
Submitted:
26 May 2023
Posted:
29 May 2023
You are already at the latest version
Abstract
The fluorinated organic compounds have superior physicochemical properties than general organic compounds due to the strong C-F single bond, and widely used in medicine, biology, pesticides, and materials science. In order to gain a deeper understanding of the physicochemical properties of fluorinated organic compounds, fluorinated aromatic compounds have been investigated by various spectroscopic techniques. 2-Fluorobenzonitrile and 3-fluorobenzonitrile are important fine chemical intermediates, and their excited state S1 and cationic ground state D0 vibrational features remain unknown. In this paper, we used two-color resonance two photon ionization (2-color REMPI) and mass analyzed threshold ionization (MATI) spectroscopy to study S1 and D0 states vibrational features of 2-fluorobenzonitrile and 3-fluorobenzonitrile. The precise excitation energy (band origin) and adiabatic ionization energy were determined to be 36028 2 cm-1 and 78650 5 cm-1 for 2-fluorobenzonitrile; and 35989 2 cm-1 and 78873 5 cm-1 for 3-fluorobenzonitrile, respectively. The density functional theory (DFT) at the levels of RB3LYP/aug-cc-pvtz, TD-B3LYP/aug-cc-pvtz, and UB3LYP/aug-cc-pvtz were used to calculate the stable structures and vibrational frequencies for the ground states S0, excited state S1, and cationic ground state D0, respectively. Franck-Condon spectral simulations for transitions of S1 S0 and D0 S1 were performed based on above DFT calculations. The theoretical and experimental results are in good agreement. The observed vibrational features in S1 and D0 states are assigned according to the simulated spectra and the comparison with structurally similar molecules. Several experimental findings and molecular features were discussed in detail.
Keywords:
fluorobenzonitrile
; vibronic spectroscopy
; cationic spectroscopy
; MATI
; Franck-Condon simulation
1. Introduction
Due to the presence of the strong C-F single bond within the molecule, fluorinated organic compounds have superior physicochemical properties than general organic compounds, such as high thermal stability, high oxidative stability, weak intermolecular interaction and weak surface tension, which make them widely used in medicine, biology, pesticides, and materials science [1,2,3,4,5]. In recent years, in order to gain a deeper understanding of the physicochemical properties of fluorinated organic compounds, a large number of fluorinated aromatic compounds have been investigated by various spectroscopic techniques. In 2017, Ling et al. used femtosecond time-resolved photoelectron imaging to study the conformation of bi-fluorophenol and bi-fluoroaniline in the excited state S1 after photoexcitation [6,7]. Wijngaarden's group used high-resolution microwave spectroscopy to measure the rotation spectra of fluorine substituted benzaldehyde, benzonitrile, phenol, and pyridine derivatives to study the molecular structure changes caused by fluorination and intramolecular hydrogen bonding interactions [8,9,10]. Many experimental groups have also studied the vibrational spectra of excited state S1 and cationic ground state D0 of fluorine substituted phenol, anisole, and aniline derivatives using laser-induced fluorescence (LIF), resonance-enhanced multiphoton ionization (REMPI), and mass analyzed threshold ionization (MATI) spectroscopy [11,12,13,14]. Mono-fluorobenzonitrile is a very important class of intermediate for organic synthesis, and their vibrational and rotational properties have been reported in many studies [15,16,17,18]. Kamaee et al. investigated the structural trends in mono-, di-, and pentafluorobenzonitriles using Fourier transform microwave spectroscopy [10]. Palmer et al. measured photoelectron spectroscopy of 2-fluorobenzonitrile (2FBN) and 3-fluorobenzonitrile (3FBN), and reported the ionization energies (IE) of 9.78 eV and 9.79 eV, respectively [19]. Jiang and Levy used laser induced fluorescence and dispersive fluorescence spectroscopy to study the vibrational relaxation of the excited state of 4-fluorobenzonitrile (4FBN) molecule [20]. In 2018, Zhao et al. [21] studied the vibrational features of the excited and cationic ground states of 4FBN by REMPI and MATI techniques. Silva et al. [16] measured the UV-Vis spectra of monofluorobenzonitriles in dichloromethane, and from the curves they measured, the approximate origins of 2FBN and 3FBN can be estimated at 283 nm. To the best of our knowledge, the vibrational properties of the excited states and cationic ground states of 2FBN and 3FBN have not been reported in the literature.
MATI and zero kinetic energy (ZEKE) spectroscopy are currently the most popular high-resolution techniques for measuring vibrational features of cationic ground states. ZEKE spectroscopy detects the electrons and MATI detects the ions yielded by field ionization of Rydberg neutrals. Due to the different masses of various ions, MATI spectroscopy has the ability of eliminating impurity interference. Kwon’s group built a vacuum ultraviolet single photon MATI system to study many cationic vibrational features [22,23,24,25,26,27,28,29]. Tzeng’s group and Ketkov’s group used two-color MATI to study the cationic spectra of many benzene derivatives and sandwich molecules [30,31,32,33,34,35,36]. Wright’s group used ZEKE technology to research cationic vibrational features of many halogenated benzene and their derivatives [37,38,39,40,41,42]. In this paper we used two-color REMPI and MATI techniques to study the vibrational features of the excited states and cationic ground states of 2FBN and 3FBN. The precise excitation energies and adiabatic ionization energies were determined. The measured vibrational features were assigned, and several experimental findings are analyzed and discussed in detail.
2. Results
The stable structures of 2- and 3-fluorobenzonitrile with atomic labels are shown in Figure 1. 2FBN and 3FBN molecules consist of 13 atoms with a total of 33 normal vibrational modes, 30 modes of which are at aromatic ring and 3 modes at CN group. The labeling convention of the vibrational modes follows the Varsanyi’s system [43]. Vibronic transitions are expressed in the Wilson notation based on benzene modes, where the v’← v” transition in the normal mode n is represented by [44], and subscript V" is omitted in present research as it is a constant 0 (The low energy level of the transition is the vibrationless or zero point energy level of the low electronic state).
2.1. Vibronic Features of 2-Fluorobenzonitrile in the S1 State
The vibronic spectrum of the S1←S0 transition of 2FBN was measured by two-color REMPI experiment with the vibration frequency range of 0 – 1350 cm-1. The experimental result is shown in Figure 2a, and its Franck-Condon simulation calculated at B3LYP/aug-cc-pvtz level is shown in Figure 2b. It can be seen that the experimental result is in good agreement with the calculated one. The obvious feature of both REMPI and its simulation in Figure 2 is that the rate of signal-to-noise in low frequency region is greater than that in high frequency region. The simulation spectrum shows that the bands in high frequency region are dense, and consist of many fundamentals, overtones, and combinations of various modes, many of which are very weak. So, dense and weak bands raise the spectral baseline and result in a bad rate of signal-to-noise in high frequency region. The band at certain frequency in the spectrum maybe come from several component (or vibration mode) contributions. For simplicity's sake, we only list the largest contributor in Table 1.
Based on DFT calculation and spectral simulation, we analyzed and assigned the vibronic spectra of 2FBN. It is very clear in Figure 2a that the band at 36028 cm-1 is assigned to the band origin of the S1←S0 transition. Many in-plane vibrational modes of the ring are active, and most of them are very strong in the REMPI spectrum. The bands at 136, 341, 424, 500, 668, 815, 946, 1171, and 1257 cm-1 are assigned to fundamental modes 15, 6b, 9b, 6a, 1, 12, 18b, 13, and 7a, respectively. One out-of-plane fundamental mode at the ring was observed, which appeared at 693 cm-1, and assigned to mode 17a. Several overtone vibrations were observed, which appeared at 170, 635, and 846 cm-1, and assigned to γCN2, 16b2, and 9b2, respectively. Other bands observed in REMPI spectrum are assigned to the combined vibrations of several modes. All the measured vibrational frequencies, calculated frequencies, and possible assignments are listed in Table 1.
From measured REMPI spectra in Figure 2a, we can find that the vibronic band 11 is much wider than other bands. From simulation calculation we knew that the band 11 consists of three components: 16a110a1 (665.2 cm-1), 11 (667.5 cm-1), and 6b2 (669.7 cm-1). The calculated dipole strength at the level of TD-B3LYP/aug-cc-pvtz for these three components are 3.365E-5, 1.254E-2, and 3.2E-3, respectively. Due to the very close vibrational energy, the resonance interactions may have played a role in the broadening of the experimental spectral line.
2.2. Photoionization Efficiency (PIE) Spectra of 2FBN
In order to measure the cationic spectra, we require first to know the ionization energy (IE). With the present experimental setup, the IE can be measured by photoionization efficiency (PIE) or MATI experiments. The PIE approach detects the prompt ions involving the field-ionization of high n Rydberg neutrals, and yield a strong signal to leads to an abruptly rising step near the ionization limit. In contrast, the MATI spectrum detects the threshold ions and yields a sharp peak at the ionization threshold and vibrational features of the cation. We have recorded both the PIE and MATI spectra by scanning the frequency of the ionization laser over a large range to determine the IE of 2FBN. Figure 3a,b show the PIE and MATI spectra via the intermediate state S100 (36028 cm-1). The adiabatic IE of 2FBN was determined to be 78647 ± 10 cm-1 by PIE and 78650 ± 5 cm-1 (9.7514 ± 0.0006 eV) by MATI including the correction of Stark effect, respectively. These results are in good agreement with the previous measured value of 9.78 eV (78881 cm-1) [19] by photoelectron spectroscopy with He I UV-light source.
2.3. Cationic Spectra of 2FBN
To investigate the molecular geometry and vibrational features of the 2FBN cation, the MATI spectra were recorded by ionizing via the S100, S16b1 (00 + 341cm-1), S111 (00 + 668 cm-1), S1121 (00 + 815 cm-1), and S118b1 (00 + 946 cm-1) intermediate states.
We first performed the theoretical calculation and spectral simulation. The Franck-Condon simulation is shown in Figure 4a, and the corresponding MATI spectrum via S100 is shown in Figure 4b. From Figure 4a,b we know that the theoretical and experimental spectra are in good agreement. The most intense peak corresponds to the origin of the D0 ← S1 transition of 2FBN. Spectral features were assigned, mainly based on DFT calculations, Franck-Condon simulation, and comparisons with the available data on substituted benzonitriles. Spectral assignment is a very tedious and error prone thing. Accurate assignments can be obtained by high dimensional or even full dimensional vibrational calculations [45,46]. For the present work, we use the Franck-Condon simulation, which greatly facilitates spectral identification work. The bands at 131, 333, 530, 571, 683, 826, 972, 1268, and 1555 cm-1 are relatively intense and assigned to ring or CN group in-plane motion modes 15, 6b, 6a, βCN, 1, 12, 18b, 7a, and 8b, respectively. Several out-of-plane bending vibrations were also observed, such as γCN and 10b appearing at 106 and 197 cm-1, respectively. Other bands are weak and assigned to overtone or combination vibrations. The measured and calculated cationic vibrational frequencies, and their possible assignments are listed in Table 2.
In order to find more vibrational modes of 2FBN cation, the different intermediate states were used to record MATI spectra. Figure 4c shows the MATI spectra via S16b1 (00 + 341cm-1). Comparing with Figure 4b, we can find that when S16b1 is used as the intermediate state, most of spectral features can be assigned to combinations of 6b and the modes found in MATI via S100. This can be verified by shifting Figure 4c to the left to align its band 6b with the 0+ band in Figure 4b. No more fundamental modes than the MATI via S100 are found.
Figure 5 shows the MATI spectra via the intermediate states of S111 (00 + 668 cm-1), S1121 (00 + 815 cm-1), and S118b1 (00 + 946 cm-1). Similarly, when S111 (00 + 668 cm-1) was used as the intermedia state, a lot of bands are assigned to the combination vibrations of the mode 1 and those found in MATI via S100. In the lower frequency region, substituent CN out-of-plane bending γCN and its overtone γCN2 were found. Aromatic ring out-of-plane bending 10a and its overtone 10a2 were also observed. Other bands are weak and assigned to combination vibrations of several modes.
