
Submitted:
30 May 2023
Posted:
31 May 2023
You are already at the latest version
Abstract
The present article describes a one-pot and cascade mode process using biocompatible/biodegradable reagents, for directly and simply obtaining surfactant compositions of mixtures of D-mannuronic acid and L-guluronic acid directly from oligoalginates or semi-refined alginates (mixtures of alginate, cellulose, hemicellulose, laminaran and fucan). Simple treatments of partial purification of the reaction crudes (elimination of the salts and/or the residual fatty alcohols) or isolation of the surfactant compositions result in derived compounds and in compositions having performance levels appropriate to applications in detergency. In addition, the challenging extension of this cascading one-pot synthesis technology to crude milled brown seaweeds was successfully carried out to provide promising surface-active compositions made up of alkyl uronate and alkyl glycoside monosaccharides.
Keywords:
oligomannuronate
; oligoguluronate
; oligoalginate
; semi-refined alginate
; brown seaweed
; one pot cascade process
; uronic acid-based surfactants
; readily biodegradability
; non-ecotoxicity
1. Introduction
100% biobased surfactants containing both a hydrophobic tail and a hydrophilic head of plant origin, currently represent 5% of the total surfactants market worldwide [1]. This is the most dynamic segment, with growth of more than 20% per year and a high penetration rate in Europe. The introduction of increasingly strict regulatory standards relating to environmental impact and effects on human health should open up new opportunities for these products. On the other hand, the increased demand of consumers for more environmentally friendly products is leading industries using surfactants to use products certified by a label (Ecolabel, COSMOS) which guarantees compliance with this requirement.
100% Biobased surfactants on the market mainly include products derived from sugars, which belong to the non-ionic family. They incorporate a glucose head for AlkylPolyGlucosides (APGs), sucrose for sucroesters and sorbitan for sorbitan esters [2-4]. Depending on the number of carbon atoms making up the lipophilic chain (4 to 22), these surfactants may have hydrotropic, foaming, degreasing, wetting, foam and viscosity boosting, emulsifying properties and may lead to good sensory properties. Sugar-derived anionic surfactants are present on the market to a much smaller extend compared with their non-ionic homologues. An alkyl polyglucoside carboxylate, Plantapon LGC Sorb (INCI name sodium lauryl glucose carboxylate (and) lauryl glucoside) has been introduced onto the market by Cognis as a new anionic surfactant for applications in bodycare formulations [4]. An industrial process based on the reaction of sodium monochloroacetate in an aqueous solution of alkyl polyglycoside (without additional solvent) has been developed in this context. Nevertheless, these marketed products do not allow to fulfil all the functions of the non-ionic surfactants derived from ethylene oxide, and even less those of the anionic surfactants which represent the most important part of the detergent market. On the other hand, the cost of renewable raw materials and the relative complexity of the manufacturing processes lead to an additional cost compared to traditional surfactants. There is a great need for creativity in transforming existing raw materials and developing new ones [5-8], with a certain number of technological obstacles to be overcome: the high oxygen content of plant-based products, which makes chemical transformation more difficult to achieve, the difficulty in identifying commercially available natural anionic substrates that can be easily transformed into the corresponding anionic surfactants, etc.
The use of starting materials based on algal polysaccharides which are characterized by an original chemical functionality compared with polysaccharides from terrestrial plants, constitutes an approach that could make it possible to broaden the fields of application of the 100% biobased surfactants. Alginates which are polysaccharides present in the cell wall of brown algae, are produced on a scale of 30,000 tonnes worldwide; their use has several advantages from the point of view of environmental impact, since their production does not require agricultural land, fresh water or fertilisers and phytosanitary products [9]. These polysaccharides are formed by (1->4) glycosidic bonds between the two monomers, D-mannuronate (M) and its C-5 epimer L-guluronate (G). These M and G units are arranged in an irregular manner, by homopolymeric blocks (MM or GG) separated by heterogenous blocks of both uronates (MG) along the alginate chain. Some studies have already shown the possibility of exploiting D-mannuronate oligomers in the surfactant field. However the preparation of compositions based on monomeric mixtures of both L-guluronate and of D-mannuronate units allowing the valorisation of all the sugars present in the biopolymer structure, has not been developed to date.
Alkyl D-mannopyranosiduronate surfactants have been produced from D-mannuronic acid oligomers through one-pot acid glycosidic bond hydrolysis, esterification and stereocontrolled Fisher glycosylation in butanol followed by transesterification/transglycosylation processes in fatty alcohols [10]. These non-ionic surfactants having identical lipophilic chains, can subsequently be saponified in order to obtain single tailed anionic surfactants including a carboxylic unit. These oligomannuronate-derived amphiphiles exhibit attractive surface tension and foaming properties. However, the exclusive use of mannuronate oligomers as starting materials for the production of anionic uronate surfactants is very restrictive and the development of new processes allowing a valorisation of the entire alginate and possibly additional polysaccharides present in the algal cell wall is essential to facilitate the industrial transfer of these innovative products.
Within this context, we investigated a novel strategy to synthesize surfactant compositions directly from less refined starting materials to reduce costs and to improve the properties expected in the surfactants field. The first challenge was to identify reaction conditions that enables the one-pot solvent-free transformation of oligoalginates (composed of both M and G units) or semi-refined alginates (mixtures mainly composed of alginate, cellulose and fucan) into compositions that contain monomeric surfactants in the form of the two uronic acids (L-guluronic acid and D-mannuronic acid), and hexoses and pentoses derived from other polysaccharides present in the algal extract (semi-refined alginates). In addition, our objective was to propose an eco-friendly process for the direct conversion of raw algae into n-alkyl glycosiduronic acids and n-alkyl glycosides without the use of organic solvents, i.e. by simultaneously carrying out the steps of extraction, depolymerisation by acid hydrolysis with a minimum of water and esterification/glycosylation. The carboxylic acid-based surfactant compositions produced through these procedures will be evaluated in terms of surface activities, biodegradability and ecotoxicity.
Figure 1.
Transformation of poly(oligo)alginates or dried milled brown seaweeds into D-mannuronate and L-guluronate-containing surfactant compositions.
Figure 1.
Transformation of poly(oligo)alginates or dried milled brown seaweeds into D-mannuronate and L-guluronate-containing surfactant compositions.
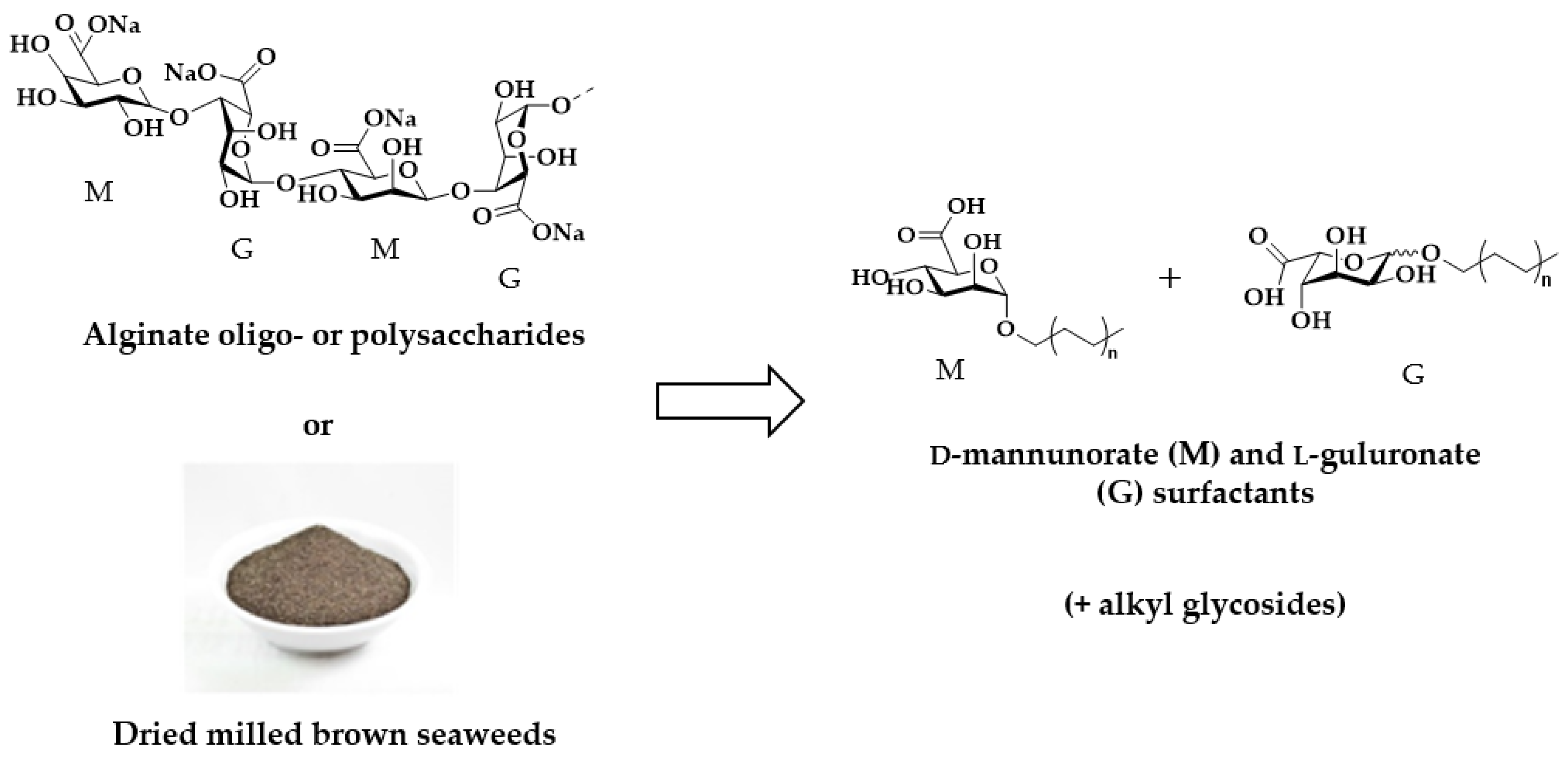
2. Results
2.1. Preparation of starting materials
Poly(oligo)mannuronates and poly(oligo)guluronates are attractive raw materials for the development of original 100% biobased surfactants. They are obtained from fresh or dry algae derived from Laminaria digitata using a process based on pre-extraction of the alginates, followed by several steps of precipitation by modulating the pH of the reaction medium in order to separate the G blocks and the M blocks constituting the alginate [10,11]. Finally, an acid hydrolysis step affords the oligomannuronate (DP = 4: Table 1) or oligoguluronate (DP = 30: Table 2).
Concerning oligoalginate (OAlg) and semi-refined alginate (s-r Alg) [12], the procedure involves acid leaching of fresh or dry algae derived from Laminaria digitata, followed by dissolution of the sodium alginates by increasing the pH of the medium and then solid/liquid separation in order to remove the algal residues. At this stage, the liquid fraction can be frozen and freeze-dried and constitutes the semi-refined alginates (Table 4) in the form of sodium alginates. In order to obtain refined oligoalginates, a purification is introduced into the preceding steps. After separation of the algal residues, the latter purification step includes or consists of precipitation of the alginic acid by reducing the pH, followed by several washes with acidic water in order to remove the co-products. Increasing the pH with Na2CO3 makes it possible to again dissolve the sodium alginates while limiting the salt, compared with the use of sodium hydroxide. Finally the alginate solution is treated with acid in order to reduce the degree of polymerisation which affords after a step of freezing and then freeze-drying the oligoalginates of DP 12.7 (Table 3).
Regarding the dried milled brown seaweeds, Asco T10 from Ascophyllum nodosum species (Thorverk, Iceland) were used.
2.2. Synthesis from oligoalginates
In order to achieve the goal of synthesising novel biobased uronic surfactants from alginates, several uronate sources have been investigated to assess their potential as biomass feedstock for the preparation of such amphiphilic molecules through green synthetic pathways. At first, oligomannuronate (OM) and oligoguluronate (OG) were used in order to define the optimal reaction conditions for the one-pot cascade synthesis of uronate monomeric surfactants. Then the optimised conditions were transposed to oligoalginate with a M/G ratio of 1.4 and a DP of 12.7.
2.2.1. Synthesis from oligomannuronate (OM)
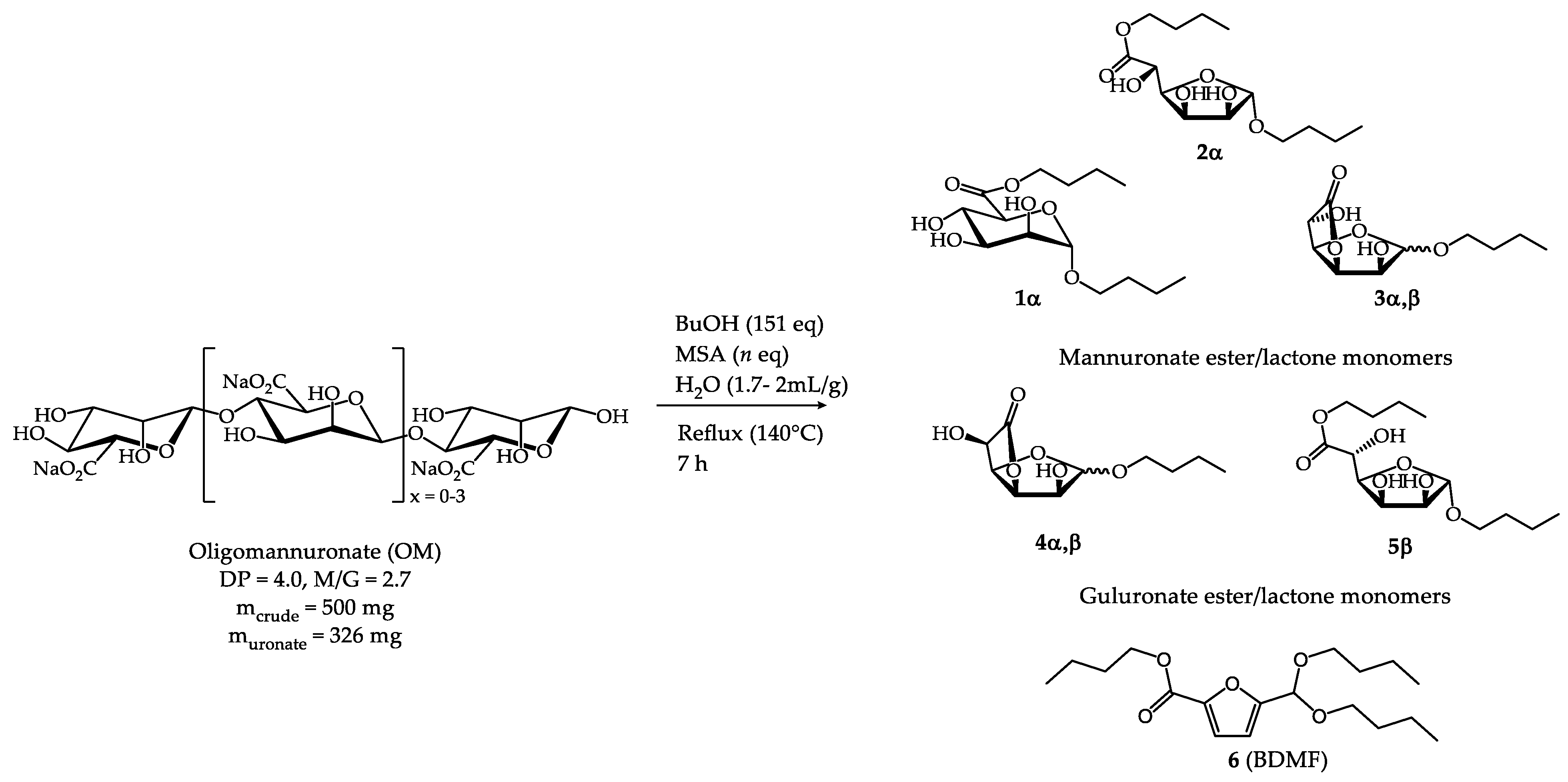
In the first stage of the study, the reactivity of sodium oligomannuronate (M/G = 2.7, DP = 4) was investigated towards acid hydrolysis, esterification and Fisher glycosidation conditions. Acid treatment of OM was performed for 7 h with various amounts of readily biodegradable methane sulfonic acid (MSA) in the presence of water and butanol (Scheme 1, Table 6). Based on previous results obtained in the laboratory [10], the quantity of butanol was set at 151 eq. Indeed, the use of larger quantities, in addition to posing a problem of oversizing the production facilities, did not allow for a significant improvement in yield. However, whereas previously the reactions were envisaged in dry butanol [10], in these new experiments it was found that the solubility of the oligomannuronate was low, thus preventing the reactions from carrying out. The addition of water at a rate of 1.7-2 mL/g of algal extract proved sufficient to allow the solubilisation of the oligomannuronate in the reaction mixture. The butanol was distilled out during heating and the water initially added in the reaction mixture and formed during the reaction was eliminated through the use of a Dean-Stark apparatus (140°C). After neutralisation and work up, the organic residue was subjected to silica gel column chromatography that allowed to isolate fractions enriched in a few compounds which facilitated the identification of the glycoside-esters present in the mixture by 1H NMR studies (Figures S1-S4, Supplementary materials). Indeed, 1H NMR analysis led to the identification of characteristic signals for each compound, as shown in Table 5. Thus, the formation of (n-butyl) n-butyl α-D-mannopyranosiduronate 1α was observed as the major monosaccharide product (52 mol%, isolated as a pure fraction, 33% yield) in addition to a mixture of isomers: (n-butyl) n-butyl α-D-mannofuranosiduronate 2α (7 mol%), n-butyl α-D-mannofuranosidurono-6,3-lactone 3α (11 mol%) and n-butyl β-D-mannofuranosidurono-6,3-lactone 3β (13 mol%). Products from the guluronate units present in smaller quantities in the raw material (M/G ratio = 2.7), were also identified, i.e. n-butyl-α-L-gulofuranosidurono-6.3-lactone 4α (5 mol%), n-butyl-β-L-gulofuranosidurono-6.3-lactone 4β (5 mol%) and (n-butyl) n-butyl-β-L-gulofuranosiduronate 5β (3 mol%). Alongside these uronate derivatives, a side product was isolated, the butyl-5-(dibutoxymethyl)-2-furoate (BDMF) 6 (5 mol%), and it was characterised by NMR (Scheme 1). This furan compound results from the dehydration of butyl uronates as reported in earlier studies [13]. Total amount of the butylated Man and Gul derivatives 1-5 is quite satisfactory but it was noticed that the increase in MSA quantity would further degrade the butyl uronates. The overall yield could not be calculated as the isomers possess different molecular weights. It should be noted that 99% or 70% MSA can be used indiscriminately since the same results were obtained in both cases.
