
Submitted:
30 May 2023
Posted:
31 May 2023
You are already at the latest version
Abstract
Fungi of the genus Aspergillus are widespread in the environment where they produce large quantities of airborne conidia. Inhalation of Aspergillus spp. conidia in immunocompromised individuals can cause a wide spectrum of diseases, ranging from hypersensitivity responses to lethal invasive infections. Upon deposition in the lung epithelial surface, conidia encounter and interact with complex microbial communities that constitute the lung microbiota. The lung microbiota has been suggested to influence the establishment and growth of Aspergillus spp. in the human airways. However, the mechanisms underlying this interaction have not yet been sufficiently investigated. In this study, we aimed to evaluate the presence of commensal bacteria antagonistic to Aspergillus in the lung. To this end, we enriched and isolated bacterial strains able to inhibit the germination of conidia from bronchoalveolar lavage fluid (BALF) samples of lung transplant recipients. We used a novel enrichment method based on a soft agar overlay plate assay in which bacteria are directly in contact with conidia and for which inhibition can be readily observed during enrichment. We isolated a total of five bacterial strains, identified as Pseudomonas aeruginosa, and able to inhibit the germination and growth of Aspergillus fumigatus in a soft agar confrontation assay, as well as in a high-throughput multiplate assay. Moreover, we also showed a strong inhibition of A. fumigatus growth on Calu-3 cell culture monolayers. However, the isolated P. aeruginosa strains were shown to cause significant damage to the cell monolayers. Overall, we validated this novel one-step enrichment approach for the isolation of bacterial strains antagonistic to A. fumigatus from BALF samples. This opens up a new venue for targeted enrichment of antagonistic bacterial strains against specific fungal pathogens.
Keywords:
lung microbiome
; bronchoalveolar lavage fluid (BALF)
; antagonistic bacteria
; biocontrol
; Pseudomonas aeruginosa
; aspergillosis
1. Introduction
Fungal pathogens are estimated to kill more than 1.5 million people worldwide every year [1]. This is notably the case of fungi belonging to the genus Aspergillus, which are ubiquitous in the environment [2], and whose conidia can reach a density of up to 108 per m3 of air [3]. Aspergillus spp. are opportunistic pathogens affecting 14 million people worldwide [4] and are associated with a wide spectrum of diseases, called aspergillosis. A. fumigatus, which is the most common causative agent of aspergillosis, has been recently classified as one of the four fungal pathogens of critical importance in the very first fungal priority pathogens list published by the WHO to guide research and public actions [5]. The clinical manifestations of aspergillosis range from hypersensitive reactions to lethal invasive infections, depending on the immune status of the host [6,7]. While conidia are cleared either via mucociliary movement, or phagocytosis by alveolar macrophages in an immunocompetent setting, this is not the case in an immunocompromised host, where conidia are not adequately cleared, thus leading to the outgrowth of Aspergillus in the lungs [6,7]. Once inside the lung, conidia are confronted to microenvironmental conditions, such as pH, iron bioavailability, or hypoxia, that can impact their germination and growth [8,9,10,11,12,13,6,14].
While aspergillosis development has been widely shown to be influenced by host and microenvironmental conditions, the specific contribution of the lung microbiota as a major factor influencing colonisation and growth of Aspergillus spp. has not yet been sufficiently addressed. Hérivaux and colleagues [15] showed that a particular lung microbiota community structure characterized by an increased abundance of the bacterial genera Staphylococcus, Escherichia, Paraclostridium, and Finegoldia, was associated with the development of invasive pulmonary aspergillosis (IPA) in a cohort of 104 critically ill immunocompromised subjects. Recently, another study from Ao et al. [16] revealed that the lung microbiota, particularly Streptococcus salivarius, Prevotella timonensis and Human betaherpesvirus 5, is strongly associated with laboratory biomarkers linked to inflammation and the immune response, such as for example total lymphocytes counts, serum albumin, or serum lactate dehydrogenase, and clinical outcomes in community-acquired pneumonia patients with IPA. However, the underlying mechanisms explaining the association between the lung microbiota and disease outcome in patients suffering from invasive aspergillosis still needs to be investigated further. The interaction with bacteria inhabiting the lungs might also act as a factor affecting conidia germination and subsequent fungal growth [7,17].
The aim of this study was to evaluate the presence of bacteria inhibiting the germination and growth of A. fumigatus conidia in the lung microbiota using bronchoalveolar lavage fluid (BALF) samples from subjects who underwent lung transplantation. To this end, we first enriched and isolated five antagonistic bacterial strains able to inhibit the germination of conidia using a novel enrichment method based on a soft agar overlay plate assay in which bacteria are directly in contact with conidia. All the bacteria were identified as Pseudomonas aeruginosa and their inhibitory effect was observed already during the enrichment by the presence of an inhibition halo. We then selected two of those for subsequent co-culture confrontation assays on Calu-3 cell culture monolayers.
2. Materials and Methods
2.1. Culturing of A. fumigatus
The A. fumigatus strains used in this study are summarized in Table 1. The strains were routinely cultured on Malt Extract Agar (MEA) medium plates, which was composed of 30 g/L malt extract (Sios Homebrewing GmbH, Wald, Switzerland), 5 g/L casein peptone (LLG Lab Logistics Group GmbH, Meckenheim, Germany), and 15 g/L technical agar (Biolife Italiana, Milano, Italy) Plates were incubated at room temperature for 10 days. The wild-type (WT) isolate was obtained from the Westerdijk Fungal Biodiversity Institue – KNAW (CBS 144.89), and the red-shifted bioluminescent strain was kindly provided by Prof. Dr. Matthias Brock from the University of Nottingham, UK.
2.2. Collection of Aspergillus spp. conidia
Conidia were collected from two 10-days old MEA plates. Briefly, 5 mL of filter-sterilized Dulbecco’s Phosphate-Buffered Saline (DPBS, PAN-Biotech GmbH, Aidenbach, Germany), supplemented with 0.16% v/v Tween® 80 (Sigma-Aldrich/Merck, Darmstadt, Germany) were added on each plate. The mycelial surface was rubbed using a delta cell spreader to dislodge and suspend conidia. The buffer containing conidia was collected in a 50 mL-Falcon tube, and the whole procedure was repeated once. Conidia suspensions were then filtered through a 40 µm-cell strainer into a new 50 mL-Falcon tube, and centrifugated at 2000 × g for 10 min. Supernatant was removed and pellet was rinsed three times by resuspending it in 5 mL DPBS. Conidia were then counted with an Improved Neubauer Counting Chamber and stored at 4°C until use. CFU counting was also perfomed in order to correct for viable conidia.
2.3. Collection and processing of bronchoalveolar lavage fluid (BALF) samples
Subjects underwent transoral bronchoscopy as described in Das, Bernasconi [20]. Briefly, Bronchoalveolar lavage fluid (BALF) was collected via the following procedure: the bronchoscope was wedged either in the middle lobe or lingula of the allograft and 100–150 ml of normal saline was instilled in 50 mL aliquots that were pooled. BALF recovery was measured, and a fraction of 3 ml was stored at 4 °C and centrifuged within 3 h at 14’000 × g for 10 min. Pellet was snap frozen, and stored at −80 °C until further processing. A negative control obtained upon washing a ready-to-use endoscope with sterile saline was prepared following the same procedure. Glycerol samples of BALF were obtained for this study and were stored at −80 °C until use.
2.4. Enrichment and isolation of antagonistic bacterial strains from BALF samples
Antagonistic bacterial strains inhibiting the germination and growth of A. fumigatus were enriched and isolated from BALF samples using a soft agar overlay plate assay. Briefly, 500 mL of filtered-sterilized, 2X RPMI 1640 liquid medium (10.4 g in 500 mL, Capricorn Scientific GmbH, Ebsdorfergrund, Germany, Cat. N° RPMI-A-P10) was mixed with 500 mL of 3% technical agar solution in order to make 1X RPMI 1640 1.5% agar medium and poured in 6 cm Petri plates. A second soft-agar (0.6%) layer of 2 mL of the same medium, in which 10 µL of A. fumigatus CEA10 (112’500 conidia/µL) were added, was poured on top of the 1.5% agar layer. While waiting for the soft agar layer to harden, 5 BALF samples (Table 2) were first thawed and pooled in a 15 mL-Flacon tube and centrifuged at 3000 × g for 10 min in order to concentrate bacterial biomass. Supernatant was removed until approx. 1 mL was left. Then, 250 µL of this BALF concentrate were spread onto the soft agar layer, the plates were incubated for 24 h in a humidified CO2 incubator. After incubation, individual strains were obtained through serial colony purification steps.
2.5. Confrontation assay on a soft agar overlay assay plate
To assess the inhibitory potential of each individual strains obtained, a similar bi-layered soft agar assay was used. A first layer of 1.5% agar RPMI medium was poured into 9 cm Petri dishes. Then, a second layer of 0.6% soft agar, containing 2×106 A. fumigatus CEA10 conidia per 5 mL of medium, was poured on top of the first layer. Five µl bacterial suspensions drops, with an OD set to 1, were spotted on top of the soft agar layer. Four different bacterial strains were spotted in a cross pattern on a single plate to maximize distance between each strain. Plates were incubated at 37°C for 24h.
2.6. High-throughput germination assay
A high-throughput germination assay was developed in order to rapidly test bacterial isolates for their ability to inhibit the germination of conidia and the growth of A. fumigatus. To do so, the five antagonistic bacterial isolates were incubated alone or in the presence of the bioluminescent A. fumigatus strain CEA10b in a white-walled 96-well plate (Costar® Corning, Ref. 3610). Each well contained a final volume of 200 µL of RPMI. For the co-culture, 80’000 A. fumigatus CEA10b conidia and 10 µL of bacterial liquid culture in RPMI (OD =1) were added per well. The controls contained the same concentration of conidia or bacteria alone. 10 µL of 20 mM D-Luciferin solution (1 mM final concentration) were added in the well inoculated with conidia [21]. As positive control of germination inhibition 2 µL of 50 mg/mL solution (in EtOH 99%) of the antifungal cycloheximide was used. Optical density (at 600 nm), as well as bioluminescence (exposition time of 10 seconds) were measured every 30 min for 48 h following the incubation of the plates at 37°C. Measurements were recorded using a Biotek Cytation 5 Cell Imaging Multimode microplate reader (Agilent, USA). All experiments were run in four independent replicates.
