Submitted:
31 May 2023
Posted:
31 May 2023
You are already at the latest version
Abstract
Hubei, Hunan and Henan Provinces are located in Central China, a region with extensive transport networks and trade. Bursaphelenchus xylophilus, the causative agent of pine wilt disease, is spreads mainly through human activities. In order to further understand the genetic structure of PWN in Central China, we studied the genetic information of PWN populations in this region and compared the genetic relationship with strains from Guangdong and Jiangsu provinces. We found that the HB (Hubei) 15, HEN (Henan) 20, HN (Hunan) 07, HN08 and HN10 had significantly more SNPs and homozygotes than other strains from Central China, and their most frequent mutant genotypes also differed from other strains. The clustering results indicated that HB15, HEN 20, HN07, HN08 and HN10 were genetically distinct from other strains and closely related to Guangdong strains. We also observed significant genetic variation among strains in Henan province, suggesting that some of them might have different transmission sources than those from Hubei and Hunan provinces. The results provide a basis for tracing the origin and spread of PWD in China.
Keywords:
Bursaphelenchus xylophilus
; SNP
; genetic diversity
; population differentiation (List three to ten pertinent keywords specific to the article yet reasonably common within the subject discipline.)
1. Introduction
Pine wood nematode (Bursaphelenchus xylophilus, PWN) is the causal agent of pine wilt disease (PWD), which poses a hazard to pine forests throughout Europe and Asia. PWN is regulated as a quarantine disease in most countries due to its ecological and economic impacts [1]. Previous studies have indicated that China is one of the most affected countries by PWD, with most provinces being suitable for PWN establishment and spread [2]. As an invasive alien species, PWN has caused significant loss to the pine forests in China. It was first detected in Nanjing, Jiangsu province in 1982, and since then it has expanded to 731 county-level administrative regions of 19 provinces (National Forestry and Grassland Administration No. 6 of 2022). This suggests that PWN has a wide distribution in China, and that human activities such as transportation of infested wood and infrastructure construction are the main pathways of its dissemination. However, the transmission routes of PWN in China are not well understood due to the difficulties and limitations of monitoring and surveillance. In recent years, many studies have focused on the early diagnosis of PWN, which provides a theoretical basis for the prevention and control of PWD [3-5].
Studies have shown that the critical time to control biological invasions is in the early stage [6,7], and studying the dispersal path of invasive organisms is crucial to achieving control. Several studies have also proved that the inference of invasion pathways provides information about the biological invasion process, which enables us to understand the ecological characteristics of invasive populations and is helpful for control or eradication [8-11]. In order to clarify the transmission path and population differentiation of PWNs, relevant scholars have used different molecular marker technologies to analyze the population genetic structure of PWNs in some geographical areas [12-15].
As early as 2007, RAPD-PCR was used to analyze the genetic variation of Spanish PWN [16]. Subsequently, Valadas et al. used ISSR molecular markers to analyze the genetic differences among 43 PWN strains from five countries: China, Japan, Korea, the United States and Portugal [17]. With the development of molecular marker technology, SNP is considered the most promising molecular marker. It is widely used in many applications with population tracking, molecular genetics and disease diagnosis [18-21]. In recent years, studies on the population diversity of PWNs using SNP have also been reported [13,22,23]. Joana Figueiredo et al. used SNP labeling technology to analyze the differences of SNP in 7 PWNs from Portugal, China, the United States and Japan. It showed that the Portugal strains were closer to those from China, and the genetic distance between the American and the Japanese was relatively wide [15]. Several studies have analyzed the population diversity of PWN in different regions of China using SNP labeling technology. The genetic structure of Guangdong Province showed that it has high genetic diversity and multiple transmission sources [13]. Population results in eastern China showed that there was some correlation between each group and geographical origin [22].
Hubei, Hunan and Henan provinces are located in the middle of China, a region with frequent economic and trade activities. Therefore, supervising infected trees poses consid-erable challenges. Some scholars speculated that Guangdong Province was the initial colonization and diffusion center of PWNs in China, and Jiangsu Province was the new diffusion center [24]. These provinces are close to Hubei, Hunan and Henan, which were classified as epidemic areas in 2000, 2003 and 2009 respectively. To better understand the genetic structure of PWNs in central China, this study used whole genome resequencing and SNP molecular marker technology to analyze the genetic diversity of PWN populations in central China and explore their genetic relationship with PWN populations in Jiangsu and Guangdong provinces. This has great significance for establishing a PWD tracing system.
2. Materials and Methods
2.1. Isolation and purification of nematodes
Infected trees were collected from Henan Province, Hubei Province, Hunan Province, Guangdong Province and Jiangsu Province. Nematodes were extracted from chopped trees using the Baermann funnel method. Morphological identification of PWNs was carried out according to the characteristics of PWNs [25]. After DNA extraction, molecular identification based on the SCAR marker was performed to ensure detection accuracy [26].
After being identified as PWN, about 30 individuals were selected and cultured on the Botrytis cinerea at 28 °C. When a sufficient sample size was obtained, nematodes were isolated using the Bermann funnel method and were cleared with 0.05% streptomycin sulfate and sterile water for storage. All the strains were stored in the PWN Strain Resource Bank of the Forest Pathology Laboratory of Nanjing Forestry University.
2.2. Genome resequencing
The DNA of PWN was extracted by the CTAB method [26] and stored in the DNA Resource Bank of PWN, Laboratory of Forest Pathology, Nanjing Forestry University. Nano-drop, Qubit (Thermo Fisher) detected the DNA concentration and quality. The qualified DNA was sent to Wuhan Future Group Biological Company for high-throughput genome sequencing on the HiSeq 4000 platform. The genome resequencing method was 150 bp paired-end sequencing, and the average sequencing depth was greater than 40 ×.
