Submitted:
31 May 2023
Posted:
01 June 2023
You are already at the latest version
Abstract
A wide variety of renal neoplasms can have cystic areas. It happens for different reasons: some tumors have an intrinsic cystic architecture, others have a pseudocystic degeneration of necrotic foci or they have cystically dilated renal tubules constrained by stromal neoplastic cells. Clear cell renal cell carcinoma (CCRCC), either solid or cystic, is the most frequent type of renal cancer. While pseudocysts are found in high-grade aggressive CCRCC, cystic growth is associated to low-grade indolent cases. The latter also form through a cyst-dependent molecular pathway and they are more frequent in patients suffering from VHL disease. The differential diagnosis with multilocular cystic renal neoplasm of low malignant potential and clear cell papillary renal cell tumor can be especially hard and it requires a focused macroscopical and microscopical pathological analysis. As every class of renal tumors include cystic forms, a knowledge of criteria for a differential diagnosis is mandatory.
Keywords:
Renal cell carcinoma
; Cystic renal neoplasm
; differential diagnosis
1. Introduction
Renal cancer is a common malignant neoplasm, whose classification has been expanding along decades [1]. There are indeed more frequent and rarer subtypes of tumors, some of which are molecularly defined [2,3]. Morphological analysis is however still at the base of pathological diagnosis and cystic areas can be present in a wide variety of renal neoplasms (whether benign or malignant) as a minor or dominant component [4,5]. They are estimated to be present in 5-15% of lesions, where they reflect an inherent architecture of the tumor [4,6]. They must be distinguished from pseudo-cystic degeneration of necrotic foci: while cystic growth is associated to a more indolent behavior, tumoral necrosis is present in aggressive masses [5]. This is especially true in cystic clear cell renal cell carcinoma (CCRCC), which is also the most frequent cystic renal cancer [4]. Cystic CCRCC is more frequent in patients with Von Hippel-Lindau (VHL) syndrome and different molecular patterns are also implicated in its development compared to solid cases [4]. Nevertheless, cystic CCRCC is not classified as separate pathologic entity. Differently, multilocular cystic renal neoplasm of low malignant potential (MCNLMP) is independently allocated in the WHO classification, despite molecular overlaps with CCRCC [5]. Cystic areas can be present in non-renal-cell neoplasms of the kidney as well, furtherly complicating the diagnostic process [4]. In this review, we address the main morphological and molecular features of cystic CCRCC, together with its main differential diagnoses
2. Macroscopic and Microscopic Features of Cystic CCRCC
According to the 2019 Bosniak Classification (BC), the term “cystic renal mass” can be applied to neoplasms with a predominant cystic pattern and less than 25% of enhancing tissue [7,8]. This term has an agnostic character, as it can imply both benign and malignant lesions [7,8]. A distinction must be made from renal cysts, which are benign, and from solid neoplasms with minor cystic components. The latter are more likely to be malignant with pseudo-cystic degenerative areas with tumoral necrosis [7,8]. Both cystic growth pattern and pseudo-cystic degeneration can occur in CCRCC [6]. Less than 5% of CCRCCs have multiple cysts as predominant architecture [4]. The minimum amount of cystic architecture necessary to define cystic CCRCC varies in the literature. Some authors mirror BC, as they require at least 75% of cystic areas [9], while others lower the threshold to 50% [10]. Interestingly, both cutoffs have proven to discriminate CCRCCs associated to a better prognosis [9,10].
Macroscopically, cystic growth appears as variably sized hollow spaces filled with clear or hemorrhagic fluid, with a clear separation from adjacent solid neoplastic tissue. Cysts can be single or multiple, with or without internal septations. When multiple cysts are predominant, the neoplasm can overall resemble a multilocular cyst. Evident solid areas have instead the typical golden-yellow color, with reddish hemorrhagic foci. Pseudo-cystic degenerative areas contain instead darker, denser, hemorrhagic material with cellular debris. They are more frequently centrally located within the lesion and they can be surrounded by soft, greyish necrotic tissue. Vital parts of the tumor, other than the typical colors, can also have whitish areas where sarcomatoid differentiation is present.
Microscopically, along with macro-cysts, even solid regions of CCRCC can reveal a micro-cystic growth pattern (Figure 1A–D). Micro-cysts arise within tumoral nests and cystic spaces are usually filled with red blood cells. Cells at the border of these micro-cysts do not have significantly different histological and immunohistochemical features, compared to solid acini. They have clear cytoplasm and variably sized nucleoli. Nuclei are usually basally located, although occasional apical alignment can be present. Positive labeling is present for carbonic anhydrase IX (CAIX) in a diffuse box-shaped fashion, together with CD10, RCC, Vimentin and pan-cytokeratin. High molecular weight cytokeratins (HMWCKs) and CK7 are usually negative.
Microscopical analysis of cystic CCRCC usually reveals bland-looking clear cells with a low-grade of differentiation (i.e. G1-G2 WHO grading). The epithelial coating of cysts, differently from solid areas, is more likely to be CK7-positive, a feature that can be misleading in small biopsy samples. Nevertheless, HMWCK is negative. In cases with a marked predominance of cystic growth, sampling of the capsule and septation can reveal clear cell clusters exceeding a 20x (1 mm) microscopic field, which is sufficient for a diagnosis of cystic CCRCC. Another criterion is the presence of an expansile growth of clear cells, large enough to alter the contours of the capsule/septum. Finally, necrosis or vascular invasion could be present. Cellular clusters below the 20x/1 mm cutoff, without expansile growth, necrosis and vascular invasion, allow instead a diagnosis of MCNLMP [5].
Pseudo-cystic degenerative areas are filled with nuclear and cytoplasmatic debris of necrotic cells, together with various amounts of red blood cells (Figure 2A–D). No epithelial lining can be identified and the surrounding tissue is necrotic as well. Vital neoplastic cells are high-grade (i.e. G3-G4 WHO grading). Blandly eosinophilic cytoplasm and hyaline globules are commonly found in high-grade CCRCC, which can become misleading if clear-cell areas cannot be identified. Moreover, CAIX tends to become positive near necrotic areas in different type of renal neoplasms as a hypoxia-induced factor [11,12]. As such, diagnosis of high-grade pseudo-cystic CCRCC can be challenging and it requires more extensive sampling.
