
Submitted:
26 May 2023
Posted:
29 May 2023
You are already at the latest version
Abstract
Nanocomposite hydrogels offer remarkable potential for applications in bone tissue engineering. They are synthesized through the chemical or physical crosslinking of polymers and nanomaterials, allowing for the enhancement of their behaviour by modifying the properties and compositions of the nanomaterials involved. However, their mechanical properties require further enhancement to meet the demands of bone tissue engineering. Here, we present an approach to improve the mechanical properties of nanocomposite hydrogels by incorporating polymer grafted silica nanoparticles into a double network inspired hydrogel (gSNP Gels). The gSNP Gels were synthesised via a graft polymerization process using a redox initiator. gSNP Gels were formed by grafting 2-acrylamido-2-methylpropanesulfonic acid (AMPS) as the first network gel followed by a sequential second network acrylamide (AAm) onto amine functionalized silica nanoparticles (ASNPs). We utilized glucose oxidase (GOx), to create an oxygen-free atmosphere during polymerization resulting in higher polymer conversion compared to argon degassing. The gSNP Gels showed excellent compressive strengths of 13.9 ± 5.5 MPa, a strain of 69.6 ± 6.4%, and a water content of 63.4% ± 1.8. The synthesis technique demonstrates a promising approach to enhance the mechanical properties of hydrogels, which can have significant implications for bone tissue engineering and other soft tissue applications.
Keywords:
Polymer grafted silica nanoparticles
; redox polymerisation
; biomaterials
; glucose oxidase
; hydrogels
; tissue engineer
1. Introduction
Hydrogels are hydrophilic 3-dimensional polymeric networks that hold large amounts of water (up to 99 %) giving them their intrinsically soft material properties. They hold numerous properties including flexibility, transparency, permeability, biocompatibility, and low friction[1,2,3]. Hydrogels can be formed through natural, synthetic and hybrid polymers, offering a wide range of biochemical and mechanical properties [4,5]. Single network hydrogels are usually soft, weak and brittle. Their applications are limited due to their tensile and compressive properties being in the sub-MPa range, and inability to withstand strains greater than 100 % compared with the hydrogel-like natural-tissues such as articular cartilage, muscle, tendon, and blood vessels[6].
Hydrogel technology has evolved beyond its initial single network systems, driven by the inherent physical and biochemical limitations they possess[7]. This progress has led to the emergence of several innovative and practical hydrogel subtypes, including interpenetrating network hydrogels (IPNs), nanocomposite hydrogels, stimuli-responsive hydrogels, and double network hydrogels (DNHGs). DNHGs consist of two contrasting polymeric networks of polyelectrolytes (rigid and brittle) and neutral polymers (soft and ductile). The opposing properties of both networks have potential for achieving synergy of the mechanical properties of the two networks, replicating the physical and biochemical properties of native human tissue such as articular cartilage and bone[8,9,10]. This improvement in mechanical properties is crucial for bone tissue engineering, where different bone types exhibit varying compressive strengths. For instance, cortical bone demonstrates an ultimate compressive strength ranging from 130 to 180 MPa, while trabecular bone typically has a compressive strength of 1 to 5 MPa[11].
DNHGs are normally synthesized using a two-step sequential free-radical polymerisation (FRP) where a high relative molecular weight neutral second polymer network is swollen within a lightly cross linked heterogeneous first network polyelectrolyte[10]. DNHGs have been developed using a wide range of pairs consisting of polyelectrolyte first networks and neutral second networks, which have shown enhanced mechanical properties when compared to their individual network hydrogels[9]. A DNHG developed by Gong[10] consisting of poly(2-acrylamido 2-methylpropanesulfonic acid)/ polyacrylamide (PAMPS/ PAAm) had a compressive fracture stress of up to 20 MPa at 92% strain whilst holding 90 wt.% water. These properties closely matched that of native articular cartilage and trabecular bone[10]. In contrast, the respective single network hydrogel components had sub-MPa fracture stresses at 80% strain for PAAm and 40% for PAMPS. However, upon bearing stress the covalent bonds in conventional DNHGs exhibit permanent damage therefore the gels cannot recover from large deformations and suffer low fatigue resistance[9,12,13]. To improve on these limitations, additives such as nanoparticles can be introduced to create microstructures that increase resistance to strain and increase stiffness.
The introduction of nanocomposite structures such as silica nanoparticles (SNPs)[14,15], copper nanopowder[16,17], laponite clay[17], superparamagnetic iron oxide nanoparticles (SPIONs)[18], nanoceria (NC)[19] and nanocellulose crystals (CNC)[20,21,22] have shown to further enhance the physical properties of hydrogels. Nanocomposite hydrogels can provide unique properties based on the type of nanoparticle introduced into the system. Properties can include internal physical reinforcement and stiffness[8,14,19], topography for cellular anchorage crucial for cell adhesion and tissue regeneration[23], conductivity[24], antibacterial, antioxidation[25,26,27], therapeutic ion release[28,29], cancer therapeutics[30], magnetic responsiveness[18,31] and electrical signals and sensing. This expands their potential applications in specialized tissues such as skeletal muscle, nerve, and cardiac tissues.
However, nanocomposite gel structures are challenging to produce due to particle agglomeration, inhomogeneous distribution during synthesis, and a lack of covalent or ionic bonds between particles and the polymer networks[32,33,34,35,36]. This could result in weaknesses and potential points for failure when the hydrogel is under mechanical stress. To avoid these issues and to ensure a more robust entanglement of polymer chains with the nanocomposites, polymers can be grafted directly onto the nanoparticles to form strong covalent bonds. In previous studies, polymers were successfully grafted on the surface of SNPs, for example: polymethyl methacrylate (PMMA) by graft photopolymerisation[37], polystyrene with living radical polymerisation[38] and with atom transfer radical polymerisation (ATRP)[39]; poly-n-isopropylacrylamide (PNIPAM) brushes by ATRP[40], polystyrene sulfonic acid sodium salt (PSSA) and PAMPS by surface initiated redox polymerisation[41]. In a previous study, PAMPS and PSSA were grafted on the surface of amine-functionalized SNP (ASNPs)[41]. The ASNPs were dispersed in acidic solution of AMPS monomers before both the initiator ceric ammonium nitrate (CAN) and stabilizer sodium dodecyl sulphate (SDS) were added. The mixture was degassed with nitrogen gas and heated to 40 ᵒC to initiate the graft polymerisation. Grafting was initiated by a redox pair of Ce (IV) (oxidant) and the alkyl amine (reductant) from the surface of the ASNPs, causing an intermediate free radical at the α-carbon atom of the alkyl amine group. However, graft percentage and polymer conversion were low at 46 % and 4.6 % for PAMPS, and 22 % and 2.2 % for PSSA, respectively. These low percentages may cause issues in hydrogel formation due to low polymer grafts and extra post processing steps to remove unreacted monomers. The study aimed at increasing polymer graft percentage, however, attempts at hydrogel synthesis were not made. An optimization of the process would be required to create a polymer-nanocomposite that has the potential to be used in the synthesis of polymer grafted silica nanoparticle gels (gSNP Gels). In our previous work, polymers were successfully grafted on the surface of cerium oxide using a novel auto-catalytic graft polymerisation to form DNHGs for articular cartilage repair[19]. The process used a self-initiating cyclical polymerisation technique that benefited from the two alternating cerium ion states (Ce3+ and Ce4+) and reaction by-product H2O2.
In this work, a method for synthesizing a double network inspired nanocomposite hydrogel is presented. The method involves using a redox initiator, ceric ammonium nitrate, in combination with glucose oxidase (GOx), an oxidoreductase enzyme that can quench oxygen in an open system, eliminating all oxygen inhibition steps that impact FRP and increase the conversion of the polymer[14]. The redox polymerization is used to graft polymers with high conversion on ASNPs, and GOx is used to enhance the kinetic profile of each polymer by degassing all saturated oxygen in the monomer solution, allowing for a more efficient graft polymerization process to take place. To demonstrate the efficacy of GOx, a comparison with argon degassing will be made for both AMPS and AAm. The gSNP Gels are produced by grafting AMPS as a first network polyelectrolyte on the surface of ASNPs, followed by a sequential graft polymerization of AAm on ASNPs as the second network. Both networks are synthesized in oxygen-free atmospheres in the presence of GOx. The goal of this approach is to achieve improved integration of the nanocomposite structure through a polymer grafting process compared to conventional mixing of nanoparticles during synthesis.
2. Results and discussion
2.1. Amine functionalized silica nanoparticles (ASNPs).
ASNPs with 100 nm diameter were successfully synthesized by post synthesis functionalization of SNPs using APTES. The spherical shapes of bare SNPs (Figure 2a) and ASNPs (Figure 2b) were confirmed using TEM imaging, and size determined by taking an average diameter of 100 individual nanoparticles using the processing program Image J (Figure 2b). The average diameter of the ASNPs was 108 ± 6 nm (PDI 0.095), using direct light scattering (DLS). Functionalisation was confirmed using zeta potential (Figure 2c). Figure 2c shows bare nanoparticles (red line) had a zeta potential of -37 mV due to the deprotonated silanol groups on the surface of the SNPs while ASNPs (black line) had a surface zeta potential of + 27 mV due to the amine groups on the surface, in accordance with literature[46]. Figure 2d shows functionalised amine SNPs (black line) had a higher absorbance compared to bare SNPs (red line).
