
Submitted:
31 May 2023
Posted:
02 June 2023
You are already at the latest version
Preprints on COVID-19 and SARS-CoV-2
Abstract
The risks for complications of severe acute respiratory syndrome coronavirus 2 (SARS-CoV2) infection are higher in obese individuals. Obesity is a state of chronic low-grade inflammation, with high leptin levels due to leptin resistance, high basal levels of other pro-inflammatory cytokines such as TNF-alpha, MCP-I and IL-6, and low adiponectin levels, thus contributing to a state of defective innate immunity as well as impaired B and T cell responses. Obesity is a risk factor for metabolic syndrome, diabetes, cardiovascular disease and hypertension. It has been observed that pre-existence of these diseases confers a higher risk of severe SARS CoV2 infection as well as the need for intensive care; even below the age of 60 years if their body mass index (BMI) is greater than 30 kg/m2, and even more so if it is > 35 kg/m2.
The metabolic factors contributing to the changes in altering the immune mechanisms in obese individuals and how this enhances the susceptibility to infection and development of serious SARS-CoV2 infection have been the subject of many debates. Future development of targeted therapy and guidelines will be benefited by greater understanding into these metabolic pathways.
Keywords:
immunity
; obesity
; insulin resistance
; innate and adaptive
; treg
; glutathione
; cytokine storm
1. Introduction
The severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), initially reported in November 2019 in Wuhan, China, has now claimed over 300,000 lives worldwide [1] and devastated the global economy.
Historically, there is evidence that previous epidemics of influenza (H1N1)[2] and influenza like illnesses have been linked to obesity as one of the risk factors.
Obesity and its related co morbid illnesses such as diabetes, cardiovascular disease and hypertension[3] and pre-existence of diseases of the lungs and kidneys; have emerged as one of the strongest risk factors for poor outcomes, especially in those individuals less than 55-60 years of age. With the World Health Organization estimating that more than 1.9 billion adults worldwide have overweight or obesity[4] any causal relationship or association between obesity and severe disease from SARS-CoV-2 has the potential to claim even more lives globally.
A meta-analysis by Ni Y et al. reported the protective factor of obesity in acute respiratory distress syndrome (ARDS) patients[5] which suggests counterintuitively that people with overweight and obesity may have a better prognosis than those with BMI values in the normal range. Since ARDS is one of the devastating clinical manifestations of COVID-19[5], this “obesity paradox” raises doubts on the impact of obesity in the disease severity and prognosis of COVID-19.
Hence it is vital to understand the immune mechanisms in an obese individual and its relationship to manifestations of COVID-19.
2.1. COVID 19 and Obesity
A recent systematic review concluded that obesity is an independent risk factor for SARS-CoV-2[6]. One of the studies included in this review by Simonnet A et al., reported that the need for invasive mechanical ventilation increased with increasing BMI categories, being greatest with BMI>35 kg/m2 with more serious disease outcomes[7].
Seidu et al conducted a meta-analysis including 8 retrospective and one prospective study and found that excess adiposity is a risk factor for severe disease and mortality in people with SARS-CoV-2 infection. BMI ≥ 25 kg/m2 was associated with an increased risk of severe illness in older age groups (≥60 years), whereas the association was weaker in younger age groups (<60 years)[8] even in Asian populations after accounting for confounders such as diabetes and hypertension[9,10].
Petrilli CM et al. studied 5279 people with COVID-19 in New York City in a prospective cohort study to determine factors associated with hospital admission and critical illness. For both variables, BMI>40 kg/m2 considerably increased the odds, besides older age and other co-morbidities[11].
Thus, in most of the studies when the two variables of age and BMI are studied with respect to severity of COVID-19 infection, there is evidence that the risk of severity and of needing intensive care is significantly increased in patients below the age of 60 years if their BMI is greater than 30 kg/m2, and even more so if it is > 35 kg/m2.
However, in a retrospective study conducted at a New York City hospital in a cohort of 3615 patients who had tested positive for the corona virus; 37% patients had a BMI>30 kg/m2. At a BMI>35 kg/m2, patients under the age of 60 years had 3.6 times higher risk of needing ICU admission[12]. Similarly, in a recent analysis of 265 patients with COVID-19, Kass et al. described a significant inverse correlation between age and BMI, in which younger individuals admitted to hospital were more likely to be obese[13]. Is it then possible that the presence of obesity seems to mitigate the protection conferred by younger age? And how can we explain these findings.
2.2. Immune Function in Obesity
There are growing pieces of evidence that support a strong relationship between adipose tissue and the immune system. For instance, white adipocytes and immune cells share the same embryonic origin, and a large number of macrophages and lymphocytes can be found in adipose tissus[16,17]. Furthermore, another study suggested that adipose tissue could be a site for the formation and maturation of immune cell precursors[18]. In general, individuals with obesity are observed with significantly high proliferation and activation of both innate and adaptive immune cells[17]. This phenomenon results from the disruption of adipocyte secreted signalling molecules called adipokines. These adipokines are classified into two groups, pro-inflammatory and anti-inflammatory. The pro-inflammatory adipokines consist of leptin, resistin, and ANGPTL2 and are primarily secreted by adipocytes in obese state[19].Conversely, the anti-inflammatory adipokines are adiponectin and adipsin, which are primarily secreted by adipocytes in the lean state[19]. Among the pro-inflammatory adipokines, leptin is extensively studied and mostly correlated to immune dysfunction involving both innate and adaptive immune cells in obesity.
Brotfain et al. found a persistent and significant elevation in chemotactic, random migration, and basal superoxide production of neutrophils in people with obesity. Intriguingly, these chronically activated neutrophils did not provide a superior phagocytosis and adherence ability comparing to that of lean individuals[20]. Furthermore, neutrophil survival is significantly enhanced in the presence of leptin due to its interference with various apoptotic mechanisms. Leptin delays the Bid and Bax’s cleavage, mitochondrial release of cytochrome C, the second mitochondria-derived activator of caspase, and inhibits the activation of caspase-3 and caspase-8[21] and enhances other anti-apoptotic pathways such as PI3K, NF-B, and MAPK[21].
Surmi et al. had found a large number of monocytes and differentiated macrophages in adipose tissues owing to a high number of macrophage attractant adipocyte-secreted cytokines, particularly leukotriene B3 (LTB3), macrophage inflammatory proteins (MIP), macrophage migration inhibitory factor (MIF), and monocyte chemotactic protein-3 (MCP30[22]. Once residing inside adipose tissues, these macrophages undergo M1-phenotype transformation, resulting in inflammatory mediators’ production, specifically nitric oxide, TNF-, and IL-1[23]. Oddly, some others have found that leptin induces macrophages to undergo M2-phenotype transformation but secrete M1-typical cytokines such as TNF-, IL-6, IL-1, IL-1R, IL-10, MCP-1, and MIP1-[24,25] to favour an inflammatory response.
Mattioli et al. found that leptin promotes the survival of dendritic cells (DCs)by modulating anti-apoptotic pathways, namely NF-B and PI3K-Akt, combined with the enhancement of Bcl-2 and Bcl-xL gene expression[26]. It also serves as an activator of DCs by upregulation of IL-1, IL-6, IL-12, TNF-, and MIP-1 production[27,28] and the converse is seen in the leptin-deficient mouse[29] resulting in a marked increase in the number of inflammatory DCs in adipose tissues culminating in a persistent inflammatory environment[30].
Leptin receptors are expressed on the cell surface of eosinophils and basophils, which speculates the leptin’s inflammatory roles on these two cell lines[31.32]. Wong et al. observed that leptin could improve eosinophil survival by activating anti-apoptotic pathways JAK, NF-B, and p38 MAPK33and promoting the release of eosinophil pro-inflammatory cytokines, primarily IL-1 and IL-6, and chemokines IL-8, MCP-1, and growth-related oncogene-. Further, it induced expression of adhesion molecules such as ICAM-1 and CD18. As a result, chronic hyperleptinemia over-intensified eosinophils functions and allergic responses[33]. Furthermore, Suzukawa et al. had noted significantly prolonged survivability of basophil in the presence of leptin, which was thought to involve similar anti-apoptotic pathways, as described in neutrophil and eosinophils[32]. Similarly, leptin promotes basophil production and secretion of type 2 cytokines such as IL-4 and IL-13, primarily involving IgE production and activation and allergic responses[32].
