
Submitted:
30 May 2023
Posted:
01 June 2023
You are already at the latest version
Abstract
Astrocytes have been implicated in a wide range of neurodegenerative diseases, with the literature indicating harmful or beneficial roles on a case-by-case basis. On such conditions, these cells are capable of secreting several inflammatory factors and also promote synapse elimination and remodeling. These responses are possible because they sense their surroundings via several receptors, including metabotropic glutamate receptor (mGluR5). However, mGluR5 activation in astrocytes under neuroinflammatory conditions can be neuroprotective or have the opposite effect. In this work, we investigated the role of mGluR5 in hiPSC-derived astrocytes subjected to pro-inflammatory stimulation by recombinant TNF-α (rTNF-α). Our results show that mGluR5 blockade by CTEP is capable of reducing pro-inflammatory cytokines (IL-6, IL-8 and TNF-α) release in rTNF-α-stimulated astrocytes, but, in contrast, leads to an augmented expression of the pan-reactive marker SERPINA3. Additionally, CTEP enhances synaptoneurosomes phagocytosis by astrocytes in both non-stimulated and TNF-α-stimulated conditions, indicating that mGluR5 blockade alone is enough to drive synaptic material engulfment. Finally, mGluR5 antagonism as well as rTNF-α stimulation leads to a reduction in the expression of synaptogenic molecules. Altogether, these data suggest a complex role for mGluR5 in human astrocytes, since its blockage may have beneficial and detrimental effects under inflammatory conditions.
Keywords:
mGluR5
; astrocyte
; hiPSC
; phagocytosis
; TNF-α
1. Introduction
Astrocytes are implicated in the pathology of a diverse array of neurological diseases [1] including Alzheimer’s disease (AD) [2], Huntington’s disease (HD) [3,4], amyotrophic lateral sclerosis (ALS) [5-7], multiple sclerosis (MS) [8-10], and Parkinson’s disease (PD) [11,12]. Astroglia is formed by a highly heterogeneous population [13]. These cells may assume a reactive phenotype in pathological contexts, releasing inflammatory factors that might impact disease outcome either in a detrimental or beneficial fashion [14,15]. One important role played by glial cells is the phagocytosis of dead cells, synapses and myelin [16,17]. Although aberrant synapse pruning was shown to contribute to dementia [18], elimination of dystrophic synapses and dendrites, as well as phagocytosis of extracellular protein aggregates, such as amyloid-β (Aβ) [19,20], is essential for proper brain function. Another important function of astrocytes is to promote synapse remodelling by secreting synaptogenic molecules, including brain derived neurotrophic factor (BDNF) [21], glypican-4 (GPC4) [22], hevin [23], and thrombospondins (TSPs) [23,24].
The metabotropic glutamate receptor type 5 (mGluR5) is a G-protein-coupled receptor (GPCR) whose involvement in neurodegenerative disorders has been widely studied [25]. As in neurons, mGluR5 stimulation in astrocytes activates the Gαq/11/PLCβ/IP3 pathway, increasing intracellular Ca2+, which facilitates glutamate release [26-29]. Moreover, stimulation of this receptor activates non-canonical cell signalling pathways, including mitogen-activated protein kinase (MAPK) and phospholipase D (PLD) [30,31]. Although mGluR5 expression in astrocytes declines postnatally [32,33], previous works show an upregulation of this receptor expression in astrocytes in neurological disorders that most commonly affect adults or the elderly, such as ALS [34], MS [35], and AD [36], suggesting a relevant role of mGluR5 in gliopathology. In the case of microglia, the activation of mGluR5 has an anti-inflammatory effect, decreasing microglial expression of tumor necrosis factor-α (TNF-α) and production of reactive oxygen species [37,38]. However, the role of mGluR5 in astrocytes is controversial, as some studies indicate that activation of astrocytic mGluR5 following injury is protective for neighbouring cells as it triggers the release of growth factors and synaptogenic molecules [39,40], whereas others have shown that mGluR5 activation is harmful as it increases the production of inflammatory mediators [41,42]. For instance, the inhibition of mGluR5 by MPEP impairs methamphetamine-induced increase in IL-6 and IL-8 in a human astroglia cell line [41]. Additionally, another work suggests that inhibition of mGluR5 in murine astrocytes could reduce the expression of IL-1β and MCP-1, although the authors do not show it directly [43]. Furthermore, it has been shown that the activation of mGluR5 by DHPG has no effect on the IL-1β-induced expression of IL-6 in human astrocytes [44]. Despite all these studies investigating the role of astrocytic mGluR5 in neurological disorders, it has been challenging to successfully replicate these findings in clinical trials. This could be partially explained by the existing molecular differences between human and murine glial cells [45,46]. For instance, it has been pointed that these cells display species-specific gene expression profiles upon poly-I:C or TNF-α stimulation, with human astrocytes showing stronger immune molecular response compared to their murine counterparts [46]. Thus, more studies are needed to investigate how mGluR5 can impact the activation of human astrocytes and the production of inflammatory and synaptogenic mediators.
In this study we used cultured astrocytes differentiated from human induced pluripotent stem cells (hiPSCs) to investigate the influence of mGluR5 on the response of these cells subjected to inflammatory stimulation with recombinant TNF-α (TNF-α). We show that the mGluR5 negative allosteric modulator (NAM) CTEP reduces the rTNF-α-induced early production of inflammatory factors, including TNF-α, IL-6 and IL-8 in astrocytes, while increasing the expression of SERPINA3, a general marker of astroglia activation. Since reactive astrogliosis involves functional alterations [47] and given that the pathology of many neurological diseases encompasses unbalances in the synaptic turnover [48], we also assessed the rate of phagocytosis of synaptoneurosomes by astrocytes and the expression of synaptogenic proteins. Treatment with the mGluR1/5 agonist, DHPG, augmented astrocytic phagocytosis in the absence of rTNF-α, whereas CTEP increased phagocytosis regardless of rTNF-α stimulation. Both CTEP and rTNF-α decreased the expression of synaptogenic molecules, with CTEP treatment reducing the expression of TSP-1 and rTNF-α decreasing the expression of GPC4, while both CTEP and rTNF-α decreased the expression of BDNF. Thereby, the results shown here point to the involvement of mGluR5 in the regulation of the expression of synaptogenic molecules and reactive astrogliosis markers, with mGluR5 blockade augmenting the phagocytosis of synaptoneurosomes by human astrocytes.
2. Materials and Methods
- Human-induced Pluripotent Stem Cell (hiPSC) line and cell culture maintenance
The 7889SA cell line was obtained from the New York Stem Cell Foundation (NYSCF ID CO0002-01-SV-003) and described previously [49]. hiPSCs were maintained on 6-well plates coated with Geltrex (Gibco, cat no. A1413302) in StemFlex Medium (Gibco, cat no. A3349301). Upon reaching 80-90% confluence, cells were incubated for 3-4 min in 500 µM EDTA (Sigma, cat no. EDS) in PBS, dissociated in clumps and seeded into new 6-well Geltrex-coated plates for expansion. Media was changed every 48 h.
- Human astrocytes differentiation
Astrocytes were differentiated from hiPSCs as previously described [50]. hiPSCs were dissociated into single cells using StemPro Accutase (Gibco, cat no. A1110501) and seeded into 6-well Geltrex-coated plates at 3x104 cells/cm2 in StemFlex Medium supplemented with 10 µM Rho-associated protein kinase (ROCK) inhibitor Thiazovivin (Sigma Aldrich, cat no. SML1045). In order to start differentiation (day 0), medium was changed to PSC Neural Induction Medium containing Neurobasal Medium (Gibco, cat no. 21103-049), Neural Induction Supplement (Gibco, cat no. A1647701) and 1% penicillin-streptomycin solution (Gibco, cat no. 15410-122). Media was changed every 48 h. On day 7, NSCs were dissociated with Accutase and plated at 1×105 cells/cm2 in 60 mm Geltrex-coated dishes in NSC Expansion Medium containing 50% Advanced DMEM/F12 (Gibco, cat no. 12634010), 50% Neurobasal Medium, Neural Induction Supplement and 1% penicillin-streptomycin solution supplemented with 10 µM ROCK inhibitor Thiazovivin. Media changes were performed every 48 h. When NSCs reached 90% confluence, cells were dissociated with Accutase and seeded at 5 × 104 cells/cm2 in 25 cm2 Geltrex-coated culture flasks in Astrocyte Induction Medium containing DMEM/F12 Medium (Gibco, cat no.12400024), N2 supplement (Gibco, cat no. 17502048), 1% fetal bovine serum (FBS) (Gibco, cat no. 12657-029) and 1% antibiotic-antimycotic solution (Gibco, cat no.15240112). Media was changed every 48 h for 21 days. During this period, upon reaching full confluence, cells were expanded at a ratio of 1:3 using Accutase to 75 cm2 Geltrex-coated culture flasks. By the end of differentiation, media was switched to Astrocyte Maturation Medium (DMEM/F12, 10% FBS and 1% antibiotic-antimycotic solution) for an additional period of at least 5 weeks. During this period, media was changed twice a week and when reaching full confluence, cells were dissociated using Trypsin/EDTA 0,125% (Gibco, cat no. 25200072) and expanded to 175 cm2 culture flasks at a ratio of 1:2 (without Geltrex coating). Cells were maintained under standard culture conditions (95% relative humidity and 5% CO2 at 37°C) and tested routinely for Mycoplasma contamination as previously described [51].
- Quantitative RT-PCR
After 9 weeks of maturation, upon reaching 100% confluence in 175 cm2 culture flasks, hiPSC-derived astrocytes were dissociated with Trypsin/EDTA 0.125% (Gibco, cat no. 25200072) and seeded at 1x104 cells/cm2 in 6-well plates. After 5 days in culture, cells were washed three times with PBS, DMEM/F12 media was replenished, and astrocytes were serum starved for 24 h. Then, cells were pre-treated for 1 h with 10 µM CTEP (Axon Medchem, cat no. Axon 1972), 10 µM DHPG (Tocris, cat no. 0805) or vehicle (DMSO; Sigma-Aldrich, cat no. 41639) and subsequently stimulated with 10 ng/mL rTNF-α for either 4h or 24 h. After treatment, the supernatant was flash frozen and stored at -80°C until further use, and cells were collected with 1 mL of TRIzol™ reagent (Invitrogen, cat no. 15596018), transferred to 1.5 mL microcentrifuge tubes and frozen at -80°C. Total RNA was isolated as per manufacturer’s instructions and resuspended in 12 μL of nuclease-free water. RNA concentration and quality was analyzed by spectrophotometer (Multiskan® GO, Thermo Scientific). cDNAs were prepared from 800 ng of total RNA extracted in a 20 µL final reverse transcription reaction. Quantitative RT-PCR (RT-qPCR) was performed with 10x diluted cDNA using Power SYBR® Green PCR Master Mix in the QuantStudio™ 7 Flex real-time PCR system platform (Applied Biosystems®). RT-qPCR assays were performed to quantify the mRNA levels of the following genes: C3 (NM_000064.3); IPO8 (NM_006390.3); RPLP0 (NM_001002.3); MERTK (NM_006343.2); SERPINA3 (NM_001085.5); VCAM1 (NM_001078.5); S100A10 (NM_002966.3); BDNF (NM_170735.6); GPC4 (NM_001448.3); TSP1 (NM_003246.4) and TNF-α (NM_000594.3). Primers were designed using the Primer3Plus Program [52]. Primer sequences are listed in Table 1.
Previous verification of undesired secondary formations or dimers between primers were performed using “OligoAnalyser 3.1” tool (Integrated DNA Technologies©), available at https://www.idtdna.com/calc/analyzer. All primers used in this work were validated by serial dilution assay and the reaction efficiency was calculated, comprising 90-110% (data not shown). Changes in gene expression were calculated by the 2−ΔCt method, using the average of the housekeeping genes IPO8 and RPLP0 for normalization.
