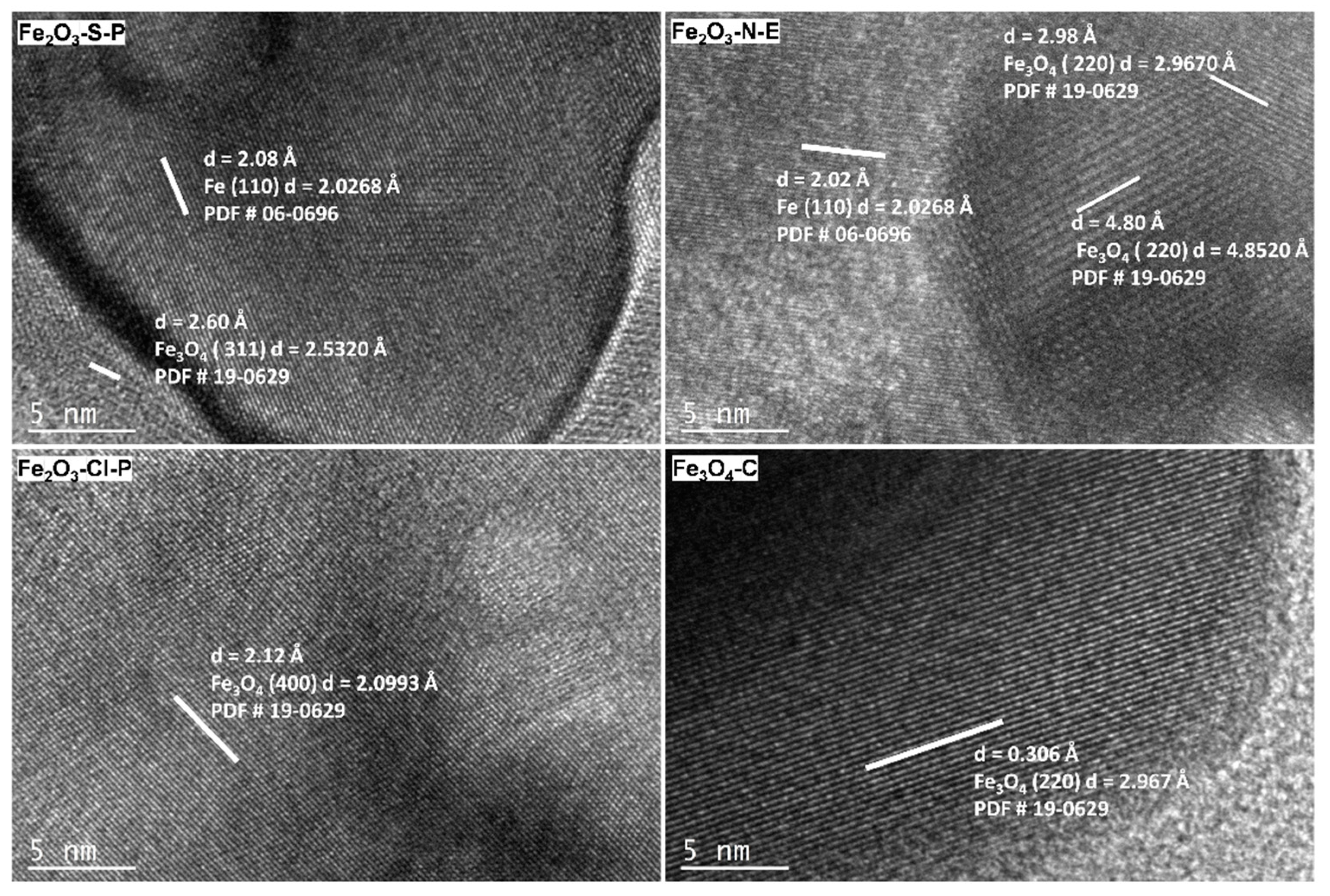
Figure 7.
The in situ PXRD patterns of the iron oxides in flowing H2.
