Submitted:
29 May 2023
Posted:
30 May 2023
You are already at the latest version
Abstract
Exercise produces oxidants from a variety of intracellular sources, including NADPH oxidases (NOX) and mitochondria. Exercise-derived ROS are beneficial, and the amount and location of these ROS are important to avoid muscle damage associated with oxidative stress. We discuss here some of the evidence that involves ROS production associated with skeletal muscle contraction and the potential oxidative stress associated with muscle contraction. We also discuss the potential role of H2O2 produced after NOX activation in the regulation of glucose transport in skeletal muscle. Finally, we propose a model based on evidence for the role of different populations of mitochondria in skeletal muscle in the regulation of ATP production upon exercise. The sub-sarcolemmal population of mitochondria has the enzymatic and metabolic components to establish a high mitochondrial membrane potential when fissioned at rest but lacks the capacity to produce ATP; calcium entry to the mitochondria will further increase the metabolic input. Upon exercise, sub-sarcolemmal mitochondria will fuse to intermyofibrillar mitochondria and will transfer the membrane potential to them. These mitochondria are rich in ATP synthase and will subsequentially produce the ATP needed for muscle contraction in long-term exercise. These events will optimize energy use and minimize mitochondria ROS production.
Keywords:
ATP production
; mitochondrial network
; mitochondria dynamics
; MCU
1. Introduction
Since the first report that muscular physiology is redox dependent, several research lines in redox biology has grown significantly. Skeletal muscle is the human buffer against aging, some metabolism pathologies as well as neuropathologies, so the understanding of how skeletal physiology process are modulated by reactive oxygen species (ROS), is a source of potential intervention based on diet supplementation or exercise protocols. Evidence suggests that exercise produces oxidants from a variety of intracellular sources, including NADPH oxidases (NOX) and mitochondria as the main sources [1,2]. Exercise-derived ROS are beneficial, and the amount and location of these ROS is important to avoid muscle damage associated with oxidative stress. In this review, we propose a model in which the role of the fusion of different subpopulations of mitochondria after exercise is relevant, increasing energy saving and decreasing H2O2 production.
2. H2O2-mediated physiological signals in skeletal muscle
2.1. Role of H2O2 in the excitation-contraction (E-C) coupling mechanism.
In the excitation-contraction (E-C) coupling mechanism, there is direct evidence pointing to hydrogen peroxide as the oxidizing agent that modulates the interaction between the Cav.1.1 and the ryanodine-1 receptor and, on the other hand, the oxidation of the contractile apparatus. In the first mechanism, Cav1.1 is an L-type Ca2+ channel located in the T tubule of skeletal muscle cells, composed of 5 subunits (α1S, α2, β, γ & δ), being α1S subunit the one carrying the transmembrane barrels responsible for its voltage sensor capacity during a change in membrane potential as occurs during an action potential [3,4]. Cav 1.1 plays a fundamental role in changing its spatial conformation after membrane depolarization which induces ryanodine receptor (RyR) channel opening, and the following massive release of Ca2+ from the sarcoplasmic reticulum to the cytoplasm, which triggers the contraction of the muscle fiber [5,6,7]. A modulatory role of H2O2 in skeletal muscle contraction has been suggested for decades; based on the idea that skeletal muscle has many redox-sensitive proteins involved in contractile apparatus and mitochondria[8]. H2O2 induces a delay in action potential associated with excitation processes in mouse myoblasts which was reverted using the antioxidant N-acetylcysteine (NAC)[9]. Indeed, a decrease in the action potential amplitude was observed in H2O2-treated fibers after high-frequency stimulation with respect to non-treated fibers [10]. Doses of H2O2 is crucial to affect E-C coupling; but contradictory information is found in literature. Some reports show that µM doses do not affect E-C coupling apparently, but mM doses have an inhibitory effect[11]. On the other hand, 10 mM H2O2 could increase the Ca2+ sensitivity of contractile machinery. Moreover, the effect of 1mM H2O2 on contractile response also depends on the fiber type, depolarization-induced contraction of slow but not fast twitch fibers [12]. One of the central stimuli that increase H2O2 production is electrical stimulation of isolated fibers or cultured muscle cells, an in vitro strategy mimicking physical activity. For example, contraction by electrical stimulation of myotubes or nude fibers can generate the intracellular increase of H2O2 and NO [13,14,15].
Exercise could be considered an acute stressor which at low doses induces the H2O2 production as an “hormetic” signal, inducing a gene expression program of defense against oxidative stress [16,17]. In this line, acute exercise significantly increased mitochondrial H2O2 and FOXO3a, a key transcription factor involved in the activation of transcription of mitochondrial superoxide dismutase and catalase, both key antioxidants enzymes[18]. Same exercise pattern upregulates the redox effector factor-1 (Ref1) and nuclear factor erythroid 2-related factor 2 (Nrf2), increases reduced glutathione (GSH) content and manganese superoxide dismutase activity, suggesting that low level of ROS production is a stimulus for antioxidant gene expression. Along the same lines, it has been observed that cultured myotubes incubated with low doses of H2O2 increase the expression of hemoxygenase 1, an enzyme related to antioxidant protection [19].
The controlled production of ROS exerted by exercise is a factor that allows the muscle to adapt to training, triggering the activation of transcription factors and expression of genes such as peroxisome proliferator-activated receptor gamma co-activator 1 α (PGC1 α) and peroxisome proliferator-activated receptor γ (PPAR γ), the first related to the induction of mitochondrial myogenesis and the second with the increase in uncoupling protein-3 (UCP3), a mitochondrial uncoupling protein, considered the first line of defense against mitochondrial ROS production [14,20]. In addition, the upregulation of UCP3 may help reduce ROS production by increase the proton gradient and protecting muscle mitochondria from oxidative stress during exercise. However, it also compromises energy coupling efficiency, as indicated by increased respiration rate[21].
Another mechanism involved in ROS production with skeletal muscle physiology is the extracellular ATP release. This mechanism has been described in macrophages where ROS increase triggers the activation of pathways needed for cytokines release [22]. When skeletal muscle fibers are electrically stimulated at low frequencies (20 Hz), ATP is released from the muscle fibers, and binds to their P2Y purinergic receptors, thereby activating a signal transduction pathway, which culminates in the regulation of the transcription of some genes through Ca2+ release via inositol 1,4,5-trisphosphate receptors (IP3) from the sarcoplasmic reticulum. Indeed, Cav1.1 is also responsible for triggering an excitation-transcription coupling (ETC) process, which is mediated by the release of ATP [23,24,25]. H2O2 may also have a role in the ETC process, based on that after contraction, ATP binds to P2Y1 receptors and then activates NOX2 through a protein kinase C (PKC)-dependent mechanism [23]. This evidence shows that exercise results in skeletal muscle exposure to H2O2, which is involved in adaptive physiological responses.
2.2. Role of H2O2 in glucose uptake in skeletal muscle
Among the physiological functions of H2O2 in skeletal muscle, besides a role as a modulator of EC and ET coupling, a metabolic participation also has been described. Muscle contraction is a process with high energy demand, so the EC coupling must have machinery associated with the constant generation of ATP. In addition, it has been shown that the production of ROS by various sources activates the entry of glucose into the cell. Myotubes treated with NAC for 24 h decreased mRNA and protein contents of glucose transporter type 4 (GLUT4), mRNA content and activity of phosphofructokinase (PFK), and lactate production and glucose uptake [26] . As for muscle contraction, exogenous stimulation with H2O2 increased 2-Deoxy-D-glucose uptake, adenosine monophosphate-activated protein kinase (AMPK) [27] and glycolytic activity [28]. These findings suggest that ROS produced after muscle contraction, play an important role increasing the glycolytic activity and glucose uptake after exercise. On the other hand, H2O2 stimulation in high doses is capable to activate the peroxisome proliferator-activated receptor γ co-activator 1 α (PGC-1 α) transcription trough AMPK activation[29]. In turn, H2O2 induces glucose uptake but this is through a way an independent of AMPK activation[30], apparently through activating a phosphatidylinositol 3-kinase (PI3K)-dependent mechanism[28,29].
