
Submitted:
25 May 2023
Posted:
30 May 2023
You are already at the latest version
Abstract
Electrochemical deposition of silicon at room temperature is problematic due to intrinsically low conductivity of the deposits. This study reports the photoelectrochemical (PEC) deposition of silicon (Si) and silicon–carbon (Si–C) layers from an ionic liquid at temperature 40 oC using silicon tetrachloride (SiCl4) as a silicon precursor. Amorphous layers are deposited on p-type silicon (p-Si), p-type gallium arsenide (p-GaAs), and aluminum–copper alloy AA2024. The semi-conductor substrates are activated by white LED illumination, which generated photoelectrons, thereby making the substrate conductive with respect to the cathodic reaction. Photoresponsive-ness of the deposits is proven by the light-induced photocurrents on an optically inactive substrate made of the alloy AA 2024. The proposed method paves the way for the electrochemical modifica-tion of semiconductors and metals with Si and Si–C structures, which are applicable in various fields, such as; batteries, anti-corrosion coatings, photovoltaics, or PEC electrodes for hydrogen production.
Keywords:
silicon
; carbon
; gallium arsenide
; semiconductor
; photoelectrochemistry
; ionic liquid
1. Introduction
Electrochemical deposition of thin silicon layers is of great importance for numerous fields of application, such as photovoltaics, hydrogen production, batteries, or platforms for sensors. Electrodeposition of thin Si layers can replace multiple Si wafer fabrication, such as production of metallurgical grade Si (MG-Si), upgrading to solar grade Si (SoG-Si) as well as ingot casting and slicing [1]. Typically, Si layers are electrodeposited in high temperature molten salts: fluorides [2,3,4,5,6,7,8,9], oxides [10,11,12], or chlorides [1,13,14].
There is a great need to produce Si layers at low temperatures; however, intrinsically low conductivity of Si prevents its electrochemical deposition. Conceptually, this paves way for the ionic liquids (ILs), known as the room temperature molten salts. However, only very thin Si structures have been deposited in ILs, which were composed of nanocrystallites and nanoislands [15,16,17,18].
Tsuyuki et al. formed thin films of Si in IL under light irradiation using SiCl4 as Si source and AlCl3 as a doping additive [19]. The authors deposited thin layers of Si on a highly doped n-type Si (n-Si) and Au substrates.
When illuminating an n-type semiconductor, minority carriers – holes – are generated, which promote the electron transfer from electrolyte to electrode. In a p-type semiconductor, photoelectrons are generated, which promote the electron transfer from electrode to electrolyte. Here, we took advantage of p-type semiconductors to generate the photoelectrons and investigated the photoelectrochemical (PEC) reduction of silicon and carbon in an IL on p-Si and p-GaAs. The photoelectrons can also be consumed in the electrochemical reaction, thereby promoting the reduction process.
We also studied the deposition process on optically inactive metallic substrate – aluminum alloy with copper AA2024. Deposition on Al substrates in aqueous electrolytes is difficult due to the presence of highly isolating alumina film. In ILs, such film can be decomposed due to the possibility of applying high polarizations. From a technical point of view, deposition of thin silicon films on technically important alloys is a challenging goal, which increases the chances of combining structural materials with silicon to harness the solar energy. Putting the silicon on thin metallic foils could expand the application fields to a wide range of subjects with flexible geometries as well as to metal-coated glass.
Silicon–carbon (Si–C) composites are promising materials for new generation batteries [20,21]. Porous Si–C structures can increase capacity of the electrode, for instance, above 2000 mA h g-1 [22]. Porous structures also increase interfacial stability of the electrode and suppress dendrite formation. Emerging area is application of the Si–C composites for post-lithium battery technologies, which are based on non-critical elements, such as sodium (Na) [20]. Si–C structures are also promising for increasing the light harvesting performance of silicon-based solar devices. Low reflectance values were determined for diamond-like carbon/porous silicon systems within the UV, the visible and the IR regions [23]. The C–SiC system improved the efficiency of solar cell up to 1.5 times [24]. A two-layer SiO2–SiC structure on Si enhanced the cell performance by 34.15% due to light trapping and improved photoelectronic properties [25]. An increased surface passivation and energy conversion efficiency was also determined for SiCxNy coatings [26].
The Si–C compositions mostly are produced by high temperature processing, such as chemical vapor deposition (CVD) or pyrolysis of organic reactants [27]. Gaseous reactants are broken down into atomic species that are deposited on the Si substrate. Various precursors can be used as a carbon source, for instance, propylene, acetylene, methane, ethanol, toluene, sucrose, etc.
Electrochemical formation of Si–C compositions is at a very early stage of investigation. Major limitation lies in low conductivity of silicon deposits at room temperatures as well as insufficient electrochemical window in aqueous electrolytes. Si–C–SiC structures were electrochemically synthesized in molten calcium chloride (CaCl2) at temperatures around 900 oC [28]. Graphite anode has been used as a carbon source, and silica (SiO2) layer on silicon substrate was employed as a silicon precursor. Zou et al. synthesized electrochemically silicon carbide (SiC) nanowires in molten CaCl2 at 900 oC [29]. SiO2 and C mixture was used as a precursor, which was prepared in the form of a pellet. The precursor was sandwiched between nickel foil electrodes and electrolyzed in the molten salt electrolyte.
In present work, we demonstrate PEC silicon codeposition with carbon in an IL. The electrolyte was composed of N-trymethyl-N-hexylamonium (TMHA) as a cation, bis(trifluoromethylsulfonyl)amide (TFSA) as an anion, and silicon tetrachloride (SiCl4) as a silicon precursor. These materials have typically been used for silicon electrodeposition in ILs, as reviewed in [30].
2. Materials and Methods
Figure 1 shows configuration of the PEC cell used for the deposition experiments. The cell body was prepared from a tube (∅ 50 mm) of polyether ether ketone (PEEK from Rochling, Germany). The working electrodes have been mounted at the bottom of the cell. The electrode/electrolyte contact area was 0.5 cm2. The back contact was prepared using highly conductive foam (Holland Shielding System BV). Pt electrode was used as a reference, whose potential was determined with respect to a standard Ag/AgCl electrode and displayed with respect to a normal hydrogen electrode (NHE). The counter electrode was prepared from Pt in the form of a wire ring, so that it did not shadow the working electrode. The electrodes were connected to a potentiostat (PARSTAT 2273 from Princeton Applied Research). The cell was supplied with the IL, which was composed of TMHA, TFSA (both from Solvionic, France), and 0.5 M or 1.0 M SiCl4 (from VWR, Austria). The total electrolyte volume was 8 mL. The electrolyte temperature was kept at 40 oC using a heating element made of nichrome (NiCr) wire wound around the cell body. The temperature was controlled by a thermocouple. A white LED illumination (4700 oK CCT) at intensity N = 50 mW cm-2 has been supplied to the working electrode through an optical window made of quartz.