Similarly, when S1121 is used as the intermediate, as shown in Figure 5b, except for the bands at 1064 and 1646 cm-1 being assigned to 6a2 and 122, other bands greater than 823 cm-1 (D0121) are assigned to combinations of 121 and other modes. In lower frequency region, some fundamental modes are active, which have been found in the MATI spectrum via S100 or S111. For the MATI via S118b1 in Figure 5c, the spectral feature is similar to the MATI via S1121, and all the assignments as well as the calculated and measured values are listed in Table 2.
2.4. Vibronic Features of 3-Fluorobenznitrile in the S1 State
The vibronic spectrum in the S1 state of 3FBN is shown in Figure 6a together with its Franck-Condon simulation shown in Figure 6b for comparing. The entire simulated spectra appear comparable to the 2-color REMPI spectra in Figure 6a. The distinct band corresponding to the transition energy of 35989 cm−1 is identified as the origin of the S1←S0 electronic transition. Table 3 lists the observed vibronic transition energies along with the energy shifts with respect to the band origin, band relative intensities, and possible assignments. The spectral assignment of 3FBN was accomplished by comparing with those of 4-fluorobenzonitrile, 3-fluorophenol, TD-B3LYP/aug-cc-pvtz calculation, and Franck-Condon simulation. The spectral features in Figure 6a mainly result from vibronic transitions related to the in-plane ring deformation and substituent sensitive bending vibrations. The bands appeared at 140, 385, 401, 470, 560, 660, 958, 1147, 1271, and 1374 cm-1 are assigned to in-plane stretching or bending vibrations 15, 9a, 6b, 6a, βCN, 1, 12, 9b, 13, and 19a, respectively. The out-of-plane overtone vibrations γ(CN)2 and 10b2 are also observed in lower frequency region. Other bands are assigned to combination vibrations of several modes.
2.5. PIE Spectra of 3FBN
Similar to the 2FBN, ionization energy is very important for the cationic spectral measurements. We first performed the PIE experiment to determine the IE of 3FBN to be 78873 ± 10 cm-1, and then measured the MATI spectra to give the precise IE of 3FBN to be 78873 ± 5 cm-1. The PIE and MATI spectra via S100 are shown in Figure 7a,b for comparing. It is obvious that they are very consistent.
2.6. MATI Spectra of 3FBN
Figure 8a,b show the calculated Franck-Condon spectrum and measured MATI spectrum via S100 state at 35989 cm-1, respectively. We can see that they are in good agreement. Many in-plane vibrations are active, such as modes 15, 6b, 6a, 1, 12, 18a, 9b, 18b, 13, and 8a appearing at 133, 371, 498, 668, 978, 1066, 1117, 1144, 1307, and 1566 cm-1, respectively. Out-of-plane bending modes 10b and 10a are also observed, but they are weak. Other bands are assigned to combinations of several modes. All the experimental and calculated cationic vibrational frequencies of 3FBN and corresponding assignments are listed in Table 4.
When measuring the MATI via S1γCN2 (Figure 8c), the distinct feature at 238 cm-1 is assigned to D0γCN2, which follows the propensity rule Δν = 0. The fundamental vibration γCN1 was also observed at 120 cm-1 with a weak intensity, which did not appear in the REMPI spectrum. Other bands are assigned to combination vibrations of γCN2 and fundamental vibrations.
When measuring the MATI via S16b1, as shown in Figure 9a, the distinct feature at 370 cm-1 is assigned to D06b1, which follows the propensity rule Δν = 0. The intense band at 399 cm-1 is assigned to 9a1. Other bands are assigned to combination vibrations of 6b1 and fundamental vibrations. Figure 9b shows the MATI spectrum via S111, where the cationic vibration 11 (668 cm-1) is most intense. The bands at 775, 803, and 1166 cm-1 are assigned to combination vibrations 4110b1, 11151, and 116b1, respectively.
3. Discussion
3.1 Breathing Vibrational Band of 2FBN
Whether the vibrational spectra of excited state S1 or cationic ground state D0, the frequencies of different vibrations in the high-frequency region maybe very close or even the same, which may come from the fundamental, overtone or combination vibrations. For example, the breathing vibration 11 of 2FBN in the REMPI spectrum (see Figure 2a) appears at 668 cm-1. The theoretical calculation shows that there are also two weaker vibrations 16a110a1 and 6b2, whose vibrational frequencies are close to that of mode 11. The calculated vibration frequencies of 16a110a1, 11 and 6b2 are 665.2 cm-1, 667.5 cm-1 and 669.7 cm-1, respectively. They are so close that the spaces between them are less than the experimental resolution, which leads to a wide spectral band in the REMPI spectrum. When using this band as the intermediate state to perform the MATI experiment, according to the propensity rule of Δν = 0, these three vibrational modes of cation may be observed with great intensity. Generally, the vibration frequency of cation is slightly different from that of the excited state for the same vibrational mode, and the frequency change can be not consistent for various vibration modes. So, these three vibration modes of cation of 2FBN may be separated in the MATI spectrum. As shown in Figure 5a, the cationic mode 11 appeared at 687 cm-1, 6b2 appeared at 672 cm-1, and 10a1 and 10a2 were also observed at 322.7 and 643.2 cm-1, respectively. The strength of the MATI signal is not only related to the Franck-Condon factor, but also to the population of the intermediate state S1, and further related to the resonance degree of each vibration mode with the excitation (S1 ← S0) photon frequency. The experimental results demonstrate that the superposition band of several vibrations can be used as an intermediate state to do the MATI experiments, and more vibrational modes of cation can be observed.
3.2. Molecular Structure in S0, S1, and D0 States and Vibrational Frequencies
Theoretical calculations show that the stable configurations of the ground state S0, excited state S1, and cationic ground state D0 of 2FBN and 3FBN molecules all have Cs symmetry, and all the atoms are in the ring plane. This is consistent with their large Franck-Condon factors, intense REMPI and MATI signals, and MATI spectra following the propensity rule of Δν = 0. However, in the transitions of S1 ← S0 and D0 ← S1, the bond length and bond angle of molecules have changed slightly. Table 5 and Table 6 show the bond lengths and bond angles of the S0, S1, and D0 states of 2FBN and 3FBN calculated at levels of RB3LYP/ang-cc-pvtz, TD-B3LYP/ang-cc -pvtz, and UB3LYP/ang-cc-pvtz, respectively. It can be seen that the bond lengths between adjacent carbon atoms of the ring of 2FBN are very close to the corresponding bond lengths of 3FBN. After the transition of S1 ← S0, each C-C bond length increased, resulting in the perimeters of ring of 2FBN and 3FBN increased by 0.160 Å and 0.158 Å, respectively. The transition of D0 ← S1 leads to the shortening of four C-C bonds and lengthening of two C-C bonds. The overall effect of D0 ← S1 transition is that the perimeters of ring of 2FBN and 3FBN decrease by 0.073 Å and 0.072 Å, respectively. Further, the ring C-C bond lengths of D0 state is averagely larger than that of S0 state. The perimeters of the aromatic ring of 2FBN and 3FBN at the cationic ground states are 0.087 Å and 0.086 Å larger than those of the neutral ground state S0, respectively. That is, the perimeters or average bond lengths of the ring in the ground state S0, excited state S1, and cationic ground state D0 meet the relationship: S0 < D0 < S1. The length of a chemical bond reflects, to some extent, the strength of that bond. The greater the bond length, the weaker the bond strength. The frequency of an ideal oscillator is proportional to the square root of the bond strength, so the larger the bond length, the lower the vibration frequency. On this basis, we can predict that, on average, the vibration frequencies of the ground state S0, the excited state S1 and the cationic ground state D0 meet the relationship: S0 > D0 > S1. The 33 normal vibration frequencies calculated at the B3LYP/ang-cc-pvtz level of 3FBN were statistically analyzed. On average, the vibration mode frequency of the ground state S0 is about 21 cm-1 greater than that of the cationic ground state D0, and the vibration frequency of D0 is about 43 cm-1 greater than that of S1. For example, the frequencies of breathing vibration mode 1 for S0, D0, and S1 of 2FBN measured in the experiment are 724 [18], 685, and 668 cm-1, respectively; for the mode 12 are 835 [18], 823, and 815 cm-1, respectively, and for the mode 18b are 1100 [18], 973, and 946 cm-1, respectively. The reported experimental and theoretical data of mFBT and mDFB [47] also indicate that most of the vibrational modes of these two molecules also follow this rule. Furthermore, from above vibration data we know that the frequency variation is larger for out-of-plane mode (such as 18b of 2FBN) than for in-plane mode (such as modes 1 and 12 of 2FBN). Our DFT theoretical results show that this law holds for most vibration modes of benzene derivative.
For 2FBN and 3FBN, the bond lengths of C-N in S0 state are equal (1.152 Å), also equal in S1 state (1.165 Å), and almost equal in D0 state (1.158 and 1.155 Å). This means that the CN bond is very strong and did not changed with substitution position (ortho- or meta-). The CF bond length yields slight change with different substitution positions.
Aromatic ring includes six bond angles of C-C-C. In the electronic transition, the bond angle of the ring also undergoes a certain degree of change. Four angles have a variation of approximately 2-3 °, and other two have relatively small changes. In the transitions of S1 ← S0 and D0 ← S1, variations of the bond angle and bond length of rings lead to the ring expansion and contraction, further activating a large number of in-plane vibration modes. Most of the observed vibronic features in experiments were assigned to in-plane vibrations, only a few of out-of-plane modes were observed. Many benzene derivative molecules exhibit such characteristics [48,49,50,51,52,53].
3.3. Substitution Effect on Ionization Energy
Molecular IE is an important parameter of molecular characteristics. In order to study the effect of fluorine and CN substitutions on ionization energy, we listed the IEs of benzene [54], fluorobenzene [55], benzonitrile [56], 2-fluorobenzonitrile, 3-fluorobenzonitrile, p-fluorobenzonitrile [21], phenol [57], o-fluorophenol [58], m-fluorophenol [59,60] and p-fluorophenol [11] in Table 7, and divided them into four groups for comparison. First, we can find that three molecular IEs reduced with respect to their parent molecules, i.e. for the fluorine substitution, the ionization energy of fluorobenzene, 4-fluorophenol, and 4-fluorobenzonitrile are reduced by 330, 490, and 48 cm-1 compared with their parent molecules, respectively, where fluorine plays a role of electron donor. However, fluorine substituted ortho and meta benzonitrile slightly increased the IEs by 160 and 383 cm-1, respectively; and fluorine substituted ortho and meta (cis and trans) phenols increased the IEs by 1381, 1563, and 1824 cm-1, respectively. For these substitutions, fluorine exhibits electron withdrawing properties. It can be seen that the role of fluorine changes with the characteristics of parent molecule and different substitution positions. Unlike fluorine substitution, CN substituted benzene and fluorobenzene at ortho, meta, and para positions increase the ionization energy by 3933, 4423, 4646, and 3773 cm-1, respectively, playing a role of strong electron withdrawing.
In addition, we can see from Table 7 that the effects of ortho and meta substitution on ionization energy are very close, while the effect of para substitution is relatively weak. Moreover, the IEs of molecules formed by ortho, meta and para substitutions meets the relative relationship: para < ortho < meta. Most benzene derivative molecules follow this rule.
4. Materials and Methods
4.1. Experimental Methods
The 2-fluorobenzonitrile and 3-fluorobenzonitrile samples were purchased from J&K Chemical and Sigma-Aldrich company, respectively. They were used without further purification. They are colorless or light brown liquid with a purity of 99%. The sample is heated to about 130 °C for 2FBN and 60°C for 3FBN to obtain sufficient vapor pressure. 3 bar krypton for 2FBN and 2.5 bar argon for 3FBN were used as the carrier gases, and they carried the sample molecules into the beam source chamber through a pulse valve of 0.5 mm diameter nozzle (0.8 mm for 3FBN). And then, the molecule beam entered the ionization chamber through a skimmer located 20 mm downstream from the nozzle orifice. The vacuum pressures of the beam source and ionization chambers are ~4 × 10-4 Pa and ~6 × 10-6 Pa, respectively.