2.2. Polymers grafts on ASNPs
Both AMPS and AAm conversion profiles were investigated using samples that were degassed using either argon or GOx. The monomer and polymer peaks were calculated relative to trioxane peaks at 0 h and 24 h samples in order to calculate the final conversion profiles. Samples were tested using AAm and AMPS monomers with the redox initiator CAN, to compare impact on the polymerisation through the addition of ASNPs, or SDS. Figure 3a compares the conversion percentage of AAm and AMPS, respectively, with both argon and GOx degassing. The results demonstrate that use of GOx has a higher conversion percentage compared to argon degassing for all scenarios. GOx has been used previously as a degassing agent, and polymerisation mediator, to prevent oxygen inhibition of free radicals[14,19]. It has also been proven that GOx provides both AAm and AMPS with better reaction kinetics when compared to argon degassing[14].
An indication that H2O2, a GOx by-product, results in free radical formation which impacts the final polymer conversion can be seen by comparing argon and GOx degassed samples of AAm/ASNP. Argon degassed samples resulted in 1 % conversion, whereas samples degassed with GOx had 58 % conversion. This can occur during the formation of H2O2 and its subsequent in-situ degradation back to water where peroxy radicals may form, potentially initiating the polymerisation[42,47]. Therefore, it can be deduced that H2O2 plays a part in initiating the polymerisation to a limited extent. This was also evident for AMPS/ASNP in Figure 3b. Monomers were reacted with CAN alone, resulting in conversion of 85 % with argon and 94 % with GOx, for AAm. Therefore, it can be concluded CAN is an effective redox initiator for grafting AAm on the ASNPs. The results for AMPS/CAN were less notable, with only 57 % conversion using argon and 66 % conversion with GOx. Conversion percentage decreased with the addition of ASNPs, however, conversion reached 98 % and 100 % with the use of SDS for AAm and AMPS, respectively. The difference between AMPS and AAm conversion numbers under the same conditions are due to their intrinsic kinetic profiles. In a previous study, the use of amine functionalised nanoceria (ANC) as a nanoparticle based redox initiator resulted in over 90 % conversion for both AAm and AMPS, using the same nanoparticle concentration in this work[19]. ANCs, however, have the ability to use their cyclic cerium ion states to form free radicals on the tertiary amine group, compared to ASNPs that lack these intrinsic redox properties[19]. In a previous study, it was determined that the kinetic profile of AAm is more sensitive to oxygen inhibition when compared to AMPS during a photopolymerisation reaction without degassing[14]. Both AAm and AMPS had 0 % conversion at 0.05 wt. % photoinitiator. The addition of GOx to AAm and AMPS with a photoinitiator concentration of 0.05 wt. % resulted in 78 % and 100 % conversion, respectively[14,48]. Here, higher conversion rates were achieved at the lowest tested photoinitiator concentration. This provides clear evidence on the impact GOx has on both its ability to eliminate oxygen inhibition and significantly increase the reaction kinetics of both polymers.
In the experiments, it was observed that samples degassed with argon resulted in inhomogeneous, runny and wet gel structures. On the other hand, samples degassed with GOx showed textural consistency and more gel-like forms. These results indicate that argon alone would not be sufficient for synthesising the grafted nanocomposite hydrogels, while GOx can be used for this purpose, therefore, only GOx was used in the next part of the study.
Thermal analysis was performed on dried and ground samples to investigate polymer grafting onto amine silica nanoparticles (ASNP). The mass loss profiles (TGA) Figure 4a of AAm-based reactions showed that AAm/ASNP had a minor increase in mass loss compared to SNPs and ASNPs, indicating very low polymer grafting on the surface of the nanoparticles. This minor increase was due to the presence of a thin polymer monolayer, initiated by free radicals on the amine groups of ASNPs caused by the inert conditions created by GOx and its H2O2 by-products[14,19,49]. On the other hand, AAm/ASNP/CAN exhibited the largest mass loss, with two sharp DTG peaks at 250 ᵒC and 420 ᵒC (Figure 4a), indicating successful polymerisation on the surface of the ASNPs through the nature of the redox initiator, CAN, under completely inert conditions using GOx. The reaction including SDS also showed successful polymer grafting, but the presence of a distinctive DTG peak at 210 ᵒC suggested incomplete removal of SDS from the sample. The best combination was found to be AAm/ASNP/CAN.
DTG analysis of AMPS/ASNP showed prominent polymer peaks and a mass loss of approximately 12.5%, indicating successful polymer grafts on the surface of the nanoparticles (Figure 4b). As with AAm, bare SNPs were unable to initiate PAMPS polymer formation, and minor mass loss and DTG peaks for AMPS/ASNP samples suggested low amounts of polymer grafts on the surface, likely due to the H2O2 by-products resulting in free radicals on the amine groups of ASNPs. The presence of SDS in the sample suggested successful polymer grafting, but incomplete removal of SDS from the sample even after several washes (Figure 4b). The best result was found with AMPS/ASNP/CAN, showing strong DTG polymer peaks and the largest mass loss based on polymer formation, similarly to AAm.
FTIR analysis was conducted to confirm the surface modification of ASNPs through graft polymerisation of PAMPS and PAAm, with GOx as a degassing agent. The results of the FTIR analysis for the different combinations of PAAm with ASNP, CAN, and SDS compared to ASNP and PAAm homopolymer are shown in Figure 5a. The main bands associated with the AAm, and AMPS grafted polymers on the surface of ASNPs can be found in Table S1 and S2 in the supplementary information.
The FTIR spectrum of AAm/ASNP showed no polymer bands, which is consistent with the TGA results indicating a low concentration of grafted polymer on the surface. This is likely due to the absence of an initiator to start the polymerisation between AAm/ASNP. In contrast, the FTIR spectrum of AAm/ASNP/CAN clearly showed polymer bands that reflect the PAAm spectra. The presence of bands between 1620 cm-1 and 1659 cm-1 confirms the presence of N-H and C=O bonds from PAAm, while 950 cm-1 and 1050 cm-1 correspond to Si-OH and Si-O-Si, respectively, which represent ASNPs.
The FTIR spectrum of AAm/ASNP/CAN/SDS in Figure 5b also showed these bands, albeit with less intensity, and an additional twin band between 2850 cm-1 and 2956 cm-1 that represents the C-H stretching found from residual SDS[50]. The inability to remove SDS from the system despite several wash and centrifuge cycles was confirmed by the TGA analysis. The results from the FTIR analysis and TGA suggest that the best combination for grafting AAm on ASNPs was with CAN alone, as evident from the presence of polymer bands in the FTIR spectrum.
Figure 5c shows AAm polymer grafts were successfully grafted on ASNPs, with polymer peaks visible between 190 nm and 250 nm. The AAm/ASNP spectra indicates low polymer grafting on the ASNPs due to the changes in absorbance compared to ASNPs. SDS can also be seen between 220 nm – 260 nm, due to the difficulty in removing the stabiliser from the final product. AAm/ASNP/CAN had the strongest polymer peak with a reduced ASNP peak. This suggests a thick layer of polymer was grafted on the surface of the ASNPs.
AMPS/ASNP absorbance spectra (Figure 5d), revealed that a reaction had taken place on the nanoparticles. The reduced absorbance for ASNPs in the AMPS/ASNP sample suggests the nanoparticles were covered in polymer, though the peak at 190 nm was very subtle. AMPS/ASNP/CAN spectra revealed graft polymerisation was successful. The addition of SDS also revealed successful polymerisation, with a peak at 220 nm – 260 nm suggesting SDS was still present in the sample. These results indicate that grafting PAMPS on the surface of silica nanoparticles was successful. These results provide an indication that the polymerisation technique was successful.
2.3. Nanocomposite polymer grafted silica nanoparticle hydrogels (gSNP Gels)
gSNP Gels were successfully synthesised using a two-step sequential thermal graft polymerisation technique adapted for this study (Figure 6a). PAMPS were grafted on the surface of ASNPs with CAN as a redox initiator, in the presence of GOx, to form the first network hydrogel. This was followed by soaking the first network in a monomer solution of AAm, suspended ASNPs, CAN and GOx. The second network was formed through a secondary graft polymerisation under the same conditions. Samples were then swollen in water at room temperature until a mass plateau was achieved, and subsequently subjected to compression studies.
gSNP gels were swollen in DI-H2O from a dried state until a plateau in water content was reached. Figure 6b shows the swelling profile over time for the gSNP gels, and the results are summarised in Table 1. The gSNP gels exhibited fast water up take of approximately 19 % at 1 h, followed by a more gradual and controlled trend to reach an average of 63.44 ± 1.76 % at 240 h. The final swelling value was 274 ± 9.21 %. The presence of ASNPs reduced the free space for water uptake due to tighter cross linking by not allowing the gels to expand as freely. Also, ASNPs in both networks take up more space in the material as opposed to having one network cross linked via nanoparticles. Ultimately, this resulted in a slower and more controlled swelling profile containing less water, allowing for a tailorable material compared to previous DNHGs and nanocomposite gels that often result in water content of + 90% [8,10,19,51]. These values are considered in the lower range of the water content found in native articular cartilage (65 – 70 %).
