
Submitted:
02 June 2023
Posted:
02 June 2023
You are already at the latest version
Abstract
Smoking have been linked to male infertility by affecting the sperm epigenome and genome. In this study, we aimed to determine possible changes in the transcript levels of the PTPRN2, PGAM5, and TYRO3 genes in heavy smokers compared with non-smokers; and to investigate their association with fundamental sperm parameters. One hundred eighteen sperm samples (63 heavy-smokers (G1) and 55 non-smokers (G2) were included in this study. Semen analysis was performed according to WHO guidelines. After total RNA extraction, RT-PCR was used to quantify the transcript levels of studied genes. In G1 a significant decrease in standard semen parameters in comparison to non-smokers has been shown (p<0.05). Moreover, PGAM5 and PTPRN2 were differentially expressed (P≤0.03 and P≤0.01, respectively), and downregulated in spermatozoa of G1 compared to G2. In contrast, no difference was observed for TYRO3 (p≤0.3). In G1, mRNA expression level of studied genes correlated negatively with motility, sperm count, normal form, vitality, and sperm membrane integrity (p<0.05). Therefore, smoking may affect gene expression and male fertility by altering DNA methylation patterns in genes associated with fertility and sperm quality, including PGAM5, PTPRN2, and TYRO3.
Keywords:
Male infertility
; heavy smoking
; transcript level
; PGAM5
; PTPRN2
; TYRO3
1. Introduction
Nearly 15% of infertile couples worldwide are attributable to the male factor [1]. Several factors contribute to male infertility. Reproductive tract infections, genetic and anatomical disorders, and immunological and endocrine disorders are among these factors [2,3,4].
Lifestyle and environmental factors such as diet, smoking, exercise, and alcohol consumption are as important to our health as our genes. These factors cause changes that affect gene expression.
Gene expression is regulated in several ways in mammals. However, DNA methylation is the most common epigenetic signaling device cells use to lock or unlock genes. DNA methylation works by adding a methyl group at position 5 of cytosine, which is found in cytosine phosphate guanine dinucleotides "CpGs" [5]. Typically, the group is added to specific sites in the DNA where it blocks proteins that bind to the DNA to "read" the gene. This group can be removed through a process called demethylation. Typically, methylation turns genes “off” and demethylation turns genes “on” [5]. In recent decades, DNA methylation has received considerable research attention due to its importance in various cellular processes. DNA methylation plays a crucial role in regulating gene expression by regulating the transcription of DNA to RNA [6,7], and is involved in protecting the genome by inhibiting the movement of DNA transposable elements Integrity [8,9]. DNA methylation processes can also be altered by many other factors, such as genetics [10], and environmental factors, smoking being a typical example [11].
Many studies have investigated the relationship between male infertility and lifestyle or habits such as alcohol consumption, smoking, diet, environmental factors, age, and stress.
Several other factors, including age, stress, and lifestyle, such as obesity, smoking, and alcohol use, have been linked to male infertility [12,13,14,15,16].
Smoking increases harmful oxidants such as reactive oxygen species (ROS) in seminal plasma. This leads to an imbalance between antioxidants and oxidants known as oxidative stress. Oxidative stress can lead to damage to sperm DNA [14,17,18,19].
Studies have shown that sperm DNA damage may also be caused by the formation of DNA adducts associated with cigarette content [20,21].
In a previous study, after applying bisulfite sequencing, we found 15 CpG sites in the amplicon associated with the PGAM5 (phosphoglycerate mutase family member 5) gene and heavy smokers DNA methylation levels were significantly increased between non-smokers and non-smokers, and there was an amplicon linked to the PTPRN2 gene (protein tyrosine phosphatase, N2-type receptor) at nine CpG sites. On the contrary, the results showed that the DNA methylation levels of three CpG sites of the TYRO3 gene-associated amplicon (tyrosine protein kinase receptor) were significantly reduced in the case group compared with the control group [25].
Furthermore, this study showed a significant correlation between changes in sperm DNA methylation levels and standard sperm parameters in the case cohort [25].
Therefore, in this study, we aimed to determine possible changes in the transcript levels of the PTPRN2, PGAM5, and TYRO3 genes in heavy smokers compared with non-smokers; and to investigate their association with fundamental sperm parameters.
2. Materials and Methods
2.1. Semen Sample Collection
Before the beginning of this study, an institutional review board approval (No. PHRC/HC/13/14) was achieved by the Ethics Committee of Helsinki. Moreover, approval consent was taken from each participant enrolled in this study. The study was conducted in the laboratory of biochemistry and molecular biology of reproductive medicine, Department of Obstetrics and Gynecology at the University Hospital-Homburg /Saar, Saarland Germany.
All participant males were in the reproductive age group (25- 45 years old) and the following parameters were excluded: cases suffering from varicocele, anti-sperm antibodies, Y chromosome microdeletions, males subjected to surgical operation in the reproductive system, and heavy body mass index.
Patients were classified into two groups. The control group includes 55 proven fertile candidates who show no previous history of smoking, and the case group includes 63 fertile heavy smokers who smoked more than 20 cigarettes per day (in the last 3 years until their enrolment in this study). Semen samples were collected by masturbation after 3 to 5 days of sexual abstinence in clean, dry, sterile and leak-proof plastic containers in a collection room attached to the laboratory. Following liquefaction of semen samples at 37oC for 30 min to 1hour, samples were analyzed according to World Health Organization guidelines [26].
2.2. Spermatozoa Purification and Analysis
The sample was kept on the heating stage for 30 minutes for liquefaction at 37ºC. After that, the sperm samples were evaluated according to the WHO laboratory manual to determine Sperm count, motility, morphology, vitality (eosin test) and, sperm membrane integrity (Hos test) [26].
Gradient centrifugation was used to purify samples. Briefly, each sample was treated by a discontinuous Puresperm gradients (40%–80%) (Nidacon International, Sweden). Then, to guaranty the elimination of somatic cells, the samples were washed with lysis buffer (0.1% SDS, 0.5% Triton X-100 in double-distilled water).
2.2.1. Assessment of Sperm Morphology
Sperm morphology was evaluated according to strict criteria as following:
Smears were prepared by spreading 20 μl of ejaculate on a glass slide. After fixation, the slides were stained using Papanicolaou method. A total of 200 spermatozoa from each slide were evaluated under oil immersion at a magnification of 1,000 x using bright field illumination. At least 10 high power fields from different areas of the slide were estimated.
2.2.2. Assessment of Sperm Vitality (Eosin Test)
On a glass slide, 10 μl of ejaculate was mixed with 10 μl of 0.5% aqueous yellowish Eosin Y solution. The mixture covered with a cover slide, then evaluated after 3-5 min by distinguishing between the dead spermatozoa (Red stained) and the live spermatozoa (not stained). 200 spermatozoa from each slide were evaluated.
