
Submitted:
26 May 2023
Posted:
29 May 2023
You are already at the latest version
Abstract
(1) Background: Breast cancer is the second leading cause of cancer death among women. The accurate prediction of survival intervals will help physicians make informed decisions on treatment strategies or the use of palliative care for patients; (2) Methods: The gene expression is predictive and correlates to patient prognosis. To establish a reliable prediction tool, we collected the RNA-seq data of breast cancer patients, a total of 1187 RNA-seq data (median age 58 years), in FPKM format from the TCGA database. Among them, 144 RNA-seq data with date of death information was selected to establish the SaBrcada-AD dataset. We first normalized the SaBrcada-AD dataset to transcripts per million (TPM) to build survival prediction model SaBrcada. After normalization and dimension raising, the differential gene expression data were used for testing eight different deep learning architectures. Among them, GoogLeNet performed the best. Considering the effect of age on prognosis, we examined all ages between the lower and upper quartiles of patient age for a stratified random sampling test; (3) Results: Stratifying by age based on a cut-off of 61 years of age improved the accuracy of SaBrcada compared to previous findings, resulting in an accuracy of 0.798. We also built a free website tool to provide 5 kinds of predicted survival period information for clinician reference; (4) Conclusions: We established a breast cancer survival analysis prediction model, SaBrcada, and a website tool with the same name. Through this highly reliable survival analysis model and website tool, information on survival intervals will be provided for clinicians as part of precision medicine.
Keywords:
breast cancer
; deep learning
; survival analysis
; data dimension raising
; age stratification
1. Introduction
Breast cancer is the most common cancer in women [1]. In 2020, approximately 2.3 million female breast cancer patients were diagnosed, accounting for 11.7% of new cancer cases. Breast cancer has not only become the main cause of global cancer but is also the fifth leading cause of cancer deaths worldwide, accounting for 1 in 6 cancer deaths [2,3]. To make matters worse, it has been predicted that the worldwide incidence of breast cancer is rising and that approximately 3.2 million new cases of female breast cancer will be diagnosed per year by 2050. These numbers indicate the urgent need for prevention and treatment strategies for breast cancer. Breast cancer commonly occurs in ducts or lobules. In addition to invading the original organs (breasts), malignant breast cancer has the ability to metastasize to distant organs such as bones, lungs, liver, and brain [4], which can lead to disease progression and eventually death in severe cases. Therefore, researchers continue to search for breakthroughs in the diagnosis, treatment and palliative care of breast cancer. Especially in palliative care, reliable and accurate prognostic prediction plays a key role in decision-making regarding medical strategies [5].
Medical treatments should be decided based on the patient's goals and expected survival time, the potential benefits and risks of treatment, and the effects on quality of life. Therefore, a comprehensive consideration of these factors determines treatment choices [6]. To predict patient survival time, many features, including pathogenesis, gene mutation, gene expression, clinical data, treatment, and general health, are typically considered for prognostic predictions [7,8]. Therefore, multiple predictors will be used in the model design and data analysis to determine the important features of the prognostic model. To date, researchers have proposed different combinations of predictors for survival analysis or death probability scoring or when developing prediction tools or analysis platforms for prognosis. These tools are often called prognostic models, predictive models, or risk scores [9,10,11,12,13,14,15,16]. Increasing the accuracy of these prognostic models or risk scores can help patients in making medical treatment decisions and providing more reliable survival analyses. In the postgenomic era, the significant features are not limited to clinical information, and the gene expression profiles of patients are also a crucial factor affecting prognosis [17,18,19].
To analyze gene expression, protein-coding RNAs (mRNAs) and noncoding RNAs, including long noncoding RNAs (lncRNAs), snRNAs, rRNAs, tRNAs, and microRNAs (miRNAs), were considered as candidates [20,21,22,23]. With the launch of the Human Genome Project [24] and the advancement of next-generation sequencing technologies, more high-throughput RNA-seq data from cancer patients has become available for bioinformatics analyses [25]. However, the analysis of such large datasets has often previously been limited by hardware capabilities [26]. With advancements of hardware and the development of deep learning architecture, more studies have applied deep learning from the information domain to bioinformatics [27] and hope to use the characteristics of deep learning to learn and extract features from genes or RNA-seq data to train and build models [28,29,30]. Compared with the complexity and diversity of genomic features, the number of samples from cancer patients from which RNA-seq data are available is limited. When the number of features is larger than the number of samples, model-overfitting tends to occur, which will reduce the accuracy of prediction in test data [31]. In addition, limited availability of clinical data also affects the effectiveness of deep learning. The hospital’s inability to actively track patients leads to loss to follow-up and censored death times for some patients. This incomplete clinical information may be the main limitation of cancer prognosis prediction [32]. For example, in the TCGA-BRCA database, the most common event date recorded is the last follow up date, not the date of death of the patient. This may be the key factor affecting the accuracy of previous studies. Therefore, we excluded this kind of data to improve the accuracy of the prediction model and then used data dimension raising and age stratification strategies to build a breast cancer patient survival analysis model SaBrcada by deep learning.
First, we downloaded the RNA-Seq and clinical data from the TCGA-BRCA database and conducted data screening. TCGA-BRCA provides the RNA-Seq data in fragments per kilobase per million (FPKM); FPKM is applicable to paired-end RNA-seq experiments only. As third-generation sequencing technologies have developed, such as single-molecule real-time sequencing (SMRT) and Oxford Nanopore’s technology, a widely applicable normalization method for different sequencing platforms is needed for survival analysis model construction. Transcripts per million (TPM) represents the relative expression level of a transcript, and the sum of all TPM values is a million in all samples. In principle, TPM should be comparable between samples; thus, we normalized the gene expression data from FPKM into TPM. Considering the correlation among gene expression levels, deep learning was selected for model construction. To process the data for CNN learning, we used a dimension raising strategy to raise the gene expression data into a matrix and then subtracted the data in pairs to generate a differential gene expression image (survival analysis image). We developed a survival analysis model by using a convolutional neural network with 8 different architectures. Among them, GoogLeNet exhibited the best performance. Patient age was also reported to be an important feature that affects survival time [3]. To test the effectiveness of the age stratification strategy, the data of breast cancer patients were grouped based on quantiles of age, from to . The results showed that the age stratification at 61 years old has the best performance, which is in agreement with the median age at the time of breast cancer diagnosis reported by the American Cancer Society [33]. For clinicians’ reference, we also established a free website tool (http://ncblab.nchu.edu.tw/SaBrcada), named SaBrcada, which provides 5 types of predicted survival intervals, including within half a year, between half and one year, between one and three years, between three and five years, and more than five years.
2. Materials and Methods
2.1. Modeling Process
The SaBrcada modeling process is shown in Figure 1. First, we downloaded the RNA-seq in FPKM format and clinical data of patients diagnosed with breast cancer from TCGA-BRCA and then excluded records with incomplete RNA-Seq expression data or without recorded clinical data, date of death, or age. The remaining RNA-seq data were converted to TPM format. Based on age stratification, we further divided the collected data into two datasets based on an age of 61. Seventy percent of the patient data in the two datasets were set aside to fit the survival model. To assess the goodness of fit of the survival model by the accuracy, survival analysis images were generated following dimension raising. The two datasets from the survival analysis images were combined as the training set for model building by deep learning architectures. The remaining 30% of the patient data were collected and processed using the same procedures to generate the test set to assess model performance.
2.2. Data Preprocessing
In this study, We are using TCGA version 27 data. RNA-seq data from breast cancer patients were collected in FPKM format. It was noted that the original counts (reads) may be different from the true values due to the sampling environment, experimental methods, or length of each RNA [34]. Although gene length was considered, FPKM uses pair-end reads as the unit, i.e., fragments, not full transcripts. On the other hand, TPM reports the relative expression level of each transcript in the sample, providing more complete data. Therefore, we chose TPM format for further study. In addition, we also collected clinical data, including information of patient age, survival time, and race, from TCGA.
Before preprocessing, we downloaded a total of 1,187 RNA-seq data to build the SaBrcada-BPP dataset. After excluding 96 samples with missing clinical data, we obtained 1,091 data records containing clinical data. We further excluded the samples that recorded the same survival time and obtained a SaBrcada-APP dataset with 807 breast cancer cases, after preprocessing. To ensure that all the samples included actual survival times, we selected 144 RNA-seq data with actual date of death information to establish the SaBrcada-AD dataset (Figure 2). Furthermore, SaBrcada-AD was classified with stratified random sampling: the samples with a patient age younger than or equal to 61 years were included in the SaBrcada-AYT61 dataset, and the remaining samples were included in the SaBrcada-AOT61 dataset, including the samples with a patient age older than 61 years. The dataset SaBrcada-train was created by combining the two training sets of SaBrcada-AYT61 and SaBrcada-AOT61. SaBrcada-test was created by combining the testing sets of these two datasets. All 7 datasets used in this study provide information on age, survival time, and race (Table 1).