NOX4 has been related to the physiological adaptation to acute exercise. Particularly, NOX4 deletion results in impaired glucose and fatty acid oxidization and decreased Mitochondrial uncoupling protein 3 (UCP3) protein expression in response to acute exercise [31]. During muscular depolarization or insulin stimulation NOX2 activation also occur increasing H2O2 production and the inhibition of this isoform decreases glucose uptake in myotubes[32]. Moreover, a specific source of ROS has been linked to exercise-stimulated glucose uptake, where H2O2 from NOX2 could be the main responsible for GLUT-4 translocation during moderate-intensity exercise[33]. Moreover, H2O2 enhances GLUT4 translocation in skeletal muscle cells, independent of insulin, and is reduced by antioxidants and NOX2 inhibition. RyR-mediated Ca2+ release and IP3-receptor-mediated mitochondrial Ca2+ uptake, alongside the canonical pathway, jointly promote glucose uptake in response to insulin[34]. Indeed, piperine administration induces intracellular Ca2+ and ROS generation through TRPV1, leading to Calcium/calmodulin-dependent protein kinase kinase β-dependent AMPK phosphorylation, and consecutively GLUT4 translocation and AMPK activation in L6 myotubes [35]. On the other hand, our research group has found an important connection between the electrical stimulation of skeletal muscle and the release of ATP into the extracellular medium. This ATP release appears to be also mediated by NOX2[23]. Also, extracellular ATP is capable to increase glucose uptake in skeletal muscle in response to exercise, through PI3K activation[31]. All of these findings point to a role of extracellular ATP, activating NOX2 which could modulate glucose uptake. Therefore, NOX2-dependent ROS production is a crucial mechanism for increasing muscle glucose uptake during exercise.
2.3. Mitochondrial ROS associated to physical activity.
Another hormetic response to physical activity is the induction of mitochondrial biogenesis, also called mitohormesis, which is mainly induced to enhance respiratory capacity and endurance [17]. Thus, exercise-induced mitochondrial ROS production might have an adaptive role at different levels, including the spread of energy between different pools of mitochondria, the fusion/fission state, and the status and location of mitochondrial Ca2+. The transient increase in ROS production resulting from fission events during exercise acts as a signaling mechanism, triggering adaptive responses in the body, orchestrating the beneficial adaptations that arise from physical activity.
2.3.1. The particular mitochondria distribution and characteristics in skeletal muscle
Skeletal muscle fibers can change from a preferentially glycolytic metabolism to oxidative metabolism upon certain types of physiological exercise, a process whose cellular mechanism needs to be better understood. In turn, the EC mechanism is connected to a network of mitochondria in skeletal muscle fiber, showing a physiological communication between contractile machinery and mitochondria, in which Ca2+ has a relevant role. Moreover, mitochondria are mobile and plastic organelles, constantly changing shape, fusing or fissioning with each other, and parallel changing their role in cellular bioenergetics [36].
Adult skeletal muscle presents two populations of mitochondria; one group comprises both perinuclear (PN) and perivascular (PV) mitochondria, both considered peripherally located mitochondria (PLM) or subsarcolemmal (SSM), and other one, called intermyofibrillar (IMF) mitochondria [37]. The SSM population comprises the mitochondria located beneath the plasma membrane of the muscle fiber (and, as nuclei have a similar location in adult skeletal muscle, mitochondria closely surrounding the myonuclei), and the IMF population of mitochondria, located regularly close to the sarcoplasmic reticulum terminal cisternae and the triad, at regular intervals along every myofibril [38,39].
IMF mitochondria form a structural arrangement characterized by the interaction of transverse mitochondrial tubules in the sarcomere, called “mitochondrial reticulum,” which has been proposed as an energetic conductive pathway from mitochondria to the contractile apparatus [37]. Specialized proteins plays such as an intermembrane linker formed by a single protein with two membrane interacting domains in sarcoplasmic reticulum [40]. This physical connectivity allows the passage of calcium from the reticulum to the mitochondria to be more effective since it allows the proximity between proteins such as voltage-dependent anion channel (VDAC) and RyR2 or IP3R. It has also been described that H2O2 would diffuse from the mitochondrial space towards these contact domains to modulate calcium release locally [40,41]. It is very interesting to note that these two populations of mitochondria contribute both to the muscle fiber oxidative capacity and bioenergetics, but they do so each in a specialized way, allowing in the end to optimize ATP production precisely where is needed for contraction, i.e., near the myofibrils, producing mitochondria membrane potential propagation. In agreement with this, it has been proposed that PML support the IMF energy based on the presence of higher oxidative enzyme activity [42].
Together with the different localization and oxidative capacity, both populations express different types of proteins and different membrane potentials. For example, SSM expresses mitochondrial calcium uniporter regulator (MICU1)[43]. The mitochondrial calcium uniporter (MCU) is a highly selective and highly regulated calcium channel inserted in the inner mitochondrial membrane that allows calcium uptake from the cytosol to mitochondria after contraction [42,44]. This complex (when regulated) is basally closed and is activated upon cytoplasmic Ca2+ increases, thus allowing the ion to enter the mitochondria. The increase in ROS production increases when the mitochondrial potential (ΔΨ) is elevated. Therefore, MICU1 expression would protect against mitochondrial H2O2-generated damage, as elevated MCU activity would dissipate mitochondrial potential [20].
This idea is consistent with the evidence showing that in neurons, MCU promoted the activity of the electron transport chain and the chemical reduction of NAD+ to NADH, which would imply that electrons are not available for ROS formation [45]. Moreover, it is important to highlight that high MCU activity, also induces a mitochondrial membrane potential dissipation, facilitating the activity of the ATP synthase [46]. It has been observed that the lower expression of MCU generates muscle atrophy, intimately relating regulation of mitochondrial Ca2+ to the size of the muscle fiber[47]. The action of MCU is crucial for ATP synthesis in aerobic conditions, because Ca2+ increase is an essential cofactor for the tricarboxylic (TCA) cycle´s enzymes such as glycerol phosphate dehydrogenase (GPDH), pyruvate dehydrogenase (PDH), isocitrate dehydrogenase (ICDH), and α-ketoglutarate dehydrogenase (α-KGDH)[48]. Thus, Ca2+ increase can elevate the efficiency of complexes I, III, and IV of OXPHOS. Ca2+ -induced activation of mitochondrial sodium calcium exchanger (NCLX) results in Na+ influx into the matrix and Na+ interacts with phospholipids in the inner leaflet of IMM, decreases its fluidity, and slows down ubiquinol (UQH2) diffusion, increasing electron flux due to major NADH availability. In simple words, calcium potentiates the establishment of the proton gradient needed for ATP synthesis, which is used for sarco/endoplasmic reticulum Ca2+-ATPase (SERCA) to pump back calcium from the cytosol to ER [49].
When we talk about muscle activity is important to remember that there are different types of muscle fibers (as different types of motor units). Slow-fatigue resistance type possesses a larger number of mitochondria, having a preferential oxidative metabolism that allows them to contract for long periods of time. The fast-fatigable types have a lower mitochondria content, relying on an anaerobic glycolytic metabolism, responsible for their poor fatigue resistance. These different types of muscle fibers can change from one phenotype to another depending on external demands. One of the main actors in maintaining or changing the muscle phenotype is the pattern of stimulation coming from motor neurons. In this way, it has been shown that low frequency electrical stimulation induces the expression of genes belonging to slow type phenotype, while high frequencies lead to expression of genes typical for fast phenotypes. Among the slow phenotype transcriptional profile, we can find slow isoforms of proteins from the contractile apparatus and of factors that lead to increase mitochondrial biogenesis as well as oxidative enzymes[50,51]. Interestingly, Quezada et al. have also shown that low frequency electrical stimulation (that allows fibers to convert to slow phenotype fibers), can decrease the expression of the MCU complex in isolated adult fibers [52].