2.2.3. Assessment of the Sperm Membrane Integrity (Hypo-Osmotic Test (HOS))
The HOS test is a sperm vitality test that predicts sperm membrane integrity. For this test, 100 μl of fresh ejaculate mixed with 1.0 ml of the hypo osmotic solution. Then, the mixture was incubated at 37°C for 30-60 minutes. Influx of the fluid due to hypo-osmotic stress causes the sperm tail to swell which indicates the presence of sperm having a functional and intact plasma membrane. A minimum of 200 swollen and/or not swollen spermatozoa were examined per slide under phase contrast microscope.
2.3. RNA Extraction and Synthesis of the cDNA (Reverse Transcription)
Total RNA was isolated from the purified semen samples according to a modified protocol of the Isolate II RNA/DNA/Protein Kit (Phenol-free) (Bioline, UK). The Nanodrop spectrophotometer ND-2000c (Thermo Scientific, USA) was used to measure the concentration and the purity of isolated RNA. The integrity of the isolated RNA was checked on an RNA Nano 6000 chip via an Agilent Bioanalyzer (Agilent technologies, Santa Clara, California, United States). The extracted RNA was stored at -80°C until the time of usage.
The reverse transcription procedure performed using miScript II RT Kit according to the standard protocol provided by the manufacturer (Cat No. 218161, Qiagen, Germany). Briefly, 300 ng extracted RNA were added to 4 μl of 5x MiScriptHiLflex buffer, 2μl of nucleotide mix, and 2 μl of reverse transcriptase mix which contains all the components required for the synthesis of the complementary DNA (cDNA), then RNA free water was added to complete the required volume. After that, the reaction buffer incubated at 37°C for 60 min, then at 95°C for 5 min to suppress the activity of the reverse transcriptase mix.
2.4. Real-Time Quantitative PCR (qPCR)
After the synthesis of cDNA, real time qPCR performed to amplify and quantify the transcript level of each of the studied genes PGAM5, TYRO3, PTPRN2, and GAPDH (as endogenous control).
Briefly, the produced cDNA used as the template for the qPCR reaction mixture. This reaction was prepared using 2x quantiTect SYBR Green PCR Mix (Cat No. 204143, Qiagen, Germany) and QuantiTect primers assay for PGAM5, TYRO3, PTPRN2, and GAPDH (as reference gene) (Cat No. QT00079247, Qiagen, Germany) according to the recommendations of the manufacturer.
At the end, the prepared mixture distributed in triplicate for each sample into 96 well plate plate. The plate placed in StepOnePlus™ System (7500 Fast Applied Biosystems, USA) and the appropriate program applied according to the instructions provided by the manufacturer.
In addition, a no template control (NTC) and a no reverse transcriptase control (NRT) were included in each run. All qPCR experiments performed in triplicate and the resulting CT values normalized to GAPDH.
2.5. Statistical Analysis
The quantification of gene expression was determined through CT value (threshold cycle) which, provided from Real-Time PCR instrument by the software when the PCR reaction reaches the start of its exponential stage and imported into a spreadsheet program such as Microsoft Excel. The average of Ct for each triple sample was calculated, the change of target gene expression Firs the Cт average of each triples sample was determined, then the expression of target and control genes were normalized to the endogenous gene by ΔCт (Cт target gene – Cт GAPDH) and (Cт control – Cт GAPDH). ΔΔCт calculated through (ΔCт target – ΔCт control) and the fold change of gene expression was calculated through the equation fold change = 2 – ΔΔCт [27].
For data analysis, IBM SPSS for Windows software package version 23.0 (SPSS Inc., USA) was used. Samples included in this study were non-normal distributed (non-parametric) according to the value of skewness test, Kurtosis test, Z-value and Shapiro test. The independent-sample t-test (Mann–Whitney test) used to compare means of quantitative variables.
In addition, Spearman’s test used to assess the correlation coefficient between expression levels in fertile heavy smokers and sperm parameters. To be qualified as statistically significant, the results should show a p-value was less than 5 % (p ≤ 0.05).
3. Results
One hundred and eighteen sperm samples were divided into two groups. The first group include 55 proven fertile candidates who show no previous history for smoking as control group with a mean age (36.33 ± 6.18). The second group include 63 fertile smokers who smoked more than 20 cigarettes per day with a mean age (37.42 ± 5.24) as case group.
3.1. Characteristics of the Study Population
Sperm parameters were compared between heavy smokers and non-smokers as shown in Table 1. Total sperm count, total motility, progressive motility, normal form, vitality, and sperm membrane integrity were significantly higher in non-smokers group in comparison to heavy smokers group: 52.38 ± 34.52 vs 64.42 ± 39.18 Mill/ml, P≤ 0.01; 44.61 ± 22.47 vs 53.67 ± 20.51%, P≤ 0.003; 33.17 ± 21.87 vs 40.96 ± 20.78%, P≤ 0.01; 17.22 ± 8.26 vs 22.98 ± 12.62%, P≤ 0.05; 59.95 ± 13.08 vs 64.87 ± 15.18 %, P≤ 0.001, and 72.19 ± 10.03 vs 78.73 ± 11.21%, P≤ 0.02 respectively. Exception was for immotile sperms that was significantly higher in heavy smokers group (51.13 ± 24.23 vs 42.21 ± 18.24%; P≤ 0.002).
3.2. Quantification of mRNA
The quantification Real-time PCR was used to quantify the expression level of the selected genes (PGAM5, TYRO3, and PTPRN2). The CT represents the threshold cycle and provides information about the cycle of the fluorescent signal that increases exponentially to cross the threshold.
Relative amounts of PGAM5, TYRO3, and PTPRN2 mRNA delta Ct (ΔCT) were differentially expressed among the compared groups (Table 2).
This difference between the group of non-smokers and the group of heavy smokers was significant for PGAM5 (P≤0.03) and PTPRN2 (p ≤0.01) but not significant for TYRO3 (p≤0.3).
3.3. Correlation between PGAM5, TYRO3, and PTPRN2 mRNA Expression and Sperm Parameters
The association between expression level of PGAM5, TYRO3 and PTPRN2 and sperm parameters was estimated.
The data included in the present study were not-normally distributed (non-parametric), therefore the spearman correlation coefficient (Spearman Roh) was used. Table 3 demonstrated that PGAM5 mRNA expression level was correlated negatively with motility (r = -0.336, P ≤ 0.005) progressive motility (r = -0.274, P ≤ 0.01) and positively with vitality (r = 0.253, P ≤ 0.02).