3. Molecular Features of Cystic CCRCC
In CCRCC, tumor-initiating molecular alterations involve deletion of the 3p chromosome [13]. Specifically, loss of the 3p25 region is observed in 85% of CCRCC [14]. As Von Hippel-Lindau (VHL) tumor-suppressor gene is located in this area of DNA, 3p25 deletion brings to the loss of one allele. The second VHL allele is instead inactivated either by mutation or methylation. Mutations of VHL are found in 64% of CCRCC [14]. VHL protein is implicated in different molecular mechanisms, including microtubular stabilization for cilia formation and inhibition of the alpha subunit of hypoxia-inducible factor (HIF) [15,16]. When VHL is not available, the accumulation of HIFα upregulates vascular endothelial growth factor (VEGF), inducing angiogenesis. After initiating factors, other molecular events drive tumoral evolution towards different neoplastic subtypes [13,17]. For examples, BAP1 and PBRM1 are two evolution-driver onco-suppressor genes (also located on chromosome 3p) mutated in 13% and 36% of CCRCC, respectively [14]. Their mutations are mutually exclusive, bringing to CCRCCs with different features. BAP1-mutated CCRCC is a high-grade neoplasm with poor vascularization, including renal cell carcinoma with sarcomatoid and rhabdoid features [18,19,20]. In these cases, also CDKN2A deletions and increased expression of MYC transcriptional programs can be present [18]. Moreover, BAP1-mutated CCRCC can composed of large tumoral cells with abundant cytoplasm and a papillary architecture (reminiscent of RCC with MITF-family rearrangement), along with IHC positivity for racemase/AMACR and CK7 [19]. Also, a rich T lymphocyte infiltration can be present, bringing to an immune-inflamed phenotype characterized by immune activation, increased cytotoxic immune infiltration with upregulation of antigen presentation machinery genes and PD-L1 expression [18]. Infiltrated tumors are also enriched for chromosomal losses of 9p21.3 [21]. PBRM1-mutated CCRCC is instead a low-grade neoplasm, with high levels of angiogenesis and fewer inflammation. Novel mutations can also be acquired by neoplasms during therapy with small molecules, bringing to an acquired resistance [22].
Different molecular patterns seem also to be implied in the formation of cystic CCRCC, for which a cyst-dependent CCRCC progression pathway have been identified [4]. As previously mentioned, VHL contributes to cilia formation through microtubules stabilization. Loss of VHL brings to an aberrant orientation of newly formed microtubules, which in turn hinders ciliogenesis. Such effect upregulates the cell-cycle, since cells without the cilium cannot rest in G0 phase, as differentiated cells would do. Therefore, cilia can be considered tumor suppressor organelles and their absence promotes the transition towards malignancy [23]. Loss of cilia is also associated to cyst development caused by impaired cellular signaling [15]. This process happens both in sporadic cystic CCRCC, as well as in inherited diseases like Polycystic Kidney Disease (PKD) and VHL disease (VHLd) [24,25]. PKD and VHL diseases are therefore both considered among so-called ciliopathies [26]. The latter is an autosomal dominant tumor syndrome: patients suffering from it develop renal cysts and CCRCC in 60% and 30% of cases, respectively [5,27]. Renal cancer in VHLd has been reported as early as 16 years of age, with a mean age of 37 years[28]. Renal cysts in VHLd are also potential precursors for CCRCC, as their epithelial lining can demonstrate dysplastic areas as well as loss of the remaining VHL non-genetically-mutated allele [4]. It follows that CCRCC in VHLd is in turn often cystic, other than bilateral. Interestingly, just as VHL is an early cancer-initiator gene that requires further downstream molecular events, also cysts formation cannot rely on VHL deficiency alone [23,29]. A critical role is played by GSK3β, a protein kinase that regulates cell proliferation, microtubule assembly, stability and dynamics [15]. Combined loss of VHL and GSK3β disrupts ciliary-maintenance and it is considered a key player in cyst-dependent CCRCC progression pathway. The role of GSK3β is however yet to be fully elucidated, as evidence has also shown higher levels of expression both in PKD and in some CCRCCs [30,31]. According to these studies, its inhibition might actually be therapeutically useful to hinder cystic expansion and progression of both PKD and CCRCCs [30,31].
4. Differential Diagnosis of Cystic CCRCC
As already mentioned, the range of renal neoplasms with cystic areas is wide. It encompasses every WHO group of tumors of the kidney (i.e. renal cell, metanephric, mixed epithelial and stromal, mesenchymal, embryonal and germ-cell tumors), including frequent and rare, adult and pediatric, inherited and sporadic forms [5]. Attention must therefore be paid to patients’ age and bilaterality of the lesions. Pathological analysis must focus on the cellular lining of cysts, as well as to the peri-cystic stroma and possible solid areas which can be focal.
Cystic areas in frequent renal neoplasms like chromophobe carcinoma, papillary carcinoma and oncocytoma are possible, but rather unusual [5]. Although rarer, the main differential diagnosis for cystic-predominant CCRCC is MNCLMP. Because the vast majority of CCRCCs harbor VHL mutation, 3p copy number loss, or both, tumors with clear cell histology lacking these alterations can often be reclassified as different established or emerging entities [32]. However, in the case of MNCLMP, there are molecular overlaps with cystic CCRCC, including deletion of the 3p chromosome and similar mutated genes which are part of the cyst-dependent pathway [5,33]. For this reason, MNCLMP might be considered a subtype of CCRCC, at the most indolent end of the spectrum. Nevertheless, it also has distinct clinical, morphological and molecular features that allow a separated classification [5,33,34]. MNCLMP accounts for less than 5% of renal tumors. It usually incidentally detected as a monolateral lesion in patients slightly younger than in CCRCC (median age 55 vs 62). Macroscopic appearance is entirely composed of variably sized cysts, with a small total diameter (usually pT1, i.e. ≤7 cm) [5]. Neither solid nodules, nor necrotic foci can be present. Even microscopical necrosis is not accepted, together with rhabdoid/sarcomatoid differentiation, lymphovascular invasion, frequent mitoses or any atypical mitosis. The epithelial lining of the cysts features one to a few layers of clear cells. Nuclei are randomly distributed, without a predilection for the apical portion of cells, and they must be low grade (G1-G2 WHO grading). The capsule and septa are fibrous and they can include clusters of clear cells, but they must be small (i.e. <1mm or <20x microscopic area). When diagnostic criteria are strictly applied, tumors identified as MNCLMPs have a benign clinical behavior [5]. IHC analysis is not of aid in the differential diagnosis with CCRCC, as they have the same profile [35]. Aside the molecular similarities between MNCLMP and CCRCC, the former has also shown to have a lower frequency of mutations. Six genes have been found significantly more frequently mutated in cystic CCRCC: SETD2, GIGYF2, FGFR3, BCR, KMT2C, and TSC2 [36]. These are potential candidate genes that could help elucidating mechanisms in the development and progression of CCRCC, as well as aiding in the differential diagnosis with MNCLMP [36].
Another benign renal cell tumor that can be nearly entirely cystic featuring bland-looking clear cells is clear cell papillary renal cell tumor (CCPRCT) (Figure 3A–D). Histologically, nuclei are oriented towards the luminal apex of the cells [37,38]. As in cystic CCRCC, CK7 is positive. However, CCPRCT expresses also HMWCK (specifically CK34βE12). CAIX signal has a cup-like pattern (i.e. with a missing luminal border), while CD10 is negative. Nevertheless, CCPRCT and low-grade CCRCC can have histologically identical areas and unequivocal diagnosis of CCPRCT on needle biopsy may not be possible [5]. Molecularly, CCPRCT have a distinct miRNA expression profile which also lacks the pattern typically associated with aggressive neoplastic behavior [39].