Higher protein levels of MCU and MICU1 per mitochondria have been observed in fast phenotypes muscles (like flexus digitorium longus) than those belonging to a slow phenotype muscle as the soleus. From these data we can propose a hypothesis where the decrease in mRNA of the MCU complex after low-frequency electrical stimulation of isolated fibers from a fast muscle would favor a lower protein level of the MCU and MICU1 per mitochondrion, being an early metabolic response to the phenotypic shift from fast to slow phenotype muscle fiber[52]. Also, a gradual increase in the number of mitochondria, together with a decrease in levels of MCU complex in response to a low-frequency electrical stimulus, could allow adapting mitochondrial Ca2+ homeostasis to finally reach that of a slow muscle. On the other hand, Mcu gen deletion produces a decrease in Ca2+ stimulated ATP synthesis, an impairment in TCA cycle substrate flux, and a turn toward fatty acid metabolism [44].
These data suggest that both mitochondria calcium transients and the total volume of mitochondria are somehow conjointly modulating metabolism to provide either a fast-fatigable or a slow-fatigue resistant response.
2.3.2. Function of the heterogeneity of mitochondria within the muscle fiber
The PLM mitochondria are quite different to the IMF mitochondria[43,53,54]. It has been shown that electron transport chain elements needed to establish the mitochondria membrane potential are differentially distributed among SSM and IMF; in particular, Complex IV and cytochrome C are located mostly in the SSM [43,54]. There are also differences in the distribution of complex V (ATP synthase), which is located mostly in the IMF mitochondria [54]. In contrast, the importin translocase of outer membrane, TOM20 has homogeneous distribution into the cell, suggesting a specific compartmentalization of different mitochondrial proteins between mitochondria subpopulations [43].
This evidence, together with the fact that there is a shift of mitochondria membrane potential (higher in SSM in resting conditions) towards the center of the muscle fiber upon electrical stimulation [43] prompted us to propose that in the SSM pool, the main proton-motive force is generated through the activation of complex I-IV. In contrast, ATP is generated in complex V which is in the IMF mitochondria and this ATP production will occur only after fusion of both mitochondria populations with the consequent spreading of mitochondria membrane potential. Mitochondrial ROS production will be minimal since the proton motive force is used for ATP synthesis when the electron transport chain works at a maximum level induced by exercise.
In fact, Mcu deletion produces a decrease in Ca2+ stimulated ATP synthesis, an impairment in TCA cycle substrate flux, and a turn toward fatty acid metabolism [44]. Stimuli, such as extracellular ATP or electrical stimulation, can increase the expression of MCU in isolated adult fibers [55]. In turn, it has been shown that the MCU-dependent increase in mitochondrial ROS is necessary for optimal skeletal muscle repair after an injury through the induction of actin polymerization dependent on RhoA [56]. It is interesting to note that, using direct measurement of superoxide by electron paramagnetic resonance; Crochemore et al. demonstrated that SSM produces more superoxide than IMF[57].
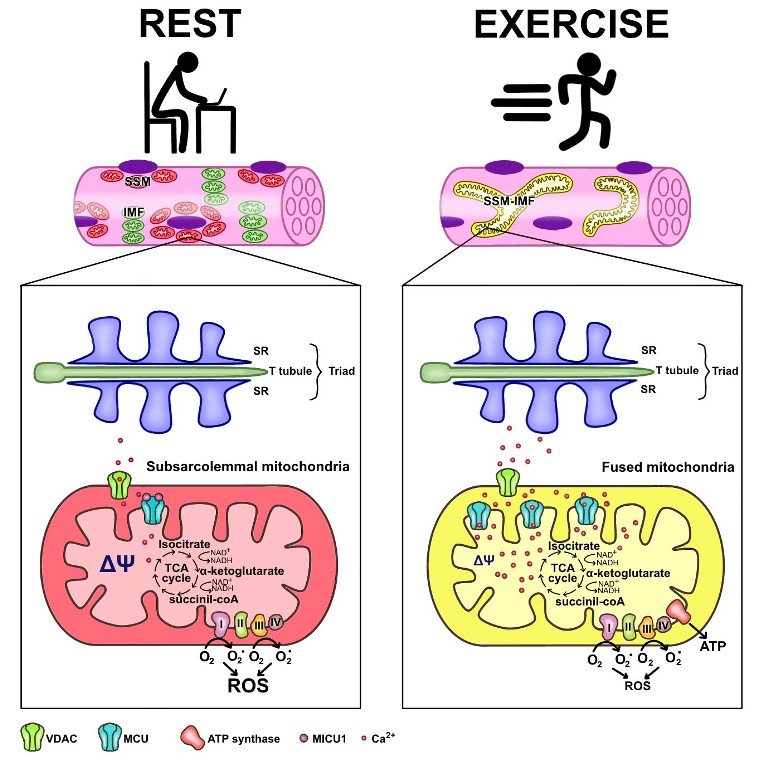
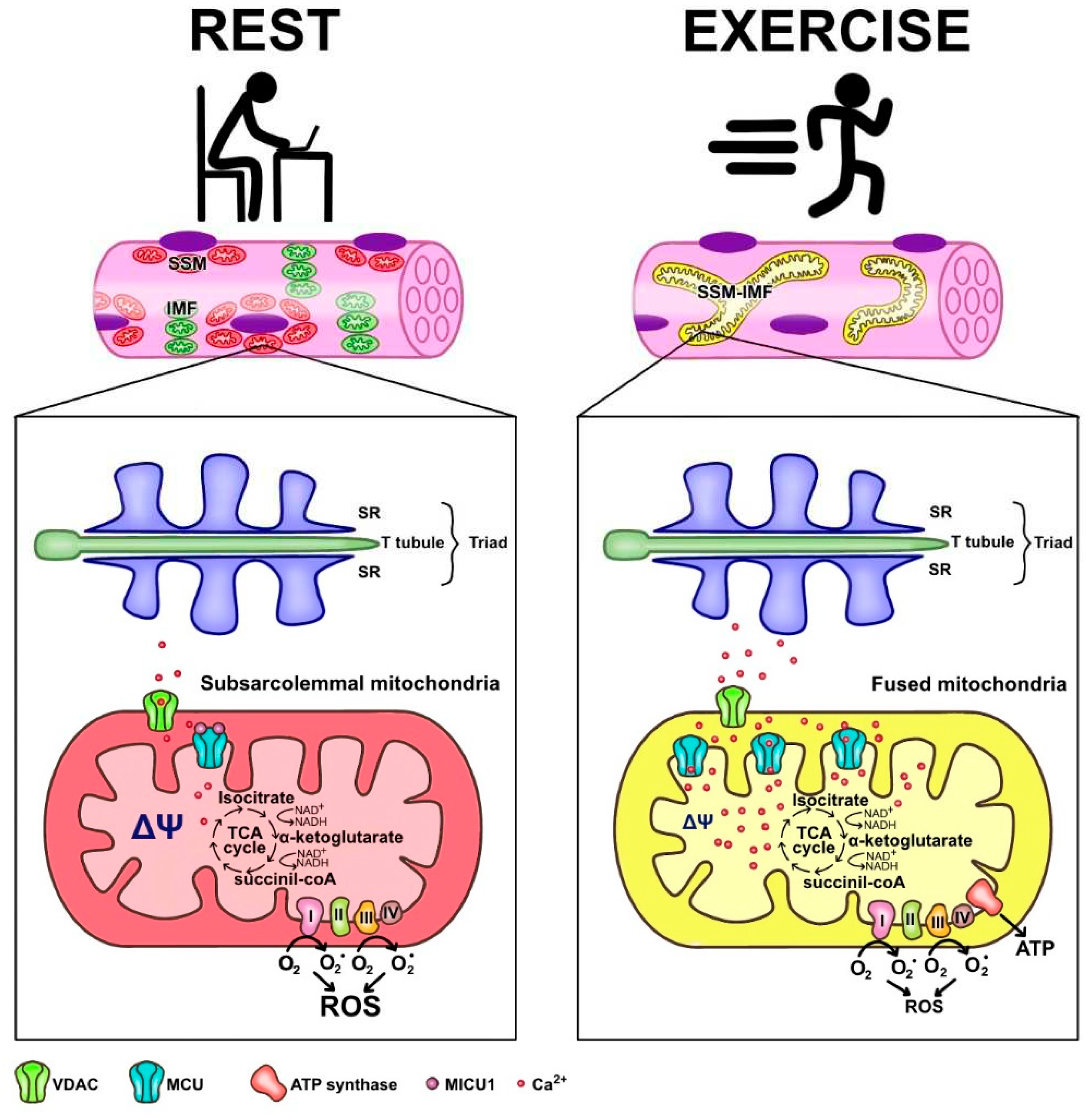
In summary, skeletal muscle mitochondria achieve this performance by separating two important functions; the first is mitochondria membrane potential generation, through calcium-sensitive oxidative phosphorylation (which occurs mainly in the subsarcolemmal population of mitochondria) and the second function is ATP production mediated by ATP synthase, which occurs in the intermyofibrillar population of mitochondria. To achieve this amazing performance, the two populations of mitochondria are not connected at rest and have a different resting protein composition. The intermyofibrillar mitochondria is enriched in ATP synthase and has a high content of mitochondrial calcium uptake 1 (MICU1) protein, MICU1 as a key regulator of mitochondrial Ca2+ uptake, which negatively regulates calcium entry to the mitochondrial matrix through the MCU calcium channel [58,59]. This protein is a Ca2+ sensor, and its functioning doesn´t impact on ΔΨ or oxygen consumption [18,58]. On the other hand, the subsarcolemmal mitochondria is enriched in the electron transport chain complex proteins and, having no MICU1, can reach a high calcium content upon muscle activation (Figure 1). The model of figure 1 shows that exercise induces mitochondrial fusion of SSM and IMF, transferring electrical properties and proteins from IMF to the mitochondrial network, decreasing the ROS generation and improving energy saving design.