Figure 7 shows the compression curves for gSNP gels, and Table 2 provides a summary of the results. An average compressive stress of 13.9 ± 5.5 MPa with fracture strain of 69.6 ± 6.4 % was achieved for gSNP gels, two orders of magnitude greater than control gels. The values for these hydrogels are comparable to the compressive strength of articular cartilage, with values ranging between 5 MPa and 20 MPa[52,53]. The compressive strength of nanocomposite hydrogels ranges from ~100 kPa to 70 MPa[12,54,55,56]. However, the results are highly dependent on testing methods and sample size during compression. Nonetheless, the improved compressive strength relative to control gels as well as other nanocomposite gels can be attributed to the covalently bonded polymer grafts on the surface of the ASNPs. The gSNP gels sustained strains up to 75 %, likely due to the internal structure of material. The polymer-ASNP composite structures are likely to interact and cross link with each other, leading to an increase in resistance to stress. This will lead to an increase in the compressive strength and allow the material to resist larger strains, as witnessed in the strain values in Figure 7. Ultimately, the gSNP Gels here show a 7 fold improvement in maximum compression strength relative to nanocomposite polymer grafted ANC Gels[19]. The synthesis method used in this work can be used to tailor the mechanical properties of these hydrogels further by varying the concentration of the two polymer-ASNP grafted networks. These gSNP Gels are intended for cyclic applications such as cartilage repair or for bone tissue regeneration.
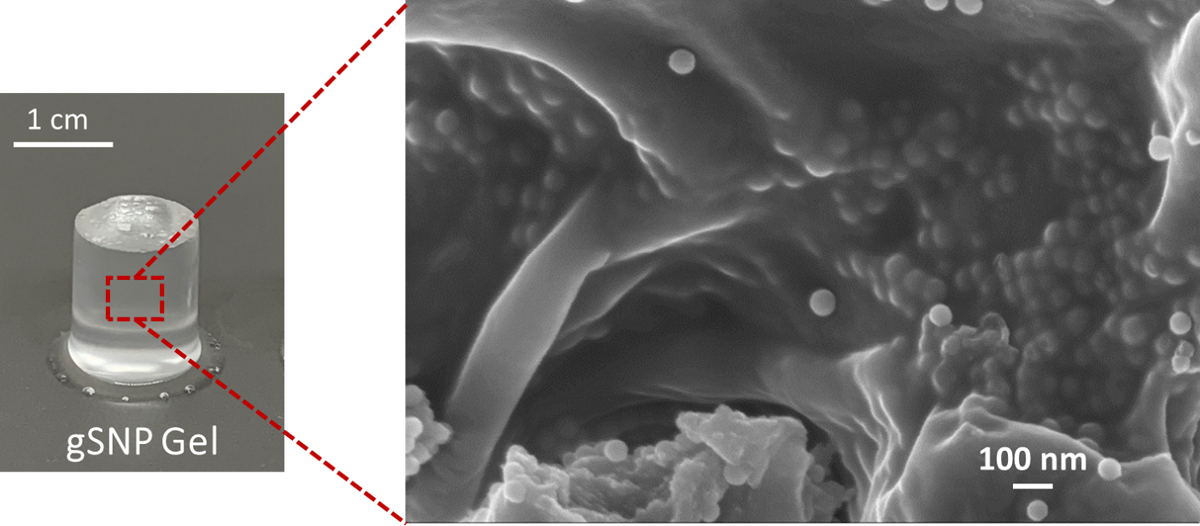
Cross sections of the gSNP Gels were freeze dried and investigated under SEM, shown in Figure 8. ASNPs were well distributed across the core structure of the hydrogel with homogenous layers, as shown by the white arrows in Figure 8a,b. The arrangement of the ASNPs within the hydrogel suggests that the synthesis technique did not hinder even distribution within the final material. The polymers on the surface of the ASNPs are likely to interact and cross link with neighbouring polymer-ASNP structures, leading to a more compact structure and an increase in resistance to stress. This compact ASNP arrangement and integration into the core structure revealed by the SEM reflects the increase seen in compressive strength of the material.
3. Conclusion
A double network inspired nanocomposite hydrogel made of first network PAMPS and second network PAAm, both grafted on amine functionalised silica nanoparticles in the presence of GOx, was successfully synthesised in this work. Polymer conversion for both PAMPs and PAAm was best for samples containing ASNP/CAN/SDS in the presence of GOx, both achieving 100 % conversion. The addition of GOx across all samples resulted in higher conversion % compared to controls degassed with argon. Ultimately, GOx proved to enhance the reaction kinetics profiles of both polymers compared to argon, resulting in more consistent gel-like polymers. GOx also showed an improvement in polymer conversion in the absence of the redox initiator CAN and allowed for open vessel synthesis due to its ability to maintain an oxygen free system. This was due to H2O2, a by-product of GOx, forming peroxy radicals that caused free radical formation on the tertiary amine group attached to the ASNPs. The grafted polymer networks were then sequentially polymerised to form a nanocomposite hydrogel with enhanced mechanical properties compared to a control without grafted ASNPs. A strong hydrogel with compressive fracture stress of 13.9 MPa at 69.6 % strain and 63.4 % water content was synthesised in an open vessel system with GOx. This compressive strength, and water content, are suitable for bone tissue engineering, as it exhibits mechanical properties comparable to soft tissues and articular cartilage. The tailorability of this hydrogel, achieved through the unique synthesis technique and the incorporation of GOx and ASNPs, opens up new possibilities for various applications in biomaterials. Future studies are needed to investigate the biological response of the nanocomposite hydrogels developed in this work. These hydrogels offer a feasible strategy to obtain tough scaffolds with enhanced mechanical properties, cell affinity, and osteoconductivity. They can also be loaded with bioactive molecules, such as growth factors or stem cells, to fabricate multifunctional hydrogels with the potential to direct bonding of the scaffold to the host bone and stimulate new bone formation.
4. Materials and Methods
Reagents were purchased from Sigma-Aldrich (UK) and used as received, unless stated otherwise: acrylamide (AAm; ≥ 99 %) and 2-acrylamido-2-methyl-1-propanesulfonic acid (AMPS; 99 %); N, N’-methylenebisacrylamide (MBIS; 99 %); photoinitiator 2-hydroxy-4′-(2-hydroxyethoxy)-2-methylpropiophenone (Irgacure 2959; 98 %), 1, 3, 5 –trioxane (≥ 99 %); (3-aminopropyl), ammonium hydroxide (28-30 % NH4OH basis), tetraethyl orthosilcate (TEOS; 98 %), triethoxysilane (APTES; ≥ 98 %), deuterium oxide (D2O; 99.9 atom % D), cerium (IV) ammonium nitrate (CAN; ≥ 99.99 % trace metals basis), sodium dodecyl sulphate (SDS; ≥ 98.5 %). No additional processing and/or purifications were performed. D-glucose (G) and glucose oxidase (GOx; from aspergillus niger as a lyophilized powder) were purchased from Sigma-Aldrich (UK) and stored in phosphate buffer saline (PBS) aliquots at -20 °C when received.
Amine functionalized silica nanoparticle (ASNP) synthesis, the procedure for polymer conversion studies of argon degassing and Gox, and the characterisation techniques used in this work can be found in the supplementary information.
4.1. Polymer grafted silica nanoparticle hydrogels (gSNP Gels) synthesis.
The first network was formed by dispersing 150 mg of ASNPs in 5 ml of H2O and sonicating the mixture until it was fully dispersed. 4.5 g AMPS was titrated to 5.4 pH using NaOH (0.25 M), to a final concentration of 0.24 M AMPS. ASNPs were then added to the AMPS monomer solution. 1 wt. % MBIS and 200 nM GOx/G[14,42] were added to the monomer/ASNP solution, followed by 0.7 g CAN and mixed for 5 minutes. The solution was then distributed into aliquots of 2 ml in polystyrene moulds and placed in a sonication bath set to 40 ᵒC to ensure homogenous ASNPs dispersion in the mixture. The optimum temperature for a GOx catalysed D-glucose oxidation is 40 ᵒC at pH 5.5, with denaturisation occurring at 60 ᵒC[43,44]. 40 ᵒC is also the temperature at which CAN becomes active to initiate the redox polymerisation[45]. Once the solutions have gelled, the moulds were placed in an oven at 40 ᵒC for 24 h followed by a further 6 h at 60 ᵒC to denature GOx.
The hydrogels were removed from the moulds and soaked in a monomer solution of 2.54 M AAm containing 150 mg ASNP, 0.1 wt. % MBIS, 0.7 g CAN and 200 nM GOx/G. Once the hydrogels were swollen, they were placed in moulds and put into a 40 ᵒC oven to form the second network. The final hydrogels are referred to as gSNP gels. The gSNP gels were then dried at 60 ᵒC and, at their dry state, placed in water to swell for a week. Figure 1 shows a schematic representation of the hydrogel synthesis route described above. A comparison control hydrogel was produced using the same concentrations of AMPS (first network) and AAm (second network) along with MBIS as a crosslinker, by means of a sequential FRP process, but without the presence of ASNPs and GOx.