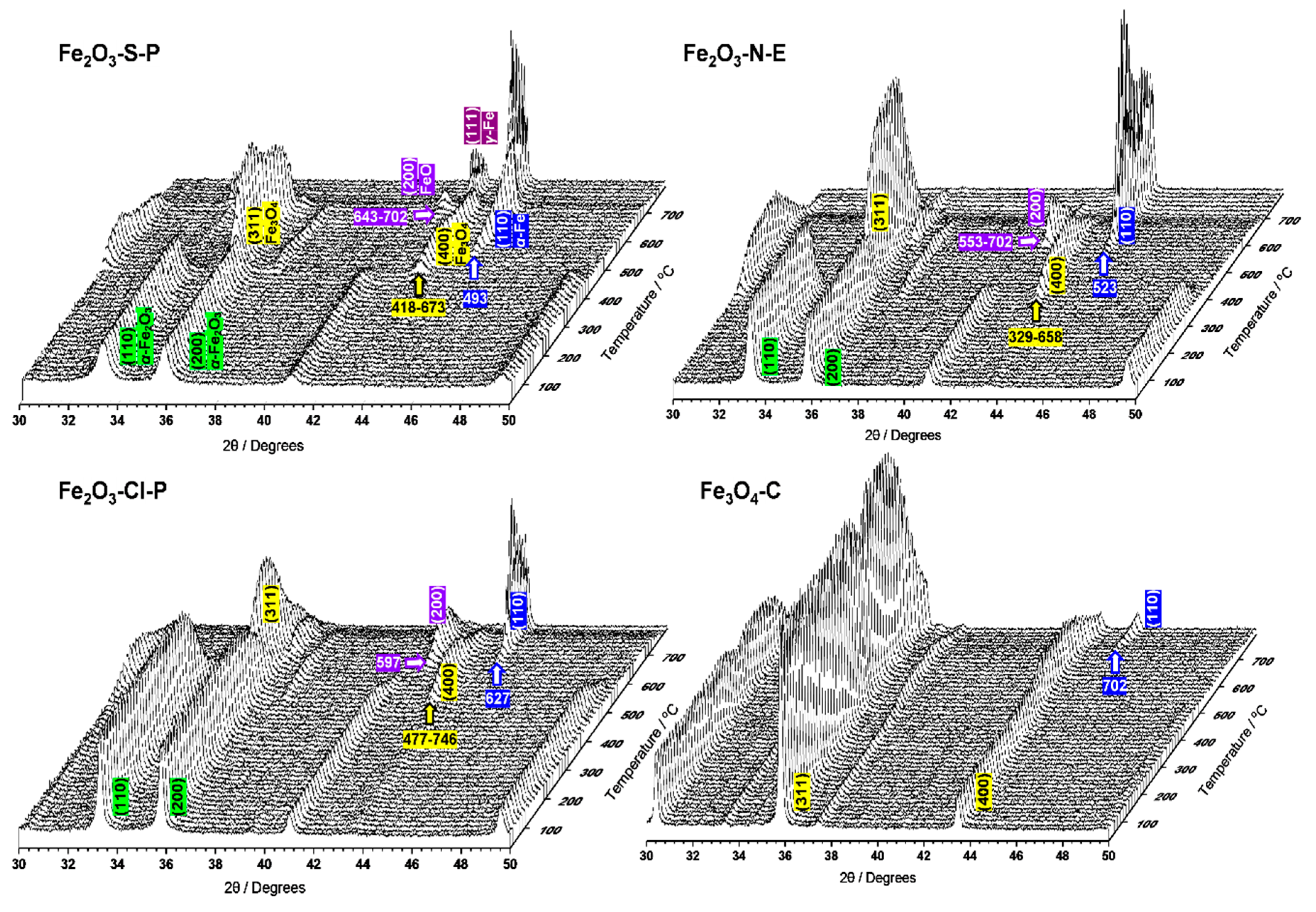
Figure 8.
The Fe 2p and O 1s XPS profiles of the iron oxides after 350 oC reduction. Ov, oxygen vacancy; OL, lattice oxygen.
Figure 8.
The Fe 2p and O 1s XPS profiles of the iron oxides after 350 oC reduction. Ov, oxygen vacancy; OL, lattice oxygen.