Adipocytes in individuals with obesity also regulate the proliferation of natural killer (NK) cells and the production of IFN- by upregulating ligands from NK-cell activating receptor 1[34] as causing the aforementioned M1-phenotype transformation of macrophages and inflammation within adipose tissues[34]. Additionally, a study had found that continuously fed high-fat diet mice had a significant elevation of numbers of NK cells and pro-inflammatory cytokines[35]. Congruently, Lo et al. had found that leptin markedly promotes the survival of immature NK cells from wild-type mice by induction of anti-apoptotic genes, Bcl2, while curtailing the pro-apoptotic genes, Bax[36]. Moreover, it enhances NK cell cytotoxicity through STAT3 activation and consequently upregulates the transcription of genes encoding IL-2 and perforin[37]. Intriguingly, leptin serves a differential function with the augmentation of NK cells in short-term stimulation and suppression of NK cell proliferation in chronic stimulation[38,39,40]. Therefore, chronic hyperleptinemia blunts the effects of NK cells in host cell immunity.
Obesity also negatively impacts the normal functions of multiple subsets of T cells despite the presence of leptin as positive effector. There is growing evidence supporting leptin’s function in improving T cells proliferation and survival in culture[41,42]. Surprisingly, Xia et al. and Martin-Romero et al. found a significant reduction in naïve CD4+ T cells with a shift of T helper cells toward pro-inflammatory CD4+ T cell subsets, particularly Th17, Th22, and Th1[43,44]. Also, the number of memory T cells in white adipose tissue of mice fed with a high-fat diet was significantly elevated[45]. Nevertheless, these memory T cells are aberrant and widely different in terms of RNA and protein expression than healthy memory T cells in the spleen[45]. Consequently, they do not confer equal immunity against foreign pathogens, but rather become harmful when activated due to massive release of lipids[45]. Another type of T-cell that is adversely impacted by the presence of chronic hyperleptinemia is the regulatory T cell, Treg[46]. Treg is accountable for anti-inflammatory response and self-tolerance; thus, it tightly regulates inflammatory response[47]. Compared to lean individuals, individuals with obesity have a depleted number of Treg. As a result, the pro-inflammatory response is favored in the obese state[48]. This finding is also valid in a murine model study, in which obese mice had higher numbers of cytotoxic CD8+ T cells with fewer suppressive Treg; hence, more significant lung inflammation and damage were observed after influenza virus exposure[49,50].
B-lymphocytes survival is greatly enhanced in the presence of chronic hyperleptinemia through inhibition of apoptotic pathways and induction of cell cycle entry via Bcl2 and cyclin D2 activation[51]. Furthermore, leptin is thought to activate peripheral B cells in a dose-dependent manner resulting in the secretion of pro-inflammatory cytokines such as TNF- and IL-6 and the anti-inflammatory cytokine IL-10 via activation of JAK-STAT and p38MAPK-ERK/12 signaling pathway[52]. Correspondingly, DeFuria et al. had found that B cells from obese mice secrete cytokines that promote the pro-inflammatory state by significantly increasing the secretion of IL-6 and INF- and impeding the secretion of anti-inflammatory IL-10. However, this inflammatory cytokine profile does not confer appropriate immunity[53]. Collectively, chronic hyperleptinemia disrupts B lymphocyte function by persistently inducing its secretion of pro-inflammatory cytokines without any additional benefit.
2.3. Covid19- the virus and how it affects immunity:
COVID19, also known as SARS-CoV-2, belongs to the family of Coronaviridae and subfamily of betacoronaviruses[54]. Like other viruses within the subfamily, COVID19 is a large-enveloped virus that comprises a single-stranded, non-segmented, positive-sense RNA genome[55]. COVID19 membrane consists of four essential structural elements, namely the spike (S), envelope (E), membrane (M), and nucleocapsid (N) protein[55]. Among the structural elements, the S glycoprotein is the single one that has the highest mutation rate[56].
Since both COVID19 and SARS-CoV are in the same subfamily, have the same origin in bats, and share 79.5% of their genetic sequencing, COVID19 likely behaves similarly to that of SARS-CoV in terms of transmission route, pathogenesis, and clinical symptoms[57]. They utilize their S glycoprotein to bind to host cell angiotensin-converting enzyme-2 (ACE2) to gain entry into host cells[58]. ACE2 protein is abundantly expressed on the surface of type 2 pneumocytes, which are responsible for producing surfactant and capable of differentiation into type 1 pneumocytes[59]. Furthermore, ACE2 is found on the surface of intestinal epithelial cells[60], upper respiratory tract, nasal mucosa, vascular endothelium, heart, and kidney[58,61]. Noteworthily, the S protein of COVID19 has a higher affinity for ACE2 than that of SARS-CoV, which accounts for its greater infectivity[62,63,64]. Additionally, COVID19 is thought to utilize CD147 to facilitate host cell invasion[65]. However, its detailed mechanism is yet to be elucidated.
Once the S protein binds to ACE2, the viral particle will be taken up into the cells via endocytosis[66]. Subsequently, the host lysosomal enzymes will degrade viral lipid bilayer and release viral RNA into the cytosol[66,67]. Then, the virus hijacks host RNA-dependent RNA polymerase to replicate, ribosomes to translate viral mRNA into viral polyproteins, and proteinases to cleave viral polyproteins into individual functional proteins[67,68]. The functional proteins will be assembled and packed in the ER-Golgi intermediate compartment[69]. Finally, the newly made viral particles will lyse the infected cells and be released into the alveolus[70](Figure 1).
Figure 1.
SAR-CoV-2 invasion mechanism of the host cell.
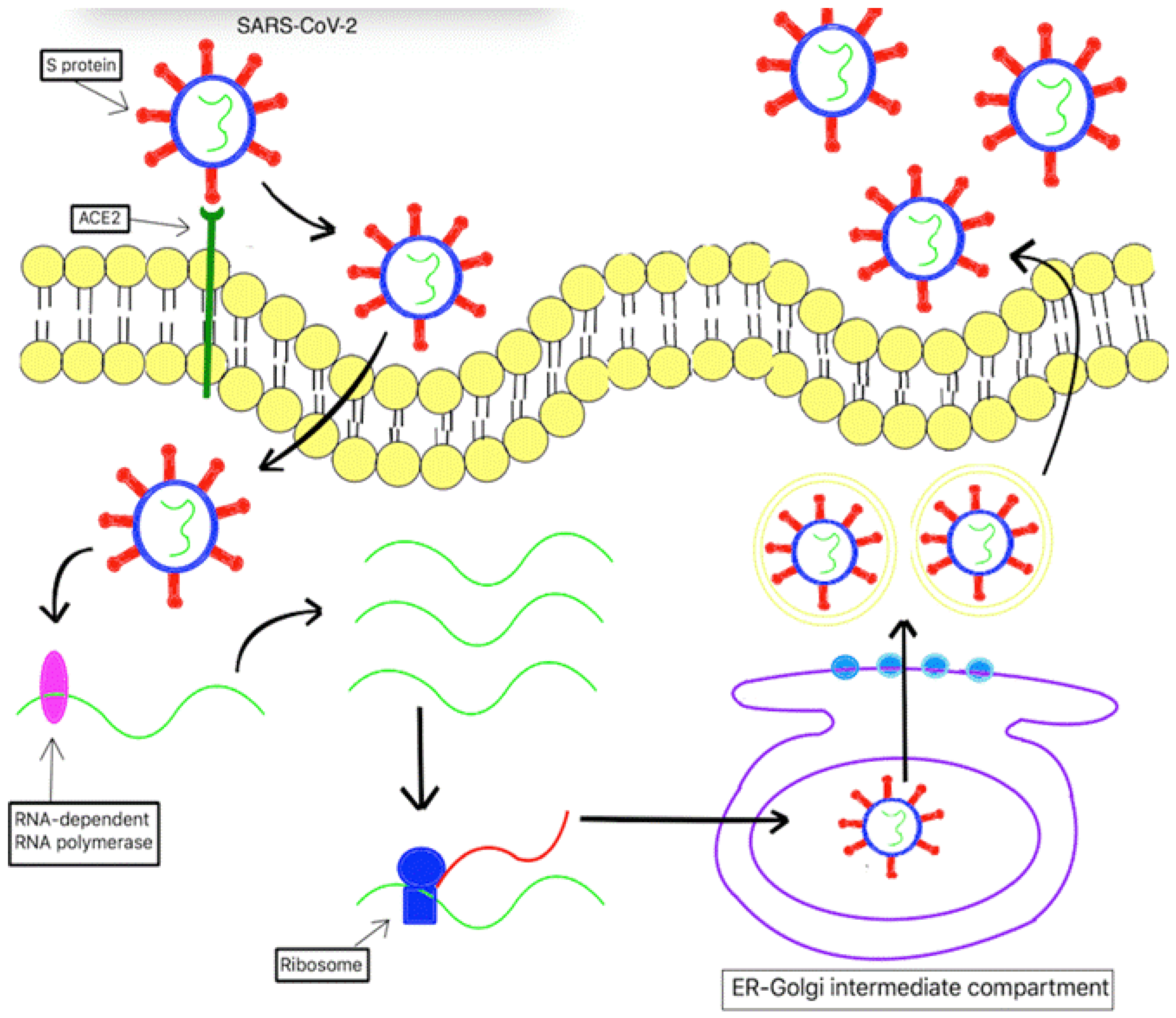
The S protein of SARS-CoV-2 binds to angiotensin-converting enzyme 2 (ACE2), a transmembrane protein found in the host cell. Subsequently, it results in endocytosis of SARS-CoV-2 into the host cell. Once located inside the host, SARS-CoV-2 uses host lysosomal enzymes to degrade its lipid bilayer, thereby releasing its viral RNA into the cytosol. Afterward, the viral RNA is replicated and translated using host RNA-dependent RNA polymerase and ribosomes, respectively. The newly produced viral polyproteins are cleaved into functional proteins. The functional proteins are assembled and packed in the ER-Golgi intermediate compartment. Finally, the newly formed viral particles lyse the cell and are releasing into the extracellular space.