4.2. Sample nomenclature
Each sample is named based on the chemicals used in its polymerization. For instance, if AMPS is polymerized using CAN and SDS on the surface of ASNPs, the sample will be named as AMPS/ASNP/CAN/SDS. If AAm is polymerized on ASNPs without any initiator or stabilizer, the sample will be named as AAm/ASNP.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
The manuscript was written by Dr Ali Mohammed. Dr. Alessandra Pinna assisted in silica nanoparticle synthesis and functionalization, Dr. Siwei Li assisted in glucose oxidase aliquots, and Dr. Tian Sang assisted in mechanical testing. All authors have given approval to the final version of the manuscript.
Acknowledgments
This work was funded by Qatar Foundation and EPSRC grant (EP/I020861/1), and the Marie Sklodowska-Curie Actions (MSCA) Individual fellowship (IF) in Horizon 2020 program of the European Union. Alessandra Pinna is currently an Imperial College Research Fellow.
References
- Hoffman, A.S. Hydrogels for biomedical applications. Advanced Drug Delivery Reviews 2012, 64, 18–23. [Google Scholar] [CrossRef]
- Zhang, Y.S.; Khademhosseini, A. Advances in engineering hydrogels. Science 2017, 356, eaaf3627. [Google Scholar] [CrossRef] [PubMed]
- Caló, E.; Khutoryanskiy, V.V. Biomedical applications of hydrogels: A review of patents and commercial products. European Polymer Journal 2015, 65, 252–267. [Google Scholar] [CrossRef]
- Tibbitt, M.W.; Anseth, K.S. Hydrogels as extracellular matrix mimics for 3D cell culture. Biotechnology and Bioengineering 2009, 103, 655–663. [Google Scholar] [CrossRef] [PubMed]
- Slaughter, B.V.; Khurshid, S.S.; Fisher, O.Z.; Khademhosseini, A.; Peppas, N.A. Hydrogels in Regenerative Medicine. Advanced Materials 2009, 21, 3307–3329. [Google Scholar] [CrossRef] [PubMed]
- Catoira, M.C.; Fusaro, L.; Di Francesco, D.; Ramella, M.; Boccafoschi, F. Overview of natural hydrogels for regenerative medicine applications. Journal of Materials Science: Materials in Medicine 2019, 30, 115. [Google Scholar] [CrossRef]
- Hoare, T.R.; Kohane, D.S. Hydrogels in drug delivery: Progress and challenges. Polymer 2008, 49, 1993–2007. [Google Scholar] [CrossRef]
- Gong, J.P. Why are double network hydrogels so tough? Soft Matter 2010, 6, 2583–2590. [Google Scholar] [CrossRef]
- Chen, Q.; Chen, H.; Zhu, L.; Zheng, J. Fundamentals of double network hydrogels. Journal of Materials Chemistry B 2015, 3, 3654–3676. [Google Scholar] [CrossRef]
- Gong, J.P.; Katsuyama, Y.; Kurokawa, T.; Osada, Y. Double-Network Hydrogels with Extremely High Mechanical Strength. Advanced Materials 2003, 15, 1155–1158. [Google Scholar] [CrossRef]
- Hart, N.H.; Nimphius, S.; Rantalainen, T.; Ireland, A.; Siafarikas, A.; Newton, R.U. Mechanical basis of bone strength: influence of bone material, bone structure and muscle action. J Musculoskelet Neuronal Interact 2017, 17, 114–139. [Google Scholar]
- Zhu, P.; Hu, M.; Deng, Y.; Wang, C. One-Pot Fabrication of a Novel Agar-Polyacrylamide/Graphene Oxide Nanocomposite Double Network Hydrogel with High Mechanical Properties Advanced Engineering Materials 2016, 18, 1799–1807. 18. [CrossRef]
- Sun, T.L.; Kurokawa, T.; Kuroda, S.; Ihsan, A.B.; Akasaki, T.; Sato, K.; Haque, M.A.; Nakajima, T.; Gong, J.P. Physical hydrogels composed of polyampholytes demonstrate high toughness and viscoelasticity. Nature Materials 2013, 12, 932–937. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, A.A.; Aviles Milan, J.; Li, S.; Chung, J.J.; Stevens, M.M.; Georgiou, T.K.; Jones, J.R. Open vessel free radical photopolymerization of double network gels for biomaterial applications using glucose oxidase. Journal of Materials Chemistry B 2019, 7, 4030–4039. [Google Scholar] [CrossRef]
- Mohammed, A.A.; Merrild, N.G.; Li, S.; Pinna, A.; Jones, J.R. Double-Network Hydrogels Reinforced with Covalently Bonded Silica Nanoparticles via 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide Chemistry. ACS Omega 2022, 7, 43904–43914. [Google Scholar] [CrossRef]
- Villanueva, M.E.; Diez, A.M.d.R.; González, J.A.; Pérez, C.J.; Orrego, M.; Piehl, L.; Teves, S.; Copello, G.J. Antimicrobial Activity of Starch Hydrogel Incorporated with Copper Nanoparticles. ACS Applied Materials & Interfaces 2016, 8, 16280–16288. [Google Scholar] [CrossRef]
- Chang, C.-W.; van Spreeuwel, A.; Zhang, C.; Varghese, S. PEG/clay nanocomposite hydrogel: a mechanically robust tissue engineering scaffold. Soft Matter 2010, 6, 5157–5164. [Google Scholar] [CrossRef]
- Mohammed, A.A.; Miao, J.; Ragaisyte, I.; Porter, A.E.; Myant, C.W.; Pinna, A. 3D printed superparamagnetic stimuli-responsive starfish-shaped hydrogels. Heliyon 2023, 9, e14682. [Google Scholar] [CrossRef]
- Mohammed, A.A.; Pinna, A.; Li, S.; Sang, T.; Jones, J.R. Auto-catalytic redox polymerisation using nanoceria and glucose oxidase for double network hydrogels. Journal of Materials Chemistry B 2020, 8, 2834–2844. [Google Scholar] [CrossRef]
- Zhou, C.; Wu, Q.; Yue, Y.; Zhang, Q. Application of rod-shaped cellulose nanocrystals in polyacrylamide hydrogels. Journal of Colloid and Interface Science 2011, 353, 116–123. [Google Scholar] [CrossRef]
- Abitbol, T.; Johnstone, T.; Quinn, T.M.; Gray, D.G. Reinforcement with cellulose nanocrystals of poly(vinyl alcohol) hydrogels prepared by cyclic freezing and thawing. Soft Matter 2011, 7, 2373–2379. [Google Scholar] [CrossRef]
- Kalossaka, L.M.; Mohammed, A.A.; Sena, G.; Barter, L.; Myant, C. 3D printing nanocomposite hydrogels with lattice vascular networks using stereolithography. Journal of Materials Research 2021, 36, 4249–4261. [Google Scholar] [CrossRef]
- Motealleh, A.; Dorri, P.; Schäfer, A.H.; Kehr, N.S. 3D bioprinting of triphasic nanocomposite hydrogels and scaffolds for cell adhesion and migration. Biofabrication 2019, 11, 035022. [Google Scholar] [CrossRef] [PubMed]
- Deng, Z.; Hu, T.; Lei, Q.; He, J.; Ma, P.X.; Guo, B. Stimuli-Responsive Conductive Nanocomposite Hydrogels with High Stretchability, Self-Healing, Adhesiveness, and 3D Printability for Human Motion Sensing. ACS Applied Materials & Interfaces 2019, 11, 6796–6808. [Google Scholar] [CrossRef]
- Pinna, A.; Torki Baghbaderani, M.; Vigil Hernández, V.; Naruphontjirakul, P.; Li, S.; McFarlane, T.; Hachim, D.; Stevens, M.M.; Porter, A.E.; Jones, J.R. Nanoceria provides antioxidant and osteogenic properties to mesoporous silica nanoparticles for osteoporosis treatment. Acta Biomaterialia 2021, 122, 365–376. [Google Scholar] [CrossRef]
- Pinna, A.; Cali, E.; Kerherve, G.; Galleri, G.; Maggini, M.; Innocenzi, P.; Malfatti, L. Fulleropyrrolidine-functionalized ceria nanoparticles as a tethered dual nanosystem with improved antioxidant properties. Nanoscale Advances 2020, 2, 2387–2396. [Google Scholar] [CrossRef] [PubMed]
- Pinna, A.; Lasio, B.; Piccinini, M.; Marmiroli, B.; Amenitsch, H.; Falcaro, P.; Tokudome, Y.; Malfatti, L.; Innocenzi, P. Combining Top-Down and Bottom-Up Routes for Fabrication of Mesoporous Titania Films Containing Ceria Nanoparticles for Free Radical Scavenging. ACS Applied Materials & Interfaces 2013, 5, 3168–3175. [Google Scholar] [CrossRef]