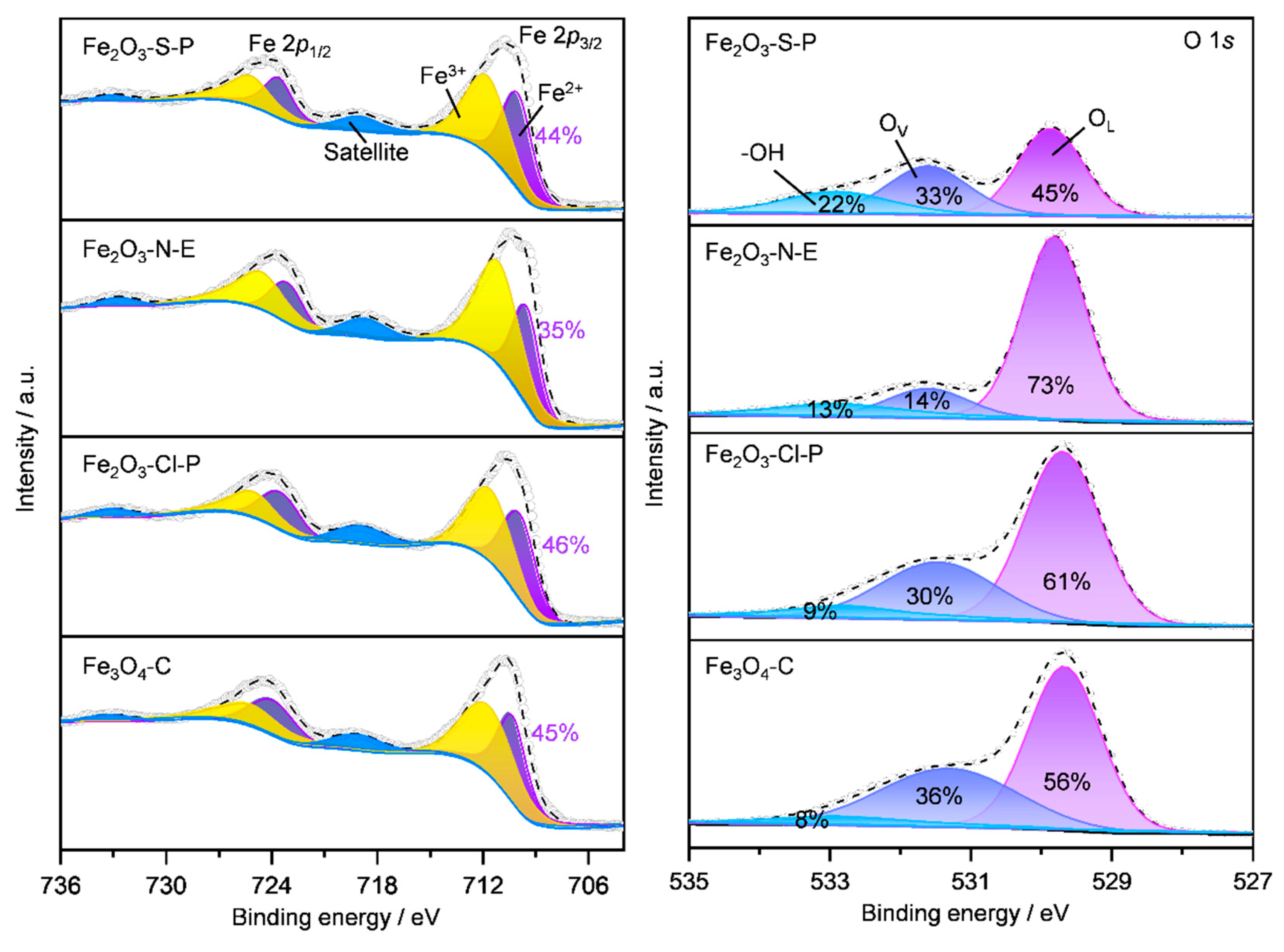
Figure 9.
(a,b) The MO decomposition activity as a function of time on different iron oxides. (c) Comparison on the initial activity of different iron oxides. Reaction conditions: wcat = 5 mg (350 oC reduced); pH = 3, V = 100 mL, CH2O2= 20 mM, a, CMO = 100 μg/mL, b, CMO = 500 μg/mL. The first three data points before the addition of H2O2 correspond the freshly prepared solution, the solutions after the adjustment of pH and after 30 min stirring, respectively.
Figure 9.
(a,b) The MO decomposition activity as a function of time on different iron oxides. (c) Comparison on the initial activity of different iron oxides. Reaction conditions: wcat = 5 mg (350 oC reduced); pH = 3, V = 100 mL, CH2O2= 20 mM, a, CMO = 100 μg/mL, b, CMO = 500 μg/mL. The first three data points before the addition of H2O2 correspond the freshly prepared solution, the solutions after the adjustment of pH and after 30 min stirring, respectively.