2.3. Identification and filtration of mutation sites
The quality of the raw data was first assessed by FastQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/). Filtered reads were aligned to the reference genome of PWN announced in 2022 [27] by BWA (http://bio-BWA.SourceForge.net/BWA.shtml). Samtools (http://samtools.sourceforge.net/samtools.shtml) and Picard were used to remove duplicates. Putative SNPs were called by Freebayes (https://github.com/ekg/freebayes) with minimum coverage (>10), and VCFtools (https://github.com/vcftools) was used for SNP site statistical analysis.
2.4. Genetic differentiation analysis
The SNPs with low allele frequency, high linkage disequilibrium and missing rate were filtered by the SNPRealte package (https://www.bioconductor.org/packages/release/bioc/html/SNPRelate.html) of RStudio software (https://www.rstudio.com/). Principal component analysis (PCA) diagram was drawn using the same package mentioned above. PLINK (v1.9) (https://www.cog-genomics.org/plink/) was used to extract the filtered site information to generate new vcf file for phylogenetic tree analysis. VCF-kit (https://vcf-kit.readthedocs.io/en/latest/) and MEGA (v11.0.11) (https://www.megasoftware.net/) was used to construct phylo-tree using the neighbour-joining method.
Treemix software (https://bitbucket.org/nygcresearch/treemix), based on allele frequency as the basis for genetic distance calculation, was used to construct phylogenetic trees and label gene exchanges. Plink was used to calculate allele frequency for all SNP loci in advance, and the parameter “-noss-global” was used to construct a maximum likelihood tree.
3. Results
3.1. Sample collection
After purification and culture, 50 PWN samples from five provinces in China were obtained: Hubei (HB), Henan (HEN), Hunan (HN), Guangdong (GD) and Jiangsu (JS). Table 1 shows the sample information of 50 B. xylophilus strains.
3.2. Statistics of SNP loci
The SNP locus information of 30 strains in Central China showed that there are 8,333,375 SNPs sites in total and the number of SNP sites varied significantly among different strains (Figure 1). HB15, HEN20, HN07, HN08 and HN10 had significantly more SNPs and homozygotes than other strains. HEN15, HEN19, HEN20, HN09, HN10 and HN13 had significantly more missing SNPs than other strains. HEN20 had the highest number of SNPs, while HN02 had the lowest. HN10 had the highest number of homozygous, missing and private SNPs, which were 1033119, 5248382 and 505849, respectively. HN02 had the lowest number of homozygous SNPs, HEN06 had the lowest number of missing SNPs, and HN09 had the lowest number of private SNPs (Table 2 and Figure 1).
3.3. Statistics of SNP genotypes
The results of SNP genotyping on strains from Central China showed that there were 12 SNP genotypes: A>C, A>G, A>T, C>A, C>G, C>T, G>A, T>A, T>C and T>G. Comparing the genotype counts among strains, it is found significant differences for some genotypes. Specifically, four genotypes (A>G, C>T, G>A, and T>C) were significantly more frequent in HB15, HEN20, HN06, HN07, HN08, and HN10 strains than in other strains. For the remaining strains, six genotypes (A>G, C>G, C>T, G>A, G>C, T>C) were significantly more frequent than the others. Meanwhile, the genotype counts of HB15, HEN20, HN06, HN07, HN08 and HN10 strains were higher than those of other strains (Figure 2).
3.4. Analysis of genetic differentiation
Principal component analysis (PCA) was conducted on 30 strains from Central China, which could be divided into four groups (Figure 3a). Group 1 consisted of HB15, HEN20, HN07, HN08 and HN10. Group 2 comprised HB08, HEN02, HEN04 and HN06. Group 3 contained 16 strains, including 8 from Hubei, 2 from Henan and 6 from Hunan. Group 4 included only Henan strains: HEN06, HEN09, HEN10, HEN14 and HEN15. The PCA results revealed genetic differences among PWN populations in Central China. Henan Province had the highest genetic diversity, as its strains were distributed across all four groups. In contrast, most of the Hubei strains clustered in group 3, and the Hunan strains were either in group 1 or group 3.
To investigate the origin of PWN strains in Central China, the principal component analysis (PCA) was performed on the strains from Central China, Jiangsu Province, and Guangdong Province (Figure 3b). The PCA results indicated that the strains from Jiangsu Province were genetically similar to group 3 in Figure 3, while the strains from Guangdong Province clustered with group 1. The neighbor-joining tree of 50 PWNs confirmed the PCA results (Figure 4). Both analyses revealed that HB15, HEN20, HN07, HN08 and HN10 were closely related to strains from Guangdong Province, while the others were closely related to Jiangsu strains. Moreover, there were significant differences between Henan strains and strains from other provinces, suggesting different sources of invasion. To understand the invasion routes of PWN in central China, the introgression analysis was conducted and identified three possible pathways: 1) Guangdong to Henan; 2) Guangdong to Hunan; 3) Jiangsu to Hubei. This implies that Guangdong Province could be a major source of PWN spread in Henan and Hunan provinces, and Jiangsu Province could be a major source of PWN spread in Hubei Province (Figure 5).