The silicon substrates were prepared from a boron-doped <100> p-Si wafer with a resistivity of 10 – 30 Ω cm. The wafer surface was polished to a mirror shiny appearance; its native oxide was removed in the solutions H2O2 + H2SO4 (1:1) and HF (1 HF : 40 H2O). The p-GaAs electrode was prepared from <100> wafer, doped with zinc (Zn) at concentration (1.2 – 1.8) × 1017 cm3. The AA2024 electrode was prepared from the alloy disc (∅ 14 mm); its surface was sequentially polished by 600, 1200, 2500 grit sandpaper as well as with 3 μm and 1 μm diamond polishing paste with waterless polishing lubricant (Struers). The samples were ultrasonically cleaned in isopropyl alcohol for 15 minutes after each polishing step.
The deposition procedures were conducted in a glow box operating in nitrogen atmosphere (N2, 0.5 ppm O2, and 0.5 ppm H2O). After deposition, the remaining electrolyte on the sample surface was removed in isopropyl alcohol at temperature 40 oC. Afterwards, the sample was dried using an argon stream and kept in a hermetic container filled with argon.
Properties of the deposits were examined by SEM Helios NanoLab 650 from FEI equipped with energy dispersive X-ray spectrometer (EDX) INCAEnergy from Oxford Instruments. Cross sections were prepared by focused ion beam of Ga+ ions with energy of 30 kV. Pt layer has been sputtered on the sample to achieve interfacial stability during the cross sectioning.
Structure of the oxide films were studied by the grazing incidence x-ray diffraction (GI-XRD) using SmartLab diffractometer from Rigaku with an X-ray tube equipped with 9 kW Cu rotating anode.
Surface micro-topography has been studied using optical profilometer Contour GT-K (Bruker) in a white light interferometry (WLI) mode.
XPS analysis was performed on Kratos Axis Supra spectrometer using monochromatic Al-Kα radiation (hν = 1486.7 eV) with X-ray gun power of 225 W at 10−8 torr pressure and room temperature. The analyzed area at the sample center was approximately 0.16 mm2 in size and high-resolution selected area scans were acquired using pass energy of 20 eV. To compensate for sample charging during the measurement, a built-in charge neutralizer was used. The samples were also electrically isolated from the sample holder to avoid preferential charging effects. Binding energy scale of recorded spectra was corrected by referencing to C 1s peak at 284.8 eV.
3. Results
First, we determined the electrochemical windows of the IL electrolyte and that supplied with the Si precursor. Figure 2(a) shows the cathodic curves obtained for p-Si electrode in the electrolyte with and without Si precursor.
The electrode is passive in the darkness – no appreciable current is observed (curve 1). Under illumination, a cathodic current appears in the precursor-free electrolyte at polarization ΔE < -3.8 V (curve 2, Figure 2a). The current increases exponentially with polarization denoting the electrochemical window of the electrolyte. A substantial current contribution is observed when Si precursor is introduced: the photocurrent appears at ~-2.0 V and reaches ~1 mA cm-2 at the potential where electrolyte decomposition begins (ΔE = ~-3.8 V). In the subsequent potential region, the difference between two curves remains approximately steady. These data indicate the potentials, at which silicon reduction is possible: SiCl4 + 4e- → Si.
Figure 2(b) identifies the voltammetry that is characteristic of the p-GaAs electrode. From a qualitative point of view, there is no significant difference between p-Si and p-GaAs electrodes: the width of the electrochemical window is similar, and the currents of silicon reduction are substantial. The reduction processes are demonstrated when precursor’s concentration for p-GaAs electrode is twice higher than the concentration for p-Si electrode (1.0 M and 0.5 M, respectively). A more intensive deposition on GaAs results in a thicker coating to the extent of slowing down the cathodic process. This is evident from the curve maximum at ΔE = ~-4.7 V and subsequent current decrease.
Figure 3 demonstrates the principal opportunity of PEC deposition of Si coating on a p-Si substrate. The deposition has been performed in a potentiodynamic mode at the scan rate of 10 mV s-1, covering wide potential region from the open circuit potential (Eocp) to the polarization ∆E = -5.0 V where electrolyte decomposition is possible (Figure 2a). SEM-EDS identified the deposition of silicon layer, which was about 270 nm thick (Figure 3). The Si deposition began when the equilibrium potential E0Si4+/Si was exceeded; the process continued to rise with the negative potential shift. In the far polarization region, simultaneous decomposition of electrolyte was possible; however, no carbon was detected in the deposit by the XPS (no C 1s peak). Carbon deposition under potentiostatic conditions will be discussed later.
The deposited sample contained some oxygen. Figure 3(b) identifies thickness of the oxygen-containing structure. The zone where content of sputtered Pt drops down signifies the Pt/Si-O interface, and the zone where Si content reaches a limit signifies thickness of the deposited layer. Note that some surface oxidation under ambient conditions is unavoidable. This is especially relevant in the case of thin layers. It is worth mentioning that the Si oxidation suggests an opportunity for electronic passivation as well as an improvement in antireflection performance due to interfacial light trapping. Porous silica (SiO2) antireflective coatings prepared by sol-gel process have been proposed. For instance, antireflection coatings based on SiO2 nanocomposites have been reported to exhibit high level of transparency, resistance to moisture, and antifogging properties [32,33].
Furthermore, we will focus on the deposition processes at high electrode polarization where electrolyte decomposition is possible (∆E = -5.7 V, Figure 4, Figure 5 and Figure 6). Figure 4 shows the topographical data obtained for the profile of the deposited layer that extends over tens of micrometers. The surface roughness characterized by the top-to-valley distance (z) is mostly less than 100 nm; in some areas, however, the distance can reach z ~300 nm (indicated in blue). The topographical data in Figure 4(b) demonstrates the surface features of the pristine p-Si substrate, on which the layer was deposited. The surface is rather smooth; its roughness is much less than that of the deposited layer. The image identifies greatest value z ~ 37 nm.