3. The Cytokine Storm
There are several defence mechanisms the host implicates to clear a viral infection. During the acute phase of infection, innate cells will be activated using Pattern Recognition Receptors (PRRs), which are responsible for recognizing highly conserved molecular structures known as Pathogen-associated molecular patterns (PAMPS) present on microbes[71]. PRRs consists of many receptor families, namely toll-like receptors (TLRs), nucleotide-binding oligomerization (NOD)-like receptors (NLRs), the retinoic acid-inducible gene I(RIG-I), RIG-I-like receptors, the V-type lectin receptors, the absent in melanoma 2(AIM2)-like receptors, and the OAS-like receptors[71]. However, among the PRRs, only cytoplasmic RNA sensors, which are endosomal TLR-3 and -7, RIG-I, and melanoma differentiation-associated protein 5 (MDA5), can recognize RNAs viruse[72,73]. Once activated, the aforementioned cytoplasmic RNA sensors will recruit their respective adaptor proteins culminating in activation and translocation of nuclear transcription factors NF-B and IRF3[74,75,76,77]. Eventually, NF-B will upregulate Type 1 IFNs, namely IFN- and IFN-, while IRF3 stimulates the generation of other pro-inflammatory cytokines, like IL-1, IL-6, and TNF-[72,78].
COVID19 can overstimulate innate immune cells yielding remarkably high levels of pro-inflammatory cytokines including IL-2, IL-6, IL-7, IL-10, G-CSF, IP-10, MCP-1, MIP-1, and TNF-, resulting in “cytokine storm”79. This “cytokine storm” accelerates disease progression to severe form with acute respiratory distress syndrome (ARDS) and multi-organ failure[79,80]. During a later stage, the activated innate cells and their inflammatory cytokines stimulate adaptive immune cells, particularly CD4+ T cells, CD8+ T cells, and B cells with their antibody production to participate in anti-viral response[73].
Due to the similarity with SARS-CoV, it was speculated that COVID19 had evolved similar strategies to circumvent host cell immune response[81]1. It was found that SARS-CoV alters its RNA components to reduce its viral RNA affinity to host MDA5[82,83]. Additionally, SARS-CoV utilizes papain-like proteases to degrade proteins associate with IFN-production pathways; thereby, significantly diminishing type 1 IFNs production[84,85]. SARS-CoV also interferes with IFN signaling cascade through inhibiting STAT1 and preventing its translocation into the nucleus[86]. Nonetheless, how COVID19 evades type I IFN response remains unknown. Interestingly, one study had found that COVID19 has a greater sensitivity to type I IFN than SARS-CoV in vitro, and COVID19 does not inhibit STAT1 protein as other studies speculated[87], High type I IFN response correlates with mild-to-moderate disease and low type I IFN response correspond to severe disease[88].
There is growing evidence to support that COVID19 infection can suppress the adaptive immune response by directly and indirectly destroying all subsets of T cell and B cells; hence, resulting in a lymphopenia state[88,89,90,91,92]. Of note, higher non-functional CD4+T cells and lower functional CD8+T cells were found in patients with severe form[93]. Wang et al. had found that COVID19 could directly destroy T cells by infecting them[94] which results in termination of the COVID19 cycle and induction of cell death[95,96]. Furthermore, the aforementioned “cytokine storm” state can negatively impact T cells. For instance, TNF-alpha can induce T cell apoptosis[97], and IL-10 is known to prevent T cell proliferation[98]. Therefore, more future studies need to be directed on the detailed mechanism of how COVID19 evades host cell defence responses.
4. Discussion
Obesity is a state of chronic low-grade inflammation, with high leptin levels due to leptin resistance, high basal levels of other pro-inflammatory cytokines such as TNF-alpha, MCP-I and IL-6, and low adiponectin levels, thus contributing to a state of defective innate immunity as well as impaired B and T cell responses.
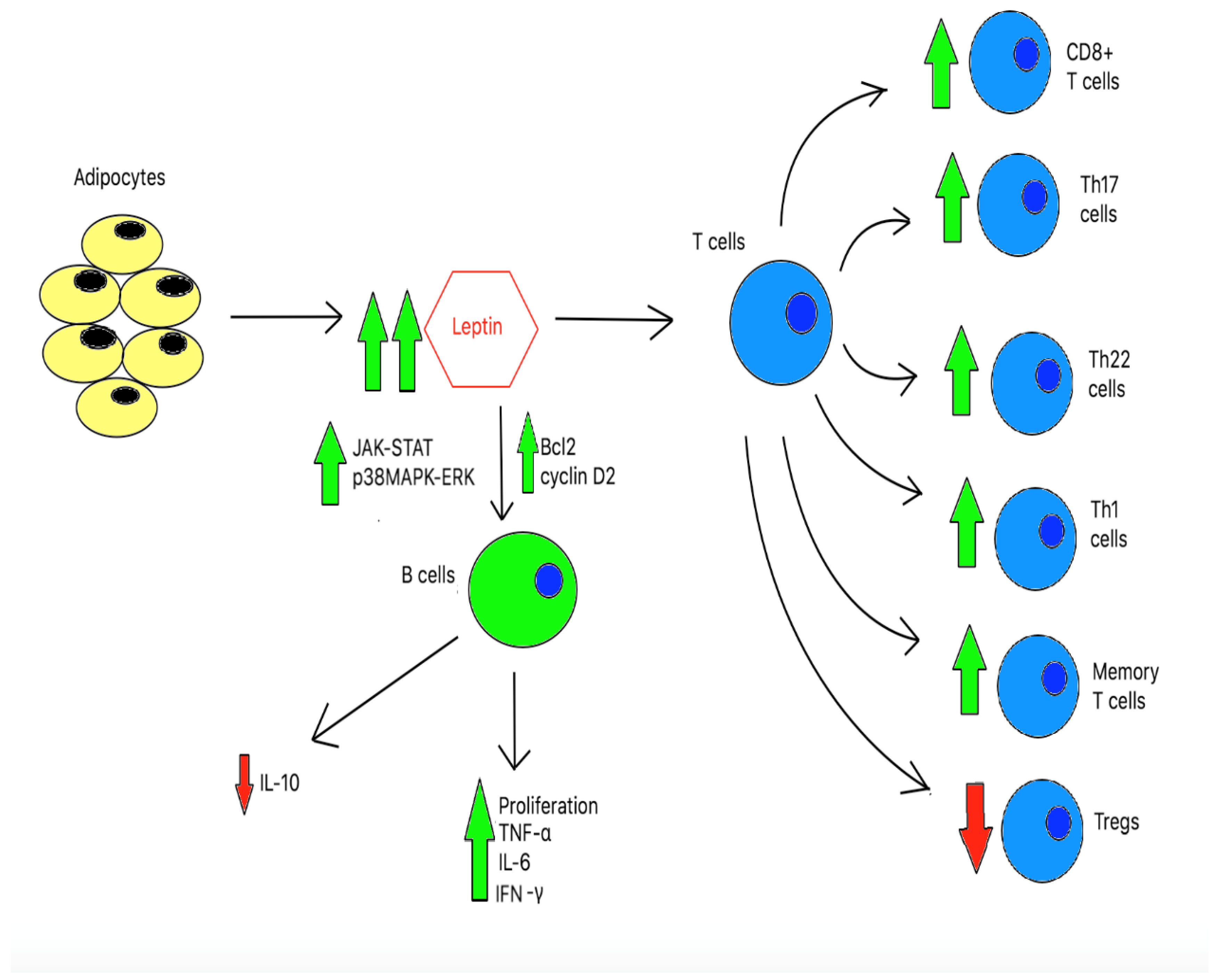
Obesity leads to a chronic hyper-inflammatory response in both innate and adaptive immune cells, as shown in Figure 2a and Figure 2b, respectively. However, there is no evidence supporting any benefit in immune function in the chronic pro-inflammatory state. In fact, studies have found that obesity adversely alters the architecture and integrity in lymphoid tissues[99] and hastens the aging process of the immune system rendering host cells vulnerable to foreign pathogens invasion[100]. Of note, recent studies found that obesity leads to inadequate vaccination responses despite being in the pro-inflammatory state[101,102].