4. Discussion
Previous studies have demonstrated that environmental factors can cause founder effects and genetic drift, leading to reduced or lost genetic diversity and population genetic differentiation in species that migrate and disperse [28-30]. Cheng et al. analyzed the genetic diversity of PWNs in different regions of China using AFLP markers and found that Chinese populations had slightly higher genetic diversity than American populations [31]. They found that Chinese populations were slightly higher than American populations in genetic diversity. However, Ding et al. used SNP markers to examine 181 PWN strains from 16 endemic areas in China and found that the Guangdong population had high genetic diversity and was genetically close to Americans, while the genetic diversity of strains in other areas tended to decrease [27]. They concluded that the invasive populations suffered from the loss of genetic diversity due to the founder effect, which was consistent with the findings of Mallez et al. [29,32]. These studies indicated that there was a specific correlation between different clusters and their geographical origin, and that SNP molecular marker technology was an effective tool to study the genetic differentiation of the PWN population [15,23]. Based on the population structure analysis of PWNs in China [27], this study revealed the finer population structure in Central China for the first time using the SNP molecular markers.
The incidence and distribution of PWD in China were mainly concentrated in Guangdong and Jiangsu provinces, located in economically developed areas. Central China has a temperate and subtropical monsoon climate with an annual average temperature higher than 15 °C, which is prone to PWD [33]. Moreover, the area is adjacent to Guangdong and Jiangsu provinces and has frequent trade activities with other parts of the country through traffic lines. It is possible that PWNs were introduced into infected pine plants and their products (cable trays, packing cases, etc.) during trade activities. Therefore, PWNs in each epidemic area of central China may have different epidemic sources, and there is a high possibility of cross-invasion. This is also consistent with our research results that there are multiple clusters in central China.
This study applied SNP molecular marker technology to analyze the genetic differences of PWN populations in central China. The results revealed that the number of SNP sites, homozygote number, and genotypes of HB15, HEN20, HN07, HN08 and HN10 were significantly higher than those of other strains, and the number of missing SNP sites differed significantly from other strains. The results showed that these strains had distinct SNP loci and genotypes, which was consistent with the clustering results. The clustering results showed that the five strains were genetically distant from other strains. Therefore, we inferred that there were different sources of transmission for the strains in Central China. Ding [27] analyzed the population genetic structure of strains from different regions of China, and the results showed that the strains from Henan, Hubei and Hunan were clustered into one group, among which the Hunan strains were distributed in several groups, showing rich genetic diversity. It was also confirmed that the insect strains in central China had different transmission sources.
Previous studies suggested that Guangdong Province was the initial colonization and dispersal center of pine wood nematode in China, and Jiangsu Province was a new dispersal center. Therefore, this study analyzed the population genetic structure of the strains from central China and those from Jiangsu and Guangdong Province. The PCA and phylogenetic tree demonstrated that HB15, HEN20, HN07, HN08 and HN10 were closely related to the strains from Guangdong Province, while other strains were closely related to those from Jiangsu Province. The introgression results also confirmed that the Henan and Hunan isolates originated from Guangdong strains. The result may be due to the genetic difference between Henan and Hubei populations in the isolated taxa. Therefore, we hypothesized that there were three main transmission routes for the strains from Central China (Guangdong to Henan; Guangdong to Hunan; Jiangsu to Hubei), and that HB15, HEN20, HN07, HN08 and HN10 strains were invasion from Guangdong. This was consistent with Ding’s results [27], indicating that the Hunan strains migrated from the Guangdong strains. This study further analyzed population genetic diversity in Central China. There was evidence of multiple invasions and cross-invasions in the epidemic areas of Hubei, Henan and Hunan, which indicated that the regulation of infected wood was insufficient.
Wang et al. used SNP to analyze the genetic differentiation of PWN populations in East China [22] and found correlations among different groups and geographical regions. However, the genetic differentiation within each group was not significant. In contrast, this study revealed that there was genetic differentiation of PWNs from central China, and the genetic diversity of Henan strains was higher. Some Henan strains were not only genetically differentiated from those in Guangdong province but also had genetic distance from those in Jiangsu province, which might result from genetic drift and founder effect during the invasion of PWNs. Huang et al. reported significant genetic differences among PWN populations in Guangdong Province [13]. Ding et al. suggested that the Guangdong strain had similar genetic variation to the American strain and speculated that it might originate from foreign invasion [15,27,34]. We hypothesized that the Henan strain might also be influenced by foreign invasion because it had a distinct genetic structure. However, there was a lack of a large number of foreign strains to verify this possibility.
5. Conclusions
By analyzing the genetic diversity of PWNs from central China, we found that some strains had higher genetic similarity to those from Guangdong. In addition, the Henan strain has rich genetic diversity and genetic differences from other strains, suggesting that there may be different transmission sources. The results provide the theoretical basis for exploring the propagation path of pine wood nematode, population genetic structure and formulating a quarantine strategy.
Author Contributions
A.-X.Y.: designed the study, conducted the experiment, performed the data analysis and wrote the article; X.-L.D.: guided the article writing and data analysis; Y.F. and T.-T.C.: collected the samples; J.-R.Y.: guaranteed the integrity of the entire study and approved the final version of the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This project is supported by the National Key Research and Development Project 2021YFD1400903 (J.Y.)
Data Availability Statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author.
Acknowledgments
We thank all forestry bureaus that kindly provided nematode samples in China.
Conflicts of Interest
The authors declare no conflict of interest.