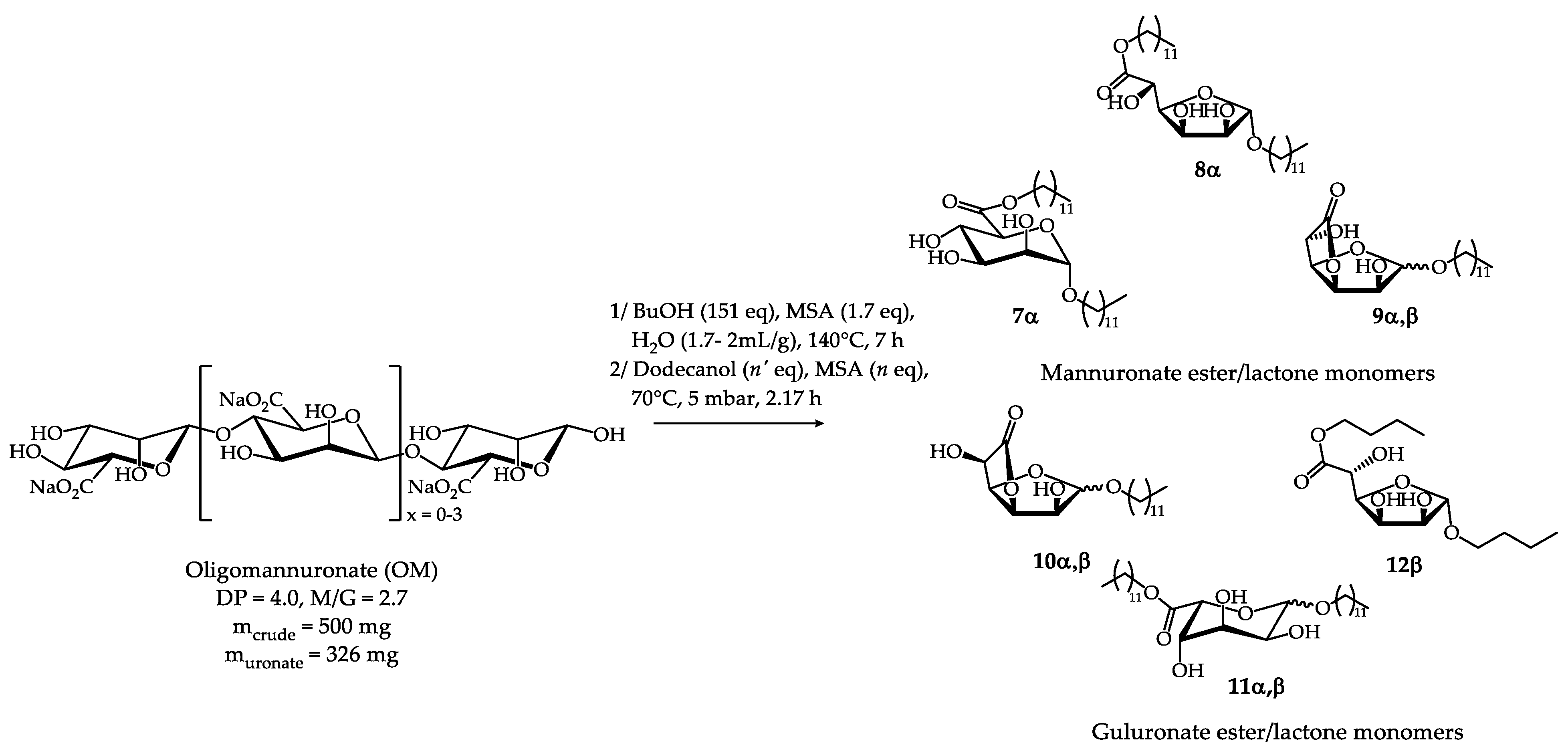
Next, seeing the possible transformation of oligomannuronates into monomeric derivatives, the optimal conditions (1.7 eq MSA, 151 eq BuOH, H2O (1.7-2 mL/g of crude OM), 140°C, 7 h) were reproduced and the reaction mixture containing the butyl ester glycoside products 1-5 was subjected directly to simultaneous transesterification and transglycosylation with dodecanol (DodOH). Then the replacement of the butyl chains by the C12 chains was performed in situ (Scheme 2) under reduced pressure (5 mbar) at 70 °C for 2.17 h after the addition of an extra equivalent of MSA (99% or 70%). The reduced pressure allows the removal of the butanol and shifts the equilibrium towards the transesterification and transglycosylation products. A variety of reaction conditions in terms of dodecanol and MSA quantities used were envisaged (Table 8). After work up of the medium and two successive purifications by column chromatography (the first one allowed to isolate the mixture of uronate monomers and the second one aimed at obtaining fractions enriched in a few compounds which facilitated the identification of the glycoside-esters present in the mixture by 1H NMR studies), (n-dodecyl) n-dodecyl α-D-mannopyranosiduronate 7α was isolated in addition to several fractions containing mixtures of isomers corresponding to (n-dodecyl) n-dodecyl α-D-mannofuranosiduronate 8α, n-dodecyl α,β-D-mannofuranosidurono-6,3-lactone 9α,β , n-dodecyl-α,β-L-gulofuranosidurono-6.3-lactone 10α,β, (n-dodecyl) n-dodecyl-α,β-L-gulopyranosiduronate 11α,β and (n-dodecyl) n-dodecyl-β-L-gulofuranosiduronate 12β (Scheme 2) (Figures S5-S8, Supplementary materials)
The comparison of entries 1 and 2 (Table 7) indicates that the use of dodecanol in large excess does not improve the yield. Finally, the comparison of entries 2 and 3 shows that the decrease in the number of equivalents of MSA introduced for the second step disadvantages the formation of (n-dodecyl) n-dodecyl α-D-mannopyranosiduronate 7α. The reaction conditions of entry 2 (4 eq DodOH, 1 eq MSA) that provided dodecyl mannuronate 7α with an overall yield of 25%, were thus selected. The molar composition of the mannuronate and guluronate mixture was determined by integrating the characteristic 1H NMR signals of each compound of the uronate mixture (Table 8) resulting from the first column chromatography (Figures S5,S6, Supplementary materials): (n-dodecyl) n-dodecyl α-D-mannopyranosiduronate 7α (48 mol%), (n-dodecyl) n-dodecyl α-D-mannofuranosiduronate 8α (9 mol%), n-dodecyl α-D-mannofuranosidurono-6,3-lactone 9α (5 mol%), n-dodecyl β-D-mannofuranosidurono-6,3-lactone 9β (22 mol%), n-dodecyl-α-L-gulofuranosidurono-6.3-lactone 10α (3 mol%), n-dodecyl-β-L-gulofuranosidurono-6.3-lactone 10β (3 mol%), (n-dodecyl) n-dodecyl-β-L-gulopyranosiduronate 11β (5 mol%) and (n-dodecyl) n-dodecyl-β-L-gulofuranosiduronate 12β (6 mol%). It is noteworthy that (n-dodecyl) n-dodecyl-α-L-gulopyranosiduronate 11α was not detectable in this mixture due to its presence in too small quantities.
The next challenge was to develop a one-pot three-step cascade for the synthesis of uronate surfactants possessing a single alkyl chain (Scheme 3). For this purpose, saponification (0.4N NaOH, 3 eq, 70°C, 1 h) was carried out directly in the reaction mixture obtained after the transesterification/transglycosylation step. Removal of dodecanol was performed at the end of the process through the removal of water by freeze-drying and the addition of EtOAc into the reaction medium followed by a filtration step. A precipitate composed of the uronate derivatives and additional salts was isolated whereas the filtrate contained the entire fatty alcohol. This solid fraction containing the desired products consists of 96-97% dry matter and 36-37% mineral matter for repeated runs (thermogravimetric analyses). The large amount of mineral matter comes from the raw material used, which already contained it (31.9% dry/crude), and from NaOH used for the saponification reaction. As the significant amount of mineral matter present in the sodium uronate composition is likely to modify the properties of the surfactants, a purification method allowing their removal was achieved through a liquid-liquid extraction using a 1N HCl aqueous phase and EtOAc. After concentration of the organic phase under reduced pressure, mannuronate and guluronate surfactants were isolated as carboxylic acids and lactones. 1H NMR analysis revealed the presence of n-dodecyl α-D-mannopyranosiduronic acid 13α (47 mol%), n-dodecyl α-D-mannofuranosidurono-6,3-lactone 9α (4 mol%), n-dodecyl β-D-mannofuranosidurono-6,3-lactone 9β (12% molar), n-dodecyl β-L-gulopyranosiduronic acid 14β (4 mol%), n-dodecyl α-L-gulofuranosidurono-6,3-lactone 10α (9 mol%), n-dodecyl β-L-gulofuranosidurono-6,3-lactone 10β (9 mol%) and n-dodecyl-β-L-gulofuranosiduronic acid 15β (4 mol%) in addition to a non-identified product X (~10 mol%) which could correspond to n-dodecyl D-mannofuranosiduronic acid (Figures S9,S10, Supplementary materials). 1H NMR analysis led to the identification of characteristic signals for each compound, as shown in Table 9. The surface tension measurements obtained with this H-C12 Man composition are presented in section 2.5.
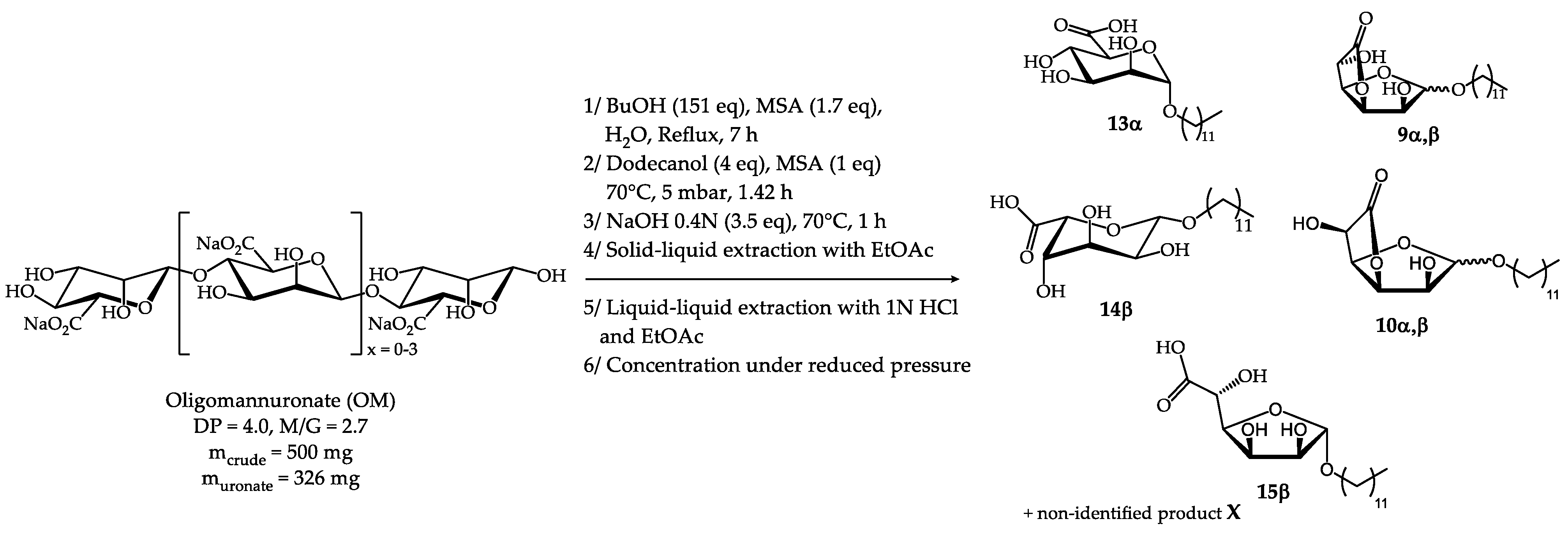
2.2.2. Synthesis from oligoguluronate (OG)
As the alginate biomass selected, either oligoalginates or semi-refined alginates, is composed of both mannuronate and guluronate units, the previously developed synthetic pathway had to be tested from oligoguluronate (OG) as well. It is noteworthy that more MSA (2.2 eq) was required (Schema 4) for OG probably due to the α-binding between guluronate units and/or their higher DP. The same workup based on a solid-liquid extraction step with EtOAc was performed to eliminate dodecanol. This uronate-based mixture was then acidified and purified by liquid-liquid extraction in order to obtain the H-C12 derivatives and to remove the salts. 1H NMR analysis revealed the presence of the following products: n-dodecyl α-D-mannopyranosiduronic acid 13α (15 mol%), n-dodecyl β-L-gulopyranosiduronic acid 14β (13 mol%), n-dodecyl α-L-gulofuranosidurono-6,3-lactone 10α (26 mol%), n-dodecyl β-L-gulofuranosidurono-6,3-lactone 10β (26 mol%) and n-dodecyl-β-L-gulofuranosiduronic acid 15β (21 mol%). The presence of n-dodecyl α,β-D-mannofuranosidurono-6,3-lactone 9α,β was also observed in trace amounts but its quantification was not possible unlike the case of synthesis from oligoalginate (See section 2.2.3). 1H NMR analysis led to the identification of characteristic signals for each compound, as shown in Table 10 (Figures S11,S12, Supplementary materials). The surface tension measurements obtained with this H-C12 Gul composition are presented in section 2.5.
2.2.3. Synthesis from oligoalginate (OAlg)
As the process has been shown to be applicable to both D-mannuronate and L-guluronate units, the same synthetic pathway has been tested with oligoalginate (OAlg) composed of D-Man and L-Gul units with the same reaction conditions used for oligoguluronate (OG) (Schema 5). The OAlg starting raw material is economically advantageous as less purification is required in comparison to OM and OG. The mixture obtained after the saponification step contains 93.4% dry matter and 41.8% mineral matter. The higher percentage of mineral matter than for oligomannuronate and oligoguluronate is explained by the higher mineral content of the raw material (44.3 %dry/crude). Further acidic treatment of the reaction medium was performed as developed for the OM and OG raw materials (Schema 5). The final H-C12 OAlg surfactant composition is characterised by the presence of n-dodecyl α-D-mannopyranosiduronic acid 13α (23 mol%), n-dodecyl α-D-mannofuranosidurono-6,3-lactone 9α (4 mol%), n-dodecyl β-D-mannofuranosidurono-6,3-lactone 9β (9 mol%), n-dodecyl β-L-gulopyranosiduronic acid 14β (9 mol%), n-dodecyl α-L-gulofuranosidurono-6,3-lactone 10α (20 mol%), n-dodecyl β-L-gulofuranosidurono-6,3-lactone 10β (20 mol%) and n-dodecyl-β-L-gulofuranosiduronic acid 15β (9 mol%). The presence of the non-identified product X (6 mol%) formed during the one-pot synthesis of the H-C12 Man composition was also observed. 1H NMR analysis led to the identification of characteristic signals for each compound, as shown in Table 11 (Figures S13,S14, Supplementary materials). The surface tension measurements obtained with this H-C12 OAlg composition are presented in section 2.5.
Scheme 5.
One pot and cascade mode synthesis of H-C12 OAlg surfactant composition.