TYRO3 mRNA expression level showed a significant negative correlation with sperm count (r = -0.331, P ≤ 0.009), and vitality (r = -0.230, P ≤ 0.04), and a negative correlation with motility (r = 0.339, P ≤ 0.008), progressive motility (r = 0.308, P ≤ 0.01), normal form (r = 0.259, P ≤ 0.04), , and sperm membrane integrity (r = 0.269, P ≤ 0.01) (Table 3).
Furthermore, PTPRN2 mRNA expression level correlated significantly negative with total sperm count (r = -0.273, P ≤ 0.01), motility (r = -0.276, P ≤ 0.01), progressive motility (r = -0.293, P ≤ 0.009), normal form (r = -0.228, P ≤ 0.04), and vitality (r = -0.344, P ≤ 0.02) (Table 3).
4. Discussion
Infertility or subfertility is the result of many pathological factors. In general, 50% of infertility cases are attributed to idiopathic subfertility. About 15 percent of these cases are due to genetic defects, with the remainder due to environmental and lifestyle factors such as diet, alcohol consumption, physical activity, and smoking.
These factors have been found to have effects on the sperm epigenome and genome [22], which may have implications for the developing embryo [23,24].
Several studies have shown an association between decreased sperm quality (decreased sperm concentration, motility, normal morphology) and heavy smoking [14,28,29,30].
In comparison to the control group, we have identified a significant decrease in standard semen parameters (count, total motility, progressive motility, normal form, vitality, and membrane integrity) in heavy smokers group (P ≤0.05).
These results are consistent with other studies showing the negative effects of smoking on sperm quality and its DNA structure [28,29,30,31]. A meta-analysis showed that that smoking negatively affect sperm parameters [32]. Conversly, other researchers have found that tobacco has no impact on standard semen parameters [33,34,35].
Thus, more research are needed to understand the molecular mechanism of how tobacco smoking influences male fecundity.
The genotoxic components of tobacco cause DNA adduct crosslinks, single- or double-strand breaks, chromosomal abnormalities and aneuploidy, and other genetic changes in spermatozoa [23].
We focused in this study on the relative quantification of PGAM5, TYRO3, and PTPRN2 genes expression and investigated the influence of tobacco smoke on the transcript level of these genes.
The relative amounts of PGAM5, TYRO3, and PTPRN2 mRNA delta Ct (ΔCT) were differentially expressed among the compared groups (Table 2).
This difference between the group of non-smokers and the group of heavy smokers was significant for PGAM5 (P≤0.03) and PTPRN2 (P ≤0.01) but not significant for TYRO3 (P ≤0.3).
PGAM5 gene were down regulated in the spermatozoa of heavy smokers compared to that of nonsmokers by 0.57 fold change (Figure 1). Similarly, for PTPRN2 gene, it was a down regulation by a fold change equal to 0.66 (Figure 2). In contrast, an upregulation by 1.21 fold change for TYRO3 gene was determined (Figure 3).
These results are in accordance to a study that demonstrated a downregulation in the transcription levels of H2BFWT, TNP1, TNP2, PRM1 and PRM2 genes in the spermatozoa of heavy smokers compared to that of nonsmokers [28]. Other studies demonstrated a significant decline in the transcription level of PRM1 and PRM2 in smokers spermatozoa compared to non-smokers [36], and that reactive oxygen species (ROS) resulting from smoking might be responsible for the decrease of the transcription level of genes like IκBα [37]. Another study showed a downregulation in the transcription levels of MAPK8IP3, GAA, ANXA2, PRRC2A, and PDE11A genes in heavy smokers compared to non-smokers and an upregulation in the MAPK8Ip3, ANXA2, PRRC2A, and PDE11A transcription levels in heavy smokers compared to non-smokers [38].
Regarding the correlation between the expression level of the studied genes and sperm parameters in the heavy smokers group (Table 3), we found that PGAM5 mRNA expression level correlated negatively with total motility, progressive motility (P ≤ 0.01) and positively with vitality (P ≤ 0.02).
TYRO3 mRNA expression correlated negatively with sperm count and vitality (P ≤ 0.01), but highly positive with total motility, progressive motility, sperm membrane integrity (P ≤ 0.01), and normal form (P ≤ 0.04)
PTPRN2 mRNA expression level showed a negative correlation with total sperm count, total motility, progressive motility ((P ≤ 0.01), normal form (P ≤ 0.04), and vitality (P ≤ 0.02) (Table 3).
The PGMA5 gene encodes two mitochondrial genes: PGAM5s and PGMA5L (phosphoglycerate mutase family member 5) [39]. Both proteins are expressed in the testis and play an important role in mitophagy, a cellular process that eliminates damaged mitochondria [40,41].
Studies have shown that mutation or downregulation of the PGMA5 gene can lead to mitochondrial dysfunction, depletion or loss of mtDNA [42,43]. mtDNA is important for energy production in cells, including sperm, which can lead to decreased sperm motility and fertility.
PTPRN2 is a gene encoding a protein called tyrosine phosphatase non-receptor type 2, which is involved in the regulation of insulin signaling and glucose metabolism [39]. A previous study found that semen parameters (semen volume, sperm concentration, motility and morphology) were reduced in diabetic patients compared with non-diabetic patients [44].
With regard to male infertility, PTPRN2 has been reported to be located near hypermethylated regions of sperm DNA from patients who failed to conceive [45]. Another study showed that the PTPRN2 gene showed highly differential methylation differences between severe oligospermia and obstructive azoospermia [46].
A case-control study published in 2019 identified candidate genes important for spermatogenesis and fertility [47]. Of the 170 genes highlighted in this study, at least 38 genes (including PTPRN2) were differentially methylated and/or exhibited high testis expression between infertility cases and controls [47].
TYRO3 is a gene encoding a receptor tyrosine kinase protein [39]. This protein is involved in cell signaling and has been shown to play a role in the development and function of the male reproductive system [48,49,50]. Research suggests that variations in the TYRO3 gene may be associated with decreased sperm motility and fertility [48,49,50].
Smoking causes the formation of free radicals, which cause oxidative stress in the body. Oxidative stress can damage DNA in sperm and lead to reduced fertility [51]. PGAM5 has been shown to play a role in protecting cells from oxidative stress [43], so downregulation of this gene may make sperm more susceptible to damage from smoking-related oxidative stress. In addition, smoking has been shown to impair mitochondrial function in various tissues, including sperm [52,53]. PGAM5 plays a role in regulating mitochondrial function, so downregulation of this gene may lead to further mitochondrial dysfunction in response to smoking.
Smoking causes inflammation in the body, and chronic inflammation has been linked to decreased fertility. PTPRN2 has been shown to play a role in regulating inflammation. Therefore, the downregulation of this gene may make sperm more vulnerable to the inflammatory damage caused by smoking.