Cystic architecture combined with prominent nucleoli in epithelial cells can be found in WHO/ISUP category 5 neoplasms: tubulocystic RCC (TcRCC), acquired cystic disease-associated RCC (ACD-RCD) and eosinophilic solid and cystic RCC [2,5]. These neoplasms have a potentially misleading nucleolar appearance, as they look high-grade despite an indolent clinical behavior. They have eosinophilic cytoplasm, which distinguish them from cystic CCRCC. Moreover, TcRCC is composed of small cystic areas, which macroscopically reminds of a sponge, rather than a multiloculated cyst. ACD-RCD are often multiple and bilateral solid masses in the setting of acquired cystic disease. Like in VHLd, cysts are possible precursor lesions and ACD-RCD is often an intracystic mass. This is however derived from a history of long-term dialysis, rather than an inherited gene mutation.
While cystic CCRCC have fibrotic septa and capsule, other neoplasms are biphasic, with specific stromal proliferations. Angiomyolipoma with epithelial cysts (AMLEC) is a rare subtype of angiomyolipoma. Along with solid areas (predominantly composed of smooth muscle and blood vessels), cystic spaces are present. They have a cuboidal-to-hobnail epithelium and a dense peri-cystic stroma, similar to the cambium layer in rhabdomyosarcoma. The epithelium is cytokeratin-positive, while the cambium-like stroma and solid areas are cytokeratin-negative and positive for melanocytic markers (HMB-45, melan-A, MiTF). Adult cystic nephromas (ACN) and mixed epithelial and stromal tumor (MEST) are two closely related biphasic neoplasms that usually arise in women [40,41,42]. Their biphasic nature is embodied by a renal cell epithelial component, along with proliferation of bland-looking spindle stromal cells (Figure 4A–C). Morphology recalls ovarian stroma, together with the expression of estrogen and progesterone receptors, and also of inhibin. While ACN is entirely cystic, MEST has solid whitish areas with different patterns of growth (e.g. glandular, papillary, thyroid-like). Pediatric cystic nephroma is a similar lesion, epidemiologically bound to children below 2 years of age and molecularly characterized by a DICER1 mutation [43]. If any immature nephroblastic element is present, the diagnosis switches to cystic partially differentiated nephroblastoma [5].
Metanephric stromal tumor is another pediatric renal neoplasm which can have cystic areas. Solid parts show a concentric peritubular growth of spindle cells expressing CD34 and with BRAF v600e mutation [5,44,45]. While tubules are more commonly unaltered by the encircling spindle cells, some become cystically dilated, rendering a cystic gross appearance.
Renal teratomas are rare, most often cystic and mature with mixed epithelial and stromal elements [5,46]. They can be pure or accompanied by a yolk-sac component. Microscopically, cystic spaces can be lined a by keratinizing squamous epithelium with skin adnexae, or alternatively by a thick fibromuscular without any lining. Generally speaking, a renal metastasis or a direct extension from a retroperitoneal germ-cell tumor, as well as a teratoid nephroblastoma, must always be ruled out [5,46].
5. Conclusions and Future Directions
Our knowledge about CCRCC pathogenesis and molecular features has been broadening along the years. This is not important only to different subtypes of CCRCC, but it also allows to track novel therapeutic targets and diagnostic markers. While there are molecular overlaps between cystic CCRCC and MCNLMP, there are also some significant differences. The validation of such data and the implementation of molecular studies in the daily pathology practice will become of great aid in challenging differential diagnoses. Moreover, furtherly clarifying the role of GSK3β in the formation and progression of cystic renal lesions could lead to a targetable protein in the treatment of cystic CCRCC, as well as PKD.
Author Contributions
G.M.P. conception, writing; R.L.: conception, M.C. conception. All authors have read and agreed to the published version of the manuscript.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| ACD-RCD | Acquired Cystic Disease-associated Renal Cell Carcinoma |
| CAN | Adult Cystic Nephroma |
| AMLEC | Angiomyolipoma with Epithelial Cyst |
| BC | Bosniak Classification |
| CAIX | Carbonic Anhydrase IX |
| CCPRCT | Clear Cell Papillary Renal Cell Tumor |
| CCRCC | Clear Cell Renal Cell Carcinoma |
| HIF | Hypoxia-Inducible Factor |
| HMWCK | High Molecular Weight Cytokeratins |
| IHC | Immunohistochemistry |
| MEST | Mixed Epithelial and Stromal Tumor |
| MiTF | Melanocyte Inducing Transcription Factor |
| MCNLMP | Multilocular Cystic Renal Neoplasm of Low Malignant Potential |
| PKD | Polycystic Kidney Disease |
| TcRCC | Tubulo-cystic Renal Cell Carcinoma |
| VEGF | Vascular endothelial growth factor |
| VHL | Von Hippel-Lindau |
| VHLd | Von Hippel-Lindau disease |
References
- Caliò A, Marletta S, Brunelli M, Martignoni G. WHO 2022 Classification of Kidney Tumors: what is relevant? An update and future novelties for the pathologist. Pathologica. 2022 Feb;115(1):23-31. Epub 2023 Jan 16. [CrossRef] [PubMed]
- Alaghehbandan R, Siadat F, Trpkov K. What’s new in the WHO 2022 classification of kidney tumours? Pathologica. 2022 Feb;115(1):8-22. Epub 2023 Jan 16. [CrossRef] [PubMed]
- Udager AM, Mehra R. Morphologic, Molecular, and Taxonomic Evolution of Renal Cell Carcinoma: A Conceptual Perspective With Emphasis on Updates to the 2016 World Health Organization Classification. Arch Pathol Lab Med. 2016 Oct;140(10):1026-37. [CrossRef] [PubMed]
- Moch, H. Cystic renal tumors: new entities and novel concepts. Adv Anat Pathol. 2010 May;17(3):209-14. [CrossRef] [PubMed]
- WHO Classification of Tumours Editorial Board. Urinary and male genital tumours. Lyon (France): International Agency for Research on Cancer; 2022. (WHO Classification of tumour series; 5th ed.; vol. 8).