2.7. Identification of the bacterial isolates
DNA samples were obtained by thermolysis. Briefly, bacterial biomass was sampled using a wooden toothpick and resuspended in 20 µL in PCR-grade water in a PCR tube. Then, PCR tubes were placed in a thermocycler for 10 min at 98°C. The 16S rRNA gene was amplified using the ALLin™ HS Red Taq Mastermix, 2X kit (highQu, Germany) with the GM3f (5´-AGAGTTTGATCMTGGC-3´)-GM4r (5´-TACCTTGTTACGACTT-3´) primer pair [22]. PCR reactions were performed in a final volume of 25 µL containing: 12.5 µL ALLin™ HS Red Taq Mastermix, 2X, 0.5 µL of each primer, and 1 µL of DNA sample. PCR reactions were run on an Arktik Thermo Cycler (Thermo Fisher Scientific, USA) with the following program: Initial denaturation step at 95°C for 1 min, followed by 40 cylces composed of a denaturation at 95°C for 15 sec, annealing at 56°C for 15 sec, and elongation at 72°C for 30 sec; and a final elongation step at 72°C for 2 min. PCR products were verified by agarose gel electrophoresis and purified using a MultiScreen® plate (Millipore Corporation, United States). DNA was quantified with a BR Qubit assay (Thermo Fisher Scientific, USA). Purified PCR amplicons were then sent to Fasteris, Life Science Genesupport SA (Plan-les-Ouates, Geneva, Switzerland) for Sanger sequencing. Nucleotide sequences were analyzed using NCBI nucleotide BLAST [23].
2.8. Confrontation on Calu-3 cell cultures on permeable Transwell® inserts
Calu-3 cells were obtained from the American Type Culture Collection (Catalog No. ATCC HTB-55, Lot No. 70037771) and used from passages 17 to 21. Calu-3 cells were expanded in T-75 cell culture flasks with vent cap in MEM Eagle (PAN-Biotech GmbH, Aidenbach, Germany, Cat. No. P04-08056) supplemented with 10% of Panexin basic (PAN-Biotech GmbH, Aidenbach, Germany, Cat. No. P04-96950, Lot No. 3430322) and 1% of 100x Penicillin-Streptomycin (PAN-Biotech GmbH, Aidenbach, Germany, Cat. No. P06-07050). The cell cultures were maintained at 37°C in a humidified incubator with 5% CO2. The culture medium was changed every other day. Cells were passaged when they reached 70-80% confluency by trypsinization with 0,05% Trypsin / 0,02% EDTA (Lifeline Cell Technology, USA), and collected after centrifugation at 125 × g for 5 min. Viable cells were counted with a hemocytometer using Trypan Blue PromoKine (Cat. No. PK-CA902-1209).
Cells were seeded onto porous membranes with a density of 108’000 cells/well in 200 μL of culture medium in the apical side of Transwell® inserts in 24-well plates (Corning, USA, Cat. No 3470). Transwell® inserts were first coated with a solution of Collagen Type I (Ibidi, Germany, Cat. No. 50203) at a final concentration of 30 μg/mL prior to the seeding of the cells in order to allow proper cell attachment onto the porous membrane, as described in Arefin, McCulloch [24]. Afterwards, 600 μL of culture medium was added to the basolateral side of Transwell® inserts in the 24-well plates. Transwell® inserts were placed in a humidified incubator at 37°C with 5% CO2 for 15 days until confluence and formation of a monolayer. Medium was changed every other day as described previously. The cultures were observed daily using an EVOS™ XL Core bright field inverted microscope (Thermo Fisher Scientific, USA).
After 15 days of incubation, Transwell® inserts were infected either with 100 conidia and/or bacterial cells per inserts. Prior to infection with the fungus, the apical medium was changed and 5 μL of IVISbrite D-Luciferin RediJect (30 mg/ml D-Luciferin, PerkinElmer, USA, Cat. No. 770504) was added per insert for bioluminescence imaging. Fungus- and bacteria-alone controls were included. Each condition was done in quintuplicate for the single and co-infections, and in octuplicate for the cell-only controls, in two independent experiments. All Transwell® inserts were incubated in a humidified incubator at 37°C with 5% CO2 for 19h.
2.9. Immunofluorescence staining and confocal imaging
Calu-3 cell cultures were fixed with 100 μL 4% paraformaldehyde in DPBS for 15 min at RT. Cells were then rinsed 3 times with 200 μL Tris Buffer Solution 1x pH 7.5 (TBS), with 5-7 min waiting time between each rinse. Cells were permeabilized with 100 μL 0.2% Triton X-100 in TBS for 15 min at RT and rinsed 3 times with 200 μL TBS, with 5-7 min waiting time between each rinse. After that, cells were blocked with 100 μL blocking solution (0.1% NaN3, 3% goat serum, 0.5% casein, 0.025% Tween 20 in TBS 1x pH 7.5, kindly provided by the Live Cell Imaging Core Facility, Department of Biomedical Research, University of Bern) for 30 min at RT. ZO-1 Mouse anti-Human conjugated with Alexa Fluor 594 (Thermo Fisher Scientific, USA, Cat. No. 339194) was prepared in the blocking solution (1/100). The actin stain (ActinGreen™ 488 ReadyProbes™ Reagent) and the nuclei counterstain (NucBlue™ Live ReadyProbes™ Reagent) were added to the same solution (2 drops/mL and 1 drop/mL, respectively). Anti-Aspergillus rabbit polyclonal primary antibody (Abcam, UK, Cat. No. ab20419) was also added (1/200) in the same solution for the conditions where A. fumigatus conidia were inoculated. Fixed cells were incubated with 100 μL solution containing the stains and antibodies overnight at 4°C. The next day, fixed cells were washed 3 times with TBS supplemented with 0.025% Tween 20, with 5-7 min waiting time between each rinse. Goat anti-Rabbit IgG secondary antibody (1/500) conjugated with Alexa Fluor 594 (Thermo Fisher Scientific, USA, Cat. No. A-11012) directed against Anti-Aspergillus antibody were prepared in TBS + 0.025% Tween 20. Fixed cells were incubated with 100 μL buffer containing the secondary antibodies for 3h at RT in the dark. Cells were then washed 3 times with TBS + 0.025% Tween 20 for 7 min. Membranes from the inserts were then finally carefully cut out with a sharp knife, and mounted on a glass slide using the EMS Shield Mount mounting medium (Electron Microscopy Sciences, USA) and imaged with a ZEISS Axio Observer Z1 LSM 980 with Airyscan 2 confocal microscope (Carl Zeiss AG, Germany), using the C-Apochromat 40x/1.20 W Korr objective and the Alexa Fluor 594 (red, λex: 590nm, λem: 610nm), Alexa Fluor 488 (green, λex: 493nm, λem: 517nm), and Alexa Fluor 405 (blue, λex: 401nm, λem: 422nm) channels.
2.10. Assessment of epithelial barrier function: TEER measurements and Lucifer Yellow permeability assay
Trans-epithelial electrical resistance (TEER) was measured before media change using an EVOM3 Epithelial Volt/Ohm Meter with STX4 “chopsticks” electrodes daily for 15 days, starting from day 2 post-seeding. Two measurements were taken per inserts. TEER values were expressed in Ohms (Ω) per insert surface area (0.33 cm2).
Permeability of the Calu-3 monolayer was assessed using the paracellular marker Lucifer Yellow (LY, Lucifer Yellow CH Lithium salt, Biotium, USA, Cat. No. 80015). A 100 μg/mL LY solution was prepared in Hanks’ Balanced Salt Solution (HBSS, PAN-Biotech GmbH, Aidenbach, Germany, Catalog No. P04-32505) and 200 μL of this LY solution was added to the apical compartment, whilst 600 μL of HBSS was added to the basal compartment. Plates were incubated for 2h30 in a humidified incubator at 37°C with 5% CO2, and 100 μL of the basal solution were added to a black flat-bottom 96-well microplate. LY fluorescence intensity was measured with λexc = 485 nm et λem = 535 using a Biotek Cytation 5 Cell Imaging Multimode microplate reader (Agilent, USA).
2.11. Bioluminescence imaging
In order to assess growth of A. fumigatus CEA10b on the Calu-3 monolayers when inoculated alone, or co-inoculated with the P. aeruginosa strains, bioluminescence imaging was performed using the Amersham Imager 680 (GE Healthcare, USA) in the chemiluminescence mode, with an exposure time of 5 min and binning of 32x.
2.12. Cytotoxicity assay
Cytotoxicity was assessed through the quantification of dead cells-associated proteases using the fluorescent kit CytoTox-Fluor™ Cytotoxicity Assay (Promega, USA, Cat. No. G9260) per the manufacturer’s instructions.
2.13. pH measurements and quantification of calcium
pH and calcium concentration were quantified in the culture medium with the iSTAT 1 Blood Analyzer (Abbott, USA) using the CG8+ cartridges (Abbott, USA, Cat. No. 10002376).
2.14. Statistical analyses
All graphical representations were performed with RStudio (Version 2022.12.0+353), and GraphPad Prism 9 (Version 9.5.1). Statistical analyses were performed on GraphPad Prism 9 using an ordinary one-way ANOVA with multiple comparisons. Significance levels used were the following: ns = P > 0.05 ; * = P ≤ 0.05 ; ** = P ≤ 0.01 ; *** = P ≤ 0.001 ; **** = P ≤ 0.0001.
3. Results
3.1. Enrichment and isolation of lung bacteria inhibiting A. fumigatus conidia germination and growth in one-step sof agar plate assay
A one-step soft agar plate assay was used to directly enrich and isolate bacterial strains able to inhibit the germination and growth of A. fumigatus CEA10 conidia from BALF samples. After 24 h of incubation, germination and growth of A. fumigatus conidia is visible in the fungus-alone control plate (Figure 1a). When the concentrated BALF was added onto the soft agar layer containing fungal spores, fungal growth is inhibited by the presence of bacteria and the fungus grew only in the area where bacteria were not present (Figure 1b). Moreover, an inhibition zone was visible in the interaction zone between bacteria and the fungus (double-headed arrow, Figure 1c).
Five macroscopically different bacterial isolates were obtained after the isolation step, which were all identified as Pseudomonas aeruginosa after Sanger sequencing of the 16S rRNA gene (Table 3).
3.2. Soft agar inhibition assay
In order to confirm their inhibitory action, the five BALF P. aeruginosa strains were first placed in confrontation with A. fumigatus using the soft agar assay in RPMI medium. All five bacterial strains showed a clear and strong inhibitory effect on A. fumigatus conidia germination and growth, which was visible by the inhibition halos around bacterial colonies (Figure 2a). Moreover, close-up images of the interaction zones between the bacteria and the fungus showed an inhibition zone where no fungal growth was observed (Figure 2c-g), compared to zones where fungal growth was visible (Figure 2b).