2.3.3. Mitochondria dynamics is altered in skeletal muscle of aging subjects and in pathological conditions.
Mitochondria fusion events in skeletal muscle fibers are hard to evidence due to the highly restricted space in which IMF mitochondria are located; fusion events nevertheless take place, and they were shown for the first time by Eisner et al. in 2014[60].
When we consider the above-described model, it is reasonable to assume that mitochondria dynamics (fission and fusion) play an essential role in skeletal muscle function and wellbeing. Maintaining optimal skeletal muscle health requires dynamic mitochondrial function, which becomes disrupted in aging and various pathological conditions. Age-related mitochondrial dysfunction is characterized by impaired fusion/fission processes and mitophagy, leading to sarcopenia and reduced exercise capacity. In addition, diseases such as muscular dystrophies, mitochondrial myopathies, and type 2 diabetes exhibit mitochondrial fusion and fission imbalances, contributing to impaired ETC coupling.
Alterations in mitochondria dynamics have been shown to occur in middle age and more advanced age in mice[61] and they parallel dramatic decreases in muscle function. This was evidenced by changes in the mitochondria orientation as seen by confocal microscopy, changes in both mitochondria size and shape as seen by electron microscopy and by changes in expression of proteins involved in the fission and fusion processes. Furthermore, mitochondria dynamics appears to be altered in skeletal muscle of a mouse model of alcohol consumption[60]. Oxidative stress and reduced antioxidants reinforce mitochondrial fragmentation, suppress fusion/fission, and impair the electron transport chain, decrease ATP production, and cause DNA damage. Mitochondrial dynamics depend on proteins such as dynamis mitofusin (MFN) 1 and 2, and atrophy protein 1(OPA1) for fusion of the outer and inner mitochondrial membranes. Mitochondrial fission involves the recruitment of cytoplasmic Drp1 to the mitochondrial outer membrane, forming a ring-like structure with adaptors Mff, MiD49, and MiD51. These protein interactions ensure a balance between fusion and fission, crucial for maintaining mitochondrial dynamics and cellular functions[62]. Deletion of OPA1 leads to mitochondrial dysfunction, reduced myogenic stem cells, decreased protein synthesis, and activation of protein breakdown. Opa1 deletion in skeletal muscle affects the entire body, causing a premature aging phenotype that ultimately leads to animal death[63]. Opa1 deficiency in myopathy engages TLR9, activating NF-κB and triggering muscle inflammation. This localized inflammation can become systemic, impacting the entire body. Altered growth hormone/IGF1 axis and enhanced FGF21 expression are observed, contributing to impaired growth. Opa1 deficiency disrupts growth-related processes and promotes inflammation through inflammatory pathway activation. These processes collectively contribute to the development and progression of myopathy[64]. Aging is associated with sarcopenia, and when an obesity condition is associated, the term sarcopenic obesity (SO) is applied. Dysfunctional adipose tissue, fatty acid excess inside the bloodstream, and low-grade systemic inflammation are combined, resulting in lipotoxicity, oxidative stress, insulin resistance, and inflammation in the skeletal muscle [59]. It has been reported that high fat diet feeding suppresses mitochondrial biogenesis in the skeletal muscle of Zebrafish [60], and decreases the number of SSM mitochondrial content, Mfn2, and Opa1. On the contrary, Fis1 and Drp1, both fission proteins, were found increased compared to non-obese conditions in mice [61]. On the other hand, mitochondrial uncoupling attenuates SO by enhancing skeletal muscle mitophagy, reducing muscle inflammation, promoting mitochondrial turnover via STAT3 signaling, and mitigating muscle degradation [62].
Figure 2.
Altered dynamics and fusion capacity between mitochondria in disease.
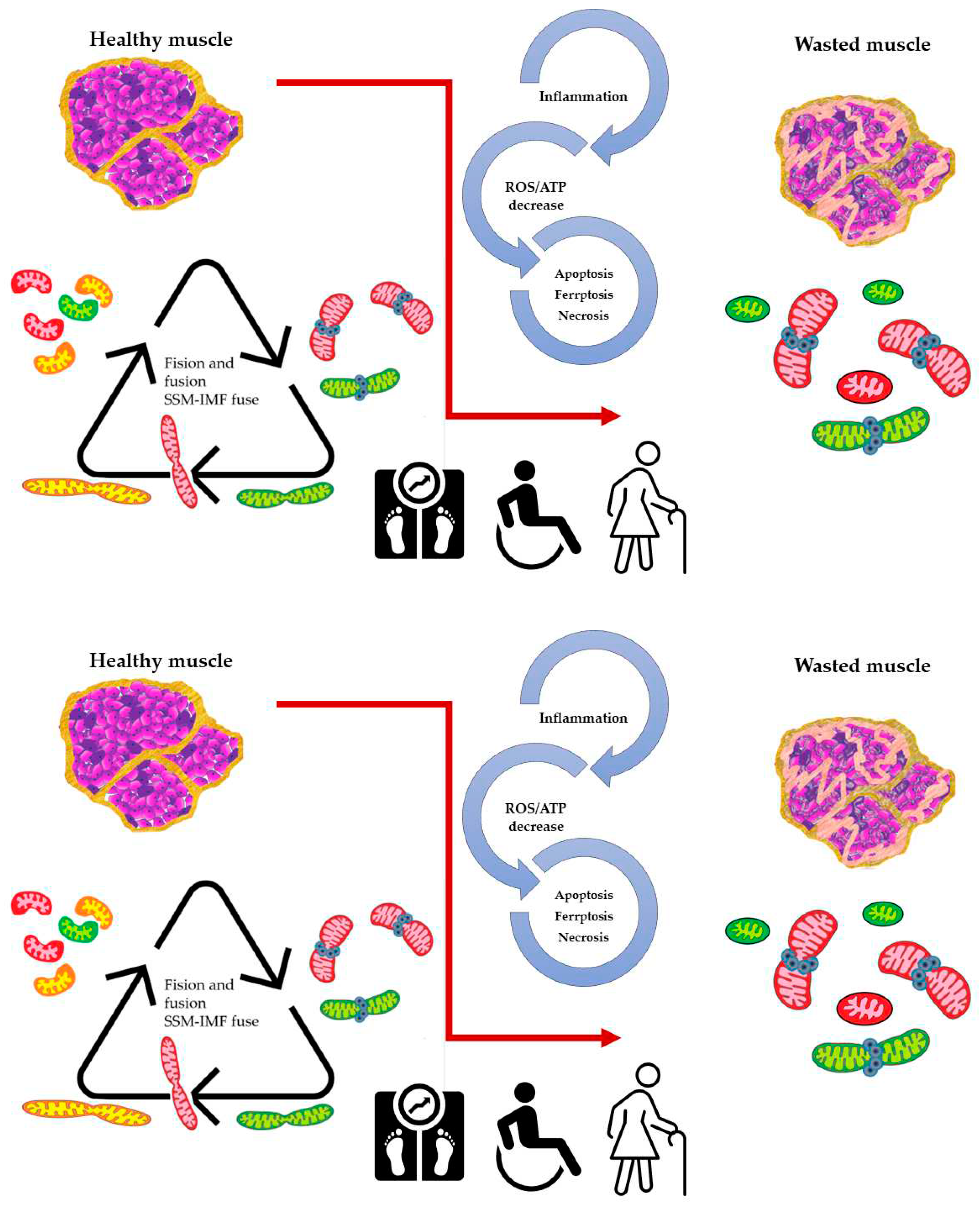
A common factor in many pathological conditions affecting skeletal muscle is an imbalance in mitochondrial dynamics. This imbalance is both cause and consequence of inflammation, increased ROS production, and cell death, leading to muscle deterioration characterized by dysfunctional mitochondria.