2.3. Age Stratification
To ensure that there was a sufficient amount of data in the two datasets after stratification, quantiles to , that is patients aged 48 to 69 years, were used as the basis for sorting the SaBrcada-AD dataset. After stratification, 70% of the patient data were extracted with the shuffle algorithm in the Random package of Python for use as the training set for the generation of survival analysis images. The other survival analysis images generated by the remaining 30% data were used to determine the most suitable age for stratification by accuracy evaluation. For example, there were 49 cases younger than or equal to 61 years old, which generated 2,352 survival analysis images as the training set. The other 20 cases generated 380 survival analysis images as the test set. For patients older than 61 years old, 53 cases generated 2756 survival analysis images as the training set, and 22 cases generated 462 survival analysis images as the test set.
2.4. Data Generation
Considering the reliability and comparability between different patients, we first normalized the 60,483 gene expression data from FPKM into TPM by fixing the total gene expression of FPKM to 1,000,000. To compare the differential gene expression between patients, we arranged the expression data of genes in the order provided by TCGA-BRCA RNA-Seq. For survival analysis, we also sorted the TPM format data according to the patient’s survival time and then subtracted the data in pairs to generate two survival analysis data types, (positive) and (negative). According to our previous study, the difference between genes improves the predictive model of survival analysis, especially its sensitivity, compared with the traditional fold change (data not shown). It may be that the fold change of gene expression overestimates the effect of gene expression differences that do not reach the activation threshold [35], but underestimates the effect of gene expression differences at high expression levels, so subtraction was chosen in this study. is the dataset containing the data with shorter survival time subtracted from the data with longer survival time to represent the differential gene expression pattern of longer survival time. In contrast, is the dataset representing a shorter survival time. Taking 5 patients as an example, the data were arranged by the length of survival time from long to short as N1 to N5, as shown in Figure 3(a). The data type is generated by subtracting the TPM data of N2, N3, N4 and N5 from that of N1 and then subtracting the TPM data of the remaining 3 samples with N2. It will generate n(n-1)/2 survival analysis data as seen in Figure 3(b). In contrast, data type is generated by subtracting the TPM data of N1, N2, N3 and N4 from that of N5 and then subtracting the TPM data of the remaining 3 samples from N4, and so on as shown in Figure 3(a).
2.5. Data Dimension Augmentation
We obtained 60,483 gene expression data from TCGA-BRCA, this RNA-seq data produces a large number of features, which are difficult to directly process by machine learning. CNNs designed to process data with a large number of features are therefore considered. However, CNN is more suitable for processing two-dimensional data with spatial structure. Therefore, the survival analysis data were arranged into a 246 × 246-matrix by dimension raising from one dimension to two dimension. Here, the 246 × 246-matrix is the smallest square matrix that can contain 60,483 differential gene expressions. All 60,483 differential gene expression levels were filled in the order from left to right and top to bottom and then were converted into grayscale pixel values ranging from 0 to 255. Zero represented the maximum negative difference in gene expression, and 255 represented the maximum positive difference. After filling the remaining 33 positions with 0, the survival analysis matrix was generated and finally saved in PNG file format to serve as the survival analysis images. The process is shown in Figure 4.
2.6. Deep Learning
We used CNN, one of the most common deep learning network architectures, implemented in PyTorch 1.9 to build a neural network framework and combined it with a Quadro GV100 32G graphics card (GPU) for model construction. The convolutional and pooling layers in the neural network architecture improve the recognition of pattern identity and the relationship between adjacent data and can learn features independently. Based on these characteristics, we used a CNN to learn features from survival analysis images. The deep learning frameworks containing 3 inceptions and 22 convolutional layers were used to learn different features and make comprehensive judgments on all features. For hyperparameter selection, we tested 3 different sets of hyperparameters by using Adam (optimizer), Cross Entropy (loss function), and a dropout value of 0.4.
2.7. Assessment of Model Performance
Accuracy is a common method to evaluate the prediction model [36]. Accuracy is calculated using equation 1 (1). Where TP, true positive, is the number of positive predictions; TN, true negative, is the number of negative predictions, and P and N are the numbers of positive and negative, respectively. The accuracy ranges between 0 and 1.0. An accuracy of 0.5 represents a random prediction, and a value of 1.0 indicates that the prediction was completely consistent with the actual value.
3. Results
3.1. Survival Analysis Image Applicability Analysis
As shown in Figure 5(a) and (b), it is difficult for the naked eye to identify the features in the survival analysis images, and . The corresponding grayscale distributions of the survival analysis images are significantly different, as shown in Figure 5(c,d). In the example shown in Figure 5, the grayscales of most pixels in are between 30 and 45, while the grayscales of are mostly between 160 and 180. These two types of images display sufficient differences to be learned from the features by a convolutional neural network for further survival interval analysis.
3.2. Deep Learning Architecture Test
Based on various features, different learning methods were selected for model construction. To detect differences between the and images, we adopted a deep learning method and used the SaBrcada-AD dataset for architecture testing, in which 70% of the data are used as the training set and 30% of the data are used as the test set. To identify the most suitable deep learning architecture, a total of 8 deep learning architectures, Resnet18, Resnet50, Resnet101, Resnet152, ResNext101, GoogLeNet, DenseNet121, DenseNet161, and 3 different hyperparameter combinations, Epoch 50 Batch size 8, Epoch 100 Batch size 16, and Epoch 150 Batch size 32, were tested (Table 2). Among them, we found that the most suitable architecture was GoogLeNet with a hyperparameter combination of a batch size of 32 and 150 epochs, which had the highest accuracy value of 0.6. Therefore, SaBrcada uses this condition for model construction.
3.3. Stratification by Age
According to the clinical data of breast cancer patients, the survival time of young patients is shorter, and the survival interval of older patients is generally longer [3], which indicates that the survival days will be affected by age. For this reason, we incorporated the age feature into the model to improve the accuracy by using age-stratified random sampling from quartiles and . That is, every age between the ages of 48 and 69 is considered as a cut-off for stratification and accuracy testing (Figure 6). The results show that the highest accuracy of 0.798 can be obtained by taking the age of 61 as the cut-off for stratification. Thus, SaBrcada used 61 years old as the cut-off for stratified random sampling to establish a model for subsequent survival analysis.
3.4. Comparison of the Previous Studies
Table 3 shows the comparison of the models constructed in this study with those from previous studies, including the accuracy, data distribution, data types, and training models. First, the SaBrcada-APP data were used to generate the survival analysis image dataset SaBrcada-APP-I for SaBrcada-APP-M model construction. SaBrcada-APP-M resulted in an accuracy of 0.5. The survival dates of most patients in the SaBrcada-APP dataset are the date of last follow-up days rather than the date of death. To improve accuracy, the records with date of death were selected from SaBrcada-APP to build the SaBrcada-AD dataset. The SaBrcada-AD data were used to generate the survival analysis image dataset SaBrcada-AD-I for SaBrcada-AD-M model construction, and an accuracy of 0.6 was obtained. Grouping by age stratification, SaBrcada-AD was divided into two datasets. The dataset SaBrcada-ASYT61 included data from patients younger than or equal to 61 years, and the dataset SaBrcada-ASOT61 included data from patients older than 61 years. The data of SaBrcada-ASYT61 and SaBrcada-ASOT61 were used to generate separate survival analysis image datasets SaBrcada-ASYT61-I and SaBrcada-ASOT61-I for the model building of SaBrcada-ASYT61-M and SaBrcada-ASOT61-M, respectively. Model accuracy was assessed, resulting in accuracy values of 0.5 and 0.681, respectively. We used stratified random sampling to build the SaBrcada model by using the survival analysis images and the SaBrcada-I dataset. To make the SaBrcada model applicable to patients of all ages for survival analysis, the training set of SaBrcada-I was integrated with the training set of SaBrcada-ASYT61-I and SaBrcada-ASOT61-I for modeling. On the other hand, the integration of SaBrcada-ASYT61-I and SaBrcada-ASOT61-I was used as the test set of SaBrcada-I. According to the above condition, SaBrcada achieved an accuracy of 0.798, which is better than SALMON [37], ConcatAE [38], and VAECox architecture [39]. Zhang et al. used the SALMON architecture and combined breast cancer patient data, gene set enrichment analysis, and age characteristics to construct a survival analysis prediction model with an accuracy of 0.7 [37]. ConcatAE integrated DNA methylation and miRNA expression data using principal component analysis features to develop a breast cancer overall survival prediction model with an accuracy of 0.641 ± 0.031 [38]. The VAECox framework was established by the common features of multiple cancers to conduct transfer learning. The average accuracy of survival analysis for 10 cancers was 0.649, and the accuracy of prediction on breast cancer was also lower than 0.7 [39].