3.3. High-Troughput screening assay
A high-throughput (HTP) germination assay was developed in order to rapidly assess inhibition potential of BALF bacterial isolates against A. fumigatus and select the best candidates for subsequent testing on Calu-3 cell monolayers. Figure 3 shows the kinetic of A. fumigatus conidia germination and growth, through the measurement of bioluminescence over 48 hours, alone or in confrontation with the five BALF P. aeruginosa isolates. When inoculated alone, A. fumigatus shows an exponential increase in bioluminescence signal after 10 h incubation, indicating its growth (Figure S1). The positive inhibition control, i.e., cycloheximide, initially inhibited the germination of A. fumigatus conidia. However, after approximately 30 h incubation, an increase in bioluminescence signal is recorded, indicating germination and growth of the fungus (Figure S1). Regarding the confrontation with the five BALF P. aeruginosa isolates, strain b1 shows a partial inhibition effect against A. fumigatus (Figure S1). On the other hand, strains b2 to b4 were able to completely inhibit the germination and growth of A. fumigatus (Figure S1), shown by a very weak bioluminescence signal. Finally, strain b5 showed a partial inhibition of A. fumigatus growth. Indeed, the bioluminescence signal increased after approximately 6 h post inoculation and reached a plateau after 24 h. Based on these results, BALF P. aeruginosa strains b1 and b2 were selected for the Calu-3 in-vitro confrontations.
3.4. In-vitro confrontation of A. fumigatus with BALF P. aeruginosa isolates on Calu-3 cell monolayers
The effect of inoculation of 100 A. fumigatus CEA10b conidia alone or in co-culture with 100 P. aeruginosa b1 or b2 cells was assessed in Calu-3 cell culture monolayers on Transwell® inserts. We first assessed fungal growth by imaging bioluminescence emission by A. fumigatus. The infection template of each of the three independent plates used is depicted on the top in Figure 4a-c, with the corresponding images taken with the bioluminescence imager on the bottom. Detection of light emission by A. fumigatus is visible by the presence of a black spot in the fungus-infected cell inserts (C+F, Figure 4). However, in the co-infected inserts, i.e., C+F+b1 and C+F+b2, no bioluminescence was detected (Figure 4), suggesting a control of A. fumigatus growth by the bacterial isolates b1 and b2.
We then performed confocal imaging in order to confirm inhibition of A. fumigatus conidia germination by both P. aeruginosa b1 and b2 strains, when co-cultured together on the Calu-3 cell monolayers. After 16 days of incubation, the Calu-3 cells in the ‘no-infection control’ displayed a polygonal shape with the actin filaments clearly visible at the edges of the cells, and expressed tight-junctions (ZO-1 protein in red) (Figure 5a). After 19h of infection, A. fumigatus conidia were able to germinate and grow, as indicated by the presence of hyphae (in red) on top of the monolayer (Figure 5b). Moreover, the orthogonal views of the Z-stacks images of the Calu-3 monolayers infected with A. fumigatus showed that the fungus forms a dense hyphal layer at the surface of the epithelial monolayer (Figure S2a), and that some hyphae penetrate the Calu-3 monolayer (Figure S2b). Infection of the Calu-3 cells with A. fumigatus did not seem to cause significant damage to the monolayer (Figure 5c). This is however not the case with both P. aeruginosa isolates b1 and b2, which caused important damage to the Calu-3 monolayer, as shown by the presence of holes in the epithelial monolayer (Figure 5d,e). Moreover, there tended to be a higher expression of tight junction proteins, and less actin visible at the edges of holes (Figure 5d,e). In the case of the co-infection conditions, we could confirm that P. aeruginosa strains b1 and b2 were able to control the germination and growth of A. fumigatus conidia, since no hyphae were visible on the Calu-3 monolayers (Figure 5f,h, respectively). Moreover, the tight junction protein ZO-1 tended to be more expressed at the edges of holes induced by the bacteria, while actin seemed to be less visible (Figure 5g,i).
We also assessed the epithelial barrier function of the Calu-3 monolayers through trans-epithelial electrical resistance (TEER) measurements and a LY permeability assay. Figure 6a depicts the evolution of TEER over 16 days. TEER showed a regular increase in all conditions until around 2500 Ω/cm2 at day 14 and stayed constant around this value until day 15, when the infection occured. Finally, 19h post-infection, TEER stayed at around 2500 Ω/cm2 for the cells-alone control and the A. fumigatus-alone treatment, and dropped at 1000 Ω/cm2 and below in the co-infection treatments.
The LY permeability assay showed similar results. Indeed, compared to the cell-alone control (C), which is at around 1.2% (Figure 6b), the measured LY permeability was significantly higher in the C+b1, C+b2, C+F+b1 and C+F+b2 conditions. Moreover, we also observed that, in the presence of bacteria b2, the permeability was higher than with bacteria b1 (Figure 6b).
Figure 7 shows the assessment of cytotoxicity upon infection with A. fumigatus alone, or co-inoculated with P. aeruginosa strains b1 or b2. Compared to the cells alone (C), A. fumigatus did not seem to induce a strong cytopathic effect (C+F). The same is true when P. aeruginosa b1 is added alone, or together with A. fumigatus, on the cells (C+b1 and C+F+b1). However, in the presence of P. aeruginosa b2 alone (C+b2), or with A. fumigatus (C+F+b2), the measured fluorescence intensity is significantly higher than in the control cells.
In order to assess the effect of the infection by A. fumigatus and co-inoculation with P. aeruginosa on lung microenvironmental parameters, pH and calcium (Ca2+) concentrations in the culture medium were measured. No difference in pH, or in Ca2+ concentrations, was measured across treatments (Figure 8a & b).
4. Discussion
In this study, we presented a novel one-step enrichment and isolation method based on a soft agar overlay plate assay in which bacteria are directly in contact with Aspergillus spp. conidia and for which inhibition can be easily observed during enrichment by the presence of an inhibition halo around antagonistic bacteria. Traditionally, the enrichment and isolation steps are completely independent from the confrontation. Indeed, antagonistic bacterial strains are first isolated, and then tested against the fungal pathogen of interest in a second step [26,27]. Here, this whole approach is performed in one single step, thus directly enriching for bacterial strains that are antagonistic to the fungal pathogen of interest. While standard confrontation experiments rely on active fungal mycelium as target for bacterial biocontrol, our approach is targeted against conidia [28], which are the infective stage in the developmental cycle of Aspergillus spp. [29]. In fact, once conidia germinate and grow as hyphae, the use of a biocontrol approach might be too late. Colonisation of the lung tissue by Aspergillus spp. is a known risk factor for the development of invasive pulmonary aspergillosis [4]. While conidia germination is known to be influenced by microenvironmental factors [8,9,10,11,12,13,14], the specific contribution of the lung microbiota in the development of Aspergillus spp. is not yet well understood.
In this study, we used BALF samples from subjects who underwent a lung transplantation and were followed up at the Lausanne University Hospital (CHUV), as source for potential bacterial isolates inhibiting the germination of Aspergillus conidia. Enrichment and isolation on bi-layered soft agar RPMI medium containing A. fumigatus conidia yielded five bacterial isolates all identified as P. aeruginosa. RPMI medium was used because of the similarity in composition with the fluid lining the lung epithelium surface [20]. P. aeruginosa is an opportunistic pathogen and is commonly isolated in lung transplanted patients [30,31]. Antibacterials, as well as antifungals, are often used as prophylactic or therapeutic treatments in lung transplanted patients [32,33]. Das, Bernasconi [20] showed that there is a negative relationship between the number of antibiotics administered to transplanted patients and the imbalance of the lung microbiota, with Pseudomonas spp. being more prevalent when increasing the number of administered antibiotics. The fact that three out of five BALF samples we used had been obtained from lung transplanted patient who has received an antibacterial treatment could explain that we only retrieved P. aeruginosa isolates. The lack of diversity in the enrichment could be the result of the composition of the medium used for enrichment and isolation. Indeed, as RPMI contains only one single carbon source, i.e., glucose [34], the restricted number of carbon sources available for growth may have favored only the most competitive strains. Actually, a recent method paper published by Ruhluel, O'Brien [35] described culture media that resembles the composition of healthy sinuses and lungs, as well as cystic fibrosis (CF) sinuses and lungs. This medium contains several carbon sources (i.e., sialic acid, galactose, N-acetyl glucosamine, and glucose) that could permit to isolate a higher diversity of bacterial isolates.
We assessed and confirmed the inhibitory potential of the five BALF P. aeruginosa isolates by using, on the one hand, the same bi-layered soft agar RPMI plate assay employed during the enrichment, and a HTP bioluminescence microplate assay. The HTP bioluminescence microplate assay permitted us to rapidly identify interesting bacterial candidates able to inhibit the germination of A. fumigatus conidia to test in subsequent Calu-3 cell monolayer models. The bioluminescent strain of A. fumigatus CEA10 used in this study was constructed by Resendiz-Sharpe, da Silva [19] by inserting a codon-optimized thermostable-red-shifted firefly luciferase. Since the expression of the red-shifted firefly luciferase is under the control of the constitutively active glyceraldehyde-3-phosphate dehydrogenase promoter (PgpdA), light emission is observed upon germination of conidia and hyphal growth Resendiz-Sharpe, da Silva [19]. P. aeruginosa strains b1 and b2 were selected because of their differential inhibitory effect: strain b1 allowed germination of conidia, while strain b2 completely inhibited conidia germination, which was easily visible in the HTP bioluminescent microplate assay. Furthermore, A. fumigatus CEA10 was confronted to P. aeruginosa strains b1 and b2 on Calu-3 cell monolayers. Inhibition of conidia germination and growth was then assessed by bioluminescence emission, as well as confocal microscopy. Moreover, the impact of these Aspergillus-Pseudomonas confrontations on epithelial barrier function, as well as cytotoxicity, pH, and calcium concentration were also investigated. We confirmed the inhibitory effect of both P. aeruginosa strains, i.e., b1 and b2, on the germination of A. fumigatus conidia and its mycelial growth on Calu-3 cell monolayers. Indeed, no bioluminescent signal was recorded when A. fumigatus conidia were co-inoculated with P. aeruginosa strains b1 and b2, as compared to when A. fumigatus was inoculated alone. To our knowledge, this is the first time a bioluminescent strain of A. fumigatus is used in a cell culture model. As light emission is proportional to fungal growth, bioluminescence measurements could be used as a semi-quantitative approach to measure A. fumigatus colonization on an in-vitro lung cell culture model. The inhibitory effect of P. aeruginosa strains b1 and b2 on A. fumigatus growth was also confirmed through confocal microscopy. While fungal hyphae were visible when A. fumigatus was inoculated alone on Calu-3 cell monolayers, no visible growth was observed in the co-cultures with P. aeruginosa b1 and b2.
