
Submitted:
27 May 2023
Posted:
30 May 2023
You are already at the latest version
Abstract
Hepatocellular carcinoma (HCC) is the most common form of liver cancer worldwide. Accumulating clinical and experimental evidence suggests the role of cyclo-oxygenase (COX) enzymes in the pathogenesis of cancers including HCC. Deuterium-enriched water (DEW) and deuterium-depleted water (DDW) play a role both in the treatment and prevention of cancers. Combination therapy using COX-inhibitors and DDW/DEW could be a rational strategy to enhance the cytotoxicity of either agent in HCC. The cytotoxicity of celecoxib or indomethacin, alone and in combination with DDW or DEW was determined in the Hep-G2 HCC cell line by MTT assay. The COX-2, MAPK pathway proteins, the anti-apoptotic Bcl2 and pro-apoptotic Bax proteins, and caspase-3 activity were determined by SDS-PAGE and western blot. Co-treatment of selective and non-selective COX-2 inhibitors with DEW led to a remarkable increase in cytotoxicity and apoptosis of Hep-G2 cells. These events were associated with the activation of p38 and JNK MAPKs and a decrease in pro-survival proteins Bcl-2, COX-2, and ERK1/2. Furthermore, the combination therapy activated caspase-3, and the apoptosis mediator, and disabled poly ADP-ribose polymerase (PARP), the key DNA repair enzyme, by cleaving it. The combination of DEW with NSAIDs might be effective against HCC cells by influencing principal cell signaling pathways, and this has a potential to become a candidate for chemotherapy.
Keywords:
Hep-G2 hepatocarcinoma cell
; Deuterium-depleted water
; Deuterium-enriched water
; Cyclooxygenase inhibitors
; MAPK pathway
; Apoptosis
1. Introduction
Liver disorders are a significant health concern worldwide, and their prevalence is increasing. These disruptions include steatosis (as a result of unhealthy lifestyles such as obesity, heavy alcohol consumption, and little physical activity) and viral infections (hepatitis B and C) that can rapidly progress to chronic hepatitis, then fibrosis, cirrhosis, and ultimately, hepatocellular carcinoma (HCC). HCC is a predominant primary liver cancer, accounting for 90% of cases [1,2,3,4]. Globally, HCC is the sixth most common cancer and the third leading cause of cancer-related deaths. Moreover, the World Health Organization (WHO) predicts that in 2030, more than 1 million people will die from liver cancer [5,6].
Using current HCC treatment chemotherapeutic agents like sorafenib, over an extended period can lead to complications such as toxicity and/or ineffectiveness and could not significantly decrease the progression of this cancer [7]. Thus, researchers have a growing focus on developing low-toxic and potent anti-cancer drugs.
The stage of fibrogenesis predominantly occurs after the inflammation caused by liver disorders. Therefore, if the inflammation is inhibited, it can prevent the progress of this stage toward HCC. [8,9]. Cyclo-oxygenase is an essential enzyme in prostanoids synthesis during the inflammation pathway and plays a crucial role in cancer progression [10,11]. COX-2 is widely expressed in liver cancer which causes tumor progression and increases the resistance of cancer cells to chemotherapy and radiation therapy [12]. Nonsteroidal anti-inflammatory drugs (NSAIDs) such as celecoxib and indomethacin are COX inhibitors that are widely used medications worldwide and are relatively safe. Primarily, NSAIDs function as antipyretic, analgesic, and anti-inflammatory medications. Although, several types of research demonstrate that NSAIDs possess protective and therapeutic properties against various cancers, like colon-rectum, breast, pancreas, prostate, head and neck, lung, ovary, and liver cancers [13,14,15,16,17,18,19,20].
NSAIDs induce apoptosis by triggering p38, c-Jun N-terminal kinase (JNK)(pro-apoptotic agents) while suppressing the extracellular-signal-regulated kinase (ERK1/2)(anti-apoptotic agent) MAPK signaling pathway [21]. NSAIDs can also assist in increasing the expression of tumor suppressor genes such as Bax, which is related to B-cell lymphoma protein 2 (Bcl-2) family, and reducing the expression of antiapoptotic genes, such as Bcl-2. Proteins associated with Bax are the primary factors that block the action of Bcl-2, leading to apoptosis through the damage of the mitochondrial membrane. This damage allows for the release of other substances involved in apoptosis, such as cytochrome C, which activates a cascade of caspase enzymes, ultimately leading to cell death [22]. Moreover, NSAIDs can cleave and deactivate poly-ADP ribose polymerase (PARP), a crucial DNA repair enzyme [23,24].
While NSAIDs have demonstrated chemotherapeutic efficacy, their significant anti-cancer effects require high doses, which dampens enthusiasm for their application due to two primary concerns: drug resistance and undesired toxicities affecting the gastrointestinal, renal, liver, and cardiovascular systems [25,26,27].
To tackle this problem, one approach is to combine NSAIDs with another antineoplastic agent. The combination of anti-cancer therapies is attractive for multiple reasons. First, combination therapy boosts treatment outcomes and improve therapeutic effects, particularly when synergistic anti-cancer activity is obtained [28]. Second, the combined methodology addresses clonal heterogeneity associated with enhanced response rates [29]. Third, the use of combined drug regimens decreases the toxicity of the regimen by enabling the administration of individual drugs at decreased dosages while preserving therapeutic effectiveness [28]. An additional benefit of combination therapies is the reduction of drug resistance emergence. In this regard, combination therapy facilitates the simultaneous targeting of multiple molecular pathways vital for cancer cell survival and eliminates cellular mechanisms connected to adaptive resistance [30]. Hereupon, several studies have reported improvements in HCC chemotherapeutic outcomes with NSAIDs when co-administered with various chemo preventive agents [31,32,33].
Deuterium-depleted water (DDW) has emerged as a potential natural option for cancer treatment [34,35,36,37,38,39] . Natural water comprises a blend of molecules containing isotopes 16O, 17O, 18O, 1H, and 2H (D: Deuterium). Deuterium is a stable (non-radioactive) hydrogen isotope with a nucleus composed of one neutron and one proton, resulting in an atomic mass approximately double that of a regular hydrogen atom [40]. It was established that the two stable hydrogen isotopes, deuterium (D) and protium (H), differ not only in their physical parameters but also in their biological and chemical properties [41,42,43,44]. More evidence suggests that D in aqueous solutions plays a crucial role in stimulating or inhibiting metabolic processes in living organisms [36]. DDW contains a lower concentration of D compared to natural sea-level occurrences (lower than 150 ppm D) [45]. It exhibits several surprising biological properties, including antidotal, antitumor, and metabolic effects [46,47,48]. Indeed, tumor cells exhibit high sensitivity to DDW, resulting in tumor shrinkage and, in some instances, necrosis. Simultaneously, healthy cells can adapt to the reduced D content in water [49,50,51,52]. The underlying mechanisms of this impact are attributed to DDW's structure, physicochemical properties, and alterations in ligand-receptor interactions within biological entities of varying hierarchical levels[53,54]. Indeed, in normal cells, the D/H ratio is determined through the balance between the activated H+ transport system and the mitochondria producing-DDW. It has been assumed that in tumor cells, this balance is disturbed, and as a result, D accumulates inside the cell which causes aneuploidy, changes in the size and function of nuclear DNA and the formation of undifferentiated blast cells. This disorder is restored back by the consumption of DDW that strongly affects the phenotype and cell proliferation [55,56]. To our knowledge, no research has been conducted on the synergistic effect of NSAIDs combined with deuterium-depleted water (DDW) and the evaluation of their cellular pathways for HCC treatment. Additionally, considering some studies highlighting the anti-cancer impact of deuterium-enriched water (DEW) [57,58,59,60], we were curious to determine if increased D concentrations in water would enhance the anti-cancer effects of NSAIDs. Hence, this study aimed to examine the cytotoxic impacts of celecoxib and indomethacin independently and in combination with DDW or DEW on the HCC cell line. Additionally, we studied alterations in apoptosis and MAPK pathways to pinpoint probable molecular pathways.
2. Materials and Methods
2.1. Materials and reagents
DDWs (31 and 127 ppm D) and DEWs (50000 and 300,000 ppm D) were prepared from Atomic Energy Organization of Iran. Dulbecco’s modified eagle medium (DMEM) (high glucose), fetal bovine serum (FBS), and penicillin/streptomycin were purchased from PAA (Australia). Trypsin–ethylenediamine tetra acetic acid (trypsin–EDTA) was prepared from Biosera Englind. C-Jun N-terminal kinase (JNK), phospho-SAPK/JNK, anti-extracellular receptor kinase 1/2 (ERK1/2), phospho-ERK1/2, p38, phospho-p38, Bax, Bcl-2, Caspase-3, COX-2, and β-actin antibodies provided from Cell Signaling Technology (USA). Poly ADP-ribose polymerase (PARP) and secondary antibodies were purchased from Roche (Germany). Coomassie blue R-250, Coomassie blue G-250, Bromophenol blue, 3- (4,5- dimethylthiazol-2-yl)- 2, 5-diphenyl tetrazolium bromide (MTT), and caspase-3 colorimetric assay kit were purchased from Sigma Chemical Company (UK). celecoxib (Cel) and indomethacin (Indo) and were kindly achieved by a collaborative lab (as 98.8% purity) and dissolved in minimal amounts of dimethyl sulfoxide (DMSO), so that the final DMSO in tests did not exceed 1%. Culture flask (25, 75 cm2) was achieved from SPL company (Korea). Western blot detection kit and poly vinylidene di fluoride (PVDF) membrane was from Roche Applied Science (Germany). Centrifuge tube (15, 50 ml), microcentrifuge tube (1.5 ml), multi well plates (6-well, 24-well, and 96-well) (microtitration) plates obtained from Nest company (China). Dithiothreitol (DTT) and all other chemicals were purchased from Merck (Germany).
2.2. Cell culture
Hep G2 (HCC cell line) was achieved from Iranian Biological Resource Center (IBRC) Tehran, Iran and grown in monolayer cultures in a complete medium (89% DMEM + 10% heat inactivated fetal bovine serum + 1% penicillin and streptomycin) at 37 ˚C in a humidified atmosphere with 5% CO2. The detachment of monolayer Hep G2 cells from the flask surface was done using 2.5% (trypsin–EDTA) solution. All the tests were performed on the cells within the logarithmic stage to lower cell reactions to stimulators and or inhibitors [61].
2.3. Preparation of media containing different concentrations of deuterium
The components of the culture medium were dissolved in DDWs (31 and 127 ppm D) and DEWs (50000 and 300,000 ppm D) to make cell culture media containing different concentration of D and then they were sterilized using 0.2 μm filters.
2.4. MTT assay for Cellular Proliferation
A cell suspension containing 8×103 Hep G2 cells in 50 µl of culture medium was prepared and then seeded into each well of a flat-bottomed 96-well plate and incubated in a humidified atmosphere of 5% CO2 for 24 hrs to reach 70-90% confluence. Afterward, Cel (2- 400 µm), Indo (2- 800 µm), and their combinations with DDW and DEW at concentrations indicated in Table 1 were added to each well. In the control group, cells were treated with an equal volume of medium (DMEM). Furthermore, the wells containing DMEM without cell were considered as blank. after 24, 48, and 72 hrs incubation, the medium was removed from each well, and after twice washing with PBS, 25 μL of MTT solution (5 mg/ml) was transferred into each well and the plates were incubated for 4 hrs at 37 ∘C in a humidified 5% CO2 atmosphere. During this period of time, the tetrazolium ring was created by selective cleavage of mitochondrial dehydrogenases in viable cells, and the blue/purple formazan crystals were produced. The formazan crystals were dissolved by adding 100 μl of DMSO and the optical density of the solution was measured at a wavelength of 570 nm by an Elisa plate reader. All experiments were performed in triplicates [62].
2.5. Protein Isolation and Western Blotting
After treatment of the Hep G2 cells for 48 hrs with the stated regimes in Table 2, the cells were harvested and then washed with ice-cold PBS. Afterward, cells were lysed by 100 µl lysis buffer (50 mM HEPES (pH: 7.4), 5 mM CHAPS, 5 mM DTT) at 4 ∘C for 15 min. Cell extracts were centrifuged at 14,000 g for 5 min, and the supernatants were poured into the fresh tubes. The protein concentration of supernatants was measured using the Bradford assay. Then, 30 µg of each protein was mixed with an equal volume of 2X SDS-sample buffer and was electrophoretically run through 10% SDS-polyacrylamide gel (SDS-PAGE). Proteins were transferred from SDS-polyacrylamide gel to PVDF membranes (Roche). Afterward, the membranes were stained with 0.1% Ponceau S to confirm equal protein loading and blocked with 0.5% blocking reagent in TBS (50 mM Tris, 150 mM NaCl) for 1 hr at room temperature. The membranes were then probed overnight at 4 ∘C with polyclonal anti-COX-2 (1: 1500), anti-Bax (1: 2500), anti-Bcl-2 (1: 1500), anti-PARP (1: 2000), anti-caspase-3 (1: 1500), anti-ERK1/2 (1: 2500), anti-phospho-ERK 1/2 (1: 2500), anti-SNAPK/JNK (1: 1000), anti-phospho-SNAPK/JNK (1: 1500), anti-P38 (1: 1000), anti-phospho-p38 (1: 1000), and anti-β-actin (1: 1500) antibodies. In the next stage, the membranes were washed four times with TBS-T (TBS containing 0.1% Tween-20) ,15 min each, and one wash with TBS and the blots were incubated with a goat anti-mouse/rabbit-antibody-HRP conjugate (Roche) for 1 hr at room temperature. the blots were washed again and immunoreactive bands became visualized by adding luminal substrate to the blots and their exposure to the (Fuji) x-ray film [63].