Figure 2.
a. The effects of chronic hyperleptinemia and SARS-CoV-2 in innate immune cells.
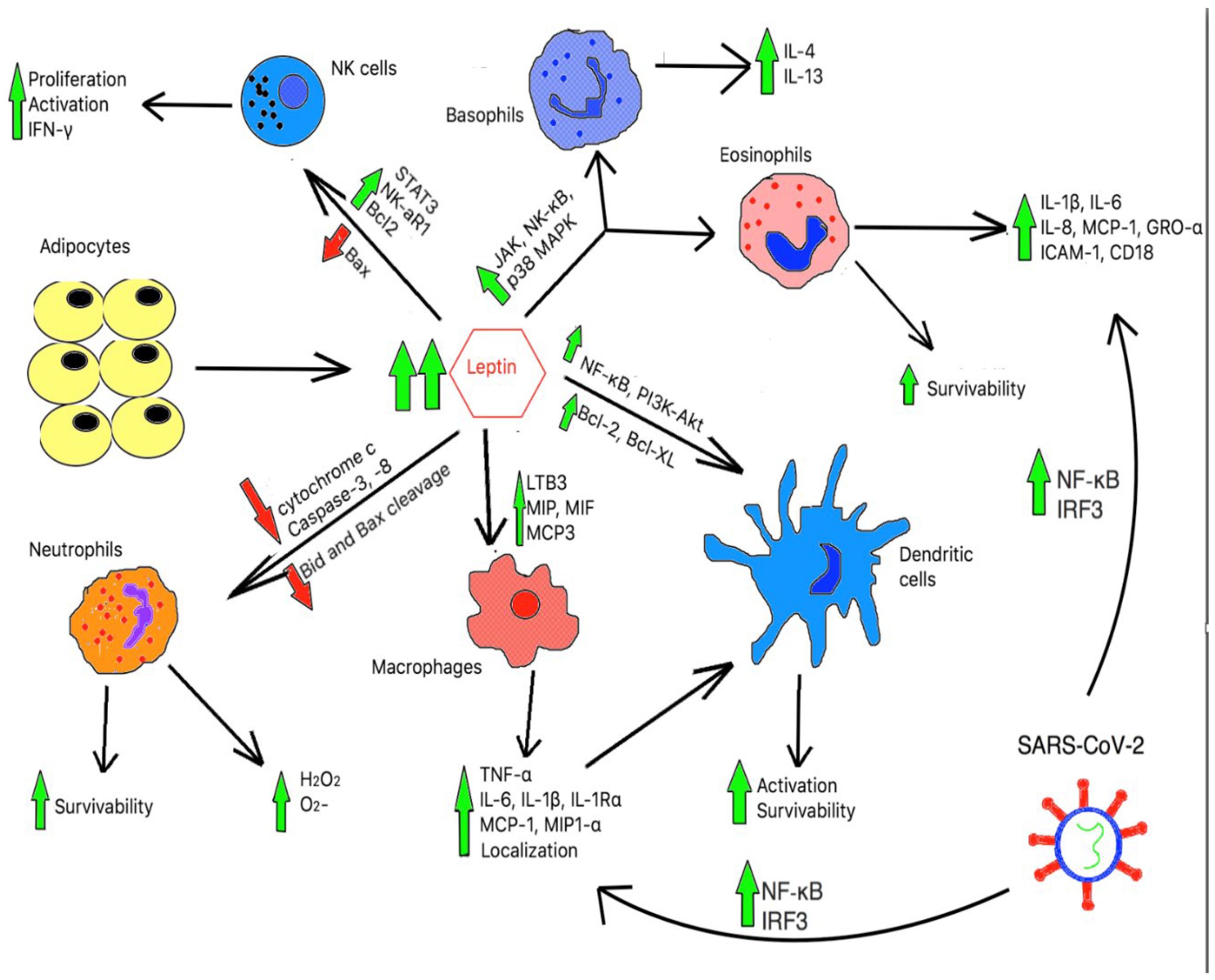
Figure 2.
b: The effects of chronic hyperleptinemia in adaptive immune cells.
High level of leptin can downregulate pro-apoptotic proteins namely, cytochrome c, caspase-3, and caspase-8, and impair cleavage of pro-apoptotic Bid and Bax proteins. As a result, it enhances neutrophils’ survivability and basal superoxide production. In addition, high hyperleptinemia upregulates LTB3, MIP, MIF, and MCP3 molecules, thereby resulting in macrophage localization and activation. In conjunction with cytokines secreted from activated macrophage, a high leptin level also supports multiple anti-apoptotic pathways in dendritic cells (DCs). Together, it enhances DCs activation and survivability. Furthermore, through the induction of JAK, NF-𝜅B, and p36 MAPK pathways, leptin induces activation of basophils and eosinophils. Finally, through the modulations of the anti-apoptotic protein, Bax, and pro-apoptotic proteins Bcl2, leptin can stimulate NK cell proliferation and survivability. Leptin also promotes NK cells activation and IFN-𝛾 production through the upregulation of NK-cell activating receptor 1 (NK-aR1) and STAT3 pathway. SARS-CoV-2, through NF-𝜅B and IRF3, induce a “cytokine storm” state with significant number of IL-2, IL-6, IL-7, G-CSF, IP-10, MCP-10, MCP-1, MIP-1, and TNF-
Hyperleptinemia induces T-cell proliferation and differentiation. As a result, it leads to an increase in numbers of aberrant pro-inflammatory T cells, particularly CD8+ T cells, Th17 cells, Th22 cells, Th1 cells, and memory T cells. Additionally, leptin suppresses the anti-inflammatory Tregs. Chronic elevation of leptin also induces anti-apoptotic proteins, specifically Bcl2 and cyclin D2. Furthermore, hyperleptinemia activates JAK-STAT and p38MAPK-ERK pathways. As a result, it leads to B-cell proliferation and activation.
It has been demonstrated that manipulating various components of the innate and/or adaptive immune system induces or ameliorates not only inflammation but also systemic insulin resistance and/or glucose intolerance. Multiple cytokines secreted by activated immune cells have been shown to impair insulin signaling via stimulation of stress kinases including IKKB and JNK1. In addition to adipose tissue, recent evidence suggests that the intestine is also a key site that becomes altered in obesity-related insulin resistance. These alterations include changes in the gastrointestinal flora, known as dysbiosis, which can impact body fat, systemic inflammation, and insulin resistance[103,104,105].
There is also data to suggest that obese subjects may be more contagious than lean subjects. Firstly, they may shed the virus longer[106]. Secondly, obesity as elucidated above can modify innate and adaptive immune responses, allowing the virus to replicate into a more virulent strain; thus, making the immune system more vulnerable to severe infections and less responsive to vaccinations, antivirals and antimicrobial drugs[107]. This has important implications for the past and any future pandemic to guide treatment of “at risk” populations. This state of low-grade chronic inflammation seen in obesity can also contribute to the onset of metabolic diseases (dyslipidemia, insulin resistance and T2DM).