3.5. Assessment of Accuracy of SaBrcada
After testing the accuracy of SaBrcada's prediction for all patients with different ages in the SaBrcada-AD database, we found that the accuracy was higher than the 0.85 for patient ages of 70, 89, and 90 years. Among them, the best performance was an accuracy of 0.92 for age of 90 years. Patient ages of 63, 84 and 88 years also obtained accuracy values higher than 0.7, with significant differences (Figure 7).
3.6. Website Tools
The purpose of developing the SaBrcada tool is to provide users guidelines for the analysis of the survival time of breast cancer patients. Combining survival analysis and clinical experience may help clinicians choose the most suitable treatment strategies to improve the quality of life of patients. The SaBrcada website interface is shown in Figure 8. The website is freely available at http://ncblab.nchu.edu.tw/SaBrcada. SaBrcada provides preprocessing tools to transfer TPM format RNA-Seq data for survival analysis image generation. After analyzing the survival analysis images uploaded by the user, SaBrcada provides the analysis information of the patient's survival period. SaBrcada obtains two modules: the first is survival analysis image creation, and the second is survival period analysis. For survival analysis image creation, the user first downloads the preprocessing program packaged by pyinstaller and then inputs the user’s TPM file with.TXT into the corresponding file according to the age of the patient. The tool compares the input data from the user and that from 4 default reference patients to generate 4 survival analysis data. The survival analysis data are then raised to a two-dimensional matrix, and 4 survival analysis images are generated by using the png package provided by Python. For survival period analysis, the user needs to upload the 4 survival analysis images generated by the preprocessing for survival analysis by using the established SaBrcada model and then obtain the results. The analysis results will show the predicted patient survival period, with possible values of less than six months, six months to one year, one year to three years, three to five years, or more than five years, as a reference for clinicians to implement treatment strategies.
4. Discussion
4.1. Comparison with Past Research Models
In this study, SaBrcada, a breast cancer survival analysis prediction model, was established by using convolutional neural networks. In brief, the SaBrcada-AD dataset was selected from TCGA-BRCA based on the completeness of RNA-seq and clinical data. The RNA-seq data in SaBrcada-AD were converted into a TPM data type to represent the relative transcript level. Using stratified random sampling based on age, and a cut-off of 61 years of age, the SaBrcada-I survival analysis image dataset was generated for prediction model construction by using GoogLeNet. SaBrcada achieved the best performance of all examined frameworks with an accuracy of 0.798.
In the past, breast cancer survival analysis models typically used deep learning to extract the nonlinear characteristics of RNA-seq data and then predicted linear Cox regression survival times [37,39]. Recently, researchers began to directly use the fully connected neural network as a survival analysis model [38], and we used a similar strategy with SaBrcada. However, we made some improvements and greatly increased its accuracy. The major difference between SaBrcada and other models is that the one-dimensional RNA-seq is augmented into a two-dimensional survival analysis image, which is beneficial to the feature learning by CNN. The second difference is, unlike in past research; GoogLeNet was used for construction of the prediction model instead of the Cox method. In addition, the SaBrcada website's prediction tool provides information on the survival interval for clinicians to refer to determine treatment strategies.
4.2. Advantages of SaBrcada
Looking at previous studies, three points were not considered in their prediction model construction. The first is the accuracy of the data collection. Whether records include the actual death time has a great impact on the accuracy of the model construction. Usually, all TCGA data are used directly. However, the last date of follow up was used to impute the time of death in TCGA, which may be not be accurate. Not excluding the records missing date of death may affect the learning capability of the model. Therefore, we specifically selected the SaBrcada-AD dataset with date of death as the basis for SaBrcada modeling. The second point is the normalization of data. We can obtain RNA-seq data in various formats. The number of reads, accounting for raw readings, may be influenced by the experimental design. FPKM counts the relative fragments per kilobase of transcript. Both may distort the comparison of gene expression between the two patients. The use of TPM, a technique based on more sophisticated bioinformatics, can improve the performance of survival prediction based on gene expression. Thus, SaBrcada converts the FPKM data provided in TCGA into a normalized TPM data type, which can more accurately present the relative expression of each gene. The third point is the impact of age on the survival risk of patients. It was reported that breast cancer patients younger than 45 years old had a worse prognosis and shorter overall survival time than older patients [3]. Young breast cancer patients usually have multiple gene mutations involved in tumor development and cancer cell metastasis, resulting in a high cancer cell metastasis rate and lower survival rate. More than 70% of breast cancer patients over 45 years old were diagnosed with luminal A and luminal B subtypes and with the best prognosis [40]. Thus, age should be considered in prognosis predictions to reflect its impact on survival risk. Consequently, SaBrcada uses age to perform stratified random sampling of the dataset to assess the effect of different age stratification cut-offs and to improve the accuracy of the model. The predictive accuracy indicated that 61 years of age is the best criterion for stratification by age, which echoes the median age of breast cancer patients reported by the American Cancer Society's Breast Cancer Statistics Report 2017-2018 [33].
4.3. Directions for Future Research
In the past, doctors analyzed the prognosis of patients by using their clinical experience, inevitably causing inconsistency in accuracy due to individual differences. In the postgenomic era, precision medicine has become a trend. In this study, we combined gene expression and clinical data to establish a reliable survival analysis model, SaBrcada. To enrich the biological information provided, we will integrate characteristics and coexpression network analyses. Based on this improvement, we may extract the determining factors from the black box of the survival analysis tool. This may provide a reliable prediction of survival intervals and an explainable result including molecular information for clinicians’ reference to determine the treatment strategy for individual patients.
5. Conclusions
In this study, we have established a breast cancer survival analysis prediction model, SaBrcada, and its same named website http://ncblab.nchu.edu.tw/SaBrcada. We downloaded the gene expression and clinical data from TCGA-BRCA. After normalization to TPM and dimension raising, survival analysis images generated by differential gene expression were subjected to deep learning architectures testing. Based on the performance, GoogLeNet was selected to build the survival prediction model, SaBrcada. After screened out the incomplete data, the performance of SaBrcada-AD-M was increased to accuracy 0.6. By adding the stratified random sampling by patients’ age of 61, the performance of SaBrcada reached the accuracy of 0.798. That indicated the accuracy of data and stratified random sampling by age will improve the performance of survival prediction model. We hope this highly reliable survival analysis model and website tool providing the information of survival interval periods for clinicians’ reference to precision medicine.
Author Contributions
S.-H. L. and M.-F. L. contributed to data collection, design of experimental processes, and system architecture. C.-H. C. set up the website. K.-P. C. supported the experimental data and data interpretation. Y.-T.C. and Y.-W.C conceived of the study goal, supervised the study, and provided advice with respect to the study direction. All authors read and approved the manuscript.
Funding
This research was supported by (1) the National Science and Technology Council, Taiwan, under grant number 109-2313-B-005-019-, 110-2221-E-005-062-MY3, 110-2320-B-039 -014, 110-2321-B-005-005 and 110-2634-F-005 -006. (2) National Chung Hsing University and Changhua Christian Hospital: NCHU-CCH 11006. (3) Smart Sustainable New Agriculture Research Center (SMARTer) 110-2634-F-005-006. (4) China Medical University Hospital: DMR-112-074.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The datasets for this study can be found in the TCGA-BRCA [https://portal.gdc.cancer.gov/projects/TCGA-BRCA].