References
- He, L.X.; Ji, J.; Qiu, X.W.; et al. Prevalence and control measures of pine wood nematode disease in the world. China For. Sci. Technol. 2014, 28, 8–13. [Google Scholar]
- Ye, J.R. Epidemic status of pine wilt disease in china and its prevention and control techniques and counter measures. Sci. Silvae Sin. 2019, 55, 1–10. [Google Scholar]
- Ni, A.S.; Yang, D.; Cheng, H.; Ye, J. Preliminary Study on Early Diagnosis and Rehabilitation Treatment of Pine Wood Nematode Disease Based on Partial Symptoms. Forests 2023, 14, 657. [Google Scholar] [CrossRef]
- Wen, T.Y.; Zhang, Y.; Wu, X.Q.; Ye, J.R.; Qiu, Y.J.; Rui, L. Studies on the Requirement of Transthyretin Protein (BxTTR-52) for the Suppression of Host Innate Immunity in Bursaphelenchus xylophilus. Int. J. Mol. Sci. 2022, 23, 15058. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.C.; Li, M.; Sheng, R.C.; Chen, F.M. Enzyme-Mediated Amplification (EMA) for Detection of the Pinewood Nematode Bursaphelenchus xylophilus. Forests 2022, 13, 1419. [Google Scholar] [CrossRef]
- Allendorf, F.W.; Lundquist, L.L. Introduction: Population biology, evolution, and control of invasive species. Conserv. Biol. 2003, 17, 24–30. [Google Scholar] [CrossRef]
- Simberloff, D.; Martin, J.; Genovesi, P.; Maris, V.; Wardle, D.A.; Aronson, J.; Vilà, M. Impacts of biological invasions: what’s what and the way forward. Trends Ecol. Evol. 2013, 28, 58–66. [Google Scholar] [CrossRef]
- Estoup, A.; Guillemaud, T. Reconstructing routes of invasion using genetic data: Why, how and so what? Mol. Ecol. 2010, 19, 4113–4130. [Google Scholar] [CrossRef]
- Papura, D.; Burban, C.; Maarten, V.H.; Giresse, X.; Nusillard, B.; Guillemaud, T.; Kerdelhué, C. Microsatellite and mitochondrial data provide evidence for a single major introduction for the Neartic leafhopper Scaphoideus titanus in Europe. PLoS ONE 2012, 7, e36882. [Google Scholar] [CrossRef]
- Boucher, A.C.; Mimee, B.; Montarry, J.; Bardou-Valette, S.; Bélair, G.; Moffett, P.; Grenier, E. Genetic diversity of the golden potato cyst nematode Globodera rostochiensis and determination of the origin of populations in Quebec, Canada. Mol. Phylogenetics Evol. 2013, 69, 75–82. [Google Scholar] [CrossRef]
- Van-Wilgen, B.W.; Moran, V.C.; Hoffmann, J.H. Some perspectives on the risks and benefits of biological control of invasive alien plants in the management of natural ecosystems. Environ. Manag. 2013, 52, 531–540. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, T.; Cotton, J.A.; Dalzell, J.J.; Hasegawa, K.; Kanzaki, N.; McVeigh, P.; Berriman, M. Genomic insights into the origin of parasitism in the emerging plant pathogen Bursaphelenchus xylophilus. PLoS Pathog. 2011, 7, e1002219. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.S.; Xi, X.T.; Ding, X.L.; Ye, J. Study on the population differentiation of Bursaphelenchus xylophilus in Guangdong province by SNP markers. J. Nanjing For. Univ. (Nat. Sci. Ed.) 2019, 43, 7. [Google Scholar]