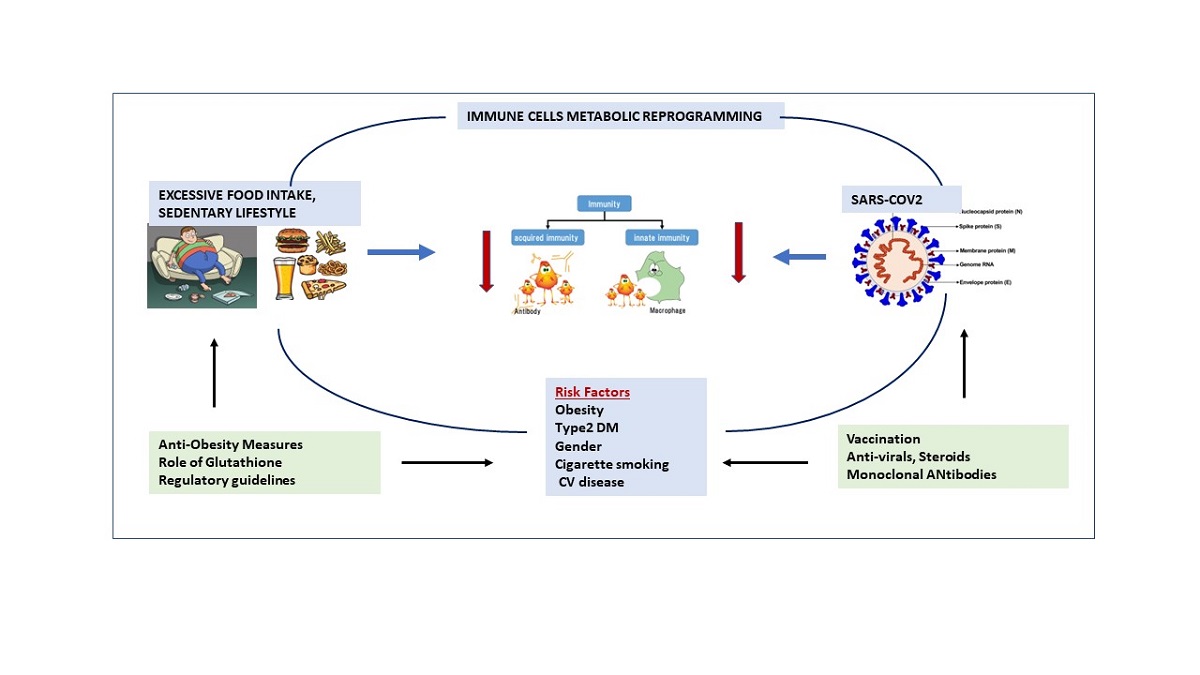
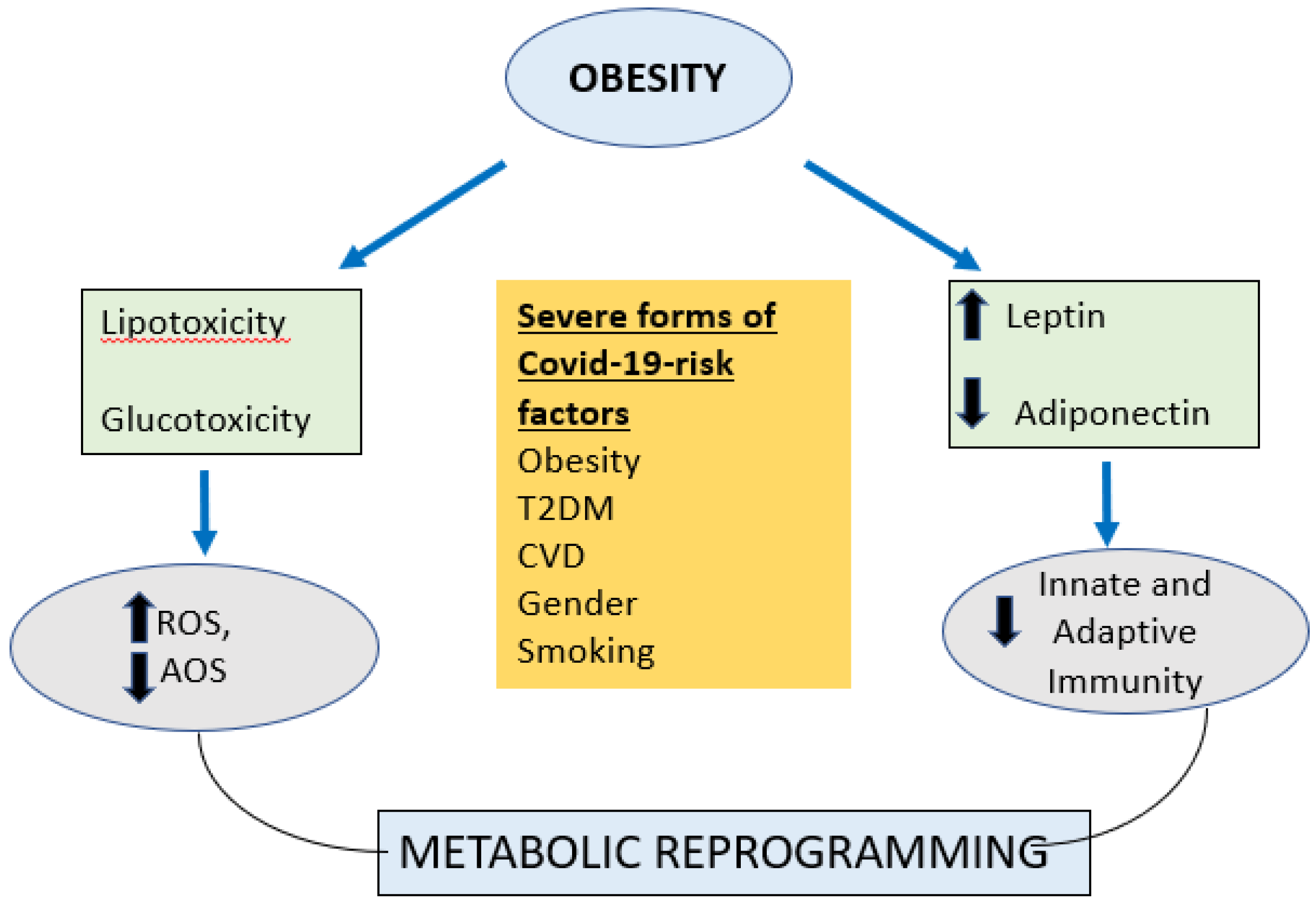
Obesity in addition to being the foremost risk factor for T2DM, cardiovascular disease, fatty liver, and hypertension all of which have insulin resistance as the common underlying mechanism; has also been linked to an increased oxidative stress [108]. Obesity is a syndrome of overnutrition and substrate excess leading to glucotoxicity and lipotoxicity. When excess glucose and lipids are presented to the cells; it leads to over production of reactive oxygen spices (ROS)which overwhelms the antioxidant systems and leads to oxidative stress [109] (see figure 3).. This elevated production of ROS in obesity is related to activation of the innate immune system in adipose tissue and subsequent low-grade chronic systemic inflammation [109].
Figure 3.
Immune Metabolic Reprogramming due to Obesity.(ROS=Reactive oxygen species, AOS=Antioxidant system, T2DM =type2 diabetes mellitus, CVD=cardiovascular disease) Obesity leads to lipotoxicity and glucotoxicity which leads to increased Reactive oxygen species (ROS) and reduced anti-oxidant systems (AOS); also leads to increased leptin and reduced adiponectin which adversely affects both innate and adaptive immunity which leads to metabolic reprogramming and severe forms of covid-19.
Figure 3.
Immune Metabolic Reprogramming due to Obesity.(ROS=Reactive oxygen species, AOS=Antioxidant system, T2DM =type2 diabetes mellitus, CVD=cardiovascular disease) Obesity leads to lipotoxicity and glucotoxicity which leads to increased Reactive oxygen species (ROS) and reduced anti-oxidant systems (AOS); also leads to increased leptin and reduced adiponectin which adversely affects both innate and adaptive immunity which leads to metabolic reprogramming and severe forms of covid-19.
Current therapeutic options that address COVID-19 illness are vaccination, monoclonal antibodies, antivirals, directed immunomodulators and corticosteroids such as dexamethasone. The goal is to decrease the viral load or address the pathophysiology of cytokine dysregulation (110). Despite their successes, each has its limitations. Vaccines have demonstrated remarkable efficacy in reducing deaths and hospitalizations; however, they are less effective in immunocompromised individuals and against new SARSCoV-2 variants [111]. Furthermore, we may have reached the limit of vaccinating the general populace given the resistance, reluctance, or contraindications to receiving vaccinations of many individuals.