- Alrumayyan M, Raveendran L, Lawson KA, Finelli A. Cystic Renal Masses: Old and New Paradigms. Urol Clin North Am. 2023 May;50(2):227-238. Epub 2023 Feb 20. [CrossRef] [PubMed]
- Silverman SG, Pedrosa I, Ellis JH, Hindman NM, Schieda N, Smith AD, Remer EM, Shinagare AB, Curci NE, Raman SS, Wells SA, Kaffenberger SD, Wang ZJ, Chandarana H, Davenport MS. Bosniak Classification of Cystic Renal Masses, Version 2019: An Update Proposal and Needs Assessment. Radiology. 2019 Aug;292(2):475-488. Epub 2019 Jun 18. PMCID: PMC6677285. [CrossRef] [PubMed]
- Krishna S, Schieda N, Pedrosa I, Hindman N, Baroni RH, Silverman SG, Davenport MS. Update on MRI of Cystic Renal Masses Including Bosniak Version 2019. J Magn Reson Imaging. 2021 Aug;54(2):341-356. Epub 2020 Oct 2. PMCID: PMC8017011. [CrossRef] [PubMed]
- Westerman ME, Cheville JC, Lohse CM, Sharma V, Boorjian SA, Leibovich BC, Thompson RH. Long-Term Outcomes of Patients With Low Grade Cystic Renal Epithelial Neoplasms. Urology. 2019 Nov;133:145-150. Epub 2019 Jul 26. [CrossRef] [PubMed]
- Tretiakova M, Mehta V, Kocherginsky M, Minor A, Shen SS, Sirintrapun SJ, Yao JL, Alvarado-Cabrero I, Antic T, Eggener SE, Picken MM, Paner GP. Predominantly cystic clear cell renal cell carcinoma and multilocular cystic renal neoplasm of low malignant potential form a low-grade spectrum. Virchows Arch. 2018 Jul;473(1):85-93. Epub 2018 May 17. [CrossRef] [PubMed]
- Pastorekova S, Gillies RJ. The role of carbonic anhydrase IX in cancer development: links to hypoxia, acidosis, and beyond. Cancer Metastasis Rev. 2019 Jun;38(1-2):65-77. PMCID: PMC6647366. [CrossRef] [PubMed]
- Genega EM, Ghebremichael M, Najarian R, Fu Y, Wang Y, Argani P, Grisanzio C, Signoretti S. Carbonic anhydrase IX expression in renal neoplasms: correlation with tumor type and grade. Am J Clin Pathol. 2010 Dec;134(6):873-9. PMCID: PMC3778911. [CrossRef] [PubMed]
- Jonasch E, Walker CL, Rathmell WK. Clear cell renal cell carcinoma ontogeny and mechanisms of lethality. Nat Rev Nephrol. 2021 Apr;17(4):245-261. Epub 2020 Nov 3. PMCID: PMC8172121. [CrossRef] [PubMed]
- Bui TO, Dao VT, Nguyen VT, Feugeas JP, Pamoukdjian F, Bousquet G. Genomics of Clear-cell Renal Cell Carcinoma: A Systematic Review and Meta-analysis. Eur Urol. 2022 Apr;81(4):349-361. Epub 2022 Jan 3. [CrossRef] [PubMed]
- Thoma CR, Frew IJ, Hoerner CR, Montani M, Moch H, Krek W. pVHL and GSK3beta are components of a primary cilium-maintenance signalling network. Nat Cell Biol. 2007 May;9(5):588-95. Epub 2007 Apr 22. [CrossRef] [PubMed]
- Rechsteiner MP, von Teichman A, Nowicka A, Sulser T, Schraml P, Moch H. VHL gene mutations and their effects on hypoxia inducible factor HIFα: identification of potential driver and passenger mutations. Cancer Res. 2011 Aug 15;71(16):5500-11. Epub 2011 Jun 29. [CrossRef] [PubMed]
- Cremona M, Espina V, Caccia D, Veneroni S, Colecchia M, Pierobon M, Deng J, Mueller C, Procopio G, Lanzi C, Daidone MG, Cho WC, Petricoin EF, Liotta L, Bongarzone I. Stratification of clear cell renal cell carcinoma by signaling pathway analysis. Expert Rev Proteomics. 2014 Apr;11(2):237-49. Epub 2014 Feb 27. [CrossRef] [PubMed]
- Bakouny Z, Braun DA, et al. Integrative molecular characterization of sarcomatoid and rhabdoid renal cell carcinoma. Nat Commun. 2021 Feb 5;12(1):808. PMCID: PMC7865061. [CrossRef] [PubMed]
- Akgul M, Williamson SR. How New Developments Impact Diagnosis in Existing Renal Neoplasms. Surg Pathol Clin. 2022 Dec;15(4):695-711. Epub 2022 Oct 13. [CrossRef] [PubMed]
- Kapur P, Rajaram S, Brugarolas J. The expanding role of BAP1 in clear cell renal cell carcinoma. Hum Pathol. 2023 Mar;133:22-31. Epub 2022 Aug 4. PMCID: PMC9898467. [CrossRef] [PubMed]
- D.A. Braun, Y. Hou, et al. Interplay of somatic alterations and immune infiltration modulates response to PD-1 blockade in advanced clear cell renal cell carcinoma., Nat. Med. 26 (2020) 909–918. [CrossRef]
- Elgendy M, Fusco JP, et al. Identification of mutations associated with acquired resistance to sunitinib in renal cell cancer. Int J Cancer. 2019 Oct 1;145(7):1991-2001. Epub 2019 Mar 30. [CrossRef] [PubMed]
- Guinot A, Lehmann H, Wild PJ, Frew IJ. Combined deletion of Vhl, Trp53 and Kif3a causes cystic and neoplastic renal lesions. J Pathol. 2016 Jul;239(3):365-73. Epub 2016 May 30. [CrossRef] [PubMed]
- Kuehn EW, Walz G, Benzing T. Von hippel-lindau: a tumor suppressor links microtubules to ciliogenesis and cancer development. Cancer Res. 2007 May 15;67(10):4537-40. [CrossRef] [PubMed]
- Seeger-Nukpezah T, Geynisman DM, Nikonova AS, Benzing T, Golemis EA. The hallmarks of cancer: relevance to the pathogenesis of polycystic kidney disease. Nat Rev Nephrol. 2015 Sep;11(9):515-34. Epub 2015 Apr 14. PMCID: PMC5902186. [CrossRef] [PubMed]
- Santoni M, Piva F, Cimadamore A, Giulietti M, Battelli N, Montironi R, Cosmai L, Porta C. Exploring the Spectrum of Kidney Ciliopathies. Diagnostics (Basel). 2020 Dec 16;10(12):1099. PMCID: PMC7766105. [CrossRef] [PubMed]
- Louise M Binderup M, Smerdel M, et al. von Hippel-Lindau disease: Updated guideline for diagnosis and surveillance. Eur J Med Genet. 2022 Aug;65(8):104538. Epub 2022 Jun 13. [CrossRef] [PubMed]
- Chahoud J, McGettigan M, Parikh N, Boris RS, Iliopoulos O, Rathmell WK, Daniels AB, Jonasch E, Spiess PE; International VHL Surveillance Guidelines Consortium-Renal Committee. Evaluation, diagnosis and surveillance of renal masses in the setting of VHL disease. World J Urol. 2021 Jul;39(7):2409-2415. Epub 2020 Sep 16. PMCID: PMC8101019. [CrossRef] [PubMed]