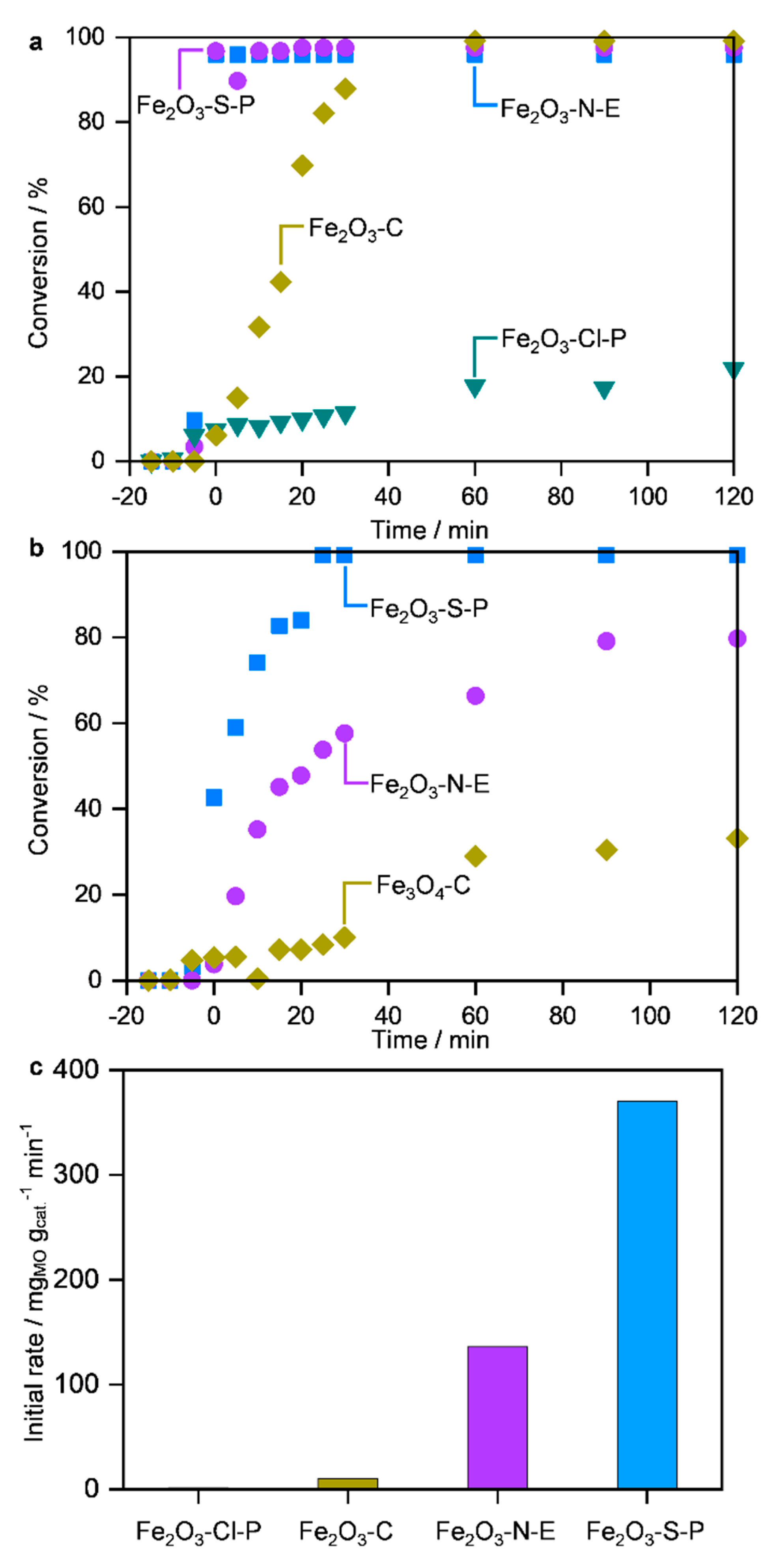
Figure 10.
The initial reaction rates of different iron oxides as a function of (a) surface Fe2+ ratio, (b) oxygen vacancy, and (c) TFe (the temperature corresponding to the appearance of Fe0 phase from in situ PXRD).
Figure 10.
The initial reaction rates of different iron oxides as a function of (a) surface Fe2+ ratio, (b) oxygen vacancy, and (c) TFe (the temperature corresponding to the appearance of Fe0 phase from in situ PXRD).