The SEM image in Figure 5 shows the cross section of the sample; it identifies thickness of the deposited layer to be ~0.65 μm.
Elemental composition of the deposited layer has been studied by EDS (Figure 6). Significant amount of carbon (~18 at. %) was co-deposited with silicon. Carbon deposition was also confirmed by XPS. The 1s energy spectrum identified 19.9 at. % of the elemental carbon on the sample surface. A characteristic feature is that carbon is distributed within the silicon in the form of micrometer-sized inclusions (red or dark spots in Figure 6). Note also that GI-XRD did not indicate any crystallinity features of the deposited layer, indicating its amorphous structure.
A thin layer (~300 nm) of Si–C structure was deposited photoelectrochemically on GaAs substrate as well (Figure 7). GI-XRD did not indicate any crystallinity features of the layer.
A challenging goal is creation of thin silicon films on technically important metallic substrates such as aluminum alloys, steel or copper. Figure 8 demonstrates a thin Si–C layer, which was deposited on the aluminum–copper alloy AA 2024. The obtained layer was about 280 nm thick and contained 17 at. % of carbon.
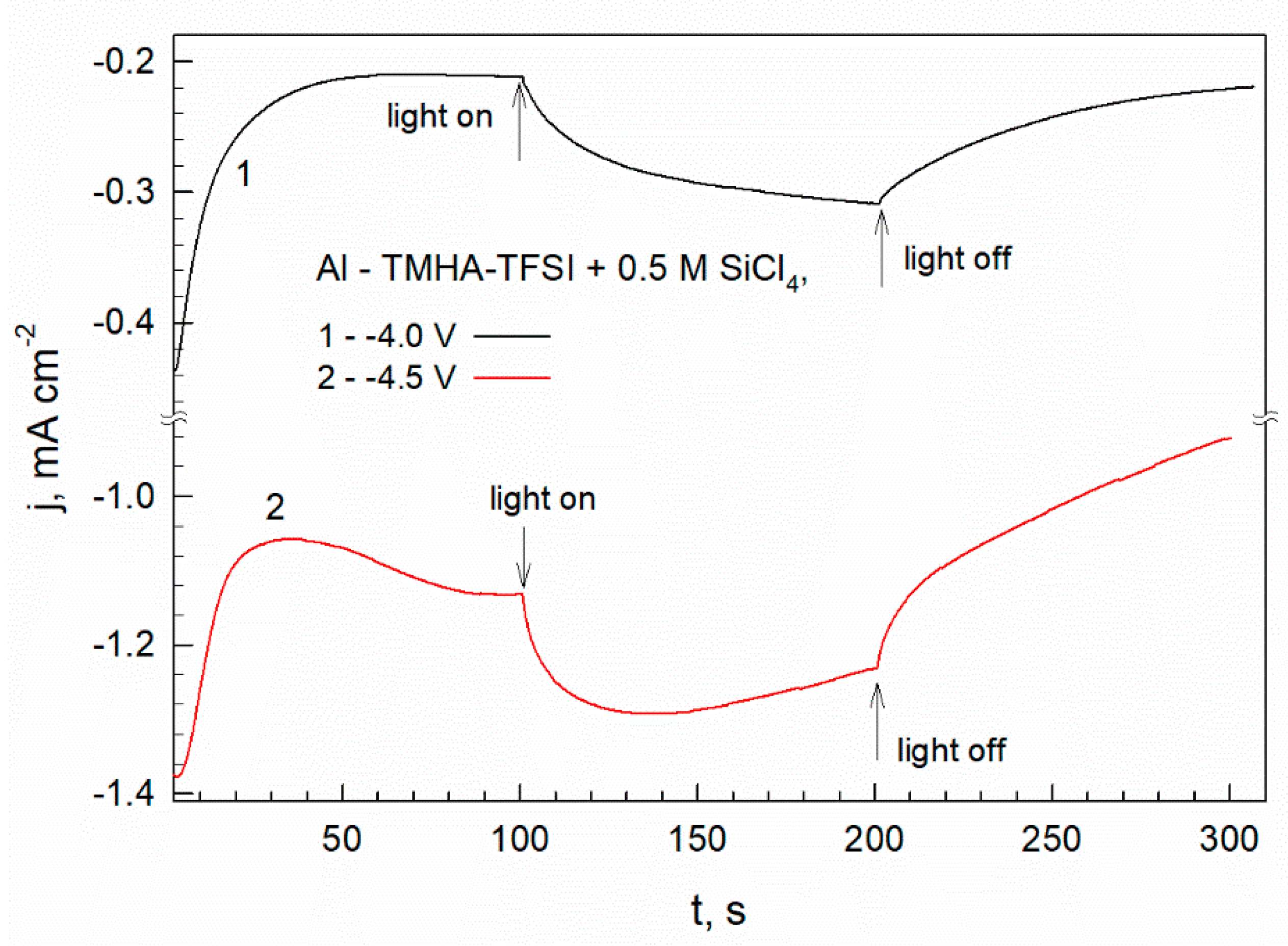
Figure 9 shows the current variation determined for the AA2024 coated with Si–C layer under potentiostatic conditions. The current drop to a limiting value is characteristic of a diffusion-limited process. A substantial current growth is observed when the electrode is illuminated. Such behavior indicates a photoresponsible surface the deposited silicon. The light-enhancing effect is also evident at higher polarization (∆E = -4.5 V, curve 2), where electrolyte decomposition is significant (Figure 2).
4. Discussion
Electrochemical deposition of Si layers is of great importance in numerous fields of application, such as photovoltaics, antireflection coatings, batteries, sensors, photoelectrodes for hydrogen and oxygen production from water, and protection of metals from corrosion. Electrodeposition of silicon at room temperatures is problematic due to low electric conductivity of this element.
When illuminating an n-type semiconductor, minority carriers – holes – are generated, which promote the electron transfer from electrolyte to electrode. An example of such generation provides the well-known photoelectrochemical generation of oxygen on an n-type Si photoelectrode. In a p-type semiconductor, photoelectrons are generated, which can promote the electron transfer from electrode to electrolyte. The photoelectrons can also be consumed in the electrochemical reaction. An example provides photoelectrochemical hydrogen generation from water on a p-type Si photoelectrode.