2.6. Statistical analysis
Statistical analysis was executed using SPSS software (version 11.0). Data were reported as Mean±SD. One-way analysis of variance (ANOVA) was used to recognize significant differences between treatment groups. The differences were considered significant when P<0.05. The IC50 was calculated using the Probit command in the SPSS.
3. Results:
3.1. Celecoxib and indomethacin inhibited Hep G2 cell proliferation in a dose- and time-dependent manner
Cel and Indo dose-response curves for cell proliferation at 24, 48, and 72 hours are shown in Figure 1 (and Figure S1 in Supplemenrary). Both medicines induced dose- and time-dependent cytotoxicity. The calculated IC50s (The half maximal growth inhibitory concentration) for Cel after 24, 48 and 72 hrs treatment were 38.71, 29.56 and 8.94 µM, respectively. These values for Indo were 233.17, 148.48 and 138.62 µM.
3.2. The combination of celecoxib and indomethacin with DEW but not DDW produces a more potent inhibitory effect on cell growth than either product alone
Since the IC50 for Cel and Indo as single agents was almost high, we decided to test combined treatment protocols including simultaneous treatment with DDW or DEW for these two drugs. Thus, cells were treated with Cel (10, 20, 50, and 70 µM) and Indo (50, 100, 175, and 250 µM) combined with DDW, or DEW (Table 1) for 24, 48, and 72 hrs, and cytotoxicity was measured. Figure 2 (and Figure S2 in Supplementary) and Figure 3 (and Figure S3 in Supplementary) show that the cytotoxicity of Cel and Indo, in combination with DDWs (31 and 127 ppm D), had no significant increment. However, combinations of these two NSAIDs with DEWs (50000 and 300,000 ppm D) remarkably decreased the viability of Hep G2 cells compared to Cel and Indo alone in a dose- and time- dependent manner.
When cells were treated with the combination of DEWs (50000 and 300,000 ppm D) plus [Cel] < IC50 (24hrs) (10, 20 µM), the viability was reduced by approximately 20 - 40% and 35 - 60% compared to lonely Cel, respectively (Figure 2). The combination of above mentioned DEWs significantly augmented the antiproliferative effect of [Indo] < IC50 (24hrs) (50, 100 µM) to almost 15 - 30% and 30 - 45%, respectively (Figure 3) (***P < 0.001, **P < 0.01, and *P < 0.05).
These results illustrated that the combination of DEWs with all concentrations of Cel and Indo significantly decreases the cell proliferation compared to NSAIDs alone (***P < 0.001, **P < 0.01, and *P < 0.05) and DEW (300000 ppm D) induced maximum synergistic toxicity effect with Cel time- and dose- dependently. In fact, at 72 hrs the combination of DEW (300000 ppm D) plus Cel (70 µM) had the cytotoxic effect even more than Cel (400 µM) (Figure S2c in Supplementary).
3.3. Western blotting analysis results
To investigate the signaling pathways induced by our agents, we evaluated the expression of proteins involved in apoptosis (Bax, phospho-p38, phospho-JNK/SAPK, and caspase-3) and survival (COX-2, Bcl- 2, phospho-ERK1/2, and PARP). For this purpose, Hep G2 cells were treated with Cel (22 µM), Indo (110 µM), and media containing various concentrations of deuterium (31, 127, 50000, and 300,000 ppm D), alone or combined, for 48 hrs and the expression of above mentioned proteins was demonstrated by western blotting method.
3.3.1. COX-2 protein expression in the cells treated with Cel, Indo, DDW, DEW and their combinations
As mentioned previously, the high amount of COX-2 protein is detected in HCC and helps cancer cells to persist against therapeutic agents [12]. Since COX-2 inhibitors have illustrated potential therapeutic effects in HCC [64,65,66], we first carried out exploratory investigations to ascertain the function of Cel and Indo and their combinations with DDWs and DEWs in COX-2 expression. Here, Cel and Indo could decrease the COX-2 protein level compared to the control and the ability of Cel was more than Indo to COX-2 inhibition. But neither DDWs (31 and 127 ppm D) nor DEWs (50000 and 300,000 ppm D) could change the COX-2 expression significantly (Figure 4). The (Cel + 31 ppm D) combination had a reasonable inhibition effect compared to the control but not better than Cel alone. However, DDW (127 ppm D) could help Cel further to improve its effectiveness. DEW (50000 ppm D) combined with Cel enhanced the COX-2 inhibitory effect similar to DDW (127 ppm D). The combination therapy of Hep G2 cells with Cel + 300,000 ppm D showed an interesting result. Indeed this combination displayed a robust synergistic effect due to complete COX-2 expression inhibition (Figure 4).
The results of Indo in combination with DDW and DEW were similar to Cel. It seems as the concentration of D (in in the water content of the culture medium) increases, the synergistic ability to reduce COX-2 expression becomes stronger.
3.3.2. Expression of Bax and Bcl-2 proteins in the cells treated with Cel, Indo, DDWs, and DEWs and their combinations
Bcl-2 family proteins have an essential role in the intrinsic apoptotic pathway and the ratio of Bax/Bcl-2 proteins determines the tendency of the cell to survive or die [67,68]. Therefore, we examined if our compounds could change the expression of Bax (pro-apoptotic) and Bcl-2 (anti-apoptotic) proteins.
As depicted in Figure 4, Cel and Indo could increase the level of the Bax significantly. Moreover, DEWs but not DDWs enhanced the anti-apoptotic Bax expression.
In combined regimes, DDW (31 ppm D) could not augment any of Cel and Indo Bax protein expression. However, DDW (127 ppm D) positively influenced the Bax protein expression ability of Indo. The combination of DEWs with Cel and Indo exhibited a strong synergistic effect on Bax expression, and this enhancement was [D] dependent. Importantly, Cel alone and combined forms had higher Bax express stimulating effects than Indo formulations.
The expression of Bcl-2 as an anti-apoptotic protein was decreased by Cel and Indo but DDWs and DEWs, except DEW (300000 ppm D), did not have a remarkable impact.
Treatments consisting of Cel + DEWs and or Indo + DEWs were able to inhibit Bcl-2 expression considerably. These results proved the enhancement of apoptotic ability by DEW that was already illustrated by impacting Bax expression.
3.3.3. Caspase-3 activation and PARP cleavage assay in the cells treated with Cel, Indo, DDWs, and DEWs and their combinations
The intrinsic or extrinsic signaling apoptotic pathways activate the ubiquitous caspase cascade. This cascade end with caspase-3. Activation of caspase-3 cleavages and inactivates crucial cellular proteins, including the DNA repair enzyme (PARP) [23]. Therefore, we assessed the involvement of caspases-3 and PARP to induce apoptosis in treated Hep G2 cells with our agents.
As shown in Figure 4, in single NSAID therapy, Cel could activate caspase-3 slightly. Also, a weak PARP cleaving occurred by Cel and Indo. DDWs did not show apoptotic effects by caspase-3 but they could cleave PARP very slightly. In contrast, DEWs activated caspase-3. Furthermore, DEWs illustrated PARP cleavage ability [D]-dependently.
The combination therapy showed that Cel + DDWs regime did not have significant caspase-3 activity enhancement compared to Cel alone. In contrast, DEWs plus Cel resulted in elevated caspase-3 activity. As expected, the results of PARP cleavage confirmed the apoptotic activity improvement of Cel by DEWs previously shown by caspase activity. Furthermore, DDW (127 ppm D) could enhance the PARP cleavage activity by Cel.
There was no increment in the activation of caspase-3 in Indo + DDWs therapy compared to Indo alone. However, DEWs, in combination with Indo, caused a more considerable caspase-3 activity effect. PARP cleavage activity of the Indo combinations demonstrated DEW (300000 ppm D) could enhance the Indo apoptotic ability.
3.3.4. Expression of MAPKinase proteins in the cells treated with Cel, Indo, DDWs, and DEWs and their combinations
The MAPKinase pathway in mammalian cells includes p38, JNK, and ERK1/2 proteins [69,70,71]. The ERK signaling is predominantly active in cancer and initiates cell cycle activity and cancer cell growth resulting in tumor progression. In contrast, P38 and JNK activity induces apoptosis [72,73]. In this study, since the changes in the expression of total ERK, JNK, and p38 proteins were not distinctive, we also investigated the phosphorylation level of these proteins. As depicted in Figure 5, Cel and at a lower grade Indo and DDWs just enhanced the p-p38. However, DEW increased the phosphorylated JNK and p38 (Figure 5).
DDW (31 ppm D) failed to enhance the apoptotic signal of Cel and Indo in MAPKinase pathways. However, Cel + DDW 127 ppm D and Indo + DDW 127 ppm D could increase p-JNK compared to Cel and Indo alone. Both Cel + DEWs and Indo + DEWs combinations had more robust apoptotic activity than Cel and Indo alone by increasing p-JNK and p-p38 and decreasing p-ERK. This issue demonstrates that [D] is critical in progressing apoptotic pathways (Figure 5).
4. Discussion
Hepatocellular carcinoma (HCC) is among the most prevalent forms of cancer and a leading cause of death in the world [6]. The most conventional methods of treating liver cancer, such as, chemotherapy, surgery, ablation and radiotherapy are ineffective to some extent [74]. Therefore, a multifaceted approach is necessary for the effective treatment of HCC. A novel HCC treatment strategy involves searching for alternative chemotherapeutic agents with minimal adverse effects or incorporating natural substances as adjuncts [75,76]. Chronic inflammation resulting from persistent damage is typically the cause of HCC. Therefore, one approach to address this cancer is by utilizing agents that have anti-inflammatory properties like NSAIDs. Recent substantial evidence from both clinical and experimental studies indicates that NSAIDs may decrease the risk of multiple types of cancer, including HCC [11,20,77,78]. This study evaluated the cytotoxicity effect of two NSAIDs, celecoxib (Cel), a selective COX-2 inhibitor, and Indomethacin (Indo), a non-selective COX-2 inhibitor, on the Hep G2 liver cancer cell line. Our results showed that Cel and Indo decreased Hep G2 cell viability dose and time-dependently (Figure 1 and Figure S1 in Supplementary). Considering this effect, Indo was a weaker cytotoxic agent (IC50 (24, 48, and 72 h) = 233.17, 148.48 and 138.62 µM) compared to Cel (IC50 (24, 48, and 72 h) = 38.71, 29.56 and 8.94 µM).
The primary mode of action of NSAIDs involves inhibiting the cyclooxygenase (COX) enzyme, which is crucial in advancing tumors. COX generates PGE2, which stimulates angiogenesis, cellular invasion, and the creation of metastasis, as well as cell survival [79,80]. Moreover, PGE2 obstructs apoptosis by promoting the production of anti-apoptotic proteins, such as Bcl-2, while hindering the production of pro-apoptotic proteins, such as Bax [81]. Several studies and a meta-analysis discovered that an elevated level of COX-2 was linked to a worse prognosis in individuals with HCC [82,83,84,85,86]. Here, Cel and Indo could decrease the COX-2 protein expression in Hep G2 cells compared to the control and the ability of Cel was more than Indo to COX -2 inhibition (Figure 4).
To make it clear how Cel and Indo cause programmed cell death in the Hep G2 cell line, we examined the levels of Bax and Bcl-2, which belong to the Bcl-2 family of proteins. The intrinsic apoptosis pathway involves mitochondria playing a role in cell death, which is regulated and mediated by the Bcl-2 family of proteins. Bax is a protein that triggers a series of events leading to cell death through the intrinsic apoptosis pathway. When Bax is activated, it causes the release of cytochrome c from the mitochondria, which then activates caspases, including caspase-3. This results in the cleavage and deactivation of essential proteins such as poly-ADP ribose polymerase (PARP), which is necessary for DNA repair, ultimately leading to cell death [87]. Our results illustrated that the Bax expression in cells treated with Cel is more than in Indo-treatment cells. Consequently, Cel's slight activation of caspase-3 and cleavage and deactivation of PARP has occurred. Furthermore, the expression of anti-apoptotic Bcl-2 was decreased by Cel more than Indo (Figure 4). Hossain et al. reported results consistent with our findings, demonstrating that aspirin, another NSAID medication, promotes apoptosis in Hep G2 cells by elevating the Bax/Bcl-2 ratio and activating the caspase cascade [24]. Also, Yoshinaka et al. found that celecoxib, a COX-2 inhibitor, effectively suppresses the growth of tumors and lung metastasis in a murine mammary cancer model by significantly increasing the activity of caspase-3 [88]. Furthermore, the enhancement of the anticancer properties of both specific and non-specific COX-2 inhibitors in human liver cancer cells is linked to the activation of caspase-3, the simultaneous cleavage of PARP, and a reduction in Bcl-2 protein expression [65]. Moreover, the COX-2 selective inhibitor, meloxicam, has been shown to induce apoptosis in Hep G2 cells by increasing the expression of pro-apoptotic proteins such as Bax [85].