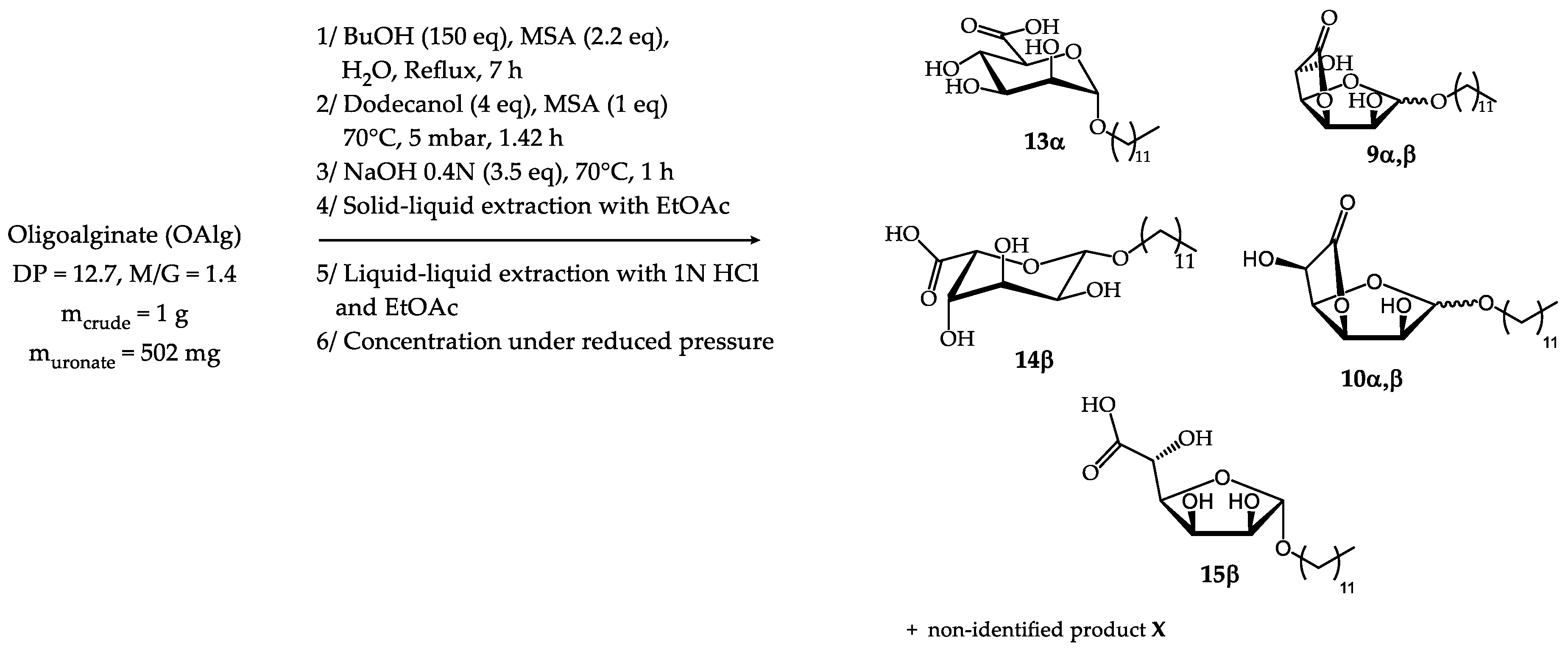
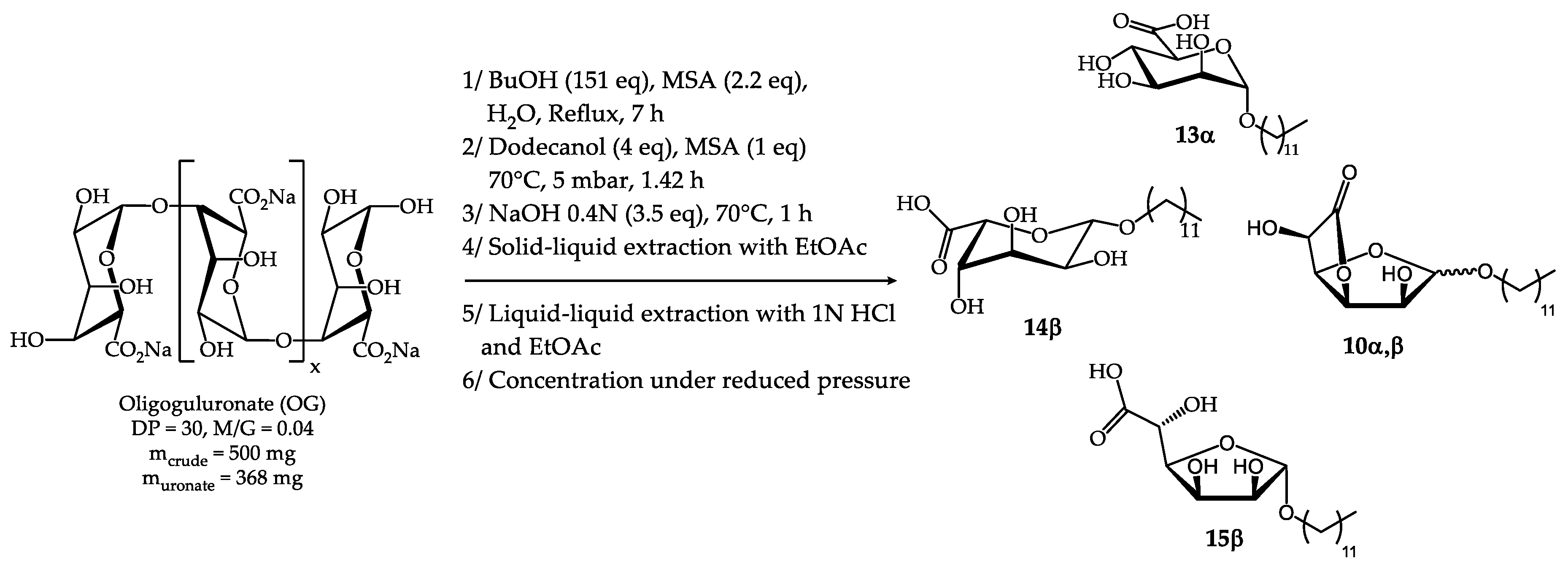
2.3. Synthesis from semi-refined alginate (s-r Alg)
In order to further reduce the cost of surfactant production, the process was applied to a semi-refined alginate (s-r Alg) extracted from Laminaria digitata species composed of D-mannuronate, L-guluronate units in addition to neutral L-fucose, D-glucose and D-xylose sugars. The first step of the synthetic scheme developed for oligosaccharides could not be directly applied. Indeed, modifications had to be made to take into account the low solubility of the mixture in butanol due to the high degree of polymerisation of the polysaccharides. Thus, the synthesis of the butyl C4-C4 derivatives was divided into two steps. First, 2 g of semi-refined alginate containing 810 mg of saccharidic materials was dispersed in water (60 mL) under reflux in the presence of acid (MSA, 5 eq) to reduce the degree of polymerisation. After 8 h reflux, butanol (152 eq) was added and the Dean-Stark set-up was used to allow the removal of water by azeotropic distillation (15 h). The overall reaction time (23 h) and the quantity of MSA (5 eq) were increased to optimise the conversion rate (Scheme 6). Then, transglycosylation and transesterification reactions with dodecanol were carried out in the same pot. The influence of the MSA amount added in the second step was studied. It was found that a better yield was obtained when no additional acid was added. Indeed, with more MSA there were more degradation products to be formed. As for the oligosaccharides, the one-pot saponification step was achieved with the mixture resulting from the second step. The concentration of the NaOH solution used was decreased (0.2N NaOH, 3 eq, 70°C, 1 h) in order to have a larger quantity of aqueous phase and thus a better dispersion of the organic phase. Water was then eliminated by freeze-drying. Solid-liquid extraction with EtOAc allowing the removal of dodecanol was evaluated from this raw material. Unfortunately, a loss of n-dodecyl α,β D-glucopyranosides 16 and n-dodecyl fucosides derivatives 17,18 was observed. As these compounds are not in salt form like the uronate derivatives, they were solubilised by EtOAc and carried along with the dodecanol in the filtrate. In order to limit the loss of these products of interest, other solvents were tested: isopropanol, anisole, acetone, methyl isobutyl ketone, methyl ethyl ketone, 2-methyltetrahydrofuran, heptane, cyclohexanone and acetonitrile. The only solvents that did not allow the non-ionic derivatives to be lost completely were acetone and acetonitrile. The best results were obtained with the latter. However, in the interest of environmental compatibility, acetone was finally chosen. The isolated mixture contains 98.5% dry matter and 40.7% mineral matter. At this stage, due to the difficulty of potential scale-up and the risk of emulsion formation, an alternative method for the final purification was developed. The mixture obtained after solid-liquid extraction with acetone was first dissolved in ice-cold water and then acidified with an oxalic acid solution to a pH of about 2. After concentration by freeze-drying, the mixture was taken up in acetone. The products were solubilised while the salts precipitated. After filtration, the products were recovered in the filtrate. Oxalic acid was chosen for its eco-compatibility and its pKa1 of 1.25, which allows the sodium carboxylate function of the uronate derivatives (pKa ≈ 4) to be protonated without the risk of also protonating the sodium methane sulfonate (pKa = - 1.92). It is also to avoid finding methane sulfonic acid in the filtrate that the pH of the aqueous solution should not be decreased below 2. This cascading one-pot method, including a final purification step is applicable on an industrial scale and it allows to eliminate the fatty alcohols in addition to the salts. 1H NMR analysis of the H-C12 s-r Alg composition revealed the presence of the following products (Table 12): n-dodecyl α-D-mannopyranosiduronic acid 13α (23 mol%), n-dodecyl α-D-mannofuranosidurono-6,3-lactone 9α (5 mol%), n-dodecyl β-D-mannofuranosidurono-6,3-lactone 9β (13 mol%), n-dodecyl β-L-gulopyranosiduronic acid 14β (11 mol%), n-dodecyl β-L-gulofuranosidurono-6,3-lactone 10β (20 mol%), n-dodecyl-α-L-fucopyranosides 17α (5 mol%), n-dodecyl-β-L-fucopyranosides 17β (8 mol%) and n-dodecyl-α,β-L-fucofuranosides 18α,β (15 mol%) (Figures S15,S16, Supplementary materials). 1H NMR study of the C12 s-r Alg composition did not allow to identify signals corresponding to n-dodecyl α,β D-glucopyranosides 16. Furthermore, a significant amount of n-dodecyl α,β D-glucopyranosides 16 and n-dodecyl fucosides derivatives 17,18 was solubilised in acetone which means that the quantity of alkyl glycosides present in the final surfactant compositions has decreased significantly following the purification steps.
2.4. Synthesis from crude brown seaweeds
The final challenge was to extend the cascading one-pot synthesis technology developed from oligo- and polysaccharide alginates to commercial milled brown seaweeds rich in alginate and fucoidan polysaccharides (Asco T10 from Ascophyllum nodosum species, Thorverk, Iceland). Indeed, the use of such crude seaweeds could contribute to lowering the production cost of surfactant compositions due to the reduced price of these raw materials compared to refined or semi-refined polysaccharides and to a simplification of the process which no longer requires prior extraction of alginates.
The process was developed on a 100 g scale of crude seaweeds in a 1 litre reactor. It was shown that it was possible to combine the depolymerisation, extraction and butanolysis steps simultaneously by immersing the milled algae in the butanol solution in the presence of the acid catalyst [14]. In this case, it was observed that the addition of water was not necessary and that the residual moisture of the algae (13 wt%) was sufficient to carry out the depolymerisation. In practice, the algae behave like sponges by absorbing the butanol. This optimisation made it possible to reduce the amount of butanol, to avoid adding water and to dispense with the Dean-Stark apparatus that was previously required. A standard distillation set-up was used, which greatly simplified the scaling up of the process. Thus, milled seaweeds (100 g) were stirred in butanol (280 mL) at 135°C for 7 h in the presence of MSA 70% (30 g). The resulting mixture composed of dibutylated uronates (alginate) and butylated fucosides (fucoidan) was then filtered on sintered glass to eliminate the insoluble materials such as salts or fibres contained in the seaweeds (Figure 2).
The second step of the process based on transesterification/transglycosylation reactions led to the replacement of butyl chains by longer chains using a fatty alcohol as well as the recycling of butanol through a vacuum distillation. Octanol was selected as the fatty alcohol to obtain C8-surfactants. The advantage of this alcohol was its liquid state at room temperature which facilitated its manipulation during the process, and also its capacity to be eliminated by distillation. The C8-C8 derivatives were then obtained after a stirring for 7 h at 70°C under reduced pressure. The anionic charge of the uronate derivatives was recovered after a saponification step with a 5N aqueous solution of NaOH during 1 h at 65°C. The salts and the excess of alcohol were then removed from the final mixture. For that purpose, residual water was first eliminated by evaporation under reduced pressure, followed by an acidification of the medium through the addition of concentrated sulfuric acid to a pH value of about 2-3. This acidification step solubilised the uronate and fucoside derivatives in the residual alcohol and precipitated the salts. Thus, the salts were easily removed by filtration in addition to other insolubles in octanol. Finally, the oily filtrate was treated by molecular distillation using a thin-film evaporator under high vacuum (2 mbar) at a temperature around 90 °C. Using this technology, octanol was recovered in a very efficient way while minimising the risk of degradation of the sugar derivatives present in the final mixture.
This overall process allowed the production of 24 g of a surfactant composition through a solvent-free process that included purification steps limited to acid/base reactions and filtration/distillation steps (Figure 2). The isolated mixture was subjected to thermogravimetric analysis which revealed the presence of 83.2% organic matter, 10.5% water and 6.75% ash. A 1H NMR analysis in CD3OD was also performed, based on the integration of the anomeric sugar proton signals in the 4.2-5.0 ppm region (Table 13), which showed that the surfactant composition contained 30 mol% n-octyl α-D-mannopyranosiduronic acid 19α, 6 mol% n-octyl β-L-gulopyranosiduronic acid 20β, 5% n-octyl α-L-gulofuranosidurono-6,3-lactone 21α, 13 mol% n-octyl-α-L-fucopyranoside 22α, 9 mol% n-octyl-β-L-fucopyranoside 22β, 9 mol% n-octyl-α-L-fucofuranoside 23α and 28 mol% n-octyl-β-L-fucofuranoside 23β (Figures S17,S18, Supplementary materials). The integration of the doublet relative to fucosyl methyl group at 1.19-1.27 ppm confirmed this percentage of fucosides in the final mixture. 1H NMR study of the surfactant composition did not allow to identify signals corresponding to n-octyl α,β D-glucopyranosides. The surface tension measurements obtained with this H-C8 surfactant composition are presented in section 2.5.
2.5. Physico-chemical properties of anionic and non-ionic surfactant compositions derived from oligoalginates, semi-refined alginates and crude brown Seaweed
Surface activities at the air-water interface were investigated for compositions H-C12 Man, H-C12 Gul, H-C12 OAlg, H-C12 s-r Alg in addition to the composition obtained from the crude seaweeds. Furthermore, biodegradability and aquatic ecotoxicity were evaluated for the H-C12 s-r Alg composition.
2.5.1. Measurements of air-water interfacial behavior
2.5.1.1. H-C12 derivatives from (OM), (OG), (OAlg) and (s-r Alg)
The different batches of H-C12 were characterised by surface tension measurements (Figure 4). The results shows that all surfactant compositions are effective in reducing the surface tension of the water, since values below 30 mN m-1 were measured for the CMCs. Pure n-dodecyl α-D-mannopyranosiduronic acid 13α [10] exhibits a CMC of 0.06 g L-1 and a γCMC of 28.9 mN m-1. Among the different surfactant compositions, it lowered the surface tension of water the least in the low concentration values. The H-C12 Man composition gives the same CMC and γCMC values (0.06 g L-1 and 29.1 mN m-1), but it lowers the surface tension much more effectively at low concentration values. This indicates, therefore, that the presence of H-C12 Man isomers allow for improved surfactant performance. The mixture from oligoguluronate gives the highest CMC with a value of 0.12 g L-1 and a γCMC of 28.8 mN m-1. However, for concentrations below 0.008 g L-1, the lowest surface tension values were measured with this H-C12 Gul composition. Similar CMC and γCMC values were measured with the batch from oligoalginate (0.11 g L-1, 27.6 mN m-1). Finally, the surfactant mixture obtained from semi-refined alginate provided the lowest CMC with a value of 0.04 g L-1 and a γCMC of 29.0 mN m-1. Tensiometry measurements were also carried out on anionic surfactant sodium laureth sulfate (SLES). The CMC was determined at 0.13 g L-1 and the γCMC at 31.9 mN m-1. Thus, all the compounds obtained from the different sources of algae extracts exhibit better performance in terms of CMC and γCMC.
2.5.1.2. H-C8 surfactant composition from crude seaweed
Surprisingly, surface tension measurements in pH 7 buffered solution from the composition derived from crude seaweed and octanol (H-C8 crude seaweed) revealed a CMC value of 0.007 g L-1 and a γCMC of 27.0 mN m-1 which demonstrated excellent surfactant properties.