- Xu, J.R.; Wu, X.Q.; Liu, Y.; Ye, J. Development of simple sequence repeats base on pine wood nematode (Bursaphelenchus xylophilus) genome sequence. J. Nanjing For. Univ. 2014, 38, 36–42. [Google Scholar]
- Figueiredo, J.; Simões, M.J.; Gomes, P.; Barroso, C.; Pinho, D.; Conceicao, L.; Egas, C. Assessment of the geographic origins of pine wood nematode isolates via single nucleotide polymorphism in effector genes. J. PLoS ONE 2013, 8, e83542. [Google Scholar] [CrossRef]
- Vieira, P.; Burgermeister, W.; Mota, M.; Metge, K.; Silva, G. Lack of genetic variation of Bursaphelenchus xylophilus in Portugal revealed by RAPD-PCR analyses. J. Nematol. 2007, 39, 118–126. [Google Scholar]
- Valadas, V.; Laranjo, M.; Barbosa, P.; Espada, M.; Mota, M.; Oliveira, S. The pine wood nematode, Bursaphelenchus xylophilus, in Portugal: Possible introductions and spread routes of a serious biological invasion revealed by molecular methods. Nematology 2012, 14, 899–911. [Google Scholar] [CrossRef]
- Zhu, H.Y.; Xu, Y.H.; Du, J.; et al. Research progress and application of SNP. Sci-Tech Innov. Product. 2019, 2, 62–65. [Google Scholar]
- Kwong, Q.B.; Teh, C.K.; Ong, A.L.; Heng, H.Y.; Lee, H.L.; Mohamed, M.; Appleton, D.R. Development and validation of a high-density SNP genotyping array for African oil palm. Mol. Plant 2016, 9, 1132–1141. [Google Scholar] [CrossRef]
- Sun, J.; Ma, D.R.; Tang, L.; Zhao, M.; Zhang, G.; Wang, W.; Chen, W. Population genomic analysis and de novo assembly reveal the origin of weedy rice as an evolutionary game. Mol. Plant 2019, 12, 632–647. [Google Scholar] [CrossRef]
- Su, T.B.; Wang, W.H.; Li, P.R.; Xin, X.; Zhang, F. A genomic variation map provides insights into the genetic basis of spring Chinese cabbage (Brassica rapa ssp. pekinensis) selection. Mol. Plant 2018, 11, 1360–1376. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.T.; Ding, X.L.; Ye, J.R.; SHI, X. Genetic differentiation of Bursaphelenchus xylophilus in east China based on single nucleotide polymorphisms (SNP) markers. J. Nanjing For. Univ. (Nat. Sci. Ed.) 2022, 46, 21–28. [Google Scholar]
- Ding, X.L.; Ye, J.R.; Lin, S.X.; Wu, X.; Li, D.; Nian, B. Deciphering the molecular variations of pine wood nematode Bursaphelenchus xylophilus with different virulence. PLoS ONE 2016, 11, e0156040. [Google Scholar] [CrossRef]
- Xie, B.Y.; Cheng, X.Y.; Shi, J.; et al. Formation and expansion mechanism of invasive populations of pine wood nematodes: Progress in “basic research on invasion mechanism and control of dangerous organisms in agriculture and forestry” of national key basic research program. Sci. China (Ser. C) 2009, 39, 333–341. [Google Scholar]