Furthermore, several studies have demonstrated that NSAIDs can impede cell growth in different types of cancer by affecting the MAPkinase pathways [21]. MAPKs, which are a collection of enzymes that add phosphate groups to serine/threonine amino acids, transfer signals from the cellular membrane to the nucleus in reaction to various stimuli. This activity alters gene transcription and leads to physiological responses. When the cell exposed to stress, the JNK and p38 pathways are usually triggered, leading to apoptosis. On the other hand, the ERK1/2 pathway is mainly activated in response to growth factors [69,71,89]. NSAIDs have been demonstrated to regulate the MAPK signaling pathway in various types of cancer, such as gastric [21], renal [90], liver [91], colorectal [73,92,93], and head and neck [94] cancers. As an example, according to research, in mice with liver cancer, celecoxib hindered ERK activity and heightened p38 and JNK signaling activation, which impeded cancer growth and triggered cancer cell apoptosis [91]. Celecoxib also was found to upregulate p38 signaling and inhibit cell growth in head and neck squamous cell carcinoma. The inhibition of celecoxib-induced cell growth was considerably reversed when the p38 signaling pathway was blocked [94]. Furthermore, Indomethacin impeded the growth and division of cells, and caused programmed cell death (apoptosis) in MKN28 human gastric cancer cell lines. This effect was achieved by blocking the ERK2/MAPK signaling pathway [21]. Upon treatment with indomethacin, p38 and JNK activity was increased and apoptosis occurred in 786-O renal carcinoma cells [90]. Here, we investigated the changes in the expression of total MAPKinase proteins and the activated (phosphorylated) form of these proteins. Our results showed that Cel could induce apoptosis by activating p38 compared to the control. Whereas Indo activated p38 but increased p-ERK1/2 as an antiapoptotic agent (Figure 5). Although ERK activation is generally linked to antiapoptotic effects, a few studies have reported that ERK activation is also essential for cytotoxic-induced apoptosis, which depends on the type of cell and therapeutic agent used. This could account for the atypical impact of indomethacin on p-ERK expression [95,96].
As previously disclosed, the IC50 (24, 48, and 72 h) of Cel and Indo were (38.71, 29.56 and 8.94 µM) and (233.17, 148.48 and 138.62 µM), respectively. It is essential to mention that this large quantity is applied directly to the cell culture medium. To achieve a lethal dose in Hep G2 cells for treating HCC, patients must consume much higher doses than the IC50 doses. Administering a high dosage of Cel and Indo may lead to drug resistance and exacerbate their adverse effects, as stated in previous studies [25,26,27].
A possible approach is to mix NSAIDs with a harmless natural anticancer substance to decrease the amount of NSAIDs required. DDW refers to a variety of water with a lower D/(D+H) ratio than typical water. Many laboratory and clinical investigations have shown DDW ability to inhibit tumor growth [35,49,97,98]. Studies have indicated that the proliferation of cells and their ability to form colonies and invade are notably diminished by DDW. Furthermore, it impacts the process of cell division by reducing the number of cells in the S phase, substantially raising the number of cells in the G1 phase, and stimulating the creation of antioxidant enzymes [99]. Wang et al. previously showed these impacts on nasopharyngeal cancer cells in a laboratory setting using D levels ranging from 50 to 100 ppm [100]. In addition, Zhang et al. cultured A549 cells in DDW for 48 hours. Their research revealed that DDW could upset the equilibrium between reactive oxygen species generation and neutralization in the mitochondria, resulting in oxidative stress within A549 cells and triggering apoptosis [39].
Considering the antineoplastic ability of DDW, we decided to use this agent as an NSAIDs adjuvant to HCC treatment. For this purpose, we treated Hep G2 cells with (Cel + DDW) or (Indo + DDW) combinations. Our MTT assay results revealed DDWs (31 and 127 ppm D) were not able to enhance the cytotoxic effect of mentioned NSAIDs in different [D] significantly, even after 72 hrs. (Figure 2 and Figure S2 in Supplementary, Figure 3 and Figure S3 in Supplementary). Similarly, Soleyman-Jahi and colleagues published a report demonstrating that exposing human cancer cell lines of the prostate, colon, breast, and stomach to varying concentrations of D in DDW did not result in any noteworthy limiting impacts. This was determined through cytotoxicity analysis based on MTT [99]. Furthermore, Kleemann showed that exposing malignant melanoma cell lines A375, SK-Mel-28, and SK-Mel-30 to water with lower-than-normal D levels did not affect their growth [57]. Moreover, these findings were consistent with our previous investigation, which revealed that DDW having 31 and 127 ppm D did not provide significant adjuvant effects to Cel and Indo in the fight against human lung cancer cells (A549) [101].
The western blotting analysis of COX-2, MAP Kinase pathway (p38, JNK, and ERK1/2), and intrinsic apoptosis pathways (Bax, Bcl-2, caspase -3, and PARP) proteins demonstrated the expression of contributed proteins in these pathways in Hep G2 treated with (Cel + DDW 31 ppm D) or (Indo + DDW 31 ppm D) for 48 hrs has not changed efficiently, whereas DDW (127 ppm D) could change the ability of Cel and Indo to express p-JNK and Bcl-2 proteins slightly in agreement with cell death. (Figure 4 and Figure 5). In contrast to our findings, Gÿongyi et al. reported that DDW containing 25 ppm D could prevent the overexpression of the Bcl-2, Kras, and Myc genes induced by DMBA in the lungs of mice. As per their conclusion, this form of water could serve as a non-toxic dietary supplement with anticancer properties that may prolong the survival of individuals with lung cancer [102]. Also, Yavari and Kooshesh investigated to evaluate the combined impact of DDWs (30, 50, 75, 100, 125 ppm D) with 5-FU in treating the MCF-7 breast cancer cell line. The results of their investigation, which involved assessing cytotoxicity, cell cycle arrest, and antioxidant enzyme levels, showed that DDWs containing 30-100 ppm D could enhance the antineoplastic impact of 5-FU [37]. Furthermore, Boros and colleagues illustrated the supplementary inhibitory D-depletion effect when combined with cisplatin in vitro on MIA-PaCa-2 pancreatic cancer cells [36].
According to our results, the combination of Cel and Indo with DDW (31 and 127 ppm D) did not have a significant synergistic impact on inhibiting the growth of Hep G2 cells. Therefore, we deduced that the decreased level of D in water did not have a notable anticancer effect on this particular cell category, at least within 72 hours. Consequently, we opted to experiment with water varieties with a D concentration higher than the standard level. We utilized DEW (50000 and 300,000 ppm D) as a combined agent to achieve this objective.
Stress caused by D disrupts the process of energy metabolism. D impacts enzymes engaged in energy metabolism, including cytochrome c oxidase in the respiratory chain of mitochondria, and ATP synthase, significantly reducing cellular ATP reserves. This impact is particularly crucial for cancerous cells [103,104]. In accordance with this hypothesis, various studies have indicated the anticancer properties of DEW [57,105,106]. Here, the addition of DEWs (50000 and 300,000 ppm D) to [Cel] < IC50 (24 hrs) (10, 20 µM) during 24, 48, and 72 hrs could improve the % Cel. Growth inhibition approximately (20 – 40%) and (35 – 60%) respectively. Also, the DEWs (50000 and 300,000 ppm D) in combination with [Indo] < IC50 (24 hrs) (50, 100 µM) after 24, 48, and 72 hrs could prevent the Hep G2 cell viability almost (15 – 30%) and (30 – 45%) compare with Indo alone. The synergistic growth inhibition of DEWs in combinations was time and dose-dependent, so the (Cel + 300,000 ppm D) and (Indo + 300,000 ppm D) induced the most growth prevention after 72 hrs. Furthermore, the combination of (70 µM Cel + 300,000 ppm D) showed cell growth inhibition even more than 400 µM Cel alone after 72 hrs. In a previous study, we demonstrated that treating Hep G2 and A549 cells with DEWs (50000, 100000, 200000, 300,000 ppm D) alone for an extended period of time (21 days) could inhibit cell growth in a [D]-dependent manner [107]. Also, Leonard and colleagues stated that PtK1 cells, when exposed to a culture medium containing as much as 500,000 ppm D, could initiate and conclude mitosis. Nonetheless, the length of the mitotic phase extended correspondingly with the amount of D2O utilized [108]. Furthermore, Bader et al. showed that when human pancreatic carcinoma cells (AsPC-1, BxPC-3, and PANC-1) were exposed to D2O and gemcitabine simultaneously, the IC50 values of gemcitabine were lowered in all studied pancreatic cancer cell lines and synergistic effects were observed in the sequential administration of D2O and gemcitabine [109].
To understand the anti-cancer molecular pathways of DEWs (50000, 300,000 ppm D) and their combination with NSAIDs, the expression of p38, JNK, and ERK1/2 (and their phosphorylated form), Bax, Bcl-2, COX-2, and caspase-3 activation and disable PARP enzyme in Hep G2 cell were evaluated. Interestingly, DEWs lonely could enhance the expression of pro-apoptotic proteins Phospho-p38, Phospho-JNK, and Bax and DEW (300000 ppm D) prohibited the expression of anti-apoptotic COX-2 and Bcl-2. Likewise, DEW (300000 ppm D) activated caspase-3 and cleaved PARP slightly stronger than DEW (50000 ppm D). According to Kalkur et al. study, it was demonstrated that DEWs (10000 – 50,000 ppm D) exhibited considerable cytotoxic effects on murine astrocytoma cells induced by the Raus sarcoma virus. The mechanism behind the DEW-mediated cytotoxicity involved the induction of apoptosis and cell cessation in the G2/M phase [106]. Moreover, it has been reported that DEW-triggered apoptosis in malignant astrocytoma cells is regulated via the caspase activation pathway, and the apoptosis rate is positively correlated with the concentration of DEW [105]. Additionally, Bahk and colleagues confirmed that DEW (at concentrations of 75% and 100%) displayed antiproliferative, anti-adhesive, and anti-invasive properties on bladder cancer cells (T-24) after exposure for more than 2.5 hours. They indicated that the antiproliferative effect of DEW resulted from the activation of the apoptosis pathway, achieved by reducing the expression of Bcl-2 and increasing the expression of Bax [59]. Likewise, Jandova and her colleagues discovered that in A375 melanoma cells, the stress response to D2O was quickly induced and activated apoptosis. This response involved changes in the levels of specific phospho-proteins, such as decreased expression of p-AKT and increased expression of p-ERK, p-JNK, peIF2α, and p-H2AX. Moreover, this group discovered that D2O exposure resulted in the inhibition of cell proliferation and the induction of apoptosis in various cultured PDAC cells. This was identified by the presence of Z-VAD-FMK (carbobenzoxy-valyl-alanyl-aspartyl-[O-methyl]-fluoromethylketone) and cleaved pro-caspase-3 and PARP-1 [110]. Another investigation demonstrated that increasing the concentration of D2O above the normal levels in the medium of malignant melanoma cells lines such as A375, SK-Mel-28, and SK-Mel-30 reduced proliferation and hindered cell migration dose-dependently. Additionally, the cell cycle analysis revealed a rise in the number of cells in the sub-G1 phase. As reported in the study, indicators for programmed cell death were stimulated, including fragments of DNA bound to histones, the protein Bax, and PARP [57].
In combined treatment, the western blotting results were in line with the MTT cytotoxicity findings. Indeed, either Cel or Indo, when co-administrated with DEWs, led to a remarkable D dose-dependent activation in apoptosis pathways of Hep G2 cells compared to their co-treatment with DDWs. In fact, DEWs could increase the Cel and Indo ability to induce the expression of proapoptotic proteins (Bax, p-JNK, and p-p38) and inhibit the expression of antiapoptotic agents (COX-2, Bcl-2, and p-ERK1/2). DEWs also could help Cel and Indo to activate Caspase-3 and inactivate PARP. Furthermore, DEWs had more synergistic effect with Cel than with Indo in most of protein expression evaluations (Figure 4 and Figure 5). Like our findings, the administration of D2O (at concentrations of 5% to 30%) along with gemcitabine resulted in a notable (statistically significant at p < 0.05) enhancement of apoptosis in AsPC-1 and PANC-1 pancreatic cancer cells. The combined treatment of D2O (at concentrations of 5% - 30%) and gemcitabine resulted in higher expression levels of the p21 tumor suppressor gene as compared to treating with gemcitabine alone [109]. Moreover, Altermatt et al. compared the antineoplastic effect of 5-FU and bleomycin alone or combined with DEW in the growth of xenotransplanted human oropharyngeal squamous-cell carcinoma and colon cancer. They illustrated DEW showed a synergistic effect in combination with 5-FU and bleomycin to delay the growth of all cancer variants and suggested DEW prolonged the tumor cell cycle time and reduced the growth fraction [58]. Additionally, we had already demonstrated that when COX inhibitors (celecoxib and indomethacin) and DEW were used together, they could increase the cytotoxicity in the A549 lung cancer cell line. This was achieved through the activation of intrinsic apoptosis pathway containing p-JNK, p-P38, Bax, and caspace-3 [101].