- Immunofluorescence staining
After 5 weeks of maturation, hiPSC-derived astrocytes were plated onto acid-etched clean glass coverslips coated with 50 µg/mL poly-D-lysine (Sigma, cat no. P6407) in 24-well plates at 1x103 cells/cm2 in Astrocytes Maturation Media. After 5 days in culture, cells were fixed for 15 min in 4% paraformaldehyde (PFA) (Sigma, cat no. 158127) diluted in PBS. Samples were permeabilized with 0.3% Triton X-100 (Labsynth, cat no. T2502) diluted in PBS (PBST) for 10 min and blocked with 2% Bovine Serum Albumin (BSA) (Sigma, cat no. A7906) diluted in PBST (blocking solution) for 1 h at room temperature. Then, cells were washed three times with PBS and incubated overnight at 4°C with the following primary antibodies diluted in blocking solution: anti-S100β (1:200, Abcam, cat no. ab52642) and anti-GFAP (1:200, Cell Signaling, cat no. 12389). After incubation, cells were washed three times with PBS and incubated for 1 h at room temperature with the following secondary antibody and staining reagents diluted in blocking solution: anti-Rabbit IgG Alexa Fluor 488 (1:400, Invitrogen, cat no. A-11008), Hoechst (1:500, Invitrogen, cat no. H3570) and Alexa Fluor 633 Phalloidin (1:1000, Invitrogen, cat no. A22284). Coverslips were washed three times as mentioned above and mounted on clean glass slides with DAKO Mounting Medium (Agilent Technologies, cat no. S302380-2). Astrocytes were imaged using a Nikon A1 Laser Confocal Microscope (CGB, UFMG, Brazil).
- Synaptoneurosomes isolation and staining
All procedures carried out in this study were approved by the Ethics Committee on Animal Use of the Federal University of Minas Gerais (CEUA #120/2017). A 12-month-old male wild-type C57BL/6 mouse was obtained from UFMG’s Central Animal Facility. Mouse brain was dissected, weighed, and cut into sections of approximately 100 mg that were individually transferred to an ice-cold sterile Dounce homogenizer with 1 mL of synaptoneurosomes isolation buffer (SIB) (10 mM HEPES, 1 mM EDTA, 2 mM EGTA, 0.5 mM DTT and protease inhibitors, pH 7.0; sterile-filtered) [53]. Tissue was broken with 10 slow strokes, homogenates were transferred to 1.5 mL microcentrifuge conical tubes and centrifuged at 1200 g for 10 min at 4°C. The supernatant was transferred to new 1.5 mL microcentrifuge tubes and centrifuged at 15000 g for 20 min at 4°C. The pelleted debris fraction and a small aliquot of the supernatant (synaptoneurosomal homogenate) were collected in SIB and stored at -80°C for later validation. The supernatant (cytosolic fraction) was collected into new 1.5 mL microcentrifuge tubes and frozen at -80°C and the pelleted synaptoneurosomes resuspended in SIB + 5% DMSO (Sigma, cat no. D8418), aliquoted and frozen at -80°C until later use.
On the day of synaptoneurosomal engulfment assay, synaptoneurosomes aliquots were thawed and their protein concentration determined via the Bradford Protein Assay (Bio-rad, cat no. 5000205). Synaptoneuromes were spun down by centrifugation at 15000 g for 20 min at 4°C, supernatant was discarded, and the pellet resuspended in 2 µM Vybrant CM-Dil solution (Invitrogen, cat no. V22888) diluted in room temperature sterile PBS. Staining was performed according to manufacturer instructions. CM-Dil-labeled synaptoneurosomes were spun down as described above, washed twice with ice-cold PBS to remove fluorescent dye excess and resuspended in ice-cold PBS adjusting its concentration to 0.25 µg/µL. Synaptoneurosomes were kept on ice and protected from light until its use in the aforementioned assay. All procedures were carried out under sterile conditions.
- Synaptoneurosomes engulfment assay and analysis
hiPSC-derived astrocytes were plated onto acid-etched clean glass coverslips coated with 50 µg/mL poly-D-lysine in 24-well plates at 5.5x103 cells/cm2 in Astrocytes Maturation Media. After 5 days in culture, cells were washed three times with PBS and stained with 10 µM CellTracker Blue CMF2HC Dye (Invitrogen, cat no. C12881) diluted in warm DMEM/F12 without FBS and incubated for 45 min in the cell culture incubator. The staining solution was removed, fresh DMEM/F12 media was replenished, and astrocytes serum starved for 22 h. On the following day, cells were pre-treated for 1 h with 10 µM CTEP, 10 µM DHPG or vehicle (DMSO) and subsequently stimulated with 10 ng/mL rTNF-α for an extra hour. Then, 1.875 µg (7.5 µL) of CM-Dil-labeled synaptoneurosomes were added into each well and incubated for 24 h. After incubation, cells were washed twice with room temperature PBS to remove non-engulfed material and fixed for 15 min with 4% PFA solution diluted in PBS. Coverslips were washed three times as mentioned above and mounted on clean glass slides with DAKO Mounting Medium. Images were acquired using a Nikon A1 Laser Confocal Microscope. All assays were carried out in duplicates and twice independently.
Images were analyzed using FIJI (v. 1.53t) and CellProfiler (v. 4.2.5) software programs. Briefly, CellTracker-labeled astrocytes and CM-Dil-labeled synaptoneurosomes corresponding channels were split and images pre-processed using File S1 and File S2 macros in FIJI, respectively, saved and exported in .tif format. Pre-processed .tif files were imported to CellProfiler and analyzed using the File S3 pipeline. Phagocytic Index (PI) was calculated for each cell using the formula below:
Orthogonal projections and 3D rendering (Figure S3 and Movie S1) of z-stacks were generated using FIJI and FluoRender (v. 2.29.2), respectively.
- Immunoblotting
Protein concentration of synaptoneurosomal preparation fractions (pelleted debris, synaptoneurosomal homogenate, cytosolic fraction and isolated synaptoneurosomes) was measured using the Bradford Protein Assay. Twenty-five µg of each fraction was diluted in Laemmli Sample Buffer, boiled at 95°C for 5 min and resolved in 10% SDS-PAGE. Proteins were transferred onto a 0.45 µm nitrocellulose membrane (Bio-Rad, cat no. 1620115), blocked with 5% BSA and 0.1% Tween-20 (Labsynth, cat no. T1028) diluted in TBS (TBST) for 1 h at room temperature, followed by overnight incubation at 4°C with the following primary antibodies in 3% BSA solution diluted in TBST: anti-syntaxin1 (1:200, Santa Cruz, cat no. sc-12736), anti-Homer (1:500, Santa Cruz, sc-8921) and anti-vinculin (1:10000, Abcam, ab129002). After incubation, primary antibodies were removed, membranes washed three times with TBST and incubated for 1 h at room temperature with secondary antibodies in 3% free-fat milk diluted in TBST: HRP-conjugated anti-mouse IgG (1:2500, Millipore, cat no. AP308P), HRP-conjugated anti-rabbit IgG (1:2500, Bio-Rad, cat no. 1706515) and HRP-conjugated anti-goat IgG (1:2500, Santa Cruz, sc-2354). Afterwards, membranes were washed three times as already described and incubated for 5 min with ECL Prime Western Blot Detection Reagent (Cytiva, cat no. RPN2232) for chemiluminescence detection using the ImageQuant LAS 4000 (GE Healthcare) platform.
- Cytokine quantification
Cytokines were quantified in cell culture supernatants by flow cytometry using the BD Cytometric Bead Array Human Inflammatory Cytokines Kit (Becton, Dickinson and company - BD Biosciences, cat no. 551811) according to manufacturer's instructions. Sample acquisition was performed on the FACS Aria Fusion (BD Biosciences). The CBA Analysis Software (BD Biosciences) was used for data analysis based on standard concentration curves and the results were expressed as pg/mL.
- Statistical analyses
Statistical analyses and data plots were performed using the GraphPad Prism (v. 8.0.1) software. Normal distribution was assumed and two-way ANOVA, followed by Tukey’s multiple comparison tests with confidence level set to 0.95 (α = 0.05) was carried out for all experiments, unless otherwise stated. For the synaptoneurosomes engulfment assay, the Kolmogorov-Smirnov test was executed, indicating the data followed a lognormal distribution. In that way, a generalized linear model fitted in the lognormal distribution, followed by Sidak’s multiple comparison test with a confidence level set to 0.99 (α = 0.01) was employed. This data was analyzed using the STATA (v. 14.0) software.
3. Results
3.1. mGluR5 Blockade Attenuates Early rTNF-α-Induced Proinflammatory Response in Human Astrocytes
The role of mGluR5 in neurodegenerative diseases and neuroinflammation, as well as its potential as target for pharmacological interventions have been widely explored in animal models [54-59]. However, little is known about the function of this receptor in human cells. In order to investigate the role of mGluR5 in human astrocytes under proinflammatory conditions, astrocytes were differentiated from hiPSCs for four weeks and matured for an additional period of five weeks until they displayed strong expression of the canonical astrocytic markers GFAP (Figure 1A) and S100β (Figure 1B). Upon stimulation with rTNF-α 10 ng/mL, astrocytes quickly responded by up-regulating TNF-α, IL-6 and IL-8 secretion (Figure 2), while IL-1β, IL-10 and IL-12p70 secretion was not detected. The increase in TNF-α protein secretion 4 h following stimulation (Figure 2A) was accompanied by an increase in its mRNA levels (Figure S1A). Nonetheless, there was a slight decline in TNF-α secretion at 24 h post-stimulation (Figure 2B), which was not mirrored by its transcript levels that remained relatively steady compared to the 4 h post-stimulation timepoint (Figure S1B). On the other hand, both IL-6 and IL-8 secreted protein levels displayed a modest elevation at 4 h (Figures 2C and 2E), followed by a marked increase at 24 h post-stimulation (Figures 2D and 2F).
Interestingly, blocking mGluR5 activity with CTEP (10 µM) led to a substantial reduction in TNF-α, IL-6 and IL-8 secretion only at 4 h post-stimulation (Figures 2A, 2C and 2E), with this effect wearing off at 24h (Figures 2B, 2D and 2F). In addition, TNF-α mRNA levels was also diminished by CTEP treatment, although this reduction was only observed at 24 h post-stimulation (Figure S1B), indicating that blocking mGluR5 activity may also decrease the pro-inflammatory response induced by rTNF-α stimulation beyond the timepoints analyzed in this study. Treatment with the mGluR1/5 agonist DHPG (10 µM) only led to a modest reduction in TNF-α secretion at 4 h post-stimulation (Figure 2A), without altering the secretion of the other cytokines (Figures 2B-2F) or the mRNA levels of TNF-α (Figures S1A-S1B) at either 4 h or 24 h post-stimulation. These data indicate that mGluR5 antagonism can prevent pro-inflammatory cytokine production by astrocytes under inflammatory conditions.
3.2. mGluR5 Modulates the Expression of the Pan-Reactive Astrocytic Marker SERPINA3 in Human Astrocytes
In the past decade, it has become widely recognized that astrocytes can be polarized to either an inflammatory and neurotoxic (A1 astrocytes) or a neuroprotective (A2 astrocytes) phenotype in response to external stimulation [60-62]. In that way, we analyzed the expression of the pan-reactive marker SERPINA3 (human homolog of murine SerpinA3n [63]), the A1-reactive markers C3 (subtype inflammatory reactive astrocyte signature cluster 1 (IRAS1)) and vascular cell adhesion molecule 1 (VCAM-1 (subtype IRAS2), and the A2-reactive marker S100A10 [60-62,64] in human astrocytes. As shown in Figures 3A-3F, rTNF-α stimulation promoted an augmentation in SERPINA3, C3 and VCAM-1 transcript levels at all tested timepoints, whereas the transcriptional levels of the A2 astrocyte marker S100A10 remained unchanged relative to non-stimulated astrocytes (Figures 3G-3H). Indeed, these data corroborate previous findings, indicating that TNF-α upregulates most pan-reactive and A1-reactive markers, while A2-reactive markers remain unchanged [62]. Notably, CTEP treatment led to an even greater up-regulation of SERPINA3 gene expression in rTNF-α-stimulated astrocytes at all tested timepoints, compared to its non-treated (NT) counterparts (Figures 3A-3B). DHPG treatment also promoted an increase in SERPINA3 expression in rTNF-α-stimulated astrocytes, but only at 4 h (Figure 3A), whereas, at 24 h post-stimulation, SERPINA3 mRNA levels were reduced in DHPG- relative to CTEP-treated astrocytes (Figure 3B). On the other hand, while there was a minor trend towards a decrease in C3 gene expression in DHPG-treated astrocytes when stimulated with rTNF-α at 24 h (Figure 3D), it did not reach statistical significance. Therefore, it can be concluded that mGluR5 blockade promotes a sustained upregulation of the pan-reactive astrocytic marker SERPINA3 gene expression, while DHPG treatment leads only to a short-lived increase in SERPINA3 gene expression under pro-inflammatory conditions (Figures 3A-3B). Additionally, neither CTEP nor DHPG treatment could drive the A2-reactive astrocyte marker S100A10 gene expression (Figure 3G-3H) or induce further alterations in the A1-reactive astrocyte markers C3 and VCAM-1 mRNA levels either in the presence or in the absence of rTNF-α stimulation (Figures 3C-3F).