Exercise enhances the healthy mitochondrial network, promoting fusion/fission markers and biogenesis. Sarcopenia diminishes mitochondrial dynamics, mitophagy markers, and network efficiency, while exercise stimulates mitochondrial biogenesis through PGC1-α activation. Also, PGC-1α overexpression mitigates age-related increases in mitophagy markers, including Fis-1 and Drp-1 proteins, improves mitochondrial function, and reduces oxidative damage in mouse muscle[65]. Moderate-intensity exercise can be a non-invasive treatment, activating pathways that regulate the mitochondrial network in skeletal muscle[66]. Reduced MICU3 expression during aging leads to decreased mitochondrial Ca2+ uptake. Studies in aged mice and senescent C2C12 cells revealed that MICU3 downregulation is associated with decreased myogenesis, increased ROS, and apoptosis. Restoring MICU3 levels increase antioxidant defenses and promotes myogenesis. These findings highlight that MICU3 is a contributing factor to ROS production and apoptotic process during aging [67]. In the same line, mitochondrial dynamics and ROS production play crucial roles in the pathogenesis of Duchenne muscular dystrophy (DMD), a genetic skeletal muscle disorder characterized by mutations in the DMD gene that encodes dystrophin. Studies have shown that DMD patients present disruptions in mitochondrial fusion and fission processes leading to mitochondrial dysfunction and generating fragmented and dysfunctional mitochondria in muscle fibers. Intracellular Ca2+ disruption also has been reported derived of both increased Ca2+ influx and altered calcium release, leading to abnormally elevated resting cytosolic Ca2+ concentration [68]. Consequently, these effects contribute to apoptosis of the muscle cells. These abnormalities increase ROS production, oxidative stress, compromised energy production, and impaired cellular signaling, exacerbating muscle weakness and degeneration in DMD. Recent studies have highlighted the involvement of ferroptosis, a form of regulated cell death involving iron-dependent lipid peroxidation, in DMD pathology[69,70]. Dysregulation of NRF2 in DMD may promote ferroptosis in muscle cells by contributing to increased susceptibility to cell death. In turn, this can exacerbate muscle degeneration and inflammation observed in DMD.
Some studies have reported cases of statin-induced muscle-related side effects, including myopathy or rhabdomyolysis; atorvastatin dose-dependently inhibits C2C12 cell viability, resulting in increased intracellular iron ions, ROS, and lipid peroxidation. These effects primarily occur in mitochondria, leading to mitochondrial dysfunction. Biomarkers of myocardial injury are elevated during atorvastatin treatment, but ferroptosis inhibitors can counteract these effects [71]. Mechanistically, GSH depletion, along with the decrease in Nrf2, GPx4, and xCT cystine-glutamate antiporter, contribute to atorvastatin-induced muscular cell ferroptosis and damage[71]. Moreover, an increase in iron overload, senescence, and muscle atrophy markers was found in old Senescence-accelerated mouse-prone 8 (SAMP8), a sarcopenia-like phenotype, suggesting that iron overload-induced ferroptosis plays an essential role in sarcopenia[72].
3. Conclusion
Many questions remain when studying both oxidative metabolism and ROS production in skeletal muscle. The aim of this review is to bring attention on one side to the beneficial role of ROS in skeletal muscle physiology as opposed to their deleterious effects. On the other side, the particular distribution of skeletal muscle mitochondria and their different composition regarding key metabolic enzymes, together with the evidences pointing to both fusion events and fast sequential changes in mitochondria membrane potential, allow to propose an energy saving model consisting of distinct compartments where mitochondria membrane potential is generated in one compartment and, when needed, is transferred to a second compartment where ATP synthesis can take place. This distribution of functions saves energy and minimizes ROS production in muscle cells. It is important to note that mitochondria dynamics i.e., the balance between mitochondria fusion and fission is essential to this process and that normal muscle functioning depends on the capacity of mitochondria to fuse and fission at the right moment. This may explain why alterations in the mitochondria dynamics machinery have been reported in conditions (obesity, aging sarcopenia) in which muscle function is altered.
References
- Wang, P.; Li, C.G.; Qi, Z.; Cui, D.; Ding, S. Acute Exercise Induced Mitochondrial H₂O₂ Production in Mouse Skeletal Muscle: Association with p(66Shc) and FOXO3a Signaling and Antioxidant Enzymes. Oxid Med Cell Longev 2015, 2015, 536456. [Google Scholar] [CrossRef] [PubMed]
- Osório Alves, J.; Matta Pereira, L.; Cabral Coutinho do Rêgo Monteiro, I.; Pontes Dos Santos, L.H.; Soares Marreiros Ferraz, A.; Carneiro Loureiro, A.C.; Calado Lima, C.; Leal-Cardoso, J.H.; Pires Carvalho, D.; Soares Fortunato, R.; et al. Strenuous Acute Exercise Induces Slow and Fast Twitch-Dependent NADPH Oxidase Expression in Rat Skeletal Muscle. Antioxidants (Basel) 2020, 9, 57. [Google Scholar] [CrossRef]
- Bannister, R.A.; Beam, K.G. Ca(V)1.1: The Atypical Prototypical Voltage-Gated Ca2+ Channel. Biochim Biophys Acta 2013, 1828, 1587–1597. [Google Scholar] [CrossRef]
- Ríos, E.; Gillespie, D.; Franzini-Armstrong, C. The Binding Interactions That Maintain Excitation-Contraction Coupling Junctions in Skeletal Muscle. J Gen Physiol 2019, 151, 593–605. [Google Scholar] [CrossRef]
- Rebbeck, R.T.; Karunasekara, Y.; Board, P.G.; Beard, N.A.; Casarotto, M.G.; Dulhunty, A.F. Skeletal Muscle Excitation-Contraction Coupling: Who Are the Dancing Partners? Int J Biochem Cell Biol 2014, 48, 28–38. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.L.-H.; Pedersen, T.H.; Fraser, J.A. Reciprocal Dihydropyridine and Ryanodine Receptor Interactions in Skeletal Muscle Activation. J Muscle Res Cell Motil 2011, 32, 171–202. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Xu, L.; Meissner, G. Phosphorylation of Dihydropyridine Receptor II-III Loop Peptide Regulates Skeletal Muscle Calcium Release Channel Function. Evidence for an Essential Role of the Beta-OH Group of Ser687. J Biol Chem 1995, 270, 18459–18464. [Google Scholar] [CrossRef] [PubMed]