The light source consists of two sets of dye lasers pumped by YAG lasers. One dye laser (CBR-D-24, Sirah) pumped by a frequency-tripled Nd: YAG laser (Qsmart 850, Quantel) was used as the excitation laser. Another dye laser (Precision Scan-D, Sirah) pumped by another frequency-tripled Nd: YAG laser (Qsmart 850, Quantel) was used as the ionization laser for two-color REMPI or probe laser for MATI experiments. The dyes of coumarin 540A and coumarin 460 or coumarin 480 were used for the excitation and ionization lasers, respectively. The dye laser wavelengths were calibrated by a wavemeter (WS7-60 UV-I). The fundamental outputs of the dye lasers were further frequency-doubled by BBO crystals.
Due to the strong electron withdrawing ability of the CN group, the transition energies of S1←S0 are lower than those of D0←S1 for 2FBN and 3FBN. Such an energy structure indicates that two sets of light sources are required for the measurement of excited state spectra. In the REMPI experiments, we fixed the ionization laser at 232 nm, then scan the excitation laser from 265 to 279 nm to obtain vibronic spectra of the first electronically excited state S1 for 2FBN and 3FBN.
In the MATI experiments, the molecules in neutral ground state S0 were resonantly excited to specific vibronic levels in the S1 state, further excited to the high Rydberg state by the probe laser, which has a scanning range of 224 – 240 nm. A -0.5 V/cm pulsed electric field was applied to remove the prompt ions. After a time delay of about 29 μs, the Rydberg molecules were ionized by a 143 V/cm pulsed electric field. Newly formed threshold ions pass through a 48 cm field-free region to be detected by a Microchannel plates (MCP) detector. The signal was collected by a multichannel scaler (SRS: SR430) and recorded by a computer. Each mass spectrum was accumulated for 300 laser shots. The time sequence of the whole system is controlled by a pulse delay generator (SRS: DG645). More details on the experimental system have been described in our previous publications [61,62,63].
4.2. Theoretical Methods
All calculations were performed using the Gaussian 16 program package [64]. The geometry optimization, vibrational frequencies of S0, S1, and D0 states are calculated at the levels of RB3LYP/aug-cc-pvtz, TD-B3LYP/aug-cc-pvtz, and UB3LYP/aug-cc-pvtz, respectively. Prior to the experiments, we also using the G4 and CBS-QB3 methods to predict IEs in order to select the appropriate dyes. The spectral simulations are performed based on above B3LYP/aug-cc-pvtz calculations. Combining with the theoretical calculations and simulated spectra, the vibrational features of 2FBN and 3FBN measured by REMPI and MATI experiments were assigned.
5. Conclusions
The high-resolution vibrational spectra of the first electronically excited state S1 and cationic ground state D0 of 2-fluorobenzonitrile and 3-fluorobenzonitrile were measured by two-color resonance enhanced multiphoton ionization and mass analyzed threshold ionization spectroscopy. The precise band origins of S1←S0 transition and adiabatic ionization energies are determined to be 36028 ± 2 cm-1 and 78650 ± 5 cm-1 for 2-fluorobenzonitrile; and 35989 ± 2 cm-1 and 78873 ± 5 cm-1 for 3-fluorobenzonitrile, respectively. DFT theory at the level of B3LYP/aug-cc-pvtz was used to calculate the molecular structure, vibrational frequency, and further perform the Franck-Condon simulations. The theoretical results are in good agreement with the experimental measurements. The vibrational features of S1 and D0 states are analyzed in detail and assigned.
The MATI spectra follow well the propensity rule Δν = 0, indicating that the molecular structures of the cationic ground states are similar to that of the excited states. The molecular structures and vibration frequencies in S0, S1, and D0 states were discussed in detail. The ring C-C bond lengths in S0, S1, and D0 states averagely obey the rule of S1 > D0 > S0. The bond length reflects the bond strength, and further the bond length is related with the vibration frequency. On average or for most vibrational modes, the vibration frequencies of the ground state S0, excited state S1 and cationic ground state D0 meet the relative relationship: S1 < D0 < S0. At the transition of S1 ← S0 and D0 ← S1, a lot of vibrational modes associated with ring in-plane distortion were active, and only a few out-of-plane fundamental vibrations were observed. The substitution effects of F and CN were discussed. Whether the electron donating group or the electron withdrawing group, the ionization energies of molecules formed by ortho, meta and para substitutions meet the relative relationship: para < ortho < meta.
Author Contributions
Conceptualization, C.L. and S.J.; investigation, S.L. and Y.Z.; writing—original draft preparation, C.L. and S.L.; writing—review and editing, C.L. and Y.Z.; funding acquisition, Y.J.; J.Z. and S.J. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by National Natural Science Foundation of China (Grants Nos. 61835007, 12241408, 61575115), PCSIRT (Grant No. IRT_17R70), 111 project (Grant No. D18001), and the Fund for Shanxi ‘‘1331 Project” Key Subjects Construction.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Conflicts of Interest
The authors declare no conflict of interest.
Sample Availability
Samples of 2-fluorobenzonitrile and 3-fluorobenzonitrile are available from commercial sources.
References
- Berger, R.; Resnati, G.; Metrangolo, P.; Weber, E.; Hulliger, J. ChemInform Abstract: Organic Fluorine Compounds: A Great Opportunity for Enhanced Materials Properties. ChemInform 2011, 42. [Google Scholar] [CrossRef]
- Ametamey, S.M.; Honer, M.; Schubiger, P.A. Molecular imaging with PET. Chem. Rev. 2008, 108, 1501–1516. [Google Scholar] [CrossRef] [PubMed]
- Müller, K.; Faeh, C.; Diederich, F. Fluorine in pharmaceuticals: looking beyond intuition. Science 2007, 317, 1881–1886. [Google Scholar] [CrossRef] [PubMed]
- Jeschke, P. The unique role of fluorine in the design of active ingredients for modern crop protection. Chembiochem 2004, 5, 571–589. [Google Scholar] [CrossRef] [PubMed]
- Da Ribeiro Silva, M.A.V.; Monte, M.J.S.; Rocha, I.M.; Cimas, A. Energetic study applied to the knowledge of the structural and electronic properties of monofluorobenzonitriles. J. Org. Chem. 2012, 77, 4312–4322. [Google Scholar] [CrossRef]
- Ling, F.; Li, S.; Song, X.; Tang, Y.; Wang, Y.; Zhang, B. Visualization of coherent nuclear motion between different geometries in photoexcited 2,4-difluorophenol. Phys. Rev. A 2017, 95. [Google Scholar] [CrossRef]
- Ling, F.; Wang, Y.; Li, S.; Wei, J.; Tang, Y.; Zhang, B. Imaging Reversible and Irreversible Structural Evolution in Photoexcited 2,4-Difluoroaniline. J. Phys. Chem. Lett. 2018, 9, 5468–5473. [Google Scholar] [CrossRef]
- Sun, W.; Lozada, I.B.; van Wijngaarden, J. Fourier Transform Microwave Spectroscopic and ab Initio Study of the Rotamers of 2-Fluorobenzaldehyde and 3-Fluorobenzaldehyde. J. Phys. Chem. A 2018, 122, 2060–2068. [Google Scholar] [CrossRef]
- Sun, W.; van Wijngaarden, J. Structural elucidation of 2-fluorothiophenol from Fourier transform microwave spectra and ab initio calculations. Journal of Molecular Structure 2017, 1144, 496–501. [Google Scholar] [CrossRef]
- Kamaee, M.; Sun, M.; Luong, H.; van Wijngaarden, J. Investigation of Structural Trends in Mono-, Di-, and Pentafluorobenzonitriles Using Fourier Transform Microwave Spectroscopy. J. Phys. Chem. A 2015, 119, 10279–10292. [Google Scholar] [CrossRef]
- Zhang, B.; Li, C.; Su, H.; Lin, J.L.; Tzeng, W.B. Mass analyzed threshold ionization spectroscopy of p-fluorophenol cation and the p-fluoro substitution effect. Chemical Physics Letters 2004, 390, 65–70. [Google Scholar] [CrossRef]
- Huang, J.; Huang, K.; Liu, S.; Luo, Q.; Tzeng, W. Vibrational spectra and theoretical calculations of p-chlorophenol in the electronically excited S1 and ionic ground D0 states. Journal of Photochemistry and Photobiology A: Chemistry 2008, 193, 245–253. [Google Scholar] [CrossRef]
- Ratzer, C.; Nispel, M.; Schmitt, M. Structure of 4-fluorophenol and barrier to internal –OH rotation in the S1-state. Phys. Chem. Chem. Phys. 2003, 5, 812–819. [Google Scholar] [CrossRef]
- Zhang, L.; Liu, S.; Cheng, M.; Du, Y.; Zhu, Q. Vibrational Spectra and Theoretical Calculations of cis- and trans-3-Fluoro-N-methylaniline in the Neutral (S(0)) and Cationic (D(0)) Ground States. J. Phys. Chem. A 2016, 120, 81–94. [Google Scholar] [CrossRef]
- Arivazhagan, M.; Meenakshi, R.; Prabhakaran, S. Vibrational spectroscopic investigations, first hyperpolarizability, HOMO-LUMO and NMR analyzes of p-fluorobenzonitrile. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2013, 102, 59–65. [Google Scholar] [CrossRef]
- Da Ribeiro Silva, M.A.V.; Monte, M.J.S.; Rocha, I.M.; Cimas, A. Energetic study applied to the knowledge of the structural and electronic properties of monofluorobenzonitriles. J. Org. Chem. 2012, 77, 4312–4322. [Google Scholar] [CrossRef]
- Varadwaj, P.R.; Jaman, A.I. Centrifugal distortion analysis of the millimeter-wave spectrum of 2-fluorobenzonitrile and ab initio DFT calculations. Journal of Molecular Spectroscopy 2006, 236, 70–74. [Google Scholar] [CrossRef]
- Kumar, A.P.; Rao, G.R. Vibrational analysis of substituted benzonitriles. I. Vibrational spectra, normal coordinate analysis and transferability of force constants of monohalogenated benzonitriles. Spectrochim. Acta A Mol. Biomol. Spectrosc. 1997, 53A, 2023–2032. [Google Scholar] [CrossRef]
- Palmer, M.H.; Moyes, W.; Spiers, M. The electronic structure of substituted benzenes: ab initio calculations and photoelectron spectra for benzonitrile, the tolunitriles, fluorobenzonitriles, dicyanobenzenes and ethynylbenzene. Journal of Molecular Structure 1980, 62, 165–187. [Google Scholar] [CrossRef]
- Jiang, S.; Levy, D.H. Supersonic Jet Studies on the Photophysics of Substituted Benzenes and Naphthalenes. J. Phys. Chem. A 2002, 106, 8590–8598. [Google Scholar] [CrossRef]
- Zhao, Y.; Jin, Y.; Hao, J.; Yang, Y.; Li, C.; Jia, S. Resonance enhanced multiphoton ionization and mass analyzed threshold ionization spectroscopy of 4-fluorobenzonitrile. Chemical Physics Letters 2018, 711, 127–131. [Google Scholar] [CrossRef]
- Eom, S.Y.; Lee, Y.R.; Kwon, C.H. Accurate conformational stability and cationic structure of piperidine determined by conformer-specific VUV-MATI spectroscopy. Phys. Chem. Chem. Phys. 2020, 22, 22823–22832. [Google Scholar] [CrossRef] [PubMed]