Changes in the transcript levels of these genes confirmed our previous study [25]. Increased DNA methylation levels in PGAM5 and PTPRN2 genes in heavy smokers lead to repression of the expression of these genes, resulting in downregulation of PGAM5 and PTPRN2 genes. Conversely, a significant decrease in DNA methylation (demethylation) levels in TYRO3 in cases compared with controls could lead to upregulation.
5. Conclusions
Overall, this study suggests that smoking may affect gene expression and male fertility by altering DNA methylation patterns in genes associated with fertility and sperm quality, including PGAM5, PTPRN2, and TYRO3. However, more research is needed to fully understand the mechanisms involved and the potential effects of smoking on these genes associated with male fertility. Quitting smoking can improve overall health and fertility and may also help improve gene expression and mitochondrial function in sperm.
Author Contributions
H.A; Conceptualization, design, writing—review and editing. Y.A.; methodology, formal analysis. RB.; software and validation. P.M.J.; investigation and resources. M.H.; supervision and project administration. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding, Saarland University.
Institutional Review Board Statement
The study was conducted in accordance with the Declaration of Helsinki and approved by the Institutional Review Board (No. PHRC/HC/13/14).
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
The data presented in this study are available on request from the corresponding author. The data are not publicly available due to ethical restrictions.
Acknowledgments
We thank the Department of Obstetrics, Gynaecology and Reproductive Medicine, University Hospital of Saarland, for their financial support.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Agarwal, A.; Mulgund, A.; Hamada, A.; Chyatte, M.R. A Unique View on Male Infertility around the Globe. Reprod. Biol. Endocrinol. 2015, 13, 1–9. [Google Scholar] [CrossRef]
- Stouffs, K.; Seneca, S.; Lissens, W. Genetic Causes of Male Infertility. Ann. Endocrinol. (Paris). 2014. [Google Scholar] [CrossRef]
- Tognon, M.; Tagliapietra, A.; Magagnoli, F.; Mazziotta, C.; Oton-Gonzalez, L.; Lanzillotti, C.; Vesce, F.; Contini, C.; Rotondo, J.C.; Martini, F. Investigation on Spontaneous Abortion and Human Papillomavirus Infection. Vaccines 2020, 8, 473. [Google Scholar] [CrossRef] [PubMed]
- Rotondo, J.C.; Lanzillotti, C.; Mazziotta, C.; Tognon, M.; Martini, F. Epigenetics of Male Infertility: The Role of DNA Methylation. Front. Cell Dev. Biol. 2021, 9, 689624. [Google Scholar] [CrossRef] [PubMed]
- Portela, A.; Esteller, M. Epigenetic Modifications and Human Disease. Nat. Biotechnol. 2010, 28, 1057–1068. [Google Scholar] [CrossRef] [PubMed]
- Shukla, S.; Kavak, E.; Gregory, M.; Imashimizu, M.; Shutinoski, B.; Kashlev, M.; Oberdoerffer, P.; Sandberg, R.; Oberdoerffer, S. CTCF-Promoted RNA Polymerase II Pausing Links DNA Methylation to Splicing. Nature 2011, 479, 74–79. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.M.; Bird, A. DNA Methylation Landscapes: Provocative Insights from Epigenomics. Nat. Rev. Genet. 2008, 9, 465–476. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.W.K.; Pausova, Z. Cigarette Smoking and DNA Methylation. Front. Genet. 2013, 4, 132. [Google Scholar] [CrossRef] [PubMed]
- Levin, H.L.; Moran, J. V Dynamic Interactions between Transposable Elements and Their Hosts. Nat. Rev. Genet. 2011, 12, 615–627. [Google Scholar] [CrossRef] [PubMed]
- Bell, J.T.; Pai, A.A.; Pickrell, J.K.; Gaffney, D.J.; Pique-Regi, R.; Degner, J.F.; Gilad, Y.; Pritchard, J.K. DNA Methylation Patterns Associate with Genetic and Gene Expression Variation in HapMap Cell Lines. Genome Biol. 2011, 12, 1–13. [Google Scholar] [CrossRef]
- Breitling, L.P.; Yang, R.; Korn, B.; Burwinkel, B.; Brenner, H. Tobacco-Smoking-Related Differential DNA Methylation: 27K Discovery and Replication. Am. J. Hum. Genet. 2011, 88, 450–457. [Google Scholar] [CrossRef]
- Kovac, J.R.; Khanna, A.; Lipshultz, L.I. The Effects of Cigarette Smoking on Male Fertility. Postgrad. Med. 2015, 127, 338–341. [Google Scholar] [CrossRef]
- Craig, J.R.; Jenkins, T.G.; Carrell, D.T.; Hotaling, J.M. Obesity, Male Infertility, and the Sperm Epigenome. Fertil. Steril. 2017, 107, 848–859. [Google Scholar] [CrossRef] [PubMed]
- Amor, H.; Hammadeh, M.E.; Mohd, I.; Jankowski, P.M. Impact of Heavy Alcohol Consumption and Cigarette Smoking on Sperm DNA Integrity. Andrologia 2022, 54, e14434. [Google Scholar] [CrossRef] [PubMed]
- Amor, H.; Jankowski, P.M.; Dahadhah, F.W.; Al Zoubi, M.S.; Hammadeh, M.E. Impact of Tobacco Smoking in Association with H2BFWT, PRM1 and PRM2 Genes Variants on Male Infertility. Andrologia 2022, 54, e14611. [Google Scholar] [CrossRef] [PubMed]
- Ilacqua, A.; Izzo, G.; Emerenziani, G. Pietro; Baldari, C.; Aversa, A. Lifestyle and Fertility: The Influence of Stress and Quality of Life on Male Fertility. Reprod. Biol. Endocrinol. 2018, 16, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Hammadeh, M.E.; Hamad, M.F.; Montenarh, M.; Fischer-Hammadeh, C. Protamine Contents and P1/P2 Ratio in Human Spermatozoa from Smokers and Non-Smokers. Hum. Reprod. 2010, 25, 2708–2720. [Google Scholar] [CrossRef] [PubMed]
- Mahat, R.K.; Kumar, S.; Arora, M.; Bhale, D. V; Mehta, R.; Batra, J. Role of Oxidative Stress and Antioxidants in Male Infertility. Int J Heal. Sci Res 2015, 5, 324–333. [Google Scholar]
- Opuwari, C.S.; Henkel, R.R. An Update on Oxidative Damage to Spermatozoa and Oocytes. Biomed Res. Int. 2016. [CrossRef] [PubMed]
- Perrin, J.; Tassistro, V.; Mandon, M.; Grillo, J.M.; Botta, A.; Sari-Minodier, I. Tobacco Consumption and Benzo(a)Pyrene-Diol-Epoxide–DNA Adducts in Spermatozoa: In Smokers, Swim-up Procedure Selects Spermatozoa with Decreased DNA Damage. Fertil. Steril. 2011, 95, 2013–2017. [Google Scholar] [CrossRef]
- Phillips, D.H.; Venitt, S. DNA and Protein Adducts in Human Tissues Resulting from Exposure to Tobacco Smoke. Int. J. Cancer 2012. [Google Scholar] [CrossRef]
- Harlev, A.; Agarwal, A.; Gunes, S.O.; Shetty, A.; du Plessis, S.S. Smoking and Male Infertility: An Evidence-Based Review. World J. Mens. Health 2015, 33, 143–160. [Google Scholar] [CrossRef]
- Beal, M.A.; Yauk, C.L.; Marchetti, F. From Sperm to Offspring: Assessing the Heritable Genetic Consequences of Paternal Smoking and Potential Public Health Impacts. Mutat. Res. - Rev. Mutat. Res. 2017. [Google Scholar]
- Donkin, I.; Barrès, R. Sperm Epigenetics and Influence of Environmental Factors. Mol. Metab. 2018. [CrossRef]
- Alkhaled, Y.; Laqqan, M.; Tierling, S.; Lo Porto, C.; Amor, H.; Hammadeh, M.E. Impact of Cigarette-Smoking on Sperm DNA Methylation and Its Effect on Sperm Parameters. Andrologia 2018, 50. [Google Scholar] [CrossRef]
- World Health Organization Laboratory Manual for the Examination and Processing of Human Semen. Cambridge Cambridge Univ. Press 2010.