In present investigation, we took advantage of p-type semiconductors to generate the photoelectrons and deposited photoelectrochemically Si as well Si – C structures from IL on p-Si and p-GaAs substrates. We activated the semiconductors using white LED illumination and deposited the structures in the far region of cathodic potentials where decomposition of IL occurs. The deposition was achieved at nearly room temperatures (40 oC).
Amorphous Si – C layers with micrometer-sized carbon inclusions in the silicon were formed. The produced layers were photo-responsive; the light-enhanced cathodic currents measured on an optically inactive electrode made of AA 2024 alloy proved this.
The proposed methodology paves the way for silicon modification by silicon-carbon structures. The proposed approach can also be applied to technically important alloys, as demonstrated by the deposition on AA 2024 alloy. The deposition of Si structures on technically important alloys is important, first of all, for their anticorrosion protection.
We have demonstrated the principal opportunity to deposit thin film silicon-based structures. There is a great margin to improve quality of the layers and optimize the deposition conditions, such as cell configuration, kind of the substrate, electrolysis parameters, precursor concentration, and temperature.
6. Patents
Patent application LT2023 519; No 107957; 19. 05. 2023.
Author Contributions
A. G., P. K., L. S., A. S. investigation, data analysis, validation; K. L. methodology, supervizion, resources; E. J. conceptualization, data analysis, writing, supervision, project administration, funding acquisition.
Funding
This research was funded by the Lithuanian Scientific Council under project agreement S-MIP-21-124.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available on request from the corresponding author.
Acknowledgments
We thank Dr. Sandra Stanionytė for assistance with the XRD measurements and Dr. Jurgis Pilipavičius with the XPS measurements.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Zou, X.; Ji, L.; Yang, X.; Lim, T.; Yu, E.T.; Bard, A. J. Electrochemical formation of a p-n junction of thin film silicon deposited in molten salt. J. Am. Chem. Soc. 2017, 139, 16060–16063. [Google Scholar] [CrossRef] [PubMed]
- Cohen, U.; Huggins, R. A. Silicon epitaxial growth by electrodeposition from molten fluorides. J. Electrochem. Soc. 1976, 123, 381–383. [Google Scholar] [CrossRef]
- Cohen, U. Some prospective applications of silicon electrodeposition from molten fluorides to solar cell fabrication. J. Electron. Mater. 1977, 6, 607–643. [Google Scholar] [CrossRef]
- Elwell, D.; Rao, G.M. Mechanism of electrodeposition of silicon from K2SiF6-flinak. Electrochim. Acta 1982, 27, 673–676. [Google Scholar] [CrossRef]
- Osen, K.S.; Martinez, A.M.; Rolseth, S.; Gudbrandsen, H.; Juel, M.; Haarberg, G.M. Electrodeposition of crystalline silicon films from alkali fluoride mixtures. ECS Trans. 2010, 33, 429–438. [Google Scholar] [CrossRef]
- Hu, Y.; Wang, X.; Xiao, J.; Hou, J.; Jiao, S.; Zhu, H. Electrochemical behavior of silicon (IV) ion in BaF2-CaF2-SiO2 melts at 1573 K. J. Electrochem. Soc. 2013, 160, D81–D84. [Google Scholar] [CrossRef]
- Zaykov, Y.P.; Isakov, A.V.; Zakiryanova, I.D.; Reznitskikh, O.G.; Chemezov, O.V.; Redkin, A.A. Interaction between SiO2 and a KF-KCl-K2SiF6 melt. J. Phys. Chem. B 2014, 118, 1584–1588. [Google Scholar] [CrossRef]
- Isakov, A. Electrodeposition of continuous silicon coatings from the KF-KCl-K2SiF6 melts. J. Electrochem. Soc. 2017, 164, H5135–H5138. [Google Scholar] [CrossRef]
- Suzuki, Y.; Inoue, Y.; Yokota, M.; Goto, T. Effects of oxide ions on the electrodeposition process of silicon in molten fluorides. J. Electrochem. Soc. 2019, 166, D564–D568. [Google Scholar] [CrossRef]
- De Mattei, R.C.; Elwell, D.; Feigelson, R.S. Electrodeposition of silicon at temperatures above its melting point. J. Electrochem. Soc. 1981, 128, 1712–1714. [Google Scholar] [CrossRef]
- Konakov, V.G.; Pivovarov, M.M. Interaction between components in the KF-SiO2 system. Glass Phys. Chem. 2003, 29, 69–74. [Google Scholar] [CrossRef]
- Suzuki, Y.; Inoue, Y.; Yokota, M.; Goto, T. Effects of oxide ions on the electrodeposition process of silicon in molten fluorides. J. Electrochem. Soc. 2019, 166, D564–D568. [Google Scholar] [CrossRef]
- Yang, X.; Ji, L.; Zou, X.; Lim, T.; Zhao, J.; Yu, E.T.; Bard, A. J. Toward cost-effective manufacturing of silicon solar cells: electrodeposition of high-quality Si films in a CaCl2-based molten salt. Angew. Chem. Int. Ed. 2017, 56, 15078–15082. [Google Scholar] [CrossRef] [PubMed]
- Zou, X.; Ji, L.; Ge, J.; Sadoway, D.R. , Yu, E.T.; Bard, A.J. Electrodeposition of crystalline silicon films from silicon dioxide for low-cost photovoltaic applications. Nat. Commun. 2019, 10, 5772. [Google Scholar] [CrossRef] [PubMed]