- Barrioni, B.R.; de Carvalho, S.M.; Naruphontjirakul, P.; Norris, E.; Kelly, N.L.; Hanna, J.V.; Jones, J.R.; Pereira, M.d.M. Cobalt-containing spherical glass nanoparticles for therapeutic ion release. Journal of the American Ceramic Society 2022, 105, 1765–1777. [Google Scholar] [CrossRef]
- Labbaf, S.; Tsigkou, O.; Müller, K.H.; Stevens, M.M.; Porter, A.E.; Jones, J.R. Spherical bioactive glass particles and their interaction with human mesenchymal stem cells in vitro. Biomaterials 2011, 32, 1010–1018. [Google Scholar] [CrossRef]
- Chen, S.; Greasley, S.L.; Ong, Z.Y.; Naruphontjirakul, P.; Page, S.J.; Hanna, J.V.; Redpath, A.N.; Tsigkou, O.; Rankin, S.; Ryan, M.P.; et al. Biodegradable zinc-containing mesoporous silica nanoparticles for cancer therapy. Materials Today Advances 2020, 6, 100066. [Google Scholar] [CrossRef]
- Kim, Y.; Yuk, H.; Zhao, R.; Chester, S.A.; Zhao, X. Printing ferromagnetic domains for untethered fast-transforming soft materials. Nature 2018, 558, 274–279. [Google Scholar] [CrossRef]
- Vashist, A.; Kaushik, A.; Ghosal, A.; Bala, J.; Nikkhah-Moshaie, R.; A Wani, W.; Manickam, P.; Nair, M. Nanocomposite Hydrogels: Advances in Nanofillers Used for Nanomedicine. Gels 2018, 4, 75. [Google Scholar] [CrossRef] [PubMed]
- Esmaeely Neisiany, R.; Enayati, M.S.; Sajkiewicz, P.; Pahlevanneshan, Z.; Ramakrishna, S. Insight Into the Current Directions in Functionalized Nanocomposite Hydrogels. Frontiers in Materials 2020, 7. [Google Scholar] [CrossRef]
- Haraguchi, K. Nanocomposite hydrogels. Current Opinion in Solid State and Materials Science 2007, 11, 47–54. [Google Scholar] [CrossRef]
- Rafieian, S.; Mirzadeh, H.; Mahdavi, H.; Masoumi, M.E. A review on nanocomposite hydrogels and their biomedical applications. Science and Engineering of Composite Materials 2019, 26, 154–174. [Google Scholar] [CrossRef]
- Zhao, H.; Liu, M.; Zhang, Y.; Yin, J.; Pei, R. Nanocomposite hydrogels for tissue engineering applications. Nanoscale 2020, 12, 14976–14995. [Google Scholar] [CrossRef]
- Kim, S.; Kim, E.; Kim, S.; Kim, W. Surface modification of silica nanoparticles by UV-induced graft polymerization of methyl methacrylate. Journal of Colloid and Interface Science 2005, 292, 93–98. [Google Scholar] [CrossRef]
- von Werne, T.; Patten, T.E. Preparation of Structurally Well-Defined Polymer−Nanoparticle Hybrids with Controlled/Living Radical Polymerizations. Journal of the American Chemical Society 1999, 121, 7409–7410. [Google Scholar] [CrossRef]
- El Harrak, A.; Carrot, G.; Oberdisse, J.; Jestin, J.; Boué, F. Atom transfer radical polymerization from silica nanoparticles using the ‘grafting from’ method and structural study via small-angle neutron scattering. Polymer 2005, 46, 1095–1104. [Google Scholar] [CrossRef]
- Wu, T.; Zhang, Y.; Wang, X.; Liu, S. Fabrication of Hybrid Silica Nanoparticles Densely Grafted with Thermoresponsive Poly(N-isopropylacrylamide) Brushes of Controlled Thickness via Surface-Initiated Atom Transfer Radical Polymerization. Chemistry of Materials 2008, 20, 101–109. [Google Scholar] [CrossRef]
- Salarizadeh, P.; Abdollahi, M.; Javanbakht, M. Modification of silica nanoparticles with hydrophilic sulfonated polymers by using surface-initiated redox polymerization. Iranian Polymer Journal 2012, 21, 661–668. [Google Scholar] [CrossRef]
- Chapman, R.; Gormley, A.J.; Herpoldt, K.-L.; Stevens, M.M. Highly Controlled Open Vessel RAFT Polymerizations by Enzyme Degassing. Macromolecules 2014, 47, 8541–8547. [Google Scholar] [CrossRef]
- Zoldák, G.; Zubrik, A.; Musatov, A.; Stupák, M.; Sedlák, E. Irreversible Thermal Denaturation of Glucose Oxidase from Aspergillus niger Is the Transition to the Denatured State with Residual Structure*. Journal of Biological Chemistry 2004, 279, 47601–47609. [Google Scholar] [CrossRef] [PubMed]
- Anjum Zia, M.; Khalil Ur, R.; M, K.S.; Andaleeb, F.; M, I.R.; M, A.S.; I, A.K.; A, I.K. Thermal Characterization of Purified Glucose Oxidase from A Newly Isolated Aspergillus Niger UAF-1. J Clin Biochem Nutr 2007, 41, 132–138. [Google Scholar] [CrossRef] [PubMed]
- Gupta, K.C.; Sahoo, S.; Khandekar, K. Graft copolymerization of ethyl acrylate onto cellulose using ceric ammonium nitrate as initiator in aqueous medium. Biomacromolecules 2002, 3, 1087–1094. [Google Scholar] [CrossRef]
- Lu, H.-T. Synthesis and characterization of amino-functionalized silica nanoparticles. Colloid Journal 2013, 75, 311–318. [Google Scholar] [CrossRef]
- Gormley, A.J.; Chapman, R.; Stevens, M.M. Polymerization Amplified Detection for Nanoparticle-Based Biosensing. Nano Letters 2014, 14, 6368–6373. [Google Scholar] [CrossRef]
- Mohammed, A. Double network hydrogels for cartilage repair and their nanocomposite structure. 2017. [CrossRef]
- Berron, B.J.; Johnson, L.M.; Ba, X.; McCall, J.D.; Alvey, N.J.; Anseth, K.S.; Bowman, C.N. Glucose Oxidase-Mediated Polymerization as a Platform for Dual-Mode Signal Amplification and Biodetection. Biotechnology and Bioengineering 2011, 108, 1521–1528. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Xie, X.; Li, C.; Wang, H.; Wang, L. The role of soft colloidal templates in the shape evolution of flower-like MgAl-LDH hierarchical microstructures. RSC Advances 2015, 5, 29757–29765. [Google Scholar] [CrossRef]
- Gu, Z.; Huang, K.; Luo, Y.; Zhang, L.; Kuang, T.; Chen, Z.; Liao, G. Double network hydrogel for tissue engineering. WIREs Nanomedicine and Nanobiotechnology 2018, 10, e1520. [Google Scholar] [CrossRef]
- Stammen, J.A.; Williams, S.; Ku, D.N.; Guldberg, R.E. Mechanical properties of a novel PVA hydrogel in shear and unconfined compression. Biomaterials 2001, 22, 799–806. [Google Scholar] [CrossRef]
- Oka, M.; Ushio, K.; Kumar, P.; Ikeuchi, K.; Hyon, S.H.; Nakamura, T.; Fujita, H. Development of artificial articular cartilage. Proceedings of the Institution of Mechanical Engineers, Part H: Journal of Engineering in Medicine 2000, 214, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Hou, R.; Cheng, Y.; Fu, J. Super-tough double-network hydrogels reinforced by covalently compositing with silica-nanoparticles. Soft Matter 2012, 8, 6048–6056. [Google Scholar] [CrossRef]
- Xu, B.; Li, H.; Wang, Y.; Zhang, G.; Zhang, Q. Nanocomposite hydrogels with high strength cross-linked by titania. RSC Advances 2013, 3, 7233–7236. [Google Scholar] [CrossRef]
- Yang, J.; Han, C.-R.; Duan, J.-F.; Ma, M.-G.; Zhang, X.-M.; Xu, F.; Sun, R.-C.; Xie, X.-M. Studies on the properties and formation mechanism of flexible nanocomposite hydrogels from cellulose nanocrystals and poly(acrylic acid). Journal of Materials Chemistry 2012, 22, 22467–22480. [Google Scholar] [CrossRef]
Figure 2.
(a) Surface functionalization of silica nanoparticles (SNPs) by APTES to produce amine functionalised silica nanoparticles (ASNPs), (b) TEM image of bare silica nanoparticles (SNPs) and (c) amine silica nanoparticles (ASNPs); (d) Zeta potential of SNPs and ASNPs using 5 mg/ ml per sample, and (e) UV-VIS of bare SNPs and ASNPs using 5 mg/ ml per sample.
Figure 2.
(a) Surface functionalization of silica nanoparticles (SNPs) by APTES to produce amine functionalised silica nanoparticles (ASNPs), (b) TEM image of bare silica nanoparticles (SNPs) and (c) amine silica nanoparticles (ASNPs); (d) Zeta potential of SNPs and ASNPs using 5 mg/ ml per sample, and (e) UV-VIS of bare SNPs and ASNPs using 5 mg/ ml per sample.