The most intense apoptosis activity was achieved by combining Cel and DEW at a concentration of 300,000 ppm D. Cel is a strong COX-2 selective inhibitor, leading to more significant inhibition of this pro-survival enzyme than Indo, which in turn inactivates anti-apoptotic pathways, such as Bcl-2, that are associated with COX-2 [111,112]. On the other hand, as previously proved, the anti-tumor effect of DEW varies depending on the D dose, and consequently, the combination of Cel and DEW (300000 ppm D) resulted in the most synergistic effect against HCC.
5. Conclusion
In conclusion, this study shows that COX inhibitor agents celecoxib and indomethacin could have anti-HCC effects as a single treatment, but this ability occurs in high doses. In order to increase the antitumor effect of these compounds, DEW can be used in combination with them. The results of cytotoxicity and investigating apoptosis and MAPKinase signaling pathways show that DEW can synergistically improve the anti-HCC effects of these two NSAID medicines [D]-dependently. The Cel + DEWs combination has stronger inhibitory effects than Indo + DEWs on Hep G2 cells and is introduced as a new candidate for the HCC treatment.
References
- Sia, D.; Villanueva, A.; Friedman, S.L.; Llovet, J.M. Liver Cancer Cell of Origin, Molecular Class, and Effects on Patient Prognosis. Gastroenterol. 2017, 152, 745–761. [Google Scholar] [CrossRef] [PubMed]
- Waller, L.P.; Deshpande, V.; Pyrsopoulos, N. Hepatocellular Carcinoma: A Comprehensive Review. World J. hepatol. 2015, 7, 2648–2663. [Google Scholar] [CrossRef] [PubMed]
- Tateishi, R.; Okanoue, T.; Fujiwara, N.; Okita, K.; Kiyosawa, K.; Omata, M.; Kumada, H.; Hayashi, N.; Koike, K. Clinical Characteristics, Treatment, and Prognosis of Non-B, Non-C Hepatocellular Carcinoma: A Large Retrospective Multicenter Cohort Study. Gastroenterol. 2015, 50, 350–360. [Google Scholar] [CrossRef] [PubMed]
- Tan, D.J.H.; Wong, C.; Ng, C.H.; Poh, C.W.; Jain, S.R.; Huang, D.Q.; Muthiah, M.D. A Meta-Analysis on the Rate of Hepatocellular Carcinoma Recurrence after Liver Transplant and Associations to Etiology, Alpha-Fetoprotein, Income and Ethnicity. J. Clin. Med. 2021, 10, 238. [Google Scholar] [CrossRef] [PubMed]
- Mathers, C. Projections of Mortality and Causes of Deaths, 2016 to 2060. Available online: https://colinmathers.com/2022/05/10/projections-of-global-deaths-from-2016-to-2060/ (Accessed on 22 May 2023).
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin., 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Anwanwan, D.; Singh, S.K.; Singh, S.; Saikam, V.; Singh, R. Challenges in Liver Cancer and Possible Treatment Approaches. Biochim. Biophys. Acta Rev. Cancer 2020, 1873, 188314. [Google Scholar] [CrossRef] [PubMed]
- Refolo, M.G.; Messa, C.; Guerra, V.; Carr, B.I.; D'Alessandro, R. Inflammatory Mechanisms of HCC Development. Cancers, 2020, 12, 641. [CrossRef]
- Yu, L.X.; Ling, Y.; Wang, H.Y. Role of Nonresolving Inflammation in Hepatocellular Carcinoma Development and Progression. NPJ Precis Oncol, 2018, 2, 6. [Google Scholar] [CrossRef]
- Kar, P.; Mishra, S. Management of Hepatitis B During Pregnancy. Expert Opin. Pharmacother. 2016, 17, 301–310. [Google Scholar] [CrossRef]
- Patel, S.; Nanavati, P.; Sharma, J.; Chavda, V.; Savjani, J. Functional Role of Novel Indomethacin Derivatives for the Treatment of Hepatocellular Carcinoma Through Inhibition of Platelet-Derived Growth Factor. Arch. Med. Res. 2021, 52, 483–493. [Google Scholar] [CrossRef]
- Hashemi Goradel, N.; Najafi, M.; Salehi, E.; Farhood, B.; Mortezaee, K. Cyclooxygenase-2 in Cancer: A Review. J. Cell. Physiol. 2019, 234, 5683–5699. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Stark, L.A. Aspirin Prevention of Colorectal Cancer: Focus on NF-κB Signalling and the Nucleolus. Biomedicines, 2017, 5, 43. [Google Scholar] [CrossRef] [PubMed]
- Bakır, E.; Çal, T.; Aydın Dilsiz, S.; Canpınar, H.; Eken, A.; Ündeğer Bucurgat, Ü. Assessment of the Cytotoxic, Genotoxic, and Apoptotic Potential of Flurbiprofen in HeLa and HepG2 Cell Lines. J. Biochem. Mol. Toxicol. 2021, 35, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Khuder, S.; Mutgi, A. Breast Cancer and NSAID Use: A meta-Analysis. Br. J. Cancer. 2001, 84, 1188–1192. [Google Scholar] [CrossRef] [PubMed]
- Andrews, P.; Zhao, X.; Allen, J.; Li, F.; Chang, M. A Comparison of the Effectiveness of Selected Non-Steroidal Anti-Inflammatory Drugs and their Derivatives Against Cancer Cells in Vitro. Cancer Chemother. Pharmacol. 2008, 61, 203–214. [Google Scholar] [CrossRef]
- Derry, S.; Wiffen, P.J.; Häuser, W.; Mücke, M.; Tölle, T.R.; Bell, R.F.; Moore, R.A. Oral NonsteroidalAnti-Inflammatory Drugs for Fibromyalgia in Adults. Cochrane Database Syst. Rev. 2017, 3, CD012332. [Google Scholar] [CrossRef]
- Rai, N.; Sarkar, M.; Raha, S. Piroxicam, a Traditional Non-Tteroidal Anti-Inflammatory Drug (NSAID) Causes Apoptosis by ROS Mediated Akt Activation. Pharmacol. Rep. 2015, 67, 1215–1223. [Google Scholar] [CrossRef]
- Hiľovská, L.; Jendželovský, R.; Fedoročko, P. Potency of Non-Steroidal Anti-Inflammatory Drugs in Chemotherapy. Mol. Clin. Oncol. 2015, 3, 3–12. [Google Scholar] [CrossRef]
- Wong, R.S.Y. Role of Nonsteroidal Anti-Inflammatory Drugs (NSAIDs) in Cancer Prevention and Cancer Promotion. Adv. Pharmacol. Sci. 2019, 3418975. [Google Scholar] [CrossRef]
- Husain, S.S.; Szabo, I.L.; Pai, R.; Soreghan, B.; Jones, M.K.; Tarnawski, A.S. MAPK (ERK2) Kinase—A Key Target for NSAIDs-Induced Inhibition of Gastric Cancer Cell Proliferation and Growth. Life Sci. 2001, 69, 3045–3054. [Google Scholar] [CrossRef]
- Porebska, I.; Wyrodek, E.; Kosacka, M.; Adamiak, J.; Jankowska, R.; Harłozińska-Szmyrka, A. Apoptotic Markers p53, Bcl-2 and Bax in Primary Lung Cancer. In Vivo, 2006, 20, 599–604. [Google Scholar]
- Wolf, B.B.; Green, D.R. Suicidal Tendencies: Apoptotic Cell Death by Caspase Family Proteinases. J Biol Chem. 1999, 274, 20049–20052. [Google Scholar] [CrossRef] [PubMed]
- Hossain, M.A.; Kim, D.H.; Jang, J.Y.; Kang, Y.J.; Yoon, J.H.; Moon, J.O.; Chung, H.Y.; Kim, G.Y.; Choi, Y.H.; Copple, B.L.; Kim, N.D. Aspirin Induces Apoptosis in Vitro and Inhibits Tumor Growth of Human Hepatocellular Carcinoma Cells in a Nude Mouse Xenograft Model. Int. J. Oncol. 2012, 40, 1298–1304. [Google Scholar] [CrossRef] [PubMed]
- Kelly, J.D.; Tan, W.S.; Porta, N.; Mostafid, H.; Huddart, R.; Protheroe, A.; Bogle, R.; Blazeby, J.; Palmer, A.; Cresswell, J.; Johnson, M.; Brough, R.; Madaan, S.; Andrews, S.; Cruickshank, C.; Burnett, S.; Maynard, L.; Hall, E.; BOXIT Investigators. BOXIT-A Randomised Phase III Placebo-controlled Trial Evaluating the Addition of Celecoxib to Standard Treatment of Transitional Cell Carcinoma of the Bladder (CRUK/07/004). Eur. Urol. 2019, 75, 593–601. [Google Scholar] [CrossRef]
- Li, Z.Y.; Yin, Y.F.; Guo, Y.; Li, H.; Xu, M.Q.; Liu, M.; Wang, J.R.; Feng, Z.; H. ; Duan, X.C.; Zhang, S.; Zhang, S.Q.; Wang, G.X.; Liao, A.; Wang, S.M.; Zhang, X. Enhancing Anti-Tumor Activity of Sorafenib Mesoporous Silica Nanomatrix in Metastatic Breast Tumor and Hepatocellular Carcinoma via the Co-Administration with Flufenamic Acid. Inte. J. Nanomedicine. 2020, 15, 1809–1821. [Google Scholar] [CrossRef] [PubMed]
- Rayburn, E.R.; Ezell, S.J.; Zhang, R. Anti-Inflammatory Agents for Cancer Therapy. Mol. Cell Pharmacol. 2009, 1, 29–43. [Google Scholar] [CrossRef]
- Bukowska, B.; Gajek, A.; Marczak, A. Two Drugs Are Better Than One. A Short History of Combined Therapy of Ovarian Cancer. Contemp. Oncol. (Pozn). 2015, 19, 350–353. [Google Scholar] [CrossRef]
- Palmer, A.C.; Chidley, C.; Sorger, P.K. A Curative Combination Cancer Therapy Achieves High Fractional Cell Killing Through Low Cross-Resistance and Drug Additivity. Elife. 2019, 8, e50036. [Google Scholar] [CrossRef]
- Narayan, R.S.; et al. A Cancer Drug Atlas Enables Synergistic Targeting of Independent Drug Vulnerabilities. Nat. Commun. 2020, 11, 2935. [Google Scholar] [CrossRef]
- Chu, T.H.; et al. Celecoxib Enhances the Therapeutic Efficacy of Epirubicin for Novikoff Hepatoma in Rats. Cancer Med. 2018, 7, 2567–2580. [Google Scholar] [CrossRef]
- Abdallah, F.M.; Helmy, M.W.; Katary, M.A.; Ghoneim, A.I. Synergistic Antiproliferative Effects of Curcumin and Celecoxib in Hepatocellular Carcinoma HepG2 Cells. Naunyn Schmiedebergs Arch. Pharmacol. 2018, 391, 1399–1410. [Google Scholar] [CrossRef] [PubMed]
- Zhong, J.; et al. Meloxicam Combined with Sorafenib Synergistically Inhibits Tumor Growth of Human Hepatocellular Carcinoma Cells via ER Stress-Related Apoptosis. Oncol. Rep. 2015, 34, 2142–2150. [Google Scholar] [CrossRef]
- Syroeshkin, A.; Levitskaya, O.; Uspenskaya, E.; Pleteneva, T.; Romaykina, D.; Ermakova, D. Deuterium Depleted Water as an Adjuvant in Treatment of Cancer. Sys. Rev. Pharm. 2019, 10, 112–117. [Google Scholar] [CrossRef]
- Cong, F.S.; Zhang, Y.R.; Sheng, H.C.; Ao, Z.H.; Zhang, S.Y.; Wang, J.Y. Deuterium-Depleted Water Inhibits Human Lung Carcinoma Cell Growth by Apoptosis. Exp. Ther. Med. 2010, 1, 277–283. [Google Scholar] [CrossRef]
- Boros, L.G.; Somlyai, I.; Kovács, B.Z.; Puskás, L.G.; Nagy, L.I.; Dux, L.; Farkas, G.; Somlyai, G. Deuterium Depletion Inhibits Cell Proliferation, RNA and Nuclear Membrane Turnover to Enhance Survival in Pancreatic Cancer. Cancer Control. 2021, 28, 1073274821999655. [Google Scholar] [CrossRef] [PubMed]
- Yavari, K.; Kooshesh, L. Deuterium Depleted Water Inhibits the Proliferation of Human MCF7 Breast Cancer Cell Lines by Inducing Cell Cycle Arrest. Nutr. Cancer. 2019, 71, 1019–1029. [Google Scholar] [CrossRef]
- Kovács, B.Z.; Somlyai, I.; Papp, A.; Somlyai, G. Deuterium-Depleted Water Delayed Hormone Therapy of Prostate Cancer. J. Clin. Rev. Case Rep. 2021, 6, 747–749. [Google Scholar]
- Zhang, X.; Gaetani, M.; Chernobrovkin, A.; Zubarev, R.A. Anticancer Effect of Deuterium Depleted Water - Redox Disbalance Leads to Oxidative Stress. Mol. Cell Proteomics. 2019, 18, 2373–2387. [Google Scholar] [CrossRef]
- Bowen, G.J.; Winter, D.A.; Spero, H.J.; Zierenberg, R.A.; Reeder, M.D.; Cerling, T.E.; Ehleringer, J.R. Stable Hydrogen andOxygen Isotope Ratios of Bottled Waters of the World. Rapid Commun. Mass Spectrom. 2005, 19, 3442–3450. [Google Scholar] [CrossRef]
- Szent-Györgyi, A. The living state and cancer. Proc. Natl. Acad. Sci. U S A. 1977, 74, 2844–2847. [Google Scholar] [CrossRef] [PubMed]
- Gat, J.R.; Gonfiantini, R. Stable Isotope Hydrology: Deuterium and Oxygen-18 in the Water Cycle. Technical Reports Series. International Atomic Energy Agency, Vienna (Austria) 1981, no. 210, 1-356.