Acknowledgments
The authors would like to acknowledge the financial support for the grants 110-2221-E-005-062-MY3, 110-2320-B-039 -014, 110-2321-B-005-005 and 110-2634-F-005 -006 from the National Science and Technology Council, Taiwan, R.O.C.; NCHU-CCH 11006 from National Chung Hsing University and Changhua Christian Hospital; and 110-2634-F-005-006 from Smart Sustainable New Agriculture Research Center (SMARTer). We would like to thank American Journal Experts for editing and proofreading this manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Nagini, S. Breast Cancer: Current Molecular Therapeutic Targets and New Players. Anticancer Agents Med. Chem. 2017, 17, 152–163. [Google Scholar] [CrossRef]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA. Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Anastasiadi, Z.; Lianos, G.D.; Ignatiadou, E.; Harissis, H.V.; Mitsis, M. Breast Cancer in Young Women: An Overview. Updat. Surg. 2017, 69, 313–317. [Google Scholar] [CrossRef] [PubMed]
- Tao, Z.; Shi, A.; Lu, C.; Song, T.; Zhang, Z.; Zhao, J. Breast Cancer: Epidemiology and Etiology. Cell Biochem. Biophys. 2015, 72, 333–338. [Google Scholar] [CrossRef]
- Morrison, R.S.; Meier, D.E. Clinical Practice. Palliative Care. N. Engl. J. Med. 2004, 350, 2582–2590. [Google Scholar] [CrossRef]
- Shachar, S.S.; Hurria, A.; Muss, H.B. Breast Cancer in Women Older Than 80 Years. J. Oncol. Pract. 2016, 12, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Milanez-Almeida, P.; Martins, A.J.; Germain, R.N.; Tsang, J.S. Cancer Prognosis with Shallow Tumor RNA Sequencing. Nat. Med. 2020, 26, 188–192. [Google Scholar] [CrossRef] [PubMed]
- Cuzick, J.; Swanson, G.P.; Fisher, G.; Brothman, A.R.; Berney, D.M.; Reid, J.E.; Mesher, D.; Speights, V.O.; Stankiewicz, E.; Foster, C.S.; et al. Prognostic Value of an RNA Expression Signature Derived from Cell Cycle Proliferation Genes for Recurrence and Death from Prostate Cancer: A Retrospective Study in Two Cohorts. Lancet Oncol. 2011, 12, 245–255. [Google Scholar] [CrossRef]
- Harrell, F.E.; Lee, K.L.; Mark, D.B. Multivariable Prognostic Models: Issues in Developing Models, Evaluating Assumptions and Adequacy, and Measuring and Reducing Errors. Stat. Med. 1996, 15, 361–387. [Google Scholar] [CrossRef]
- Altman, D.G.; Royston, P. What Do We Mean by Validating a Prognostic Model? Stat. Med. 2000, 19, 453–473. [Google Scholar] [CrossRef]
- Concato, J. Challenges in Prognostic Analysis. Cancer 2001, 91, 1607–1614. [Google Scholar] [CrossRef] [PubMed]
- McShane, L.M.; Altman, D.G.; Sauerbrei, W.; Taube, S.E.; Gion, M.; Clark, G.M. REporting Recommendations for Tumour MARKer Prognostic Studies (REMARK). Br. J. Cancer 2005, 93, 387–391. [Google Scholar] [CrossRef]
- Reilly, B.M.; Evans, A.T. Translating Clinical Research into Clinical Practice: Impact of Using Prediction Rules To Make Decisions. Ann. Intern. Med. 2006, 144, 201–209. [Google Scholar] [CrossRef] [PubMed]
- Royston, P.; Moons, K.G.M.; Altman, D.G.; Vergouwe, Y. Prognosis and Prognostic Research: Developing a Prognostic Model. BMJ 2009, 338, b604. [Google Scholar] [CrossRef]
- Huang, Z.; Zhan, X.; Xiang, S.; Johnson, T.S.; Helm, B.; Yu, C.Y.; Zhang, J.; Salama, P.; Rizkalla, M.; Han, Z.; et al. SALMON: Survival Analysis Learning With Multi-Omics Neural Networks on Breast Cancer. Front. Genet. 2019, 10, 166. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Kim, K.; Choe, J.; Lee, I.; Kang, J. Improved Survival Analysis by Learning Shared Genomic Information from Pan-Cancer Data. Bioinforma. Oxf. Engl. 2020, 36, i389–i398. [Google Scholar] [CrossRef]
- Gascard, P.; Bilenky, M.; Sigaroudinia, M.; Zhao, J.; Li, L.; Carles, A.; Delaney, A.; Tam, A.; Kamoh, B.; Cho, S.; et al. Epigenetic and Transcriptional Determinants of the Human Breast. Nat. Commun. 2015, 6, 6351. [Google Scholar] [CrossRef]
- Sun, C.-C.; Li, S.-J.; Hu, W.; Zhang, J.; Zhou, Q.; Liu, C.; Li, L.-L.; Songyang, Y.-Y.; Zhang, F.; Chen, Z.-L.; et al. Comprehensive Analysis of the Expression and Prognosis for E2Fs in Human Breast Cancer. Mol. Ther. 2019, 27, 1153–1165. [Google Scholar] [CrossRef]
- Sanchez-Vega, F.; Mina, M.; Armenia, J.; Chatila, W.K.; Luna, A.; La, K.C.; Dimitriadoy, S.; Liu, D.L.; Kantheti, H.S.; Saghafinia, S.; et al. Oncogenic Signaling Pathways in The Cancer Genome Atlas. Cell 2018, 173, 321–337. [Google Scholar] [CrossRef]
- Cooper, T.A.; Wan, L.; Dreyfuss, G. RNA and Disease. Cell 2009, 136, 777–793. [Google Scholar] [CrossRef]
- Matera, A.G.; Wang, Z. A Day in the Life of the Spliceosome. Nat. Rev. Mol. Cell Biol. 2014, 15, 108–121. [Google Scholar] [CrossRef]
- Bracken, C.P.; Scott, H.S.; Goodall, G.J. A Network-Biology Perspective of MicroRNA Function and Dysfunction in Cancer. Nat. Rev. Genet. 2016, 17, 719–732. [Google Scholar] [CrossRef] [PubMed]
- Wickramasinghe, V.O.; Laskey, R.A. Control of Mammalian Gene Expression by Selective MRNA Export. Nat. Rev. Mol. Cell Biol. 2015, 16, 431–442. [Google Scholar] [CrossRef] [PubMed]
- Venter, J.C.; Adams, M.D.; Myers, E.W.; Li, P.W.; Mural, R.J.; Sutton, G.G.; Smith, H.O.; Yandell, M.; Evans, C.A.; Holt, R.A.; et al. The Sequence of the Human Genome. Science 2001, 291, 1304–1351. [Google Scholar] [CrossRef] [PubMed]
- Phan, J.H.; Quo, C.F.; Cheng, C.; Wang, M.D. Multiscale Integration of -Omic, Imaging, and Clinical Data in Biomedical Informatics. IEEE Rev. Biomed. Eng. 2012, 5, 74–87. [Google Scholar] [CrossRef] [PubMed]
- O’Driscoll, A.; Daugelaite, J.; Sleator, R.D. “Big Data”, Hadoop and Cloud Computing in Genomics. J. Biomed. Inform. 2013, 46, 774–781. [Google Scholar] [CrossRef] [PubMed]
- Gujar, R.; Panwar, B.; Dhanda, S.K. Bioinformatics Drives Discovery in Biomedicine. Bioinformation 2020, 16, 13–16. [Google Scholar] [CrossRef]
- Chaudhary, K.; Poirion, O.B.; Lu, L.; Garmire, L.X. Deep Learning Based Multi-Omics Integration Robustly Predicts Survival in Liver Cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2018, 24, 1248–1259. [Google Scholar] [CrossRef]
- Ching, T.; Zhu, X.; Garmire, L.X. Cox-Nnet: An Artificial Neural Network Method for Prognosis Prediction of High-Throughput Omics Data. PLoS Comput. Biol. 2018, 14, e1006076. [Google Scholar] [CrossRef]
- Katzman, J.L.; Shaham, U.; Cloninger, A.; Bates, J.; Jiang, T.; Kluger, Y. DeepSurv: Personalized Treatment Recommender System Using a Cox Proportional Hazards Deep Neural Network. BMC Med. Res. Methodol. 2018, 18, 24. [Google Scholar] [CrossRef]
- Cox, D.R.; Oakes, D. Analysis of Survival Data; Chapman and Hall/CRC: Boca Raton, 2017; ISBN 978-1-315-13743-8. [Google Scholar]
- Concato, J. Challenges in Prognostic Analysis. Cancer 2001, 91, 1607–1614. [Google Scholar] [CrossRef] [PubMed]
- Street, W. Breast Cancer Facts & Figures 2017-2018. 44.
- Wagner, G.P.; Kin, K.; Lynch, V.J. Measurement of MRNA Abundance Using RNA-Seq Data: RPKM Measure Is Inconsistent among Samples. Theory Biosci. 2012, 131, 281–285. [Google Scholar] [CrossRef] [PubMed]
- Richelle, A.; Joshi, C.; Lewis, N.E. Assessing Key Decisions for Transcriptomic Data Integration in Biochemical Networks. PLOS Comput. Biol. 2019, 15, e1007185. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Zhang, Y.; Zou, L.; Wang, M.; Li, A. A Gene Signature for Breast Cancer Prognosis Using Support Vector Machine. In Proceedings of the 2012 5th International Conference on BioMedical Engineering and Informatics; October 2012; pp. 928–931. [Google Scholar]
- Kim, S.; Kim, K.; Choe, J.; Lee, I.; Kang, J. Improved Survival Analysis by Learning Shared Genomic Information from Pan-Cancer Data. Bioinformatics 2020, 36, i389–i398. [Google Scholar] [CrossRef]
- Tong, L.; Mitchel, J.; Chatlin, K.; Wang, M.D. Deep Learning Based Feature-Level Integration of Multi-Omics Data for Breast Cancer Patients Survival Analysis. BMC Med. Inform. Decis. Mak. 2020, 20, 225. [Google Scholar] [CrossRef]
- Gascard, P.; Bilenky, M.; Sigaroudinia, M.; Zhao, J.; Li, L.; Carles, A.; Delaney, A.; Tam, A.; Kamoh, B.; Cho, S.; et al. Epigenetic and Transcriptional Determinants of the Human Breast. Nat. Commun. 2015, 6, 6351. [Google Scholar] [CrossRef]
- Colak, D.; Nofal, A.; AlBakheet, A.; Nirmal, M.; Jeprel, H.; Eldali, A.; AL-Tweigeri, T.; Tulbah, A.; Ajarim, D.; Malik, O.A.; et al. Age-Specific Gene Expression Signatures for Breast Tumors and Cross-Species Conserved Potential Cancer Progression Markers in Young Women. PLoS ONE 2013, 8, e63204. [Google Scholar] [CrossRef]
Figure 1.