3.3. CTEP Treatment Leads to Enhanced Synaptic Material Engulfment by Human Astrocytes
It has been shown that astrocyte reactivity affects its capacity to uptake synaptic material [62]. Then, we decided to develop an assay to assess synaptic material phagocytosis by our hiPSC-derived astrocytes. Firstly, we isolated synaptoneurosomes from mouse brain and evaluated if this procedure was effective in enriching both pre- and post-synaptic proteins. As it can be seen in Figure S3, both syntaxin-1 and Homer, pre- and postsynaptic markers respectively, were indeed enriched in the synaptoneurosomes fraction, while vinculin, a cytoskeleton protein, was mostly confined to the cytosolic compartment. Afterwards, we investigated whether our hiPSC-derived astrocytes could engulf fluorescently-labeled synaptoneurosomes. In fact, human astrocytes were capable to engulf synaptic material, as several red puncta, corresponding to fluorescently-labeled synaptoneurosomes, were observed inside the astrocytes cell body (Figure S3 and Movie S1).
rTNF-α stimulation led to an increase in astrocytic phagocytosis, as compared to non-stimulated cells (Figure 4A, 4D and 4G). Taking in consideration that both rTNF-α and mGluR5 pharmacological manipulation modified the expression of reactive astrocytic markers, we decided to analyze whether either blocking or activating mGluR5 could impact phagocytosis by astrocytes. Interestingly, both CTEP and DHPG treatment led to enhanced synaptoneurosomal phagocytosis in human astrocytes in basal (no rTNF-α) conditions (Figures 4A-4C and 4G). Remarkably, CTEP-treated astrocytes stimulated with rTNF-α showed higher synaptic material engulfment level than their NT and DHPG-treated counterparts (Figures 4D-4G). In addition, both non-stimulated and rTNF-α-stimulated astrocytes that were pre-treated with 10 µM CTEP displayed similar synaptoneurosomal phagocytic level (Figures 4B, 4E and 4G). These data suggest that blocking mGluR5 activity is enough to enhance synaptic material engulfment by human astrocytes, even in the absence of proinflammatory stimulation.
Since the tyrosine-protein kinase mer (MERTK) has been identified as one of the main phagocytic receptors responsible for promoting synapse engulfment by astrocytes [19,65], we analyzed its expression level in human astrocytes upon mGluR5 pharmacological activation or inhibition. Remarkably, only CTEP treatment increased MERTK gene expression in human astrocytes in basal conditions (Figure 4H and 4I). Contrariwise, rTNF-α stimulation provoked the reduction in MERTK mRNA levels in all treatment groups at both 4 h and 24 h post-stimulation (Figures 4H and 4I). Therefore, this phagocytic receptor may be implicated in the augmented synaptoneurosomal engulfment observed when mGluR5 receptor activity is antagonized only in the absence of inflammatory stimulation.
3.4. rTNF-α Stimulation and mGluR5 Blockade Decrease the Expression of Synaptogenic Molecules in Human Astrocytes
It has been shown that mGluR5 can induce the expression of synaptogenic factors by astrocytes and promote synapse remodeling [40,66,67]. To investigate whether mGluR5 could modulate the expression of synaptogenic molecules in an inflammatory context, hiPSC-derived astrocytes were subjected to rTNF-α stimulation for 4 h and 24 h and gene expression of the synaptogenic factors BDNF, GPC4 and TSP1 was analyzed. rTNF-α-stimulated astrocytes displayed reduced BDNF and GPC4 mRNA levels at both timepoints analyzed (Figures 5A-5D). TSP1 expression, on the other hand, was not affected by rTNF-α stimulation (Figures 5E-5F). Furthermore, neither CTEP nor DHPG treatment had any impact in BDNF and GPC4 expression, when human astrocytes were subjected to rTNF-α stimulation (Figures 5A-5D). Notably, CTEP treatment led to a reduction in BDNF expression only under basal conditions and at the 4 h timepoint, with no differences being observed at a later timepoint (Figures 5A-5B). In addition, CTEP drove a reduction in TSP1 gene expression, but only at the 24 h timepoint (Figure 5F). Interestingly, this effect was observed regardless of rTNF-α-stimulation, indicating that mGluR5 antagonism alone is responsible for decreasing TSP1 expression (Figures 5E-5F). DHPG treatment had no effect in the gene expression of these three synaptogenic factors. Altogether, these data indicate that both pro-inflammatory stimulation and mGluR5 negative allosteric modulation can dampen the production of synaptogenic molecules, which may contribute to the synaptic deficits often seen in neurodegenerative disorders [68,69].
4. Discussion
It is well known that mGluR5 is involved in neuroinflammation and neurodegeneration and has hence been pointed as a potential pharmacological target for neuroprotection in a variety of neurodegenerative diseases [58]. However, the role of mGluR5 in neurodegenerative processes is controversial, as the literature provides conflicting information concerning whether activation of neuronal mGluR5 is either neuroprotective or neurotoxic and whether mGluR5 stimulation in astrocytes would elicit protective or toxic effects on neighbouring cells. While some studies have reported positive effects of astrocytic mGluR5 activation following injury through the actions of growth factors and synaptogenic molecules [39,40], others have shown that activation of this receptor may elicit harmful effects through the production of cytokines and inflammatory mediators [41,42]. It is possible that the utility of agonists or antagonists of the receptor may vary based on the underlying disease. For instance, it has been shown that stimulation of mGluR5 in astrocytes leads to BDNF release, which supports myelin protein synthesis in the cuprizone-induced demyelination mouse model [40]. On the other hand, in the case of AD, it was demonstrated that Aβ induces an increase in intracellular Ca2+ levels, which can be explained, at least in part, by an increase in mGluR5 expression in astrocytes [36,70-74]. Thus, in this case, mGluR5 antagonism is efficient to reverse Ca2+ rise, helping to avoid Aβ-induced astrocytic Ca2+ signalling perturbation [70,71,73]. Moreover, mGluR5 blockade in cultured astrocytes derived from hSOD1G93A, a transgenic mouse model of amyotrophic lateral sclerosis, was shown to prevent cell death [42]. The results presented here may shed some light on this dichotomy, as we show that mGluR5 blockade by CTEP decreased the expression of inflammatory factors at the same time that it reduced the expression of trophic and synaptogenic molecules, such as BDNF and TSP1. It has been demonstrated that astrocytic mGluR5 antagonism prevents the secretion of IL-8 and IL-6 in an astrocytic cell line [41]. The data shown here corroborate these findings, as CTEP was effective to lessen the levels of secreted TNF-α, IL-6 and IL-8 induced by short stimulation (4 h) of astrocytes with rTNF-α. However, it is odd that CTEP decreased the levels of these inflammatory cytokines while it augmented the gene expression of SERPINA3, which is a marker of pan-reactive astrocytes. In addition, CTEP treatment did not modify the expression of A1 and A2 reactive astrocytic markers. Further studies will be needed to dissect the mGluR5-stimulated cell signalling pathways associated with these events, which will help to clarify whether mGluR5 pharmacological manipulation in astrocytes would be beneficial in specific disease contexts.
Astrocyte in vitro stimulation with rTNF-α to generate a reactive phenotype is well described in the scientific literature [50]. Herein, astrocytes were stimulated with rTNF-α for 4h or 24h. However, TNF-α was also used as a functional readout since secretion of this cytokine by the reactive astrocytes was quantified in cell culture supernatants to evaluate the proinflammatory status of these cells, along with the secretion of IL-6 and IL-8. One possible pitfall of this analysis is that remaining rTNF-α could interfere with the detection of secreted TNF-α. Nonetheless, the half-life of TNF-α is very short, less than 20 minutes [75,76]. Thus, by the time supernatants are collected, 4h after stimulation, rTNF-α is probably fully degraded. Moreover, TNF-α mRNA transcripts were also evaluated and the results show an increase in TNF-α gene expression following rTNF-α stimulation, corroborating the protein quantification results.
Astrocytes aid neuronal survival and induce synapse remodelling by secreting trophic and synaptogenic factors, including BDNF [21], GPC4 [22], hevin [23], and TSP [24]. The results presented here show that rTNF-α stimulation decreased the levels of BDNF and GPC4, without modifying TSP1 mRNA levels. A previous study has shown that murine A1 astrocytes, induced by IL-1α (3 ng/mL), TNF-α (30 ng/mL), and C1q (400 ng/mL) for 24 h, exhibit increased levels of GPC4 and TSP1/2 [62]. These contrasting results could be explained by the difference in inflammatory stimuli employed, as we stimulated astrocytes with only one inflammatory factor: TNF-α (10 ng/mL). However, it is also possible that murine and human iPSC-derived astrocytes respond differently to inflammatory insult, highlighting the importance of employing cell models relevant to humans. Interestingly, although CTEP was effective to decrease TNF-α levels, mGluR5 blockade did not result in an increase in the expression of the aforementioned synaptogenic factors. In fact, CTEP decreased BDNF and TSP-1 expression levels, which could contribute to further attenuation of synaptogenesis. These results were anticipated, as it has been shown that astrocytic mGluR5 is necessary for BDNF and TSP1 secretion [40,66,67]. In the case of BDNF, it has been shown that mGluR5 activation enhances the expression of this trophic factor by increasing the phosphorylation of CREB [77-80]. Regarding glypican-4, it has been demonstrated that astrocytic mGluR5 can trigger its expression [67], although these findings were not corroborated by the data shown here. In addition, previous data from our laboratory showed that mGluR5 genetic ablation leads to reduced dendritic spine numbers in a Huntington’s Disease mouse model [69], while this receptor positive allosteric modulation via VU0409551 treatment is capable of rescuing this phenotype [79], indicating that mGluR5 stimulation is synaptogenic. Thus, both rTNF-α and mGluR5 blockade may impair synaptogenesis and future studies will be important to determine whether either astrocyte conditioned media or co-culture of these astrocytes with neurons would lead to decreased number of synapses.
Phagocytosis is important to eliminate dead cells in both a healthy and diseased central nervous system (CNS). Synapses and myelin are also eliminated by phagocytosis to maintain or refine neural networks during development and adulthood [16,17]. However, aberrant synapse pruning by microglia is suggested to cause undesired synapse loss in AD [18]. Although microglia play a major role in phagocytosis, astrocytes also prune synapses by phagocytosis in the developing brain and take up extracellular protein aggregates, such as Aβ [19,20]. In fact, recent findings indicate that hippocampal synapses are preferentially phagocytosed by astrocytes [81]. In a mouse model of AD (APP/PS1 mice), dysfunctional synapses are engulfed by Aβ-associated astrocytes, but not microglia [82]. However, this beneficial effect is limited, as Aβ impairs the phagocytosis of dystrophic synapses by astrocytes and decrease the expression of the phagocytic receptors MERTK and MEGF10 [83,84]. Even though healthy synapses should be preserved, accumulation of faulty synapses could result in an unhealthy synaptic environment, causing alterations in circuit connectivity and memory loss. The results shown here demonstrate that rTNF-α decreased MERTK expression and enhanced phagocytosis by astrocytes derived from hiPSCs. In contrast, it has been shown that murine A1 astrocytes, induced by IL-1α, TNF-α, and C1q, exhibit suppressed phagocytic activity for synapses and myelin, concomitantly with downregulation of the phagocytic receptors MERTK and MEGF10 [62]. These data suggest that the combination of different inflammatory factors might produce contrasting results on the phagocytic activity of astrocytes, adding to the debate of whether this simplified dichotomic classification of astrocytes into A1 and A2 is enough to fully describe the myriad of phenotypes astrocytes can display [47]. We also hypothesize that other factors and even other phagocytic receptors could play a role in rTNF-α-induced phagocytosis by astrocytes derived from human cells.
Not many studies have addressed the role of mGluR5 on astrocytic phagocytosis. A recent study shows that a silent allosteric modulator (SAM) of mGluR5 prevents synaptic localization of the complement component C1q and synaptic engulfment by astrocytes in an AD mouse model [68]. Here we show that CTEP treatment enhanced phagocytosis in the presence and in the absence of rTNF-α. However, although CTEP increased the expression of MERTK in the absence of rTNF-α, in the presence of this inflammatory factor, MERTK expression remained reduced. This increase in MERTK expression by CTEP is in line with the enhanced phagocytosis observed in astrocytes treated with CTEP. Regarding mGluR5 activation, it is intriguing that DHPG had a similar effect as CTEP on astrocytic phagocytosis. For that matter, DHPG, as CTEP, also augmented the gene expression of SERPINA3 and lessened the secreted protein levels of TNF-α. It is relevant to mention that DHPG activates both mGluR5 and mGluR1, whereas CTEP is an inverse agonist that blocks mGluR5 specifically [85]. Although the expression of mGluR5 in astrocytes declines after birth and mGluR1 is not normally expressed by astrocytes, both mGluR5 and mGluR1 were shown to be upregulated in response to injury [34-36,86,87]. Thus, it is possible that the increase in phagocytosis and the changes in SERPINA3 and TNF-α levels induced by DHPG are due to mGluR1 activation and that these two group I mGluRs have opposite roles in astrocytes. Although more experiments will be needed to investigate this hypothesis, one important conclusion from these set of experiments is that CTEP has a prominent effect to induce phagocytosis, regardless of the presence of rTNF-α. Considering that phagocytosis by microglia [88] and astrocytes [84] decline in certain diseases, including AD, CTEP could be an option to compensate for impaired phagocytic clearance of Aβ and dystrophic synapses in AD and in other brain disorders caused by protein aggregates.