Figure 3.
Conversion profiles of (a) AAm and (b) AMPS in combination with amine functionalised silica nanoparticles (ASNP), ASNP/CAN, and ASNP/CAN/SDS, degassed with argon (black) and GOx (red).
Figure 3.
Conversion profiles of (a) AAm and (b) AMPS in combination with amine functionalised silica nanoparticles (ASNP), ASNP/CAN, and ASNP/CAN/SDS, degassed with argon (black) and GOx (red).
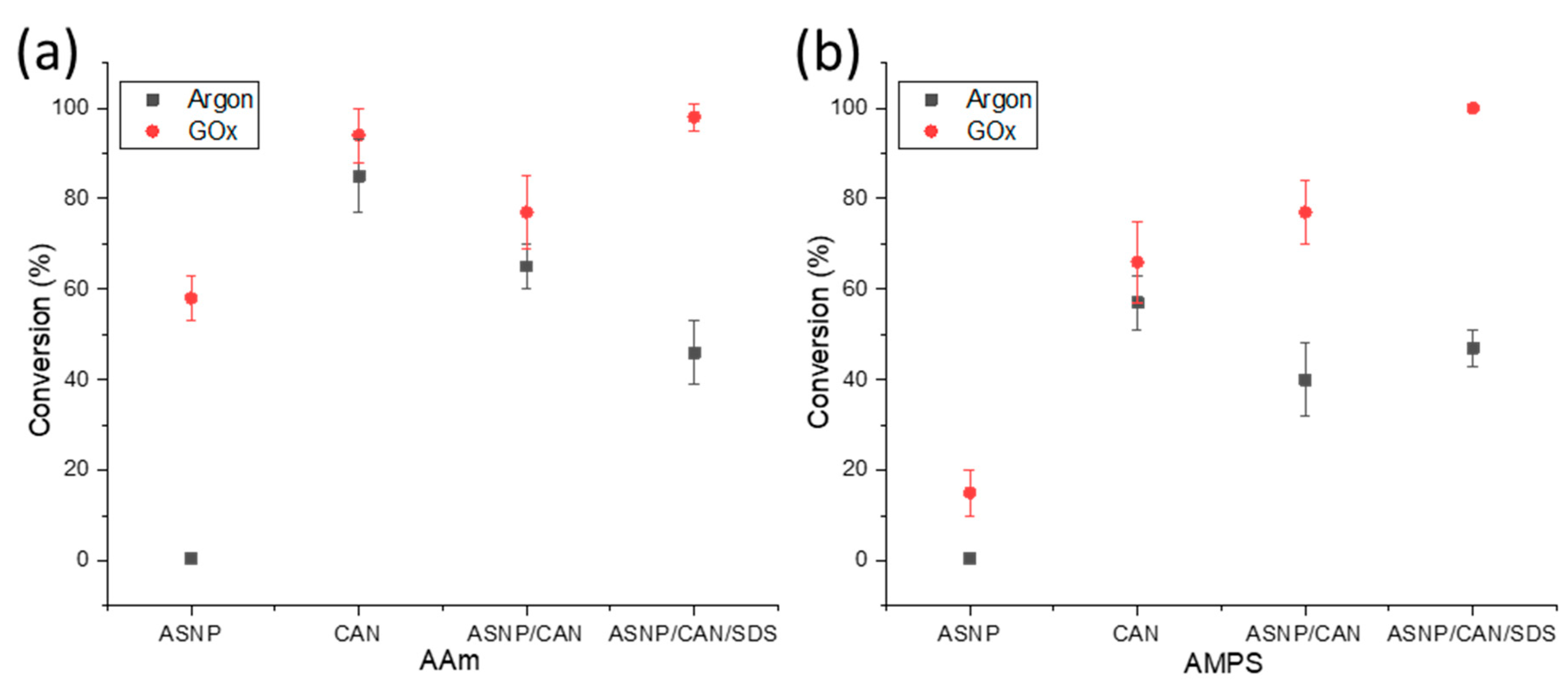
Figure 4.
DTG and TGA profiles for (a) AAm and (b) AMPS degassed using GOx with ASNP, ASNP/CAN and ASNP/CAN/SDS.
Figure 4.
DTG and TGA profiles for (a) AAm and (b) AMPS degassed using GOx with ASNP, ASNP/CAN and ASNP/CAN/SDS.
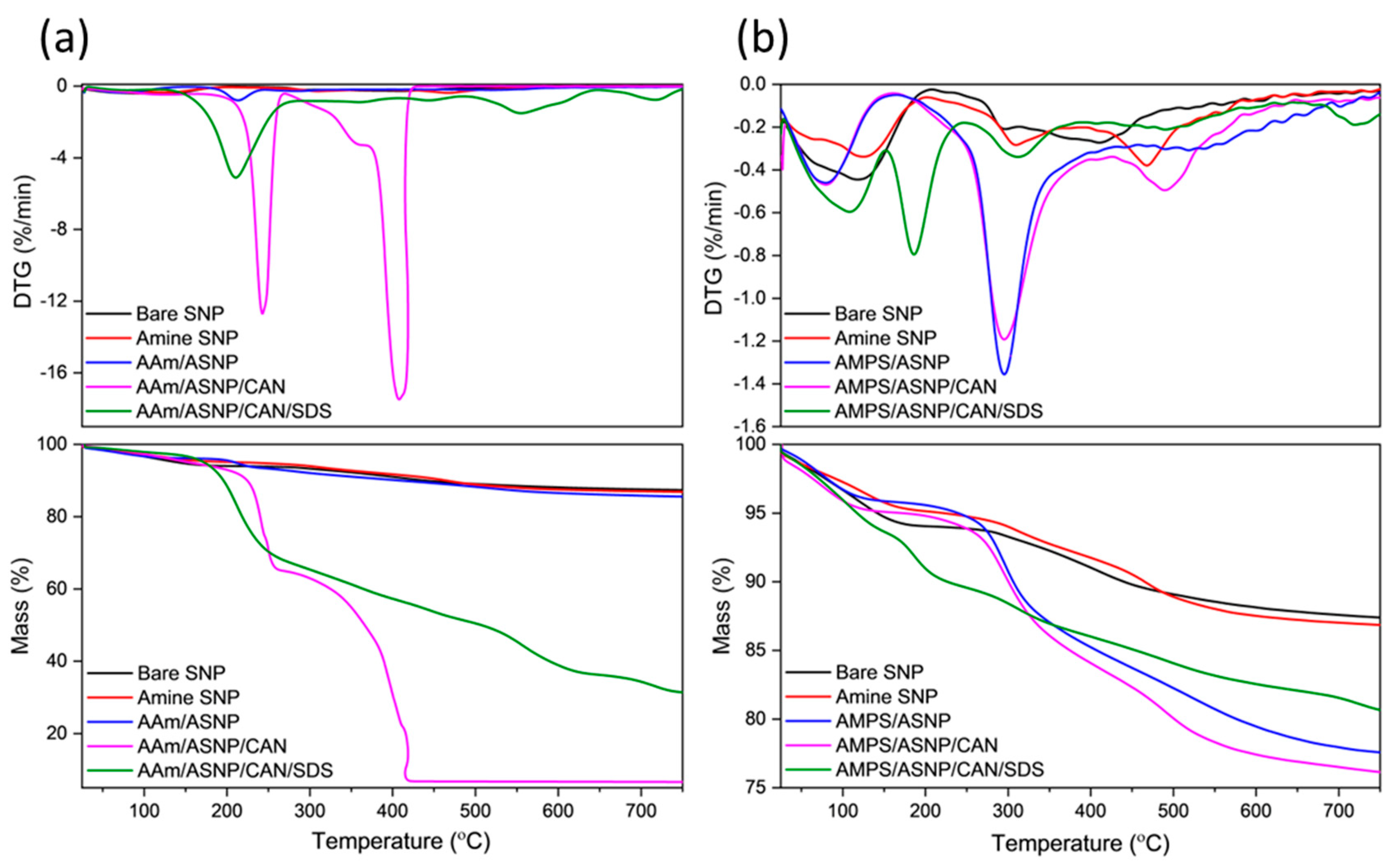
Figure 5.
FTIR spectra post graft polymerisation of (a) AAm and (b) AMPS on the surface of amine silica nanoparticles (ASNP) compared to ASNPs, PAAm and PAMPS, degassed using GOx. Band indexing A – J shown in Supplementary information Table S1 and S2. UV-Vis of (c) AAm and (d) AMPS in combination with amine functionalised silica nanoparticles (ASNP), ASNP/CAN, and ASNP/CAN/SDS compared to ASNP.
Figure 5.
FTIR spectra post graft polymerisation of (a) AAm and (b) AMPS on the surface of amine silica nanoparticles (ASNP) compared to ASNPs, PAAm and PAMPS, degassed using GOx. Band indexing A – J shown in Supplementary information Table S1 and S2. UV-Vis of (c) AAm and (d) AMPS in combination with amine functionalised silica nanoparticles (ASNP), ASNP/CAN, and ASNP/CAN/SDS compared to ASNP.