- Altman, L.J.; Laungani, P.; Gunnarsson, G.; Wennerstorm, H.; Forsen, S. Proton, Deuterium, and Tritium Nuclear Magnetic Resonance of Intramolecular Hydrogen Bonds. Isotope Effects and the Shape of the Potential Energy Function. J. Am. Chem. Soc. 1978, 100, 8264–8266. [Google Scholar] [CrossRef]
- Westheimer, F.H. The Magnitude of thePrimary Kinetic Isotope Effect for Compounds of Hydrogen and Deuterium. Chem. Rev. 1961, 61, 265–273. [Google Scholar] [CrossRef]
- Goncharuk, V.V.; Kavitskaya, A.A.; Romanyukina, I.Y.; Loboda, O.A. Revealing Water’s Secrets: Deuterium Depleted Water. Chem. Cent. J. 2013, 7, 103. [Google Scholar] [CrossRef]
- Goncharuk; et al. Determination of Biological Activity of Water Having a Different Isotope Ratio of Protium and Deuterium. J. Water Chem. Technol. 2018, 40, 27–34. [Google Scholar] [CrossRef]
- Syroeshkin, A.V.; Antipova, N.V.; Zlatska, A.V.; Zlatskiy, I.A.; Skylska, M.D.; Grebennikova, T.V.; Goncharuk, V.V. The Effect of the Deuterium Depleted Water on the Biological Activity of the Eukaryotic Cells. J. Trace Elem. Med. Biol. 2018, 50, 629–633. [Google Scholar] [CrossRef] [PubMed]
- Zlatska, A.; Gordiienko, I.; Vasyliev, R.; Zubov, D.; Gubar, O.; Rodnichenko, A.; Syroeshkin, A.; Zlatskiy, I. In Vitro Study of Deuterium Effect on Biological Properties of Human Cultured Adipose-Derived Stem Cells. ScientificWorldJournal. 2018, 5454367. [Google Scholar] [CrossRef]
- Wang, H.; Zhu, B.; He, Z.; Fu, H.; Dai, Z.; Huang, G.; Li, B.; Qin, D.; Zhang, X.; Tian, L.; Fang, W.; Yang, H. Deuterium-Depleted Water (DDW) Inhibits the Proliferation and Migration of Nasopharyngeal Carcinoma Cells in Vitro. Biomed. Pharmacother. 2013, 67, 489–496. [Google Scholar] [CrossRef] [PubMed]
- Mirică, R.E. , Deuterium-Depleted Water in Cancer Therapy. Environ. Eng. Manag. J. 2010, 9, 1543–1545. [Google Scholar] [CrossRef]
- Krempels, K.; Somlyai, I.; Somlyai, G. Molecular and Clinical Effects of Deuterium Depleted Water in Treatment and Prevention of Cancer. PositiveHealthOnline [serial on the Internet]. 2013, issue 203.
- Krempels, K.; Abonyi, O.; Balog, K.; Somlyai, I. Deuterium Depletion in Cancer Treatment and Prevention. PositiveHealthOnline [serial on the Internet]. 2013, issue 209.
- Goncharuk, V.V.; Syroeshkin, A.V.; Zlatskiy, I.A.; Uspenskaya, E.V.; Orekhova, A.V.; Levitskaya, O.V.; Dobrovolskiy, V.I.; Pleteneva, T.V. Quasi-Chemical Description of the Kinetics of Cell Death Spirostomum ambiguum Biosensor for Biological Activity of Aqueous Solutions. J. Water Chem. Technol. 2017, 39, 97–102. [Google Scholar] [CrossRef]
- Syroeshkin, A.; Pleteneva, T.; Uspenskaya, E.; Zlatskiy, I.; Antipova, N.; Grebennikova, T.; Levitskaya, O. D/H Control of Chemical Kinetics in Water Solutions under low Deuterium Concentrations. Chem. Eng. J. 2019, 377, 119827. [Google Scholar] [CrossRef]
- Boros, L.G.; D'Agostino, D.P.; Katz, H.E.; Roth, J.P.; Meuillet, E.J.; Somlyai, G. Submolecular Regulation of Cell Transformation by Deuterium Depleting Water Exchange Reactions in the Tricarboxylic Acid Substrate Cycle. Med. hypotheses. 2016, 87, 69–74. [Google Scholar] [CrossRef]
- Boros, L.G.; et al. Fumarate Hydratase and Deuterium Depletion Control Oncogenesis via NADPH-Dependent Reductive Synthesis: Mitochondrial Matrix Water, DNA Deuteration and Epigenetic Events. Cancer Res. 2014, 74(19_Supplement), 1426. [CrossRef]
- Kleemann, J.; Reichenbach, G.; Zöller, N.; Jäger, M.; Kaufmann, R.; Meissner, M.; Kippenberger, S. Heavy Water Affects Vital Parameters of Human Melanoma Cells in Vitro. Cancer. Manag. Res. 2020, 12, 1199–1209. [Google Scholar] [CrossRef]
- Altermatt, H.J.; Gebbers, J.O.; Laissue, J.A. Heavy Water Enhances the Antineoplastic Effect of 5-Fluoro-uracil and Bleomycin in Nude Bice Bearing Human Carcinoma. Int. J. Cancer. 1990, 45, 475–480. [Google Scholar] [CrossRef]
- Bahk, J.Y.; Lee, J.H.; Chung, H.S.; Lee, H.Y.; Chung, B.C.; Park. M.S.; Min, S.K.; Kim, M.O. Anticancer Effect of Deuterium Oxide on a Bladder Cancer Cell Related to Bcl-2 and Bax. J. Ind. Eng. Chem. 2007, 13, 501–507. [Google Scholar]
- Jandova, J.; Hua, A.B.; Fimbres, J.; Wondrak, G.T. Deuterium Oxide (D2O) Induces Early Stress Response Gene Expression and Impairs Growth and Metastasis of Experimental Malignant Melanoma. Cancers (Basel). 2021, 13, 605. [Google Scholar] [CrossRef] [PubMed]
- Capes-Davis, A.; Freshney, R.I. Culture of Animal Cells: A Manual of Basic Technique and Specialized Applications. 7th ed.; Wiley-Blackwell, 2016.
- Cree, I.A. Principles of Cancer Cell Culture. Methods Mol. Biol. 2011, 731, 13–26. [Google Scholar] [CrossRef] [PubMed]
- Kurien, B.T.; Scofield, R.H. Western Blotting: Methods and Protocols. Springer: New York, USA, 2015.
- Lampiasi, N.; Azzolina, A.; Umezawa, K.; Montalto, G.; McCubrey, J.A.; Cervello, M. The Novel NF-κB Inhibitor DHMEQ Synergizes with Celecoxib to Exert Antitumor Effects on Human Liver Cancer Cells by a ROS-Dependent Mechanism. Cancer Lett. 2012, 322, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Cusimano, A.; Foderà, D.; D'Alessandro, N.; Lampiasi, N.; Azzolina, A.; Montalto, G.; Cervello, M. Potentiation of the Antitumor Effects of Both Selective Cyclooxygenase-1 and Cyclooxygenase-2 Inhibitors in Human Hepatic Cancer Cells by Inhibition of the MEK/ERK Pathway. Cancer biol. ther. 2007, 6, 1457–1464. [Google Scholar] [CrossRef]
- Lampiasi, N.; et al. The Selective Cyclooxygenase-1 Inhibitor SC-560 Suppresses Cell Proliferation and Induces Apoptosis in Human Hepatocellular Carcinoma Cells. Int. J. Mol. Med. 2006, 17, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Oltval, Z.N.; Milliman, C.L.; Korsmeyer, S.J. Bcl-2 Heterodimerizes in Vivo with a Conserved Homolog, Bax, That Accelerates Programed Cell Death. cell. 1993, 74, 609–619. [Google Scholar] [CrossRef]
- Zhang, L.; Yu, J.; Park, B.H.; Kinzler, K.W.; Vogelstein, B. Role of BAX in the Apoptotic Response to Anticancer Agents. Science. 2000, 290, 989–992. [Google Scholar] [CrossRef] [PubMed]
- Raman, M.; Chen, W.; Cobb, M. Differential Regulation and Properties of MAPKs. Oncogene. 2007, 26, 3100–3112. [Google Scholar] [CrossRef]
- Cargnello, M.; Roux, P.P. Activation and Function of the MAPKs and Their Substrates, the MAPK-Activated Protein Kinases. Microbiol. Mol. Biol. Rev. 2011, 75, 50–83. [Google Scholar] [CrossRef] [PubMed]
- Dhillon, A.S.; Hagan, S.; Rath, O.; Kolch, W. MAP Kinase Signalling Pathways in Cancer. Oncogene. 2007, 26, 3279–3290. [Google Scholar] [CrossRef]
- Torii, S.; Yamamoto, T.; Tsuchiya, Y.; Nishida, E. ERK MAP Kinase in G1 Cell Cycle Progression and Cancer. Cancer Sci. 2006, 97, 697–702. [Google Scholar] [CrossRef] [PubMed]
- Setia, S.; Nehru, B.; Sanyal, S.N. Upregulation of MAPK/Erk and PI3K/Akt Pathways in Ulcerative Colitis-Associated Colon Cancer. Biomed. Pharmacother. 2014, 68, 1023–1029. [Google Scholar] [CrossRef] [PubMed]
- Ho, P.H.; Teng, W.; Lin, C.C.; Jeng, W.J.; Chen, W.T.; Lin, C.Y.; Lin, S.M.; Sheen, I.S. Prolonged Post-Ablation Fever May Predict One-Year Tumor Recurrence in Hepatocellular Carcinoma After Radiofrequency Ablation. Int. J. Hyperthermia. 2020, 37, 1008–1015. [Google Scholar] [CrossRef]
- Roy, A.; Bharadvaja, N. Venom-Derived Bioactive Compounds as Potential Anticancer Agents: A Review. Int. J. Pept. Res. Ther. 2021, 27, 129–147. [Google Scholar] [CrossRef]
- Mansour, G.H.; El-Magd, M.A.; Mahfouz, D.H.; Abdelhamid, I.A.; Mohamed, M.F.; Ibrahim, N.S.; Hady, A. Abdel Wahab, A.; Elzayat, E.M. Bee Venom and its Active Component Melittin Synergistically Potentiate the Anticancer Effect of Sorafenib Against HepG2 Cells. Bioorg. Chem. 2021, 116, 105329. [Google Scholar] [CrossRef]
- Narożna, M.; Krajka-Kuźniak, V.; Bednarczyk-Cwynar, B.; Kleszcz, R.; Kujawski, J.; Baer-Dubowska, W. The Effect of Novel Oleanolic Acid Oximes Conjugated with Indomethacin on the Nrf2-ARE And NF-κB Signaling Pathways in Normal Hepatocytes and Human Hepatocellular Cancer Cells. Pharmaceuticals (Basel). 2020, 14, 32. [Google Scholar] [CrossRef]
- Hossain, M.A.; et al. Aspirin Enhances Doxorubicin-Induced Apoptosis and Reduces Tumor Growth in Human Hepatocellular Carcinoma Cells in Vitro and in Vivo. Int. J. Oncol. 2012, 40, 1636–1642. [Google Scholar] [CrossRef] [PubMed]