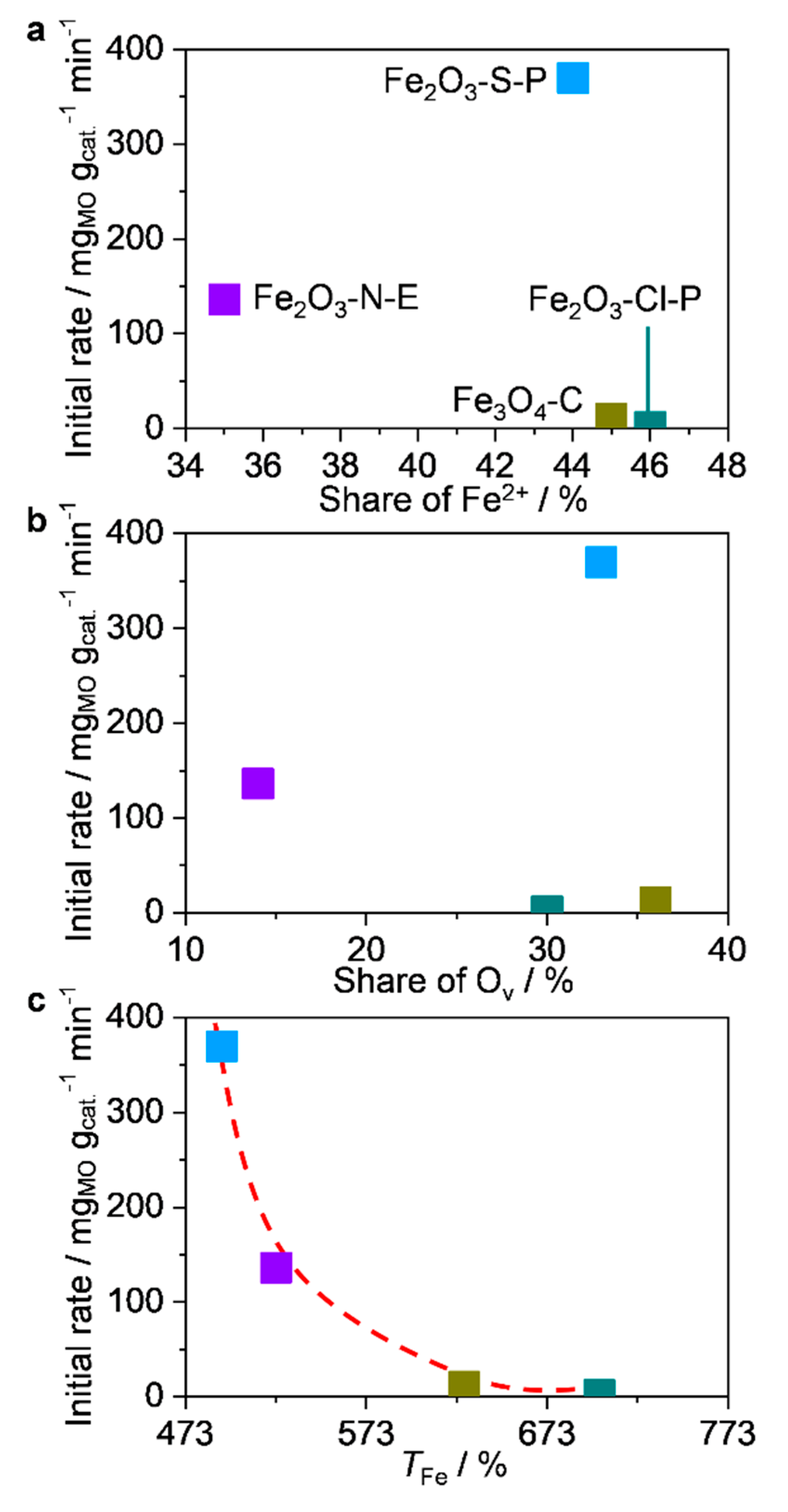
Figure 11.
The PXRD patterns of Fe2O3-S-P after reduction in H2 at 300-450 oC. Vertical lines are the reference standards.
Figure 11.
The PXRD patterns of Fe2O3-S-P after reduction in H2 at 300-450 oC. Vertical lines are the reference standards.