Hence, nutritional interventions, caloric restriction, physical activity, and other newer emerging therapies may emerge as a cornerstone for prevention of COVID or future viral infections especially in obese/immunocompromised individuals; in addition to the strategies mentioned above. Recently there has been interest in using glutathione (GSH) to manage COVID 19 especially to limit severity of the disease [112]. As our master antioxidant, GSH plays an integral role in maintaining our health. GSH deficiency is present in individuals with co-morbidities that are associated with greater severity of COVID-19 illness such as advanced age, hypertension, diabetes, ischemic heart disease, and obesity [113]. GSH seems to offer protection against the cytokine storm. This is achieved by its action as a cellular reducing substrate to modulate the unchecked cytokine cascade that results in multi-organ damage through direct tissue injury and a hypercoagulable state [114]. GSH both modulates the inflammatory response and augments innate adaptive cellular immunity favorable to the host [115]; and thus, should be used early in SARS-CoV2 infection to prevent the deadly complications associated with it.
5. CONCLUSIONS
Obesity is tightly linked to insulin resistance and oxidative stress. This underlying metabolic milieu leads to a reprogramming of the immune mechanisms predisposing the individual to an increased susceptibility to infection and also increased severity of the same. The eventual control or resolution of pandemics such as the current COVID-19 pandemic will require a greater understanding of these metabolic perturbations that occur at the host level. In order to contain or prevent future pandemics, we need to develop better treatments for reducing severity of the illness, long term sequelae and the development of new variants. Reducing the severity is addressed with vaccines, and antivirals. Early mitigation of a maladaptive immune response is where agents such as glutathione may be promising. Additionally, management of obesity, diabetes and underlying metabolic diseases more effectively will be the cornerstone of future therapies.
Author Contributions
RJM, HN ND ; writing—original draft preparation , VV; writing—review All authors have read and agreed to the published version of the manuscript.”.
Funding
We appreciate the funding support from National Institutes of Health (NIH) award [RHL143545-01A1].
References
- Coronavirus disease (COVID-19). Available at: https://www.who.int/docs/default-source/coronaviruse/situation-reports/20201012-weekly-epi-update-9.pdf. (Accessed: 14th October 2020).
- Morgan, O. W. et al. Morbid Obesity as a Risk Factor for Hospitalization and Death Due to 2009 Pandemic Influenza A(H1N1) Disease. PLoS ONE 5, (2010). [CrossRef]
- Gupta, R., Ghosh, A., Singh, A. K. & Misra, A. Clinical considerations for patients with diabetes in times of COVID-19 epidemic. Diabetes & Metabolic Syndrome: Clinical Research & Reviews 14, 211–212 (2020). [CrossRef]
- Obesity and overweight. World Health Organization Available at: https://www.who.int/news-room/fact-sheets/detail/obesity-and-overweight. (Accessed: 14th October 2020).
- Ni, Y.-N. et al. Can body mass index predict clinical outcomes for patients with acute lung injury/acute respiratory distress syndrome? A meta-analysis. Critical Care 21, (2017). [CrossRef]
- Tamara, A. & Tahapary, D. L. Obesity as a predictor for a poor prognosis of COVID-19: A systematic review. Diabetes & Metabolic Syndrome: Clinical Research & Reviews 14, 655–659 (2020). [CrossRef]
- Simonnet, A. et al. High Prevalence of Obesity in Severe Acute Respiratory Syndrome Coronavirus-2 (SARS-CoV-2) Requiring Invasive Mechanical Ventilation. Obesity 28, 1195–1199 (2020).
- Seidu, S. et al. The impact of obesity on severe disease and mortality in people with SARS-CoV-2: A systematic review and meta-analysis. Endocrinology, Diabetes & Metabolism (2020). doi:10.1002/edm2.176. [CrossRef]
- Cai, Q. et al. Obesity and COVID-19 Severity in a Designated Hospital in Shenzhen, China. Diabetes Care 43, 1392–1398 (2020). [CrossRef]
- Gao, F. et al. Obesity Is a Risk Factor for Greater COVID-19 Severity. Diabetes Care 43, (2020). [CrossRef]
- Petrilli, C. M. et al. Factors associated with hospital admission and critical illness among 5279 people with coronavirus disease 2019 in New York City: prospective cohort study. Bmj m1966 (2020). doi:10.1136/bmj.m1966. [CrossRef]
- Lighter, J. et al. Obesity in Patients Younger Than 60 Years Is a Risk Factor for COVID-19 Hospital Admission. Clinical Infectious Diseases 71, 896–897 (2020). [CrossRef]
- Kass, D. A., Duggal, P. & Cingolani, O. Obesity could shift severe COVID-19 disease to younger ages. The Lancet 395, 1544–1545 (2020).
- Onder, G., Rezza, G. & Brusaferro, S. Case-Fatality Rate and Characteristics of Patients Dying in Relation to COVID-19 in Italy. Jama (2020). doi:10.1001/jama.2020.4683. [CrossRef]
- Hu, C. & Jia, W. Diabetes in China: Epidemiology and Genetic Risk Factors and Their Clinical Utility in Personalized Medication. Diabetes 67, 3–11 (2017). [CrossRef]
- Caspar-Bauguil, S. et al. Adipose tissues as an ancestral immune organ: Site-specific change in obesity. FEBS Letters 579, 3487–3492 (2005). [CrossRef]
- Saely, C. H., Geiger, K. & Drexel, H. Brown versus White Adipose Tissue: A Mini-Review. Gerontology 58, 15–23 (2012). [CrossRef]
- Poglio, S. et al. Adipose Tissue as a Dedicated Reservoir of Functional Mast Cell Progenitors. Stem Cells 28, 2065–2072 (2010). [CrossRef]
- Wensveen, F. M., Valentić, S., Šestan, M., Wensveen, T. T. & Polić, B. Interactions between adipose tissue and the immune system in health and malnutrition. Seminars in Immunology 27, 322–333 (2015).
- Brotfain, E. et al. Neutrophil functions in morbidly obese subjects. Clinical & Experimental Immunology 181, 156–163 (2015). [CrossRef]
- Bruno, A., Conus, S., Schmid, I. & Simon, H.-U. Apoptotic Pathways Are Inhibited by Leptin Receptor Activation in Neutrophils. The Journal of Immunology 174, 8090–8096 (2005). [CrossRef]
- Surmi, B. K. & Hasty, A. H. The role of chemokines in recruitment of immune cells to the artery wall and adipose tissue. Vascular Pharmacology 52, 27–36 (2010). [CrossRef]
- Torres-Castro, I. et al. Corrigendum to ‘Human monocytes and macrophages undergo M1-type inflammatory polarization in response to high levels of glucose’ [Immunol. Lett. 176 (2016) 81–89]. Immunology Letters 192, 106 (2017).
- Acedo, S. C., Gambero, S., Cunha, F. G. P., Lorand-Metze, I. & Gambero, A. Participation of leptin in the determination of the macrophage phenotype: an additional role in adipocyte and macrophage crosstalk. In Vitro Cellular & Developmental Biology - Animal 49, 473–478 (2013). [CrossRef]
- Santos-Alvarez, J., Goberna, R. & Sánchez-Margalet, V. Human Leptin Stimulates Proliferation and Activation of Human Circulating Monocytes. Cellular Immunology 194, 6–11 (1999). [CrossRef]
- Mattioli, B., Giordani, L., Quaranta, M. G. & Viora, M. Leptin exerts an anti-apoptotic effect on human dendritic cells via the PI3K-Akt signaling pathway. FEBS Letters 583, 1102–1106 (2009). [CrossRef]
- Mattioli, B. et al. Leptin as an immunological adjuvant: enhanced migratory and CD8 + T cell stimulatory capacity of human dendritic cells exposed to leptin. The FASEB Journal 22, 2012–2022 (2008). [CrossRef]
- Al-Hassi, H. O. et al. A mechanistic role for leptin in human dendritic cell migration: differences between ileum and colon in health and Crohn’s disease. Mucosal Immunology 6, 751–761 (2012). [CrossRef]
- Lam, Q. L. K., Liu, S., Cao, X. & Lu, L. Involvement of leptin signaling in the survival and maturation of bone marrow-derived dendritic cells. European Journal of Immunology 36, 3118–3130 (2006). [CrossRef]
- Bertola, A. et al. Identification of Adipose Tissue Dendritic Cells Correlated With Obesity-Associated Insulin-Resistance and Inducing Th17 Responses in Mice and Patients. Diabetes 61, 2238–2247 (2012). [CrossRef]
- Conus, S., Bruno, A. & Simon, H. Leptin is an eosinophil survival factor. Journal of Allergy and Clinical Immunology 116, 1228–1234 (2005).