- Greenhough, A.; Smartt, H.J.; Moore, A.E.; Roberts, H.R.; Williams, A.C.; Paraskeva, C.; Kaidi, A. The COX-2/PGE 2 Pathway: Key Roles in the Hallmarks of Cancer and Adaptation to the Tumour Microenvironment. Carcinogenesis. 2009, 30, 377–386. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.-T.; Honn, K.V. Nie, D. Cyclooxygenases, Prostanoids, and Tumor Progression. Cancer. Metastasis. Rev. 2007, 26, 525–534. [Google Scholar] [CrossRef]
- Poligone, B.; Baldwin, A.S. Positive and Negative Regulation of NF-κB by COX-2: Roles of Different Prostaglandins. J. Biol. Chem. 2001, 276, 38658–38664. [Google Scholar] [CrossRef]
- Chen, G.; Li, X.; Yang, J.; Li, J.; Wang, X.; He, J.; Huang, Z. Prognostic Significance of Cyclooxygenase-2 Expression in Patients With Hepatocellular Carcinoma: A Meta-Analysis. Arch. Med. Sci. 2016, 12, 1110–1117. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Zhu, J.; Gou, H.; Cao, D.; Jiang, M.; Hou, M. Clinical Significance of Cox-2, Survivin and Bcl-2 Expression in Hepatocellular Carcinoma (HCC). Med. Oncol. 2011, 28, 796–803. [Google Scholar] [CrossRef] [PubMed]
- Tai, Y.; Zhang, L.H.; Gao, J.H.; Zhao, C.; Tong, H.; Ye, C.; Huang, Z.Y.; Liu, R.; Tang, C.W. Suppressing Growth and Invasion of Human Hepatocellular Carcinoma Cells by Celecoxib Through Inhibition of Cyclooxygenase-2. Cancer Manag. Res. 2019, 11, 2831–2848. [Google Scholar] [CrossRef]
- Dong, X.; et al. Meloxicam Executes its Antitumor Effects Against Hepatocellular Carcinoma in COX-2-Dependent and -Independent Iathways. PloS One. 2014, 9, e92864. [Google Scholar] [CrossRef]
- Hu, J.W.; Chen, B.; Zhang, J.; Qi, Y.P.; Liang, J.H.; Zhong, J.H.; Xiang, B.D. Novel Combination of Celecoxib and Metformin Improves the Antitumor Effect by Inhibiting the Growth of Hepatocellular Carcinoma. J. Cancer. 2020, 11, 6437–6444. [Google Scholar] [CrossRef]
- Hardwick, J.M.; Soane, L. Multiple Functions of BCL-2 Family Proteins. Cold Spring Harb. Perspect. Biol. 2013, 5, a008722. [Google Scholar] [CrossRef] [PubMed]
- Yoshinaka, R.; Shibata, M.A.; Morimoto, J.; Tanigawa, N.; Otsuki, Y. COX-2 Inhibitor Celecoxib Suppresses Tumor Growth and Lung Metastasis of a Murine Mammary Cancer. Anticancer Res. 2006, 26, 4245–4254. [Google Scholar] [PubMed]
- Yue, J.; López, J.M. Understanding MAPK Signaling Pathways in Apoptosis. Int. J. Mol. Sci. 2020, 21, 2346. [Google Scholar] [CrossRef] [PubMed]
- Ou, Y.C.; Yang, C.R.; Cheng, C.L.; Raung, S.L.; Hung, Y.Y.; Chen, C.J. Indomethacin Induces Apoptosis in 786-O Renal Cell Carcinoma Cells by Activating Mitogen-Activated Protein Kinases and AKT. Euro. J. Pharmacol. 2007, 563, 49–60. [Google Scholar] [CrossRef]
- Jia, Z.; Zhang, H.; Ma, C.; Li, N.; Wang, M. Celecoxib Enhances Apoptosis of the Liver Cancer Cells via Regulating ERK/JNK/P38 Pathway. J. Buon. 2021, 26, 875–881. [Google Scholar] [PubMed]
- Elder, D.J.; Halton, D.E.; Playle, L.C.; Paraskeva, C. The MEK/ERK Pathway Mediates COX-2-Selective NSAID-Induced Apoptosis and Induced COX-2 Protein Expression in Colorectal Carcinoma Cells. Int. J. Cancer. 2002, 99, 323–327. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Sinicrope, F.A. Selective Inhibitors of MEK1/ERK44/42 and p38 Mitogen-Activated Protein Kinases Potentiate Apoptosis Induction by Sulindac Sulfide in Human Colon Carcinoma Cells. Mol. Cancer Ther. 2005, 4, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Park, S.W.; Kim, H.S.; Hah, J.W.; Jeong, W.J.; Kim, K.H.; Sung, M.W. Celecoxib Inhibits Cell Proliferation Through the Activation of ERK and p38 MAPK in Head and Neck Squamous Cell Carcinoma Cell Lines. Anticancer drugs. 2010, 21, 823–830. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Martindale, J.L.; Holbrook, N.J. Requirement for ERK Activation in Cisplatin-Induced Apoptosis. J. Biol. Chem. 2000, 275, 39435–39443. [Google Scholar] [CrossRef]
- Mandegary, A.; Hosseini, R.; Ghaffari, S.H.; Alimoghaddam, K.; Rostami, S.; Ghavamzadeh, A.; Ghahremani, M.H. The Expression of p38, ERK1 and Bax Proteins Has Increased During the Treatment of Newly Diagnosed Acute Promyelocytic Leukemia With Arsenic Trioxide. Ann. Oncol. 2010, 21, 1884–1890. [Google Scholar] [CrossRef]
- Somlyaia, G.; Javaherib, B.; Davaric, H.; Gyöngyid, Z.; Somlyaia, I.; Tamaddone, K.A.; Boros, L.G. Pre-Clinical and Clinical Data Confirm the Anticancer Effect of Deuterium Depletion. Biomacromol. J. 2016, 2, 1–7. [Google Scholar] [CrossRef]
- Bayrak, B.B.; Kulak, G.Y.; Yanardag, R.; Yarat, A. Short Term Deuterium Depletion in Drinking Water Reduced Tumor Induced Oxidative Stress in Mice Liver. Pathol. Res. Prac. 2022, 240, 154186. [Google Scholar] [CrossRef] [PubMed]
- Soleyman-Jahi, S.; Zendehdel, K.; Akbarzadeh, K.; Haddadi, M.; Amanpour, S.; Muhammadnejad, S. In Vitro Assessment of Antineoplastic Effects of Deuterium Depleted Water. Asian Pac. J. Cancer Prev. 2014, 15, 2179–2183. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhu, B.; Liu, C.; Fang, W.; Yang, H. [Deuterium-Depleted Water Selectively Inhibits Nasopharyngeal Carcinoma Cell Proliferation in Vitro]. Nan Fang Yi Ke Da Xue Xue Bao ( J. South. Med. Univ.). 2012, 32, 1394–1399. [Google Scholar]
- Hassanzade, A.; Mandegary, A.; Sharif, E.; Rasooli, R.; Mohammadnejad, R.; Masoumi-Ardekani, Y. Cyclooxygenase Inhibitors Combined With Deuterium-Enriched Water Augment Cytotoxicity in A549 Lung Cancer Cell Line via Activation of Apoptosis and MAPK Pathways. Iran J. Basic Med. Sci. 2018, 21, 508–516. [Google Scholar] [CrossRef] [PubMed]
- Gyöngyi, Z.; Budán, F.; Szabó, I.; Ember, I.; Kiss, I.; Krempels, K.; Somlyai, I.; Somlyai, G. Deuterium Depleted Water Effects on Survival of Lung Cancer Patients and Expression of Kras, Bcl2, and Myc Genes in Mouse Lung. Nutr. Cancer. 2013, 65, 240–246. [Google Scholar] [CrossRef] [PubMed]
- Salomonsson, L.; Branden, G.; Brzezinski, P. Deuterium Isotope Effect of Proton Pumping in Cytochrome c Oxidase. Biochim. Biophys. Acta. 2008, 1777, 343–350. [Google Scholar] [CrossRef] [PubMed]
- Olgun, A. , Biological Effects of Deuteronation: ATP Synthase as an Example. Theor. Biol. Med. Model. 2007, 4, 9. [Google Scholar] [CrossRef] [PubMed]
- Uemura, T.; Moritake, K.; Akiyama, Y.; Kimura, Y.; Shingu, T.; Yamasaki, T. Experimental Validation of Deuterium Oxide-Mediated Antitumoral Activity as it Relates to Apoptosis in Murine Malignant Astrocytoma Aells. J. Neurosurg. 2002, 96, 900–908. [Google Scholar] [CrossRef]
- Kalkur, R.S.; Ballast, A.C.; Triplett, A.R.; Spendier, K. Effects of Deuterium Oxide on Cell Growth and Vesicle Speed in RBL-2H3 Cells. PeerJ. 2014, 2, e553. [Google Scholar] [CrossRef]
- Mandegary, A.; Sharif, E.; Rasooli, R.; Hassanzadeh, A. The Effect of Deuterium Depleted/Enriched Water on the Growth of A549 and HepG2 Cell Lines. J. Kerman. Univ. Med. Sci. 2019, 26, 357–367. [Google Scholar] [CrossRef]
- Leonard, P.J.; Mullins, J.M. D2O Induced Alterations of Mitosis in PtK1 Cells. Exp. Cell Res. 1987, 172, 204–211. [Google Scholar] [CrossRef]
- Bader, Y.; et al. Synergistic Effects of Deuterium Oxide and Gemcitabine in Human Pancreatic Cancer Cell Lines. Cancer Lett. 2008, 259, 231–239. [Google Scholar] [CrossRef] [PubMed]
- Jandova, J.; Hua, A.B.; Fimbres, J.; Wondrak, G.T. Deuterium Oxide (D2O) Induces Early Stress Response Gene Expression and Impairs Growth and Metastasis of Experimental Malignant Melanoma. Cancers (Basel). 2021, 13, 605. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, C.P.; Kadara, H.; Lotan, D.; Woo, J.K.; Lee, H.Y.; Hong, W.K.; Lotan, R. Involvement of Mitochondrial and Akt Signaling Pathways in Augmented Apoptosis Induced by a Combination of Low Doses of Celecoxib and N-(4-Hydroxyphenyl) Retinamide in Premalignant Human Bronchial Epithelial Cells. Cancer Res. 2006, 66, 9762–9770. [Google Scholar] [CrossRef]
- Liggett, J.L.; Min, K.W.; Smolensky, D.; Baek, S.J. A Novel COX-Independent Mechanism of Sulindac Sulfide Involves Cleavage of Epithelial Eell Adhesion Molecule Protein. Exp. Cell Res. 2014, 326, 1–9. [Google Scholar] [CrossRef]
Figure 1.
The (logarithmic) growth inhibitory effect of celecoxib (a) and indomethacin (b) on the Hep G2 cell line. The viability of treated cells was evaluated through mitochondrial activity using the MTT assay, and data were presented as a percentage of control (n = 8) ±SD; SD: standard deviation.
Figure 1.
The (logarithmic) growth inhibitory effect of celecoxib (a) and indomethacin (b) on the Hep G2 cell line. The viability of treated cells was evaluated through mitochondrial activity using the MTT assay, and data were presented as a percentage of control (n = 8) ±SD; SD: standard deviation.
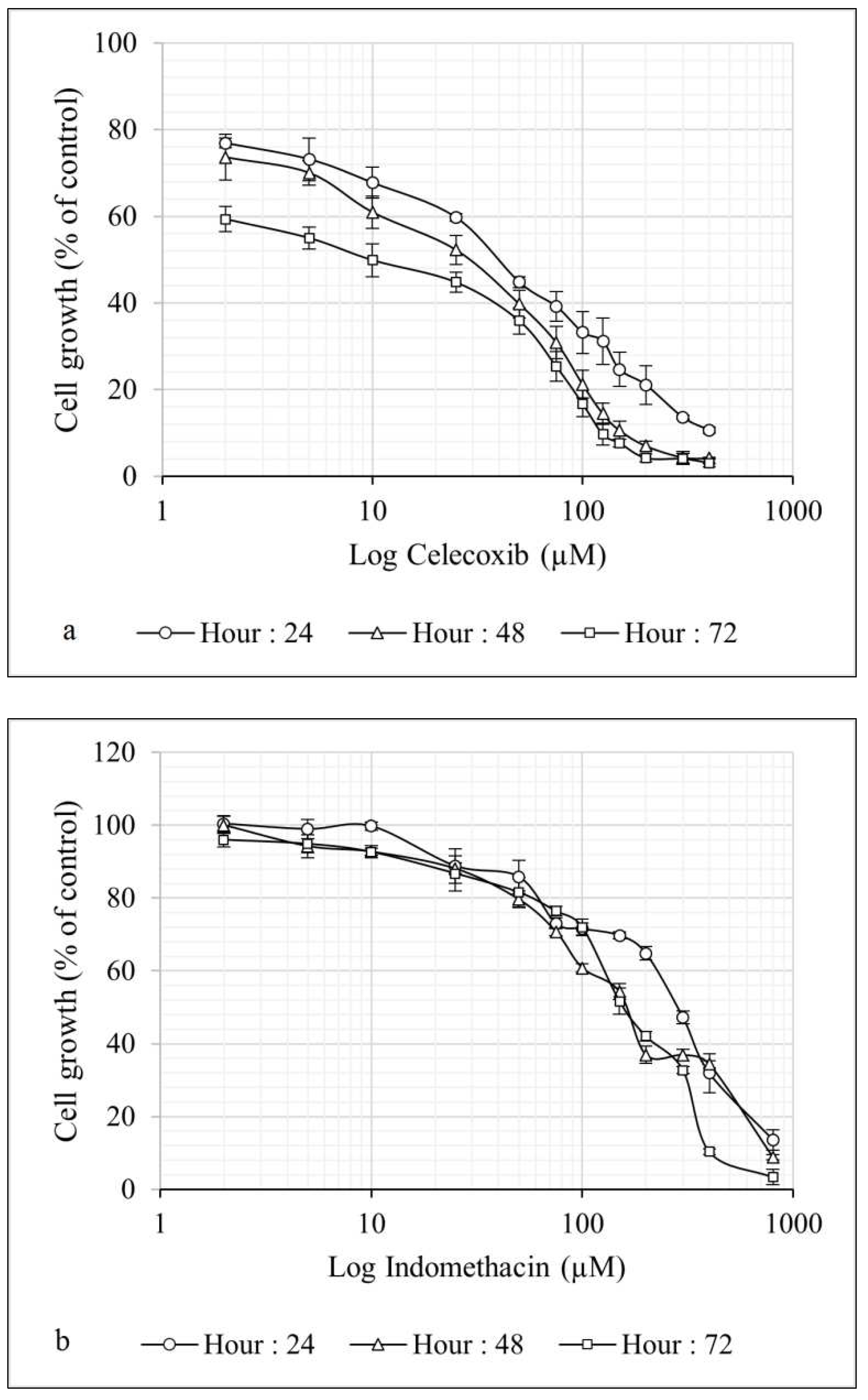
Figure 2.