The interaction between A. fumigatus and P. aeruginosa is well established and has been reviewed extensively [36,37]. The Aspergillus-Pseudomonas interaction is common in CF patients and relies mainly on iron [38]. Among other mechanisms, Aspergillus and Pseudomonas mutually inhibit each other through the secretion of gliotoxin or siderophores (hydroxamate), and pyoverdine or dirhamnolipids by A. fumigatus and P. aeruginosa, respectively [36,37].
Moreover, P. aeruginosa strains b1 and b2, either inoculated alone, or co-inoculated with A. fumigatus, were found to cause important damage to the Calu-3 cell monolayers. Holes were visible in the Calu-3 monolayers, especially in the presence of P. aeruginosa b2. These observations were confirmed by the cytotoxicity assay, which showed a high fluorescent signal in the presence of P. aeruginosa b2 alone, or co-inoculated with A. fumigatus, suggesting the release of high numbers of dead-cell proteases. P. aeruginosa is known to have a strong cytotoxicity effect in lung epithelial cells, notably through the induction of various forms of cell death, leading to cell lysis [39]. Epithelial barrier integrity was assessed through TEER measurements over 16 days, as well as LY permeability assay as an end-point measurement.
TEER was found to increase regularly until reaching a value of around 2500 Ω/cm2 at day 15, when the infection was done. TEER values in the cell-control only and A. fumigatus-alone infected Calu-3 monolayers stayed constant around 2500 Ω/cm2. However, a strong decrease in TEER was observed when both P. aeruginosa strains were added to Calu-3 cells alone, or co-inoculated with A. fumigatus. P. aeruginosa has a strong cytotoxicity effect [39], and is known to have a disruptive effect on TEER [40]. Permeability assay using the paracellular marker LY showed similar results. However, TEER was found to be more sensitive. TEER is a strong indicator of the integrity of the barrier, even before the ability to measure it using permeability markers [41]. Finally, pH and calcium concentration in the cell culture medium were assessed as important microenvironmental parameters for cellular health. pH was found to be constant at a value of 8 across all conditions tested. The same was true for calcium concentration in the cell culture medium, which was at around 1.5 mM in all conditions tested.
To conclude, we were able to confirm the presence of bacteria inhibiting the germination and growth of A. fumigatus conidia in the lung microbiota with the isolation of five P. aeruginosa isolates from BALF samples coming from subjects who underwent a lung transplantation. These bacteria were able to inhibit the germination and growth of A. fumigatus conidia in different experimental set-ups. Although P. aeruginosa is a known opportunistic lung pathogen, the results presented here validate the experimental pipeline of one-step enrichment and isolation using a soft agar bi-layered plate assay described in this manuscript. This approach opens up a new venue for targeted enrichment of antagonistic bacterial strains against specific fungal pathogens.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Figure S1. Single graphics of the evolution of A. fumigatus conidia germination and growth in the HTP assay in confrontation with the five Pseudomonas aeruginosa strains (b1 – b5); Figure S2. Orthogonal views of confocal microscopic Z-stacks pictures of Calu-3 cells infected by A. fumigatus.
Author Contributions
For research articles with several authors, a short paragraph specifying their individual contributions must be provided. The following statements should be used “Conceptualization, F.P. and P.J.; methodology, F.P. and P.J.; software, F.P. and J.D.; validation, F.P., N.U. and P.J.; formal analysis, F.P., M.M., J.D. and M.G.; investigation, F.P., M.M., J.D. and M.G.; resources, C.VG., N.U. and P.J.; data curation, F.P. and J.D.; writing—original draft preparation, F.P., M.M., J.D. and M.G.; writing—review and editing, All authors; visualization, F.P., M.M., J.D. and M.G.; supervision, P.J.; project administration, P.J.; funding acquisition, P.J., M.P., A.K., E.B. and F.P. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Swiss National Science Foundation through the BRIDGE Discovery program under the grant agreement 194701 (P.J., M.P., A.K.), the Novartis Foundation through the FreeNovation program (P.J.), the Gebert Rüf Stiftung under the grant agreement GRS-064/18 (P.J.), the “Fondation Professeur Placide Nicod” (E.B.), and by the “Ligue Pulmonaire Neuchâteloise” (F.P.).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
All data is available in the main text or the supplementary materials. The R code used for the generation of kinetic bioluminescence graphic in the high-throughput assay can be found at: https://github.com/palmierif/htp-biolum-afum.
Acknowledgments
The authors kindly acknowledge Prof. Dr. Matthias Brock for providing the red-shifted bioluminescent strain of A. fumigatus CEA10, as well as Mr. Carlos Wotzkow and Dr. Fabian Blank from the Live Cell Imaging Core Facility at the University of Bern for assistance during confocal images acquisition.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- Bongomin, F., et al., Global and multi-national prevalence of fungal diseases—estimate precision. Journal of Fungi, 2017. 3. [CrossRef]
- Paulussen, C., et al., Ecology of aspergillosis: insights into the pathogenic potency of Aspergillus fumigatus and some other Aspergillus species. Microbial Biotechnology, 2017. 10(2): p. 296-322.
- Latgé, J.P. and G. Chamilos, Aspergillus fumigatus and Aspergillosis in 2019. Clin Microbiol Rev, 2019. 33(1).
- Gago, S., D.W. Denning, and P. Bowyer, Pathophysiological aspects of Aspergillus colonization in disease. Medical Mycology, 2019. 57(2): p. S219-S227.
- WHO fungal priority pathogens list to guide research, development and public health action. 2022.
- Arastehfar, A., et al., Aspergillus fumigatus and aspergillosis: From basics to clinics. Studies in Mycology, 2021. 100: p. 100115.
- Palmieri, F., et al., Recent Advances in Fungal Infections: From Lung Ecology to Therapeutic Strategies With a Focus on Aspergillus spp. Frontiers in Medicine, 2022. 9. [CrossRef]
- Ortiz, S.C., et al., Novel Insights into Aspergillus fumigatus Pathogenesis and Host Response from State-of-the-Art Imaging of Host-Pathogen Interactions during Infection. Journal of Fungi, 2022. 8(3): p. 264. [CrossRef]
- Barda, O., et al., The pH-Responsive Transcription Factor PacC Governs Pathogenicity and Ochratoxin A Biosynthesis in Aspergillus carbonarius. Frontiers in Microbiology, 2020. 11.
- Bertuzzi, M., et al., The pH-responsive PacC transcription factor of Aspergillus fumigatus governs epithelial entry and tissue invasion during pulmonary aspergillosis. PLoS Pathogens, 2014. 10(10): p. e1004413.
- Bignell, E., et al., The Aspergillus pH-responsive transcription factor PacC regulates virulence. Molecular Microbiology, 2005. 55(4): p. 1072-84.
- Blatzer, M., et al., SidL, an Aspergillus fumigatus transacetylase involved in biosynthesis of the siderophores ferricrocin and hydroxyferricrocin. Applied and Environmental Microbiology, 2011. 77(14): p. 4959-66. [CrossRef]
- Haas, H., M. Eisendle, and B.G. Turgeon, Siderophores in fungal physiology and virulence. Annual Review of Phytopathology, 2008. 46: p. 149-87. [CrossRef]
- Gresnigt, M.S., et al., Reducing hypoxia and inflammation during invasive pulmonary aspergillosis by targeting the Interleukin-1 receptor. Scientific Reports, 2016. 6(1): p. 26490. [CrossRef]
- Hérivaux, A., et al., Lung microbiota predict invasive pulmonary aspergillosis and its outcome in immunocompromised patients. Thorax, 2021. [CrossRef]
- Ao, Z., et al., Clinical characteristics, diagnosis, outcomes and lung microbiome analysis of invasive pulmonary aspergillosis in the community-acquired pneumonia patients. BMJ Open Respir Res, 2023. 10(1). [CrossRef]
- Kolwijck, E. and F.L. van de Veerdonk, The potential impact of the pulmonary microbiome on immunopathogenesis of Aspergillus-related lung disease. European Journal of Immunology, 2014. 44(11): p. 3156-3165.
- Bowyer, P., et al., Telomere-to-telomere genome sequence of the model mould pathogen Aspergillus fumigatus. Nature Communications, 2022. 13(1): p. 5394. [CrossRef]
- Resendiz-Sharpe, A., et al., Longitudinal multimodal imaging-compatible mouse model of triazole-sensitive and -resistant invasive pulmonary aspergillosis. Dis Model Mech, 2022. 15(3). [CrossRef]
- Das, S., et al., A prevalent and culturable microbiota links ecological balance to clinical stability of the human lung after transplantation. 2020: p. 0-3. [CrossRef]
- Brock, M., et al., Bioluminescent Aspergillus fumigatus, a new tool for drug efficiency testing and in vivo monitoring of invasive aspergillosis. Applied and Environmental Microbiology, 2008. 74(22): p. 7023-7035. [CrossRef]
- Muyzer, G., et al., Phylogenetic relationships of Thiomicrospira species and their identification in deep-sea hydrothermal vent samples by denaturing gradient gel-electrophoresis of 16S rDNA fragments. Archives of Microbiology, 1995. 164(3): p. 165-172.
- Altschul, S.F., et al., Basic local alignment search tool. J Mol Biol, 1990. 215(3): p. 403-10.
- Arefin, A., et al., Micromachining of Polyurethane Membranes for Tissue Engineering Applications. ACS Biomaterials Science and Engineering, 2018. 4(10): p. 3522-3533. [CrossRef]
- Quast, J., ggplate: Create Layout Plots of Biological Culture Plates and Microplates. 2023.
- Anith, K.N., N.S. Nysanth, and C. Natarajan, Novel and rapid agar plate methods for in vitro assessment of bacterial biocontrol isolates’ antagonism against multiple fungal phytopathogens. Letters in Applied Microbiology, 2021. 73(2): p. 229-236. [CrossRef]
- Muñoz, C.Y., et al., Biocontrol properties from phyllospheric bacteria isolated from Solanum lycopersicum and Lactuca sativa and genome mining of antimicrobial gene clusters. BMC Genomics, 2022. 23(1): p. 152. [CrossRef]
- Estoppey, A., et al., Improved methods to assess the effect of bacteria on germination of fungal spores. FEMS Microbiology Letters, 2022. 369(1). [CrossRef]
- Verburg, K., et al., Novel Treatment Approach for Aspergilloses by Targeting Germination. J Fungi (Basel), 2022. 8(8). [CrossRef]
- Stjärne Aspelund, A., et al., Microbiological findings in bronchoalveolar lavage fluid from lung transplant patients in Sweden. Transpl Infect Dis, 2018. 20(6): p. e12973. [CrossRef]
- Zeglen, S., et al., Frequency of Pseudomonas aeruginosa colonizations/infections in lung transplant recipients. Transplant Proc, 2009. 41(8): p. 3222-4. [CrossRef]
- Okamoto, K. and C.A.Q. Santos, Management and prophylaxis of bacterial and mycobacterial infections among lung transplant recipients. Ann Transl Med, 2020. 8(6): p. 413. [CrossRef]
- Patel, T.S., et al., Antifungal Prophylaxis in Lung Transplant Recipients. Transplantation, 2016. 100(9): p. 1815-26. [CrossRef]
- Moore, G.E., R.E. Gerner, and H.A. Franklin, Culture of Normal Human Leukocytes. JAMA, 1967. 199(8): p. 519-524.