- Katayama, Y.; Yokomizo, M.; Miura, T.; Kishi, T. Preparation of novel fluorosilicate salt for electrodeposition of silicon at low temperature. Electrochemistry, 2001, 69, 834–836. [Google Scholar] [CrossRef]
- Zein El Abedin, S.; Borissenko, N.; Endres, F. Electrodeposition of nanoscale silicon in a room temperature ionic liquid. Electrochem. Commun. 2004, 6, 510–514. [Google Scholar] [CrossRef]
- Borisenko, N.; Zein El Abedin, S.; Endres, F. In situ STM investigation of gold reconstruction and of silicon electrodeposition on Au(111) in the room temperature ionic liquid 1-butyl-1-methylpyrrolidinium bis(trifluoromethylsulfonyl)imide. J. Phys. Chem. B, 2006, 110, 6250–6256. [Google Scholar] [CrossRef]
- Schmuck, M.; Balducci, A.; Rupp, B.; Kern, W.; Passerini, S.; Winter, M. Alloying of electrodeposited silicon with lithium – a principal study of applicability as anode material for lithium ion batteries. J. Solid State Electrochem. 2010, 14, 2203–2207. [Google Scholar] [CrossRef]
- Tsuyuki, Y.; Takai, H.; Fukunaka, Y.; Homma, T. Formation of Si thin films by electrodeposition in ionic liquids for solar cell applications. Jpn. J. Appl. Phys. 2018, 57, 08RB11. [Google Scholar] [CrossRef]
- Wang, J.J.; Xu, T.T.; Huang, X.; Li, H.; Ma, T.L. Recent progress of silicon composites as anode materials for secondary batteries. RSC Advances 2016, 6, 87778–87790. [Google Scholar] [CrossRef]
- Dou, F.; Shi, L.; Chen, G.; Zhang, D. Silicon/Carbon composite anode materials for lithium-ion batteries. Electrochem. Energ. Rev. 2019, 2, 149–198. [Google Scholar] [CrossRef]
- Vrankovic, D.; Graczyk-Zajac, M.; Kalcher, C.; Rohrer, J.; Becker, M.; Stabler, C.; Trykowski, G.; Albe, K.; Riedel, R. ACS Nano. 2017, 11, 11409–11416. [Google Scholar] [CrossRef] [PubMed]
- Aroutiounian, V.M.; Martirosyan, Kh. S.; Soukiassian, P. G. Reflectance spectrum of diamond-like carbon/porous silicon double-layer antireflection coating for silicon solar cells. Phys. Status Solidi C 2007, 4, 2107–2110. [Google Scholar] [CrossRef]
- Klyui, N.I.; Litovchenko, V.G.; Rozhin, A. G.; Dikusha, V. N.; Kittler, M.; Seifert, W. Silicon solar cells with antireflection diamond-like carbon and silicon carbide films. Sol. Energy Mater. Sol. Cells, 2002, 72, 597–603. [Google Scholar] [CrossRef]
- Zhao, Y.; Chen, F.; Shen, O.; Zhang, L. Optimal structure of light trapping in thin-film solar cells: dielectric nanoparticles or multilayer antireflection coatings? Applied Optics, 2014, 53, 5222–5229. [Google Scholar] [CrossRef] [PubMed]
- Kang, M.H.; Kim, D.S.; Ebong, A.; Rounsaville, B.; Rohatgi, A.; Okoniewska, G.; Hong, J. The study of silane-free SiCxNy film for crystalline silicon solar cells, J. Electrochem. Soc. 2009, 156, H495–H499. [Google Scholar] [CrossRef]
- Shi,Q. ; Zhou, J.; Ullah, S.; Yang, X.; Tokarska, K.; Trzebicka, B.; Ta, H. Q., Rümmeli, M. H. A review of recent developments in Si/C composite materials for Li-ion batteries. Energy Storage Mater. 2021, 34, 735–754. [Google Scholar] [CrossRef]
- Juzeliūnas, E.; Fray, D.J.; Kalinauskas, P.; Valsiūnas, I.; Niaura, G.; Selskis, A.; Jasulaitienė, V. Electrochemical synthesis of photoactive carbon-carbide structure on silicon in molten salt. Electrochem. Commun. 2018, 90, 6–10. [Google Scholar] [CrossRef]
- Zou, X.; Ji, L.; Lu, X.; Zhou, Z. Facile electrosynthesis of silicon carbide nanowires from silica/carbon precursors in molten salt. Scientific reports 2017, 7, 9978. [Google Scholar] [CrossRef]
- Juzeliūnas, E. Silicon. Electrochemistry, production, purification and applications; Wiley: New York, NY, USA, 2023; ISBN 9783527831913. [Google Scholar]
- Zhang, X. G. Electrochemistry of silicon and its oxide; Springer: New York, NY, USA, 2001; p. 45. ISBN 978-0-306-47921-2. [Google Scholar]
- Zhang, X.; Lan, P.; Lu, Y.; Li, J.; Xu, H.; Zhang, J.; Lee, Y.; Rhee, J.Y.; Choy, K-L. ; Song, W. Multifunctional antireflection coatings based on novel hollow silica – silica nanocomposites. ACS Appl. Mater. Interfaces 2014, 6, 1415–1423. [Google Scholar] [CrossRef]
- Cai, S.; Zhang, Y.; Zhang, H.; Yan, H.; Lv, H.; Jiang, B. Sol-gel preparation of hydrophobic silica antireflective coatings with low refractive index by base/acid two-step catalysis. ACS Appl. Mater. Interfaces, 2014, 6, 11470–11475. [Google Scholar] [CrossRef] [PubMed]
- Bard, A.J.; Faulkner, L.R. Electrochemical Methods: Fundamentals and Applications, 2nd ed.; Wiley: New York, NY, USA, 2001; ISBN 978-0-471-04372-0. [Google Scholar]
- Kalinauskas, P.; Staišiūnas, L.; Grigucevičienė, A.; Leinartas, K.; Selskis, A.; E. Juzeliūnas. E. Photoelectrochemical and nanogravimetric study of electrolytic transformation of silicon-oxide interface. J. Electrochem. Soc. 2022, 169, 036508. [Google Scholar] [CrossRef]
Figure 1.
The PEC cell used for deposition of Si–C coatings.