- Suzukawa, M. et al. Leptin Enhances Survival and Induces Migration, Degranulation, and Cytokine Synthesis of Human Basophils. The Journal of Immunology 186, 5254–5260 (2011). [CrossRef]
- Wong, C. K., Cheung, P. F.-Y. & Lam, C. W. K. Leptin-mediated cytokine release and migration of eosinophils: Implications for immunopathophysiology of allergic inflammation. European Journal of Immunology 37, 2337–2348 (2007). [CrossRef]
- Wensveen, F. M. et al. NK cells link obesity-induced adipose stress to inflammation and insulin resistance. Nature Immunology 16, 376–385 (2015). [CrossRef]
- Lee, B.-C. et al. Adipose Natural Killer Cells Regulate Adipose Tissue Macrophages to Promote Insulin Resistance in Obesity. Cell Metabolism 23, 685–698 (2016). [CrossRef]
- Lo, C. K. C. et al. Leptin Signaling Protects NK Cells from Apoptosis During Development in Mouse Bone Marrow. Cellular & Molecular Immunology 6, 353–360 (2009). [CrossRef]
- Zhao, Y., Sun, R., You, L., Gao, C. & Tian, Z. Expression of leptin receptors and response to leptin stimulation of human natural killer cell lines. Biochemical and Biophysical Research Communications 300, 247–252 (2003). [CrossRef]
- Wrann, C. D. et al. Short-term and long-term leptin exposure differentially affect human natural killer cell immune functions. American Journal of Physiology-Endocrinology and Metabolism 302, (2012). [CrossRef]
- Laue, T. et al. Altered NK cell function in obese healthy humans. BMC Obesity 2, (2015). [CrossRef]
- Nave, H. et al. Resistance of Janus Kinase-2 Dependent Leptin Signaling in Natural Killer (NK) Cells: A Novel Mechanism of NK Cell Dysfunction in Diet-Induced Obesity. Endocrinology 149, 3370–3378 (2008). [CrossRef]
- Papathanassoglou, E. et al. Leptin Receptor Expression and Signaling in Lymphocytes: Kinetics During Lymphocyte Activation, Role in Lymphocyte Survival, and Response to High Fat Diet in Mice. The Journal of Immunology 176, 7745–7752 (2006). [CrossRef]
- Procaccini, C. et al. Leptin as immune mediator: Interaction between neuroendocrine and immune system. Developmental & Comparative Immunology 66, 120–129 (2017). [CrossRef]
- Xia, C., Rao, X. & Zhong, J. Role of T Lymphocytes in Type 2 Diabetes and Diabetes-Associated Inflammation. Journal of Diabetes Research 2017, 1–6 (2017). [CrossRef]
- Martín-Romero, C., Santos-Alvarez, J., Goberna, R. & Sánchez-Margalet, V. Human Leptin Enhances Activation and Proliferation of Human Circulating T Lymphocytes. Cellular Immunology 199, 15–24 (2000). [CrossRef]
- Misumi, I. et al. Obesity Expands a Distinct Population of T Cells in Adipose Tissue and Increases Vulnerability to Infection. Cell Reports 27, (2019). [CrossRef]
- Rosa, V. D. et al. A Key Role of Leptin in the Control of Regulatory T Cell Proliferation. Immunity 26, 241–255 (2007).
- Plitas, G. & Rudensky, A. Y. Regulatory T Cells: Differentiation and Function. Cancer Immunology Research 4, 721–725 (2016). [CrossRef]
- Kucharska, A. M., Pyrżak, B. & Demkow, U. Regulatory T Cells in Obesity. Advances in Experimental Medicine and Biology Noncommunicable Diseases 35–40 (2015). doi:10.1007/5584_2015_147. [CrossRef]
- Milner, J. J. et al. Diet-Induced Obese Mice Exhibit Altered Heterologous Immunity during a Secondary 2009 Pandemic H1N1 Infection. The Journal of Immunology 191, 2474–2485 (2013). [CrossRef]
- Milner, J. J. et al. Obesity Increases Mortality and Modulates the Lung Metabolome during Pandemic H1N1 Influenza Virus Infection in Mice. The Journal of Immunology 194, 4846–4859 (2015). [CrossRef]
- Lam, Q. L. K., Wang, S., Ko, O. K. H., Kincade, P. W. & Lu, L. Leptin signaling maintains B-cell homeostasis via induction of Bcl-2 and Cyclin D1. Proceedings of the National Academy of Sciences 107, 13812–13817 (2010). [CrossRef]
- Agrawal, S., Gollapudi, S., Su, H. & Gupta, S. Leptin Activates Human B Cells to Secrete TNF-α, IL-6, and IL-10 via JAK2/STAT3 and p38MAPK/ERK1/2 Signaling Pathway. Journal of Clinical Immunology 31, 472–478 (2011).
- Defuria, J. et al. B cells promote inflammation in obesity and type 2 diabetes through regulation of T-cell function and an inflammatory cytokine profile. Proceedings of the National Academy of Sciences 110, 5133–5138 (2013). [CrossRef]
- Chan, J. F.-W. et al. Genomic characterization of the 2019 novel human-pathogenic coronavirus isolated from a patient with atypical pneumonia after visiting Wuhan. Emerging Microbes & Infections 9, 221–236 (2020). [CrossRef]
- Fehr, A. R. & Perlman, S. Coronaviruses: An Overview of Their Replication and Pathogenesis. Coronaviruses Methods in Molecular Biology 1–23 (2015). doi:10.1007/978-1-4939-2438-7_1. [CrossRef]
- Yin, C. Genotyping coronavirus SARS-CoV-2: methods and implications. Genomics 112, 3588-3596 (2020).
- Shah, A., Kashyap, R., Tosh, P., Sampathkumar, P. & O’Horo, J. C. Guide to Understanding the 2019 Novel Coronavirus. Mayo Clinic Proceedings 95, 646–652 (2020). [CrossRef]
- Hoffmann, M. et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 181, (2020).
- Hamming, I. et al. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. The Journal of Pathology 203, 631–637 (2004). [CrossRef]
- Xu, H. et al. High expression of ACE2 receptor of 2019-nCoV on the epithelial cells of oral mucosa. International Journal of Oral Science 12, (2020). [CrossRef]
- Donoghue, M. et al. A Novel Angiotensin-Converting Enzyme–Related Carboxypeptidase (ACE2) Converts Angiotensin I to Angiotensin 1-9. Circulation Research 87, (2000). [CrossRef]
- Wrapp, D. et al. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 367, 1260–1263 (2020). [CrossRef]
- Wang, Q. et al. Structural and Functional Basis of SARS-CoV-2 Entry by Using Human ACE2. Cell 181, (2020).
- Rabaan, A. et al. SARS-CoV-2, SARS-CoV, and MERS-COV: A comparative overview. Infez Med 28, 174-184 (2020).
- Wang, K. et al. SARS-CoV-2 invades host cells via a novel route: CD147-spike protein. (2020). doi:10.1101/2020.03.14.988345. [CrossRef]
- Sawicki, S. G. & Sawicki, D. L. Coronavirus Transcription: A Perspective. Current Topics in Microbiology and Immunology Coronavirus Replication and Reverse Genetics 31–55 (2005). doi:10.1007/3-540-26765-4_2. [CrossRef]
- Hussain, S. et al. Identification of Novel Subgenomic RNAs and Noncanonical Transcription Initiation Signals of Severe Acute Respiratory Syndrome Coronavirus. Journal of Virology 79, 5288–5295 (2005). [CrossRef]
- Perrier, A. et al. The C-terminal domain of the MERS coronavirus M protein contains a trans-Golgi network localization signal. Journal of Biological Chemistry 294, 14406–14421 (2019). [CrossRef]
- Tang, T., Bidon, M., Jaimes, J. A., Whittaker, G. R. & Daniel, S. Coronavirus membrane fusion mechanism offers a potential target for antiviral development. Antiviral Research 178, 104792 (2020). [CrossRef]
- Guo, Y.-R. et al. The origin, transmission and clinical therapies on coronavirus disease 2019 (COVID-19) outbreak – an update on the status. Military Medical Research 7, (2020). [CrossRef]
- Thaiss, C. A., Levy, M., Itav, S. & Elinav, E. Integration of Innate Immune Signaling. Trends in Immunology 37, 84–101 (2016). [CrossRef]
- Prompetchara, E., Ketloy, C. & Palaga, T. Immune responses in COVID-19 and potential vaccines: Lessons learned from SARS and MERS epidemic. Asian Pacific Journal of Allergy and Immunology (2020). doi:10.12932/ap-200220-0772. [CrossRef]
- Felsenstein, S., Herbert, J. A., Mcnamara, P. S. & Hedrich, C. M. COVID-19: Immunology and treatment options. Clinical Immunology 215, 108448 (2020). [CrossRef]
- Yamato, M. et al. Role of adaptor TRIF in the MyD88-indepenent toll-like receptor signaling pathway. Science 301, 640-643 (2003).