- Park, S.M.; Lee, Y.R.; Kwon, C.H. Conformational Structures of Neutral and Cationic Pivaldehyde Revealed by IR-Resonant VUV-MATI Mass Spectroscopy. Int. J. Mol. Sci. 2022, 23. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.R.; Kwon, C.H. Valence molecular orbitals and cationic structures of 2-fluoropyridine by high-resolution ion spectroscopy and Franck-Condon fitting. J. Chem. Phys. 2022, 157, 154306. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.R.; Kwon, C.H. Innovative mass spectrometer for high-resolution ion spectroscopy. J. Chem. Phys. 2021, 155, 164203. [Google Scholar] [CrossRef]
- Eom, S.Y.; Lee, Y.R.; Park, S.M.; Kwon, C.H. Determination of the highest occupied molecular orbital and conformational structures of morpholine based on its conformer-specific photoionization dynamics. Phys. Chem. Chem. Phys. 2022, 24, 28477–28485. [Google Scholar] [CrossRef] [PubMed]
- Eom, S.Y.; Kang, D.W.; Kwon, C.H. Conformational structure of cationic tetrahydropyran by one-photon vacuum ultraviolet mass-analyzed threshold ionization spectroscopy. Phys. Chem. Chem. Phys. 2021, 23, 1414–1423. [Google Scholar] [CrossRef]
- Lee, Y.R.; Kim, H.L.; Kwon, C.H. Determination of the cationic conformational structure of tetrahydrothiophene by one-photon MATI spectroscopy and Franck-Condon fitting. Phys. Chem. Chem. Phys. 2020, 22, 6184–6191. [Google Scholar] [CrossRef]
- Kang, D.W.; Yoon, D.K.; Kwon, C.H. Conformational potential energy surfaces and cationic structure of 3,4-dihydro-2H-pyran by VUV-MATI spectroscopy and Franck-Condon fitting. Phys. Chem. Chem. Phys. 2020, 22, 27673–27680. [Google Scholar] [CrossRef]
- Ketkov, S.Y.; Tzeng, S.-Y.; Rychagova, E.A.; Markin, G.V.; Makarov, S.G.; Tzeng, W.-B. Laser spectroscopic and computational insights into unexpected structural behaviours of sandwich complexes upon ionization. Dalton Trans. 2021, 50, 10729–10736. [Google Scholar] [CrossRef]
- Ketkov, S.; Tzeng, S.-Y.; Rychagova, E.; Tzeng, W.-B. Ionization of Decamethylmanganocene: Insights from the DFT-Assisted Laser Spectroscopy. Molecules 2022, 27. [Google Scholar] [CrossRef] [PubMed]
- Ketkov, S.Y.; Tzeng, S.Y.; Rychagova, E.A.; Kalakutskaya, L.V.; Fuss, M.; Braunschweig, H.; Tzeng, W.-B. Rydberg state mediated multiphoton ionization of (η7-C7H7)(η5-C5H5)Cr: DFT-supported experimental insights into the molecular and electronic structures of excited sandwich complexes. Phys. Chem. Chem. Phys. 2019, 21, 9665–9671. [Google Scholar] [CrossRef] [PubMed]
- Ketkov, S.Y.; Rychagova, E.A.; Tzeng, S.-Y.; Tzeng, W.-B. TD DFT insights into unusual properties of excited sandwich complexes: structural transformations and vibronic interactions in Rydberg-state bis(η6-benzene)chromium. Phys. Chem. Chem. Phys. 2018, 20, 23988–23997. [Google Scholar] [CrossRef] [PubMed]
- Ketkov, S.Y.; Tzeng, S.-Y.; Wu, P.-Y.; Markin, G.V.; Tzeng, W.-B. DFT-Supported Threshold Ionization Study of Chromium Biphenyl Complexes: Unveiling the Mechanisms of Substituent Influence on Redox Properties of Sandwich Compounds. Chemistry 2017, 23, 13669–13675. [Google Scholar] [CrossRef] [PubMed]
- Ketkov, S.Y.; Markin, G.V.; Tzeng, S.Y.; Tzeng, W.B. Fine Substituent Effects in Sandwich Complexes: A Threshold Ionization Study of Monosubstituted Chromium Bisarene Compounds. Chemistry 2016, 22, 4690–4694. [Google Scholar] [CrossRef] [PubMed]
- Tzeng, S.Y.; Takahashi, K.; Tzeng, W.B. Two-Color Resonant Two-Photon Mass-Analyzed Threshold Ionization of 2,4-Difluoroanisole and the Additivity Relation of Ionization Energy. J. Phys. Chem. A 2020, 124, 10517–10526. [Google Scholar] [CrossRef] [PubMed]
- Kemp, D.J.; Whalley, L.E.; Tuttle, W.D.; Gardner, A.M.; Speake, B.T.; Wright, T.G. Vibrations of the p-chlorofluorobenzene cation. Phys. Chem. Chem. Phys. 2018, 20, 12503–12516. [Google Scholar] [CrossRef] [PubMed]
- Davies, A.R.; Kemp, D.J.; Wright, T.G. Electronic, vibrational, and torsional couplings in N-methylpyrrole: Ground, first excited, and cation states. J. Chem. Phys. 2021, 154, 224305. [Google Scholar] [CrossRef]
- Kemp, D.J.; Gardner, A.M.; Tuttle, W.D.; Midgley, J.; Reid, K.L.; Wright, T.G. Identifying complex Fermi resonances in p-difluorobenzene using zero-electron-kinetic-energy (ZEKE) spectroscopy. J. Chem. Phys. 2018, 149, 94301. [Google Scholar] [CrossRef] [PubMed]
- Davies, A.R.; Kemp, D.J.; Wright, T.G. Comment on “Electronic, vibrational and torsional couplings in N-methylpyrrole: Ground, first excited and cation states” J. Chem. Phys. 154, 224305 (2021). J. Chem. Phys. 2021, 155, 117101. [Google Scholar] [CrossRef]
- Kemp, D.J.; Fryer, E.F.; Davies, A.R.; Wright, T.G. Vibration-modified torsional potentials and vibration-torsion (“vibtor”) levels in the m-fluorotoluene cation. J. Chem. Phys. 2019, 151, 84311. [Google Scholar] [CrossRef] [PubMed]
- Davies, A.R.; Kemp, D.J.; Warner, L.G.; Fryer, E.F.; Rees, A.; Wright, T.G. Variations in Duschinsky rotations in m-fluorotoluene and m-chlorotoluene during excitation and ionization. J. Chem. Phys. 2020, 152, 214303. [Google Scholar] [CrossRef]
- Varsányi, G. Assignments for vibrational spectra of seven hundred benzene derivatives; Adam Hilger: London, UK, 1974; ISBN 0852742835. [Google Scholar]
- Wilson, E.B. The Normal Modes and Frequencies of Vibration of the Regular Plane Hexagon Model of the Benzene Molecule. Phys. Rev. 1934, 45, 706–714. [Google Scholar] [CrossRef]
- Asmis, K.R.; Yang, Y.; Santambrogio, G.; Brümmer, M.; Roscioli, J.R.; McCunn, L.R.; Johnson, M.A.; Kühn, O. Gas-phase infrared spectroscopy and multidimensional quantum calculations of the protonated ammonia dimer N2H7+. Angew. Chem. Int. Ed Engl. 2007, 46, 8691–8694. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Kuhn, O. A concise method for kinetic energy quantisation. Mol. Phys. 2008, 106, 2445–2457. [Google Scholar] [CrossRef]
- Davies, A.R.; Kemp, D.J.; Wright, T.G. Unpicking vibration-vibration and vibration-torsion interactions in m-fluorotoluene. Journal of Molecular Spectroscopy 2021, 381, 111522. [Google Scholar] [CrossRef]
- Tzeng, S.Y.; Takahashi, K.; Tzeng, W.B. Two-Color Resonant Two-Photon Mass-Analyzed Threshold Ionization of 2,4-Difluoroanisole and the Additivity Relation of Ionization Energy. J. Phys. Chem. A 2020, 124, 10517–10526. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Tzeng, S.Y.; Shivatare, V.; Takahashi, K.; Zhang, B.; Tzeng, W.B. Identification of four rotamers of m-methoxystyrene by resonant two-photon ionization and mass analyzed threshold ionization spectroscopy. J. Chem. Phys. 2015, 142, 124314. [Google Scholar] [CrossRef]
- Xu, Y.; Tzeng, S.Y.; Zhang, B.; Tzeng, W.B. Rotamers of 3,4-difluoroanisole studied by two-color resonant two-photon mass-analyzed threshold ionization spectroscopy. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2013, 102, 365–370. [Google Scholar] [CrossRef]
- Huang, W.C.; Huang, P.S.; Hu, C.H.; Tzeng, W.B. Vibronic and cation spectroscopy of 2,4-difluoroaniline. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2012, 93, 176–179. [Google Scholar] [CrossRef]
- Shivatare, V.S.; Kundu, A.; Patwari, G.N.; Tzeng, W.B. Studies of structural isomers o-, m-, and p-fluorophenylacetylene by two-color resonant two-photon mass-analyzed threshold ionization spectroscopy. J. Phys. Chem. A 2014, 118, 8277–8286. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Lin, J.L.; Tzeng, W.B. Mass-analyzed threshold ionization spectroscopy of the rotamers of p-n-propylphenol cations and configuration effect. J. Chem. Phys. 2005, 122, 44311. [Google Scholar] [CrossRef] [PubMed]
- Neuhauser, R.G.; Siglow, K.; Neusser, H.J. High n Rydberg spectroscopy of benzene: Dynamics, ionization energy and rotational constants of the cation. J. Chem. Phys. 1997, 106, 896–907. [Google Scholar] [CrossRef]
- Lembach, G.; Brutschy, B. Fragmentation energetics and dynamics of fluorobenzene⋅Arn (n=1–3) clusters studied by mass analyzed threshold ionization spectroscopy. J. Chem. Phys. 1997, 107, 6156–6165. [Google Scholar] [CrossRef]
- Araki, M.; Sato, S.; Kimura, K. Two-Color Zero Kinetic Energy Photoelectron Spectra of Benzonitrile and Its van der Waals Complexes with Argon. Adiabatic Ionization Potentials and Cation Vibrational Frequencies. J. Phys. Chem. 1996, 100, 10542–10546. [Google Scholar] [CrossRef]
- Dopfer, O.; Müller-Dethlefs, K. S1 excitation and zero kinetic energy spectra of partly deuterated 1:1 phenol–water complexes. J. Chem. Phys. 1994, 101, 8508–8516. [Google Scholar] [CrossRef]
- Yuan, L.; Li, C.; Lin, J.L.; Yang, S.C.; Tzeng, W.B. Mass analyzed threshold ionization spectroscopy of o-fluorophenol and o-methoxyphenol cations and influence of the nature and relative location of substituents. Chemical Physics 2006, 323, 429–438. [Google Scholar] [CrossRef]
- Oikawa, A.; Abe, H.; Mikami, N.; Ito, M. Electronic spectra and ionization potentials of rotational isomers of several disubstituted benzenes. Chemical Physics Letters 1985, 116, 50–54. [Google Scholar] [CrossRef]
- Yosida, K.; Suzuki, K.; Ishiuchi, S.; Sakai, M.; Fujii, M.; Dessent, C.E.H.; Müller-Dethlefs, K. The PFI-ZEKE photoelectron spectrum of m-fluorophenol and its aqueous complexes: Comparing intermolecular vibrations in rotational isomers. Phys. Chem. Chem. Phys. 2002, 4, 2534–2538. [Google Scholar] [CrossRef]
- Zhao, Y.; Jin, Y.; Hao, J.; Yang, Y.; Wang, L.; Li, C.; Jia, S. Rotamers of p-isopropylphenol studied by hole-burning resonantly enhanced multiphoton ionization and mass analyzed threshold ionization spectroscopy. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2019, 207, 328–336. [Google Scholar] [CrossRef]
- Li, N.; Li, S.; Wang, L.; Wang, H.; Zhao, J.; Li, C. Vibrational spectra of 2-cyanophenol cation studied by the mass analyzed threshold ionization technique. Chemical Physics Letters 2022, 792, 139402. [Google Scholar] [CrossRef]
- Hao, J.; Duan, C.; Yang, Y.; Li, C.; Jia, S. Resonance enhanced two-photon ionization and mass analyzed threshold ionization spectroscopy of 4-ethylanisole. Journal of Molecular Spectroscopy 2020, 369, 111258. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 16; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
Figure 1.
The stable structures of 2- and 3-fluorobenzonitrile with atomic labels.
Figure 2.
REMPI spectrum of 2-fluorobenzonitrile (a) and its Franck-Condon simulation (b).