- Zuo, L.; Pannell, B.K. Redox Characterization of Functioning Skeletal Muscle. Front Physiol 2015, 6, 338. [Google Scholar] [CrossRef]
- Luin, E.; Giniatullin, R.; Sciancalepore, M. Effects of H₂O₂ on Electrical Membrane Properties of Skeletal Myotubes. Free Radic Biol Med 2011, 50, 337–344. [Google Scholar] [CrossRef]
- Oba, T.; Ishikawa, T.; Takaishi, T.; Aoki, T.; Yamaguchi, M. Hydrogen Peroxide Decelerates Recovery of Action Potential after High-Frequency Fatigue in Skeletal Muscle. Muscle Nerve 2000, 23, 1515–1524. [Google Scholar] [CrossRef]
- Posterino, G.S.; Lamb, G.D. Effects of Reducing Agents and Oxidants on Excitation-Contraction Coupling in Skeletal Muscle Fibres of Rat and Toad. J Physiol 1996, 496 Pt 3, 809–825. [Google Scholar] [CrossRef] [PubMed]
- Plant, D.R.; Lynch, G.S.; Williams, D.A. Hydrogen Peroxide Increases Depolarization-Induced Contraction of Mechanically Skinned Slow Twitch Fibres from Rat Skeletal Muscles. J Physiol 2002, 539, 883–891. [Google Scholar] [CrossRef]
- Espinosa, A.; Leiva, A.; Peña, M.; Müller, M.; Debandi, A.; Hidalgo, C.; Carrasco, M.A.; Jaimovich, E. Myotube Depolarization Generates Reactive Oxygen Species through NAD(P)H Oxidase; ROS-Elicited Ca2+ Stimulates ERK, CREB, Early Genes. J Cell Physiol 2006, 209, 379–388. [Google Scholar] [CrossRef] [PubMed]
- Silveira, L.R.; Pereira-Da-Silva, L.; Juel, C.; Hellsten, Y. Formation of Hydrogen Peroxide and Nitric Oxide in Rat Skeletal Muscle Cells during Contractions. Free Radic Biol Med 2003, 35, 455–464. [Google Scholar] [CrossRef]
- Palomero, J.; Pye, D.; Kabayo, T.; Spiller, D.G.; Jackson, M.J. In Situ Detection and Measurement of Intracellular Reactive Oxygen Species in Single Isolated Mature Skeletal Muscle Fibers by Real Time Fluorescence Microscopy. Antioxid Redox Signal 2008, 10, 1463–1474. [Google Scholar] [CrossRef] [PubMed]
- Goto, S.; Radák, Z. Hormetic Effects of Reactive Oxygen Species by Exercise: A View from Animal Studies for Successful Aging in Human. Dose Response 2009, 8, 68–72. [Google Scholar] [CrossRef]
- Musci, R.V.; Hamilton, K.L.; Linden, M.A. Exercise-Induced Mitohormesis for the Maintenance of Skeletal Muscle and Healthspan Extension. Sports (Basel) 2019, 7, 170. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Li, C.G.; Qi, Z.; Cui, D.; Ding, S. Acute Exercise Stress Promotes Ref1/Nrf2 Signalling and Increases Mitochondrial Antioxidant Activity in Skeletal Muscle. Exp Physiol 2016, 101, 410–420. [Google Scholar] [CrossRef]
- McArdle, F.; Spiers, S.; Aldemir, H.; Vasilaki, A.; Beaver, A.; Iwanejko, L.; McArdle, A.; Jackson, M.J. Preconditioning of Skeletal Muscle against Contraction-Induced Damage: The Role of Adaptations to Oxidants in Mice. J Physiol 2004, 561, 233–244. [Google Scholar] [CrossRef]
- Mailloux, R.J.; Harper, M.-E. Uncoupling Proteins and the Control of Mitochondrial Reactive Oxygen Species Production. Free Radic Biol Med 2011, 51, 1106–1115. [Google Scholar] [CrossRef]
- Jiang, N.; Zhang, G.; Bo, H.; Qu, J.; Ma, G.; Cao, D.; Wen, L.; Liu, S.; Ji, L.L.; Zhang, Y. Upregulation of Uncoupling Protein-3 in Skeletal Muscle during Exercise: A Potential Antioxidant Function. Free Radic Biol Med 2009, 46, 138–145. [Google Scholar] [CrossRef]
- Cruz, C.M.; Rinna, A.; Forman, H.J.; Ventura, A.L.M.; Persechini, P.M.; Ojcius, D.M. ATP Activates a Reactive Oxygen Species-Dependent Oxidative Stress Response and Secretion of Proinflammatory Cytokines in Macrophages. J Biol Chem 2007, 282, 2871–2879. [Google Scholar] [CrossRef] [PubMed]
- Díaz-Vegas, A.; Campos, C.A.; Contreras-Ferrat, A.; Casas, M.; Buvinic, S.; Jaimovich, E.; Espinosa, A. ROS Production via P2Y1-PKC-NOX2 Is Triggered by Extracellular ATP after Electrical Stimulation of Skeletal Muscle Cells. PLoS One 2015, 10, e0129882. [Google Scholar] [CrossRef] [PubMed]
- Jorquera, G.; Altamirano, F.; Contreras-Ferrat, A.; Almarza, G.; Buvinic, S.; Jacquemond, V.; Jaimovich, E.; Casas, M. Cav1.1 Controls Frequency-Dependent Events Regulating Adult Skeletal Muscle Plasticity. J Cell Sci 2013, 126, 1189–1198. [Google Scholar] [CrossRef]
- Casas, M.; Buvinic, S.; Jaimovich, E. ATP Signaling in Skeletal Muscle: From Fiber Plasticity to Regulation of Metabolism. Exerc Sport Sci Rev 2014, 42, 110–116. [Google Scholar] [CrossRef]
- Pinheiro, C.H. da J.; Silveira, L.R.; Nachbar, R.T.; Vitzel, K.F.; Curi, R. Regulation of Glycolysis and Expression of Glucose Metabolism-Related Genes by Reactive Oxygen Species in Contracting Skeletal Muscle Cells. Free Radical Biology and Medicine 2010, 48, 953–960. [Google Scholar] [CrossRef] [PubMed]
- Bouviere, J.; Fortunato, R.S.; Dupuy, C.; Werneck-de-Castro, J.P.; Carvalho, D.P.; Louzada, R.A. Exercise-Stimulated ROS Sensitive Signaling Pathways in Skeletal Muscle. Antioxidants 2021, 10, 537. [Google Scholar] [CrossRef]
- Higaki, Y.; Mikami, T.; Fujii, N.; Hirshman, M.F.; Koyama, K.; Seino, T.; Tanaka, K.; Goodyear, L.J. Oxidative Stress Stimulates Skeletal Muscle Glucose Uptake through a Phosphatidylinositol 3-Kinase-Dependent Pathway. Am J Physiol Endocrinol Metab 2008, 294, E889–897. [Google Scholar] [CrossRef]
- Irrcher, I.; Ljubicic, V.; Hood, D.A. Interactions between ROS and AMP Kinase Activity in the Regulation of PGC-1alpha Transcription in Skeletal Muscle Cells. Am J Physiol Cell Physiol 2009, 296, C116–123. [Google Scholar] [CrossRef]
- Jensen, T.E.; Schjerling, P.; Viollet, B.; Wojtaszewski, J.F.P.; Richter, E.A. AMPK Alpha1 Activation Is Required for Stimulation of Glucose Uptake by Twitch Contraction, but Not by H2O2, in Mouse Skeletal Muscle. PLoS One 2008, 3, e2102. [Google Scholar] [CrossRef]
- Specht, K.S.; Kant, S.; Addington, A.K.; McMillan, R.P.; Hulver, M.W.; Learnard, H.; Campbell, M.; Donnelly, S.R.; Caliz, A.D.; Pei, Y.; et al. Nox4 Mediates Skeletal Muscle Metabolic Responses to Exercise. Mol Metab 2021, 45, 101160. [Google Scholar] [CrossRef]