- Schönenberger D, Harlander S, Rajski M, Jacobs RA, Lundby AK, Adlesic M, Hejhal T, Wild PJ, Lundby C, Frew IJ. Formation of Renal Cysts and Tumors in Vhl/Trp53-Deficient Mice Requires HIF1α and HIF2α. Cancer Res. 2016 Apr 1;76(7):2025-36. Epub 2016 Jan 12. [CrossRef] [PubMed]
- Tao S, Kakade VR, Woodgett JR, Pandey P, Suderman ED, Rajagopal M, Rao R. Glycogen synthase kinase-3β promotes cyst expansion in polycystic kidney disease. Kidney Int. 2015 Jun;87(6):1164-75. Epub 2015 Jan 28. PMCID: PMC4449797. [CrossRef] [PubMed]
- Bilim V, Ougolkov A, Yuuki K, Naito S, Kawazoe H, Muto A, Oya M, Billadeau D, Motoyama T, Tomita Y. Glycogen synthase kinase-3: a new therapeutic target in renal cell carcinoma. Br J Cancer. 2009 Dec 15;101(12):2005-14. Epub 2009 Nov 17. PMCID: PMC2795437. [CrossRef] [PubMed]
- Favazza L, Chitale DA, Barod R, Rogers CG, Kalyana-Sundaram S, Palanisamy N, Gupta NS, Williamson SR. Renal cell tumors with clear cell histology and intact VHL and chromosome 3p: a histological review of tumors from the Cancer Genome Atlas database. Mod Pathol. 2017 Nov;30(11):1603-1612. Epub 2017 Jul 21. [CrossRef] [PubMed]
- Halat S, Eble JN, Grignon DJ, Lopez-Beltran A, Montironi R, Tan PH, Wang M, Zhang S, MacLennan GT, Cheng L. Multilocular cystic renal cell carcinoma is a subtype of clear cell renal cell carcinoma. Mod Pathol. 2010 Jul;23(7):931-6. Epub 2010 Mar 26. [CrossRef] [PubMed]
- Gong K, Zhang N, He Z, Zhou L, Lin G, Na Y. Multilocular cystic renal cell carcinoma: an experience of clinical management for 31 cases. J Cancer Res Clin Oncol. 2008 Apr;134(4):433-7. Epub 2007 Sep 1. [CrossRef] [PubMed]
- Williamson SR, Halat S, et al. Multilocular cystic renal cell carcinoma: similarities and differences in immunoprofile compared with clear cell renal cell carcinoma. Am J Surg Pathol. 2012 Oct;36(10):1425-33. [CrossRef] [PubMed]
- Kim SH, Park WS, Chung J. SETD2, GIGYF2, FGFR3, BCR, KMT2C, and TSC2 as candidate genes for differentiating multilocular cystic renal neoplasm of low malignant potential from clear cell renal cell carcinoma with cystic change. Investig Clin Urol. 2019 May;60(3):148-155. Epub 2019 Apr 1. PMCID: PMC6495037. [CrossRef] [PubMed]
- Diolombi ML, Cheng L, Argani P, Epstein JI. Do Clear Cell Papillary Renal Cell Carcinomas Have Malignant Potential? Am J Surg Pathol. 2015 Dec;39(12):1621-34. [CrossRef] [PubMed]
- Williamson, SR. Clear cell papillary renal cell carcinoma: an update after 15 years. Pathology. 2021 Jan;53(1):109-119. Epub 2020 Nov 19. [CrossRef] [PubMed]
- Munari E, Marchionni L, Chitre A, Hayashi M, Martignoni G, Brunelli M, Gobbo S, Argani P, Allaf M, Hoque MO, Netto GJ. Clear cell papillary renal cell carcinoma: micro-RNA expression profiling and comparison with clear cell renal cell carcinoma and papillary renal cell carcinoma. Hum Pathol. 2014 Jun;45(6):1130-8. Epub 2014 Jan 31. PMCID: PMC4332813. [CrossRef] [PubMed]
- Mohanty SK, Parwani AV. Mixed epithelial and stromal tumors of the kidney: an overview. Arch Pathol Lab Med. 2009 Sep;133(9):1483-6. [CrossRef] [PubMed]
- Turbiner J, Amin MB, Humphrey PA, Srigley JR, De Leval L, Radhakrishnan A, Oliva E. Cystic nephroma and mixed epithelial and stromal tumor of kidney: a detailed clinicopathologic analysis of 34 cases and proposal for renal epithelial and stromal tumor (REST) as a unifying term. Am J Surg Pathol. 2007 Apr;31(4):489-500. [CrossRef] [PubMed]
- Zhou M, Kort E, Hoekstra P, Westphal M, Magi-Galluzzi C, Sercia L, Lane B, Rini B, Bukowski R, Teh BT. Adult cystic nephroma and mixed epithelial and stromal tumor of the kidney are the same disease entity: molecular and histologic evidence. Am J Surg Pathol. 2009 Jan;33(1):72-80. [CrossRef] [PubMed]
- Vanecek T, Pivovarcikova K, Pitra T, Peckova K, Rotterova P, Daum O, Davidson W, Montiel DP, Kalusova K, Hora M, Ondic O, Dubova M, Michal M, Hes O. Mixed Epithelial and Stromal Tumor of the Kidney: Mutation Analysis of the DICER 1 Gene in 29 Cases. Appl Immunohistochem Mol Morphol. 2017 Feb;25(2):117-121. [CrossRef] [PubMed]
- Argani P, Beckwith JB. Metanephric stromal tumor: report of 31 cases of a distinctive pediatric renal neoplasm. Am J Surg Pathol. 2000 Jul;24(7):917-26. [CrossRef] [PubMed]
- Kacar A, Azili MN, Cihan BS, Demir HA, Tiryaki HT, Argani P. Metanephric stromal tumor: a challenging diagnostic entity in children. J Pediatr Surg. 2011 Dec;46(12):e7-e10. [CrossRef] [PubMed]
- Idrissi-Serhrouchni K, El-Fatemi H, El madi A, Benhayoun K, Chbani L, Harmouch T, Bouabdellah Y, Amarti A. Primary renal teratoma: a rare entity. Diagn Pathol. 2013 Jun 25;8:107. PMCID: PMC3751105. [CrossRef] [PubMed]
Figure 1.
Cystic Clear Cell Renal Cell Carcinoma. A The gross specimen features a multiloculated predominantly-cystic nodule with variably thin walls and greyish solid areas. B (H&E) Low-power view of the lesion shows multiple scattered blood-filled cystic spaces, along with solid whitish areas. C (H&E, 10x) Cysts are delimited by an epithelial lining with the same features of solid peri-cystic tissue. Around bigger cysts, higher magnification reveals the presence of micro-cystic spaces within neoplastic acini. No prominent nucleoli are evident at 10x. D (H&E, 40x) In this low-grade lesion, nucleoli are either very bland or absent even with a high-power view.