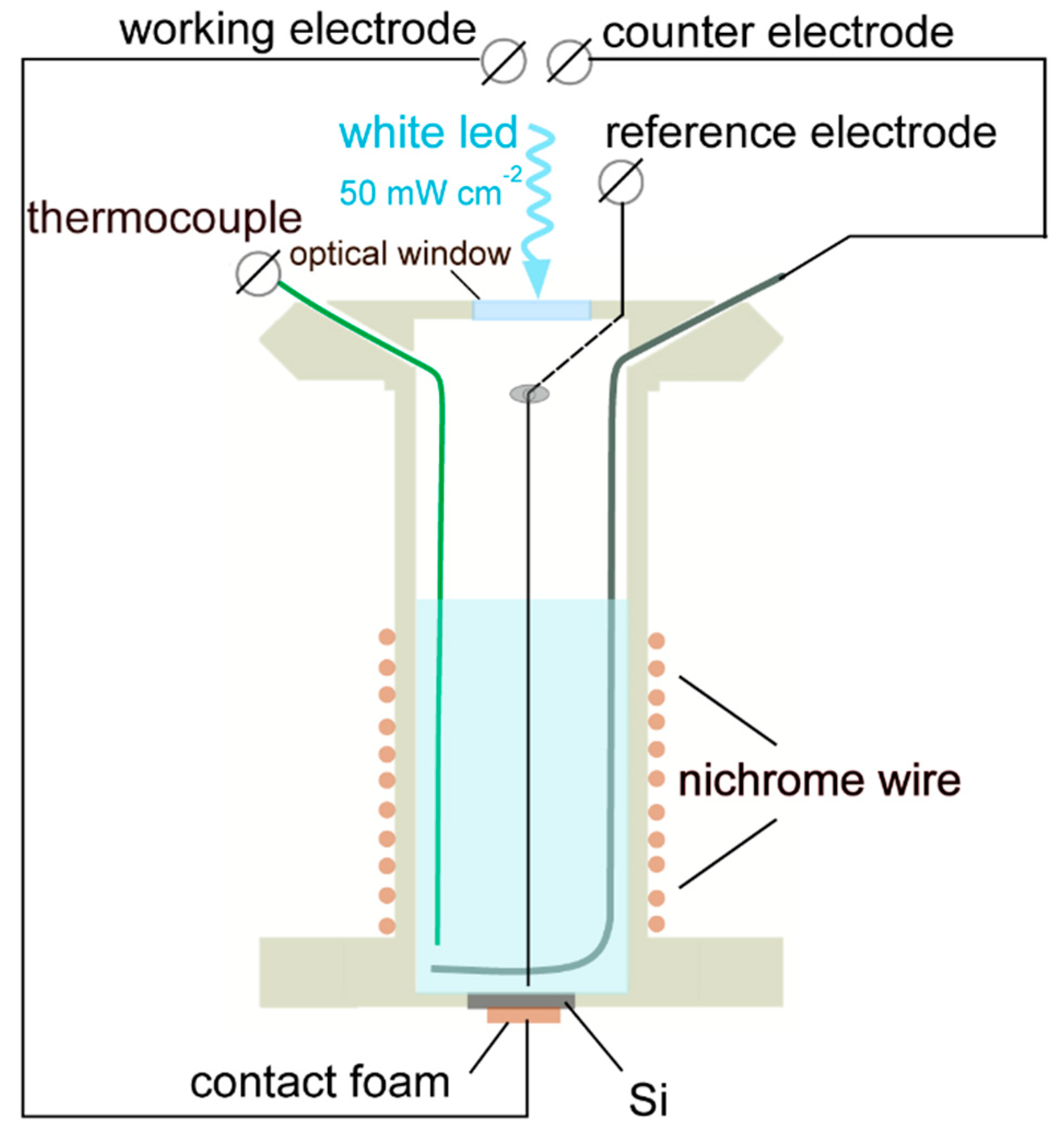
Figure 2.
(a) Cathodic polarization curves obtained at the potential sweep rate of 25 mV s-1 for p-Si in the electrolyte TMHA– TFSI + 0.5 M SiCl4 in the dark (1) and under illumination (3). Curve 2 shows the photocurrents for the illuminated electrode in the precursor-free electrolyte. Figure (b) shows the curves analogous to (a) determined for p-GaAs electrode in the electrolyte with 1.0 M SiCl4.
Figure 2.
(a) Cathodic polarization curves obtained at the potential sweep rate of 25 mV s-1 for p-Si in the electrolyte TMHA– TFSI + 0.5 M SiCl4 in the dark (1) and under illumination (3). Curve 2 shows the photocurrents for the illuminated electrode in the precursor-free electrolyte. Figure (b) shows the curves analogous to (a) determined for p-GaAs electrode in the electrolyte with 1.0 M SiCl4.
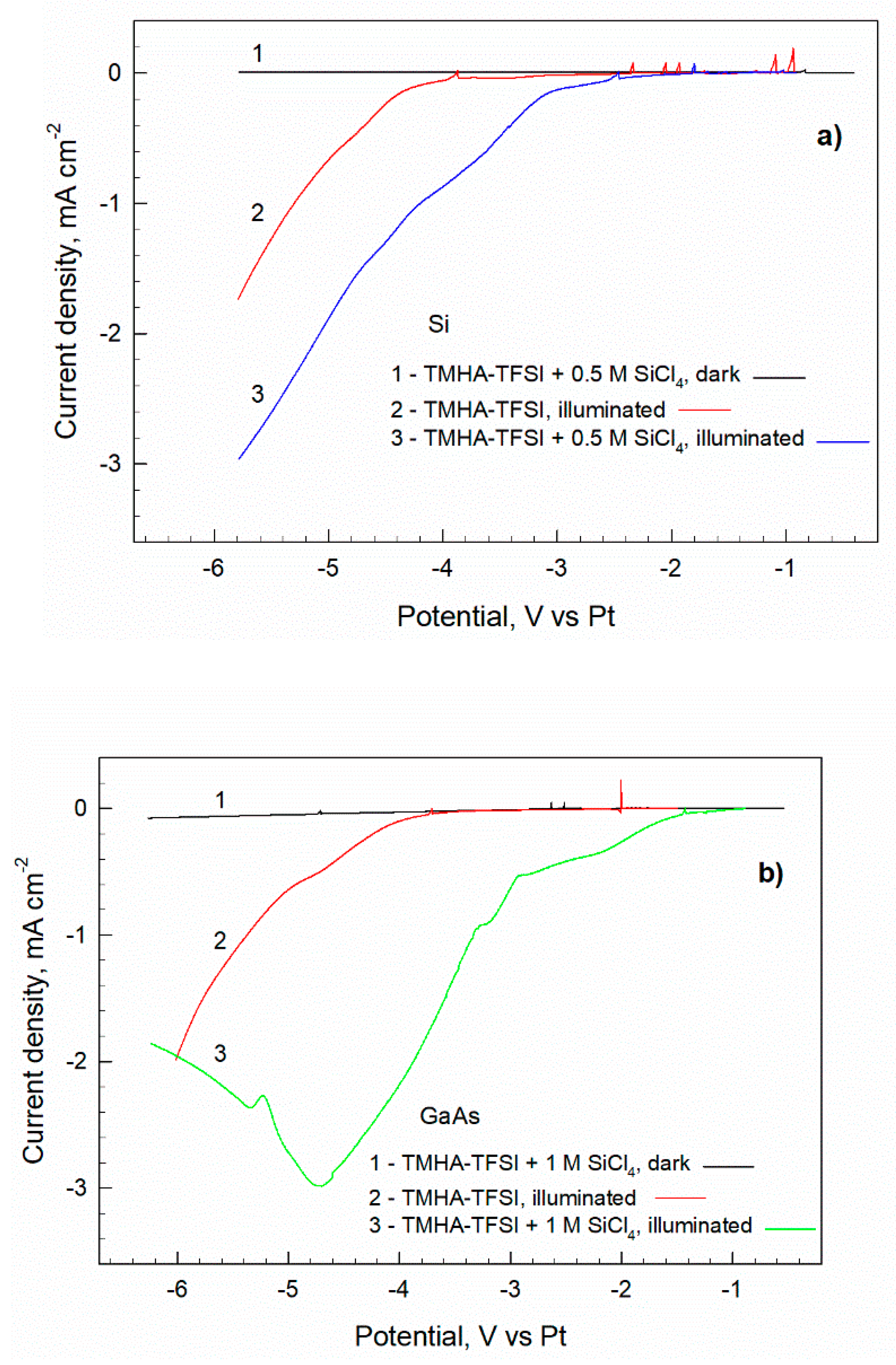
Figure 3.