- Xie, H. Taxonomy of Plant Nematodes, 2nd ed.; Higher Education Press: Beijing, China, 2005; pp. 305–310. [Google Scholar]
- Chen, F.M.; Ye, J.R.; Wu, X.Q.; et al. SCAR Marker and Detection Technique of Bursaphelenchus xylophilus. Sci. Silvae Sin. 2012, 48, 88–94. [Google Scholar]
- Ding, X.L.; Guo, Y.F.; Ye, J.R.; et al. Population differentiation and epidemic tracking of Bursaphelenchus xylophilus in China based on chromosome-level assembly and whole-genome sequencing data. Pest Manag. Sci. 2022, 78, 1213–1226. [Google Scholar] [CrossRef]
- Huang, Z.H.; Liu, N.F. Advances in Population Genetics. J. Anhui Agric. Sci. 2008, 36, 13490–13491,13499. [Google Scholar]
- Nei, M.; Maruyama, T.; Chakraborty, R. The Bottleneck Effect and Genetic Variability in Populations. Evolution 1975, 29, 1–10. [Google Scholar] [CrossRef]
- Li, Y.X.; Zhang, X.Y. Analysis on the trend of invasion and expansion of Bursaphelenchus xylophilus. For. Pest Dis. 2018, 37, 1–4. [Google Scholar]
- Cheng, X.Y.; Cheng, F.X.; Xu, R.M.; Xie, B.Y. Genetic variation in the invasive process of Bursaphelenchus xylophilus (Aphelenchida: Aphelenchoididae) and its possible spread routes in China. Heredity 2008, 100, 356–365. [Google Scholar] [CrossRef]
- Mallez, S.; Castagnone, C.; Espada, M.; Vieira, P.; Eisenback, J.D.; Harrell, M.; Guillemaud, T. Worldwide invasion routes of the pinewood nematode: What can we infer from population genetics analyses? Biol. Invasions 2015, 17, 1199–1213. [Google Scholar] [CrossRef]
- Pan, H.W. Study on the Potential Geographic Distribution of Bursaphelenchus xylophilus in China. Ph.D Thesis, Chinese Academy of Forestry, Beijing, China, 2009. [Google Scholar]
- Robinet, C.; Roques, A.; Pan, H.Y.; Fang, G.; Ye, J.; Zhang, Y.; Sun, J. Role of human-mediated dispersal in the spread of the pine wood nematode in China. PLoS ONE 2009, 4, e4646. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Homozygosity, SNP count, missing SNPs and private SNPs distributions of the SNPs found in 30 strains. Note: HB is Hubei strain, HEN is Henan strain, HN is Hunan strain. The same is below.
Figure 1.
Homozygosity, SNP count, missing SNPs and private SNPs distributions of the SNPs found in 30 strains. Note: HB is Hubei strain, HEN is Henan strain, HN is Hunan strain. The same is below.

Figure 2.
Box plots of SNP genotypes among 30 B. xylophilus strains.
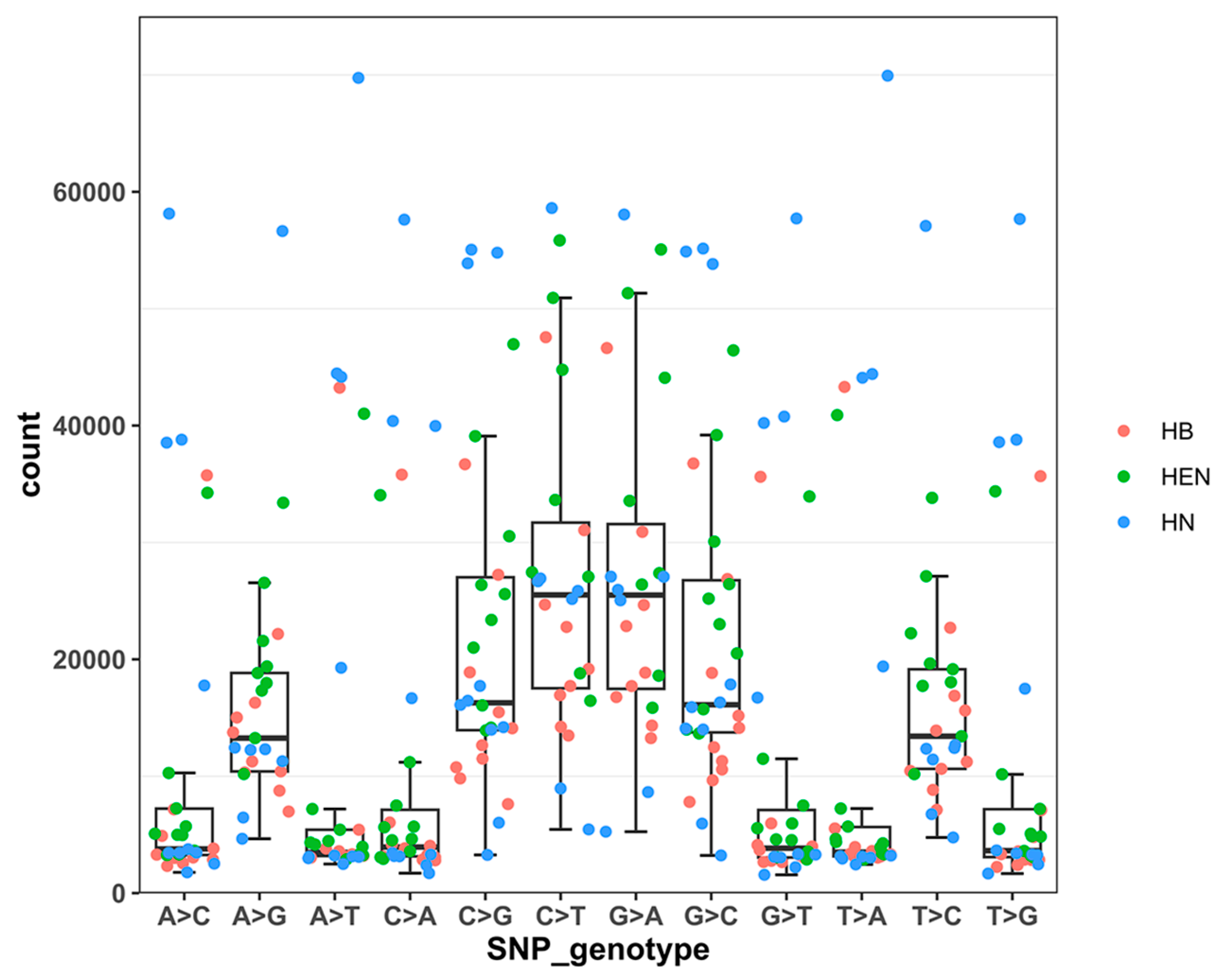
Figure 3.
PCA of population genetic structure: (a) PCA results of 30 strains based on 1312 SNP markers; (b) PCA results of 50 strains based on 2244 SNP markers. Note: GD is Guangdong strain, JS is Jiangsu strain.
Figure 3.
PCA of population genetic structure: (a) PCA results of 30 strains based on 1312 SNP markers; (b) PCA results of 50 strains based on 2244 SNP markers. Note: GD is Guangdong strain, JS is Jiangsu strain.
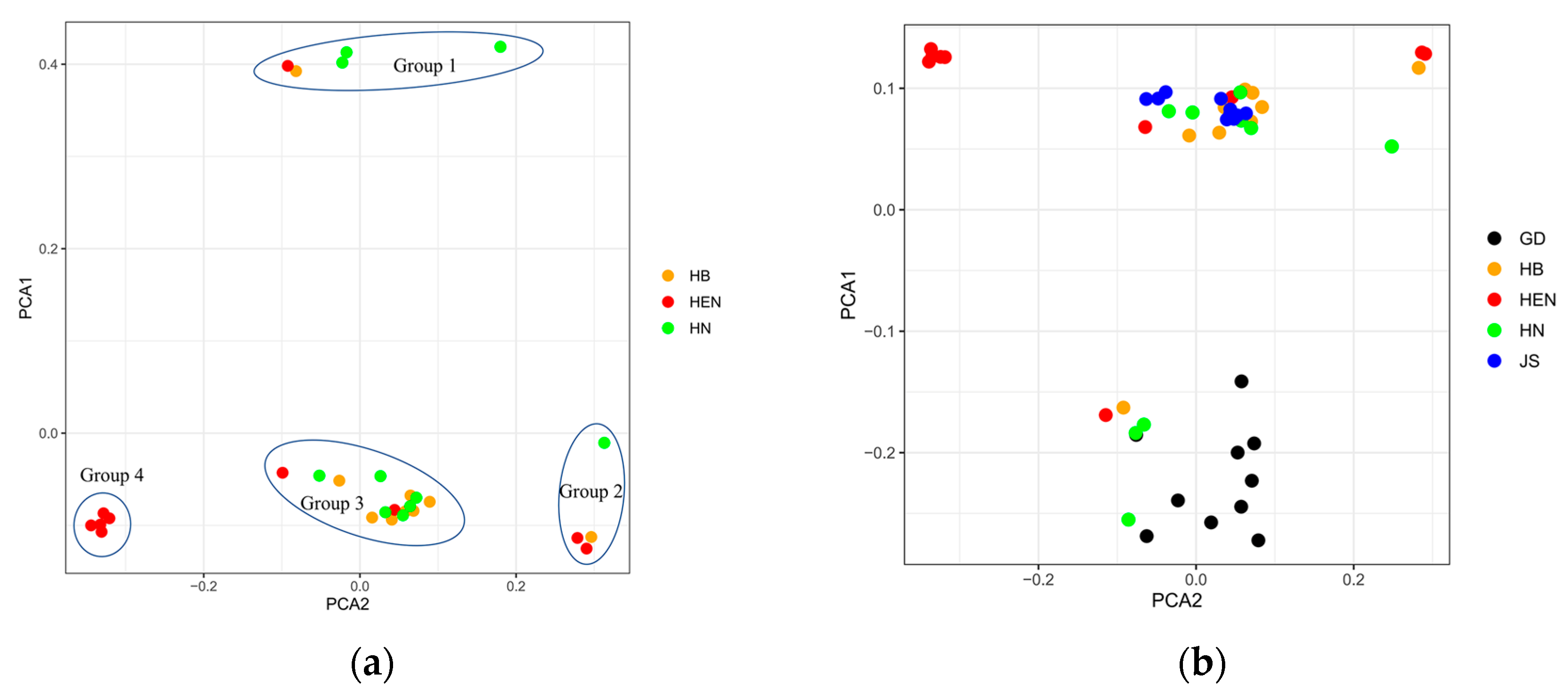
Figure 4.