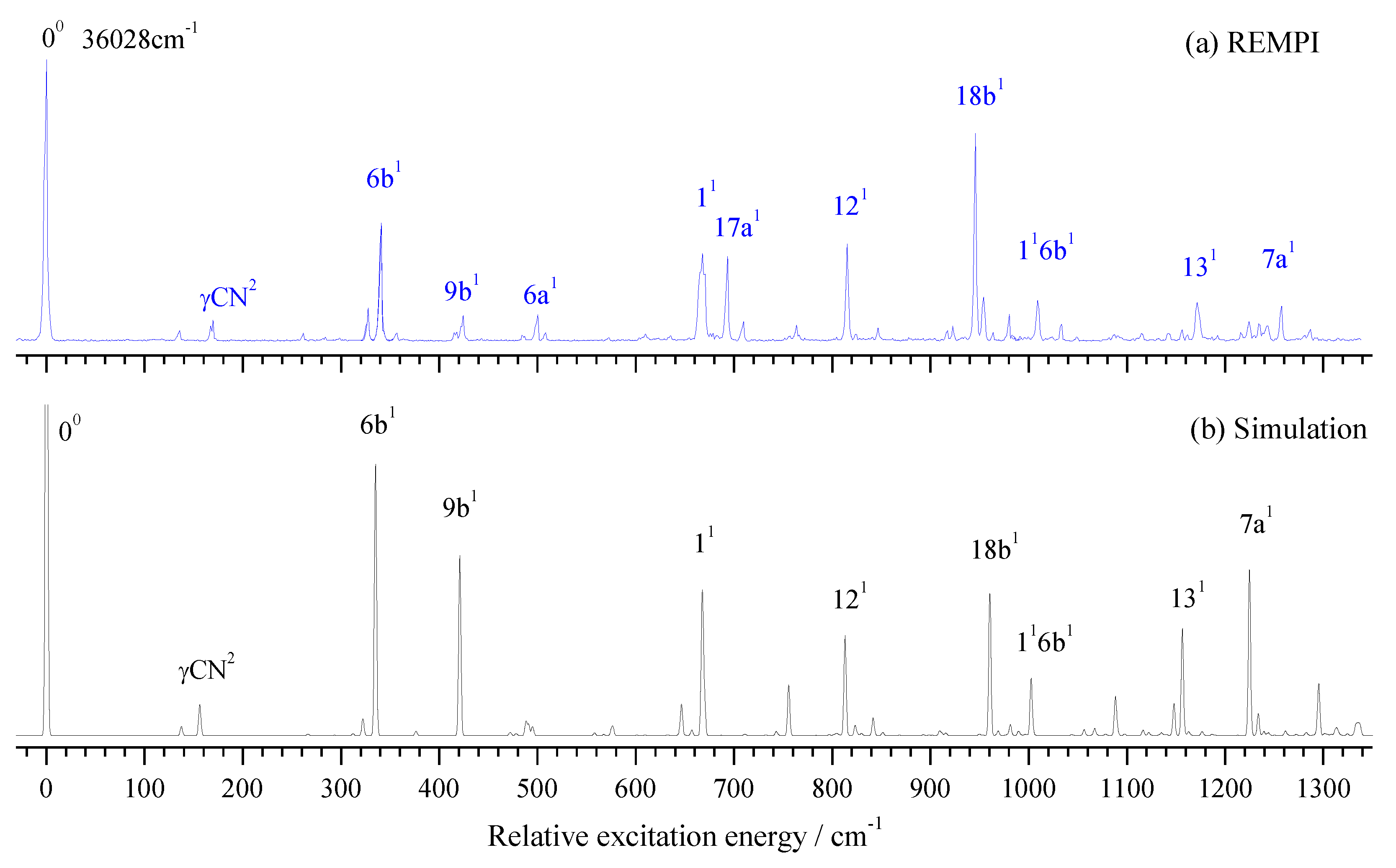
Figure 3.
PIE spectrum of 2-fluorobenzonitrile recorded by ionizing via S100 intermediate state at 36028 cm-1 (a) and MATI spectrum near the cationic origin 0+ for comparison (b).
Figure 3.
PIE spectrum of 2-fluorobenzonitrile recorded by ionizing via S100 intermediate state at 36028 cm-1 (a) and MATI spectrum near the cationic origin 0+ for comparison (b).
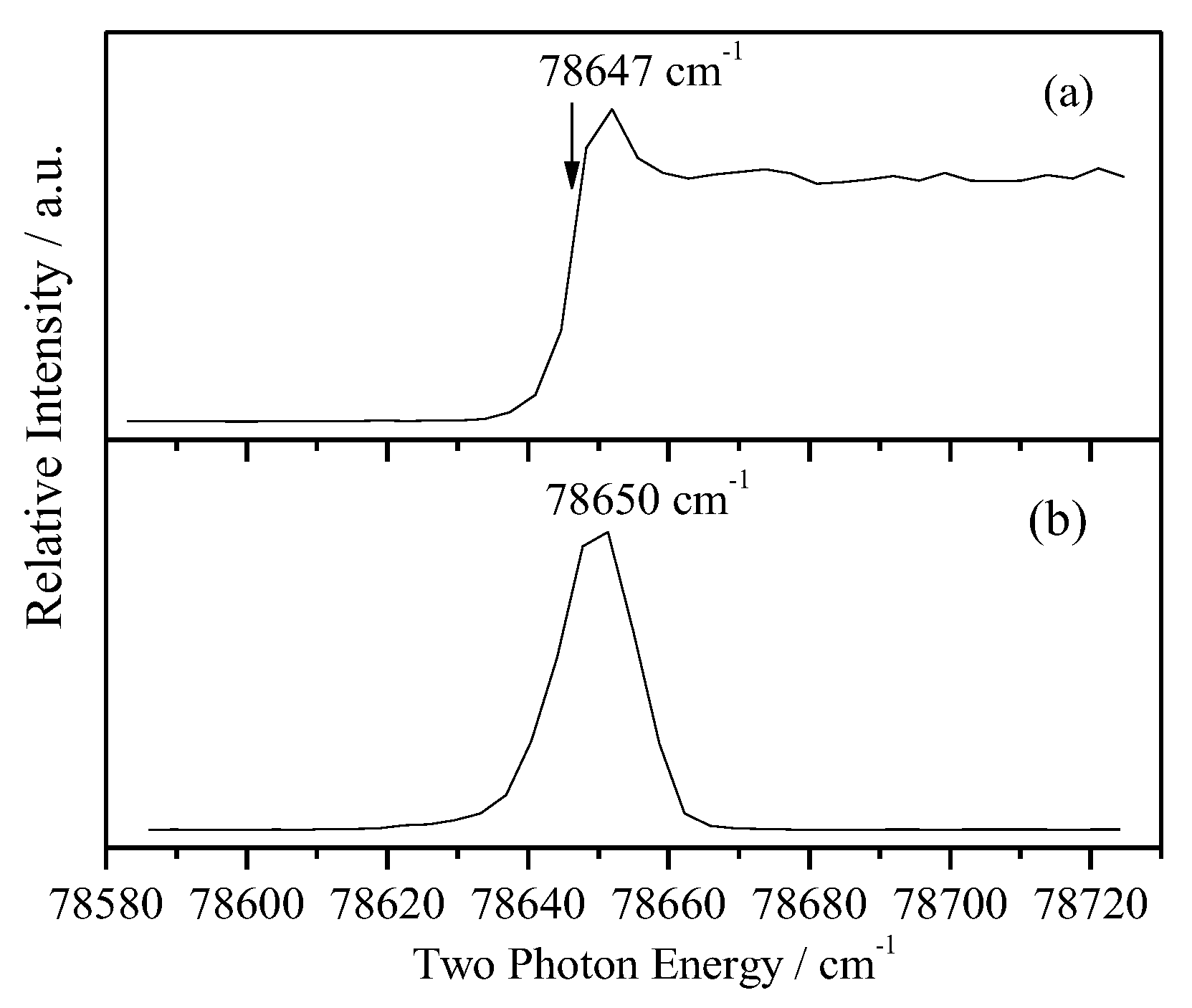
Figure 4.
Franck-Condon simulation of the D0 ← S100 transition (a) and the MATI spectra of 2-fluorobenzonitrile via S100 (b) and S16b1 (c) intermediate states.
Figure 4.
Franck-Condon simulation of the D0 ← S100 transition (a) and the MATI spectra of 2-fluorobenzonitrile via S100 (b) and S16b1 (c) intermediate states.
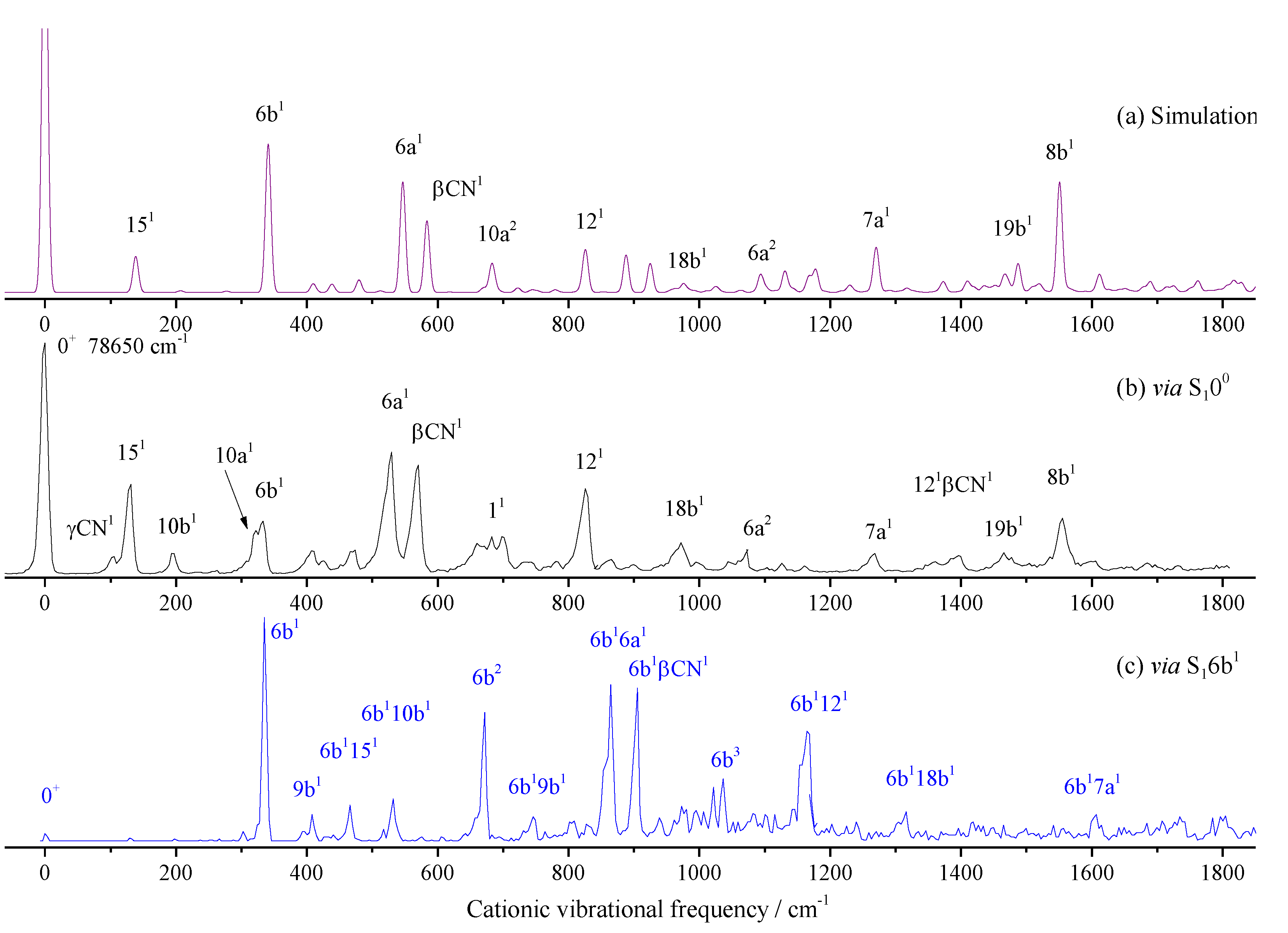
Figure 5.