SEM-EDS study of the surface layer deposited when sweeping the electrode potential negatively at 10 mV s-1 rate from the open circuit potential to ∆E = -5.0 V: (a) cross section of the deposited layer on a p-Si substrate prepared by Ga+ beam; (b) EDS data for distribution of the elements within the cross-section: oxygen, silicon, and Pt sputtered on top of the sample.
Figure 3.
SEM-EDS study of the surface layer deposited when sweeping the electrode potential negatively at 10 mV s-1 rate from the open circuit potential to ∆E = -5.0 V: (a) cross section of the deposited layer on a p-Si substrate prepared by Ga+ beam; (b) EDS data for distribution of the elements within the cross-section: oxygen, silicon, and Pt sputtered on top of the sample.
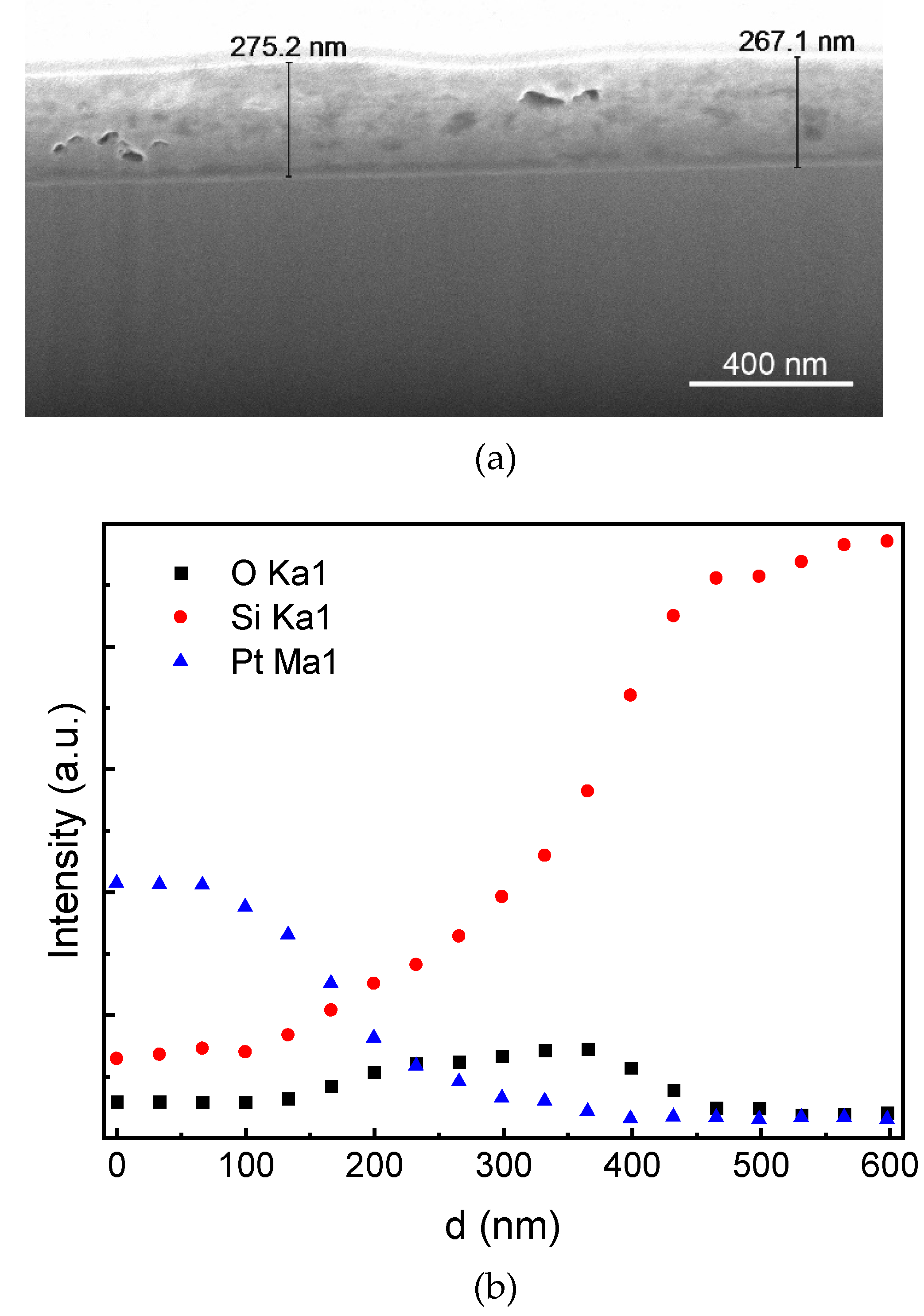
Figure 4.