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2− ΔΔCT Method. methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Amor, H.; Zeyad, A.; Hammadeh, M.E. Tobacco Smoking and Its Impact on the Expression Level of Sperm Nuclear Protein Genes: H2BFWT, TNP1, TNP2, PRM1 and PRM2. Andrologia 2021, 53, e13964. [Google Scholar] [CrossRef] [PubMed]
- Hamad, M.F.; Shelko, N.; Kartarius, S.; Montenarh, M.; Hammadeh, M.E. Impact of Cigarette Smoking on Histone (H2B) to Protamine Ratio in Human Spermatozoa and Its Relation to Sperm Parameters. Andrology 2014. [Google Scholar] [CrossRef] [PubMed]
- Hammadeh, M.E.; Hamad, M.F.; Montenarh, M.; Fischer-Hammadeh, C. Protamine Contents and P1/P2 Ratio in Human Spermatozoa from Smokers and Non-Smokers. Hum. Reprod. 2010, 25, 2708–2720. [Google Scholar] [CrossRef]
- Amor, H.; Nyaz, S.; Hammadeh, M.E. Paternal Smoking in Relation to Sperm Quality and Intracytoplasmic Sperm Injection Outcomes. Int. J. Women’s Heal. Reprod. Sci. 2019, 7, 451–460. [Google Scholar] [CrossRef]
- Sharma, R.; Harlev, A.; Agarwal, A.; Esteves, S.C. Cigarette Smoking and Semen Quality: A New Meta-Analysis Examining the Effect of the 2010 World Health Organization Laboratory Methods for the Examination of Human Semen. Eur. Urol. 2016, 70, 635–645. [Google Scholar] [CrossRef] [PubMed]
- Sepaniak, S.; Forges, T.; Gerard, H.; Foliguet, B.; Bene, M.C.; Monnier-Barbarino, P. The Influence of Cigarette Smoking on Human Sperm Quality and DNA Fragmentation. Toxicology 2006, 223, 54–60. [Google Scholar] [CrossRef] [PubMed]
- Aghamohammadi, A.; Zafari, M. The Impact of Cigarette Smoking on Sperm Parameters: A Cross-Sectional Study. In Proceedings of the International Conference on Environmental, Biomedical and Biotechnology Web site. http://www. ipcbee. com/vol16/17-E20004. pdf; 2011. [Google Scholar]
- Cinar, O.; Dilbaz, S.; Terzioglu, F.; Karahalil, B.; Yücel, C.; Turk, R.; Taskin, L.; Kose, S.K. Does Cigarette Smoking Really Have Detrimental Effects on Outcomes of IVF? Eur. J. Obstet. Gynecol. Reprod. Biol. 2014, 174, 106–110. [Google Scholar] [CrossRef]
- Hamad, M.; Shelko, N.; Montenarh, M.; Hammadeh, M.E. The Impact of Cigarette Smoking on Protamines 1 and 2 Transcripts in Human Spermatozoa. Hum. Fertil. 2019. [Google Scholar] [CrossRef]
- Yu, B.; Ding, Q.; Zheng, T.; Jiang, L.; Li, Q.; Sun, X.; Bai, C.; Huang, Z. Smoking Attenuated the Association between IκBα Rs696 Polymorphism and Defective Spermatogenesis in Humans. Andrologia 2015, 47, 987–994. [Google Scholar] [CrossRef] [PubMed]
- Laqqan, M.M.; Yassin, M.M. Cigarette Heavy Smoking Alters DNA Methylation Patterns and Gene Transcription Levels in Humans Spermatozoa. Environ. Sci. Pollut. Res. 2022, 29, 26835–26849. [Google Scholar] [CrossRef]
- Consortium, E.P. An Integrated Encyclopedia of DNA Elements in the Human Genome. Nature 2012, 489, 57. [Google Scholar] [CrossRef]
- Chen, G.; Han, Z.; Feng, D.; Chen, Y.; Chen, L.; Wu, H.; Huang, L.; Zhou, C.; Cai, X.; Fu, C.; et al. A Regulatory Signaling Loop Comprising the PGAM5 Phosphatase and CK2 Controls Receptor-Mediated Mitophagy. Mol. Cell 2014, 54, 362–377. [Google Scholar] [CrossRef]
- Wu, H.; Xue, D.; Chen, G.; Han, Z.; Huang, L.; Zhu, C.; Wang, X.; Jin, H.; Wang, J.; Zhu, Y. The BCL2L1 and PGAM5 Axis Defines Hypoxia-Induced Receptor-Mediated Mitophagy. Autophagy 2014, 10, 1712–1725. [Google Scholar] [CrossRef]