MATI spectra of 2-fluorobenzonitrile via S111 (a), S1121 (b), and S118b1 intermediate states.
Figure 5.
MATI spectra of 2-fluorobenzonitrile via S111 (a), S1121 (b), and S118b1 intermediate states.
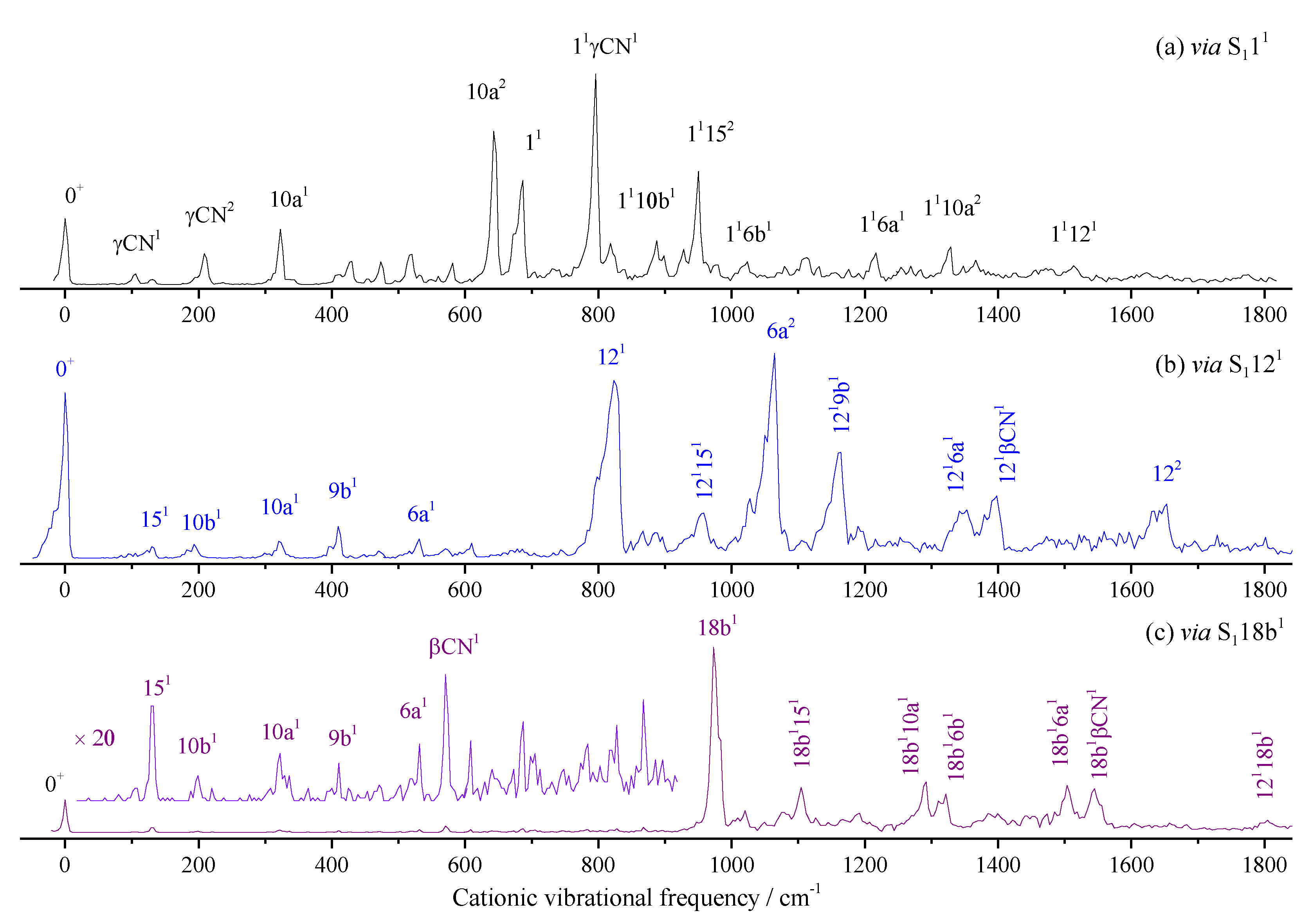
Figure 6.
REMPI spectrum of 3-fluorobenzonitrile (a) and its Franck-Condon simulation (b).
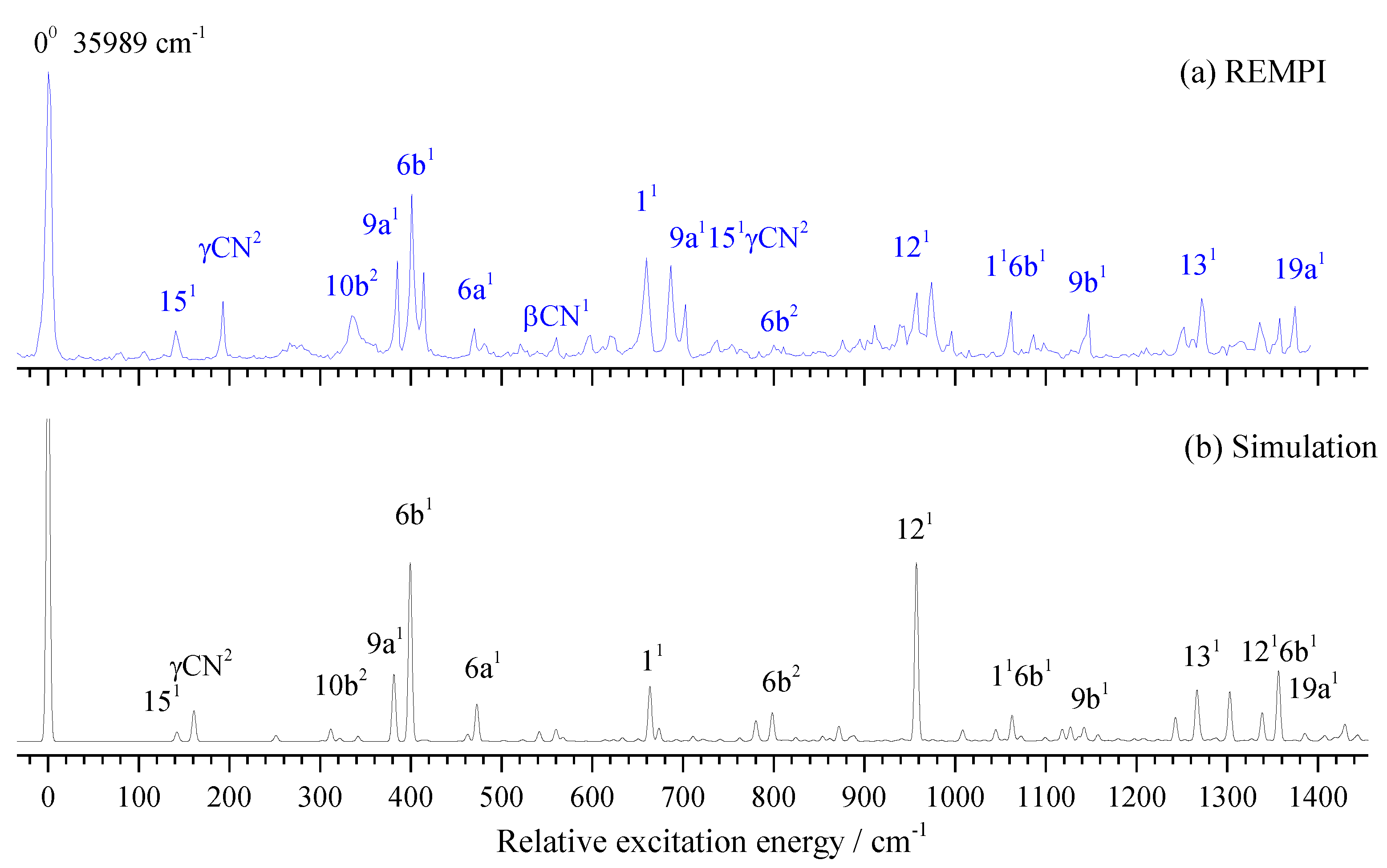
Figure 7.
PIE spectrum of 3-fluorobenzonitrile recorded by ionizing via its S100 state at 35989 cm-1 (a) and MATI spectrum near the cationic origin 0+ for comparison (b).
Figure 7.
PIE spectrum of 3-fluorobenzonitrile recorded by ionizing via its S100 state at 35989 cm-1 (a) and MATI spectrum near the cationic origin 0+ for comparison (b).
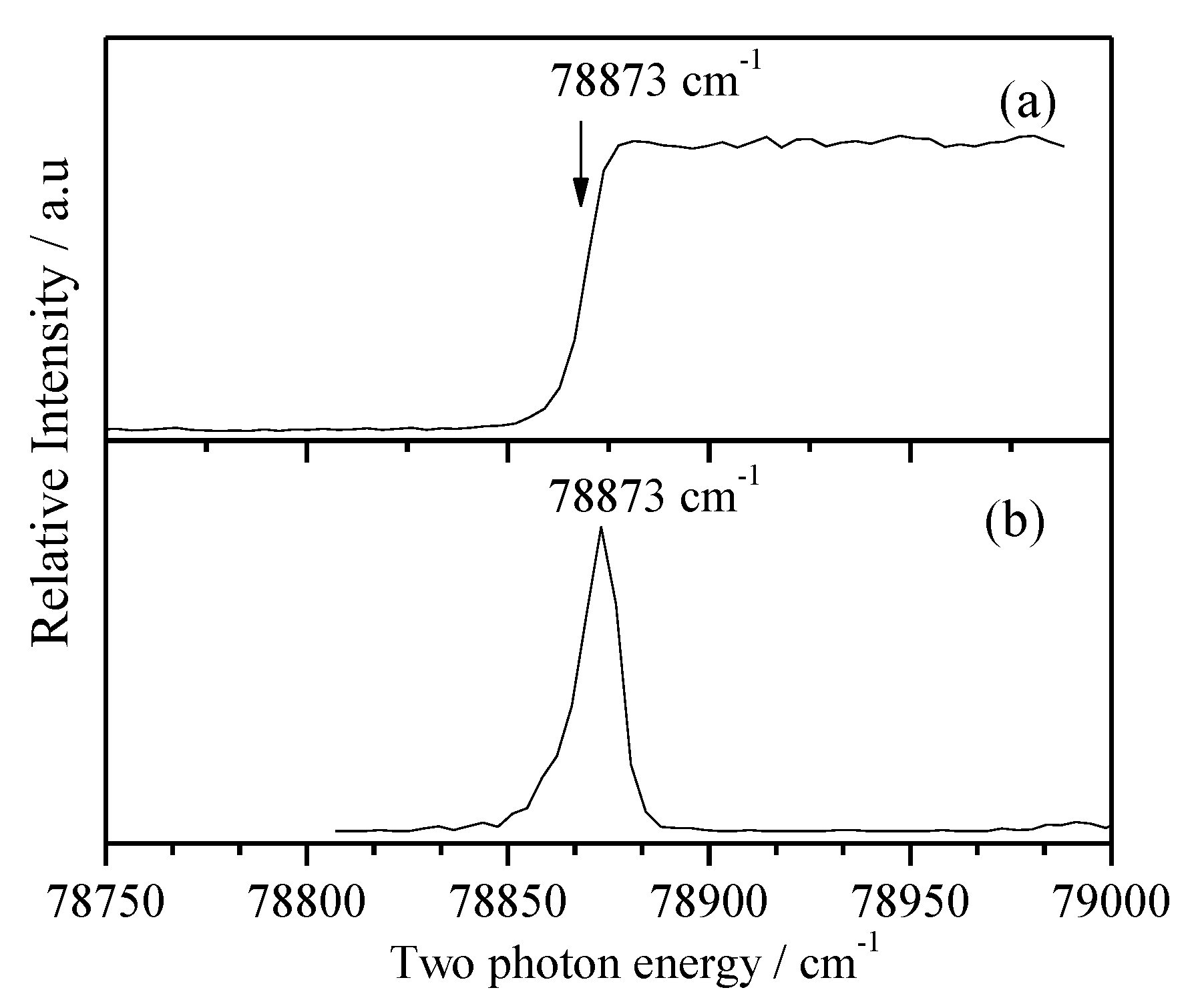
Figure 8.