SaBrcada modeling process. RNA-seq and clinical data of breast cancer patients from downloaded from TCGA-BRCA has first been filtered to exclude records with incomplete RNA-Seq expression data or missing clinical data, death dates, or age information. After converting the RNA-seq data into TPM format, it was split into two subsets based on the age of 61, and 70% of the data in each subset was used for training. Through dimension raising, survival analysis images were generated and used for deep learning modeling. Finally, the remaining 30% of the data was used as test data to verify the accuracy of the model.
Figure 1.
SaBrcada modeling process. RNA-seq and clinical data of breast cancer patients from downloaded from TCGA-BRCA has first been filtered to exclude records with incomplete RNA-Seq expression data or missing clinical data, death dates, or age information. After converting the RNA-seq data into TPM format, it was split into two subsets based on the age of 61, and 70% of the data in each subset was used for training. Through dimension raising, survival analysis images were generated and used for deep learning modeling. Finally, the remaining 30% of the data was used as test data to verify the accuracy of the model.
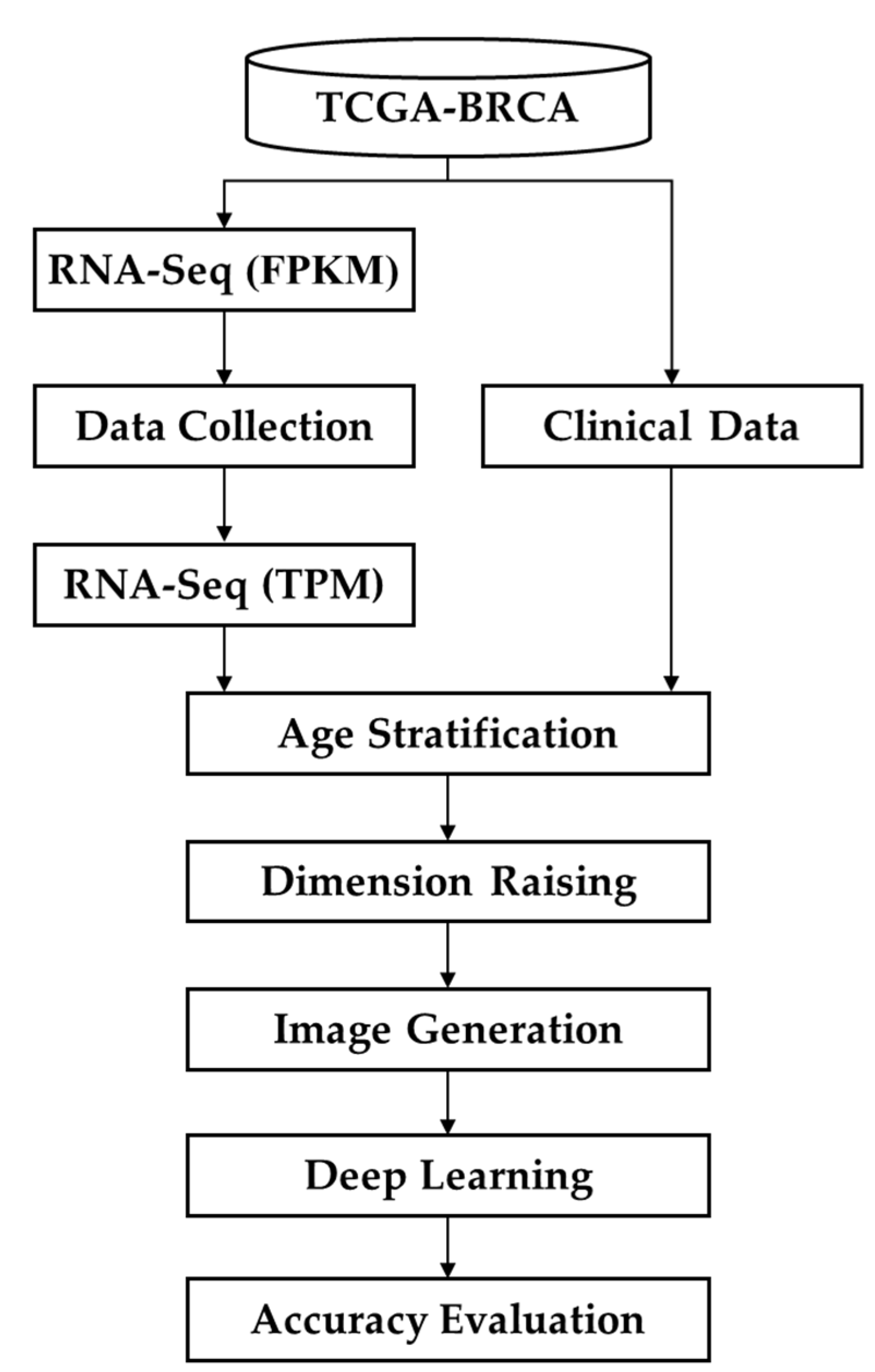
Figure 2.
Data screening flowchart. The flowchart details how much data were deleted at each stage and why. From TCGA, 1187 samples were downloaded to construct SaBrcada-BPP, before preprocessing. After excluding 96 samples lacking clinical data, and further excluding 284 samples with the same survival time as other samples, we then built SaBrcada-APP dataset containing 807 breast cancer cases after preprocessing. Finally, 663 samples without death date were removed, and we obtained 144 samples with actual death date to build the SaBrcada-AD dataset.
Figure 2.
Data screening flowchart. The flowchart details how much data were deleted at each stage and why. From TCGA, 1187 samples were downloaded to construct SaBrcada-BPP, before preprocessing. After excluding 96 samples lacking clinical data, and further excluding 284 samples with the same survival time as other samples, we then built SaBrcada-APP dataset containing 807 breast cancer cases after preprocessing. Finally, 663 samples without death date were removed, and we obtained 144 samples with actual death date to build the SaBrcada-AD dataset.
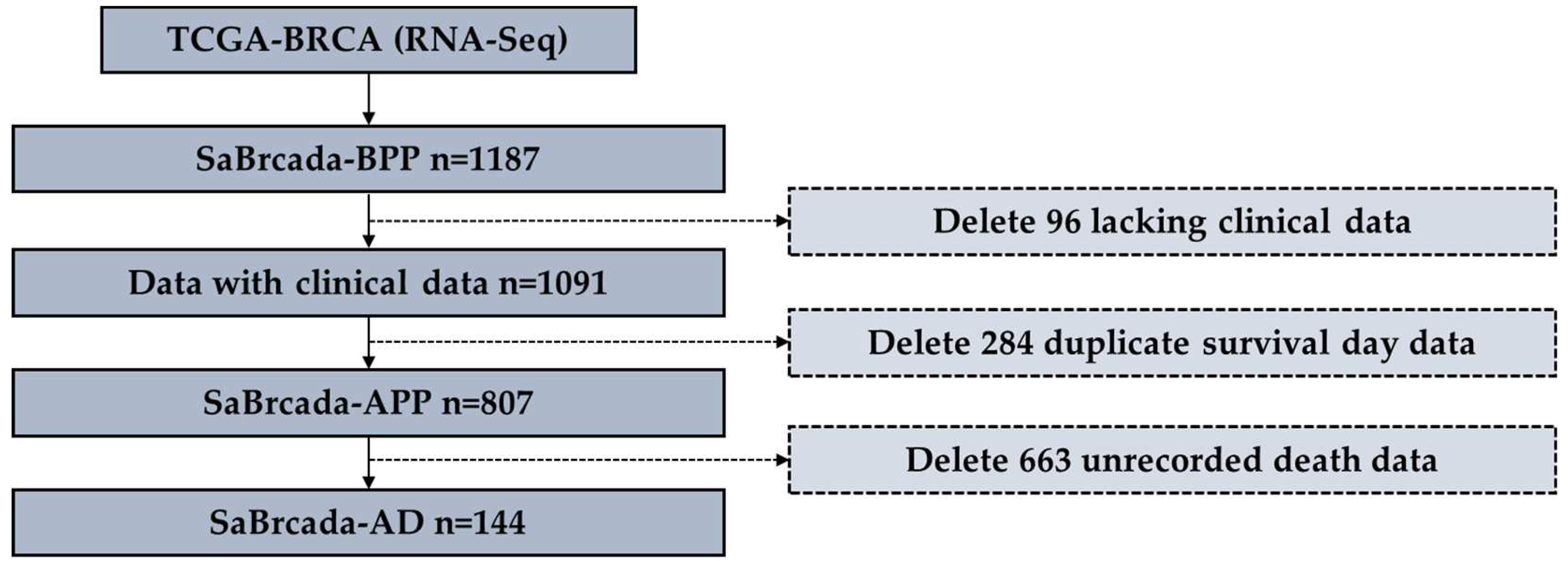
Figure 3.