- Chen, Y.; Gong, K.; Xu, Q.; Meng, J.; Long, T.; Chang, C.; Wang, Z.; Liu, W. Phosphoglycerate Mutase 5 Knockdown Alleviates Neuronal Injury after Traumatic Brain Injury through Drp1-Mediated Mitochondrial Dysfunction. Antioxid. Redox Signal. 2021, 34, 154–170. [Google Scholar] [CrossRef]
- Villavicencio Tejo, F.; Quintanilla, R.A. Contribution of the Nrf2 Pathway on Oxidative Damage and Mitochondrial Failure in Parkinson and Alzheimer’s Disease. Antioxidants 2021, 10, 1069. [Google Scholar] [CrossRef]
- La Vignera, S.; Condorelli, R.A.; Balercia, G.; Vicari, E.; Calogero, A.E. Does Alcohol Have Any Effect on Male Reproductive Function? A Review of Literature. Asian J. Androl. 2013, 15, 221. [Google Scholar] [CrossRef]
- Jenkins, T.G.; Aston, K.I.; Meyer, T.D.; Hotaling, J.M.; Shamsi, M.B.; Johnstone, E.B.; Cox, K.J.; Stanford, J.B.; Porucznik, C.A.; Carrell, D.T. Decreased Fecundity and Sperm DNA Methylation Patterns. Fertil. Steril. 2016, 105, 51–57. [Google Scholar] [CrossRef]
- Li, Z.; Zhuang, X.; Zeng, J.; Tzeng, C.-M. Integrated Analysis of DNA Methylation and MRNA Expression Profiles to Identify Key Genes in Severe Oligozoospermia. Front. Physiol. 2017, 8, 261. [Google Scholar] [CrossRef]
- Sarkar, S.; Sujit, K.M.; Singh, V.; Pandey, R.; Trivedi, S.; Singh, K.; Gupta, G.; Rajender, S. Array-Based DNA Methylation Profiling Reveals Peripheral Blood Differential Methylation in Male Infertility. Fertil. Steril. 2019, 112, 61–72. [Google Scholar] [CrossRef]
- Godowski, P.J.; Mark, M.R.; Chen, J.; Sadick, M.D.; Raab, H.; Hammonds, R.G. Reevaluation of the Roles of Protein S and Gas6 as Ligands for the Receptor Tyrosine Kinase Rse/Tyro 3. Cell 1995, 82, 355–358. [Google Scholar] [CrossRef]
- Joseph, D.R. Sequence and Functional Relationships between Androgen-Binding Protein/Sex Hormone-Binding Globulin and Its Homologs Protein S, Gas6, Laminin, and Agrin. Steroids 1997, 62, 578–588. [Google Scholar] [CrossRef] [PubMed]
- Richard, I.; Broux, O.; Chiannilkulchai, N.; Fougerousse, F.; Allamand, V.; Bourg, N.; Brenguier, L.; Devaud, C.; Pasturaud, P.; Roudaut, C. Regional Localization of Human Chromosome 15 Loci. Genomics 1994, 23, 619–627. [Google Scholar] [CrossRef] [PubMed]
- Bisht, S.; Faiq, M.; Tolahunase, M.; Dada, R. Oxidative Stress and Male Infertility. Nat. Rev. Urol. 2017. [CrossRef] [PubMed]
- Tawadrous, G.A.; Aziz, A.A.; Mostafa, T. Effect of Smoking Status on Seminal Parameters and Apoptotic Markers in Infertile Men. J. Urol. 2011, 186, 1986–1990. [Google Scholar] [CrossRef] [PubMed]
- La Maestra, S.; De Flora, S.; Micale, R.T. Effect of Cigarette Smoke on DNA Damage, Oxidative Stress, and Morphological Alterations in Mouse Testis and Spermatozoa. Int. J. Hyg. Environ. Health 2015. [Google Scholar] [CrossRef] [PubMed]
- Hamad, M.F.; Dayyih, W.A.A.; Laqqan, M.; AlKhaled, Y.; Montenarh, M.; Hammadeh, M.E. The Status of Global DNA Methylation in the Spermatozoa of Smokers and Non-Smokers. Reprod. Biomed. Online 2018, 37, 581–589. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Expression levels of the PGAM5 gene in Heavy smokers compared to non-smokers males (P-value ≤ 0.03). * P ≤0.05: significant.
Figure 1.
Expression levels of the PGAM5 gene in Heavy smokers compared to non-smokers males (P-value ≤ 0.03). * P ≤0.05: significant.
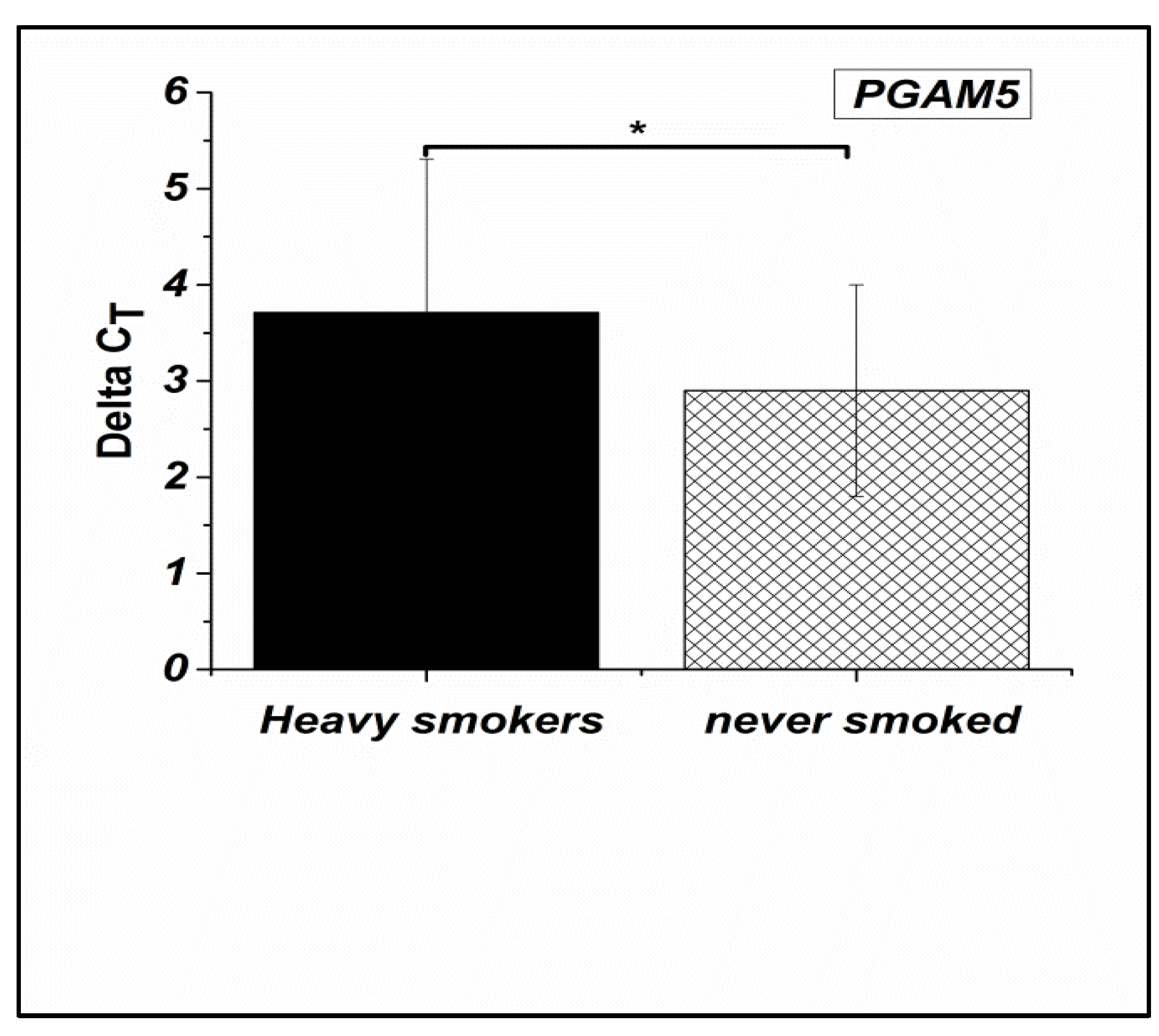
Figure 2.