- Espinosa, A.; Campos, C.; Díaz-Vegas, A.; Galgani, J.; Juretic, N.; Osorio-Fuentealba, C.; Bucarey, J.; Tapia, G.; Valenzuela, R.; Contreras-Ferrat, A.; et al. Insulin-Dependent H2O2 Production Is Higher in Muscle Fibers of Mice Fed with a High-Fat Diet. IJMS 2013, 14, 15740–15754. [Google Scholar] [CrossRef] [PubMed]
- Henríquez-Olguin, C.; Knudsen, J.R.; Raun, S.H.; Li, Z.; Dalbram, E.; Treebak, J.T.; Sylow, L.; Holmdahl, R.; Richter, E.A.; Jaimovich, E.; et al. Cytosolic ROS Production by NADPH Oxidase 2 Regulates Muscle Glucose Uptake during Exercise. Nat Commun 2019, 10, 4623. [Google Scholar] [CrossRef] [PubMed]
- Contreras-Ferrat, A.; Llanos, P.; Vásquez, C.; Espinosa, A.; Osorio-Fuentealba, C.; Arias-Calderon, M.; Lavandero, S.; Klip, A.; Hidalgo, C.; Jaimovich, E. Insulin Elicits a ROS-Activated and an IP₃-Dependent Ca2+ Release, Which Both Impinge on GLUT4 Translocation. J Cell Sci 2014, 127, 1911–1923. [Google Scholar] [CrossRef]
- Maeda, A.; Shirao, T.; Shirasaya, D.; Yoshioka, Y.; Yamashita, Y.; Akagawa, M.; Ashida, H. Piperine Promotes Glucose Uptake through ROS-Dependent Activation of the CAMKK/AMPK Signaling Pathway in Skeletal Muscle. Mol Nutr Food Res 2018, 62, e1800086. [Google Scholar] [CrossRef] [PubMed]
- Favaro, G.; Romanello, V.; Varanita, T.; Andrea Desbats, M.; Morbidoni, V.; Tezze, C.; Albiero, M.; Canato, M.; Gherardi, G.; De Stefani, D.; et al. DRP1-Mediated Mitochondrial Shape Controls Calcium Homeostasis and Muscle Mass. Nat Commun 2019, 10, 2576. [Google Scholar] [CrossRef]
- Willingham, T.B.; Ajayi, P.T.; Glancy, B. Subcellular Specialization of Mitochondrial Form and Function in Skeletal Muscle Cells. Front. Cell Dev. Biol. 2021, 9, 757305. [Google Scholar] [CrossRef]
- Takahashi, M.; Hood, D.A. Protein Import into Subsarcolemmal and Intermyofibrillar Skeletal Muscle Mitochondria. Differential Import Regulation in Distinct Subcellular Regions. J Biol Chem 1996, 271, 27285–27291. [Google Scholar] [CrossRef]
- Ferreira, R.; Vitorino, R.; Alves, R.M.P.; Appell, H.J.; Powers, S.K.; Duarte, J.A.; Amado, F. Subsarcolemmal and Intermyofibrillar Mitochondria Proteome Differences Disclose Functional Specializations in Skeletal Muscle. Proteomics 2010, 10, 3142–3154. [Google Scholar] [CrossRef]
- Csordás, G.; Weaver, D.; Hajnóczky, G. Endoplasmic Reticulum-Mitochondrial Contactology: Structure and Signaling Functions. Trends Cell Biol 2018, 28, 523–540. [Google Scholar] [CrossRef]
- Booth, D.M.; Enyedi, B.; Geiszt, M.; Várnai, P.; Hajnóczky, G. Redox Nanodomains Are Induced by and Control Calcium Signaling at the ER-Mitochondrial Interface. Molecular Cell 2016, 63, 240–248. [Google Scholar] [CrossRef] [PubMed]
- Elander, A.; Sjöström, M.; Lundgren, F.; Scherstén, T.; Bylund-Fellenius, A.C. Biochemical and Morphometric Properties of Mitochondrial Populations in Human Muscle Fibres. Clin Sci (Lond) 1985, 69, 153–164. [Google Scholar] [CrossRef]
- Díaz-Vegas, A.R.; Cordova, A.; Valladares, D.; Llanos, P.; Hidalgo, C.; Gherardi, G.; De Stefani, D.; Mammucari, C.; Rizzuto, R.; Contreras-Ferrat, A.; et al. Mitochondrial Calcium Increase Induced by RyR1 and IP3R Channel Activation After Membrane Depolarization Regulates Skeletal Muscle Metabolism. Front. Physiol. 2018, 9, 791. [Google Scholar] [CrossRef] [PubMed]
- Kwong, J.Q.; Huo, J.; Bround, M.J.; Boyer, J.G.; Schwanekamp, J.A.; Ghazal, N.; Maxwell, J.T.; Jang, Y.C.; Khuchua, Z.; Shi, K.; et al. The Mitochondrial Calcium Uniporter Underlies Metabolic Fuel Preference in Skeletal Muscle. JCI Insight 2018, 3, e121689–121689. [Google Scholar] [CrossRef] [PubMed]
- Groten, C.J.; MacVicar, B.A. Mitochondrial Ca2+ Uptake by the MCU Facilitates Pyramidal Neuron Excitability and Metabolism during Action Potential Firing. Commun Biol 2022, 5, 900. [Google Scholar] [CrossRef]
- Patron, M.; Raffaello, A.; Granatiero, V.; Tosatto, A.; Merli, G.; De Stefani, D.; Wright, L.; Pallafacchina, G.; Terrin, A.; Mammucari, C.; et al. The Mitochondrial Calcium Uniporter (MCU): Molecular Identity and Physiological Roles. Journal of Biological Chemistry 2013, 288, 10750–10758. [Google Scholar] [CrossRef]
- Mammucari, C.; Gherardi, G.; Zamparo, I.; Raffaello, A.; Boncompagni, S.; Chemello, F.; Cagnin, S.; Braga, A.; Zanin, S.; Pallafacchina, G.; et al. The Mitochondrial Calcium Uniporter Controls Skeletal Muscle Trophism in Vivo. Cell Rep 2015, 10, 1269–1279. [Google Scholar] [CrossRef] [PubMed]
- Li, A.; Yi, J.; Li, X.; Zhou, J. Physiological Ca2+ Transients Versus Pathological Steady-State Ca2+ Elevation, Who Flips the ROS Coin in Skeletal Muscle Mitochondria. Front Physiol 2020, 11, 595800. [Google Scholar] [CrossRef]
- Alevriadou, B.R.; Patel, A.; Noble, M.; Ghosh, S.; Gohil, V.M.; Stathopulos, P.B.; Madesh, M. Molecular Nature and Physiological Role of the Mitochondrial Calcium Uniporter Channel. Am J Physiol Cell Physiol 2021, 320, C465–C482. [Google Scholar] [CrossRef]
- Schiaffino, S.; Reggiani, C. Fiber Types in Mammalian Skeletal Muscles. Physiol Rev 2011, 91, 1447–1531. [Google Scholar] [CrossRef]
- Khodabukus, A.; Baar, K. Contractile and Metabolic Properties of Engineered Skeletal Muscle Derived from Slow and Fast Phenotype Mouse Muscle. J Cell Physiol 2015, 230, 1750–1757. [Google Scholar] [CrossRef]
- Loucif, H.; Dagenais-Lussier, X.; Beji, C.; Telittchenko, R.; Routy, J.-P.; van Grevenynghe, J. Plasticity in T-Cell Mitochondrial Metabolism: A Necessary Peacekeeper during the Troubled Times of Persistent HIV-1 Infection. Cytokine Growth Factor Rev 2020, 55, 26–36. [Google Scholar] [CrossRef] [PubMed]
- Picard, M.; Shirihai, O.S.; Gentil, B.J.; Burelle, Y. Mitochondrial Morphology Transitions and Functions: Implications for Retrograde Signaling? Am J Physiol Regul Integr Comp Physiol 2013, 304, R393–406. [Google Scholar] [CrossRef] [PubMed]
- Glancy, B.; Hartnell, L.M.; Malide, D.; Yu, Z.-X.; Combs, C.A.; Connelly, P.S.; Subramaniam, S.; Balaban, R.S. Mitochondrial Reticulum for Cellular Energy Distribution in Muscle. Nature 2015, 523, 617–620. [Google Scholar] [CrossRef] [PubMed]
- Quezada, E.R.; Díaz-Vegas, A.; Jaimovich, E.; Casas, M. Changes in Gene Expression of the MCU Complex Are Induced by Electrical Stimulation in Adult Skeletal Muscle. Front Physiol 2020, 11, 601313. [Google Scholar] [CrossRef]