Survival analysis data generation. (a) The survival analysis data generation method. (positive) is the data type that was generated by subtracting the TPM data of patients with shorter survival times from that of patients with longer survival times (negative) was generated by subtracting the TPM data of patients with longer survival times from that of patients with shorter survival times. (b) Schematic diagram of survival analysis data example. N1 and N2 indicate the gene expression of patients N1 and N2 in TPM format, respectively. Data Type is the survival analysis data generated by subtracting the TPM data of patient N2 from that of N1.
Figure 3.
Survival analysis data generation. (a) The survival analysis data generation method. (positive) is the data type that was generated by subtracting the TPM data of patients with shorter survival times from that of patients with longer survival times (negative) was generated by subtracting the TPM data of patients with longer survival times from that of patients with shorter survival times. (b) Schematic diagram of survival analysis data example. N1 and N2 indicate the gene expression of patients N1 and N2 in TPM format, respectively. Data Type is the survival analysis data generated by subtracting the TPM data of patient N2 from that of N1.
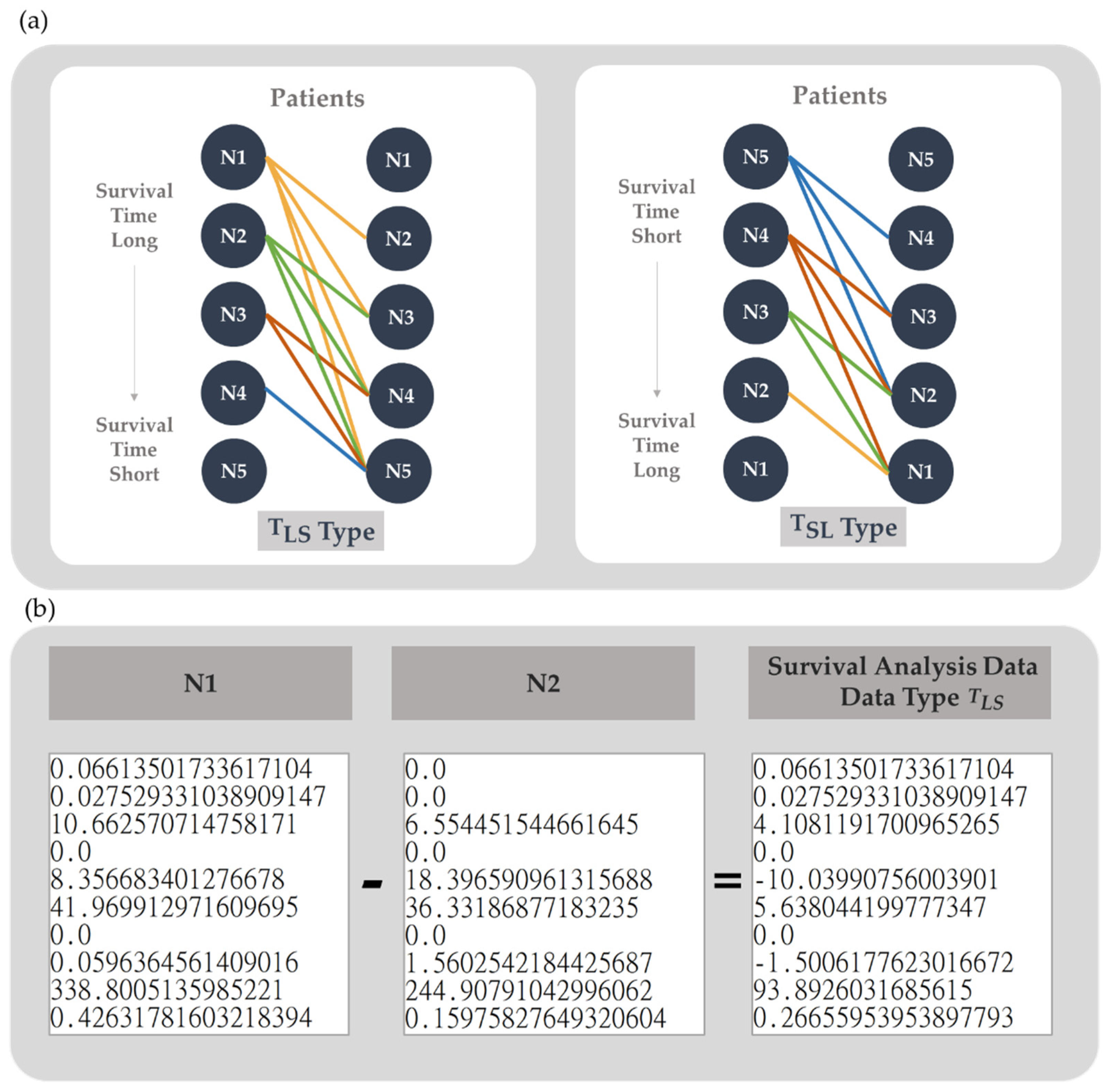
Figure 4.
Schematic diagram of survival analysis images. By dimension raising and scaling the survival analysis data in the range from 0 to 255, a survival analysis matrix was generated for further survival analysis image conversion.
Figure 4.
Schematic diagram of survival analysis images. By dimension raising and scaling the survival analysis data in the range from 0 to 255, a survival analysis matrix was generated for further survival analysis image conversion.
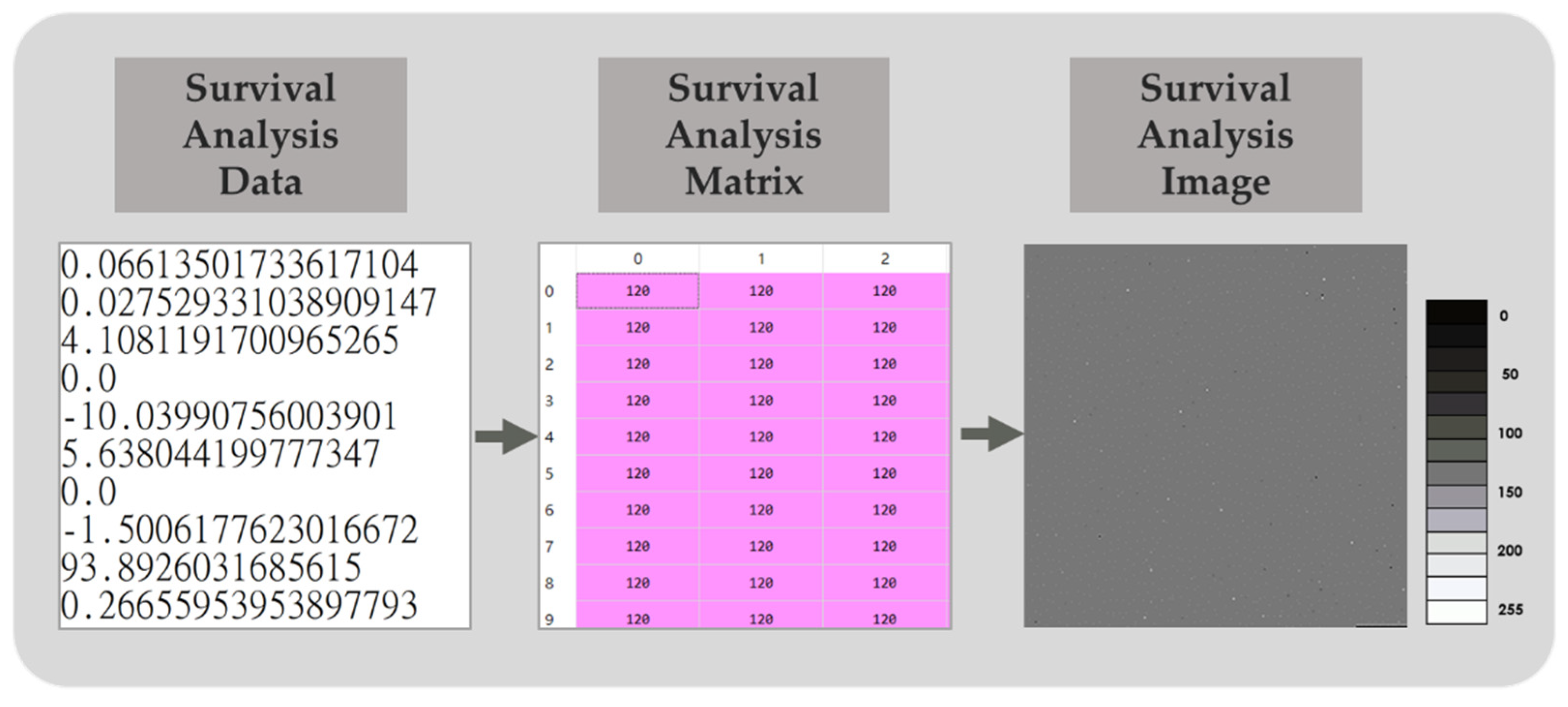
Figure 5.