Figure 1.
Cystic Clear Cell Renal Cell Carcinoma. A The gross specimen features a multiloculated predominantly-cystic nodule with variably thin walls and greyish solid areas. B (H&E) Low-power view of the lesion shows multiple scattered blood-filled cystic spaces, along with solid whitish areas. C (H&E, 10x) Cysts are delimited by an epithelial lining with the same features of solid peri-cystic tissue. Around bigger cysts, higher magnification reveals the presence of micro-cystic spaces within neoplastic acini. No prominent nucleoli are evident at 10x. D (H&E, 40x) In this low-grade lesion, nucleoli are either very bland or absent even with a high-power view.
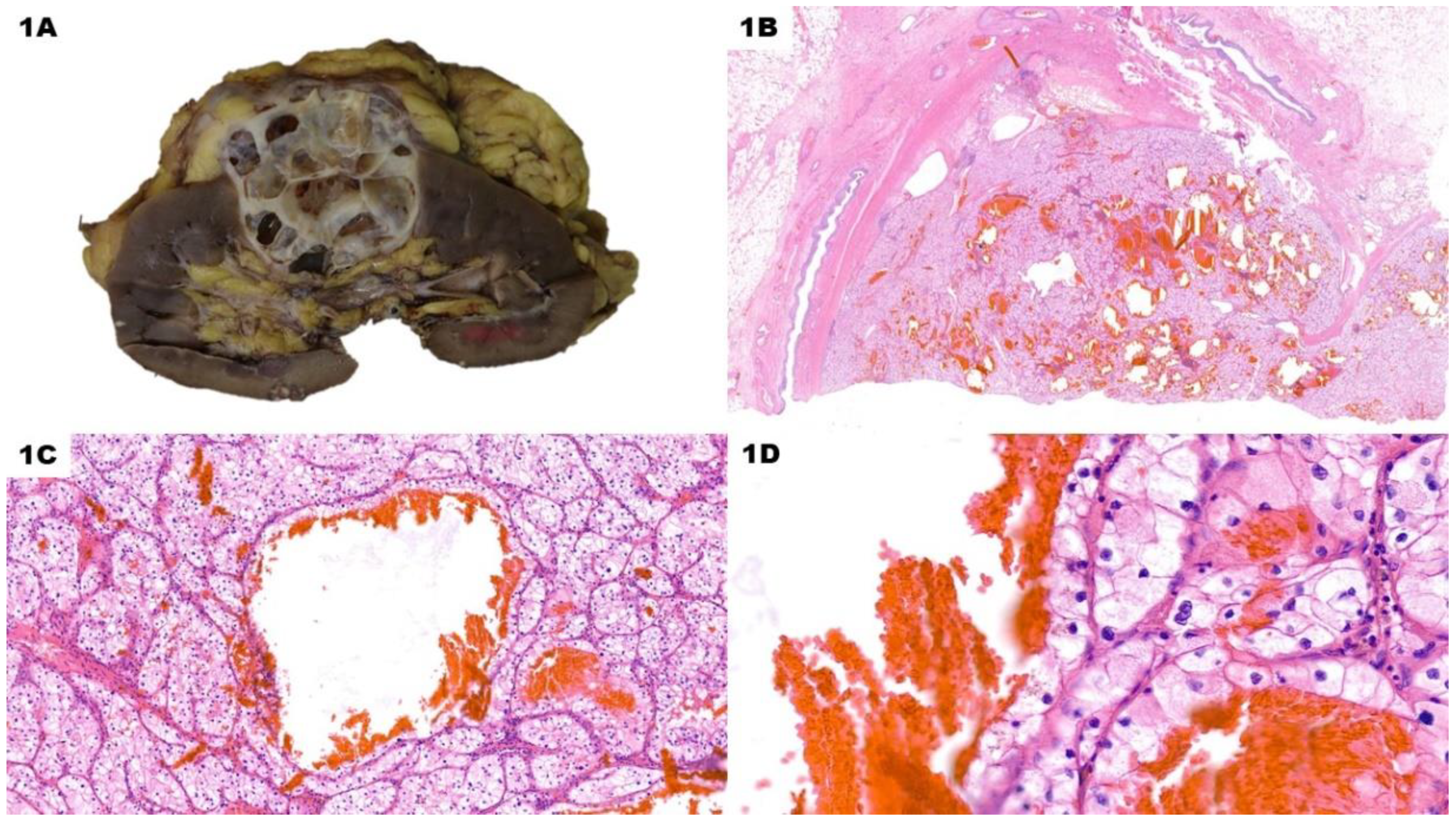
Figure 2.
Pseudo-cystic Clear Cell Renal Cell Carcinoma. (A) Macroscopically, the nodule has a large hemorrhagic area surrounded by whitish and yellowish solid tissue. (B) (H&E) Low-power view shows a blood-filled area with blueish material at the border with the adjacent solid whitish neoplastic tissue. (C) (H&E, 10x) The cystic area at the bottom of the picture, along with red blood cells, also contains blueish necrotic debris. Vital neoplastic cells at the top show prominent nucleoli already at this magnification, as it is a high-grade lesion. Other areas also showed rhabdoid cells. (D) (H&E, 40x) There is no clear-cut boundary between the necrotic debris on the right and vital solid neoplastic tissue on the left of the picture. This must be considered a pseudo-cyst rather than a cyst.
Figure 2.
Pseudo-cystic Clear Cell Renal Cell Carcinoma. (A) Macroscopically, the nodule has a large hemorrhagic area surrounded by whitish and yellowish solid tissue. (B) (H&E) Low-power view shows a blood-filled area with blueish material at the border with the adjacent solid whitish neoplastic tissue. (C) (H&E, 10x) The cystic area at the bottom of the picture, along with red blood cells, also contains blueish necrotic debris. Vital neoplastic cells at the top show prominent nucleoli already at this magnification, as it is a high-grade lesion. Other areas also showed rhabdoid cells. (D) (H&E, 40x) There is no clear-cut boundary between the necrotic debris on the right and vital solid neoplastic tissue on the left of the picture. This must be considered a pseudo-cyst rather than a cyst.