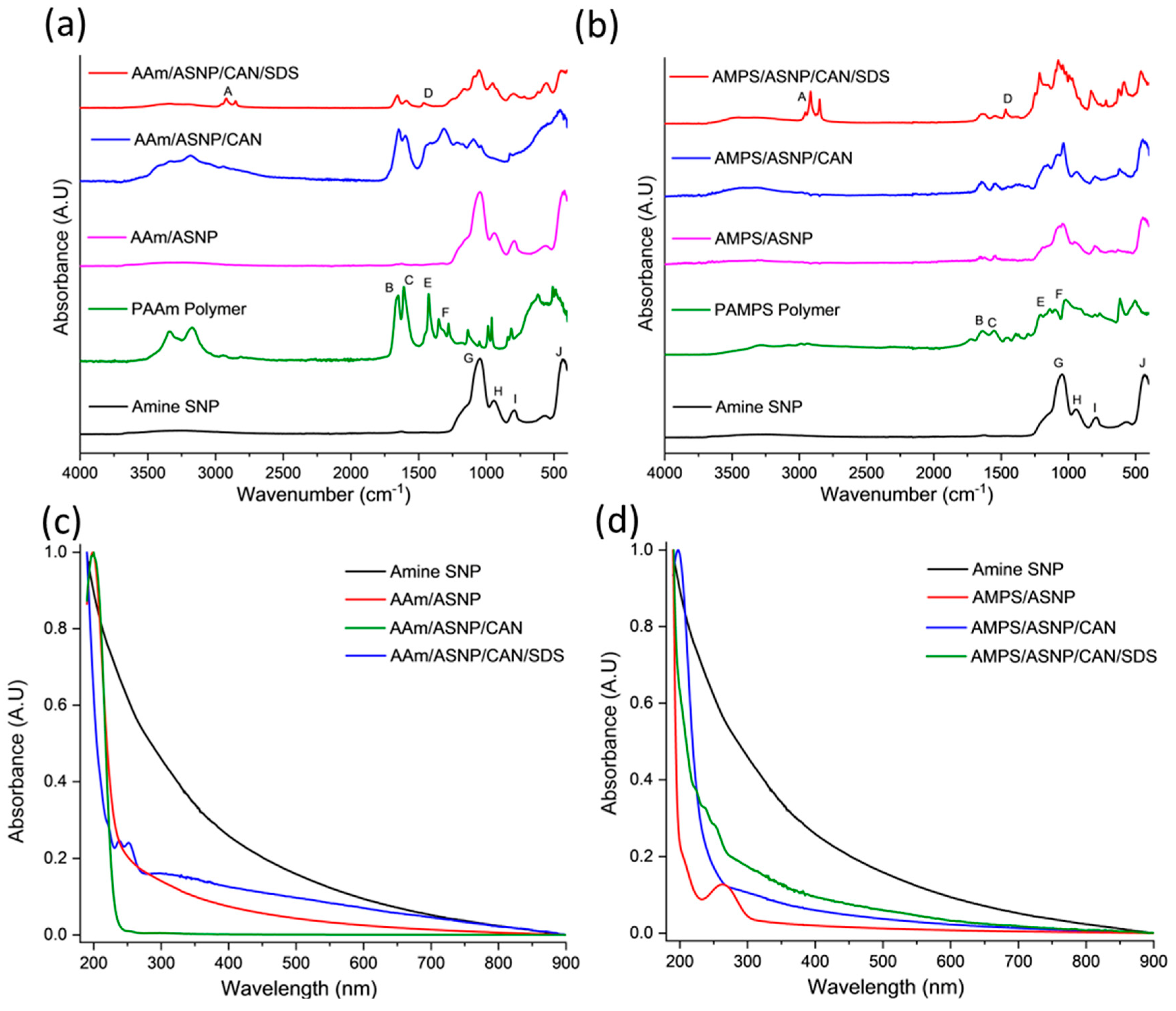
Figure 6.
(a) Images of the amine silica nanoparticle hydrogels with PAMPS and PAAm polymer grafts (gSNP gels). (b) Swelling profile for the amine silica nanoparticle hydrogel with PAMPS and PAAm polymer grafts (gSNP gels), and Control (gels without ASNPs/ GOx.).
Figure 6.
(a) Images of the amine silica nanoparticle hydrogels with PAMPS and PAAm polymer grafts (gSNP gels). (b) Swelling profile for the amine silica nanoparticle hydrogel with PAMPS and PAAm polymer grafts (gSNP gels), and Control (gels without ASNPs/ GOx.).
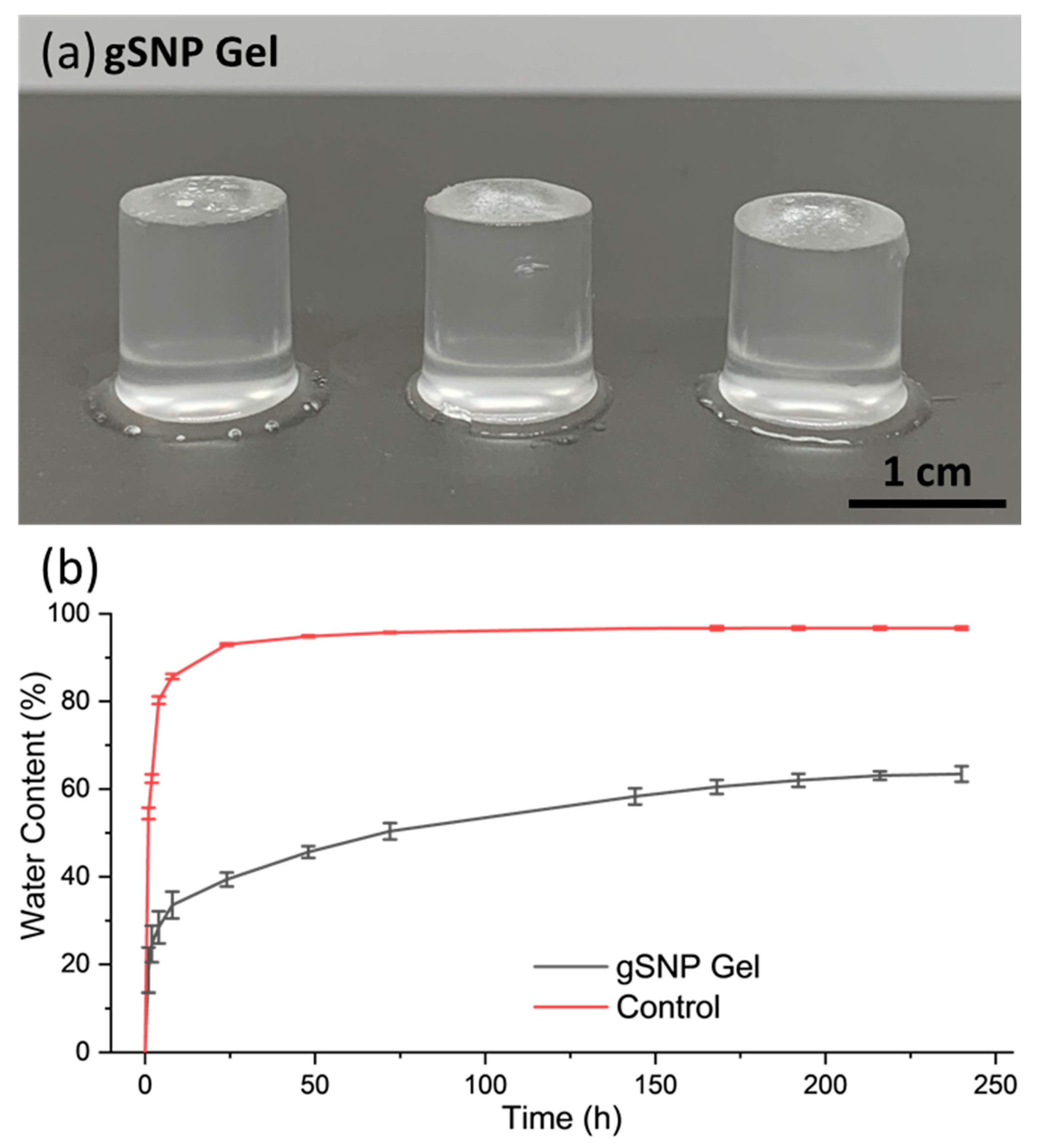
Figure 7.
(a) Images of a nanocomposite PAMPS and PAAm grafted silica nanoparticle hydrogel (gSNP Gel) under compression at increasing strains. (b) Compression curves of the gSNP Gel, compared to a control hydrogel without ASNPs/ GOx.
Figure 7.
(a) Images of a nanocomposite PAMPS and PAAm grafted silica nanoparticle hydrogel (gSNP Gel) under compression at increasing strains. (b) Compression curves of the gSNP Gel, compared to a control hydrogel without ASNPs/ GOx.