2.5.2. Ecotoxicity studies
A series of ecotoxicity studies have been performed with the uronic surfactant compositions derived from semi-refined alginate and dodecanol (H-C12 s-r Alg) in addition to standard SLES. Experiments to determine the ecotoxicity of products include effects on aquatic organisms, from microalgae to fish. These different pollutant-sensitive organisms serve as controls. Three standardized tests were conducted: an algal growth inhibition test, a microcrustaceous immobilization test and a lethal toxicity test on a freshwater fish. The microalgae used, called Pseudokirchneriella subcapitata, are ubiquitous in the environment and are sensitive to toxic substances. The test is conducted according to the OECD 201 method [15], which corresponds to the percentage of inhibition of the algal growth rate after a 72-hour incubation period (CEr50). The microcrustaceans used are Daphnia Magna. They are freshwater crustaceans that are an important nutritional source for many aquatic organisms, their presence in sufficient numbers and in good health helps to maintain a certain balance in their ecosystem. The Daphnia Magna have the distinction of being extremely susceptible to changes, even minor, in the composition of their aquatic environment. The OECD 202 test [16] is based on the determination of the CE50 concentration, which, in 24 h and/or 48 h, immobilizes 50% of the microcrustaceans experimented. Finally, the test of freshwater fish, conducted according to the OECD 203 method [17], is to determine the concentration for which the sample has lethal toxicity for 50% of a Brachydanio rerio test population after a 96-hour exposure period (CL50). Brachydanio rerio is one of the model organisms commonly encountered in research laboratories for fish behaviour studies [18]. These tests are based on acute aquatic toxicity tests, i.e. adverse effects on aquatic organisms during short-term exposure. The results observed based on the concentration of the samples to be analyzed allow the substances to be categorized into different categories, as shown in Table 14.
As shown in Table 15, SLES can be considered toxic to algae and is borderline toxic to Daphnia and fish. In comparison, H-C12 s-r Alg has a reduced ecotoxicity since it is not very toxic to algae and fish and not-toxic to Daphnia.
2.5.3. Biodegradability studies
Comparative aerobic readily biodegradability of compounds H-C12 s-r Alg, and SLES was finally studied using the OECD 301 B method [19]. The objective of this test is to determine the release of carbon dioxide by microbial digestion in the aerobic environment of the compound to be analyzed. During the test, the compound is placed in a watery medium to which is added a mixed seeding of a plant dealing with urban wastewater. The surfactant studied is the only source of carbon and energy and it is introduced at a theoretical concentration of 10 mg L-1 of dissolved organic carbon (COD). The CO2 formed during degradation is trapped in external containers. The tests are conducted during 28 days during which the evolution of the biodegradation rate is determined. OECD 301 B method considers a product to be readily biodegradable if the biodegradation rate has reached at least 60% after 28 days and readily biodegradable with respect for the 10 day period if the biodegradation rate has reached at least 60% rate 10 days after the rate has reached 10%.
As the limit of 60% biodegradation was reached, all two products are readily biodegradable but H-C12 s-r Alg, exhibited lower biodegradability rates after 28 days (72%) than SLES (94%). Under the experimental conditions of the test, H-C12 s-r Alg, (Figures S19, Supplementary materials) is considered to be readily biodegradable without respecting the 10-day interval. Indeed, the CO2 release threshold of 60% of theoretical CO2 was not reached within the 10-day interval, but within the first 28 days of testing (threshold reached on day 21). In contrast, for SLES the CO2 release threshold of 60% of the theoretical CO2 was reached within 10 days (Figures S20, Supplementary materials).
3. Discussion
The main anionic surfactants on the market belong to the family of carboxylates, sulfates and sulfonates. The vast majority of them are of petrochemical origin and are irritating and highly ecotoxic. Carboxylate surfactants derived from natural anionic carbohydrates are not, or only to a limited extent, developed on an industrial scale. However, several polysaccharides incorporating negatively charged functional groups in their chemical structure are commercially available and could constitute a natural anionic source for the production of surfactants. In particular alginates which are derived from the cell wall of brown algae, are made up of 100% carboxylate monosaccharides. These biopolymers can be engaged in esterification or transesterification reactions with alcohols for the introduction of the lipophilic chains and then saponified to release the anionic function. A final acidification allows the surfactant to be isolated in its more easily purified carboxylic acid form.
To facilitate the industrial development of innovative surfactants based on carboxylate saccharides, we have proposed a new approach to produce these surfactants without sulfate and without ethylene oxide according to a smart strategy based on a multi-step process carried out in a cascading single pot mode without separating and isolating any reaction intermediates and applicable to oligomeric or polymeric alginates in a more or less refined form. The innovation of this research is also based on its positioning in green/blue chemistry (solvent-free reactions, with little waste, biodegradable reagents, valorisation of marine plant biomass, eco-compatible products). The syntheses involve a succession of chemical reactions such as depolymerisation by acid hydrolysis of oligo or polysaccharides, esterification and glycosylation with n-butanol, transesterification and transglycosylation with fatty alcohols and saponification. A final treatment involving mainly solid-liquid extraction, acidification, precipitation in an organic solvent (EtOAc or acetone) and filtration steps allows the final compositions to be enriched with active ingredients by removing (at least partially) residual salts and fatty alcohols. In terms of performance, the carboxylic acid-based surfactant compositions allow surface tension to be lowered to values ≤ 30 mN m-1 at concentrations equivalent to or lower than that of sodium laureth sulfate. In particular, the compositions derived from semi-refined alginate (s-r Alg) or oligomannuronate (OM) lead to CMC values two to three times lower than that obtained from sodium laureth sulfate. In addition, a significant improvement in ecotoxicity profile was observed for the biodegradable surfactant composition H-C12 s-r Alg, especially towards Daphnia.
In order to further simplify the process and reduce costs, a novel strategy has been successfully tested based on the direct use of plant materials in their raw state, i.e. without any chemical transformation, in particular solvent extraction or enzymatic extraction, but at most a mechanical and/or physical transformation, such as washing, grinding and/or drying. A notable advantage of this approach is the use of plant materials with a water content of up to 13% by weight, thus avoiding the addition of water during the depolymerisation step by hydrolysis of the polysaccharides. The process has been applied to brown seaweed containing alginates and fucans and/or fucoidans. The process incorporates the same reaction sequences as for polysaccharides (acid hydrolysis, esterification/glycosylation, transesterification/transglycosylation, saponification) with the addition of a preliminary extraction step carried out in situ as well as filtration, centrifugation or distillation steps under reduced pressure allowing the elimination of the residues of plant material not involved in the syntheses and the recycling of the excess alcohols. The composition H-C8 crude Alg obtained is rich in carboxylic acid sugars but they also contain alkyl glycosides derived from neutral sugars representative of the structure of the polysaccharides present in the starting plant material (D-fucose) which modify the physicochemical properties of the final compositions and optimise their surface-active performance [14]. Indeed, low values of surface tension (27.0 mN m-1) and critical micellar concentration (0.007 g L-1) were obtained in the case of these mixtures of uronic acid- and fucose-based surfactants.
4. Materials and Methods
4.1. Chemistry
Oligomannnuronate, oligoguluronate, oligoalginate and semi-refined alginate were produced from fresh or dry seaweeds (CEVA, 83 Rue de Pen Lan, 22610 Pleubian, France) and dried milled brown seaweeds (Asco T10) were purchased from Thorverk (Iceland); all other commercially available chemicals were used without further purification. All reactions were monitored by thin layer chromatography (Kieselgel 60F254 Merck). Compounds were visualized using a H2SO4 solution (5 % H2SO4 in EtOH) or a vanillin solution (15 g of vanillin in 250 mL of EtOH and 2.5 mL of conc. H2SO4) followed by heating. Geduran 60 (40-63 µm, Merck) was used for column chromatography. NMR spectra were recorded on a Bruker Avance III 400 spectrometer operating at 400.13 MHz for 1H, equipped with a BBFO probe with a Z-gradient coil and a GREAT 1/10 gradient unit. The standard temperature was adjusted to 298 K. The zg30 Bruker pulse program was used for 1D 1H NMR, with a TD of 64k, a relaxation delay d1 = 2 s and 8 scans. The spectrum width was set to 18 ppm. Fourier transform of the acquired FID was performed with an apodization of 0.3 Hz in most of the cases. Chemical shifts are mentioned in parts per million (ppm) with tetramethylsilane as an internal standard. Coupling constants were expressed in Hertz (Hz) and the following abbreviations were used to indicate the multiplicity: s (singulet), d (doublet), t (triplet), q (quadruplet), m (multiplet), dd (doublet of doublets), dt (doublet of triplets) and br (broad signal).
Preparation of C4-C4 Man from OM
Oligomannuronate (500 mg, 1.81 mmol CO2-, 1 eq.) was dispersed in water (0.87 mL) and n-butanol (25 mL, 273 mmol, 151 eq.) in a round-bottom flask with a Dean-Stark apparatus. Methanesulfonic acid technical grade 70% wt (422 mg, 3.08 mmol, 1.7 eq.) was added and the mixture was refluxed under vigorous stirring. The water formed in the medium was gradually removed by azeotropic distillation. After 7 h, the mixture was cooled to ambient temperature. The mixture was then neutralised with 1N NaOH solution (400 µL) and concentrated. The resulting mixture was dissolved in diethyl ether (20 mL) and then filtered using celite. The celite was finally rinsed with diethyl ether (100 mL) and the filtrate was concentrated. The products obtained (495 mg) were purified by silica gel chromatography with CH2Cl2:CH3OH, (97/3–96/4, v/v) to help determine the molar composition. The molar composition of this mixture of products before silica gel chromatography is as follows: 52% (n-butyl) n-butyl-α-D-mannopyranosiduronate (1α), 7% (n-butyl) n-butyl-α-D-mannofuranosiduronate (2α), 11% n-butyl-α-D-mannofuranosidurono-6.3-lactone (3α), 13% n-butyl-β-D-mannofuranosidurono-6.3-lactone (3β), 5% BDMF (6), 5% n-butyl-α-L-gulofuranosidurono-6.3-lactone (4α), 5% n-butyl-β-L-gulofuranosidurono-6.3-lactone (4β), 3% (n-butyl) n-butyl-β-L-gulofuranosiduronate (5β). 1H NMR (400 MHz, CDCl3) δ 7.11 (d, J = 3.4 Hz, BDMF), 6.51 (dd, J = 3.5, 0.8 Hz, BDMF), 5.53 (s, BDMF), 5.14 (dd, J = 7.4, 4.7 Hz, H3-4β), 5.06 (s, H1-4β), 5.05 (d, J = 2.0 Hz, H1-3α), 5.03 (d, J = 4.6 Hz, H1-3β), 5.00 (t, J = 4.8 Hz, H3-3α), 4.95 (t, J = 4.5 Hz, H1-4α), 4.91 (d, J = 1.7 Hz, H1-1α), 4.90 (t, J = 5.0 Hz, H3-3β), 4.79 (dd, J = 6.8, 4.8 Hz, H4-3β), 4.76 (dd, J = 6.0, 4.5 Hz, H4-3α), 4.72 (dd, J = 7.6, 4.0 Hz, H4-4β), 4.69 (d, J = 5.5 Hz, H4-4α), 4.43 (d, J = 2.9 Hz, H5-2α),4.31 – 4.15 (m, OCH2), 4.08 (d, J = 9.5 Hz, H5-1α), 4.00 (t, J = 9.3 Hz, H4-1α), 3.94 (m, H2-1α), 3.87 (dd, J = 8.9, 3.5 Hz, H3-1α), 3.83 – 3.69 (m, OCH2), 3.65 (t, J = 6.6 Hz, BDMF), 3.60 – 3.38 (m, OCH2) 1.76 – 1.62 (m, CH2), 1.62 – 1.49 (m, CH2), 1.46 – 1.29 (m, CH2), 0.99 – 0.88 (m, CH3). After column chromatography, five fractions F1-F5 were isolated and characterised by 1H NMR. F1: 57 mg of BDMF (6). (Rf=0.94, CH2Cl2/MeOH (95/5, v/v)): 1H NMR (400 MHz, CDCl3) δ 7.11 (d, J = 3.4 Hz), 6.51 (dd, J = 3.5, 0.8 Hz), 5.53 (s), 4.36 (t, J = 6.7 Hz), 3.65 (t, J = 6.6 Hz), 1.80 – 1.63 (m, CH2), 1.63 – 1.50 (m, CH2), 1.48 – 1.31 (m, CH2), 1.00 – 0.86 (m, CH3); F2: 12 mg of n-butyl-α-L-gulofuranosidurono-6.3-lactone (4α) and (n-butyl) n-butyl-β-L-gulofuranosiduronate (5β). (Rf=0.42, CH2Cl2/MeOH (95/5, v/v)) : 1H NMR (400 MHz, CDCl3) δ 4.98 (t, J = 5.6 Hz, H3-4α), 4.95 (d, J = 4.5 Hz, H1-4α), 4.91 (s, H1-5β), 4.69 (dd, J = 5.5, 0.8 Hz, H4-4α), 4.52 (dd, J = 8.0, 1.3 Hz, H4-5β), 4.38 (d, J = 1.3 Hz, H5-5β), 4.28 (d, J = 0.7 Hz, H5-4α), 4.26 – 4.18 (m, OCH2), 4.15 (t, J = 5.1 Hz, H2-4α), 3.91 (d, J = 5.4 Hz, H2-5β), 3.71 (dt, J = 9.6, 6.7 Hz, OCH2), 3.58 (dt, J = 9.7, 6.7 Hz, OCH2), 3.43 (dt, J = 9.6, 6.7 Hz, OCH2), 1.73 – 1.59 (m, CH2), 1.58 – 1.47 (m, CH2), 1.46 – 1.24 (m, CH2), 0.99 – 0.86 (m, CH3); F3: 65 mg of n-butyl-β-D-mannofuranosidurono-6.3-lactone (3β), (n-butyl) n-butyl-α-D-mannofuranosiduronate (2α), n-butyl-α-L-gulofuranosidurono-6.3-lactone (4α), n-butyl-β-L-gulofuranosidurono-6.3-lactone (4β) and (n-butyl) n-butyl-β-L-gulofuranosiduronate (5β). (Rf=0.38, CH2Cl2/MeOH (95/5, v/v)): 1H NMR (400 MHz, CDCl3) δ 5.14 (dd, J = 7.4, 4.7 Hz, H3-4β), 5.06 (s, H1-4β), 5.03 (d, J = 4.6 Hz, H1-3β), 4.97 (s, H1-2α), 4.95 (d, J = 4.5 Hz, H1-4α), 4.90 (t, J = 5.0 Hz, H3-3β), 4.79 (dd, J = 6.8, 4.8 Hz, H4-3β), 4.72 (dd, J = 7.6, 4.0 Hz, H4-4β), 4.69 (d, J = 5.5 Hz, H4-4α), 4.56 (d, J = 4.0 Hz, H5-4β), 4.52 (d, J = 8.0 Hz, H4-5β), 4.43 (d, J = 2.9 Hz, H5-2α), 4.39 (d, J = 7.4 Hz, H5-3β), 4.30 – 4.19 (m, OCH2), 3.93 (d, J = 4.7 Hz, H2-2α), 3.79 (dt, J = 9.5, 6.7 Hz, OCH2), 3.66 (dt, J = 9.6, 6.7 Hz, OCH2), 3.54 – 3.38 (m, OCH2), 1.73 – 1.47 (m, CH2), 1.45 – 1.24 (m, CH2), 0.93 (m, CH3); F4: 49 mg of n-butyl-β-D-mannofuranosidurono-6.3-lactone (3β), n-butyl-α-D-mannofuranosidurono-6.3-lactone (3α) and n-butyl-β-L-gulofuranosidurono-6.3-lactone(4β). (Rf=0.29, CH2Cl2/MeOH (95/5, v/v)): 1H NMR (400 MHz, CDCl3) δ 5.13 (dd, J = 7.4, 4.7 Hz, H3-4β), 5.05 (d, J = 2.0 Hz, H1-3α), 5.03 (d, J = 4.6 Hz, H1-3β), 5.00 (t, J = 4.8 Hz, H3-3α), 4.90 (t, J = 4.8 Hz, H3-3β), 4.79 (dd, J = 6.8, 4.8 Hz, H4-3β), 4.76 (dd, J = 6.0, 4.5 Hz, H4-3α), 4.72 (dd, J = 7.6, 4.0 Hz, H4-4β), 4.27 – 4.17 (m, OCH2), 3.74 – 3.61 (m, OCH2), 3.50 – 3.35 (m, OCH2), 1.70 – 1.46 (m, CH2), 1.42 – 1.27 (m, CH2), 0.96 – 0.86 (m, CH3); F5: 183 mg of (n-butyl) n-butyl-α-D-mannopyranosiduronate (1α). (Rf=0.22, CH2Cl2/MeOH (95/5, v/v)) : 1H NMR (400 MHz, CDCl3) δ 4.90 (d, J = 1.7 Hz, H1-1α), 4.28 – 4.15 (m, OCH2), 4.28 – 4.15 (m, OCH2), 4.08 (d, J = 9.5 Hz, H5-1α), 4.00 (t, J = 9.3 Hz, H4-1α), 3.93 (dd, J = 3.4, 1.8 Hz, H2-1α), 3.86 (dd, J = 9.0, 3.4 Hz, H3-1α), 3.72 (dt, J = 9.7, 6.7 Hz, OCH2), 3.50 – 3.43 (m, OCH2), 1.72 – 1.62 (m, CH2), 1.62 – 1.51 (m, CH2), 1.44 – 1.30 (m, CH2), 0.93 (m, CH3).