Phylogenetic trees of all 50 rains by based on Neighbor Joining method.
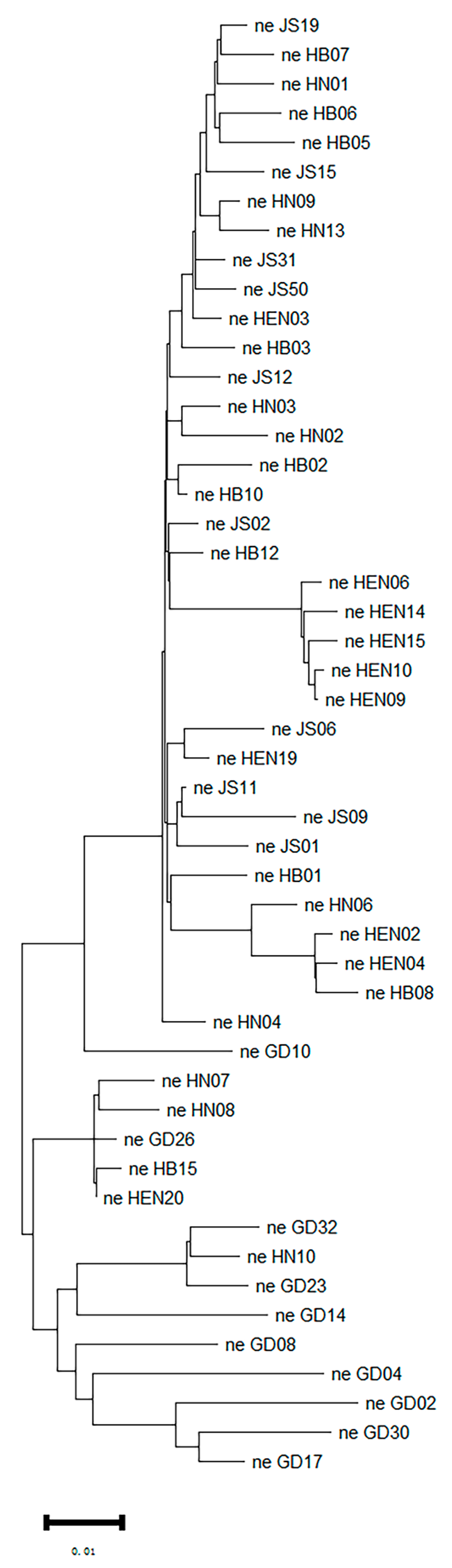
Figure 5.
Introgression analysis revealed possible B. xylophilus migration routes.

Table 1.
Sample information of 50 B. xylophilus strains.
| Strains No. | Origin | Host | Sampling time |
|---|---|---|---|
| GD02 | Qingcheng District, Qingyuan City, Guangdong Province | Pinus massoniana | 2015.01 |
| GD04 | Huiyang District, Huizhou City, Guangdong Province | P. massoniana | 2015.01 |
| GD08 | Boluo County, Huizhou City, Guangdong Province | P. massoniana | 2015.01 |
| GD10 | Huangpu District, Guangzhou City, Guangdong Province | P. massoniana | 2015.01 |
| GD14 | Dongguan, Guangdong Province | P. massoniana | 2015.01 |
| GD17 | Tianhe District, Guangzhou City, Guangdong Province | P. massoniana | 2015.01 |
| GD23 | Meijiang District, Meizhou City, Guangdong Province | P. massoniana | 2015.11 |
| GD26 | Fengshun County, Meizhou City, Guangdong Province | P. massoniana | 2017.08 |
| GD30 | Haifeng County, Shanwei City, Guangdong Province | P. massoniana | 2017.08 |
| GD32 | Dongyuan County, Heyuan City, Guangdong Province | P. massoniana | 2017.08 |
| HB01 | Chibi, Xianning City, Hubei Province | P. massoniana | 2015.04 |
| HB02 | Changyang County, Yichang City, Hubei Province | P. massoniana | 2015.04 |
| HB03 | Enshi City, Enshi Prefecture, Hubei Province | P. massoniana | 2015.04 |
| HB05 | Huangpi District, Wuhan City, Hubei Province | P. massoniana | 2015.04 |
| HB06 | Huangpi District, Wuhan City, Hubei Province | P. massoniana | 2015.04 |
| HB07 | Huangpi District, Wuhan City, Hubei Province | P. massoniana | 2015.04 |
| HB08 | Yiling District, Yichang City, Hubei Province | P. massoniana | 2015.11 |
| HB10 | Yidu District, Yichang City, Hubei Province | P. massoniana | 2015.11 |
| HB12 | Zengdu District, Suizhou City, Hubei Province | P. massoniana | 2017.08 |
| HB15 | Luotian County, Huanggang City, Hubei Province | P. massoniana | 2017.01 |
| HEN02 | Xin County, Xinyang City, Henan Province | P. massoniana | 2015.08 |
| HEN03 | Xin County, Xinyang City, Henan Province | P. massoniana | 2015.08 |
| HEN04 | Xin County, Xinyang City, Henan Province | P. massoniana | 2015.08 |
| HEN06 | Xichuan County, Nanyang City, Henan Province | P. massoniana | 2017.01 |
| HEN09 | Xichuan County, Nanyang City, Henan Province | P. massoniana | 2017.11 |
| HEN10 | Xichuan County, Nanyang City, Henan Province | P. massoniana | 2017.11 |
| HEN14 | Xixia County, Nanyang City, Henan Province | Pinus tabuliformis | 2018.01 |
| HEN15 | Xixia County, Nanyang City, Henan Province | P. massoniana | 2018.01 |
| HEN19 | Xin County, Xinyang City, Henan Province | P. massoniana | 2018.01 |
| HEN20 | Xin County, Xinyang City, Henan Province | P. massoniana | 2018.01 |
| HN01 | Cili County, Zhangjiajie City, Hunan Province | P. massoniana | 2015.03 |
| HN02 | Linxiang City, Yueyang City, Hunan Province | P. massoniana | 2015.03 |
| HN03 | Yunxi District, Yueyang City, Hunan Province | P. massoniana | 2015.03 |