In conclusion, the data shown here indicate that mGluR5 blockade by CTEP attenuates the rTNF-α-induced secretion of inflammatory factors, including TNF-α, IL-6 and IL-8. At the same time, CTEP treatment did not modify either A1 or A2 astrocytic markers, while rTNF-α led to an increase in the A1 markers, C3 and VCAM-1. SERPINA3 expression and astrocyte phagocytosis were enhanced by both CTEP and rTNF-α, whereas the expression of the synaptogenic factors was decreased. Thus, CTEP treatment could be an option when augmented phagocytosis is desired, although it might lead to increased synaptic pruning and diminished synaptogenesis. These data illustrate well the complexity of mGluR5 pharmacology and show that the simplified classification of reactive astrocytes into A1 and A2 falls short of capturing their phenotypic diversity.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Figure S1: CTEP decreases the mRNA levels of TNF-α in astrocytes stimulated with rTNF-α; Figure S2: Synaptoneurosomes isolated from mouse brain are enriched in pre- and post-synaptic markers; Figure S3: hiPSC-derived astrocytes phagocytose synaptoneurosomes; Figure S4: Original, uncropped and unadjusted western blot images shown on Fig. S2; Video S1: 3D renderization of astrocytes engulfing CM-Dil-labeled synaptoneurosomes.
Author Contributions
Conceptualization, F.M.R., I.Q.L. and P.L.C.; methodology, I.Q.L., P.L.C., J.S.F. and J.P.S.L.; validation, I.Q.L., P.L.C., J.S.F. and J.P.S.L.; formal analysis, I.Q.L., P.L.C., J.S.F., J.P.S.L. and F.M.R.; investigation, I.Q.L., P.L.C., J.S.F., J.P.S.L. and F.M.R.; resources, F.M.R.; data curation, F.M.R.; writing—original draft preparation, I.Q.L., P.L.C. and J.S.F.; writing—review and editing, F.M.R.; supervision, F.M.R.; project administration, F.M.R.; funding acquisition, F.M.R. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by CNPq, grant number 441719/2020-1 and FAPEMIG, grant numbers APQ-03921-22 and BPD-00067-22.
Institutional Review Board Statement
Animal experiments were approved by the Ethics Committee on Animal Use of the Federal University of Minas Gerais (CEUA #120/2017).
Data Availability Statement
Data is contained within the article or supplementary material. Raw data are available from the corresponding authors upon reasonable request.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- Lee, H.G.; Wheeler, M.A.; Quintana, F.J. Function and therapeutic value of astrocytes in neurological diseases. Nat Rev Drug Discov 2022, 21, 339–358. [Google Scholar] [CrossRef]
- Olabarria, M.; Noristani, H.N.; Verkhratsky, A.; Rodriguez, J.J. Concomitant astroglial atrophy and astrogliosis in a triple transgenic animal model of Alzheimer's disease. Glia, 2010, 58, 831–838. [Google Scholar] [CrossRef]
- Huang, S.; Tong, H.; Lei, M. , et al. Astrocytic glutamatergic transporters are involved in Abeta-induced synaptic dysfunction. Brain Res. 2018, 1678, 129–137. [Google Scholar] [CrossRef]
- de Lima, I.B.Q.; Ribeiro, F.M. The Implication of Glial Metabotropic Glutamate Receptors in Alzheimer's Disease. Curr. Neuropharmacol. 2023, 21, 164–182. [Google Scholar] [CrossRef]
- Matos, M.; Augusto, E.; Machado, N.J. , et al. Astrocytic adenosine A2A receptors control the amyloid-beta peptide-induced decrease of glutamate uptake. J Alzheimers Dis. 2012, 31, 555–567. [Google Scholar] [CrossRef] [PubMed]
- Reichenbach, N.; Delekate, A.; Plescher, M., et al. Inhibition of Stat3-mediated astrogliosis ameliorates pathology in an Alzheimer's disease model. EMBO Mol Med. 2019, 11. [CrossRef]
- Habib, N.; McCabe, C.; Medina, S.; Varshavsky, M.; Kitsberg, D.; Dvir-Szternfeld, R.; Green, G.; Dionne, D.; Nguyen, L.; Marshall, J.L.; et al. Disease-associated astrocytes in Alzheimer’s disease and aging. Nat. Neurosci. 2020, 23, 701–706. [Google Scholar] [CrossRef]
- Bradford, J.; Shin, J.Y.; Roberts, M.; Wang, C.E.; Li, X.J.; Li, S. Expression of mutant huntingtin in mouse brain astrocytes causes age-dependent neurological symptoms. Proceedings of the National Academy of Sciences of the United States of America 2009, 106, 22480–22485. [Google Scholar] [CrossRef] [PubMed]
- Wood, T.E.; Barry, J.; Yang, Z.; Cepeda, C.; Levine, M.S.; Gray, M. Mutant huntingtin reduction in astrocytes slows disease progression in the BACHD conditional Huntington's disease mouse model. Human molecular genetics 2019, 28, 487–500. [Google Scholar] [CrossRef]
- Nagai, M.; Re, D.B.; Nagata, T.; Chalazonitis, A.; Jessell, T.M.; Wichterle, H.; Przedborski, S. Astrocytes expressing ALS-linked mutated SOD1 release factors selectively toxic to motor neurons. Nature neuroscience 2007, 10, 615–622. [Google Scholar] [CrossRef]
- Yamanaka, K.; Komine, O. The multi-dimensional roles of astrocytes in ALS. Neurosci Res 2018, 126, 31–38. [Google Scholar] [CrossRef]
- Izrael, M.; Slutsky, S.G.; Revel, M. Rising Stars: Astrocytes as a Therapeutic Target for ALS Disease. Frontiers in neuroscience 2020, 14, 824. [Google Scholar] [CrossRef]
- Rothhammer, V.; Mascanfroni, I.D.; Bunse, L.; Takenaka, M.C.; Kenison, J.E.; Mayo, L.; Chao, C.C.; Patel, B.; Yan, R.; Blain, M.; et al. Type I interferons and microbial metabolites of tryptophan modulate astrocyte activity and central nervous system inflammation via the aryl hydrocarbon receptor. Nat Med 2016, 22, 586–597. [Google Scholar] [CrossRef] [PubMed]
- Rothhammer, V.; Borucki, D.M.; Tjon, E.C.; Takenaka, M.C.; Chao, C.C.; Ardura-Fabregat, A.; de Lima, K.A.; Gutierrez-Vazquez, C.; Hewson, P.; Staszewski, O.; et al. Microglial control of astrocytes in response to microbial metabolites. Nature 2018, 557, 724–728. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, M.A.; Clark, I.C.; Tjon, E.C.; Li, Z.; Zandee, S.E.J.; Couturier, C.P.; Watson, B.R.; Scalisi, G.; Alkwai, S.; Rothhammer, V.; et al. MAFG-driven astrocytes promote CNS inflammation. Nature 2020, 578, 593–599. [Google Scholar] [CrossRef] [PubMed]
- Gu, X.L.; Long, C.X.; Sun, L.; Xie, C.; Lin, X.; Cai, H. Astrocytic expression of Parkinson's disease-related A53T alpha-synuclein causes neurodegeneration in mice. Molecular brain 2010, 3, 12. [Google Scholar] [CrossRef] [PubMed]
- Yun, S.P.; Kam, T.I.; Panicker, N.; Kim, S.; Oh, Y.; Park, J.S.; Kwon, S.H.; Park, Y.J.; Karuppagounder, S.S.; Park, H.; et al. Block of A1 astrocyte conversion by microglia is neuroprotective in models of Parkinson's disease. Nat Med 2018, 24, 931–938. [Google Scholar] [CrossRef]
- Verkhratsky, A.; Nedergaard, M. Physiology of Astroglia. Physiological reviews 2018, 98, 239–389. [Google Scholar] [CrossRef]
- Verkhratsky, A.; Li, B.; Scuderi, C.; Parpura, V. Principles of Astrogliopathology. Adv Neurobiol 2021, 26, 55–73. [Google Scholar] [CrossRef]
- Moulson, A.J.; Squair, J.W.; Franklin, R.J.M.; Tetzlaff, W.; Assinck, P. Diversity of Reactive Astrogliosis in CNS Pathology: Heterogeneity or Plasticity? Frontiers in cellular neuroscience 2021, 15, 703810. [Google Scholar] [CrossRef]
- Paolicelli, R.C.; Bolasco, G.; Pagani, F.; Maggi, L.; Scianni, M.; Panzanelli, P.; Giustetto, M.; Ferreira, T.A.; Guiducci, E.; Dumas, L.; et al. Synaptic pruning by microglia is necessary for normal brain development. Science 2011, 333, 1456–1458. [Google Scholar] [CrossRef] [PubMed]
- Trachtenberg, J.T.; Chen, B.E.; Knott, G.W.; Feng, G.; Sanes, J.R.; Welker, E.; Svoboda, K. Long-term in vivo imaging of experience-dependent synaptic plasticity in adult cortex. Nature 2002, 420, 788–794. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.; Beja-Glasser, V.F.; Nfonoyim, B.M.; Frouin, A.; Li, S.; Ramakrishnan, S.; Merry, K.M.; Shi, Q.; Rosenthal, A.; Barres, B.A.; et al. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 2016, 352, 712–716. [Google Scholar] [CrossRef] [PubMed]
- Chung, W.S.; Clarke, L.E.; Wang, G.X.; Stafford, B.K.; Sher, A.; Chakraborty, C.; Joung, J.; Foo, L.C.; Thompson, A.; Chen, C.; et al. Astrocytes mediate synapse elimination through MEGF10 and MERTK pathways. Nature 2013, 504, 394–400. [Google Scholar] [CrossRef]
- Koistinaho, M.; Lin, S.; Wu, X.; Esterman, M.; Koger, D.; Hanson, J.; Higgs, R.; Liu, F.; Malkani, S.; Bales, K.R.; et al. Apolipoprotein E promotes astrocyte colocalization and degradation of deposited amyloid-beta peptides. Nat Med 2004, 10, 719–726. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Casati, M.E.; Murtie, J.C.; Rio, C.; Stankovic, K.; Liberman, M.C.; Corfas, G. Nonneuronal cells regulate synapse formation in the vestibular sensory epithelium via erbB-dependent BDNF expression. Proc Natl Acad Sci U S A 2010, 107, 17005–17010. [Google Scholar] [CrossRef]
- Allen, N.J.; Bennett, M.L.; Foo, L.C.; Wang, G.X.; Chakraborty, C.; Smith, S.J.; Barres, B.A. Astrocyte glypicans 4 and 6 promote formation of excitatory synapses via GluA1 AMPA receptors. Nature 2012, 486, 410–414. [Google Scholar] [CrossRef]
- Kucukdereli, H.; Allen, N.J.; Lee, A.T.; Feng, A.; Ozlu, M.I.; Conatser, L.M.; Chakraborty, C.; Workman, G.; Weaver, M.; Sage, E.H.; et al. Control of excitatory CNS synaptogenesis by astrocyte-secreted proteins Hevin and SPARC. Proc Natl Acad Sci U S A 2011, 108, E440–E449. [Google Scholar] [CrossRef]