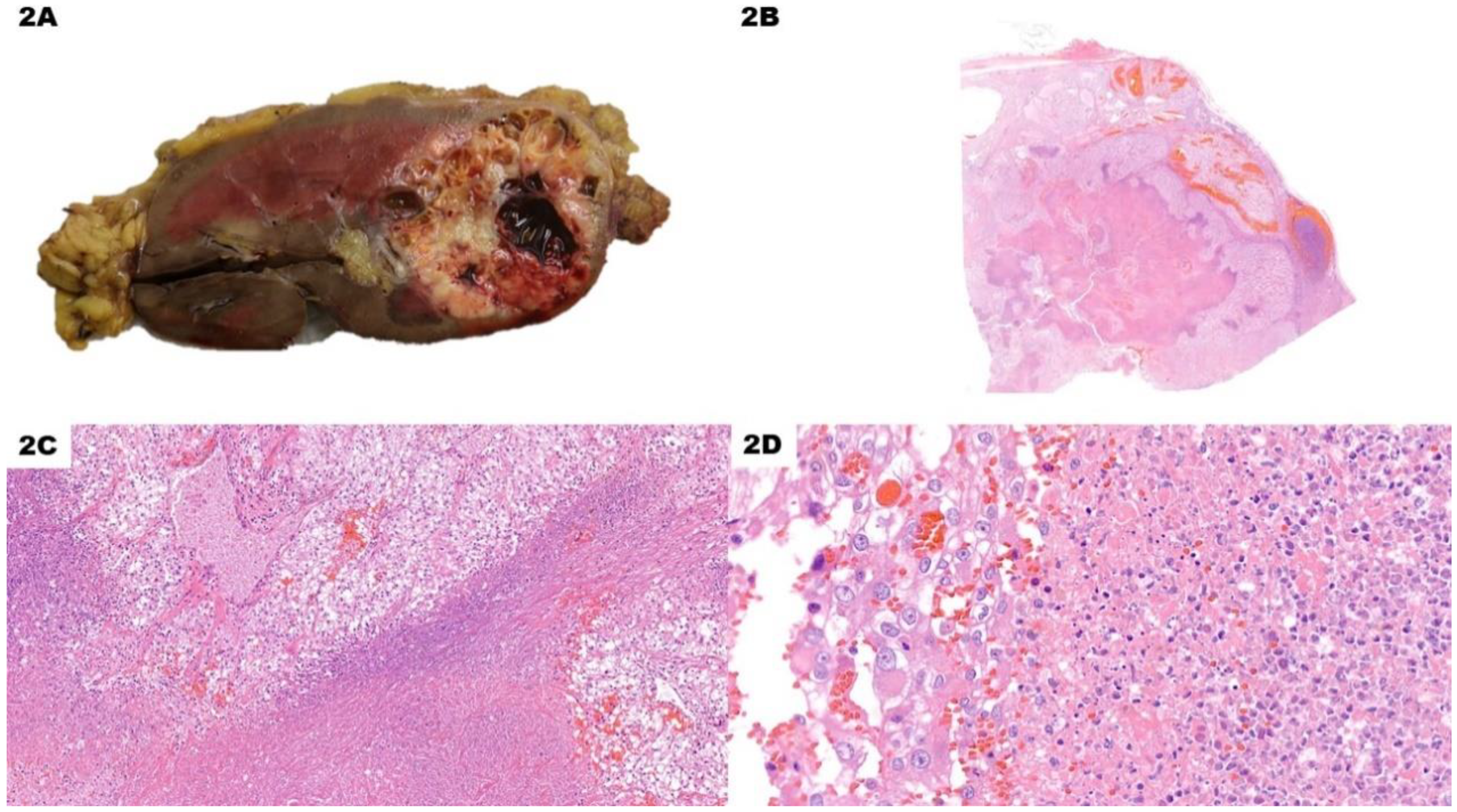
Figure 3.
Clear Cell Papillary Renal Cell Tumor. (A) (H&E) Low-power view shows multiple blood-filled areas with fibrotic walls. (B) (H&E, 10x) The epithelial lining is composed of cuboidal to low-columnar clear cells. Nuclei in low-columnar cells tends to be oriented towards the cellular luminal apex. Nucleoli are not prominent. (C) (H&E) As the name of the tumor implies, also papillary areas can be present alongside cystic areas and they can protrude inside the cystic lumen. (D) (CAIX, 40x) Immunohistochemistry for CAIX signal has a cup-like pattern (i.e. with a missing luminal border). This pattern is typical of Clear Cell Papillary Renal Cell Tumor. The neoplasm is also CK7 and HMWCK positive, while CD10 is negative.
Figure 3.
Clear Cell Papillary Renal Cell Tumor. (A) (H&E) Low-power view shows multiple blood-filled areas with fibrotic walls. (B) (H&E, 10x) The epithelial lining is composed of cuboidal to low-columnar clear cells. Nuclei in low-columnar cells tends to be oriented towards the cellular luminal apex. Nucleoli are not prominent. (C) (H&E) As the name of the tumor implies, also papillary areas can be present alongside cystic areas and they can protrude inside the cystic lumen. (D) (CAIX, 40x) Immunohistochemistry for CAIX signal has a cup-like pattern (i.e. with a missing luminal border). This pattern is typical of Clear Cell Papillary Renal Cell Tumor. The neoplasm is also CK7 and HMWCK positive, while CD10 is negative.