Preparation of C12-C12 Man from OM
Oligomannuronate (500 mg, 1.81 mmol CO2-, 1 eq.) was dispersed in water (0.9 mL) and n-butanol (25 mL, 273 mmol, 151 eq.) in a round-bottom flask with a Dean-Stark apparatus. Methanesulfonic acid technical grade 70% wt (419 mg, 3.05 mmol, 1.7 eq.) was added and the mixture was refluxed under vigorous stirring. The water formed in the medium was gradually removed by azeotropic distillation. After 7 h, the mixture was cooled to ambient temperature. Dodecanol (1.6 mL, 7.2 mmol, 4 eq.) and the 70% methanesulfonic acid solution (246 mg, 1.8 mmol, 1 eq.) were added. The mixture was stirred at 70° C under reduced pressure (up to 5 mbar) using distillation apparatus. Once the butanol had been completely removed (2.17 h), water (16 mL) was added and the mixture was then neutralised with 1N NaOH solution (0.5 mL). The whole mixture was left to stir vigorously at 80° C for 1 h and the mixture was cooled to ambient temperature. The mixture was cooled at 0°C to solidify the organic phase which was isolated by solid-liquid extraction and dried under vacuum. The resulting mixture (1.684 g) was purified by a first silica gel chromatography with CH2Cl2/CH3OH, (100:0–96:4) to give crude mixture C12-C12 Man (504 mg). The molar composition of this mixture is as follows: 48% (n-dodecyl) n-dodecyl-α-D-mannopyranosiduronate (7α), 9% (n-dodecyl) n-dodecyl-α-D-mannofuranosiduronate (8α), 5% n-dodecyl-α-D-mannofuranosidurono-6.3-lactone (9α), 22% n-dodecyl-β-D-mannofuranosidurono-6.3-lactone (9β), 3% n-dodecyl-α-L-gulofuranosidurono-6.3-lactone (10α), 3% n-dodecyl-β-L-gulofuranosidurono-6.3-lactone (10β), 5% (n-dodecyl) n-dodecyl-β-L-gulopyranosiduronate (11β), 6% (n-dodecyl) n-dodecyl-β-L-gulofuranosiduronate (12β). 1H NMR (400 MHz, CDCl3) δ 5.07 (s, H1-10β), 5.05 (d, J = 2.0 Hz, H1-9α), 5.03 (d, J = 4.5 Hz, H1-9β), 4.99 (t, J = 4.7 Hz, H3-9α), 4.97 (t, J = 5.7 Hz, H3-10α), 4.97 (s, H1-8α, H2-9α), 4.95 (d, J = 4.5 Hz, H1-10α), 4.92 (d, J = 1.7 Hz, H1-7α), 4.91 (s, H1-12β), 4.90 (t, J = 5.0 Hz, H3-9β), 4.79 (dd, J = 6.8, 4.8 Hz, H4-9β), 4.72 (dd, J = 7.4, 4.0 Hz, H4-10β), 4.69 (d, J = 5.6 Hz, H4-10α), 4.58 (d, J = 4.0 Hz, H5-10β), 4.55 (d, J = 1.0 Hz, H5-11β), 4.53 (dd, J = 7.9, 1.4 Hz, H4-12β), 4.43 (d, J = 2.9 Hz, H5-8α), 4.39 (d, J = 7.0 Hz, H5-9β), 4.29 – 4.14 (m, OCH2), 4.09 (d, J = 9.6 Hz, H5-7α), 4.00 (t, J = 9.2 Hz, H4-7α), 3.95 (dd, J = 3.4, 1.7 Hz, H2-7α), 3.89 (dd, J = 8.9, 3.4 Hz, H3-7α), 3.82 – 3.68 (m, OCH2), 3.53 – 3.37 (m, OCH2), 1.74 – 1.64 (m, CH2), 1.63 – 1.53 (m, CH2), 1.38 – 1.20 (m, CH2), 0.91 – 0.85 (m, CH3). A second silica gel chromatography with CH2Cl2/CH3OH, (97:3) was made to help determine the molar composition. Five fractions F1-F5 were isolated and characterised by 1H NMR. F1 : 74 mg of (n-dodecyl) n-dodecyl-α-D-mannofuranosiduronate (8α), n-dodecyl-β-D-mannofuranosidurono-6.3-lactone (9β), n-dodecyl-α-L-gulofuranosidurono-6.3-lactone (10α) and (n-dodecyl) n-dodecyl-β-L-gulopyranosiduronate (12β). (Rf=0.47, CH2Cl2/MeOH (95/5, v/v)) : 1H NMR (400 MHz, CDCl3) δ 5.03 (d, J = 4.5 Hz, H1-9β), 4.98 (t, J = 5.8 Hz, H3-10α), 4.97 (s, H1-8α), 4.96 (d, J = 4.5 Hz, H1-10α), 4.91 (s, H1-12β), 4.89 (t, J = 5.0 Hz, H3-9β), 4.79 (dd, J = 6.8, 4.8 Hz, H4-9β), 4.70 (dd, J = 5.7, 0.8 Hz, H4-10α), 4.71 – 4.65 (m, H3-12β), 4.65 – 4.61 (m, H3-H4-8α), 4.52 (dd, J = 7.9, 1.3 Hz, H4-12β), 4.43 (d, J = 2.8 Hz, H5-8α), 4.37 (d, J = 1.3 Hz, H5-12β), 4.27 – 4.18 (m, OCH2), 3.92 (d, J = 4.5 Hz, H2-8α), 3.81 – 3.54 (m, OCH2), 1.76 – 1.63 (m, CH2), 1.62 – 1.48 (m, CH2), 1.39 – 1.20 (m, CH2), 0.91 – 0.85 (m, CH3); F2 : 64 mg of n-dodecyl-α-D-mannofuranosiduronate (8α), n-dodecyl-β-D-mannofuranosidurono-6.3-lactone (9β). (Rf=0.45, CH2Cl2/MeOH (95/5, v/v)) : 1H NMR (400 MHz, CDCl3) δ 5.03 (d, J = 4.6 Hz, H1-9β), 4.97 (s, H1-8α), 4.90 (t, J = 5.0 Hz, H3-9β), 4.79 (dd, J = 6.8, 4.8 Hz, H4-9β), 4.62 (m, H3-H4-8α), 4.42 (d, J = 2.9 Hz, H5-8α), 4.38 (d, J = 6.6 Hz, H5-9β), 4.29 – 4.18 (m, OCH2), 3.92 (d, J = 4.7 Hz, H2-8α), 3.78 (dt, J = 9.5, 6.8 Hz, OCH2), 3.65 (dt, J = 9.5, 7.3 Hz, OCH2), 3.53 – 3.35 (m, OCH2) ), 1.74 – 1.63 (m, CH2), 1.63 – 1.50 (m, CH2), 1.38 – 1.19 (m, CH2), 0.91 – 0.85 (m, CH3); F3: 30 mg of (n-dodecyl) n-dodecyl-α-D-mannopyranosiduronate (7α), (n-dodecyl) n-dodecyl-α-D-mannofuranosiduronate (8α), n-dodecyl-α-D-mannofuranosidurono-6.3-lactone (9α), n-dodecyl-β-D-mannofuranosidurono-6.3-lactone (9β), n-dodecyl−β-L-gulofuranosidurono-6.3-lactone (10β), (n-dodecyl) n-dodecyl-α-L-gulopyranosiduronate (11α), (n-dodecyl) n-dodecyl-β-L-gulopyranosiduronate (11β), and (n-dodecyl) n-dodecyl-β-L-gulofuranosiduronate (12β). (Rf=0.38, CH2Cl2/MeOH (95/5, v/v)): 1H NMR (400 MHz, CDCl3) δ 5.14 (dd, J = 7.4, 4.7 Hz, H3-10β), 5.07 (s, H1-10β), 5.05 (d, J = 2.0 Hz, H1-9α), 5.03 (d, J = 4.5 Hz, H1-9β), 4.99 (t, J = 4.7 Hz, H3-7α), 4.97 (s, H2-9α), 4.92 (d, J = 1.7 Hz, H5-7α), 4.91 (s, H1-12β), 4.89 (t, J = 5.0 Hz, H3-9β), 4.78 (dt, J = 5.8, 4.6 Hz, H4-9α,β), 4.72 (dd, J = 7.5, 4.0 Hz, H4-10β), 4.65 – 4.61 (m, H3-H4-8α), 4.63 (d, J = 7.8 Hz, H1-11β), 4.60 (d, J = 1.7 Hz, H5-11α), 4.57 (d, J = 4.0 Hz, H5-10β), 4.55 (d, J = 1.5 Hz, H5-11β), 4.52 (dd, J = 7.9, 1.0 Hz, H4-12β), 4.43 (d, J = 2.9 Hz, H5-8α), 4.40 (d, J = 1.3 Hz, H5-12β), 4.39 (d, J = 6.3 Hz, H5-9β), 4.27 – 4.18 (m, OCH2), 4.13 (dd, J = 4.0, 1.7 Hz, H4-11β), 4.09 (d, J = 9.5 Hz, H5-7α), 3.80 – 3.64 (m, OCH2), 3.55 – 3.37 (m, OCH2), 1.73 – 1.48 (m, CH2), 1.42 – 1.18 (m, CH2), 0.93 – 0.84 (m, CH3); F4 : 240 mg of (n-dodecyl) n-dodecyl-α-D-mannopyranosiduronate (7α). (Rf=0.3, CH2Cl2/MeOH (95/5, v/v)) : 1H NMR (400 MHz, CDCl3) δ 4.91 (d, J = 1.7 Hz, H1-7α), 4.21 (m, OCH2), 4.08 (d, J = 9.6 Hz, H5-7α), 4.01 (t, J = 9.3 Hz, H4-7α), 3.94 (dd, J = 3.4, 1.8 Hz, H2-7α), 3.88 (dd, J = 8.9, 3.4 Hz, H3-7α), 3.72 (dt, J = 9.7, 6.8 Hz, OCH2), 3.47 (dt, J = 9.7, 6.5 Hz, OCH2), 1.73 – 1.64 (m, CH2), 1.64 – 1.52 (m, CH2), 1.38 – 1.22 (m, CH2), 0.92 – 0.84 (m, CH3); F5 : 9 mg of a mixture of products (numerous 1H NMR signals).
Preparation of H-C12 Man from OM
Oligomannuronate (500 mg, 1.81 mmol CO2-, 1 eq.) was dispersed in water (1 mL) and n-butanol (25 mL, 273 mmol, 151 eq.) in a round-bottom flask with a Dean-Stark apparatus. Methanesulfonic acid technical grade 70% wt (422 mg, 3.08 mmol, 1.7 eq.) was added and the mixture was refluxed under vigorous stirring. The water formed in the medium was gradually removed by azeotropic distillation. After 7 h, the mixture was cooled to ambient temperature. Dodecanol (1.6 mL, 7.2 mmol, 4 eq.) and the 70% methanesulfonic acid solution (248 mg, 1.81 mmol, 1 eq.) were added. The mixture was stirred at 70° C under reduced pressure (up to 5 mbar) using distillation apparatus. Once the butanol had been completely removed (1.42 h), a 0.4N NaOH solution (13.5 mL, 5.4 mmol, 3.0 eq.) was added and the mixture was left to stir vigorously at 70° C. for 1 h. The water was then removed by freeze-drying or by azeotropic distillation with butanol. The excess dodecanol present in the crude product was remove by solid-liquid extraction with EtOAc. At the end of this treatment, the mixture of products was dissolved in ice-cold water (15 mL) and EtOAc (22.5 mL), then a 1N hydrochloric acid solution (3.1 mL) was added. The aqueous solution was extracted with EtOAc (5x7.5 mL). The organic phases were combined and washed with a saturated NaC1 solution (30 mL) and a 1N hydrochloric acid solution (150 µL). The organic phase was dried with MgSO4 and then concentrated under vacuum. A mixture of products H-C12 Man was obtained (341 mg), the molar composition of which is: 47% n-dodecyl-α-D-mannopyranosiduronic (13α), 4% n-dodecyl-α-D-mannofuranosidurono-6.3-lactone (9α), 12% n-dodecyl-β-D-mannofuranosidurono-6.3-lactone (9β), 4% n-dodecyl-β-L-gulopyranosiduronic (14β), 9% n-dodecyl-α-L-gulofuranosidurono-6,3-lactone (10α), 9% n-dodecyl-β-L-gulofuranosidurono-6,3-lactone (10β), 4% n-dodecyl-β-L-gulofuranosiduronic acid (15β), 10% non-identified molecule X. 1H NMR (400 MHz, CD3OD) δ 5.11 (d, J = 6.5 Hz, X), 5.07 (dd, J = 7.6, 4.7 Hz, H3-10β), 5.00 (d, J = 1.6 Hz, H1-9α), 4.98 (s, H1-10β), 4.97 (t, J = 5.7 Hz, H3-10α), 4.96 (d, J = 3.8 Hz, H1-9β), 4.92 (d, J = 4.4 Hz, H1-10α), 4.80 (d, J = 2.1 Hz, H1-13α), 4.73 (dt, J = 5.0, 2.3 Hz, H4-9α,β), 4.62 (d, J = 8.3 Hz, H1-14β), 4.62 – 4.56 (m, H4-10α,β), 4.55 (d, J = 4.9 Hz, H-X), 4.49 (s, H5-14β), 4.47 (d, J = 6.6 Hz, H5-9β), 4.43 – 4.38 (m), 4.35 (d, J = 5.1 Hz, H-X), 4.29 (d, J = 2.2 Hz, H5-15β), 4.18 (t, J = 3.9 Hz, H3-14β), 4.14 – 4.08 (m, H2-10β/H2-10α/H4-14β), 4.05 (t, J = 4.8 Hz, X), 4.00 (d, J = 9.4 Hz, H5-13α), 3.89 (t, J = 9.2 Hz, H4-13α), 3.79 (dd, J = 3.4, 2.1 Hz, H2-13α), 3.76 – 3.63 (m, OCH2), 3.56 – 3.38 (m, OCH2), 1.67 – 1.51 (m, CH2), 1.46 – 1.22 (m, CH2), 0.94 – 0.87 (m, CH3).
Preparation of H-C12 Gul from OG
Oligoguluronate (500 mg, 2.1 mmol CO2-, 1 eq.) was dispersed in water (1.5 mL) and n-butanol (29 mL, 317 mmol, 151 eq.) in a round-bottom flask with a Dean-Stark apparatus. Methanesulfonic acid technical grade 70% wt (634 mg, 4.62 mmol, 2.2 eq.) was added and the mixture was refluxed under vigorous stirring. The water formed in the medium was gradually removed by azeotropic distillation. After 7 h, the mixture was cooled to ambient temperature. Dodecanol (1.8 mL, 8.3 mmol, 4 eq.) and the 70% methanesulfonic acid solution (288 mg, 2.1 mmol, 1 eq.) were added. The mixture was stirred at 70° C under reduced pressure (up to 5 mbar) using distillation apparatus. Once the butanol had been completely removed (1.42 h), a 0.4N NaOH solution (18.5 mL, 7.4 mmol, 3.5 eq.) was added and the mixture was left to stir vigorously at 70° C. for 1 h. The water was then removed by freeze-drying or by azeotropic distillation with butanol. The excess dodecanol present in the crude product was removed by solid-liquid extraction with EtOAc. At the end of this treatment, the mixture of products was dissolved in ice-cold water (27.5 mL) and EtOAc (27.5 mL), then a 1N hydrochloric acid solution (3.6 mL) was added. The aqueous solution was extracted with EtOAc (3x15 mL). The organic phases were combined and washed with a saturated NaC1 solution (35 mL). The organic phase was dried with MgSO4 and then concentrated under vacuum. A mixture of products H-C12 Gul was obtained (356 mg), the molar composition of which is: 15% n-dodecyl α-D-mannopyranosiduronic acid (13α), 13% n-dodecyl β-L-gulopyranosiduronic acid (14β), 26% n-dodecyl α-L-gulofuranosidurono-6,3-lactone (10α), 26% n-dodecyl β-L-gulofuranosidurono -6,3 –lactone (10β), 21% n-dodecyl-β-L-gulofuranosiduronic acid (15β). 1H NMR (400 MHz, CD3OD) δ 5.07 (dd, J = 7.5, 4.7 Hz, H3-10β), 4.98 (s, H1-10β), 4.96 (t, J = 5.5 Hz, H3-10α), 4.92 (d, J = 4.5 Hz, H1-10α), 4.80 (d, J = 2.1 Hz, H1-13α), 4.62 (d, J = 8.4 Hz, H1-14β), 4.51 (dd, J = 7.6, 2.1 Hz, H4- 15β), 4.49 (d, J = 1.7 Hz, H5-14β), 4.39 (d, J = 4.3 Hz, H5-10β), 4.29 (d, J = 2.1 Hz, H5- 15β), 4.02 (dd, J = 3.6, 1.4 Hz, H4-14β), 4.00 (d, J = 9.7 Hz, H5-13α), 3.89 (t, J = 9.2 Hz, H4-13α), 3.84 (d, J = 5.0 Hz, H2- 15β), 3.79 (dd, J = 3.4, 2.1 Hz, H2-13α), 3.76 – 3.59 (m, OCH2), 3.56 – 3.36 (m, OCH2), 1.69 – 1.50 (m, CH2), 1.45 – 1.25 (m, CH2), 0.97 – 0.87 (m, CH3).