- Christopherson, K.S.; Ullian, E.M.; Stokes, C.C.; Mullowney, C.E.; Hell, J.W.; Agah, A.; Lawler, J.; Mosher, D.F.; Bornstein, P.; Barres, B.A. Thrombospondins are astrocyte-secreted proteins that promote CNS synaptogenesis. Cell 2005, 120, 421–433. [Google Scholar] [CrossRef]
- Ribeiro, F.M.; Vieira, L.B.; Pires, R.G.; Olmo, R.P.; Ferguson, S.S. Metabotropic glutamate receptors and neurodegenerative diseases. Pharmacological research 2017, 115, 179–191. [Google Scholar] [CrossRef]
- Biber, K.; Laurie, D.J.; Berthele, A.; Sommer, B.; Tolle, T.R.; Gebicke-Harter, P.J.; van Calker, D.; Boddeke, H.W. Expression and signaling of group I metabotropic glutamate receptors in astrocytes and microglia. Journal of neurochemistry 1999, 72, 1671–1680. [Google Scholar] [PubMed]
- Miller, S.; Romano, C.; Cotman, C.W. Growth factor upregulation of a phosphoinositide-coupled metabotropic glutamate receptor in cortical astrocytes. The Journal of neuroscience : the official journal of the Society for Neuroscience 1995, 15, 6103–6109. [Google Scholar] [CrossRef]
- Pasti, L.; Volterra, A.; Pozzan, T.; Carmignoto, G. Intracellular calcium oscillations in astrocytes: a highly plastic, bidirectional form of communication between neurons and astrocytes in situ. The Journal of neuroscience : the official journal of the Society for Neuroscience 1997, 17, 7817–7830. [Google Scholar] [CrossRef]
- Paquet, M.; Ribeiro, F.M.; Guadagno, J.; Esseltine, J.L.; Ferguson, S.S.; Cregan, S.P. Role of metabotropic glutamate receptor 5 signaling and homer in oxygen glucose deprivation-mediated astrocyte apoptosis. Molecular brain 2013, 6, 9. [Google Scholar] [CrossRef] [PubMed]
- Servitja, J.M.; Masgrau, R.; Sarri, E.; Picatoste, F. Group I metabotropic glutamate receptors mediate phospholipase D stimulation in rat cultured astrocytes. Journal of neurochemistry 1999, 72, 1441–1447. [Google Scholar] [CrossRef] [PubMed]
- Peavy, R.D.; Conn, P.J. Phosphorylation of mitogen-activated protein kinase in cultured rat cortical glia by stimulation of metabotropic glutamate receptors. Journal of neurochemistry 1998, 71, 603–612. [Google Scholar] [CrossRef]
- Cai, Z.; Schools, G.P.; Kimelberg, H.K. Metabotropic glutamate receptors in acutely isolated hippocampal astrocytes: developmental changes of mGluR5 mRNA and functional expression. Glia 2000, 29, 70–80. [Google Scholar] [CrossRef]
- Sun, W.; McConnell, E.; Pare, J.F.; Xu, Q.; Chen, M.; Peng, W.; Lovatt, D.; Han, X.; Smith, Y.; Nedergaard, M. Glutamate-dependent neuroglial calcium signaling differs between young and adult brain. Science 2013, 339, 197–200. [Google Scholar] [CrossRef]
- Aronica, E.; Catania, M.V.; Geurts, J.; Yankaya, B.; Troost, D. Immunohistochemical localization of group I and II metabotropic glutamate receptors in control and amyotrophic lateral sclerosis human spinal cord: upregulation in reactive astrocytes. Neuroscience 2001, 105, 509–520. [Google Scholar] [CrossRef]
- Geurts, J.J.; Wolswijk, G.; Bo, L.; van der Valk, P.; Polman, C.H.; Troost, D.; Aronica, E. Altered expression patterns of group I and II metabotropic glutamate receptors in multiple sclerosis. Brain 2003, 126, 1755–1766. [Google Scholar] [CrossRef]
- Shrivastava, A.N.; Kowalewski, J.M.; Renner, M.; Bousset, L.; Koulakoff, A.; Melki, R.; Giaume, C.; Triller, A. beta-amyloid and ATP-induced diffusional trapping of astrocyte and neuronal metabotropic glutamate type-5 receptors. Glia 2013, 61, 1673–1686. [Google Scholar] [CrossRef] [PubMed]
- Byrnes, K.R.; Stoica, B.; Loane, D.J.; Riccio, A.; Davis, M.I.; Faden, A.I. Metabotropic glutamate receptor 5 activation inhibits microglial associated inflammation and neurotoxicity. Glia 2009, 57, 550–560. [Google Scholar] [CrossRef]
- Loane, D.J.; Stoica, B.A.; Pajoohesh-Ganji, A.; Byrnes, K.R.; Faden, A.I. Activation of metabotropic glutamate receptor 5 modulates microglial reactivity and neurotoxicity by inhibiting NADPH oxidase. The Journal of biological chemistry 2009, 284, 15629–15639. [Google Scholar] [CrossRef] [PubMed]
- Fulmer, C.G.; VonDran, M.W.; Stillman, A.A.; Huang, Y.; Hempstead, B.L.; Dreyfus, C.F. Astrocyte-derived BDNF supports myelin protein synthesis after cuprizone-induced demyelination. The Journal of neuroscience : the official journal of the Society for Neuroscience 2014, 34, 8186–8196. [Google Scholar] [CrossRef]
- Saitta, K.S.; Lercher, L.D.; Sainato, D.M.; Patel, A.; Huang, Y.; McAuliffe, G.; Dreyfus, C.F. CHPG enhances BDNF and myelination in cuprizone-treated mice through astrocytic metabotropic glutamate receptor 5. Glia 2021, 69, 1950–1965. [Google Scholar] [CrossRef] [PubMed]
- Shah, A.; Silverstein, P.S.; Singh, D.P.; Kumar, A. Involvement of metabotropic glutamate receptor 5, AKT/PI3K signaling and NF-kappaB pathway in methamphetamine-mediated increase in IL-6 and IL-8 expression in astrocytes. J Neuroinflammation 2012, 9, 52. [Google Scholar] [CrossRef]
- Rossi, D.; Brambilla, L.; Valori, C.F.; Roncoroni, C.; Crugnola, A.; Yokota, T.; Bredesen, D.E.; Volterra, A. Focal degeneration of astrocytes in amyotrophic lateral sclerosis. Cell Death Differ 2008, 15, 1691–1700. [Google Scholar] [CrossRef]
- Kim, H.; Woo, J.H.; Lee, J.H.; Joe, E.H.; Jou, I. 22(R)-hydroxycholesterol induces HuR-dependent MAP kinase phosphatase-1 expression via mGluR5-mediated Ca(2+)/PKCalpha signaling. Biochim Biophys Acta 2016, 1859, 1056–1070. [Google Scholar] [CrossRef]
- Aronica, E.; Gorter, J.A.; Rozemuller, A.J.; Yankaya, B.; Troost, D. Activation of metabotropic glutamate receptor 3 enhances interleukin (IL)-1beta-stimulated release of IL-6 in cultured human astrocytes. Neuroscience 2005, 130, 927–933. [Google Scholar] [CrossRef]
- Degl'Innocenti, E.; Dell'Anno, M.T. Human and mouse cortical astrocytes: a comparative view from development to morphological and functional characterization. Front Neuroanat 2023, 17, 1130729. [Google Scholar] [CrossRef]
- Li, J.; Pan, L.; Pembroke, W.G.; Rexach, J.E.; Godoy, M.I.; Condro, M.C.; Alvarado, A.G.; Harteni, M.; Chen, Y.W.; Stiles, L.; et al. Conservation and divergence of vulnerability and responses to stressors between human and mouse astrocytes. Nat Commun 2021, 12, 3958. [Google Scholar] [CrossRef] [PubMed]
- Escartin, C.; Galea, E.; Lakatos, A.; O'Callaghan, J.P.; Petzold, G.C.; Serrano-Pozo, A.; Steinhauser, C.; Volterra, A.; Carmignoto, G.; Agarwal, A.; et al. Reactive astrocyte nomenclature, definitions, and future directions. Nat Neurosci 2021, 24, 312–325. [Google Scholar] [CrossRef] [PubMed]
- Cardozo, P.L.; de Lima, I.B.Q.; Maciel, E.M.A.; Silva, N.C.; Dobransky, T.; Ribeiro, F.M. Synaptic Elimination in Neurological Disorders. Curr Neuropharmacol 2019, 17, 1071–1095. [Google Scholar] [CrossRef]
- Kwart, D.; Gregg, A.; Scheckel, C.; Murphy, E.A.; Paquet, D.; Duffield, M.; Fak, J.; Olsen, O.; Darnell, R.B.; Tessier-Lavigne, M. A Large Panel of Isogenic APP and PSEN1 Mutant Human iPSC Neurons Reveals Shared Endosomal Abnormalities Mediated by APP beta-CTFs, Not Abeta. Neuron 2019, 104, 256–270. [Google Scholar] [CrossRef] [PubMed]
- Trindade, P.; Loiola, E.C.; Gasparotto, J.; Ribeiro, C.T.; Cardozo, P.L.; Devalle, S.; Salerno, J.A.; Ornelas, I.M.; Ledur, P.F.; Ribeiro, F.M.; et al. Short and long TNF-alpha exposure recapitulates canonical astrogliosis events in human-induced pluripotent stem cells-derived astrocytes. Glia 2020, 68, 1396–1409. [Google Scholar] [CrossRef] [PubMed]
- Molla Kazemiha, V.; Shokrgozar, M.A.; Arabestani, M.R.; Shojaei Moghadam, M.; Azari, S.; Maleki, S.; Amanzadeh, A.; Jeddi Tehrani, M.; Shokri, F. PCR-based detection and eradication of mycoplasmal infections from various mammalian cell lines: a local experience. Cytotechnology 2009, 61, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Untergasser, A.; Nijveen, H.; Rao, X.; Bisseling, T.; Geurts, R.; Leunissen, J.A. Primer3Plus, an enhanced web interface to Primer3. Nucleic Acids Res 2007, 35, 71–74. [Google Scholar] [CrossRef]
- Villasana, L.E.; Klann, E.; Tejada-Simon, M.V. Rapid isolation of synaptoneurosomes and postsynaptic densities from adult mouse hippocampus. J Neurosci Methods 2006, 158, 30–36. [Google Scholar] [CrossRef]
- Berger, J.V.; Dumont, A.O.; Focant, M.C.; Vergouts, M.; Sternotte, A.; Calas, A.G.; Goursaud, S.; Hermans, E. Opposite regulation of metabotropic glutamate receptor 3 and metabotropic glutamate receptor 5 by inflammatory stimuli in cultured microglia and astrocytes. Neuroscience 2012, 205, 29–38. [Google Scholar] [CrossRef]
- Budgett, R.F.; Bakker, G.; Sergeev, E.; Bennett, K.A.; Bradley, S.J. Targeting the Type 5 Metabotropic Glutamate Receptor: A Potential Therapeutic Strategy for Neurodegenerative Diseases? Front Pharmacol 2022, 13, 893422. [Google Scholar] [CrossRef]
- Kumar, A.; Dhull, D.K.; Mishra, P.S. Therapeutic potential of mGluR5 targeting in Alzheimer's disease. Front Neurosci 2015, 9, 215. [Google Scholar] [CrossRef] [PubMed]
- Planas-Fontanez, T.M.; Dreyfus, C.F.; Saitta, K.S. Reactive Astrocytes as Therapeutic Targets for Brain Degenerative Diseases: Roles Played by Metabotropic Glutamate Receptors. Neurochem Res 2020, 45, 541–550. [Google Scholar] [CrossRef]