| HN04 | Hengnan County, Hengyang City, Hunan Province | P. massoniana | 2015.03 |
| HN06 | Cili County, Zhangjiajie City, Hunan Province | P. massoniana | 2015.08 |
| HN07 | Taoyuan County, Changde City, Hunan Province | P. massoniana | 2016.08 |
| HN08 | Taoyuan County, Changde City, Hunan Province | P. massoniana | 2016.08 |
| HN09 | Lingling District, Yongzhou City, Hunan Province | P. massoniana | 2019.03 |
| HN10 | Shaoyang County, Shaoyang City, Hunan Province | P. massoniana | 2019.03 |
| HN13 | Lingling District, Yongzhou City, Hunan Province | P. massoniana | 2019.03 |
| JS01 | Liuhe District, Nanjing City, Jiangsu Province | P. massoniana | 2014.12 |
| JS02 | Runzhou District, Zhenjiang City, Jiangsu Province | P. massoniana | 2014.12 |
| JS06 | Binhu District, Wuxi City, Jiangsu Province | P. massoniana | 2014.12 |
| JS09 | Xuyi County, Huai‘an City, Jiangsu Province | P. massoniana | 2015.01 |
| JS11 | Haizhou District, Lianyungang City, Jiangsu Province | Pinus densiflora | 2015.01 |
| JS12 | Yizheng, Yangzhou City, Jiangsu Province | P. massoniana | 2015.01 |
| JS15 | Changshu City, Suzhou City, Jiangsu Province | P. massoniana | 2015.02 |
| JS19 | Runzhou District, Zhenjiang City, Jiangsu Province | P. massoniana | 2014.12 |
| JS31 | Lishui District, Nanjing City, Jiangsu Province | P. massoniana | 2017.01 |
| JS50 | Jintan District, Changzhou City, Jiangsu Province | P. massoniana | 2017.01 |
Table 2.
A summary of SNPs found in 30 B. xylophilus strains.
| Strains NO. | SNP count | Homozygous | Missing | Specific SNP count |
|---|---|---|---|---|
| HB01 | 81,723 | 46,927 | 1,367,096 | 6,644 |
| HB02 | 110,859 | 75,239 | 1,189,888 | 6,547 |
| HB03 | 128,236 | 34,233 | 957,159 | 2,695 |
| HB05 | 90,192 | 45,461 | 1,318,256 | 2,291 |
| HB06 | 91,540 | 57,049 | 965,177 | 2,290 |
| HB07 | 121,555 | 69,319 | 1,038,514 | 9,634 |
| HB08 | 85,121 | 33,889 | 1,277,353 | 2,195 |
| HB10 | 230,409 | 66,327 | 954,380 | 7,499 |
| HB12 | 156,058 | 92,696 | 978,007 | 8,018 |
| HB15 | 747,217 | 620,622 | 1,364,706 | 10,220 |
| HEN02 | 179,367 | 39,985 | 890,120 | 6,229 |
| HEN03 | 115,598 | 73,932 | 1,032,402 | 13,361 |
| HEN04 | 161,298 | 59,004 | 863,933 | 6,837 |
| HEN06 | 387,158 | 74,788 | 670,407 | 31,052 |
| HEN09 | 265,742 | 52,417 | 848,798 | 23,215 |
| HEN10 | 215,398 | 41,954 | 816,941 | 6,011 |
| HEN14 | 111,679 | 61,536 | 918,903 | 4,289 |
| HEN15 | 125,089 | 47,713 | 5,169,332 | 1,586 |
| HEN19 | 220,318 | 37,791 | 5,087,640 | 5,128 |
| HEN20 | 748,317 | 620,041 | 5,223,813 | 3,968 |
| HN01 | 123,168 | 86,716 | 946,562 | 1,316 |
| HN02 | 38,277 | 14,055 | 1,572,950 | 716 |
| HN03 | 124,574 | 91,984 | 928,412 | 1,906 |
| HN04 | 58,334 | 25,275 | 1,484,882 | 2,413 |
| HN06 | 373,296 | 26,536 | 1,482,689 | 80,083 |
| HN07 | 894,621 | 704,750 | 747,078 | 24,547 |
| HN08 | 878,428 | 611,007 | 837,408 | 12,192 |
| HN09 | 130,567 | 23,176 | 5,151,805 | 396 |
| HN10 | 1,210,980 | 1,033,119 | 5,248,382 | 505,849 |
| HN13 | 128,256 | 64,496 | 5,140,161 | 1,855 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