Franck-Condon simulation of the transition D0 ← S100 (a) and the MATI spectra of 3-fluorobenzonitrile via S100 (b) and S1γCN2 (c) intermediate states.
Figure 8.
Franck-Condon simulation of the transition D0 ← S100 (a) and the MATI spectra of 3-fluorobenzonitrile via S100 (b) and S1γCN2 (c) intermediate states.
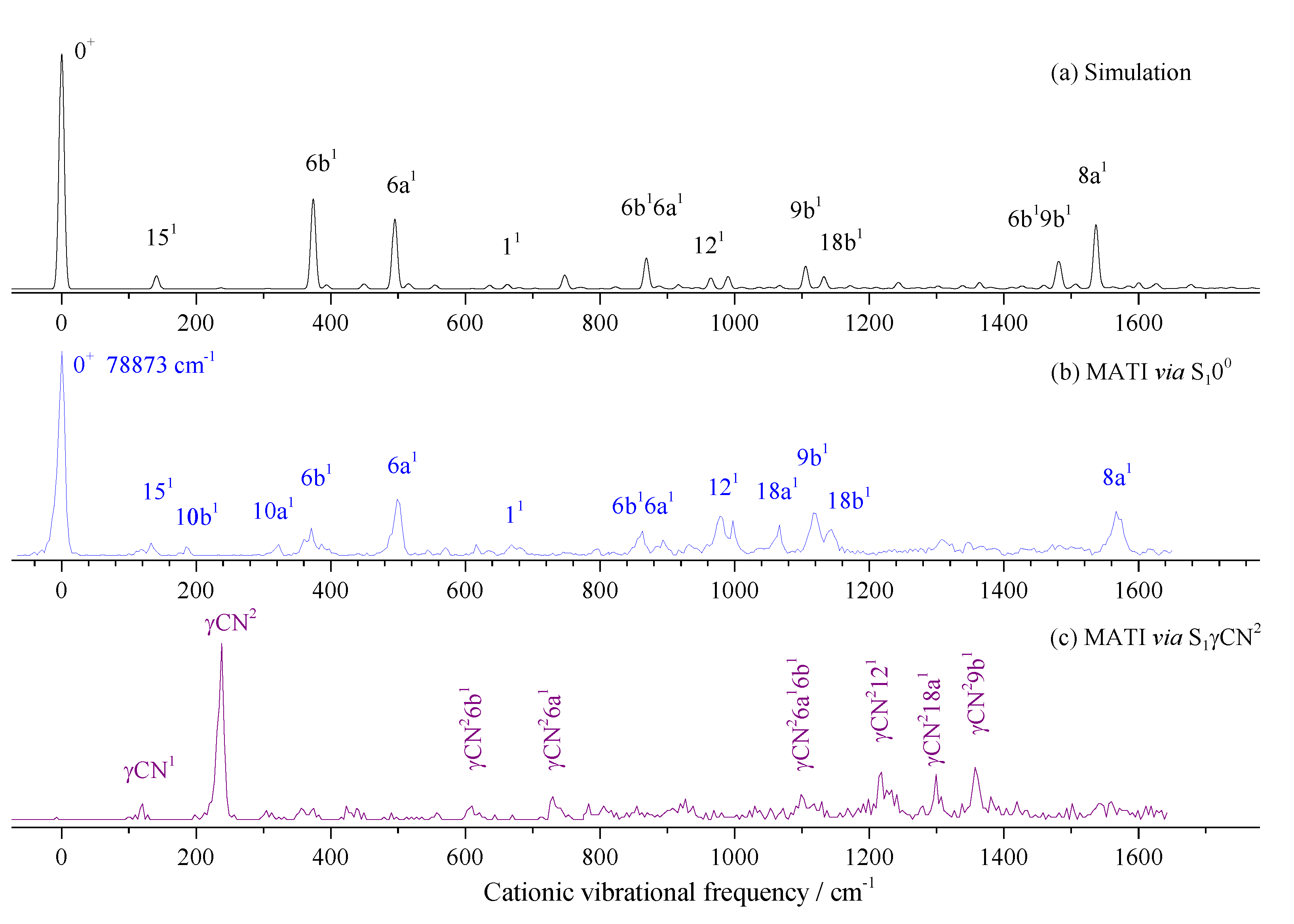
Figure 9.
The MATI spectra of 3-fluorobenzonitrile via S16b1 (a) and S111 (b) intermediate states.
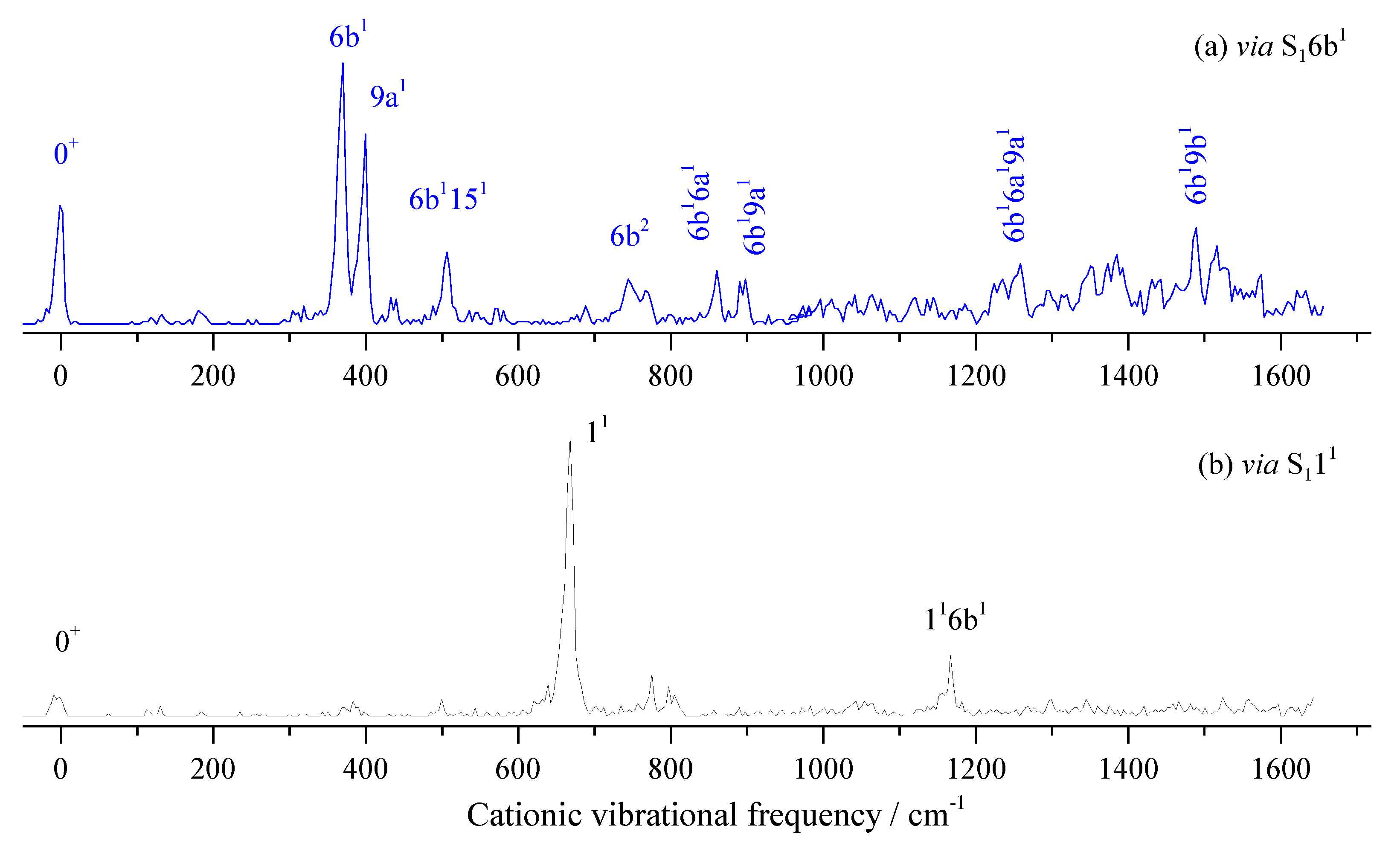

Table 1.
Observed bands in the vibronic spectrum of 2FBN and their possible assignments a.
| Transition energy (cm-1) |
Relative intensity |
Shift (cm-1) |
Calc. (cm-1) |
Assignment b |
|---|---|---|---|---|
| 36028 | 100 | 0 | 0 | 00 |
| 36164 | 4 | 136 | 137 | 151 |
| 36198 | 7 | 170 | 156 | γCN2 |
| 36290 | 3 | 262 | 10b1γCN1 | |
| 36356 | 12 | 328 | 10a1γCN1 | |
| 36369 | 42 | 341 | 335 | 6b1 |
| 36452 | 9 | 424 | 421 | 9b1 |
| 36528 | 9 | 500 | 495 | 6a1 |
| 36638 | 3 | 610 | 16a110b1 | |
| 36663 | 31 | 635 | 646 | 16b2 |
| 36696 | 30 | 668 | 668 | 11 |
| 36721 | 30 | 693 | 698 | 17a1 |
| 36738 | 7 | 710 | 711 | 6b110b2 |
| 36792 | 6 | 764 | 755 | 9b16b1 |
| 36843 | 34 | 815 | 813 | 121 |
| 36874 | 2 | 846 | 841 | 9b2 |
| 36945 | 4 | 917 | 910 | βCN16b1 |
| 36950 | 5 | 922 | 916 | 6a19b1 |
| 36974 | 74 | 946 | 960 | 18b1 |
| 36982 | 15 | 954 | 969 | 121γCN2 |
| 37008 | 10 | 980 | 981 | 6b116b2 |
| 37037 | 14 | 1009 | 1003 | 116b1 |
| 37062 | 6 | 1034 | 1022 | 16b210b2 |
| 37199 | 14 | 1171 | 1156 | 131 |
| 37285 | 12 | 1257 | 1224 | 7a1 |
| 37315 | 4 | 1287 | 1295 | 18b16b1 |
a Experimental values are shifts from 36028 cm-1, and the calculated ones (scaled by 0.9649) are obtained from the TD-B3LYP/aug-cc-pvtz calculations. b β, in-plane bending; γ, out-of-plane bending.
Table 2.
Assignment of the observed bands (cm−1) in the MATI spectra of 2FBN.a
| Intermediate levels in the S1 state | Calc. | Assignment b | ||||
|---|---|---|---|---|---|---|
| 00 | 6b1 | 11 | 121 | 18b1 | ||
| 106 | 106 | 104 | γCN1 | |||
| 131 | 130 | 132 | 139 | 151 | ||
| 197 | 193 | 199 | 205 | 10b1 | ||
| 209 | γCN2 | |||||
| 323 | 321 | 322 | 335 | 10a1 | ||
| 333 | 335 | 341 | 6b1 | |||
| 407 | 408 | 409 | 410 | 410 | 9b1 | |
| 430 | 6b1γCN1 | |||||
| 466 | 6b1151 | |||||
| 474 | 473 | 152γCN2 | ||||
| 520 | 10a110b1 | |||||
| 530 | 531 | 532 | 547 | 6a1 | ||
| 532 | 6b110b1 | |||||
| 582 | 6b1151γCN1 | |||||
| 643 | 10a2 | |||||
| 571 | 570 | 571 | 584 | βCN1 | ||
| 672 | 672 | 6b2 | ||||
| 683 | 687 | 687 | 696 | 11 | ||
| 698 | 16a110b1 | |||||
| 746 | 6b19b1 | |||||
| 796 | 11γCN1 | |||||
| 818 | 11151 | |||||
| 826 | 823 | 826 | 121 | |||
| 865 | 6b16a1 | |||||
| 888 | 1110b1 | |||||
| 906 | 6b1βCN1 | |||||
| 950 | 11152 | |||||
| 955 | 121151 | |||||
| 972 | 973 | 976 | 18b1 | |||
| 1023 | 116b1 | |||||
| 1036 | 6b3 | |||||
| 1059 | 1064 | 6a2 | ||||
| 1104 | 18b1151 | |||||
| 1164 | 1160 | 6b1121 | ||||
| 1217 | 116a1 | |||||
| 1268 | 1271 | 7a1 | ||||
| 1292 | 18b110a1 | |||||
| 1316 | 1321 | 6b118b1 | ||||
| 1329 | 1110a2 | |||||
| 1349 | 1216a1 | |||||
| 1394 | 121βCN1 | |||||
| 1396 | 1410 | 121βCN1 | ||||
| 1466 | 1466 | 19b1 | ||||
| 1503 | 18b16a1 | |||||
| 1513 | 11121 | |||||
| 1545 | 18b1βCN1 | |||||
| 1555 | 1551 | 8b1 | ||||
| 1607 | 6b17a1 | |||||
| 1646 | 122 | |||||
a The experimental values are shifts from 78650 cm−1, whereas the calculated ones are obtained from the B3LYP/aug-cc-pVDZ calculations, scaled by 0.9849. b β, in-plane bending; γ, out-of-plane bending.