- Spampinato, S.F.; Copani, A.; Nicoletti, F.; Sortino, M.A.; Caraci, F. Metabotropic Glutamate Receptors in Glial Cells: A New Potential Target for Neuroprotection? Front Mol Neurosci 2018, 11, 414. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.N.; Fan, J.K.; Gu, L.; Yang, H.M.; Zhan, S.Q.; Zhang, H. Metabotropic glutamate receptor 5 inhibits alpha-synuclein-induced microglia inflammation to protect from neurotoxicity in Parkinson's disease. J Neuroinflammation 2021, 18, 23. [Google Scholar] [CrossRef] [PubMed]
- Zamanian, J.L.; Xu, L.; Foo, L.C.; Nouri, N.; Zhou, L.; Giffard, R.G.; Barres, B.A. Genomic analysis of reactive astrogliosis. J Neurosci 2012, 32, 6391–6410. [Google Scholar] [CrossRef]
- Fan, Y.Y.; Huo, J. A1/A2 astrocytes in central nervous system injuries and diseases: Angels or devils? Neurochem Int 2021, 148, 105080. [Google Scholar] [CrossRef]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Munch, A.E.; Chung, W.S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef]
- Heit, C.; Jackson, B.C.; McAndrews, M.; Wright, M.W.; Thompson, D.C.; Silverman, G.A.; Nebert, D.W.; Vasiliou, V. Update of the human and mouse SERPIN gene superfamily. Hum Genomics 2013, 7, 22. [Google Scholar] [CrossRef]
- Leng, K.; Rose, I.V.L.; Kim, H.; Xia, W.; Romero-Fernandez, W.; Rooney, B.; Koontz, M.; Li, E.; Ao, Y.; Wang, S.; et al. CRISPRi screens in human iPSC-derived astrocytes elucidate regulators of distinct inflammatory reactive states. Nat Neurosci 2022, 25, 1528–1542. [Google Scholar] [CrossRef]
- Konishi, H.; Koizumi, S.; Kiyama, H. Phagocytic astrocytes: Emerging from the shadows of microglia. Glia 2022, 70, 1009–1026. [Google Scholar] [CrossRef]
- Kim, S.K.; Hayashi, H.; Ishikawa, T.; Shibata, K.; Shigetomi, E.; Shinozaki, Y.; Inada, H.; Roh, S.E.; Kim, S.J.; Lee, G.; et al. Cortical astrocytes rewire somatosensory cortical circuits for peripheral neuropathic pain. J Clin Invest 2016, 126, 1983–1997. [Google Scholar] [CrossRef] [PubMed]
- Danjo, Y.; Shigetomi, E.; Hirayama, Y.J.; Kobayashi, K.; Ishikawa, T.; Fukazawa, Y.; Shibata, K.; Takanashi, K.; Parajuli, B.; Shinozaki, Y.; et al. Transient astrocytic mGluR5 expression drives synaptic plasticity and subsequent chronic pain in mice. J Exp Med 2022, 219. [Google Scholar] [CrossRef]
- Spurrier, J.; Nicholson, L.; Fang, X.T.; Stoner, A.J.; Toyonaga, T.; Holden, D.; Siegert, T.R.; Laird, W.; Allnutt, M.A.; Chiasseu, M.; et al. Reversal of synapse loss in Alzheimer mouse models by targeting mGluR5 to prevent synaptic tagging by C1Q. Sci Transl Med 2022, 14, eabi8593. [Google Scholar] [CrossRef] [PubMed]
- de Souza, J.M.; Ferreira-Vieira, T.H.; Maciel, E.M.A.; Silva, N.C.; Lima, I.B.Q.; Doria, J.G.; Olmo, I.G.; Ribeiro, F.M. mGluR5 ablation leads to age-related synaptic plasticity impairments and does not improve Huntington's disease phenotype. Sci Rep 2022, 12, 8982. [Google Scholar] [CrossRef] [PubMed]
- Ronco, V.; Grolla, A.A.; Glasnov, T.N.; Canonico, P.L.; Verkhratsky, A.; Genazzani, A.A.; Lim, D. Differential deregulation of astrocytic calcium signalling by amyloid-beta, TNFalpha, IL-1beta and LPS. Cell Calcium 2014, 55, 219–229. [Google Scholar] [CrossRef]
- Casley, C.S.; Lakics, V.; Lee, H.G.; Broad, L.M.; Day, T.A.; Cluett, T.; Smith, M.A.; O'Neill, M.J.; Kingston, A.E. Up-regulation of astrocyte metabotropic glutamate receptor 5 by amyloid-beta peptide. Brain Res 2009, 1260, 65–75. [Google Scholar] [CrossRef]
- Lim, D.; Iyer, A.; Ronco, V.; Grolla, A.A.; Canonico, P.L.; Aronica, E.; Genazzani, A.A. Amyloid beta deregulates astroglial mGluR5-mediated calcium signaling via calcineurin and Nf-kB. Glia 2013, 61, 1134–1145. [Google Scholar] [CrossRef]
- Grolla, A.A.; Fakhfouri, G.; Balzaretti, G.; Marcello, E.; Gardoni, F.; Canonico, P.L.; DiLuca, M.; Genazzani, A.A.; Lim, D. Abeta leads to Ca(2)(+) signaling alterations and transcriptional changes in glial cells. Neurobiol Aging 2013, 34, 511–522. [Google Scholar] [CrossRef]
- Grolla, A.A.; Sim, J.A.; Lim, D.; Rodriguez, J.J.; Genazzani, A.A.; Verkhratsky, A. Amyloid-beta and Alzheimer's disease type pathology differentially affects the calcium signalling toolkit in astrocytes from different brain regions. Cell Death Dis 2013, 4, e623. [Google Scholar] [CrossRef]
- Oliver, J.C.; Bland, L.A.; Oettinger, C.W.; Arduino, M.J.; McAllister, S.K.; Aguero, S.M.; Favero, M.S. Cytokine kinetics in an in vitro whole blood model following an endotoxin challenge. Lymphokine Cytokine Res 1993, 12, 115–120. [Google Scholar]
- Liu, C.; Chu, D.; Kalantar-Zadeh, K.; George, J.; Young, H.A.; Liu, G. Cytokines: From Clinical Significance to Quantification. Adv Sci (Weinh) 2021, 8, e2004433. [Google Scholar] [CrossRef] [PubMed]
- Mao, L.; Wang, J.Q. Phosphorylation of cAMP response element-binding protein in cultured striatal neurons by metabotropic glutamate receptor subtype 5. Journal of neurochemistry 2003, 84, 233–243. [Google Scholar] [CrossRef] [PubMed]
- Doria, J.G.; de Souza, J.M.; Andrade, J.N.; Rodrigues, H.A.; Guimaraes, I.M.; Carvalho, T.G.; Guatimosim, C.; Dobransky, T.; Ribeiro, F.M. The mGluR5 positive allosteric modulator, CDPPB, ameliorates pathology and phenotypic signs of a mouse model of Huntington's disease. Neurobiology of disease 2015, 73, 163–173. [Google Scholar] [CrossRef] [PubMed]
- Doria, J.G.; de Souza, J.M.; Silva, F.R.; Olmo, I.G.; Carvalho, T.G.; Alves-Silva, J.; Ferreira-Vieira, T.H.; Santos, J.T.; Xavier, C.Q.S.; Silva, N.C.; et al. The mGluR5 positive allosteric modulator VU0409551 improves synaptic plasticity and memory of a mouse model of Huntington's disease. Journal of neurochemistry 2018, 147, 222–239. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhuo, M. Group I metabotropic glutamate receptor-mediated gene transcription and implications for synaptic plasticity and diseases. Front Pharmacol 2012, 3, 189. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Kim, J.Y.; Noh, S.; Lee, H.; Lee, S.Y.; Mun, J.Y.; Park, H.; Chung, W.S. Astrocytes phagocytose adult hippocampal synapses for circuit homeostasis. Nature 2021, 590, 612–617. [Google Scholar] [CrossRef]
- Gomez-Arboledas, A.; Davila, J.C.; Sanchez-Mejias, E.; Navarro, V.; Nunez-Diaz, C.; Sanchez-Varo, R.; Sanchez-Mico, M.V.; Trujillo-Estrada, L.; Fernandez-Valenzuela, J.J.; Vizuete, M.; et al. Phagocytic clearance of presynaptic dystrophies by reactive astrocytes in Alzheimer's disease. Glia 2018, 66, 637–653. [Google Scholar] [CrossRef]
- Sanchez-Mico, M.V.; Jimenez, S.; Gomez-Arboledas, A.; Munoz-Castro, C.; Romero-Molina, C.; Navarro, V.; Sanchez-Mejias, E.; Nunez-Diaz, C.; Sanchez-Varo, R.; Galea, E.; et al. Amyloid-beta impairs the phagocytosis of dystrophic synapses by astrocytes in Alzheimer's disease. Glia 2021, 69, 997–1011. [Google Scholar] [CrossRef]
- Iram, T.; Trudler, D.; Kain, D.; Kanner, S.; Galron, R.; Vassar, R.; Barzilai, A.; Blinder, P.; Fishelson, Z.; Frenkel, D. Astrocytes from old Alzheimer's disease mice are impaired in Abeta uptake and in neuroprotection. Neurobiology of disease 2016, 96, 84–94. [Google Scholar] [CrossRef]
- Lindemann, L.; Jaeschke, G.; Michalon, A.; Vieira, E.; Honer, M.; Spooren, W.; Porter, R.; Hartung, T.; Kolczewski, S.; Buttelmann, B.; et al. CTEP: a novel, potent, long-acting, and orally bioavailable metabotropic glutamate receptor 5 inhibitor. J Pharmacol Exp Ther 2011, 339, 474–486. [Google Scholar] [CrossRef]
- Gwak, Y.S.; Hulsebosch, C.E. Upregulation of Group I metabotropic glutamate receptors in neurons and astrocytes in the dorsal horn following spinal cord injury. Exp Neurol 2005, 195, 236–243. [Google Scholar] [CrossRef] [PubMed]
- Iyer, A.M.; van Scheppingen, J.; Milenkovic, I.; Anink, J.J.; Lim, D.; Genazzani, A.A.; Adle-Biassette, H.; Kovacs, G.G.; Aronica, E. Metabotropic glutamate receptor 5 in Down's syndrome hippocampus during development: increased expression in astrocytes. Curr Alzheimer Res 2014, 11, 694–705. [Google Scholar] [CrossRef] [PubMed]
- Krabbe, G.; Halle, A.; Matyash, V.; Rinnenthal, J.L.; Eom, G.D.; Bernhardt, U.; Miller, K.R.; Prokop, S.; Kettenmann, H.; Heppner, F.L. Functional impairment of microglia coincides with Beta-amyloid deposition in mice with Alzheimer-like pathology. PLoS One 2013, 8, e60921. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Characterization of astrocytes derived from human induced pluripotent stem cells (hiPSCs). (A) Shown are representative laser scanning confocal micrographs from hiPSC-derived astrocytes immunolabeled for phalloidin (magenta), GFAP (green), and Hoechst (blue). (B) Shown are representative laser scanning confocal micrographs from hiPSC-derived astrocytes immunolabeled for phalloidin (magenta), S100β (green), and Hoechst (blue). Scale bar=200 μm.
Figure 1.
Characterization of astrocytes derived from human induced pluripotent stem cells (hiPSCs). (A) Shown are representative laser scanning confocal micrographs from hiPSC-derived astrocytes immunolabeled for phalloidin (magenta), GFAP (green), and Hoechst (blue). (B) Shown are representative laser scanning confocal micrographs from hiPSC-derived astrocytes immunolabeled for phalloidin (magenta), S100β (green), and Hoechst (blue). Scale bar=200 μm.
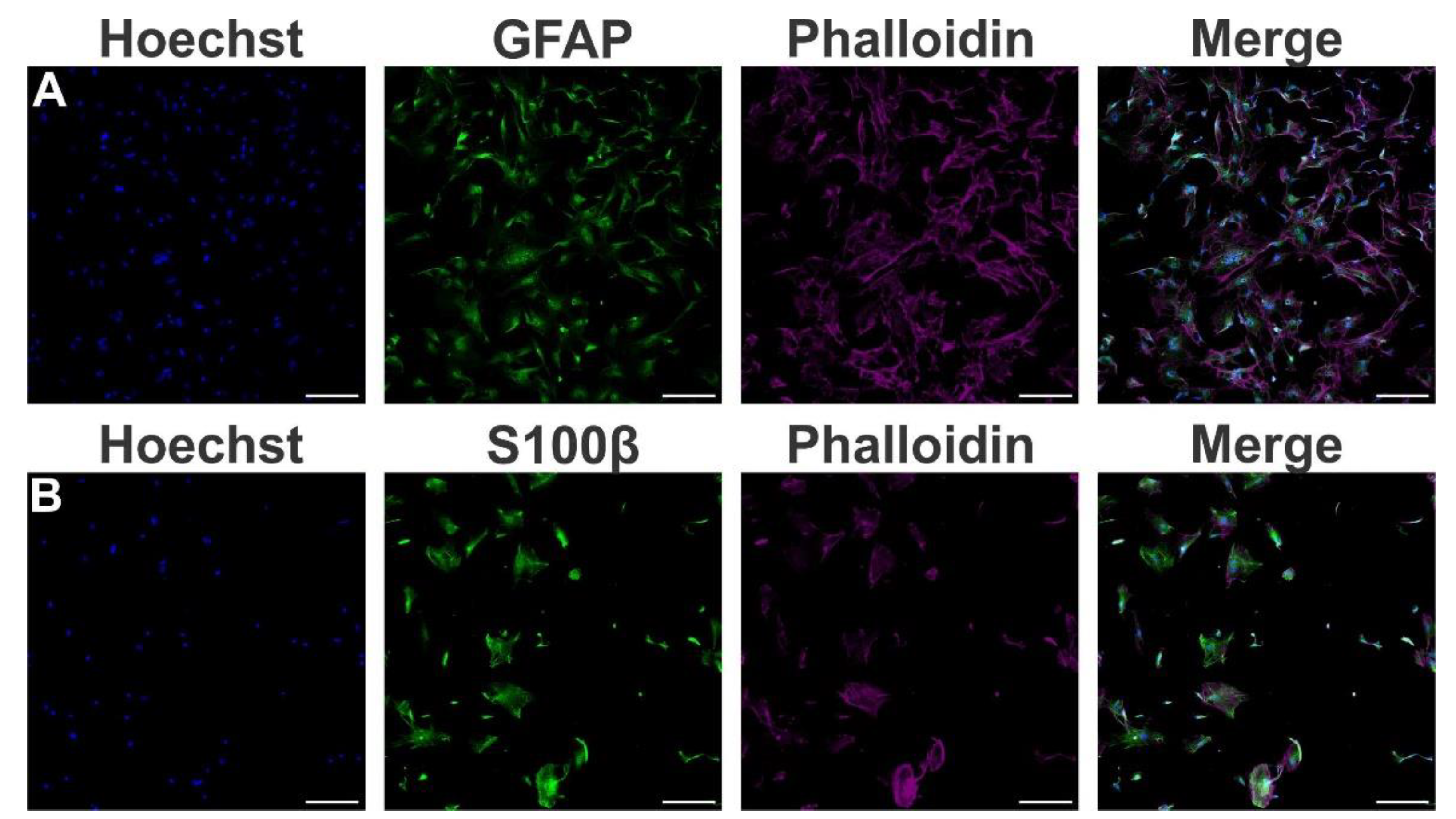
Figure 2.