Preparation of H-C12 OAlg from OAlg
Oligoalginate (1.0 g, 2.84 mmol CO2-, 1 eq.) was dispersed in water (2.0 mL) and n-butanol (39 mL, 426 mmol, 150 eq.) in a round-bottom flask with a Dean-Stark apparatus. Methanesulfonic acid technical grade 70% wt (857 mg, 6.24 mmol, 2.2 eq.) was added and the mixture was refluxed under vigorous stirring. The water formed in the medium was gradually removed by azeotropic distillation. After 7 h, the mixture was cooled to ambient temperature. Dodecanol (2.5 mL, 11.2 mmol, 4 eq.) and the 70% methanesulfonic acid solution (391 mg, 2.85 mmol, 1 eq.) were added. The mixture was stirred at 70° C under reduced pressure (up to 5 mbar) using distillation apparatus. Once the butanol had been completely removed (1.42 h), a 0.4N NaOH solution (25 mL) was added and the mixture was left to stir vigorously at 70° C. for 1 h. The water was then removed by freeze-drying or by azeotropic distillation with butanol. The excess dodecanol present in the crude product was removed by solid-liquid extraction with EtOAc. At the end of this treatment, the mixture of products was dissolved in ice-cold water (30 mL) and EtOAc (45 mL), then a 1N hydrochloric acid solution (6 mL) was added. The aqueous solution was extracted with EtOAc (8x15 mL). The organic phases were combined and washed with a saturated NaC1 solution (60 mL) and a 1N hydrochloric acid solution (300 µL). The organic phase was dried with MgSO4 and then concentrated under vacuum. A mixture of products H-C12 OAlg was obtained (759 mg), the molar composition of which is: 23% n-dodecyl-α-D-mannopyranosiduronic (13α), 9% n-dodecyl-β-L-gulopyranosiduronic (14β), 20% n-dodecyl-α-L-gulofuranosidurono-6,3-lactone (10α), 20% n-dodecyl-β-L-gulofuranosidurono -6,3 –lactone (10β), 9% n-dodecyl-β-L-gulofuranosiduronic acid (15β), 4% n-dodecyl-α-D-mannofuranosidurono-6.3-lactone (9α), 9% n-dodecyl-β-D-mannofuranosidurono-6.3-lactone (9β), 6% non-identified molecule X. 1H NMR (400 MHz, CD3OD) δ 5.11 (d, J = 6.5 Hz, X), 5.07 (dd, J = 7.5, 4.7 Hz, H3-10β), 4.97 (s, H1-10β), 4.95 (t, J = 5.6 Hz, H3-10α), 4.95 (d, J = 4.1 Hz, H1-9β), 4.91 (d, J = 4.5 Hz, H1-10α), 4.80 (d, J = 2.1 Hz, H1-13α), 4.72 (td, J = 4.7, 1.5 Hz, H4-9α,β), 4.61 (d, J = 8.2 Hz, H1-14β), 4.60 – 4.55 (m, H4-10α,β), 4.54 (d, J = 5.0 Hz, X), 4.51 (s (br), H4-15β), 4.48 (d, J = 1.6 Hz, H5-14β), 4.46 (d, J = 6.5 Hz, H5-9β), 4.38 (d, J = 4.3 Hz, H5-10β), 4.35 (d, J = 5.0 Hz, X), 4.29 (d, J = 2.0 Hz, H5- 15β), 4.12 – 4.08 (m, H2-10β/H4-10α), 3.88 (t, J = 9.3 Hz, H4-13α), 3.78 (dd, J = 3.4, 2.1 Hz, H2-13α), 3.75 – 3.61 (m, OCH2), 3.55 – 3.35 (m, OCH2), 1.70 – 1.49 (m, CH2), 1.45 – 1.23 (m, CH2), 0.95 – 0.86 (m, CH3).
Preparation of H-C12 s-r Alg from s-r Alg
The semi-refined alginate derived from Laminaria digitata (2.0 g, 4.3 mmol sugar units, 1 eq.) was dispersed in water (60 mL) and the 70% methane-sulfonic acid solution (2.95 g, 21.5 mmol, 5 eq.) was added in a round-bottom flask with a Dean-Stark apparatus. The mixture was refluxed with vigorous stirring. At 8 h of reaction, butanol (60 mL, 656 mmol, 152 eq.) was added and the mixture was left at reflux with vigorous stirring. The water present in the medium was gradually removed by azeotropic distillation. After a further 15 h of reaction, and once the mixture had returned to ambient temperature, dodecanol (3.8 mL, 17 mmol, 4 eq.) was added. The mixture was stirred at 70° C under reduced pressure (up to 5 mbar) using distillation apparatus. Once the butanol had been completely removed (1.42 h), a 0.2N NaOH solution (60 mL) was added and the mixture was left to stir vigorously at 70° C. for 1 h. The water was then removed by freeze-drying or by azeotropic distillation with butanol. The excess dodecanol present in the crude product was removed by solid-liquid extraction with acetone. At the end of this treatment, the mixture of products was dissolved in ice-cold water (75 mL) and then a 0.5M oxalic acid solution (8.0 mL) was added. The water was then removed by freeze-drying. The crude product was purified by solid-liquid extraction with acetone (20 mL + 8x10 mL). The filtrate was concentrated under vacuum. At the end of this treatment, a mixture of products was obtained (628 mg), the weight composition of which is: 23% n-dodecyl-α-D-mannopyranosiduronic (13α), 5% n-dodecyl-α-D-mannofuranosidurono-6.3-lactone (9α), 13% n-dodecyl-β-D-mannofuranosidurono-6.3-lactone (9β), 11% n-dodecyl-β-L-gulopyranosiduronic (14β), 20% n-dodecyl-β-L-gulofuranosidurono -6,3 –lactone (10β), 5% n-dodecyl-α−L-fucopyranosides 17α, 8% n-dodecyl-β-L-fucopyranosides 17β and 15% n-dodecyl-α,β-L-fucofuranosides 18α,β. 1H NMR (400 MHz, acetone-d6) δ 5.08 (dd, J = 7.5, 4.7 Hz, H3-10β), 5.03 (s, H1-10β), 4.95 (d, J = 4.7 Hz, H1-9β), 4.89 (t, J = 4.8 Hz, H3-9β), 4.85 (d, J = 2.0 Hz, H1-13α), 4.83 (d, J = 3.8 Hz, H1-17α), 4.80 (td, J = 4.9, 1.4 Hz, H4-9α,β), 4.75 (d, J = 3.6 Hz, H1-18α,β), 4.65 (dd, J = 7.3, 4.4 Hz, H4-10β), 4.56 (d, J = 5.4 Hz), 4.51 (d, J = 1.7 Hz, H5-14β), 4.43 (d, J = 4.4 Hz, H5-10β), 4.27 (d, J = 7.7 Hz, H1-17β), 4.25 (t, J = 4.4 Hz), 4.18 (d, J = 4.7 Hz, H2-10β), 4.09 (dd, J = 3.6, 1.6 Hz, H4-14β), 3.83 (dd, J = 3.4, 2.0 Hz, H2-13α), 3.79 – 3.63 (m, OCH2), 3.51 – 3.31 (m, OCH2), 1.68 – 1.49 (m, CH2), 1.46 – 1.26 (m, CH2), 1.24 (d, J = 6.6 Hz, CH3 17 or 18), 1.22 (d, J = 6.8 Hz, CH3-17 or 18), 1.21 (d, J = 6.7 Hz, CH3-17 or 18), 0.97 – 0.86 (m, CH3).
Preparation of H-C8 crude Alg from crude Alg
The dried milled Ascophyllum nodosum seaweed (100 g) was introduced into the reactor (1 L). Butanol (140 ml) and the methane-sulfonic acid solution (30 g of 70% MSA in 140 mL of butanol) were added. The mixture was refluxed and stirred with a four-blade Teflon paddle at 400 rpm. For the double envelope, the temperature was fixed at 135°C. After 7 h of reaction, the mixture was recovered hot through the lower tap and then filtered or centrifuged to remove residues. The presence of products in the reaction medium was monitored by TLC with CH2Cl2/MeOH (95/5, v/v) as the eluent and vanillin solution as the staining reagent. The filtrate/supernatant was transferred into the reactor (1 L) and octanol (100 ml) was added. The reaction mixture is pulled under vacuum at 70° C for 5-7 h to evaporate butanol and perform the transesterification/transglycosylation reactions. The mixture was made alkaline with a concentrated sodium hydroxide solution (5N NaOH, 20 mL). After 1-2 h at 70°C, only nonionic surfactants were visible by TLC (eluent: CH2Cl2/MeOH (95/5, v/v)). The anionic molecules were revealed by TLC (AcOEt/iPrOH/H2O (6/3/1, v/v/v)). The mixture was pulled under vacuum to evaporate the water. At room temperature, 95% of H2SO4 was added to acidify to pH 2-3 and precipitate the salts (counter-ions of the anionic surfactants). Surfactants were miscible in octanol. This oil phase was filtered to remove the salts which were insoluble. The oily filtrate was injected into a molecular distillation apparatus to eliminate octanol. At 100° C under reduced pressure (1 mbar), 24 g of the surfactant composition was recovered in the form of a viscous syrup, and consequently in a mass yield of approximately 24% relative to the starting algae. At room temperature, the product is a pasty and hygroscopic solid. The mixture was submitted to thermogravimetric analysis, which showed that it consisted of 83.2% organic matter; 10.5% water; and 6.75% ash. 1H NMR analysis revealed the presence of 30 mol% n-octyl α-D-mannopyranosiduronic acid 19α , 6 mol% n-octyl β-L-gulopyranosiduronic acid 20β, n-octyl α-L-gulofuranosidurono-6,3-lactone 21α, 13 mol% n-octyl-α-L-fucopyranoside 22α, 9 mol% n-octyl-β-L-fucopyranosides 22β, 9 mol% n-ocyl-α-L-fucofuranosides 23α and 28 mol% n-octyl-β-L-fucofuranoside 23β. 1H NMR (400 MHz, CD3OD) δ 5.05 (d, J = 3.2 Hz), 4.97 (t, J = 5.5 Hz, H5-21α), 4.93 (d, J = 4.6 Hz, H1-21α), 4.82 (d, J = 2.0 Hz, H1-19α), 4.78 (d, J = 3.8 Hz, H1-22α), 4.75 (d, J = 2.2 Hz, H1-23β), 4.72 (d, J = 3.7 Hz, H1-23α), 4.64 (d, J = 8.1 Hz, H1-20β), 4.61 (d, J = 4.6 Hz), 4.50 (d, J = 1.6 Hz, H5-20β), 4.43 (t, J = 4.8 Hz), 4.26 (d, J = 7.8 Hz, H1-22β), 3.88 (t, J = 9.4 Hz, H4-19α), 3.80 (dd, J = 3.5, 1.9 Hz, H2-19α), 3.77 – 3.63 (m, OCH2), 3.53 – 3.35 (m, OCH2), 1.72 – 1.50 (m, CH2), 1.48 – 1.30 (m, CH2), 1.27 (d, J = 6.4 Hz, CH3 22 or 18), 1.26 (d, J = 6.5 Hz, CH3-22 or 23), 1.22 (d, J = 6.6 Hz, CH3-22 or 23), 1.19 (d, J = 6.5 Hz, CH3-22 or 23), 1.01 – 0.88 (m, CH3).
4.2. Physico-Chemistry
Critical micelle concentrations (CMC) and interfacial tensions (IFT) were measured on a force tensiometer Krüss K100. The critical micelle concentrations (CMC) were determined using the du Noüy ring method and the Krüss Laboratory Desktop software with the Surfactant Characteristics program. A water solution of surfactant was prepared around 10 g/L. Only 10 ml of this solution was used for the CMC determination. The deionized water was added thanks to an automatic burette controlled by the software. The concentration and the tension determination were automatically determined by the software. The CMC was measured at 25°C.
5. Conclusions
In this study, it has been proven for the first time that the conversion of oligo- and polysaccharide alginates in refined or semi-refined forms into biodegradable alkyl uronate surfactants can be carried out by one-pot acid hydrolysis, butanolysis, transesterification, transacetalisation, saponification reactions, thus avoiding the isolation of any reaction intermediates. In addition, an in situ process was developed to manufacture surfactant compositions directly from crude milled brown seaweeds without requiring the standard steps of polysaccharide extraction and purification. These alkyl uronate monosaccharides as isomeric mixtures, with or without alkyl glycoside co-products, exhibit attractive surface activity and reduced aquatic ecotoxicity compared to commercial sodium laureth sulphate. Further physicochemical studies to evaluate the potential of these innovative surfactant compositions in cosmetic formulations are currently under investigation.
Supplementary Materials
The following are available online Preprints.org, Figure S1: 1H NMR spectrum of C4-C4 Man from OM (δ: 7.2-0.6 ppm) (400 MHz, CDCl3); Figure S2: Zooms of 1H NMR spectrum of C4-C4 Man from OM in the zones: (a) 7.2-5.5 ppm; (b) 5.16-4.68 ppm; (c) 4.50-3.60 ppm (400 MHz, CDCl3); Figure S3: Zooms of 1H NMR spectra of fractions F1, F2 and F3 isolated after column chromatography of C4-C4 Man from OM (400 MHz, CDCl3); Figure S4: Zooms of 1H NMR spectra of fractions F4 and F5 isolated after column chromatography of C4-C4 Man from OM (400 MHz, CDCl3); Figure S5: 1H NMR spectrum of C12-C12 Man from OM (δ: 0.6 -2.2 ppm) (400 MHz, CDCl3); Figure S6: Zooms of 1H NMR spectrum of C12-C12 Man from OM in the zones: (a) 5.15-4.50 ppm; (b) 4.50-3.86 ppm (400 MHz, CDCl3); Figure S7: Zooms of 1H NMR spectra of fractions F1, F2 and F3 isolated after the second column chromatography of C12-C12 Man from OM (400 MHz, CDCl3); Figure S8: Zoom of 1H NMR spectrum of fraction F4 isolated after column chromatography of C12-C12 Man from OM (400 MHz, CDCl3); Figure S9: 1H NMR spectrum of H-C12 Man from OM (δ: 5.2-0.6 ppm) (400 MHz, CD3OD); Figure S10: Zooms of 1H NMR spectrum of H-C12 Man from OM in the zones: (a) 5.15-4.35 ppm; (b) 4.35-3.77ppm (400 MHz, CD3OD); Figure S11: 1H NMR spectrum of H-C12 Gul from OG (δ: 5.2-0.6 ppm) (400 MHz, CD3OD); Figure S12: Zooms of 1H NMR spectrum of H-C12 Gul from OG in the zones: (a) 5.15-4.35 ppm; (b) 4.32-3.76 ppm (400 MHz, CD3OD); Figure S13: 1H NMR spectrum of H-C12 OAlg from OAlg (δ: 5.2-0.6 ppm) (400 MHz, CD3OD); Figure S14: Zooms of 1H NMR spectrum of H-C12 OAlg from OAlg in the zones: (a) 5.15-4.35 ppm; (b) 4.32-3.74 ppm (400 MHz, CD3OD); Figure S15: 1H NMR spectrum of H-C12 s-r Alg from s-r Alg (δ: 5.2-0.6 ppm) (400 MHz, acetone-d6); Figure S16: Zooms of 1H NMR spectrum of H-C12 s-r Alg from s-r Alg in the zones: (a) 5.10-4.40 ppm; (b) 4.34-3.81 ppm; (c) 1.75-0.80 ppm (400 MHz, acetone-d6); Figure S17: 1H NMR spectrum of H-C8 crude Alg from crude Alg (δ: 5.20-3.80 ppm) (400 MHz, CD3OD); Figure S18: Zoom of 1H NMR spectrum of H-C8 crude Alg from crude Alg in the zone 1.76-0.85 (400 MHz, CD3OD); Figure S19: Biodegradability results (H-C12 s-r Alg) according to the OCDE 301 B method; Figure S20: Biodegradability results (SLES) according to the OCDE 301 B method.
Author Contributions
F.P., C.B., S.P.A., T.W., G.N., L.D. and T.B conceived and designed the experiments; F.P., G. N. and C.B. performed the experiments and/or analysed the data; T.B., S.P.A., T.W., G.N and L.D. wrote the paper. All authors have read and agreed to the published version of the manuscript.
Acknowledgments
This work was funded by ADEME and SurfactGreen.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Hayes, D. G.; Smith, G. A. in Biobased Surfactants (Second Edition): Synthesis, Properties, and Applications. Hayes, D. G.Solaiman, K. Y. & Ashby, R. D. Eds, Academic Press and AOCS Press, 2019, pp. 3-38.
- Ribinsky, W.; Hill, K. Alkyl polyglycosides - Properties and applications of a new class of surfactants. Ang. Chem. Int. Edit. 1998, 37, 1328–1345. [Google Scholar] [CrossRef]
- Hill, K.; von Rybinsky, W.; Stoll, G. Eds, Alkyl Polyglucosides, Technology, Properties and Applications, Weinheim, 1996; Hill, K. In Surfactants from renewable Resources, Kjellin M & Johansson, J. Eds, John Wiley & Sons, ltd, 2010.
- Sugar-based Surfactants: fundamental and applications, C. C. Ruiz Ed., Surfactant Science Series, CRC Press Taylor & Francis Group, 2009, vol. 143.
- Jérôme, F.; Marinkovic, S.; Estrine, B. Transglycosylation: A key reaction to access alkylpolyglycosides from lignocellulosic biomass. ChemSusChem 2018, 11, 1395-1409.