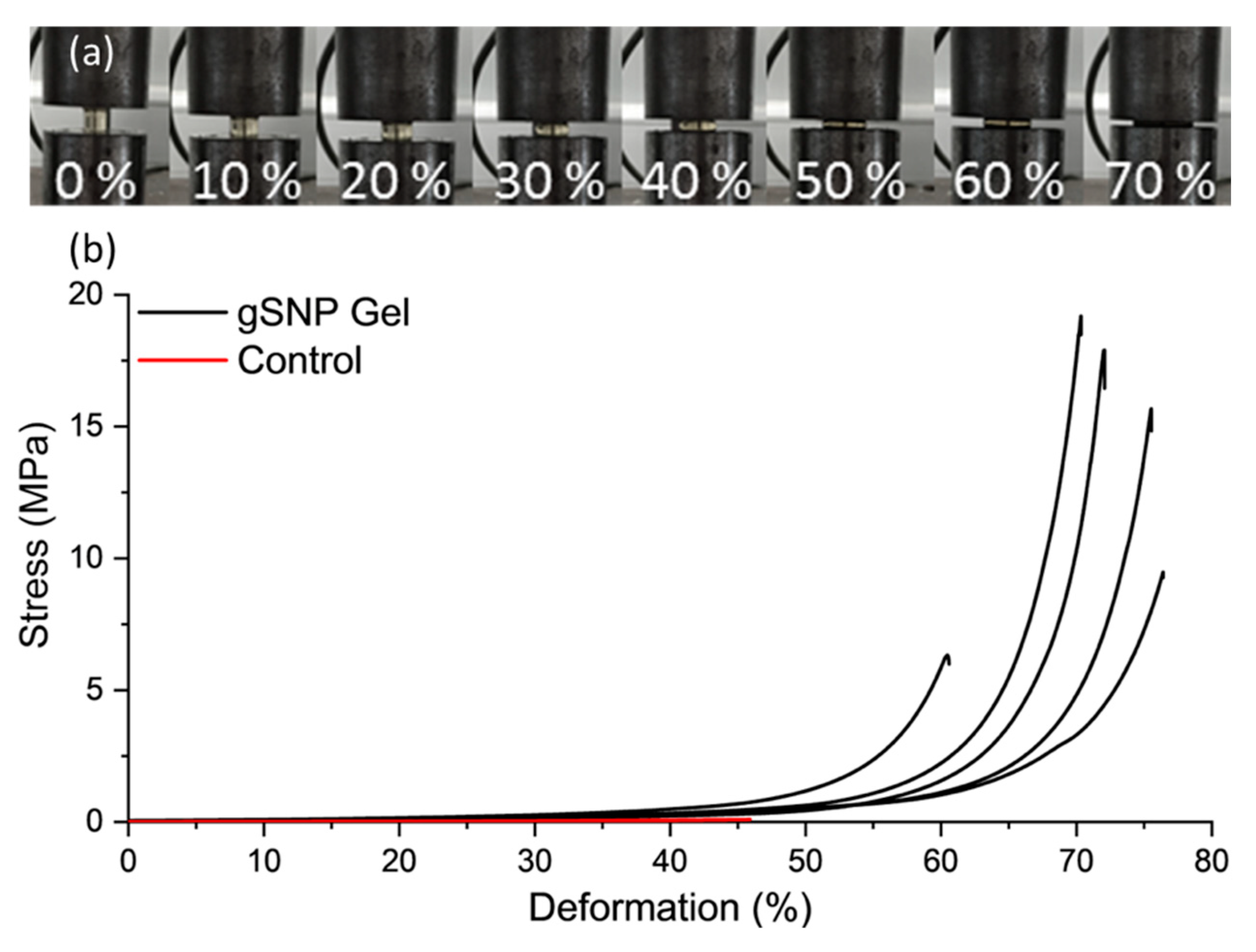
Figure 8.
SEM images of nanocomposite PAMPS and PAAm grafted silica nanoparticle hydrogels (gSNP Gel) at varying magnifications: (a) 30K X, (b) 50 K X. ASNPs are highlighted with white arrows.
Figure 8.
SEM images of nanocomposite PAMPS and PAAm grafted silica nanoparticle hydrogels (gSNP Gel) at varying magnifications: (a) 30K X, (b) 50 K X. ASNPs are highlighted with white arrows.

Figure 1.
Schematic representation showing the synthesis route for polymer grafted silica nanoparticle gels (gSNP Gels).
Figure 1.
Schematic representation showing the synthesis route for polymer grafted silica nanoparticle gels (gSNP Gels).
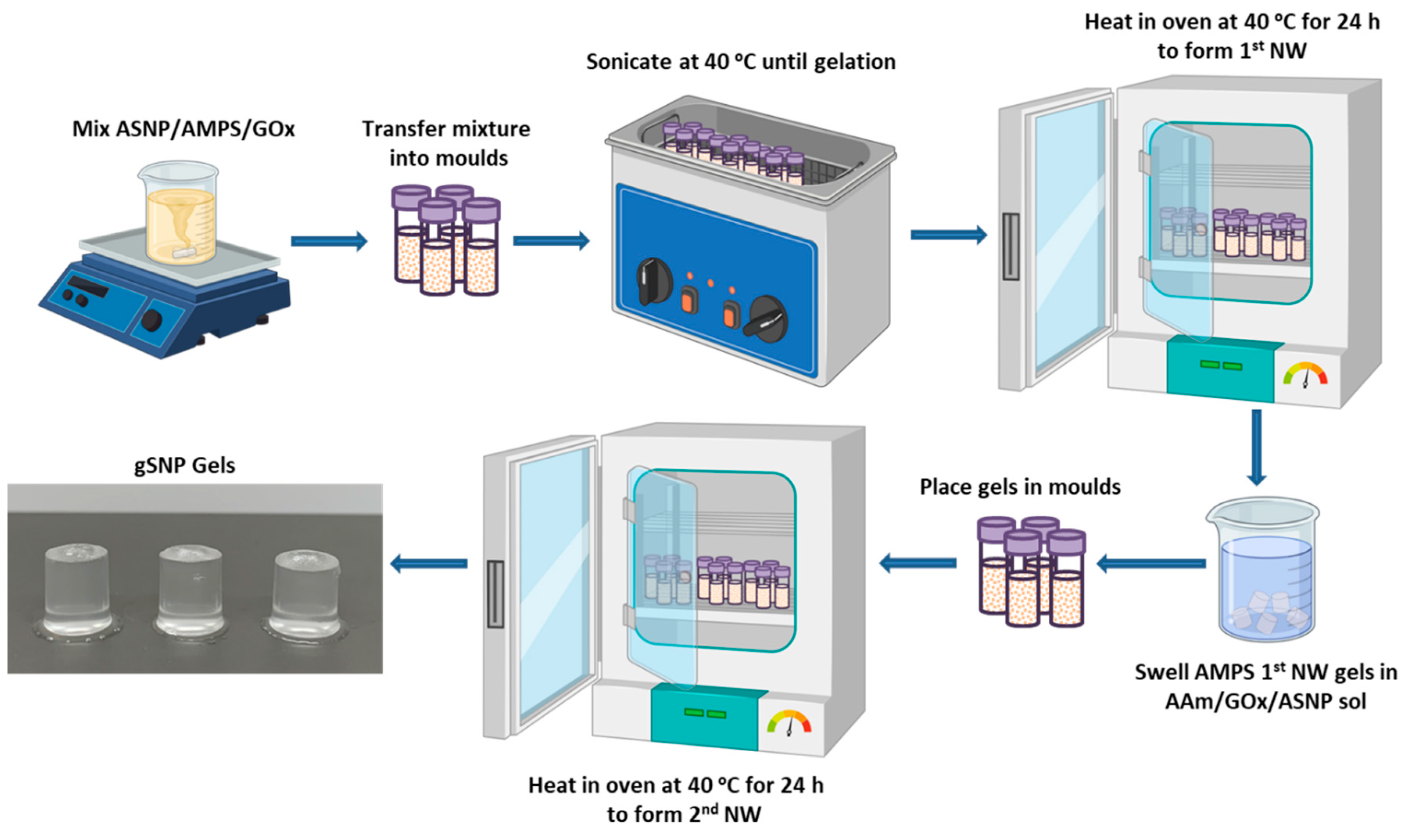

Table 1.
Swelling and water content for the amine silica nanoparticle hydrogel with PAMPS and PAAm polymer grafts (GSNP gel).
Table 1.
Swelling and water content for the amine silica nanoparticle hydrogel with PAMPS and PAAm polymer grafts (GSNP gel).
| Control Gel (no ASNPs/ GOx) | gSNP gel | |
|---|---|---|
| Water Content (%) | 96.7 % ± 0.4 | 63.4 % ± 1.8 |
| Swelling (%) | 2757 % ± 157 | 274 % ± 9 |
Table 2.
Fracture compressive stress and strain values for amine silica nanoparticles hydrogels with PAMPS and PAAm polymers grafts (gSNP Gel).
Table 2.
Fracture compressive stress and strain values for amine silica nanoparticles hydrogels with PAMPS and PAAm polymers grafts (gSNP Gel).
| Control Gel | gSNP Gel | |
|---|---|---|
| Compressive Fracture Stress (MPa) | 0.10 ± 0.06 | 13.9 ± 5.5 |
| Fracture Strain (%) | 45.9 ± 2.1 | 69.6 ± 6.4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