CTEP treatment reduces rTNF-α-induced expression of inflammatory factors. Graphs show protein quantification of TNF-α (A), IL-6 (C), and IL-8 (E) in the supernatant of hiPSC-derived astrocytes that were either unstimulated (NS) or stimulated with rTNF-α 10 ng/mL and treated with either vehicle (NT), CTEP 10 µM or DHPG 10 µM for 4 h. Graphs show protein quantification of TNF-α (B), IL-6 (D), and IL-8 (F) in the supernatant of hiPSC-derived astrocytes that were either unstimulated (NS) or stimulated with rTNF-α 10 ng/mL and treated with either vehicle (NT), CTEP 10 µM or DHPG 10 µM for 24 h. Protein levels were assessed by CBA, which was performed in duplicates. Data represents the means ± SEM. * (p<0.05) indicates significant differences.
Figure 2.
CTEP treatment reduces rTNF-α-induced expression of inflammatory factors. Graphs show protein quantification of TNF-α (A), IL-6 (C), and IL-8 (E) in the supernatant of hiPSC-derived astrocytes that were either unstimulated (NS) or stimulated with rTNF-α 10 ng/mL and treated with either vehicle (NT), CTEP 10 µM or DHPG 10 µM for 4 h. Graphs show protein quantification of TNF-α (B), IL-6 (D), and IL-8 (F) in the supernatant of hiPSC-derived astrocytes that were either unstimulated (NS) or stimulated with rTNF-α 10 ng/mL and treated with either vehicle (NT), CTEP 10 µM or DHPG 10 µM for 24 h. Protein levels were assessed by CBA, which was performed in duplicates. Data represents the means ± SEM. * (p<0.05) indicates significant differences.
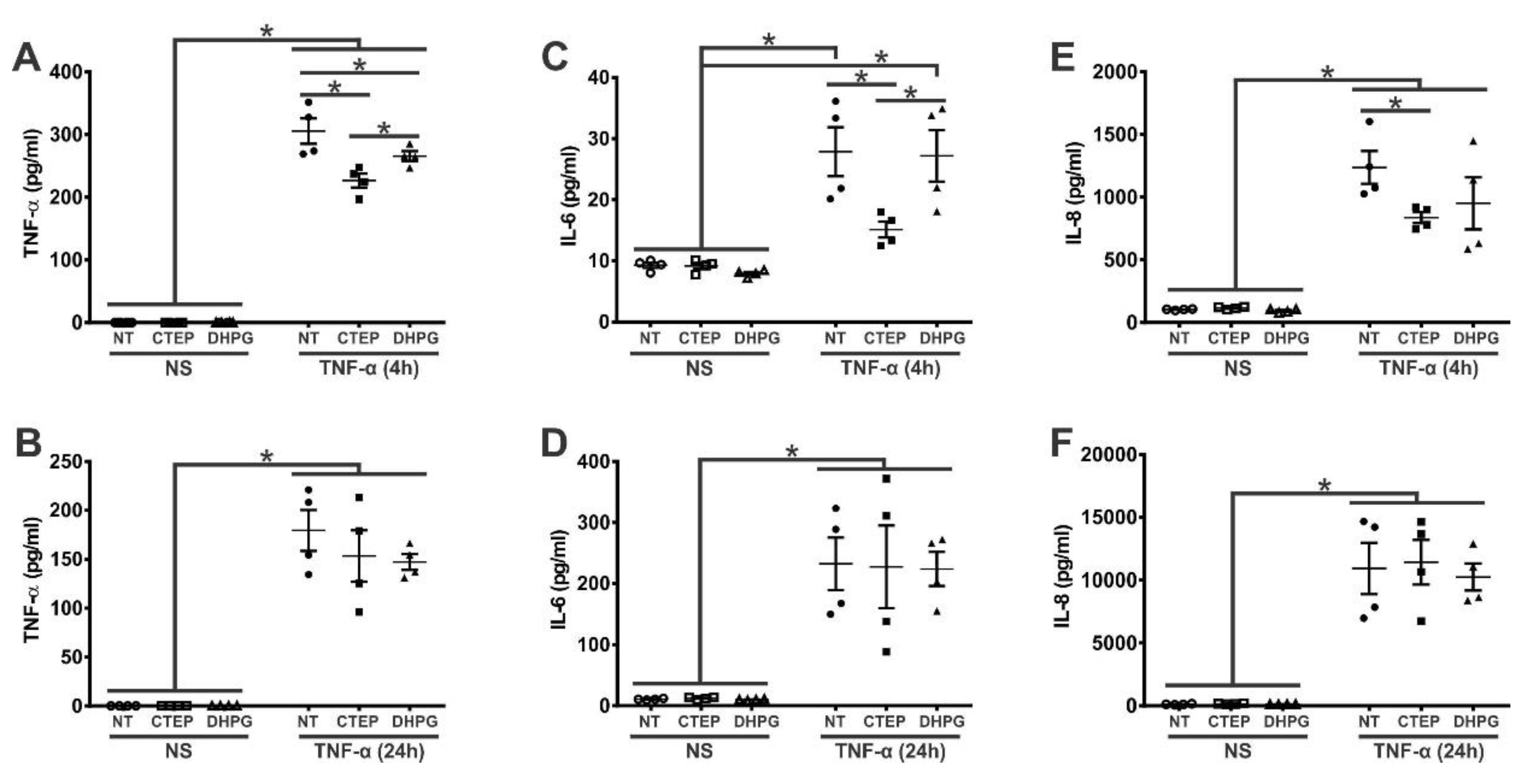
Figure 3.
mGluR5 modulates the expression of the pan-reactive astrocytic marker SERPINA3 without modifying the expression of A1 and A2 markers. Graphs show mRNA levels of SERPINA3 (A), C3 (C), VCAM-1 (E), and S100A10 (G) in hiPSC-derived astrocytes that were either unstimulated (NS) or stimulated with rTNF-α 10 ng/mL and treated with either vehicle (NT), CTEP 10 µM or DHPG 10 µM for 4 h. Graphs show mRNA levels of SERPINA3 (B), C3 (D), VCAM-1 (F), and S100A10 (H) in hiPSC-derived astrocytes that were either unstimulated (NS) or stimulated with rTNF-α 10 ng/mL and treated with either vehicle (NT), CTEP 10 µM or DHPG 10 µM for 24 h. mRNA levels were assessed by quantitative RT-PCR, which was performed in triplicates and normalized to the average of RPLP0 and IPO8 mRNA levels. Data represents the means ± SEM. * (p<0.05) indicates significant differences.
Figure 3.
mGluR5 modulates the expression of the pan-reactive astrocytic marker SERPINA3 without modifying the expression of A1 and A2 markers. Graphs show mRNA levels of SERPINA3 (A), C3 (C), VCAM-1 (E), and S100A10 (G) in hiPSC-derived astrocytes that were either unstimulated (NS) or stimulated with rTNF-α 10 ng/mL and treated with either vehicle (NT), CTEP 10 µM or DHPG 10 µM for 4 h. Graphs show mRNA levels of SERPINA3 (B), C3 (D), VCAM-1 (F), and S100A10 (H) in hiPSC-derived astrocytes that were either unstimulated (NS) or stimulated with rTNF-α 10 ng/mL and treated with either vehicle (NT), CTEP 10 µM or DHPG 10 µM for 24 h. mRNA levels were assessed by quantitative RT-PCR, which was performed in triplicates and normalized to the average of RPLP0 and IPO8 mRNA levels. Data represents the means ± SEM. * (p<0.05) indicates significant differences.
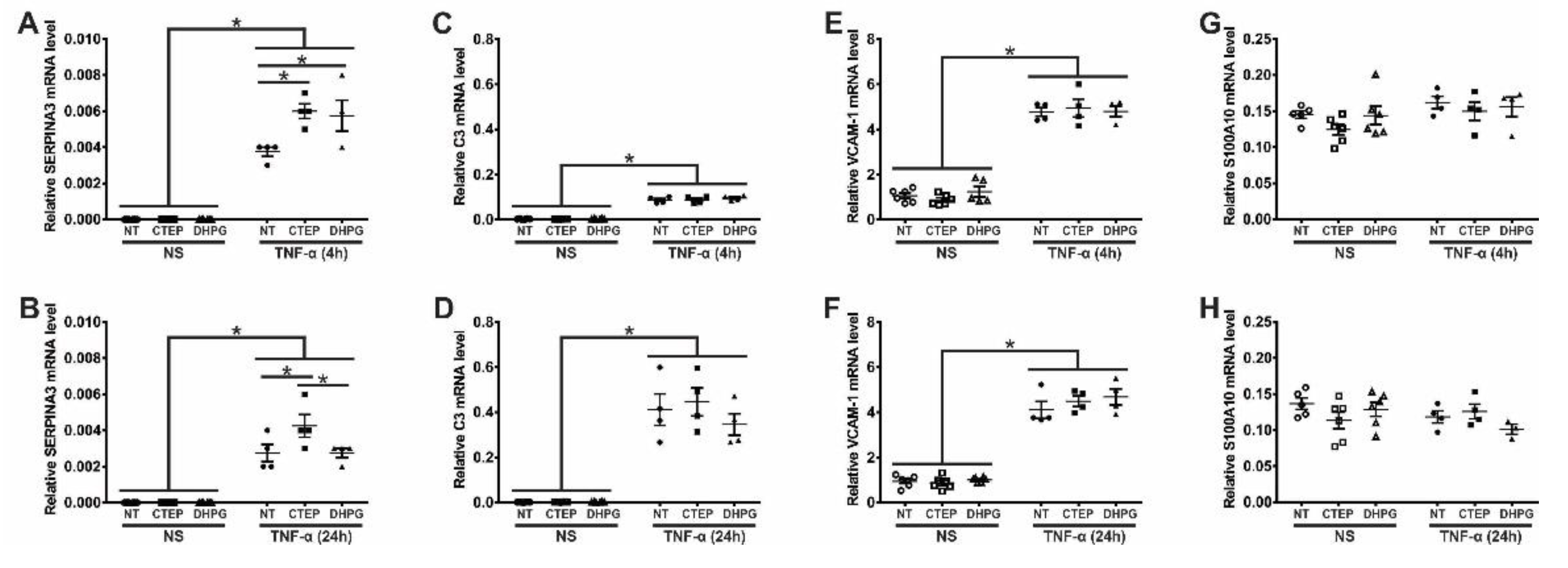
Figure 4.
Both CTEP and rTNF-α increase astrocytic phagocytosis. Shown are representative laser scanning confocal micrographs from hiPSC-derived astrocytes labelled with CellTracker blue and synaptoneurosomes (SNS) labelled with Vybrant CM-Dil (red). hiPSC-derived astrocytes were either unstimulated (NS), and treated with either vehicle (NT) (A), CTEP 10 µM (B) or DHPG 10 µM (C), or stimulated with rTNF-α 10 ng/mL, and treated with either vehicle (NT) (D), CTEP 10 µM (E) or DHPG 10 µM (F) for 24 h. Scale bar=200 μm. (G) Graph shows phagocytic index of hiPSC-derived astrocytes that were either unstimulated (NS) or stimulated with rTNF-α 10 ng/mL and treated with either vehicle (NT), DHPG 10 µM or CTEP 10 µM for 24 h. Dashed line represents median and dotted lines represent interquartile interval. n = 258-414. Graphs show mRNA levels of MERTK in hiPSC-derived astrocytes that were either unstimulated (NS) or stimulated with rTNF-α 10 ng/mL and treated with either vehicle (NT), CTEP 10 µM or DHPG 10 µM for either 4 h (H) or 24 h (I). mRNA levels were assessed by quantitative RT-PCR, which was performed in triplicates and normalized to the average of RPLP0 and IPO8 mRNA levels. Data represents the means ± SEM. * (p<0.05) indicates significant differences.