The growth inhibitory effect of celecoxib in combination with DDWs and DEWs for 24 (a), 48 (b), and 72 (c) hours on the Hep G2 cell line. The viability of treated cells evaluated using MTT assay. Data are shown as Mean ± SD of triplicate wells for two independent experiments. Statistical significance of the differences was determined by one way ANOVA. * P<0.05, ** P<0.01. Celecoxib alone treated cells considered as referent.
Figure 2.
The growth inhibitory effect of celecoxib in combination with DDWs and DEWs for 24 (a), 48 (b), and 72 (c) hours on the Hep G2 cell line. The viability of treated cells evaluated using MTT assay. Data are shown as Mean ± SD of triplicate wells for two independent experiments. Statistical significance of the differences was determined by one way ANOVA. * P<0.05, ** P<0.01. Celecoxib alone treated cells considered as referent.
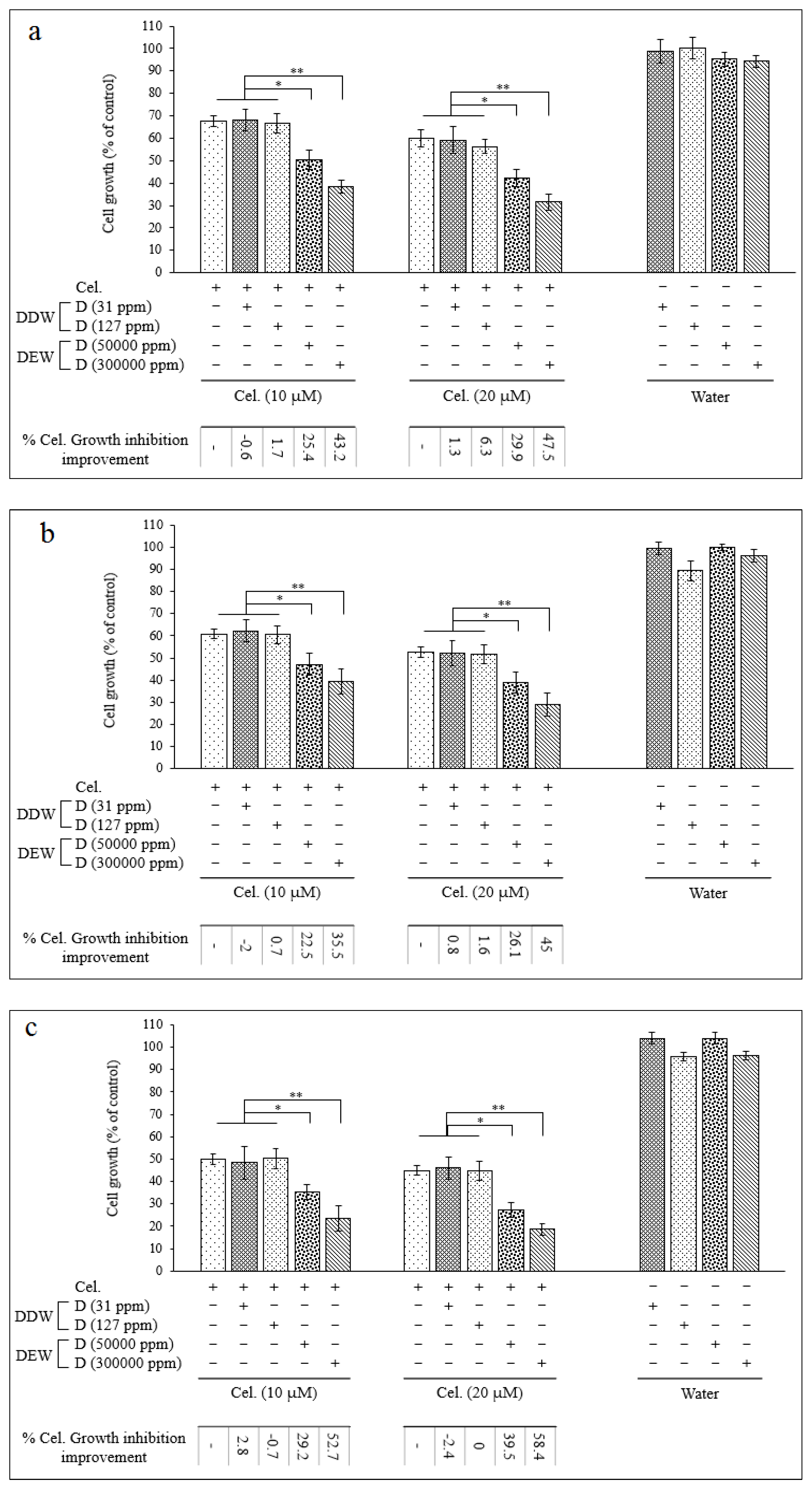
Figure 3.
The growth inhibitory effect of indomethacin in combination with DDWs and DEWs for 24 (a), 48 (b), and 72 (c) hours on the Hep G2 cell line. The viability of treated cells evaluated using MTT assay. Data are shown as Mean ± SD of triplicate wells for two independent experiments. Statistical significance of the differences was determined by one way ANOVA. * P<0.05, ** P<0.01. Indomethacin alone treated cells considered as referent.
Figure 3.
The growth inhibitory effect of indomethacin in combination with DDWs and DEWs for 24 (a), 48 (b), and 72 (c) hours on the Hep G2 cell line. The viability of treated cells evaluated using MTT assay. Data are shown as Mean ± SD of triplicate wells for two independent experiments. Statistical significance of the differences was determined by one way ANOVA. * P<0.05, ** P<0.01. Indomethacin alone treated cells considered as referent.
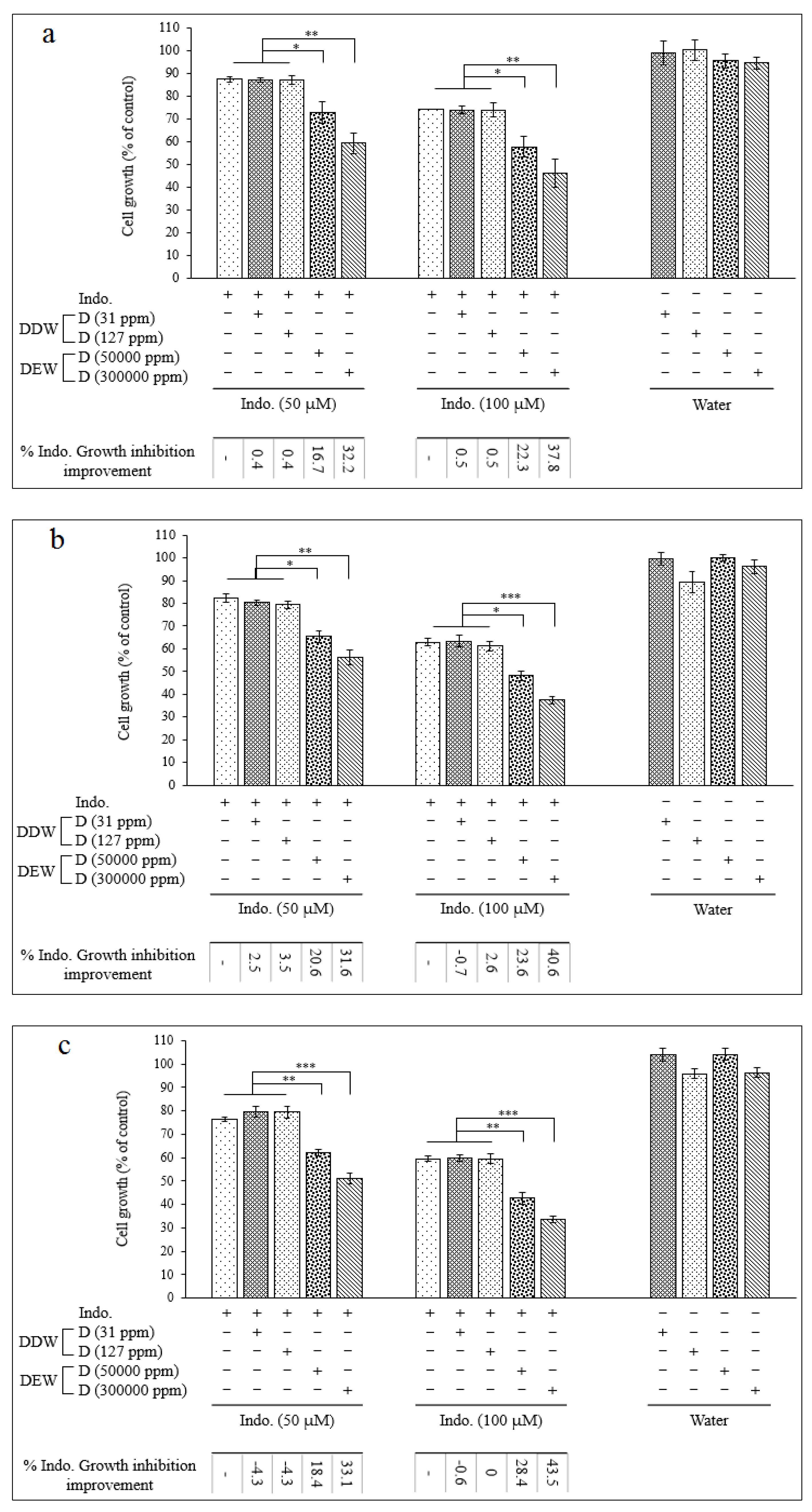
Figure 4.
Western blot analysis of COX-2, Bax, Bcl-2, poly ADP-ribose polymerase (PARP) and Caspase-3 proteins in the Hep G2 cells treated with celecoxib and indomethacin alone or in combination with different concentrations of deuterium.
Figure 4.
Western blot analysis of COX-2, Bax, Bcl-2, poly ADP-ribose polymerase (PARP) and Caspase-3 proteins in the Hep G2 cells treated with celecoxib and indomethacin alone or in combination with different concentrations of deuterium.
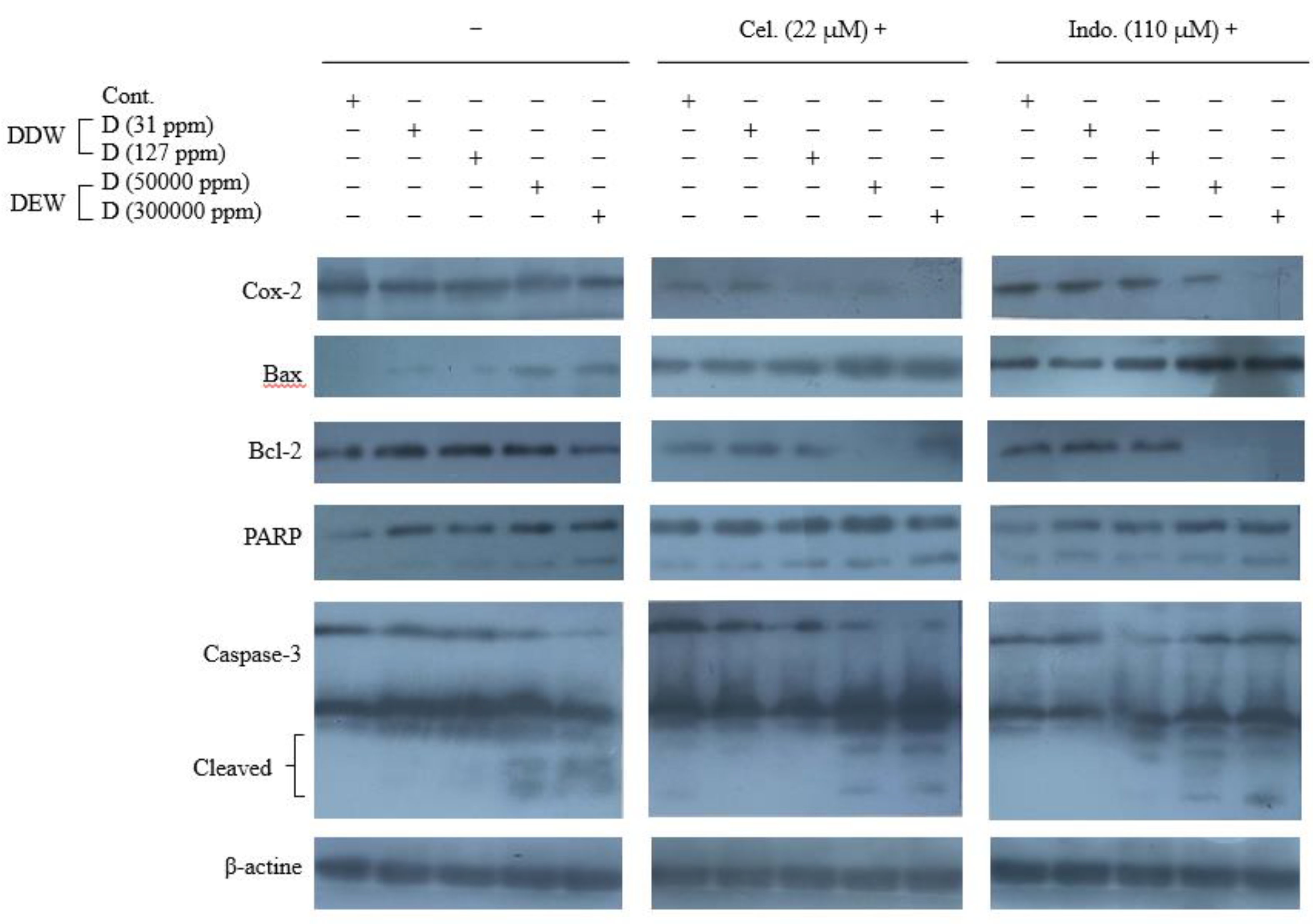
Figure 5.