Table 3.
Assignment of observed bands (cm-1) in the 2-color REMPI spectrum of 3FBN. a
| Transition energy | Exp. | Relative Intensity | Calc. a | Assignment b |
|---|---|---|---|---|
| 35989 | 0 | 100 | 00, band origin | |
| 36129 | 140 | 11 | 142 | 151 |
| 36182 | 193 | 21 | 171 | γ(CN)2 |
| 36255 | 266 | 7 | 251 | 10b1γ(CN)1 |
| 36324 | 335 | 16 | 342 | 10b2 |
| 36374 | 385 | 35 | 381 | 9a1, β(C-F) |
| 36390 | 401 | 58 | 399 | 6b1, β(CCC) |
| 36403 | 414 | 31 | 417 | 10b1γ(CN)3 |
| 36459 | 470 | 11 | 473 | 6a1, β(CCC) |
| 36469 | 480 | 6 | 484 | 10b2151 |
| 36509 | 520 | 6 | 523 | 9a1151 |
| 36549 | 560 | 9 | 567 | βCN |
| 36584 | 595 | 9 | 593 | 10b3γ(CN)1 |
| 36611 | 622 | 9 | 623 | 10a2γ(CN)2 |
| 36649 | 660 | 36 | 663 | 11, breather |
| 36675 | 686 | 33 | 684 | 9a1151γ(CN)2 |
| 36692 | 703 | 20 | 709 | 151β(CN)1 |
| 36727 | 738 | 8 | 741 | 6b110b2 |
| 36789 | 800 | 6 | 798 | 6b2 |
| 36864 | 875 | 8 | 872 | 6a16b1 |
| 36900 | 911 | 13 | 915 | 1110b1γ(CN)1 |
| 36927 | 938 | 13 | 940 | 6b2151 |
| 36947 | 958 | 24 | 957 | 121 |
| 36963 | 974 | 27 | 975 | 1110a1γ(CN)1 |
| 36985 | 996 | 11 | 996 | 6a19a1151 |
| 37051 | 1062 | 17 | 1063 | 116b1 |
| 37075 | 1086 | 9 | 1087 | 416a1γ(CN)1 |
| 37136 | 1147 | 17 | 1142 | 9b1 |
| 37241 | 1252 | 12 | 1253 | 6a16b19a1 |
| 37260 | 1271 | 22 | 1266 | 131 |
| 37304 | 1315 | 7 | 1314 | 1116b116b1 |
| 37324 | 1335 | 14 | 1338 | 1219a1 |
| 37347 | 1358 | 15 | 1356 | 1216b1 |
| 37363 | 1374 | 19 | 1385 | 19a1 |
a The experimental values are shifts from 35989 cm−1, whereas the calculated ones are obtained from the TD-B3LYP/aug-cc-pVDZ calculations, scaled by 0.9722. b β, in-plane bending; γ, out-of-plane bending.
Table 4.
Assignment of observed bands (in cm−1) in the MATI spectra of 3FBN.a
| Intermediate levels in the S1 state | Calc. | Assignment b | |||
|---|---|---|---|---|---|
| 00 | γ(CN)2 | 6b1 | 11 | ||
| 120 | 118 | γ(CN)1 | |||
| 133 | 141 | 151 | |||
| 238 | 237 | γ(CN)2 | |||
| 185 | 188 | 10b1 | |||
| 322 | 331 | 10a1 | |||
| 371 | 370 | 374 | 6b1, β(CCC) | ||
| 399 | 394 | 9a1 | |||
| 498 | 495 | 6a1, β(CCC) | |||
| 506 | 6b1151 | ||||
| 609 | γ(CN)26b1 | ||||
| 615 | 605 | 16a1 | |||
| 668 | 668 | 679 | 11, breathing | ||
| 688 | 6b110b1151 | ||||
| 730 | γ(CN)26a1 | ||||
| 744 | 6b2 | ||||
| 775 | 4110b1 | ||||
| 803 | 11151 | ||||
| 863 | 860 | 6b16a1 | |||
| 893 | 894 | 6a19a1 | |||
| 978 | 965 | 121, β(CCC) | |||
| 997 | 990 | 6a2 | |||
| 1066 | 1067 | 18a1, β(CH) | |||
| 1098 | γ(CN)26a16b1 | ||||
| 1117 | 1105 | 9b1, β(CH) | |||
| 1144 | 1133 | 18b1, β(CH) | |||
| 1166 | 116b1 | ||||
| 1218 | γ(CN)2121 | ||||
| 1235 | 6b26a1 | ||||
| 1258 | 6b16a19a1 | ||||
| 1299 | γ(CN)218a1 | ||||
| 1307 | 1302 | 131, β(CH) | |||
| 1350 | 6b1121 | ||||
| 1357 | γ(CN)29b1 | ||||
| 1374 | 6b16a2 | ||||
| 1385 | 1382 | 19a1 | |||
| 1489 | 6b19b1 | ||||
| 1566 | 1537 | 8a1, ν(CC) | |||
| 1516 | 6b118b1 | ||||
| 1574 | 6b118a1151 | ||||
a The experimental values are shifts from 78873 cm−1, whereas the calculated ones are obtained from the B3LYP/aug-cc-pVDZ calculations, scaled by 0.9704. b β, in-plane bending; γ, out-of-plane bending.
Table 5.
Bond length and bond angle of electronic ground state S0, first excited state S1 and cationic ground state D0 of 2-fluorobenzonitrile calculated at B3LYP/aug-cc-pvtz level.
Table 5.
Bond length and bond angle of electronic ground state S0, first excited state S1 and cationic ground state D0 of 2-fluorobenzonitrile calculated at B3LYP/aug-cc-pvtz level.
| S0 | S1 | D0 | Δ(S1-S0) | Δ(D0-S1) | Δ(D0-S0) | |
|---|---|---|---|---|---|---|
| Bond length (Å) | ||||||
| C1-C2 | 1.396 | 1.436 | 1.455 | 0.040 | 0.019 | 0.059 |
| C2-C3 | 1.381 | 1.406 | 1.393 | 0.025 | -0.013 | 0.012 |
| C3-C4 | 1.389 | 1.409 | 1.372 | 0.020 | -0.037 | -0.017 |
| C4-C5 | 1.392 | 1.408 | 1.434 | 0.016 | 0.026 | 0.042 |
| C5-C6 | 1.385 | 1.421 | 1.386 | 0.036 | -0.035 | 0.001 |
| C6-C1 | 1.401 | 1.424 | 1.391 | 0.023 | -0.033 | -0.010 |
| C1-C11 | 1.427 | 1.395 | 1.407 | -0.032 | 0.012 | -0.020 |
| C11-N12 | 1.152 | 1.165 | 1.158 | 0.013 | -0.007 | 0.006 |
| C2-F13 | 1.341 | 1.324 | 1.296 | -0.017 | -0.028 | -0.045 |
| Band angle (°) | ||||||
| C1-C2-C3 | 122.0 | 124.7 | 122.5 | 2.7 | -2.2 | 0.5 |
| C2-C3-C4 | 118.8 | 119.0 | 117.5 | 0.2 | -1.5 | -1.3 |
| C3-C4-C5 | 120.5 | 118.1 | 121.1 | -2.4 | 3.0 | 0.6 |
| C4-C5-C6 | 120.0 | 122.7 | 121.4 | 2.7 | -1.3 | 1.4 |
| C5-C6-C1 | 120.4 | 120.6 | 119.0 | 0.2 | -1.6 | -1.4 |
| C6-C1-C2 | 118.3 | 114.9 | 118.4 | -3.4 | 3.5 | 0.1 |
Table 6.
Bond length and bond angle of the electronic ground state S0, first excited state S1 and cationic ground state D0 of 3-fluorobenzonitrile calculated at B3LYP/aug-cc-pvtz level.
Table 6.
Bond length and bond angle of the electronic ground state S0, first excited state S1 and cationic ground state D0 of 3-fluorobenzonitrile calculated at B3LYP/aug-cc-pvtz level.
| S0 | S1 | D0 | Δ(S1-S0) | Δ(D0-S1) | Δ(D0-S0) | |
|---|---|---|---|---|---|---|
| Bond length (Å) | ||||||
| C1-C2 | 1.398 | 1.425 | 1.380 | 0.026 | -0.045 | -0.018 |
| C2-C3 | 1.380 | 1.413 | 1.393 | 0.033 | -0.019 | 0.013 |
| C3-C4 | 1.384 | 1.406 | 1.433 | 0.022 | 0.027 | 0.049 |
| C4-C5 | 1.390 | 1.404 | 1.374 | 0.014 | -0.029 | -0.016 |
| C5-C6 | 1.387 | 1.411 | 1.395 | 0.024 | -0.016 | 0.008 |
| C6-C1 | 1.398 | 1.438 | 1.449 | 0.039 | 0.010 | 0.051 |
| C1-C11 | 1.430 | 1.397 | 1.415 | -0.033 | 0.017 | -0.015 |
| C11-N12 | 1.152 | 1.165 | 1.155 | 0.013 | -0.009 | 0.003 |
| C2-F13 | 1.346 | 1.330 | 1.300 | -0.016 | -0.030 | -0.046 |
| Band angle (°) | ||||||
| C1-C2-C3 | 118.2 | 118.4 | 116.9 | 0.2 | -1.4 | -1.3 |
| C2-C3-C4 | 122.5 | 125.2 | 123.4 | 2.7 | -1.7 | 0.9 |
| C3-C4-C5 | 118.5 | 115.9 | 118.8 | -2.6 | 2.9 | 0.3 |
| C4-C5-C6 | 120.6 | 121.1 | 119.2 | 0.4 | -1.9 | -1.4 |
| C5-C6-C1 | 119.6 | 122.2 | 120.9 | 2.6 | -1.3 | 1.3 |
| C6-C1-C2 | 120.4 | 116.9 | 120.5 | -3.4 | 3.5 | 0.1 |
Table 7.
Ionization energy of benzene, phenol, and their F and CN substituted molecules (cm-1).
| molecule | IE | ΔIE | molecule | IE | ΔIE | |
| Benzene a | 74557 | 0 | Benzonitrile c | 78490 | 0 | |
| Fluorobenzene b | 74227 | -330 | 2-Fluorobenzonitrile d | 78650 | 160 | |
| Benzonitrile c | 78490 | 3933 | 3-Fluorobenzonitrile d | 78873 | 383 | |
| 4-Fluorobenzonitrile e | 78000 | -490 | ||||
| Phenol f | 68625 | 0 | ||||
| 2-Fluorophenol g | 70006 | 1381 | Fluorobenzene b | 74227 | 0 | |
| 3-Fluorophenol, cis h,i | 70188 | 1563 | 2-fluorobenzonitrile d | 78650 | 4423 | |
| 3-Fluorophenol, trans h,i | 70449 | 1824 | 3-fluorobenzonitrile d | 78873 | 4646 | |
| 4-Fluorophenol j | 68577 | -48 | 4-fluorobenzonitrile e | 78000 | 3773 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