Figure 4.
Both CTEP and rTNF-α increase astrocytic phagocytosis. Shown are representative laser scanning confocal micrographs from hiPSC-derived astrocytes labelled with CellTracker blue and synaptoneurosomes (SNS) labelled with Vybrant CM-Dil (red). hiPSC-derived astrocytes were either unstimulated (NS), and treated with either vehicle (NT) (A), CTEP 10 µM (B) or DHPG 10 µM (C), or stimulated with rTNF-α 10 ng/mL, and treated with either vehicle (NT) (D), CTEP 10 µM (E) or DHPG 10 µM (F) for 24 h. Scale bar=200 μm. (G) Graph shows phagocytic index of hiPSC-derived astrocytes that were either unstimulated (NS) or stimulated with rTNF-α 10 ng/mL and treated with either vehicle (NT), DHPG 10 µM or CTEP 10 µM for 24 h. Dashed line represents median and dotted lines represent interquartile interval. n = 258-414. Graphs show mRNA levels of MERTK in hiPSC-derived astrocytes that were either unstimulated (NS) or stimulated with rTNF-α 10 ng/mL and treated with either vehicle (NT), CTEP 10 µM or DHPG 10 µM for either 4 h (H) or 24 h (I). mRNA levels were assessed by quantitative RT-PCR, which was performed in triplicates and normalized to the average of RPLP0 and IPO8 mRNA levels. Data represents the means ± SEM. * (p<0.05) indicates significant differences.
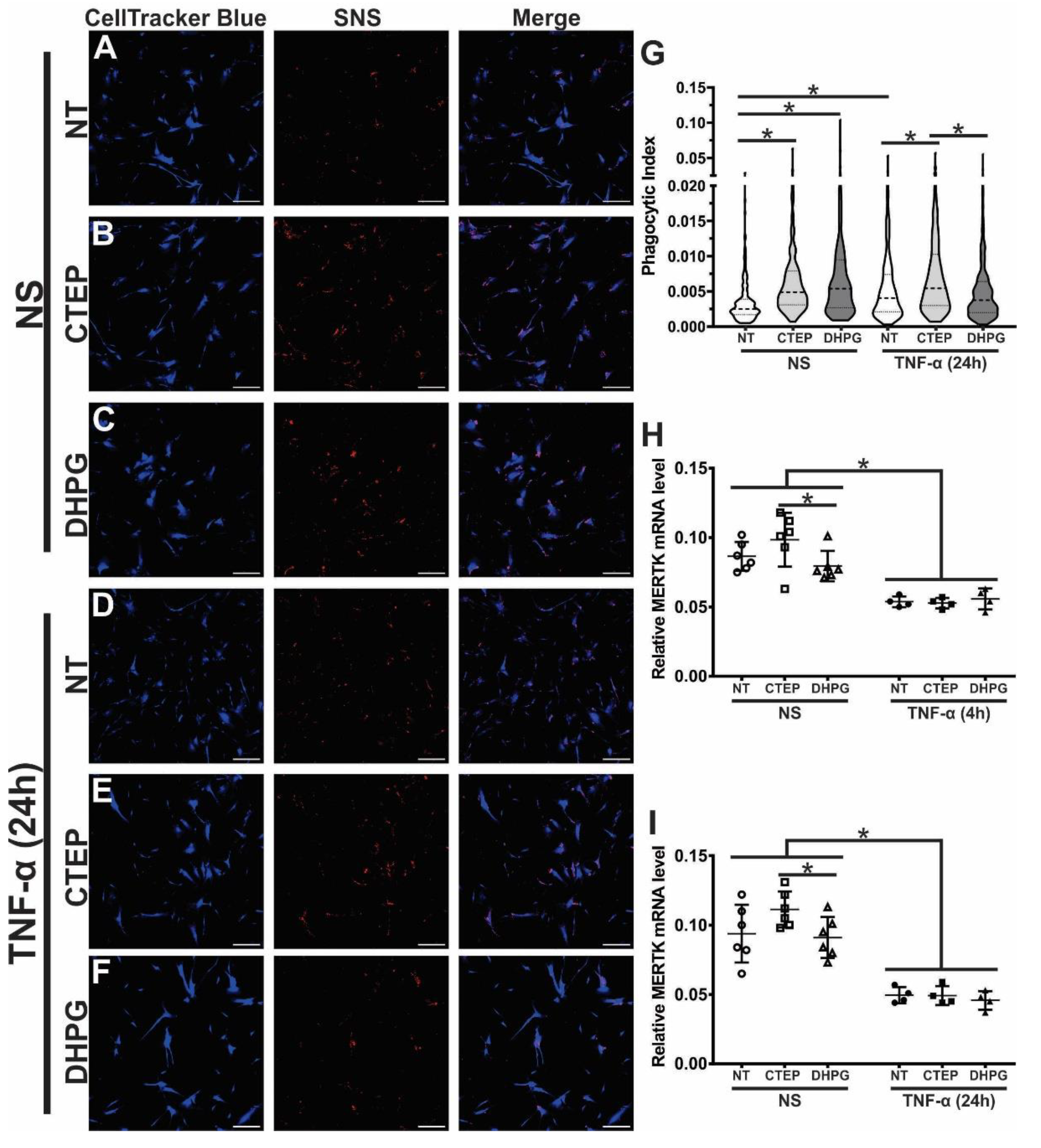
Figure 5.
Both CTEP and rTNF-α decrease the expression of synaptogenic molecules. Graphs show mRNA levels of BDNF (A), glypican-4 (GPC4) (C), and trombospoindin-1 (TSP1) (E) in hiPSC-derived astrocytes that were either unstimulated (NS) or stimulated with rTNF-α 10 ng/mL and treated with either vehicle (NT), CTEP 10 µM or DHPG 10 µM for 4 h. Graphs show mRNA levels of BDNF (B), GPC4 (D), and TSP1 (F) in hiPSC-derived astrocytes that were either unstimulated (NS) or stimulated with rTNF-α 10 ng/mL and treated with either vehicle (NT), CTEP 10 µM or DHPG 10 µM for 24 h. mRNA levels were assessed by quantitative RT-PCR, which was performed in triplicates and normalized to the average of RPLP0 and IPO8 mRNA levels. Data represents the means ± SEM. * (p<0.05) indicates significant differences.
Figure 5.
Both CTEP and rTNF-α decrease the expression of synaptogenic molecules. Graphs show mRNA levels of BDNF (A), glypican-4 (GPC4) (C), and trombospoindin-1 (TSP1) (E) in hiPSC-derived astrocytes that were either unstimulated (NS) or stimulated with rTNF-α 10 ng/mL and treated with either vehicle (NT), CTEP 10 µM or DHPG 10 µM for 4 h. Graphs show mRNA levels of BDNF (B), GPC4 (D), and TSP1 (F) in hiPSC-derived astrocytes that were either unstimulated (NS) or stimulated with rTNF-α 10 ng/mL and treated with either vehicle (NT), CTEP 10 µM or DHPG 10 µM for 24 h. mRNA levels were assessed by quantitative RT-PCR, which was performed in triplicates and normalized to the average of RPLP0 and IPO8 mRNA levels. Data represents the means ± SEM. * (p<0.05) indicates significant differences.
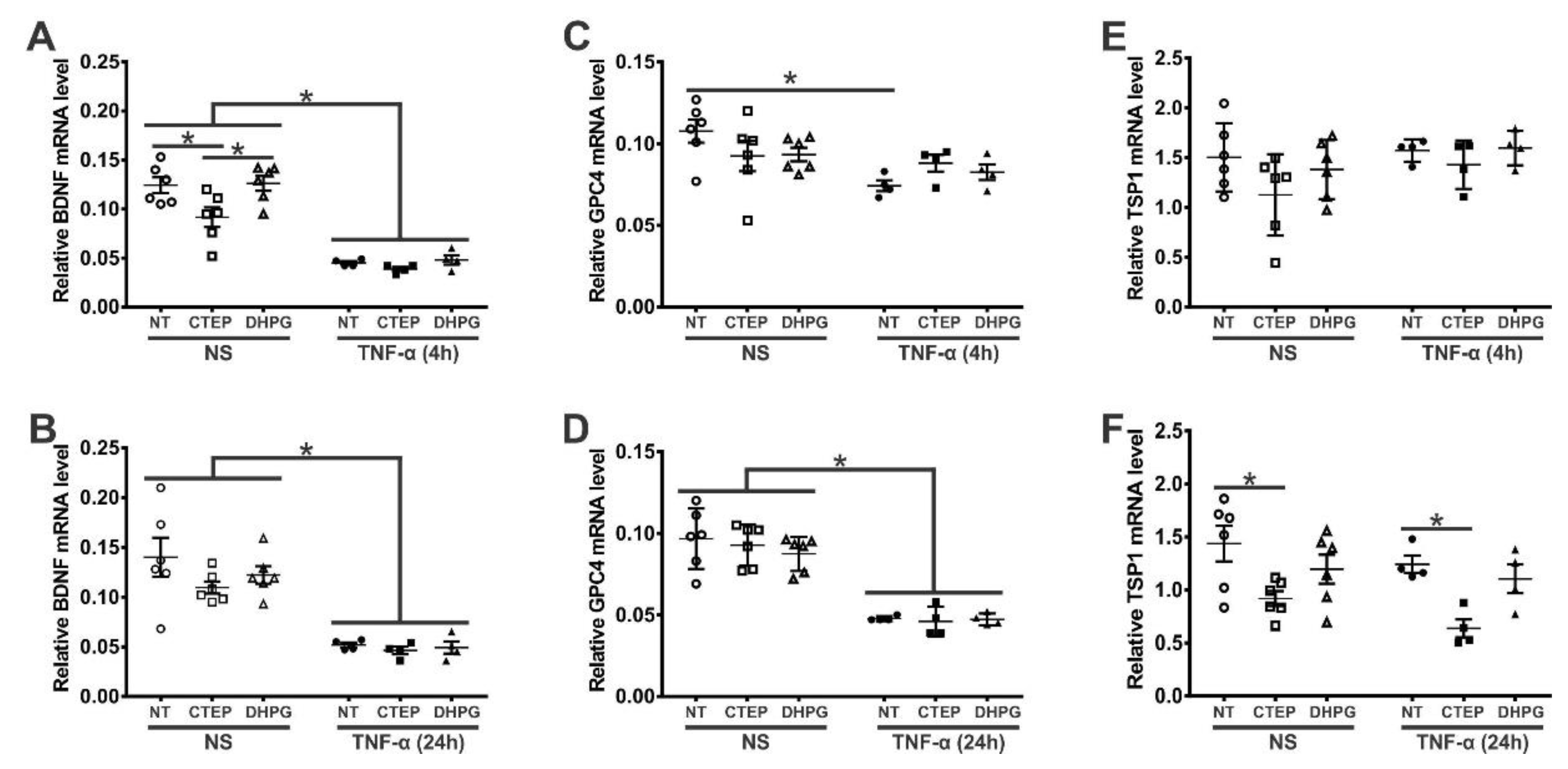

Table 1.
qPCR primer sequences.
| Gene | Forward primer (5’-3’) | Reverse primer (5’-3’) |
|---|---|---|
| C3 | CTGCCCAGTTTCGAGGTCAT | CGAGCCATCCTCAATCGGAA |
| IPO8 | TCCGAACTATTATCGACAGGACC | GTTCAAAGAGCCGAGCTACAA |
| RPLP0 | TTAAACCCTGCGTGGCAATC | ATCTGCTTGGAGCCCACATT |
| MERTK | TGGCGTAGAGCTATCACTG | CTGGCGTGAGGAAGGGATAA |
| SERPINA3 | CCTGAGGCAGAGTTGAGAATGG | TCAAGTGGGCTGTTAGGGTG |
| VCAM1 | CGAACCCAAACAAAGGCAGA | ACAGGATTTTCGGAGCAGGA |
| S100A10 | AACAAAGGAGGACCTGAGAGTAC | CTTTGCCATCTCTACACTGGTCC |
| BDNF | AGTTGGGAGCCTGAAATAGTGG | AGGATGCTGGTCCAAGTGGT |
| GPC4 | GTCAGCGAACAGTGCAATCAT | ACATTTCCCACCACGTAGTAAC |
| TSP1 | GCCAACAAACAGGTGTGCAA | GCAGATGATGCCATTGCCAG |
| TNF-α | CTGCACTTTGGAGTGATCGG | TGAGGGTTTGCTACAACATGGG |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