Expression levels of the PTPRN2 gene in Heavy smokers compared to non-smokers males (P-value ≤ 0.01). * P ≤0.05: significant.
Figure 2.
Expression levels of the PTPRN2 gene in Heavy smokers compared to non-smokers males (P-value ≤ 0.01). * P ≤0.05: significant.
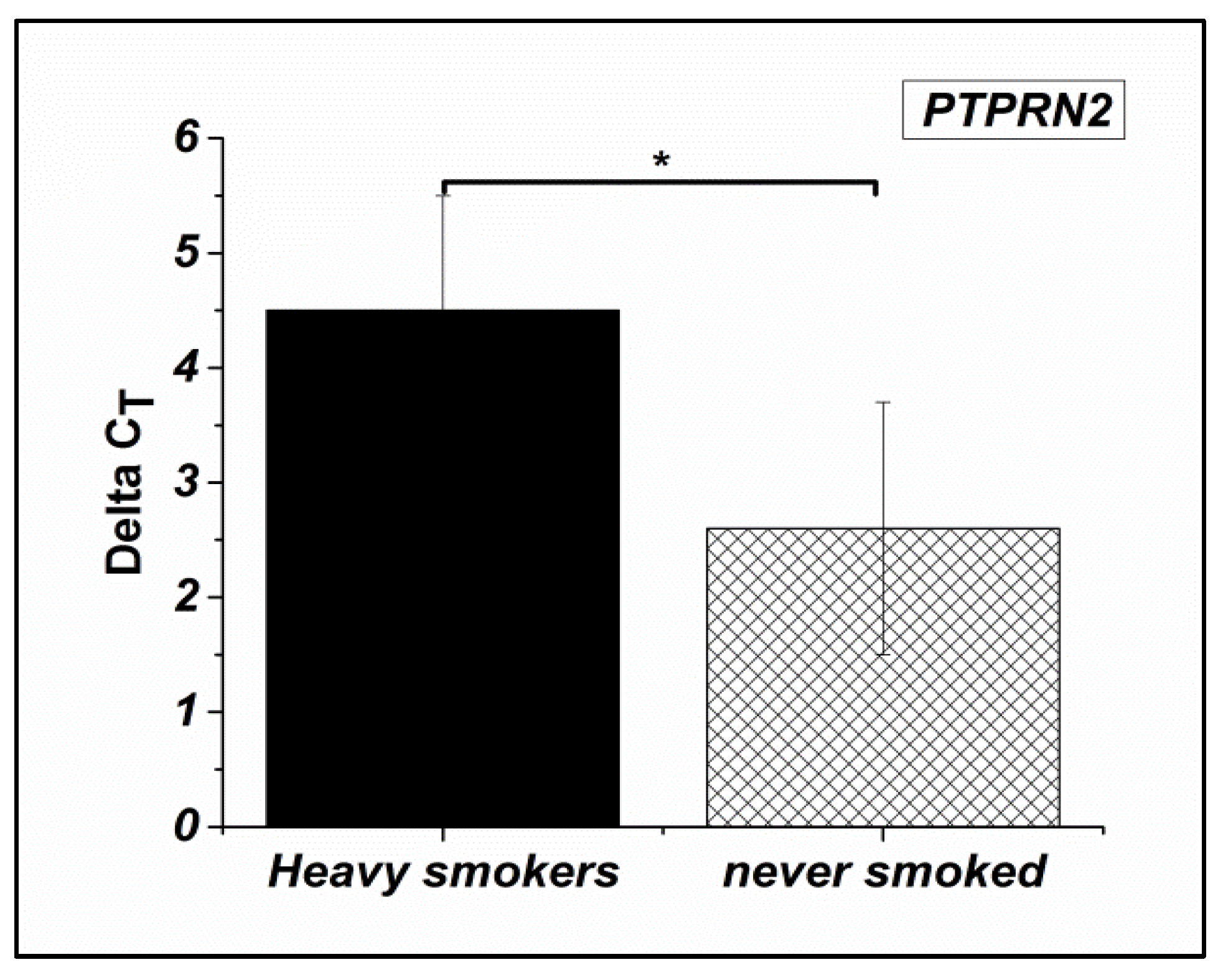
Figure 3.
Expression levels of the TYRO3 gene in Heavy smokers compared to non-smokers males (P-value ≤ 0.3).
Figure 3.
Expression levels of the TYRO3 gene in Heavy smokers compared to non-smokers males (P-value ≤ 0.3).