- Ruhluel, D., et al., Development of liquid culture media mimicking the conditions of sinuses and lungs in cystic fibrosis and health [version 2; peer review: 2 approved]. F1000Research, 2022. 11(1007).
- Sass, G., et al., Aspergillus-Pseudomonas interaction, relevant to competition in airways. Med Mycol, 2019. 57(Supplement_2): p. S228-S232.
- Margalit, A., J.C. Carolan, and K. Kavanagh, Bacterial Interactions with Aspergillus fumigatus in the Immunocompromised Lung. Microorganisms, 2021. 9(2). [CrossRef]
- Keown, K., et al., Coinfection with Pseudomonas aeruginosa and Aspergillus fumigatus in cystic fibrosis. European Respiratory Review, 2020. 29(158): p. 200011.
- Wood, S.J., et al., Pseudomonas aeruginosa Cytotoxins: Mechanisms of Cytotoxicity and Impact on Inflammatory Responses. Cells, 2023. 12(1). [CrossRef]
- Li, J., et al., Pseudomonas aeruginosa Exoprotein-Induced Barrier Disruption Correlates With Elastase Activity and Marks Chronic Rhinosinusitis Severity. Frontiers in Cellular and Infection Microbiology, 2019. 9. [CrossRef]
- Srinivasan, B., et al., TEER Measurement Techniques for In Vitro Barrier Model Systems. Journal of Laboratory Automation, 2015. 20(2): p. 107-126. [CrossRef]
Figure 1.
One-step soft agar enrichment and isolation of A. fumigatus conidia germination-inhibiting bacterial strains from BALF samples. (a) Fungus-alone control plate after 24h incubation. (b) BALF-Aspergillus co-culture plate after 24h incubation. (c) Close-up stereoscopic image of the co-culture plate where an inhibition zone is visible (double-headed arrow) in-between the bacterial growth zone, which is delineated with a yellow-dashed lined, and the fungal growth zone, which is delineated with a purple-dashed line.
Figure 1.
One-step soft agar enrichment and isolation of A. fumigatus conidia germination-inhibiting bacterial strains from BALF samples. (a) Fungus-alone control plate after 24h incubation. (b) BALF-Aspergillus co-culture plate after 24h incubation. (c) Close-up stereoscopic image of the co-culture plate where an inhibition zone is visible (double-headed arrow) in-between the bacterial growth zone, which is delineated with a yellow-dashed lined, and the fungal growth zone, which is delineated with a purple-dashed line.
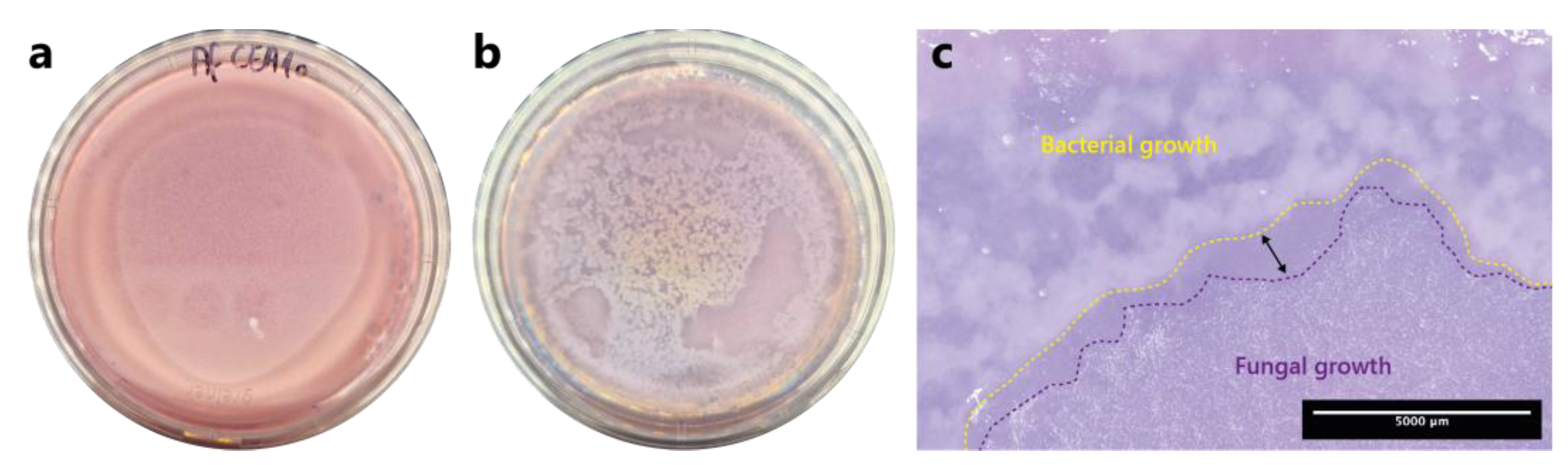
Figure 2.
Soft agar confrontation assay between A. fumigatus CEA10 and the five Pseudomonas aeruginosa isolates. (a) Macroscopic image of the confrontation plates where A. fumigatus conidia germination inhibition halos are visible around bacterial colonies. Af1 = P. aeruginosa b1, Af2 = P. aeruginosa b2, Af3 = P. aeruginosa b3, Af4 = P. aeruginosa b4, Af5 = P. aeruginosa b5. Images (b) to (g) were taken with a stereoscope. (b) Fungal growth control. (c) P. aeruginosa b1. (d) P. aeruginosa b2. (e) P. aeruginosa b3. (f) P. aeruginosa b4. (g) P. aeruginosa b5.
Figure 2.
Soft agar confrontation assay between A. fumigatus CEA10 and the five Pseudomonas aeruginosa isolates. (a) Macroscopic image of the confrontation plates where A. fumigatus conidia germination inhibition halos are visible around bacterial colonies. Af1 = P. aeruginosa b1, Af2 = P. aeruginosa b2, Af3 = P. aeruginosa b3, Af4 = P. aeruginosa b4, Af5 = P. aeruginosa b5. Images (b) to (g) were taken with a stereoscope. (b) Fungal growth control. (c) P. aeruginosa b1. (d) P. aeruginosa b2. (e) P. aeruginosa b3. (f) P. aeruginosa b4. (g) P. aeruginosa b5.
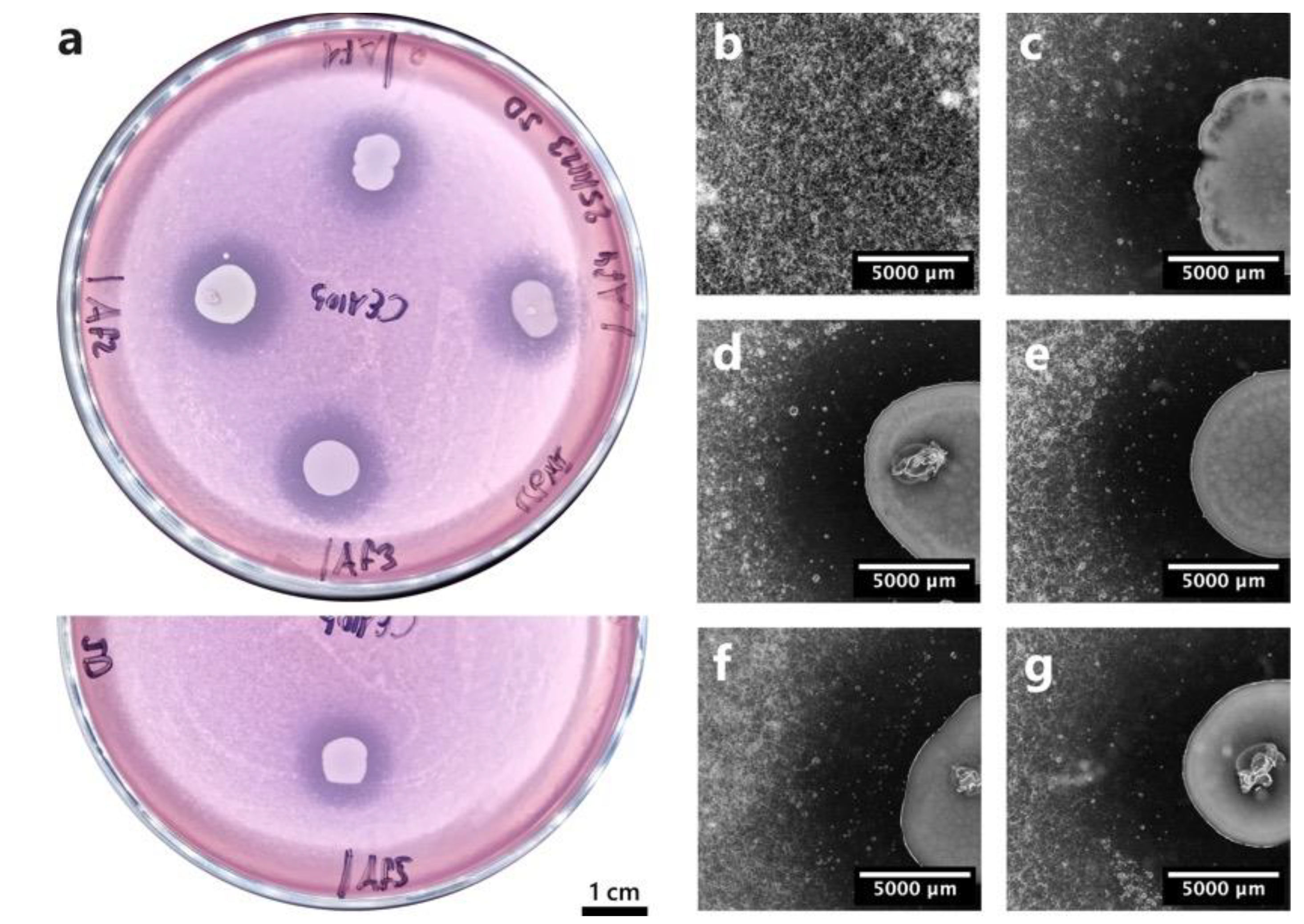
Figure 3.