- Akira, S., Uematsu, S. & Takeuchi, O. Pathogen Recognition and Innate Immunity. Cell 124, 783–801 (2006). [CrossRef]
- Kell, A. M. & Gale, M. RIG-I in RNA virus recognition. Virology 479-480, 110–121 (2015). [CrossRef]
- Loo, Y.-M., Galem M. Immune signaling by RIG-I-like receptors. Immunity 34, 680-692 (2011).
- Wit, E. D., Doremalen, N. V., Falzarano, D. & Munster, V. J. SARS and MERS: recent insights into emerging coronaviruses. Nature Reviews Microbiology 14, 523–534 (2016).
- Gabriella, D. M., Cristina, S., Concetta, R., Francesco, R. & Annalisa, C. SARS-Cov-2 infection: Response of human immune system and possible implications for the rapid test and treatment. International Immunopharmacology 84, 106519 (2020).
- Mehta, P. et al. COVID-19: consider cytokine storm syndromes and immunosuppression. The Lancet 395, 1033–1034 (2020). [CrossRef]
- Jamilloux, Y. et al. Should we stimulate or suppress immune responses in COVID-19? Cytokine and anti-cytokine interventions. Autoimmunity Reviews 19, 102567 (2020). [CrossRef]
- Decroly, E. et al. Coronavirus Nonstructural Protein 16 Is a Cap-0 Binding Enzyme Possessing (Nucleoside-2′O)-Methyltransferase Activity. Journal of Virology 82, 8071–8084 (2008). [CrossRef]
- Züst, R. et al. Ribose 2′-O-methylation provides a molecular signature for the distinction of self and non-self mRNA dependent on the RNA sensor Mda5. Nature Immunology 12, 137–143 (2011). [CrossRef]
- Sun, L. et al. Coronavirus Papain-like Proteases Negatively Regulate Antiviral Innate Immune Response through Disruption of STING-Mediated Signaling. PLoS ONE 7, (2012). [CrossRef]
- Menachery, V. D. et al. Pathogenic Influenza Viruses and Coronaviruses Utilize Similar and Contrasting Approaches To Control Interferon-Stimulated Gene Responses. mBio 5, (2014). [CrossRef]
- Frieman, M. et al. Severe Acute Respiratory Syndrome Coronavirus ORF6 Antagonizes STAT1 Function by Sequestering Nuclear Import Factors on the Rough Endoplasmic Reticulum/Golgi Membrane. Journal of Virology 81, 9812–9824 (2007). [CrossRef]
- Lokugamage, K. G. et al. Type I interferon susceptibility distinguishes SARS-CoV-2 from SARS-CoV. (2020). doi:10.1101/2020.03.07.982264. [CrossRef]
- Hadjadj, J. et al. Impaired type I interferon activity and inflammatory responses in severe COVID-19 patients. Science 369, 718–724 (2020). [CrossRef]
- Diao, B. et al. Reduction and Functional Exhaustion of T Cells in Patients With Coronavirus Disease 2019 (COVID-19). Frontiers in Immunology 11, (2020). [CrossRef]
- Qin, C. et al. Dysregulation of Immune Response in Patients With Coronavirus 2019 (COVID-19) in Wuhan, China. Clinical Infectious Diseases 71, 762–768 (2020). [CrossRef]
- Wang, F. et al. Characteristics of Peripheral Lymphocyte Subset Alteration in COVID-19 Pneumonia. The Journal of Infectious Diseases 221, 1762–1769 (2020). [CrossRef]
- Wang, D. et al. Clinical Characteristics of 138 Hospitalized Patients With 2019 Novel Coronavirus–Infected Pneumonia in Wuhan, China. Jama 323, 1061 (2020). [CrossRef]
- Zheng, H.-Y. et al. Elevated exhaustion levels and reduced functional diversity of T cells in peripheral blood may predict severe progression in COVID-19 patients. Cellular & Molecular Immunology 17, 541–543 (2020). [CrossRef]
- Wang, X. et al. Retraction Note to: SARS-CoV-2 infects T lymphocytes through its spike protein-mediated membrane fusion. Cellular & Molecular Immunology 17, 894–894 (2020).
- Tan, Y.-X. et al. Induction of Apoptosis by the Severe Acute Respiratory Syndrome Coronavirus 7a Protein Is Dependent on Its Interaction with the Bcl-XL Protein. Journal of Virology 81, 6346–6355 (2007).
- Yue, Y. et al. SARS-Coronavirus Open Reading Frame-3a drives multimodal necrotic cell death. Cell Death & Disease 9, (2018). [CrossRef]
- Mehta, A. K., Gracias, D. T. & Croft, M. TNF activity and T cells. Cytokine 101, 14–18 (2018). [CrossRef]
- Brooks, D. G. et al. Interleukin-10 determines viral clearance or persistence in vivo. Nature Medicine 12, 1301–1309 (2006). [CrossRef]
- Andersen, C. J., Murphy, K. E. & Fernandez, M. L. Impact of Obesity and Metabolic Syndrome on Immunity. Advances in Nutrition 7, 66–75 (2016). [CrossRef]
- Hunsche, C., Hernandez, O. & Fuente, M. D. L. Impaired Immune Response in Old Mice Suffering from Obesity and Premature Immunosenescence in Adulthood. The Journals of Gerontology Series A: Biological Sciences and Medical Sciences 71, 983–991 (2015). [CrossRef]
- Painter, S. D., Ovsyannikova, I. G. & Poland, G. A. The weight of obesity on the human immune response to vaccination. Vaccine 33, 4422–4429 (2015). [CrossRef]
- Park, H.-L. et al. Obesity-induced chronic inflammation is associated with the reduced efficacy of influenza vaccine. Human Vaccines & Immunotherapeutics 10, 1181–1186 (2014). [CrossRef]
- Backhed, F. et al. The gut microbiota as an environmental factor that regulates fat storage. Proceedings of the National Academy of Sciences 101, 15718–15723 (2004). [CrossRef]
- Membrez, M. et al. Gut microbiota modulation with norfloxacin and ampicillin enhances glucose tolerance in mice. The FASEB Journal 22, 2416–2426 (2008). [CrossRef]
- Turnbaugh, P. J. et al. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 444, 1027–1031 (2006). [CrossRef]
- Maier, H. E. et al. Obesity Increases the Duration of Influenza A Virus Shedding in Adults. The Journal of Infectious Diseases 218, 1378–1382 (2018). [CrossRef]
- Dhurandhar, N. V., Bailey, D. & Thomas, D. Interaction of obesity and infections. Obesity Reviews 16, 1017–1029 (2015). [CrossRef]
- Panic A, Stanimirovic J, Sudar-Milovanovic E, Isenovic ER. Oxidative stress in obesity and insulin resistance. Explor Med. 2022;3:58–70. [CrossRef]
- Hensley K, Robinson KA, Gabbita SP, Salsman S, Floyd RA. Reactive oxygen species, cell signaling, and cell injury. Free Radic Biol Med. 2000;28:1456–62.
- National Institutes of Health (NIH). Coronavirus Disease 2019 (COVID-19) Treatment Guidelines. http://www.covid29treatmentguidelines.nih.gov/. Accessed 8/3/2022.
- Andrews N, Stowe N, Kirsebom F, et al. Covid-19 Vaccine Effectiveness against the Omicron (B1.1.529) Variant. N Engl J Med. 2022;2119451(March):•••. doi:10.1056/NEJMoa. [CrossRef]
- Ray Yutani, Viswanath Venketaraman. The COVID-19 Illness: Addressing the Current Treatment Limitations and Care Gaps with a Novel Alternative and Complementary Agent-the Glutathione-Cyclodextrin Complex. Altern Ther Health Med. 2023 May;29(4):28-35.
- Khanfar A, Al Qaroot B. Could glutathione depletion be the Trojan horse of COVID-19 mortality? Eur Ver Med Pharmacol Sci. 2020;202012:24046. 10.26355’eurrev.
- 7. Ghezzi P. Role of glutathione in immunity and inflammation in the lung. Int J Gen Med. 2011;4:105-113. doi:10.2147/IJGM.S15618. [CrossRef]
- Pena LR, Hill DB, McClain CJ. Treatment with glutathione precursor decreases cytokine activity. JPEN J Parenter Enteral Nutr. 1999;23(1):1-6. doi:10.1177/014860719902300101. [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