- Ji, S.; Shen, W.; Chen, L.; Zhang, Y.; Wu, X.; Fan, Y.; Fu, F.; Chen. G. Synthesis and properties of sugar-b ased surfactants alkoxyethyl -D-glucopyranoside. Colloids Surf. A: Physicochem. Eng. Asp. 2019, 564, 59-68.
- Stubbs, S.; Yousaf, S.; Khan, I. A review on the synthesis of bio-based surfactants using green chemistry principles. DARU J. Pharm.Sci. 2022, 30, 407–426.
- Abdellahi, B.; Bois, R.; Golonu, S.; Pourceau, G.; Lesur, D.; Chagnault, V.; Drelich, A.; Pezron, I.; Nesterenko, A. Wadouachi, A. Synthesis and interfacial properties of new 6-sulfate sugar-based anionic surfactants. Tetrahedron Lett. 2021, 74, 153113.
- Abka-khajouei, R.; Tounsi, L.; Shahabi, N.; Patel, A. K.; Abdelkafi, S.; Michaud, P. Structures, properties and applications of alginates, Mar. Drugs 2022, 20, 364. [Google Scholar]
- Roussel, M.; Benvegnu, T.; Lognoné, V.; Le Deit, H.; Soutrel, I.; Laurent, I.; Plusquellec, D. Synthesis and physico-chemical properties of novel biocompatible alkyl D-mannopyranosiduronate surfactants derived from alginate. Eur. J. Org. Chem. 2005, 3085–3094. [Google Scholar] [CrossRef]
- Brault, D.; Heyraud, A.; Lognoné, V. . Roussel, M. Methods for obtaining oligomannuronates and guluronates, products obtained and use thereof, WO03099870 (A2), 2003.
- Perez, R. The culture of marine algae throughout the world, IFREMER, 1992, 613.
- Renault, L.; Marchal, R.; Le Guennic, B.; Roussel, X.; Divet, P.-Y.; Benvegnu, T. Direct conversion of alginate oligo- and polysaccharides into biodegradable and non-ecotoxic anionic furanic surfactants- An experimental and mechanistic study. Adv. Sustainable Syst. 2021, 2100108. [Google Scholar] [CrossRef]
- Boyère, C.; Galle, F.; Pessel, F.; Roussel, X. Method fot obtaining surfactant compositions from raw plant materials, WO2022/013500 A1, 2022.
- OECD guideline for testing of chemicals. Freshwater Alga and Cyanobacteria, Growth Inhibition Test. OECD/OCDE 201, July 2011. 20 July.
- OECD guideline for testing of chemicals. Daphnia sp., Acute Immobilisation Test. OECD/OCDE 202, April 2004.
- OECD guideline for testing of chemicals. Fish, Acute Toxicity Test. OECD/OCDE 203, July 1992.
- Spitsbergen, J. M.; Kent, M. L. The state of the art of the zebrafish model for toxicology and toxicologic pathology research-advantages and current limitations. Toxicol. Pathol. 2003, 31, 62–87. [Google Scholar] [PubMed]
- OECD guideline for testing of chemicals. Readily Biodegradability, OECD/OCDE 301, July 1992.
Scheme 1.
Synthesis of butylated mannuronate monomers 1-3 in addition to side-product 5 (BDMF) and butylated guluronate monomers 4-5.
Scheme 1.
Synthesis of butylated mannuronate monomers 1-3 in addition to side-product 5 (BDMF) and butylated guluronate monomers 4-5.
Scheme 2.
Synthesis of dodecyl mannuronate 7-9 and guluronate 10-12 ester/lactone monomers.
Scheme 3.
One pot and cascade mode synthesis of H-C12 Man-based surfactant composition.
Scheme 4.
One pot and cascade mode synthesis of H-C12 Gul-based surfactant composition.
Scheme 6.
One pot and cascade mode synthesis of H-C12 s-r Alg surfactant composition.
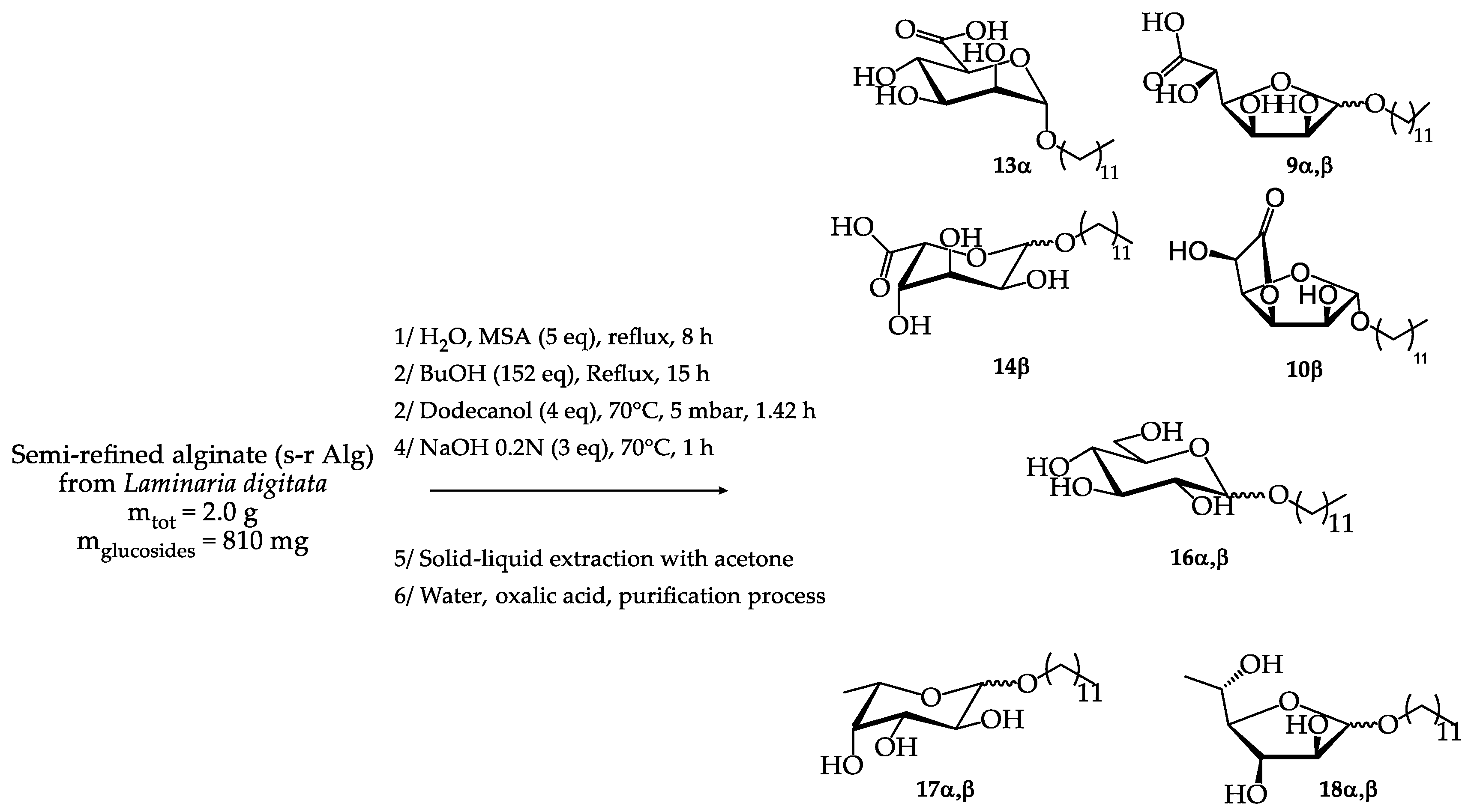
Figure 2.
Cascading one-pot process for the production of surfactant composition from crude seaweeds.
Figure 2.
Cascading one-pot process for the production of surfactant composition from crude seaweeds.
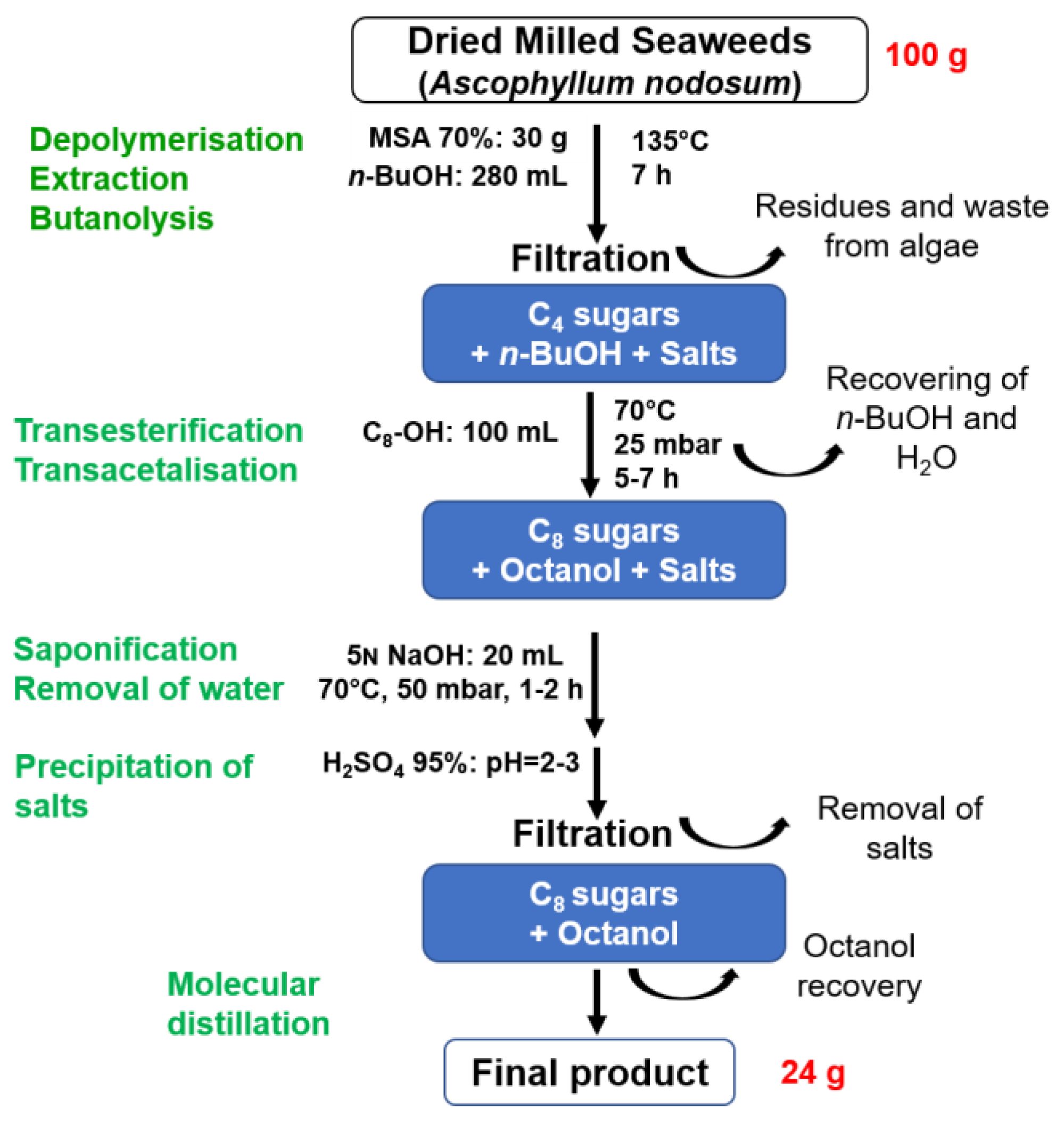
Figure 3.
Proposed surfactant composition H-C8 crude Alg based on 1H NMR analysis.
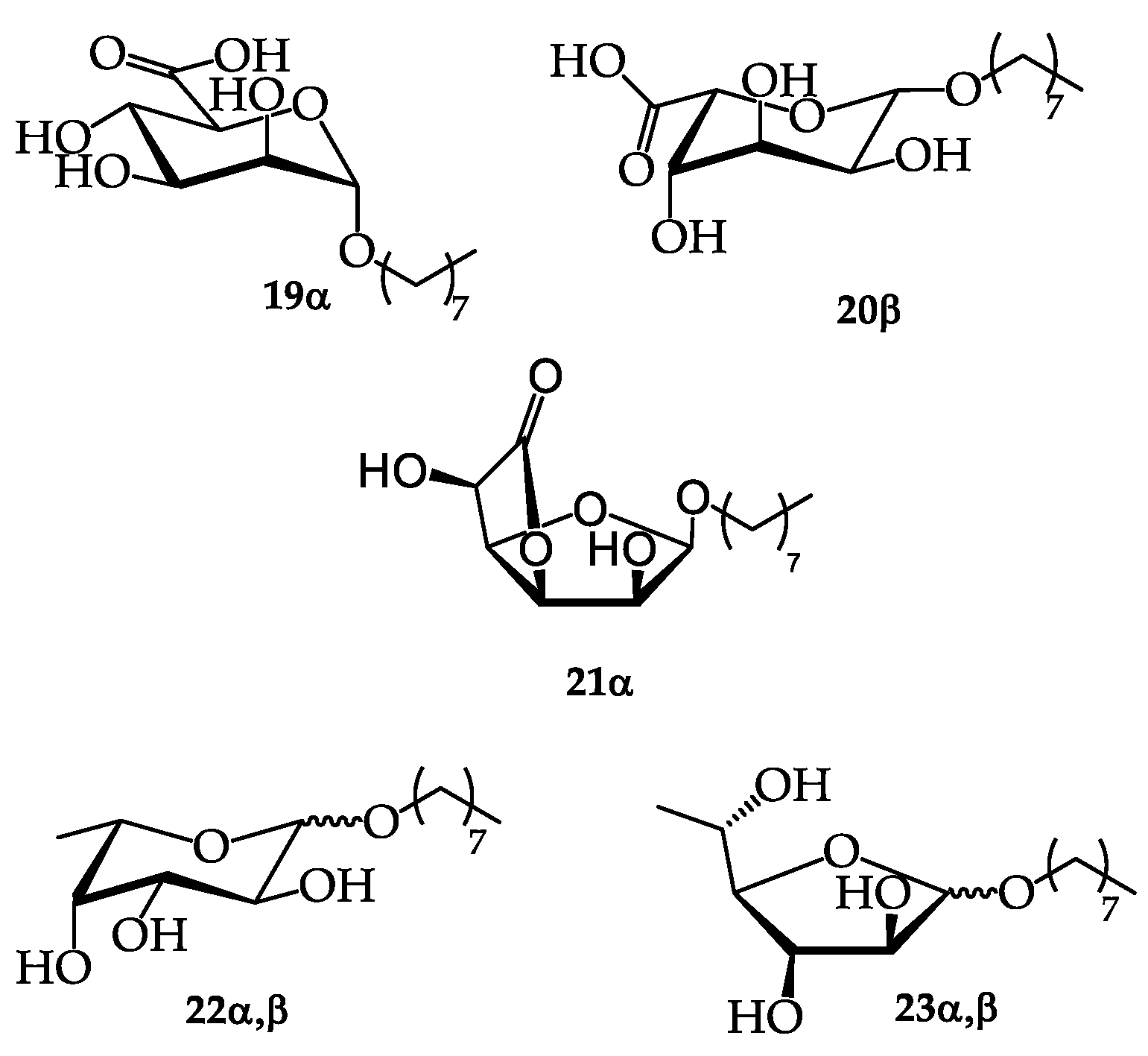
Figure 4.
Values of the surface tension of H-C12-based surfactant compositions derived from dodecanol.
Figure 4.
Values of the surface tension of H-C12-based surfactant compositions derived from dodecanol.