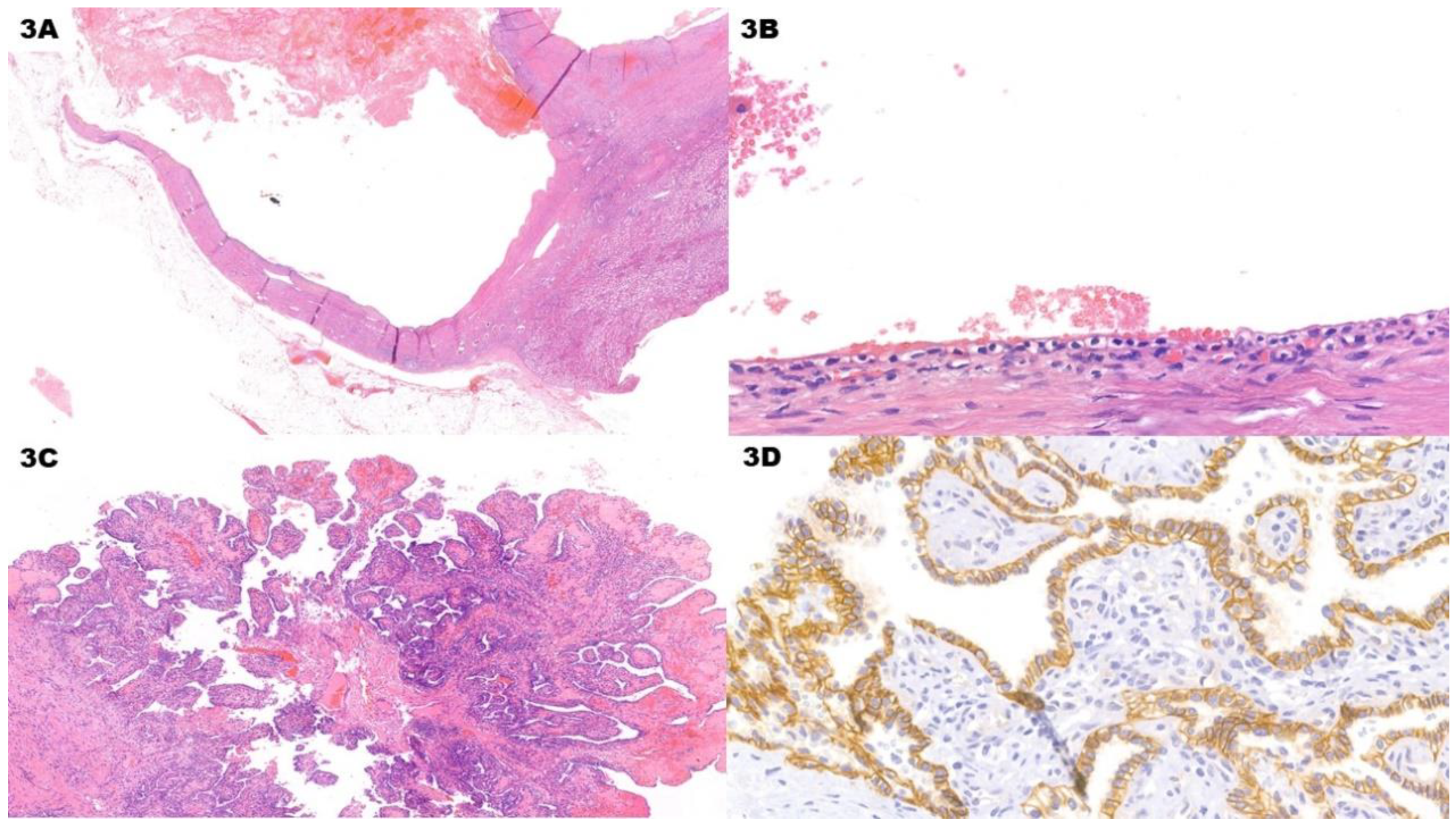
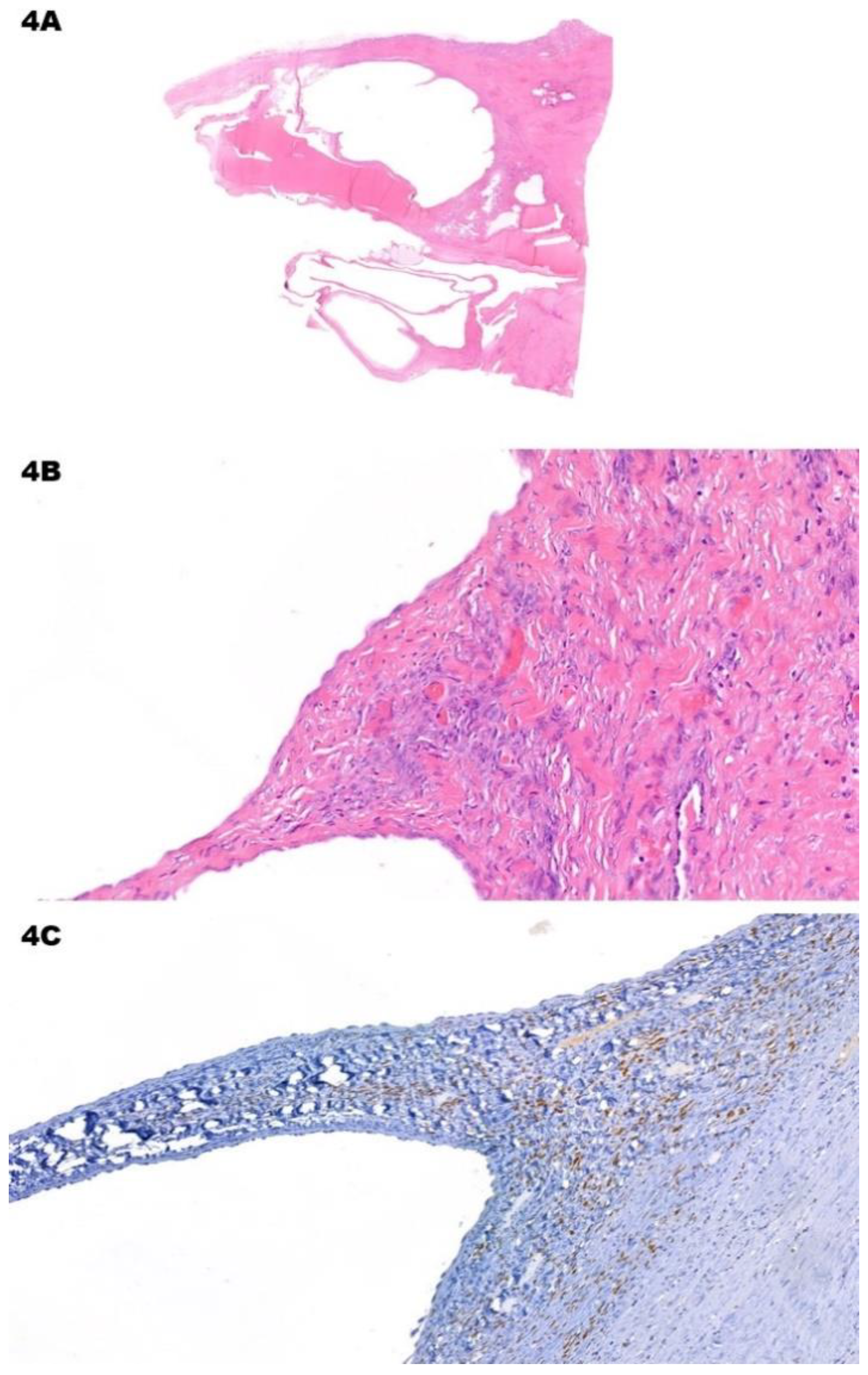
Figure 4.
Mixed Epithelial and Stromal Tumor. (A) (H&E) Low-power view shows multiple empty cystic areas with fibrotic walls. (B) (H&E, 10x) The epithelial lining is flat and bland, while the peri-cystic stroma has foci with higher cellularity. Stromal cells are spindled and bland. These foci can be focal and hard to find. (C) (Estrogen Receptor, 40x) Immunohistochemistry for estrogen receptor is positive in stromal cells. There are also reactive for progesterone receptors and inhibin.
Figure 4.
Mixed Epithelial and Stromal Tumor. (A) (H&E) Low-power view shows multiple empty cystic areas with fibrotic walls. (B) (H&E, 10x) The epithelial lining is flat and bland, while the peri-cystic stroma has foci with higher cellularity. Stromal cells are spindled and bland. These foci can be focal and hard to find. (C) (Estrogen Receptor, 40x) Immunohistochemistry for estrogen receptor is positive in stromal cells. There are also reactive for progesterone receptors and inhibin.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