Evolution of A. fumigatus conidia germination and growth in the HTP assay in confrontation with the five Pseudomonas aeruginosa strains (b1 – b5). This kinetic graph shows the mean bioluminescence signal [photon/sec] of four replicates emitted by A. fumigatus over 48h. Measurements were taken every 30 min. Panel (b) is a close-up view of (a). A. fumigatus alone (in red) was used as a control for conidia germination and growth. A. fumigatus + Cycloheximide (in green) was used as a positive control of A. fumigatus inhibition. A. fumigatus + b1-b5 represent the five confrontation treatments.
Figure 3.
Evolution of A. fumigatus conidia germination and growth in the HTP assay in confrontation with the five Pseudomonas aeruginosa strains (b1 – b5). This kinetic graph shows the mean bioluminescence signal [photon/sec] of four replicates emitted by A. fumigatus over 48h. Measurements were taken every 30 min. Panel (b) is a close-up view of (a). A. fumigatus alone (in red) was used as a control for conidia germination and growth. A. fumigatus + Cycloheximide (in green) was used as a positive control of A. fumigatus inhibition. A. fumigatus + b1-b5 represent the five confrontation treatments.
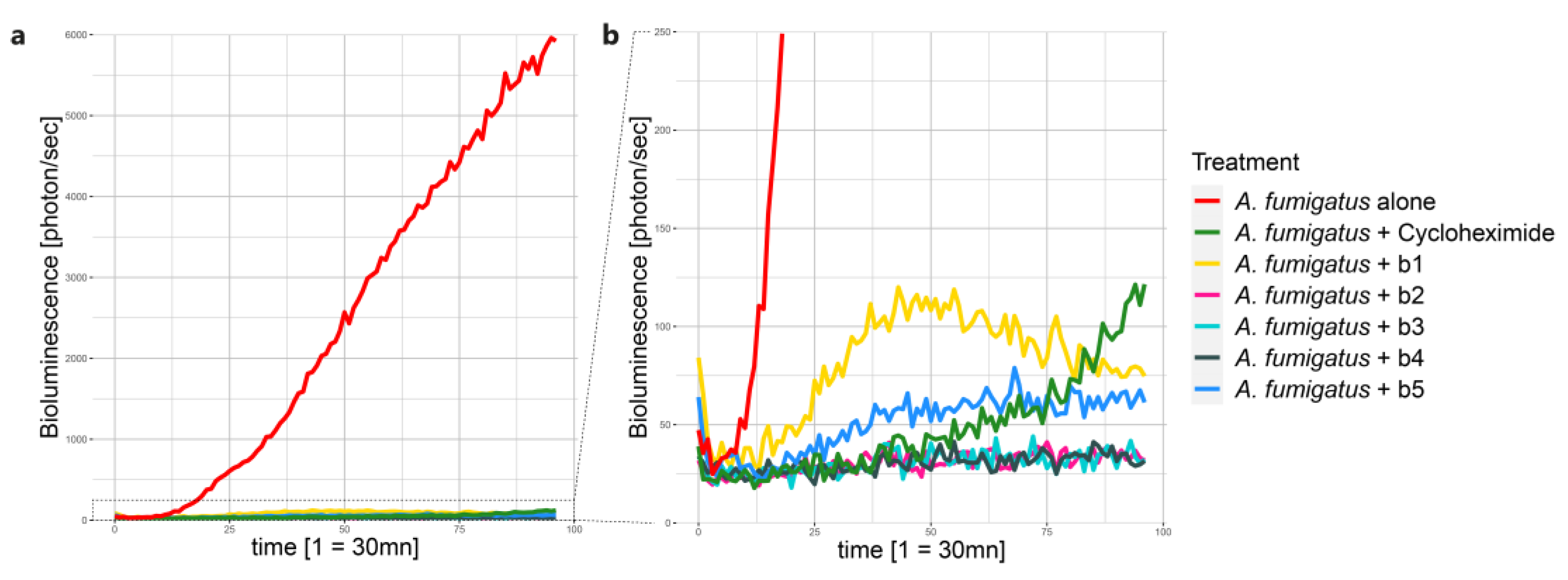
Figure 4.
Bioluminescence imaging of infected plates. Plate layout is depicted on the top of each panel. Bioluminescence emission by A. fumigatus CEA10b alone or in the presence of the five BALF P. aeruginosa isolates was recorded and imaged using an Amersham Imager 680 (GE Healthcare, USA) in the chemiluminescence mode. Exposure time was set to 5 min and binning was set to 32x. (a) Plate 1. (b) Plate 2. (c) Plate 3. The black spots visible in the fungal infection condition, i.e., C+F, show bioluminescence emission by A. fumigatus CEA10b. Moreover, no bioluminescence emission was detected in the co-infections, i.e., C+F+b1 and C+F+b2, suggesting a biocontrol of A. fumigatus by the BALF isolates. The plate layout schemes were done in RStudio using the “ggplate” package [25]. C = cells alone. C+F = cells + A. fumigatus. C+b1 = cells + strain b1. C+b2 = cells + strain b2. C+F+b1 = cells + A. fumigatus + strain b1. C+F+b2 = cells + A. fumigatus + strain b2.
Figure 4.
Bioluminescence imaging of infected plates. Plate layout is depicted on the top of each panel. Bioluminescence emission by A. fumigatus CEA10b alone or in the presence of the five BALF P. aeruginosa isolates was recorded and imaged using an Amersham Imager 680 (GE Healthcare, USA) in the chemiluminescence mode. Exposure time was set to 5 min and binning was set to 32x. (a) Plate 1. (b) Plate 2. (c) Plate 3. The black spots visible in the fungal infection condition, i.e., C+F, show bioluminescence emission by A. fumigatus CEA10b. Moreover, no bioluminescence emission was detected in the co-infections, i.e., C+F+b1 and C+F+b2, suggesting a biocontrol of A. fumigatus by the BALF isolates. The plate layout schemes were done in RStudio using the “ggplate” package [25]. C = cells alone. C+F = cells + A. fumigatus. C+b1 = cells + strain b1. C+b2 = cells + strain b2. C+F+b1 = cells + A. fumigatus + strain b1. C+F+b2 = cells + A. fumigatus + strain b2.
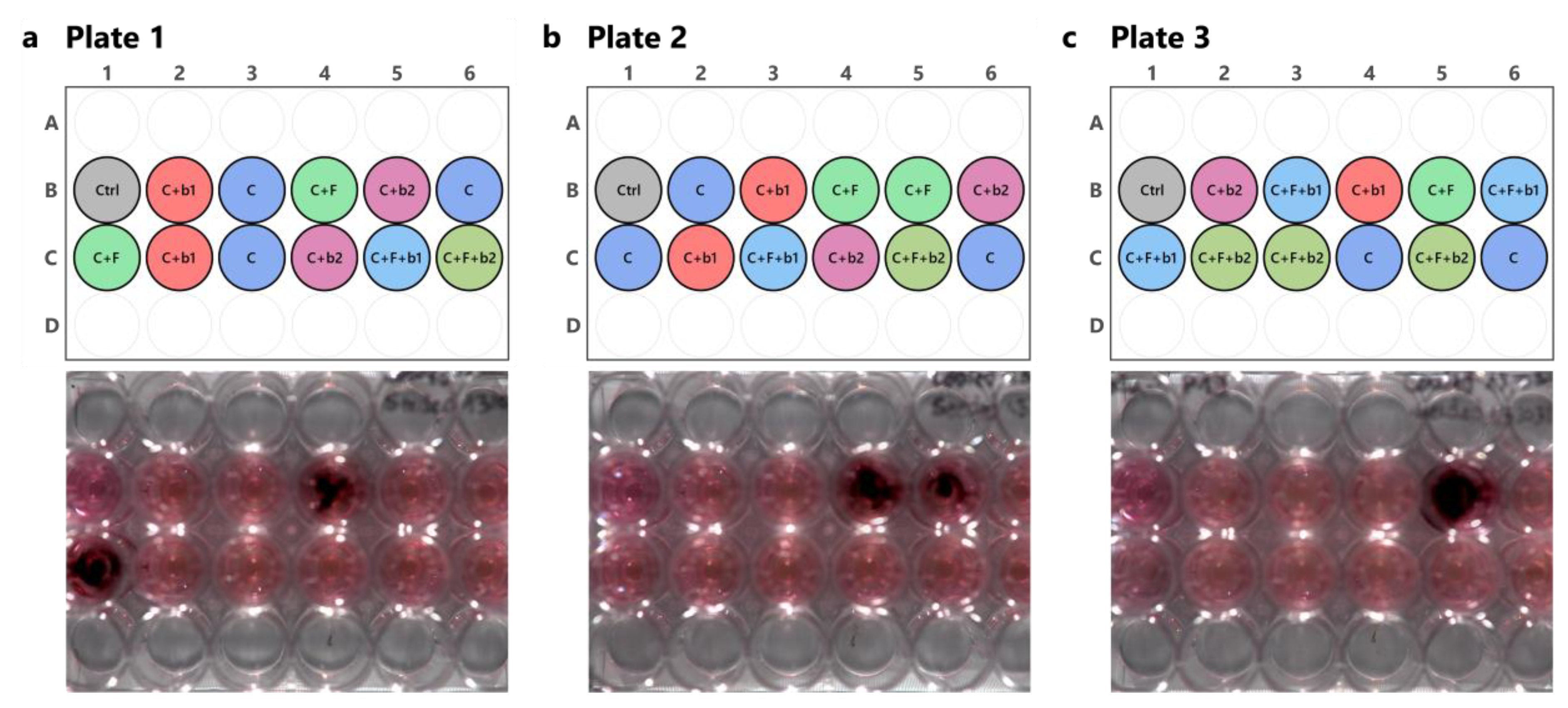
Figure 5.
Confocal microscopic imaging. Actin is indicated in green, and nuclei in blue. (a) Cells alone control (ZO-1 antibody). (b) Cells + A. fumigatus (anti-Aspergillus antibody). (c) Cells + A. fumigatus (ZO-1 antibody). (d) Cells + b1 (ZO-1 antibody). (e) Cells + b2 (ZO-1 antibody). (f) Cells + A. fumigatus + b1 (anti-Aspergillus antibody). (g) Cells + A. fumigatus + b1 (ZO-1 antibody). (h) Cells + A. fumigatus + b2 (anti-Aspergillus antibody). (i) Cells + A. fumigatus + b2 (ZO-1 antibody).
Figure 5.
Confocal microscopic imaging. Actin is indicated in green, and nuclei in blue. (a) Cells alone control (ZO-1 antibody). (b) Cells + A. fumigatus (anti-Aspergillus antibody). (c) Cells + A. fumigatus (ZO-1 antibody). (d) Cells + b1 (ZO-1 antibody). (e) Cells + b2 (ZO-1 antibody). (f) Cells + A. fumigatus + b1 (anti-Aspergillus antibody). (g) Cells + A. fumigatus + b1 (ZO-1 antibody). (h) Cells + A. fumigatus + b2 (anti-Aspergillus antibody). (i) Cells + A. fumigatus + b2 (ZO-1 antibody).