Pixel distribution diagram after image generation. (a) type image; (b) pixel value distribution of type image; (c) type image; (d) pixel value distribution of type image.
Figure 5.
Pixel distribution diagram after image generation. (a) type image; (b) pixel value distribution of type image; (c) type image; (d) pixel value distribution of type image.
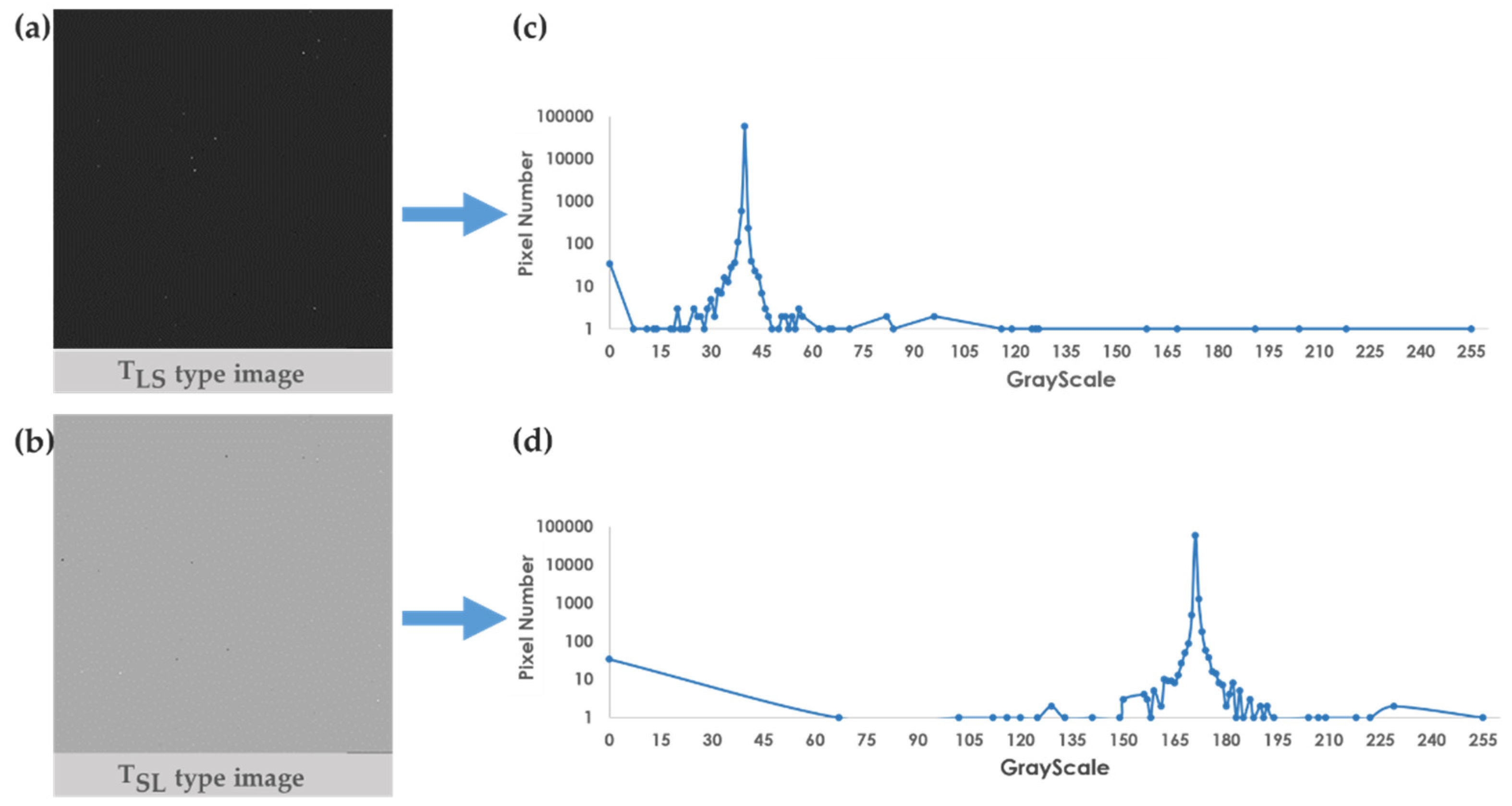
Figure 6.
Performance of stratified random sampling by age. The X axis is the age cut-off, and the Y axis is the accuracy.
Figure 6.
Performance of stratified random sampling by age. The X axis is the age cut-off, and the Y axis is the accuracy.
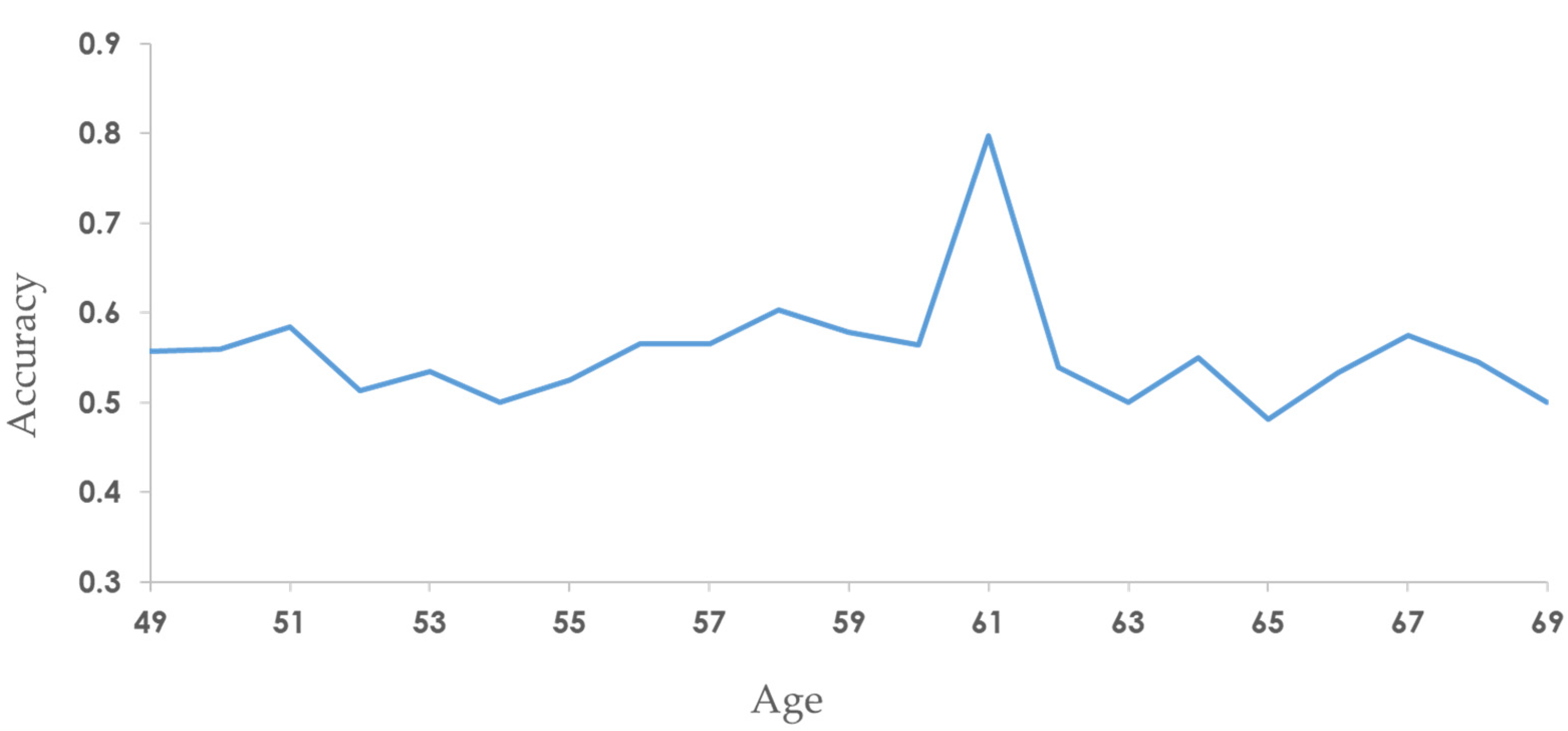
Figure 7.