(a) Topographical data obtained by the optical profilometry from the surface of Si–C layer deposited at ∆E = -5.7 V (t = 1200 s) in the electrolyte composed of TMHA, TFSA and 0.5 M SiCl4. (b) Topographical data obtained for the pristine substrate.
Figure 4.
(a) Topographical data obtained by the optical profilometry from the surface of Si–C layer deposited at ∆E = -5.7 V (t = 1200 s) in the electrolyte composed of TMHA, TFSA and 0.5 M SiCl4. (b) Topographical data obtained for the pristine substrate.
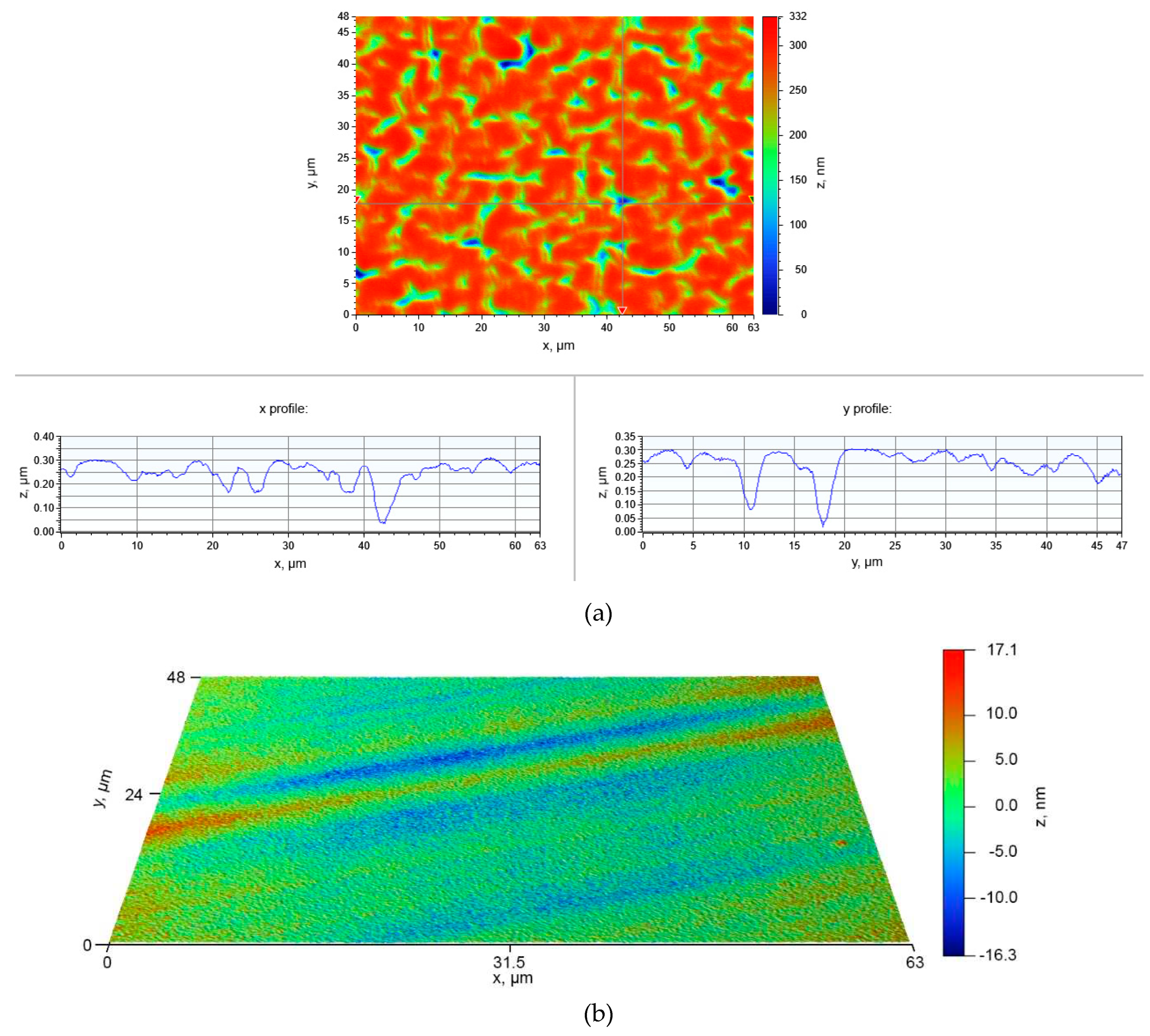
Figure 5.
Cross section of the surface layer deposited on p-Si substrate at ∆E = -5.7 V (t = 1200 s) in the electrolyte composed of TMHA, TFSA and 0.5 M SiCl4.
Figure 5.
Cross section of the surface layer deposited on p-Si substrate at ∆E = -5.7 V (t = 1200 s) in the electrolyte composed of TMHA, TFSA and 0.5 M SiCl4.

Figure 6.
SEM-EDS analysis of Si–C structure deposited at ∆E = -5.7 V, t = 1200 s: carbon (red), silicon (blue) and oxygen (green).
Figure 6.
SEM-EDS analysis of Si–C structure deposited at ∆E = -5.7 V, t = 1200 s: carbon (red), silicon (blue) and oxygen (green).

Figure 7.
Silicon layer (dark and green) deposited photoelectrochemically on GaAs substrate from IL at polarization at ∆E = -2.7 V.
Figure 7.
Silicon layer (dark and green) deposited photoelectrochemically on GaAs substrate from IL at polarization at ∆E = -2.7 V.

Figure 8.
Si–C layer deposited on AA 2024 substrate at ∆E = -0.37 V. EDS identified in the layer 17 at. % of carbon.
Figure 8.
Si–C layer deposited on AA 2024 substrate at ∆E = -0.37 V. EDS identified in the layer 17 at. % of carbon.

Figure 9.
Photocurrents measured under potentiostatic polarization (∆E = -3.7 V and -4.3 V) in the dark and when illumination was switched on and off. The measurement has been performed using AA 2024 substrate in the electrolyte composed of TMHA, TFSA and 0.5 M SiCl4.
Figure 9.
Photocurrents measured under potentiostatic polarization (∆E = -3.7 V and -4.3 V) in the dark and when illumination was switched on and off. The measurement has been performed using AA 2024 substrate in the electrolyte composed of TMHA, TFSA and 0.5 M SiCl4.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