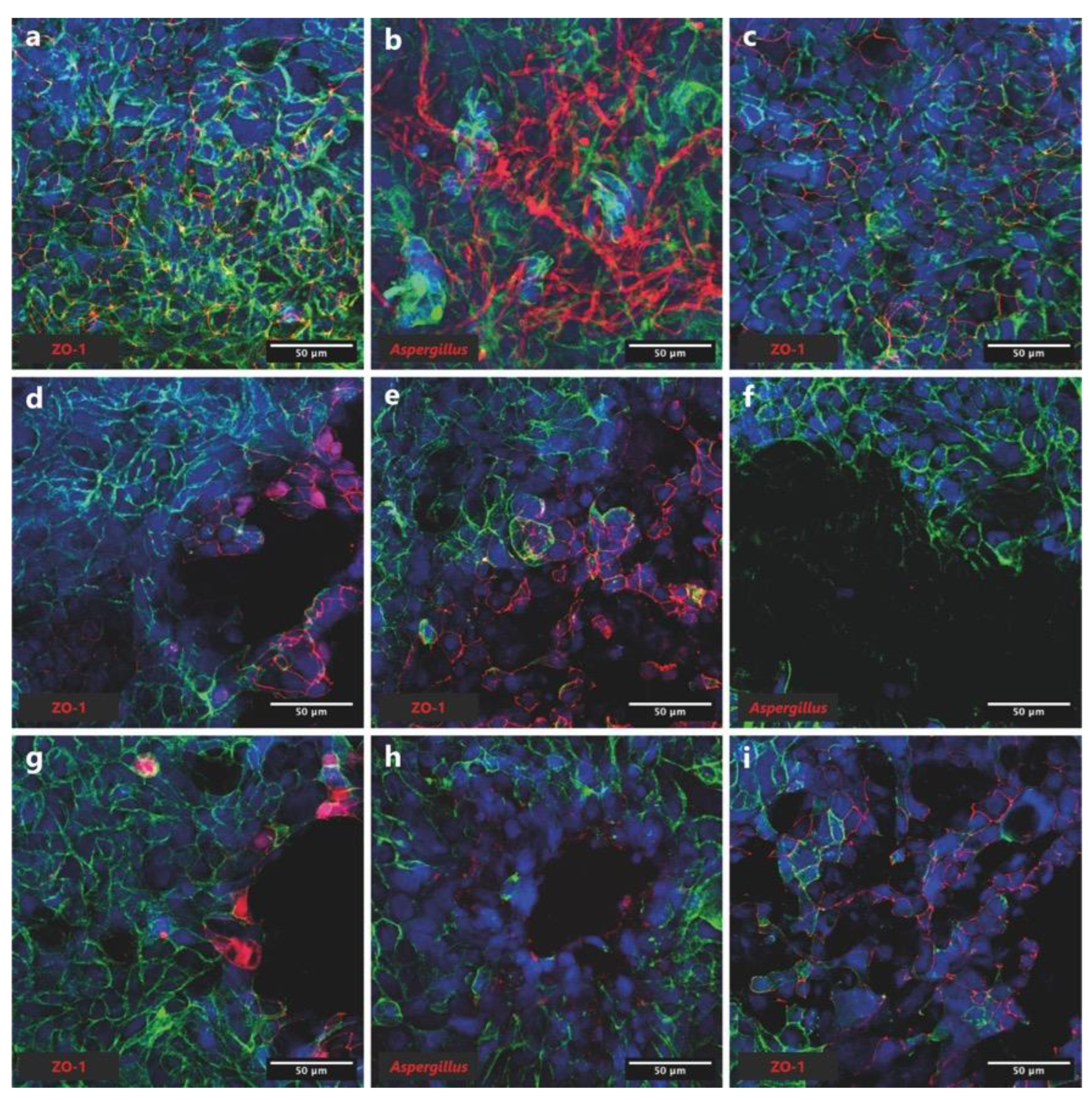
Figure 6.
Epithelial barrier function. (a) TEER measurements. TEER was measured daily across Calu-3 monolayers for 15 days. TEER shows a constant augmentation until around 2500 Ω/cm2 at day 15. Infection was performed at day 16. TEER stayed at around 2500 Ω/cm2 for the cells-alone control and the A. fumigatus-alone treatment, and dropped at 1000 Ω/cm2 and below in the co-infection treatments 19h post-infection. (b) Lucifer Yellow (LY) permeability assay. Permeability is expressed as a percentage compared to the no-cell control, which is set to 100%. Compared to the cell-alone control, which is at around 1.2%, the measured LY permeability was significantly higher in the C+b1, C+b2, C+F+b1 and C+F+b2 conditions. In the presence of bacteria b2, the permeability was higher than with bacteria b1. C = cells alone. C+F = cells + A. fumigatus. C+b1 = cells + strain b1. C+b2 = cells + strain b2. C+F+b1 = cells + A. fumigatus + strain b1. C+F+b2 = cells + A. fumigatus + strain b2. Significance levels: ns = P > 0.05 ; * = P ≤ 0.05 ; ** = P ≤ 0.01 ; *** = P ≤ 0.001 ; **** = P ≤ 0.0001.
Figure 6.
Epithelial barrier function. (a) TEER measurements. TEER was measured daily across Calu-3 monolayers for 15 days. TEER shows a constant augmentation until around 2500 Ω/cm2 at day 15. Infection was performed at day 16. TEER stayed at around 2500 Ω/cm2 for the cells-alone control and the A. fumigatus-alone treatment, and dropped at 1000 Ω/cm2 and below in the co-infection treatments 19h post-infection. (b) Lucifer Yellow (LY) permeability assay. Permeability is expressed as a percentage compared to the no-cell control, which is set to 100%. Compared to the cell-alone control, which is at around 1.2%, the measured LY permeability was significantly higher in the C+b1, C+b2, C+F+b1 and C+F+b2 conditions. In the presence of bacteria b2, the permeability was higher than with bacteria b1. C = cells alone. C+F = cells + A. fumigatus. C+b1 = cells + strain b1. C+b2 = cells + strain b2. C+F+b1 = cells + A. fumigatus + strain b1. C+F+b2 = cells + A. fumigatus + strain b2. Significance levels: ns = P > 0.05 ; * = P ≤ 0.05 ; ** = P ≤ 0.01 ; *** = P ≤ 0.001 ; **** = P ≤ 0.0001.
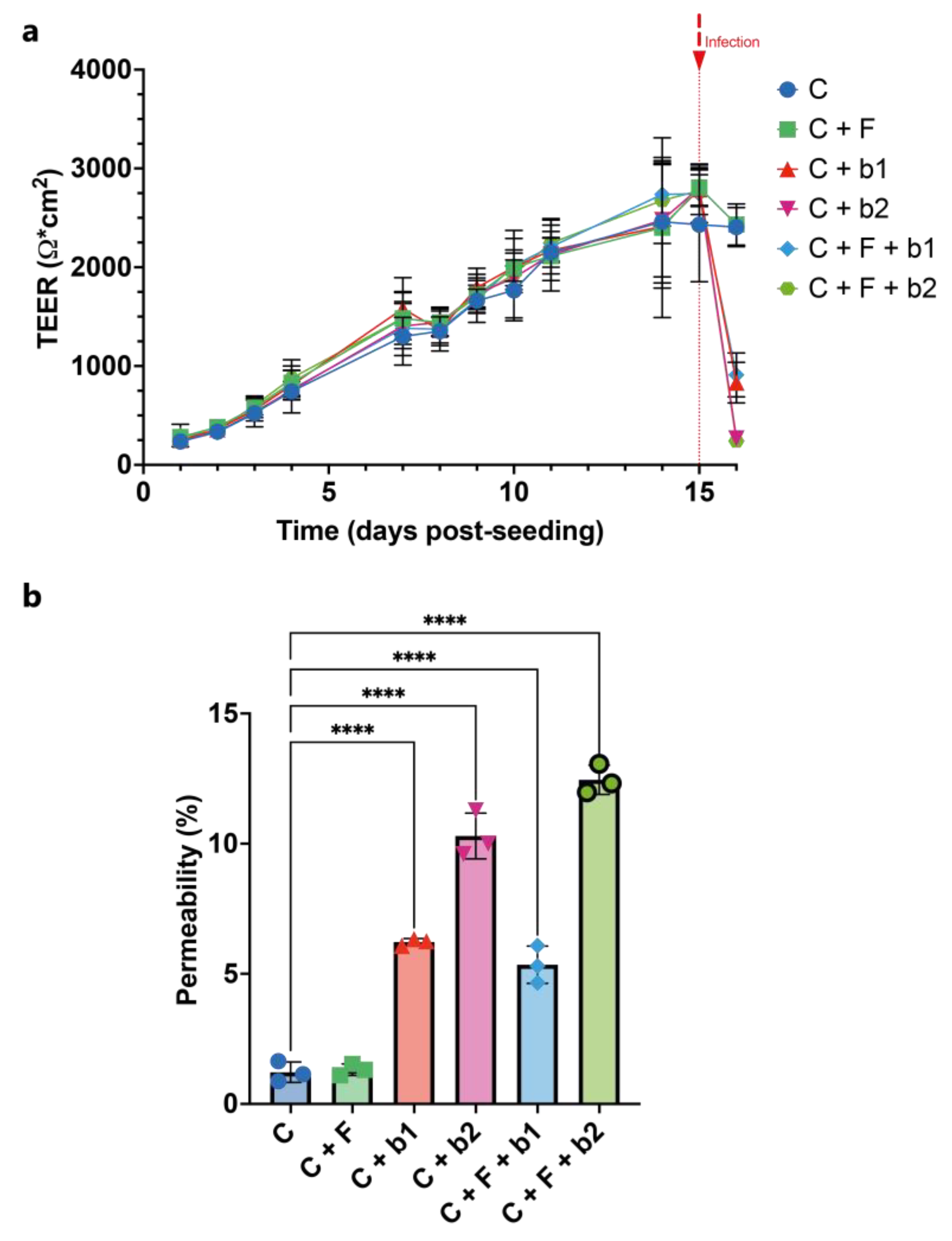
Figure 7.
Cytotoxicity assay. Significant fluorescence intensity detected in the conditions with b2 compared to the control cells. C = cells alone. C+F = cells + A. fumigatus. C+b1 = cells + strain b1. C+b2 = cells + strain b2. C+F+b1 = cells + A. fumigatus + strain b1. C+F+b2 = cells + A. fumigatus + strain b2. Significance levels: ns = P > 0.05 ; * = P ≤ 0.05 ; ** = P ≤ 0.01 ; *** = P ≤ 0.001 ; **** = P ≤ 0.0001.