The prediction accuracy for breast cancer patients using SaBrcada by age. The X-axis is the age of the patient, and the Y-axis is the accuracy. The red dots indicate that the accuracy is greater than 0.7.
Figure 7.
The prediction accuracy for breast cancer patients using SaBrcada by age. The X-axis is the age of the patient, and the Y-axis is the accuracy. The red dots indicate that the accuracy is greater than 0.7.
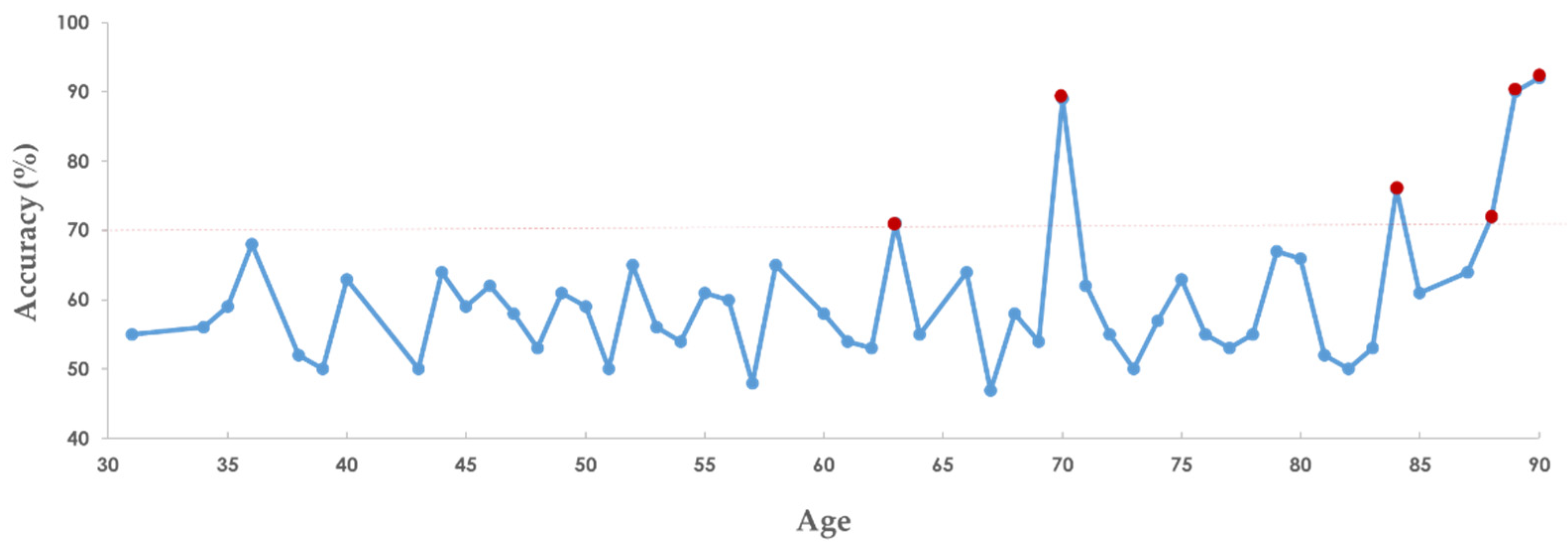
Figure 8.
SaBrcada website tool interface. The tool is freely available at http://ncblab.nchu.edu.tw/SaBrcada. It provides a tool for generating survival analysis images and online analysis of survival time. The outcome of the analysis is the patient's predicted survival time, which can be classified as less than six months, six months to one year, one to three years, three to five years, or more than five years.
Figure 8.
SaBrcada website tool interface. The tool is freely available at http://ncblab.nchu.edu.tw/SaBrcada. It provides a tool for generating survival analysis images and online analysis of survival time. The outcome of the analysis is the patient's predicted survival time, which can be classified as less than six months, six months to one year, one to three years, three to five years, or more than five years.

Table 1.
List of the datasets used in this study.
| Dataset | No. | Age at index, median (range) | Survival day, median (range) | Race no. (%) (W, BAA, A, AIAN, NR) * |
|---|---|---|---|---|
| SaBrcada-BPPa | 1187 | 58 (26,90) | 912 (-7,8605) | 753 (68%), 182 (16%), 61 (5%), 1 (0.09%), 94 (9%) |
| SaBrcada-APPb | 807 | 57 (26,90) | 1026 (0,8605) | 583 (72%), 141 (17%), 34 (4%), 1 (0.1%), 48 (5%) |
| SaBrcada-ADc | 144 | 58 (25,90) | 1163 (0,7455) | 106 (74%), 30 (21%), 2 (1%), 0 (0%), 6 (4%) |
| SaBrcada-AYT61d | 84 | 46 (25,58) | 1439 (227, 7455) | 51 (74%), 15 (22%), 1 (1%), 0 (0%), 2 (3%) |
| SaBrcada-AOT61e | 60 | 69 (54,90) | 1004 (0,4267) | 55 (73%), 15 (18%), 1 (3%), 0 (0%), 4 (5%) |
| SaBrcada -trainf | 103 | 58 (25,90) | 1032 (0, 7455) | 77 (74%), 19 (18%), 2 (2%), 0 (0%), 5 (7%) |
| SaBrcada -testg | 41 | 58 (27,85) | 1692 (158, 3926) | 29 (71%), 11 (27%), 0 (0%), 0 (0%), 1 (2%) |
aBefore preprocessing, incomplete data were included; bAfter preprocessing; cAll data with actual death interval recorded; dPatients age younger than 61 years old; ePatients age older than 61 years old; fCombination of AYT61 and AOT61 training sets; gCombination of AYT61 and AOT61 testing sets. *W, White; BAA, Black or African American; A, Asian; AIAN, American Indian or Alaska Native; NR, Not Reported.
Table 2.
Comparison among different Convolutional Neural Network Architecture.
| Architecture | Accuracy | Batch Size | Epoch |
|---|---|---|---|
| Resnet18 | 0.50 | 8 | 50 |
| 0.49 | 16 | 100 | |
| 0.50 | 32 | 150 | |
| Resnet50 | 0.50 | 8 | 50 |
| 0.50 | 16 | 100 | |
| 0.50 | 32 | 150 | |
| Resnet101 | 0.50 | 8 | 50 |
| 0.50 | 16 | 100 | |
| 0.50 | 32 | 150 | |
| Resnet152 | 0.50 | 8 | 50 |
| 0.49 | 16 | 100 | |
| 0.50 | 32 | 150 | |
| ResNext101 | 0.50 | 8 | 50 |
| 0.50 | 16 | 100 | |
| 0.50 | 32 | 150 | |
| GoogLeNet* | 0.55 | 8 | 50 |
| 0.50 | 16 | 100 | |
| 0.60 | 32 | 150 | |
| DenseNet121 | 0.55 | 8 | 50 |
| 0.54 | 16 | 100 | |
| 0.54 | 32 | 150 | |
| DenseNet161 | 0.55 | 8 | 50 |
| 0.55 | 16 | 100 | |
| 0.53 | 32 | 150 |
Optimizer: Adam; Loss Function: CrossEntropyLoss. *SaBrcada adopted the architecture of GoogLeNet with Epoch 150 and Batch size 32.
Table 3.
Comparison of SaBrcada with other breast cancer survival analyses.
| Model | Number of Cancer | Type of Data | Patient Number | Method | C-index* /Accuracy† |
|---|---|---|---|---|---|
| SaBrcada-APP-M | 1a | mRNA | 807c | GoogLeNet | 0.500† |
| SaBrcada-AD-M | 1a | mRNA | 144c | GoogLeNet | 0.600† |
| SaBrcada-ASYT61-M | 1a | mRNA | 84c | GoogLeNet | 0.500† |
| SaBrcada-ASOT61-M | 1a | mRNA | 60c | GoogLeNet | 0.681† |
| SaBrcada | 1a | mRNA | 144c | GoogLeNet | 0.798† |
| VAECox (2019) | 10b | mRNA | 6127d | VAE, Cox | 0.649* |
| SALMON (2020) | 1a | mRNA, miRNA–target interactions | 626c | Cox | 0.700* |
| ConcatAE (2020) | 1 a | DNA methylation, miRNA | 1060e | ConcatAE | 0.641* |
aOnly one cancer type, breast cancer; b10 cancer types; c70% for training, 30% for testing; d80% for training, 20% for testing; e60% for training, 15% for validation, 25% for testing; *the performance was evaluated by C-index;†the performance was verified by accuracy.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