Western blot analysis of the EK1/2 and p- ERK1/2 proteins, JNK and p- JNK proteins, and p38 and p- p38 MAPK proteins expression in the Hep G2 cells treated with celecoxib and indomethacin, alone or in combination with different concentrations of deuterium.
Figure 5.
Western blot analysis of the EK1/2 and p- ERK1/2 proteins, JNK and p- JNK proteins, and p38 and p- p38 MAPK proteins expression in the Hep G2 cells treated with celecoxib and indomethacin, alone or in combination with different concentrations of deuterium.
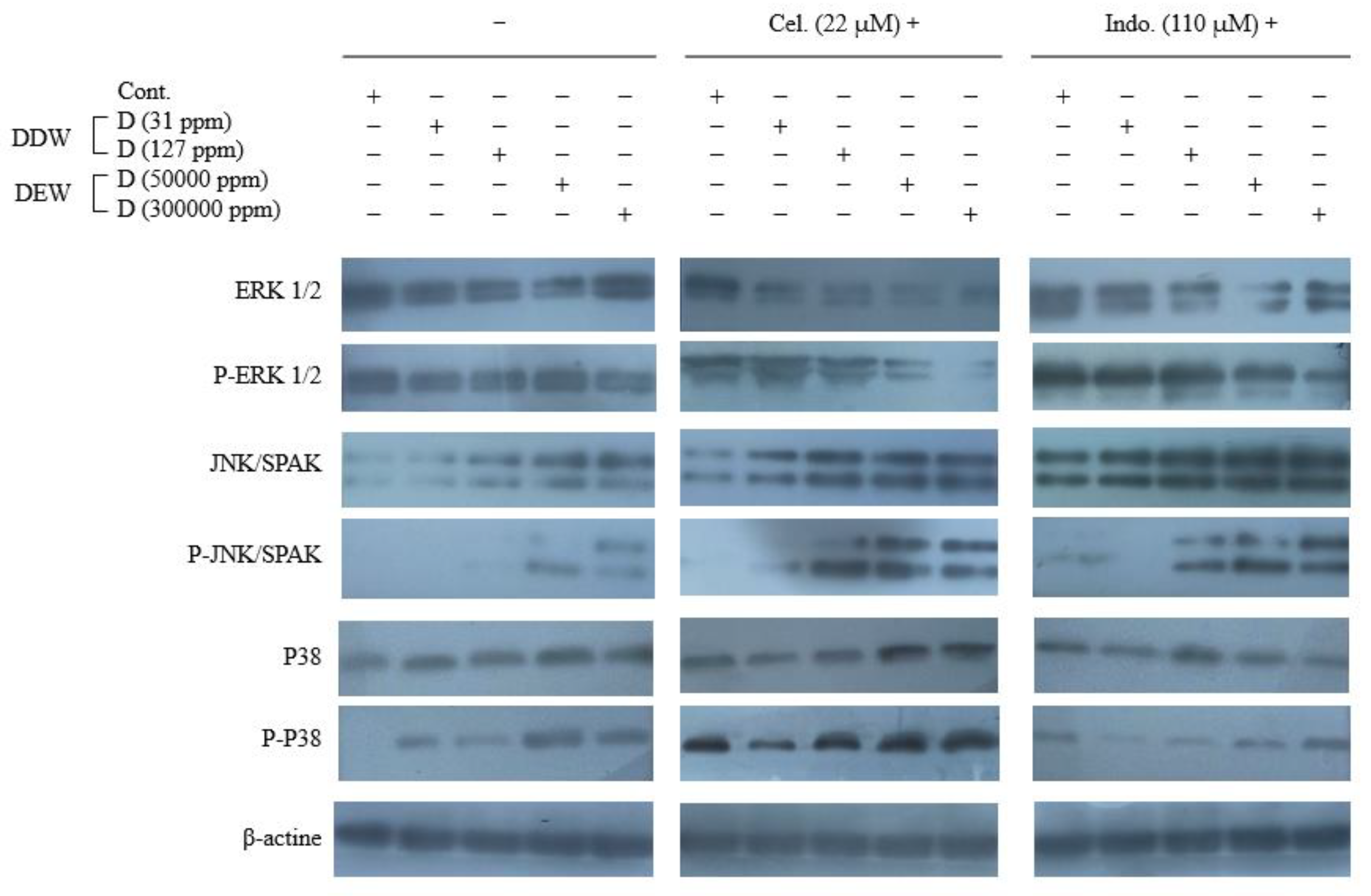

Table 1.
The concentration of celecoxib and indomethacin alone or in combination with various
concentrations of deuterium utilized to treat the Hep G2 cells for cytotoxicity assay.
Table 1.
The concentration of celecoxib and indomethacin alone or in combination with various
concentrations of deuterium utilized to treat the Hep G2 cells for cytotoxicity assay.
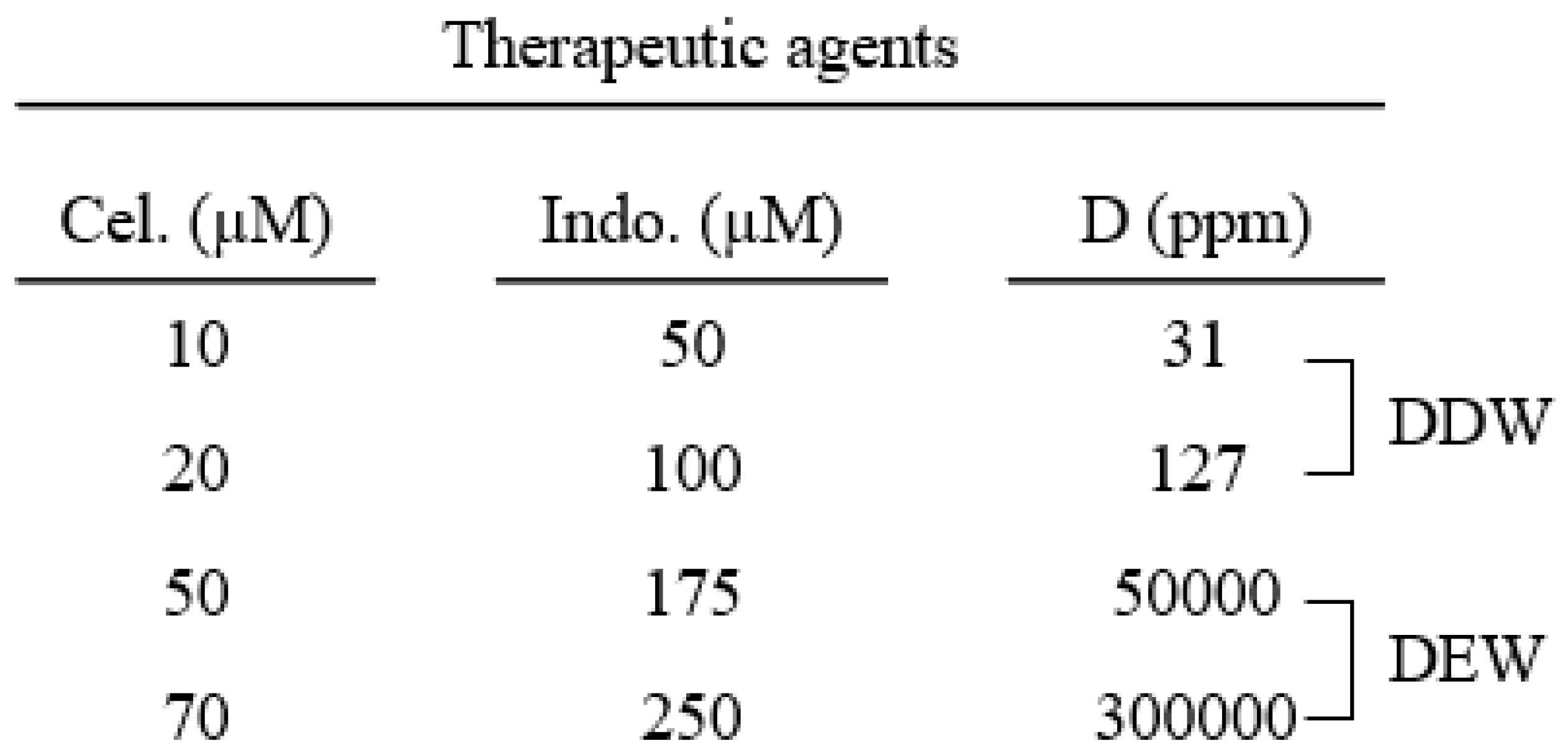
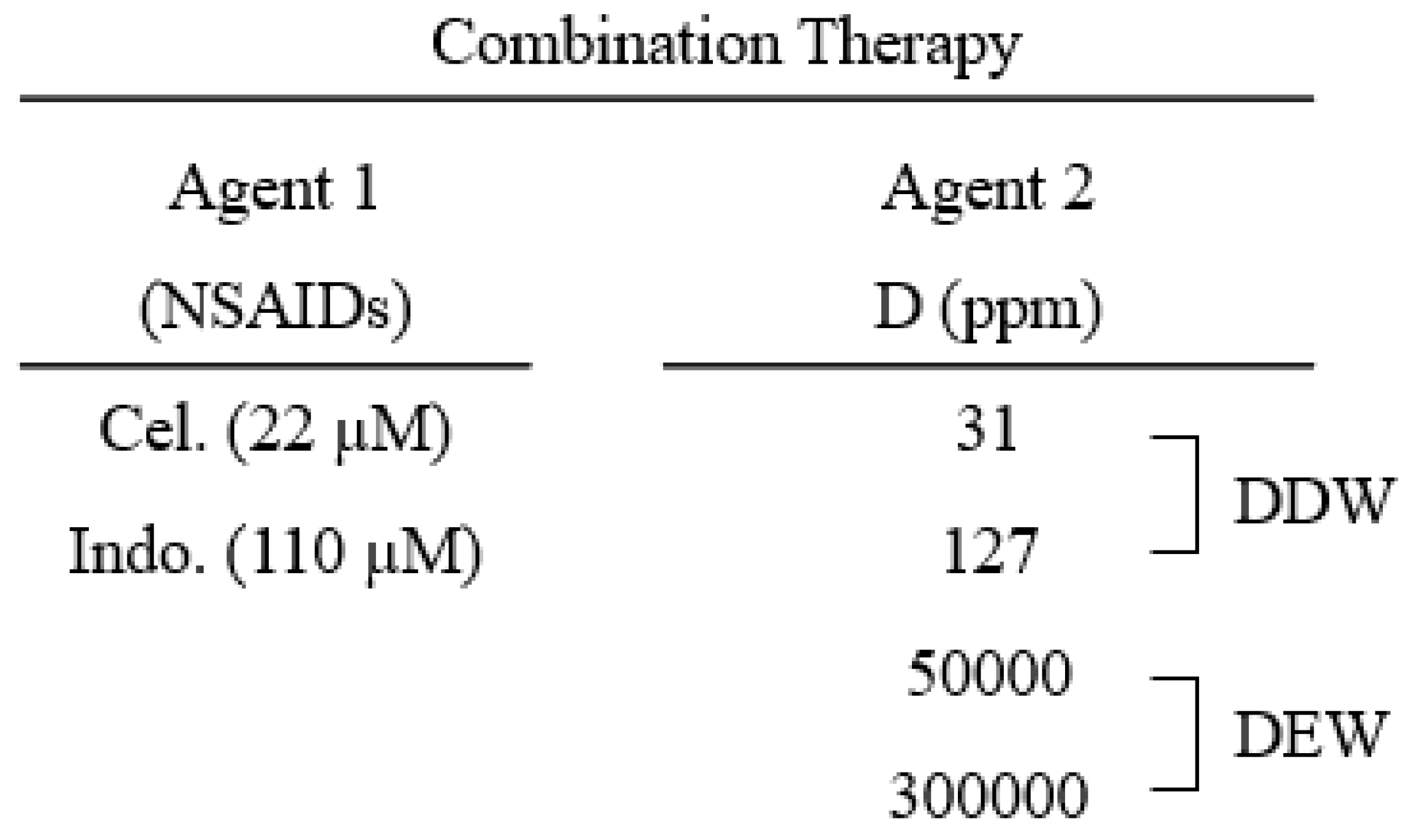
Table 2.
Celecoxib, indomethacin, and deuterium concentration to treat the Hep G2 cells for western
blot analysis.
Table 2.
Celecoxib, indomethacin, and deuterium concentration to treat the Hep G2 cells for western
blot analysis.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