Figure 7.
Cytotoxicity assay. Significant fluorescence intensity detected in the conditions with b2 compared to the control cells. C = cells alone. C+F = cells + A. fumigatus. C+b1 = cells + strain b1. C+b2 = cells + strain b2. C+F+b1 = cells + A. fumigatus + strain b1. C+F+b2 = cells + A. fumigatus + strain b2. Significance levels: ns = P > 0.05 ; * = P ≤ 0.05 ; ** = P ≤ 0.01 ; *** = P ≤ 0.001 ; **** = P ≤ 0.0001.
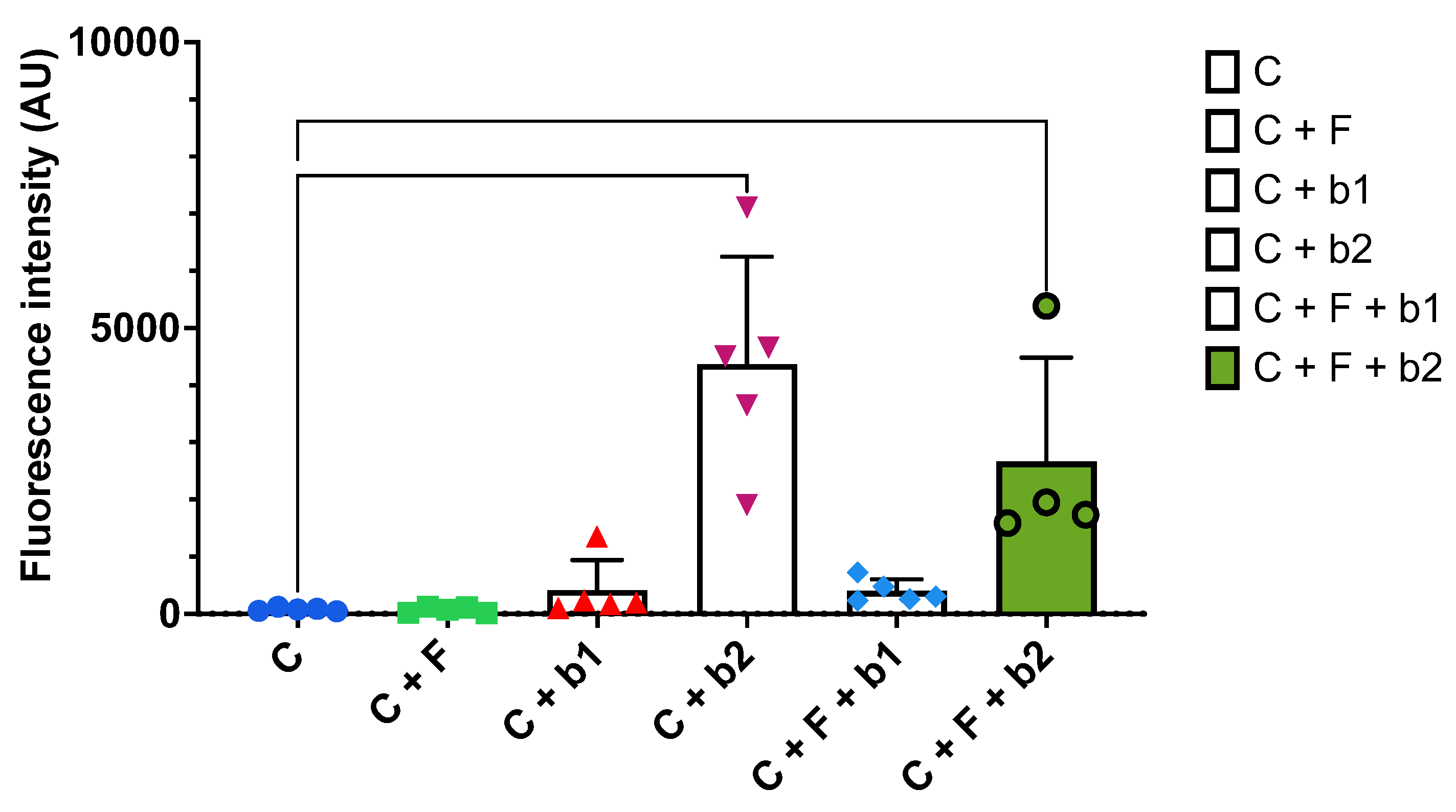
Figure 8.
pH and calcium measurements. (a) pH of the culture supernatant. pH was constant across all conditions tested (around 8). (b) Calcium concentration of the culture supernatant. Calcium concentrations were constant across all conditions tested (around 1.5 mM). C = cells alone. C+F = cells + A. fumigatus. C+b1 = cells + strain b1. C+b2 = cells + strain b2. C+F+b1 = cells + A. fumigatus + strain b1. C+F+b2 = cells + A. fumigatus + strain b2.
Figure 8.
pH and calcium measurements. (a) pH of the culture supernatant. pH was constant across all conditions tested (around 8). (b) Calcium concentration of the culture supernatant. Calcium concentrations were constant across all conditions tested (around 1.5 mM). C = cells alone. C+F = cells + A. fumigatus. C+b1 = cells + strain b1. C+b2 = cells + strain b2. C+F+b1 = cells + A. fumigatus + strain b1. C+F+b2 = cells + A. fumigatus + strain b2.
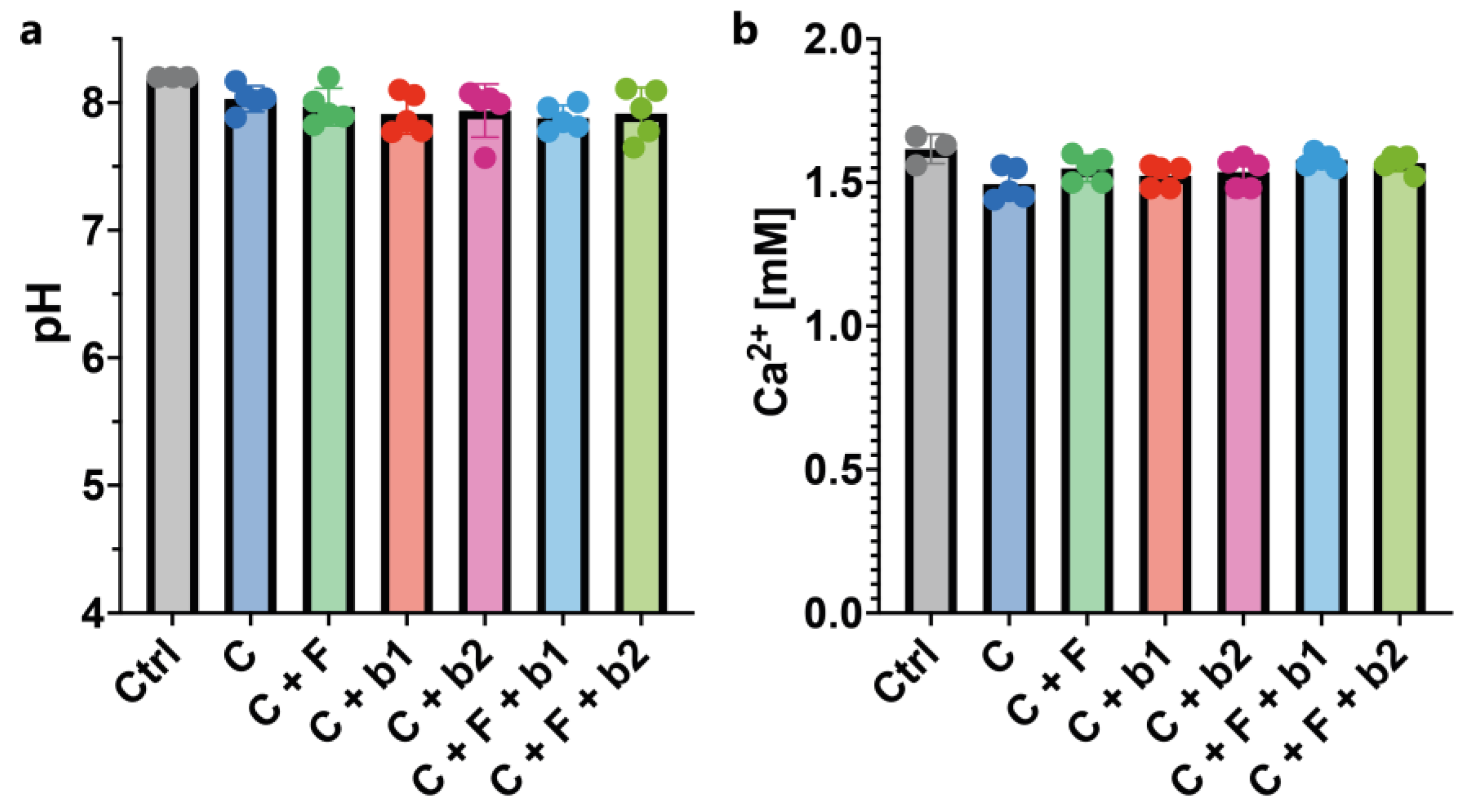

Table 1.
A. fumigatus strains used in this study.
| ID | Strain name | Reference |
|---|---|---|
| CEA10 | Aspergillus fumigatus CEA10, WT | [18] |
| CEA10b | Aspergillus fumigatus CEA10, red-shifted bioluminescent strain | [19] |
Table 2.
BALF samples used in this study. BALF samples from lung transplanted patients have been obtained from the Lausanne University Hospital (CHUV).
Table 2.
BALF samples used in this study. BALF samples from lung transplanted patients have been obtained from the Lausanne University Hospital (CHUV).
| ID | Antibacterial exposure | Antibacterial/Antifungal drugs administered |
|---|---|---|
| BAL1 | yes | Colifin (colistimethate) |
| BAL2 | yes | Bactrim (sulfamethoxazole + trimethoprim) |
| BAL3 | yes | Colifin (colistimethate) |
| BAL4 | no | Ambisome (amphotericin B) |
| BAL5 | no | Ambisome (amphotericin B) |
Table 3.
Identity of the isolated strains.
| Strain code | Species | Query cover | Per. ident |
|---|---|---|---|
| b1 | Pseudomonas aeruginosa | 100% | 99.21% |
| b2 | Pseudomonas aeruginosa | 100% | 97.40% |
| b3 | Pseudomonas aeruginosa | 100% | 98.64% |
| b4 | Pseudomonas aeruginosa | 100% | 98.72% |
| b5 | Pseudomonas aeruginosa | 100% | 99.48% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