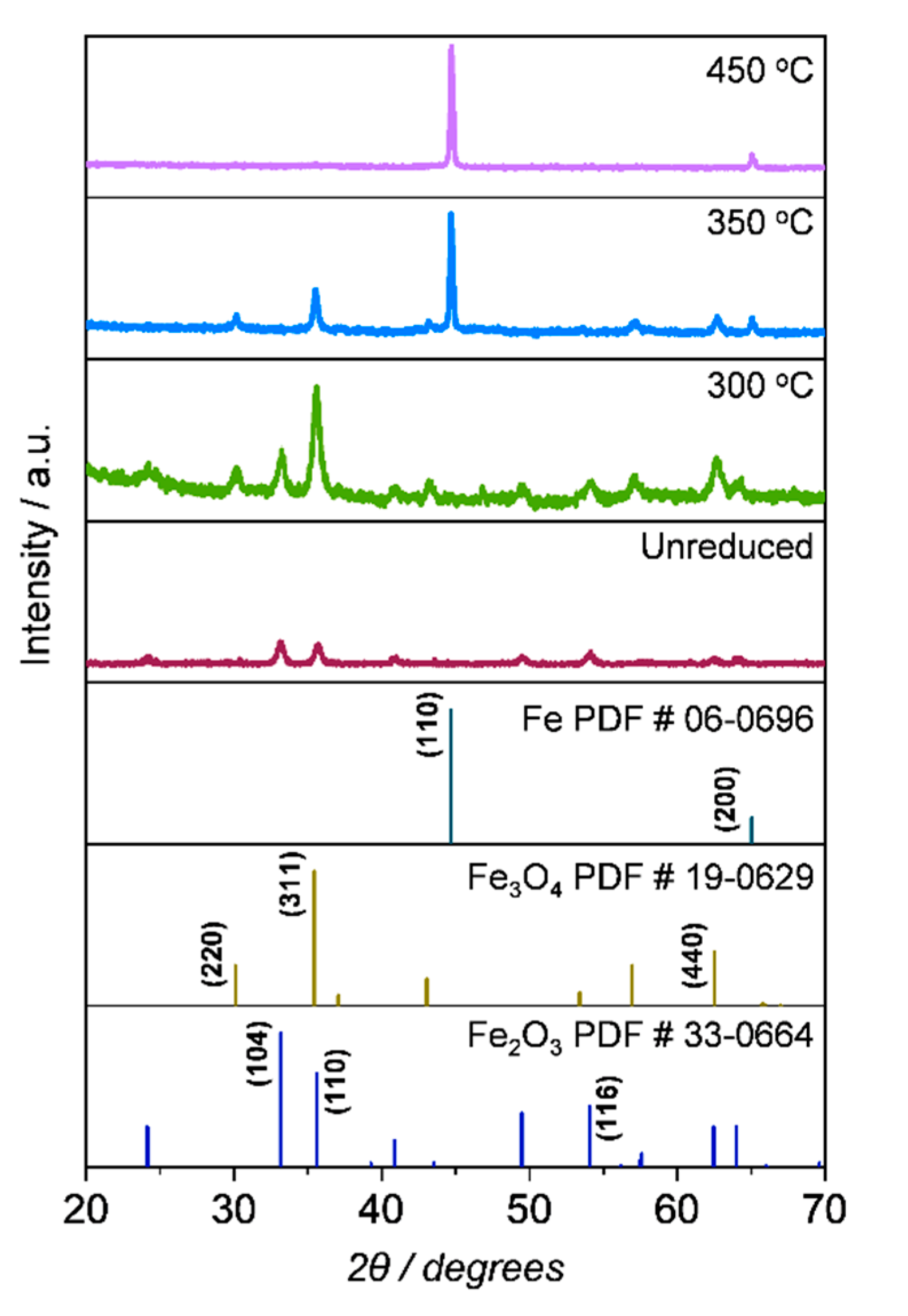
Figure 12.
The MO decomposition activity as a function of time on Fe2O3-S-P subject to H2 reduction at different temperatures. Reaction conditions: wcat = 5 mg; pH = 3, V = 100 mL, CH2O2= 20 mM, CMO = 500 μg/mL. The first three data points before the addition of H2O2 correspond the freshly prepared solution, the solutions after the adjustment of pH and after 30 min stirring, respectively.
Figure 12.
The MO decomposition activity as a function of time on Fe2O3-S-P subject to H2 reduction at different temperatures. Reaction conditions: wcat = 5 mg; pH = 3, V = 100 mL, CH2O2= 20 mM, CMO = 500 μg/mL. The first three data points before the addition of H2O2 correspond the freshly prepared solution, the solutions after the adjustment of pH and after 30 min stirring, respectively.

Figure 13.
The Fe 2p XPS depth profiles of Fe2O3-S-P after 450 oC reduction.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