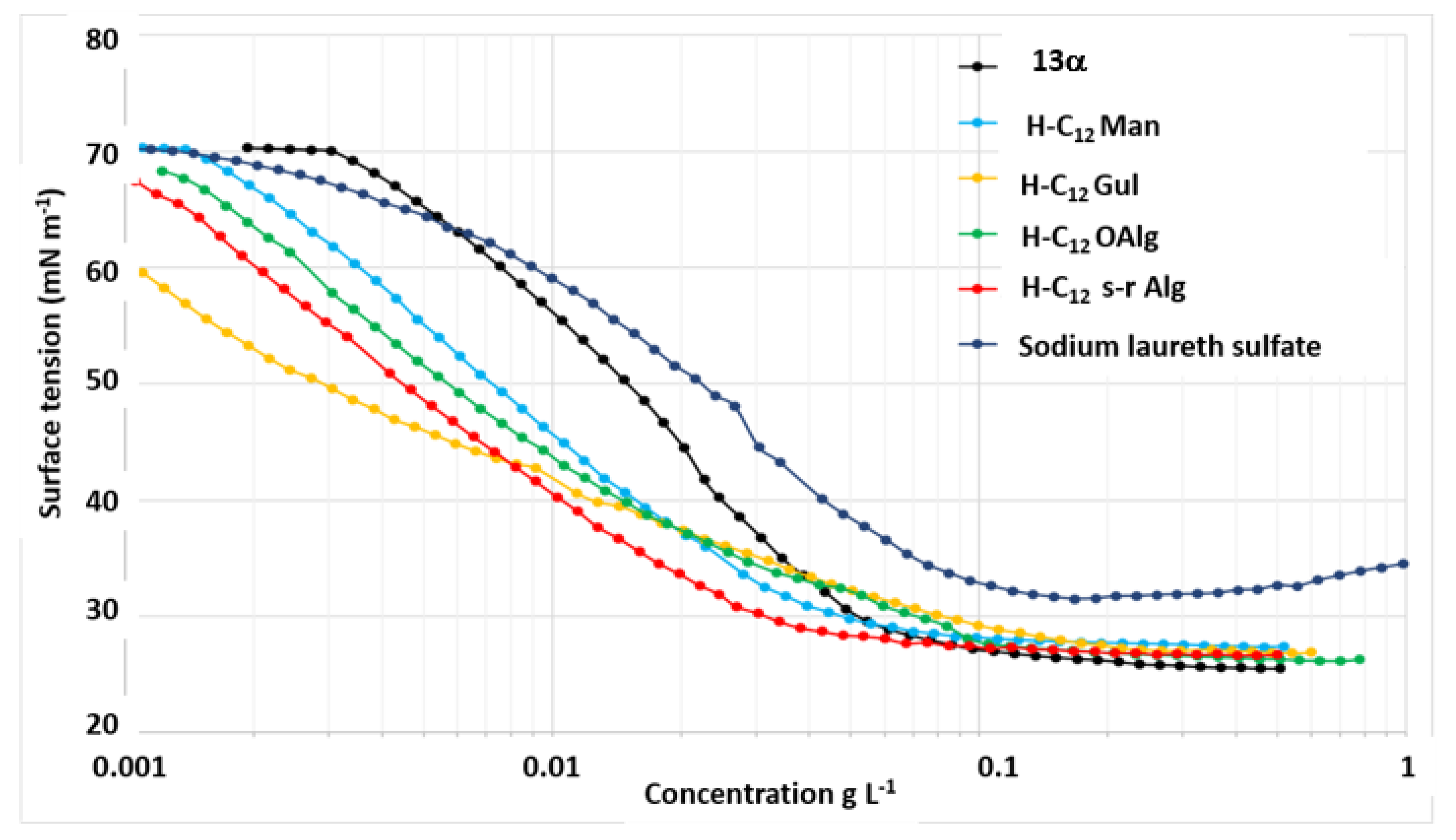

Table 1.
Characteristics of oligomannuronate (OM).
| Description | Method | Result | Units |
| Solids | Constant weight at 103°C | 95.6 | %dry/crude |
| Mineral matter | 12 h, 550°C | 31.9 | %dry/crude |
| Ratio (M/G) | By calculation, proton NMR | 2.7 | |
| DP | By calculation | 4.0 |
Table 2.
Characteristics of oligoguluronate (OG).
| Description | Method | Result | Units |
| Solids | Constant weight at 103°C | 100.0 | %dry/crude |
| Mineral matter | 12 h, 550°C | 26.3 | %dry/crude |
| Ratio (M/G) | By calculation, proton NMR | 0.04 | |
| DP | By calculation | 30.0 |
Table 3.
Characteristics of oligoalginate (OAlg).
| Description | Method | Result | Units |
| Solids | Constant weight at 103°C | 90.1 | %dry/crude |
| Mineral matter | 12 h, 550°C | 44.3 | %dry/crude |
| Ratio (M/G) | By calculation, proton NMR | 1.4 | |
| DP | By calculation | 12.7 |
Table 4.
Characteristics of semi-refined alginates (s-r Alg).
| Description | Method | Result | Units |
| Solids | Constant weight at 103°C | 94.9 | %dry/crude |
| Mineral matter | 12 h, 550°C | 47.0 | %dry/crude |
| Mannuronic and guluronic content | Methanolysis | 29.2 | %dry/crude |
| Glucose content | Methanolysis | 10.9 | %dry/crude |
| Xylose content | Methanolysis | <0.5 | %dry/crude |
| Fucose content | Methanolysis | 2.1 | %dry/crude |
| Ratio (M/G) | By calculation, proton NMR | 2.6 |
Table 5.
1H NMR (CDCl3, 400 MHz) characteristics of the butylated lactone, furanose and pyranose mannuronate 1-3 and guluronate 4-5 monomers identified on the crude mixture C4-C4 Man and/or in the fractions isolated after column chromatography.
Table 5.
1H NMR (CDCl3, 400 MHz) characteristics of the butylated lactone, furanose and pyranose mannuronate 1-3 and guluronate 4-5 monomers identified on the crude mixture C4-C4 Man and/or in the fractions isolated after column chromatography.
| Compounds | δH1 (ppm) (multiplicity) | J1,2(Hz) | Form |
| 1α(Man) | 4.91 (d) | 1.7 | α-Pyranose |
| 2α(Man) | 4.97 (s) | - | α-Furanose |
| 3α(Man) | 5.05 (d) | 2.0 | α-Lactone |
| 3β(Man) | 5.03 (d) | 4.6 | β-Lactone |
| 4α(Gul) | 4.95 (d) | 4.4 | α-Lactone |
| 4β(Gul) | 5.06 (s) | - | β-Lactone |
| 5β(Gul) | 4.91 (s) | - | β-Furanose |
Table 6.
Yield and/or mass of butyl mannuronate 1-3 and guluronate 4-5 monomers, as well as furan derivative 6 isolated after column chromatography, depending on MSA quantity. Reaction conditions: crude OM = 500 mg (OM = 326 mg), nuronate unit = 1.81 mmol, BuOH = 151 eq., H2O (1.7-2 mL/g of crude OM) 140°C, 7 h.
Table 6.
Yield and/or mass of butyl mannuronate 1-3 and guluronate 4-5 monomers, as well as furan derivative 6 isolated after column chromatography, depending on MSA quantity. Reaction conditions: crude OM = 500 mg (OM = 326 mg), nuronate unit = 1.81 mmol, BuOH = 151 eq., H2O (1.7-2 mL/g of crude OM) 140°C, 7 h.
| MSA (n eq) | ||||
| Compounds | 1.7 | 2.2 | 2.6 | 4.7 |
| 1α | 183 mg(yld = 33%) | 132 mg(yld = 24%) | 150 mg (yld = 27%) | 69 mg (yld = 12%) |
| Mannuronates 2,3 + Guluronates 4,5 | 126 mg | 118 mg | 117 mg | 31 mg |
| 6 | 57 mg | 166 mg | 148 mg | 380 mg |
Table 7.
Yield and/or mass of dodecyl mannuronate and guluronate monomers 7-12 isolated after two successive column chromatographies, depending on DodOH and MSA quantities. Reaction conditions: (i) crude OM = 500 mg (OM = 326 mg), nuronate unit = 1.81 mmol, MSA = 1.7 eq, BuOH = 151 eq, H2O (1.7-2 mL/g of crude OM), 140°C, 7 h; (ii) DodOH (n’ eq), MSA (n eq), 5 mbar, 2.17 h. a Reaction time for the second step = 1.75 h.
Table 7.
Yield and/or mass of dodecyl mannuronate and guluronate monomers 7-12 isolated after two successive column chromatographies, depending on DodOH and MSA quantities. Reaction conditions: (i) crude OM = 500 mg (OM = 326 mg), nuronate unit = 1.81 mmol, MSA = 1.7 eq, BuOH = 151 eq, H2O (1.7-2 mL/g of crude OM), 140°C, 7 h; (ii) DodOH (n’ eq), MSA (n eq), 5 mbar, 2.17 h. a Reaction time for the second step = 1.75 h.
| Entries | DodOH (n’ eq) | MSA (n eq) | 7α | Mannuronates 8,9 + Guluronates 10-12 |
| 1a | 16 | 2 | 229 mg(yld = 24%) | 153 mg |
| 2 | 4 | 1 | 240 mg(yld = 25%) | 168 mg |
| 3 | 4 | 0.5 | 200 mg(yld = 21%) | 184 mg |
Table 8.
1H NMR (CDCl3, 400 MHz) characteristics of the dodecyl lactone, furanose and pyranose mannuronate 7-9 and guluronate 10-12 monomers identified on the crude mixture C12-C12 Man (after first column chromatography). n. d. = not detectable/determined.
Table 8.
1H NMR (CDCl3, 400 MHz) characteristics of the dodecyl lactone, furanose and pyranose mannuronate 7-9 and guluronate 10-12 monomers identified on the crude mixture C12-C12 Man (after first column chromatography). n. d. = not detectable/determined.
| Compounds | δH1 (ppm) (multiplicity) | J1,2(Hz) | Form |
| 7α(Man) | 4.92 (d) | 1.7 | α-Pyranose |
| 8α(Man) | 4.97 (s) | - | α-Furanose |
| 9α(Man) | 5.05 (d) | 2.0 | α-Lactone |
| 9β(Man) | 5.03 (d) | 4.5 | β-Lactone |
| 10α(Gul) | 4.95 (d) | 4.5 | α-Lactone |
| 10β(Gul) | 5.07 (s) | - | β-Lactone |
| 11β(Gul)* | n.d. | n.d. | β-Pyranose |
| 12β(Gul) | 4.91 (s) | - | β-Furanose |
*Characteristic signal: δ H5 = 4.52 ppm (d), J=1.0 Hz.
Table 9.
1H NMR (CD3OD, 400 MHz) characteristics of the C12 lactone, furanose and pyranose mannuronate 9,13 and guluronate 10,14,15 monomers present in the crude mixture H-C12 Man. n. d. = not detectable/determined.
Table 9.
1H NMR (CD3OD, 400 MHz) characteristics of the C12 lactone, furanose and pyranose mannuronate 9,13 and guluronate 10,14,15 monomers present in the crude mixture H-C12 Man. n. d. = not detectable/determined.
| Compounds | δH1 (ppm) (multiplicity) | J1,2(Hz) | Form |
| 13α(Man) | 4.80 (d) | 2.1 | α-Pyranose |
| 9α(Man) | 5.00 (d) | 1.6 | α-Lactone |
| 9β(Man) | 4.96 (d) | 3.8- | β-Lactone |
| 14β(Gul) | 4.62 (d) | 8.3 | β-Pyranose |
| 10α(Gul) | 4.92 (d) | 4.4 | α-Lactone |
| 10β(Gul) | 4.98 (s) | - | β-Lactone |
| 15β(Gul)* | n.d. | n.d. | β-Furanose |
*Characteristic signal: δ H5 = 4.29 ppm (d), J=2.2 Hz; **Characteristic signal: δ H = 5.11 ppm (d), J=6.5 Hz.
Table 10.
1H NMR (CD3OD, 400 MHz) characteristics of the C12 lactone, furanose and pyranose mannuronate 13 and guluronate 10,14,15 monomers identified on the crude mixture H-C12 Gul. n. d. = not detectable/determined.
Table 10.
1H NMR (CD3OD, 400 MHz) characteristics of the C12 lactone, furanose and pyranose mannuronate 13 and guluronate 10,14,15 monomers identified on the crude mixture H-C12 Gul. n. d. = not detectable/determined.
| Compounds | δH1 (ppm) (multiplicity) | J1,2(Hz) | Form |
| 13α(Man) | 4.80 (d) | 2.1 | α-Pyranose |
| 14β(Gul) | 4.62 (d) | 8.4 | β-Pyranose |
| 10α(Gul) | 4.92 (d) | 4.5 | α-Lactone |
| 10β(Gul) | 4.98 (s) | - | β-Lactone |
| 15β(Gul)* | n.d. | n.d. | β-Furanose |
*Characteristic signal: δ H5 = 4.29 ppm (d), J=2.0 Hz.
Table 11.
1H NMR (CD3OD, 400 MHz) characteristics of the C12 lactone, furanose and pyranose mannuronate 9,13 and guluronate 10,14,15 monomers identified on the crude mixture H-C12 OAlg. n. d. = not detectable/determined.
Table 11.
1H NMR (CD3OD, 400 MHz) characteristics of the C12 lactone, furanose and pyranose mannuronate 9,13 and guluronate 10,14,15 monomers identified on the crude mixture H-C12 OAlg. n. d. = not detectable/determined.
| Compounds | δH1 (ppm) (multiplicity) | J1,2(Hz) | Form |
| 13α(Man) | 4.80 (d) | 2.1 | α-Pyranose |
| 9α(Man)* | n.d. | n.d. | α-Lactone |
| 9β(Man) | 4.95 (d) | 4.1- | β-Lactone |
| 14β(Gul) | 4.61 (d) | 8.2 | β-Pyranose |
| 10α(Gul) | 4.91 (d) | 4.5 | α-Lactone |
| 10β(Gul) | 4.97 (s) | - | β-Lactone |
| 15β(Gul)** | n.d. | n.d. | β-Furanose |
* Characteristic signal: δ H4 = 4.72 ppm (td), J=4.7, 1.5 Hz; **Characteristic signal: δ H5 = 4.29 ppm (d), J=2.0 Hz.
Table 12.
1H NMR (acetone-d6, 400 MHz) characteristics of the C12 lactone, furanose and pyranose mannuronate 9,13 and guluronate 10,14 monomers in addition to C12 fucosides 17,18 identified on the crude mixture H-C12 s-r Alg. n. d. = not detectable/determined.
Table 12.
1H NMR (acetone-d6, 400 MHz) characteristics of the C12 lactone, furanose and pyranose mannuronate 9,13 and guluronate 10,14 monomers in addition to C12 fucosides 17,18 identified on the crude mixture H-C12 s-r Alg. n. d. = not detectable/determined.
| Compounds | δ H1 (ppm) (multiplicity) | J1,2(Hz) | Form |
| 13α(Man) | 4.85 (d) | 2.0 | α-Pyranose |
| 9α(Man)* | n.d. | n.d. | α-Lactone |
| 9β(Man) | 4.95 (d) | 4.7- | β-Lactone |
| 14β(Gul)** | n.d. | n.d. | β-Pyranose |
| 10β(Gul) | 5.03 (s) | - | β-Lactone |
| 17α(Fuc) | 4.83 (d) | 3.8 | α-Pyranose |
| 17β(Fuc) | 4.27 (d) | 7.7 | β-Pyranose |
| 18α (Fuc) | 4.75 (d) | 3.6 | α-Furanose |
| 18β(Fuc) | 4.75 (d) | n.d. | β-Furanose |
* Characteristic signal: δ H4 = 4.80 ppm (td), J= 4.9, 1.5 Hz; **Characteristic signal: δ H5 = 4.51 ppm (d), J=1.7 Hz.
Table 13.
1H NMR (CD3OD, 400 MHz) characteristics of the C8 lactone, furanose and pyranose mannuronate 19 and guluronate 20,21 monomers in addition to C8 fucosides 22,23 identified on the mixture H-C8 crude Alg derived from brown seaweeds. n. d. = not detectable/determined.
Table 13.
1H NMR (CD3OD, 400 MHz) characteristics of the C8 lactone, furanose and pyranose mannuronate 19 and guluronate 20,21 monomers in addition to C8 fucosides 22,23 identified on the mixture H-C8 crude Alg derived from brown seaweeds. n. d. = not detectable/determined.
| Compounds | δ H1 (ppm) (multiplicity) | J1,2(Hz) | Form |
| 19α(Man) | 4.82 (d) | 2.0 | α-Pyranose |
| 20β(Gul) | 4.64 (d) | 8.1 | β-Pyranose |
| 21α(Gul) | 4.93 (d) | 4.6 | α-Lactone |
| 22α(Fuc) | 4.78 (d) | 3.8 | α-Pyranose |
| 22β(Fuc) | 4.26 (d) | 7.8 | β-Pyranose |
| 23α(Fuc) | 4.72 (d) | 3.7 | α-Furanose |
| 23β(Fuc) | 4.75 (d) | 2.2 | β-Furanose |
Table 14.
Classification of substances that may have acute toxicity to the aquatic environment.
| Category Acute 1 (very toxic) | |
| CL50 96h (for the fish) | ≤ 1 mg L-1 and/or |
| CE50 48h (for the shellfish) | ≤ 1 mg L-1 and/or |
| CEr50 72h (for the seaweed) | ≤ 1 mg L-1 |
| Category Acute 2 (toxic) | |
| CL50 96h (for the fish) | > 1 but ≤ 10 mg L-1 and/or |
| CE50 48h (for the shellfish) | > 1 but ≤ 10 mg L-1 and/or |
| CEr50 72h (for the seaweed) | > 1 but ≤ 10 mg L-1 |
| Category Acute 3 (poorly toxic) | |
| CL50 96h (for the fish) | > 10 but ≤ 100 mg L-1 and/or |
| CE50 48h (for the shellfish) | > 10 but ≤ 100 mg L-1 and/or |
| CEr50 72h (for the seaweed) | > 10 but ≤ 100 mg L-1 |
| Above 100 mg L-1, the substance is considered as non-toxic | |
Table 15.
Results of ecotoxicity tests on SLES, H-C12 s-r Alg.
| Test | Effect | Toxicological Descriptor* | SLES | H-C12 s-r Alg |
| Microalgae | Rate of growth | CEr50 - 72 h | 7.0 mg L-1Toxic | 45.8 mg L-1Poorly toxic |
| Daphnia | Immobilization | CE50 - 24 h | mg L-1Poorly toxic | > 100 mg L-1Non-toxic |
| Fish | Mortality | CL50 - 96 h | 11.2 mg L-1Poorly toxic | 67.0 mg L-1Poorly toxic |
*The toxicological descriptors C X - T correspond to the concentrations causing an effect on X % of the population after a time T. NOEC (No observed effect concentration) is the highest concentration in the range of tests performed that do not cause significant effects on organisms.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