- Horn, A.; Van der Meulen, J.H.; Defour, A.; Hogarth, M.; Sreetama, S.C.; Reed, A.; Scheffer, L.; Chandel, N.S.; Jaiswal, J.K. Mitochondrial Redox Signaling Enables Repair of Injured Skeletal Muscle Cells. Sci Signal 2017, 10, eaaj1978. [Google Scholar] [CrossRef] [PubMed]
- Crochemore, C.; Mekki, M.; Corbière, C.; Karoui, A.; Noël, R.; Vendeville, C.; Vaugeois, J.-M.; Monteil, C. Subsarcolemmal and Interfibrillar Mitochondria Display Distinct Superoxide Production Profiles. Free Radic Res 2015, 49, 331–337. [Google Scholar] [CrossRef]
- Debattisti, V.; Horn, A.; Singh, R.; Seifert, E.L.; Hogarth, M.W.; Mazala, D.A.; Huang, K.T.; Horvath, R.; Jaiswal, J.K.; Hajnóczky, G. Dysregulation of Mitochondrial Ca2+ Uptake and Sarcolemma Repair Underlie Muscle Weakness and Wasting in Patients and Mice Lacking MICU1. Cell Rep 2019, 29, 1274–1286. [Google Scholar] [CrossRef]
- Baughman, J.M.; Perocchi, F.; Girgis, H.S.; Plovanich, M.; Belcher-Timme, C.A.; Sancak, Y.; Bao, X.R.; Strittmatter, L.; Goldberger, O.; Bogorad, R.L.; et al. Integrative Genomics Identifies MCU as an Essential Component of the Mitochondrial Calcium Uniporter. Nature 2011, 476, 341–345. [Google Scholar] [CrossRef]
- Eisner, V.; Lenaers, G.; Hajnóczky, G. Mitochondrial Fusion Is Frequent in Skeletal Muscle and Supports Excitation-Contraction Coupling. J Cell Biol 2014, 205, 179–195. [Google Scholar] [CrossRef] [PubMed]
- Del Campo, A.; Contreras-Hernández, I.; Castro-Sepúlveda, M.; Campos, C.A.; Figueroa, R.; Tevy, M.F.; Eisner, V.; Casas, M.; Jaimovich, E. Muscle Function Decline and Mitochondria Changes in Middle Age Precede Sarcopenia in Mice. Aging (Albany NY) 2018, 10, 34–55. [Google Scholar] [CrossRef] [PubMed]
- Fealy, C.E.; Grevendonk, L.; Hoeks, J.; Hesselink, M.K.C. Skeletal Muscle Mitochondrial Network Dynamics in Metabolic Disorders and Aging. Trends Mol Med 2021, 27, 1033–1044. [Google Scholar] [CrossRef] [PubMed]
- Tezze, C.; Romanello, V.; Desbats, M.A.; Fadini, G.P.; Albiero, M.; Favaro, G.; Ciciliot, S.; Soriano, M.E.; Morbidoni, V.; Cerqua, C.; et al. Age-Associated Loss of OPA1 in Muscle Impacts Muscle Mass, Metabolic Homeostasis, Systemic Inflammation, and Epithelial Senescence. Cell Metab 2017, 25, 1374–1389. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Nuevo, A.; Díaz-Ramos, A.; Noguera, E.; Díaz-Sáez, F.; Duran, X.; Muñoz, J.P.; Romero, M.; Plana, N.; Sebastián, D.; Tezze, C.; et al. Mitochondrial DNA and TLR9 Drive Muscle Inflammation upon Opa1 Deficiency. EMBO J 2018, 37, e96553. [Google Scholar] [CrossRef] [PubMed]
- Yeo, D.; Kang, C.; Gomez-Cabrera, M.C.; Vina, J.; Ji, L.L. Intensified Mitophagy in Skeletal Muscle with Aging Is Downregulated by PGC-1alpha Overexpression in Vivo. Free Radic Biol Med 2019, 130, 361–368. [Google Scholar] [CrossRef] [PubMed]
- Alizadeh Pahlavani, H.; Laher, I.; Knechtle, B.; Zouhal, H. Exercise and Mitochondrial Mechanisms in Patients with Sarcopenia. Front Physiol 2022, 13, 1040381. [Google Scholar] [CrossRef]
- Yang, Y.-F.; Yang, W.; Liao, Z.-Y.; Wu, Y.-X.; Fan, Z.; Guo, A.; Yu, J.; Chen, Q.-N.; Wu, J.-H.; Zhou, J.; et al. MICU3 Regulates Mitochondrial Ca2+-Dependent Antioxidant Response in Skeletal Muscle Aging. Cell Death Dis 2021, 12, 1115. [Google Scholar] [CrossRef]
- Altamirano, F.; López, J.R.; Henríquez, C.; Molinski, T.; Allen, P.D.; Jaimovich, E. Increased Resting Intracellular Calcium Modulates NF-ΚB-Dependent Inducible Nitric-Oxide Synthase Gene Expression in Dystrophic Mdx Skeletal Myotubes. J Biol Chem 2012, 287, 20876–20887. [Google Scholar] [CrossRef]
- Mechler, F.; Imre, S.; Dioszeghy, P. Lipid Peroxidation and Superoxide Dismutase Activity in Muscle and Erythrocytes in Duchenne Muscular Dystrophy. J Neurol Sci 1984, 63, 279–283. [Google Scholar] [CrossRef]
- Łoboda, A.; Dulak, J. Nuclear Factor Erythroid 2-Related Factor 2 and Its Targets in Skeletal Muscle Repair and Regeneration. Antioxid Redox Signal 2023, 38, 619–642. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Qu, H.; Chen, Y.; Luo, X.; Chen, C.; Xiao, B.; Ding, X.; Zhao, P.; Lu, Y.; Chen, A.F.; et al. Atorvastatin Induces Mitochondria-Dependent Ferroptosis via the Modulation of Nrf2-XCT/GPx4 Axis. Front Cell Dev Biol 2022, 10, 806081. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Wu, B.; Shen, D.; Chen, J.; Yu, Z.; Chen, C. Ferroptosis in a Sarcopenia Model of Senescence Accelerated Mouse Prone 8 (SAMP8). Int J Biol Sci 2021, 17, 151–162. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Energy saving design of mitochondria activation of ATP that minimizes mitochondria ROS production. Under resting conditions, skeletal muscle has different pools of mitochondria, which have a high mitochondrial potential and a basal rate of ROS generation from superoxide anion generated by the electron transport chain. SSM have high expression of MICU1, which prevents calcium from massively entering the mitochondrial space. Thus, the transporter chain allows the "leakage of electrons" and generates ROS. A condition of exercise implies that the SSM and the ITM fuse, generating a network characterized by lower density of MICU1, high density of MCU, and a maximal proton gradient needed to generate ATP, which finally allows a low ROS generation.
Figure 1.
Energy saving design of mitochondria activation of ATP that minimizes mitochondria ROS production. Under resting conditions, skeletal muscle has different pools of mitochondria, which have a high mitochondrial potential and a basal rate of ROS generation from superoxide anion generated by the electron transport chain. SSM have high expression of MICU1, which prevents calcium from massively entering the mitochondrial space. Thus, the transporter chain allows the "leakage of electrons" and generates ROS. A condition of exercise implies that the SSM and the ITM fuse, generating a network characterized by lower density of MICU1, high density of MCU, and a maximal proton gradient needed to generate ATP, which finally allows a low ROS generation.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