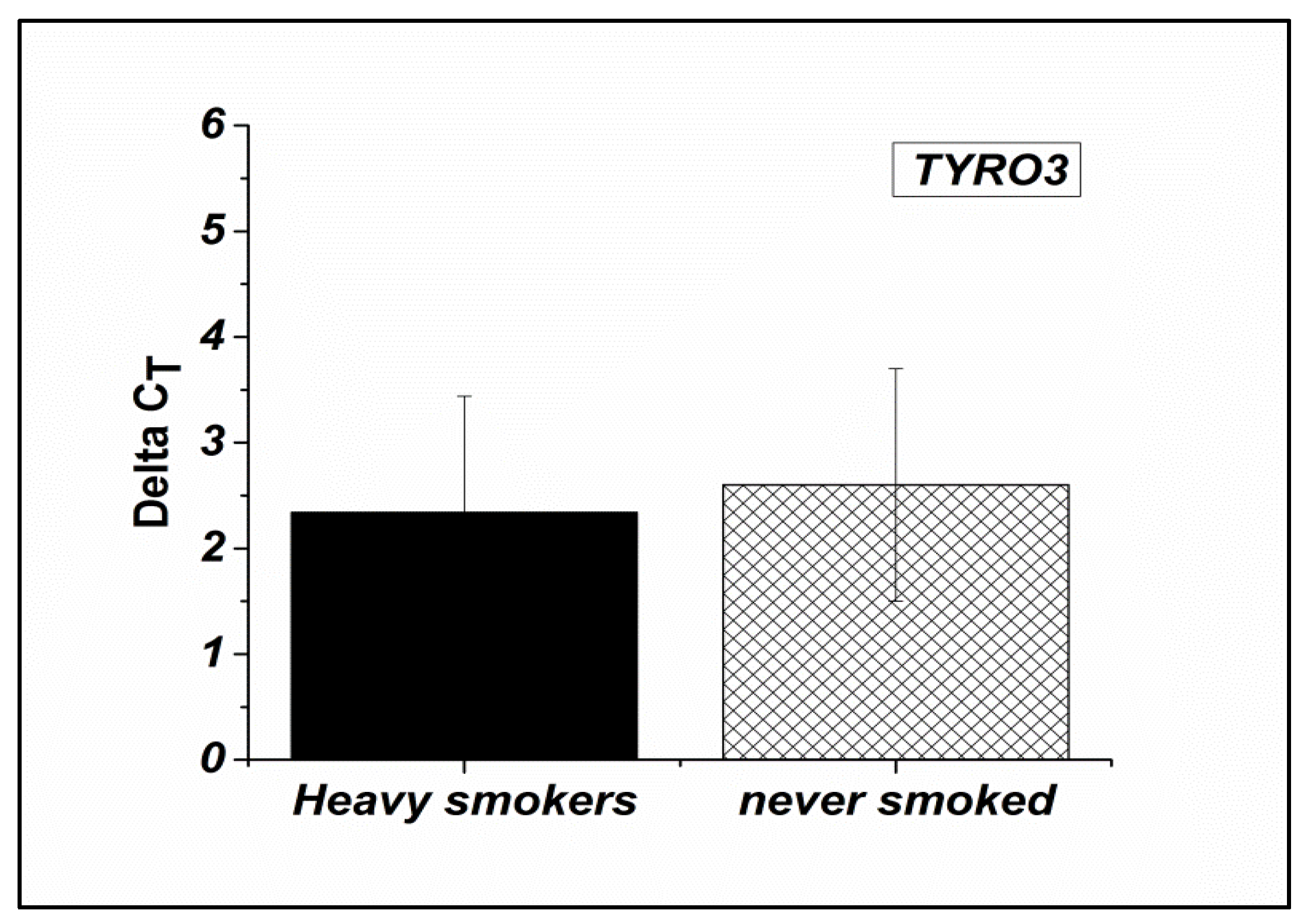

Table 1.
Descriptive characteristics of the study population (N=118): comparison between heavy smokers (N=63) and non-smokers (N=55).
Table 1.
Descriptive characteristics of the study population (N=118): comparison between heavy smokers (N=63) and non-smokers (N=55).
| Variables | Heavy Smokers (Mean ± SD) |
Non-smokers (Mean ± SD) |
P-value |
|---|---|---|---|
| Age | 37.42 ± 5.24 | 36.33 ± 6.18 | ≤ 0.4* |
| Count [mill/ml] | 52.38 ± 34.52 | 64.42 ± 39.18 | ≤ 0.01* |
| Total motility (%) | 44.61 ± 22.47 | 53.67 ± 20.51 | ≤ 0.003* |
| Progressive motility (%) | 33.17 ± 21.87 | 40.96 ± 20.78 | ≤ 0.01* |
| Immotile (%) | 51.13 ± 24.23 | 42.21 ± 18.24 | ≤0.002* |
| Normal form (%) | 17.22 ± 8 .26 | 22.98 ± 12.62 | ≤ 0.05* |
| Vitality (%) | 59.95 ± 13.08 | 64.87 ± 15.18 | ≤ 0.001* |
| Membrane integrity (HOS) (%) | 72.19 ± 10.03 | 78.73± 11.21 | ≤ 0.02* |
| SD, stander deviation. *Mann-Whitney test. P> 0.05: not significant. P ≤0.05: significant. ≤0.01 highly significant | |||
Table 2.
Expression levels of PGAM5, TYRO3 and PTPRN2 genes from spermatozoa in Heavy smokers compared to non-smokers controls groups (n = 118).
Table 2.
Expression levels of PGAM5, TYRO3 and PTPRN2 genes from spermatozoa in Heavy smokers compared to non-smokers controls groups (n = 118).
|
Target |
ΔCT Heavy smokers |
ΔCT Non-smokers |
ΔΔCT |
Fold Change 2- ΔΔCT |
Regulation |
P-value |
|---|---|---|---|---|---|---|
| PGAM5 | 3.7 ± 1.6 |
2.9 ± 1.1 |
0.804 |
0.573 |
Down |
≤0.03* |
| TYRO3 |
2.3 ± 1.1 |
2.4 ± 1.0 |
-0.275 |
1.209 |
Up |
≤0.3* |
| PTPRN2 |
4.5 ± 1.0 |
3.9 ± 0.9 |
0.587 |
0.666 |
Down |
≤0.01* |
*Mann-Whitney test. P> 0.05: not significant. P ≤0.05: significant. ≤0.01 highly significant.
Table 3.
Correlation coefficient between the gene expression level and semen parameters for heavy smokers cases group (n = 63).
Table 3.
Correlation coefficient between the gene expression level and semen parameters for heavy smokers cases group (n = 63).
| Total sperm count (mill/ml) |
Motility (%) | Progressive (%) | Immotile (%) |
Normal form (%) | Vitality (%) | Membrane integrity (HOS) (%) |
||
|---|---|---|---|---|---|---|---|---|
|
PGAM5 |
R | 0.078 | -0.336** | -0.274* | -0.001 | -0.017 | 0.253* | 0.020 |
| P | 0.668 | 0.005 | 0.015 | 0.994 | 0.925 | 0.026 | 0.913 | |
|
TYRO3 |
R | -0.331** | 0.239** | 0.308* | -0.218 | 0.259* | -0.230* | 0.269* |
| P | 0.009 | 0.008 | 0.016 | 0.057 | 0.045 | 0.044 | 0.018 | |
|
PTPRN5 |
R | -0.273* | -0.276* | -0.293** | 0.268* | -0.228* | -0.344* | -0.194 |
| P | 0.016 | 0.015 | 0.009 | 0.018 | 0.046 | 0.021 | 0.202 |
Spearman’s test, r: Correlation coefficient. *P ≤0.05: significant. **<0.01 highly significant.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



