
Submitted:
03 June 2023
Posted:
05 June 2023
You are already at the latest version
Abstract
Co/TiO2 catalysts with different cobalt loadings (3.8, 7.5 and 15 wt%) were prepared by impreg-nation method of Co(NO3)2 .6H2O over titania. Samples containing Co(NO3)2 .6H2O and TiO2 in stoichiometric proportions in order to obtain CoTiO3 and Co2 TiO4 phases were also synthesized. The effect of the calcination treatment at two different temperatures, 550 and 1150 °C was investigated. Characterizations by several techniques, such as XRD UV-vis-NIR, DRS, Raman and XPS were carried out. XRD showed the coexistence of three phases: CoTiO3; Co2TiO4 and Co3O4 after calci-nation at 550°C, while the calcination at high temperature (1150°C) leads to single-phase systems (CoTiO3 or Co2TiO4). Diffuse reflection and XPS spectroscopy showed that divalent cobalt occupies octahedral sites in the ilmenite phase and both tetrahedral and octahedral sites in the spinel phase.
The catalytic performances of the prepared catalysts, calcined at the two different temperatures, were evaluated in the oxidative dehydrogenation reaction (ODH) of ethane to ethylene. The con-version values were almost comparable for all the samples, calcined at 550 °C, and comparable the ethylene selectivity were recorded, slightly higher only in the case of CoTiO3. The calcination at 1150°C did not modify the overall activity (ethane conversion values around 20%), however, the production of COx becomes more significant.
The study examined the influence of the reaction mixture composition, specifically the presence of water, and reveals a slight increase in the yield of ethylene accompanied by a decrease in the overall catalyst activity. This behavior is likely attributed to an increase in the surface concentration of hydroxyl species (OH), resulting in heightened surface acidity.
Keywords:
ODH
; cobalt titanate
; ethane
; oxidative dehydrogenation
1. Introduction
The chemical industry heavily relies on unsaturated hydrocarbons as a raw material for various industrial processes. However, the current industrial capacity for light alkenes such as ethene, propene and butene is expected to be insufficient due to the increasing demands of the petrochemical industry [1,2]. Traditional methods for producing light alkenes, such as catalytic or steam cracking of naphtha and fluid catalytic cracking (FCC) of petroleum crude oil, have negative impacts on the environment [3,4,5]. With the increasing concern for climate change, there is a growing need for new, environmentally friendly methods of producing these chemicals [4,6]. It should be noted that in recent years, several commercial and academic proposals have been put forth as alternatives, promising to produce high-value chemicals from sustainable raw materials [7]. The revolution of shale gas and the prevalence of liquefied natural gas (LNG) technologies have increased the production of light alkanes, thus enhancing the interest in their dehydrogenation as a potential way to apply liquefied natural gas. Alkane dehydrogenation can be mainly achieved by non-oxidative or oxidative dehydrogenation [8,9,10]. It should be noted that thermodynamic dehydrogenation at high temperatures can result in the formation of undesirable by-products. While the oxidation process can improve propylene yield, however, it can also increase undesirable deep oxidation. Various catalytic formulations have been tested in oxidative dehydrogenation of alkanes, often consisting of complex mixtures of oxides. Thus, Catalysts often contain oxides of vanadium, molybdenum, and niobium. For example, the Mo-V-Nb-O system has been shown to have a 100% selectivity at 10% conversion for ODH from ethane to ethylene [11], however, the best results have been obtained using boron oxides on different supports. On alumina catalysts containing 30% weight of B2O3, the yield of ethylene was 14.6% at 38% conversion [12]. Additionally, a system composed of oxides of Co, Zr, P, Fe and K was found to have a selectivity of 74% at 53% conversion [12]. Oxidative dehydrogenation of ethane (ODH-E) is regarded as a highly promising approach for the conversion of ethane into valuable products [13]. When sustainable and easily accessible sources of ethane are employed in this process, the resulting ethylene production has the potential to satisfy both industrial production standards and environmental requirements. It is important to note that such processes should achieve an ethylene yield of greater than 60% in a single pass, with minimal formation of carbon oxides as a byproduct. Others transition metals have also demonstrated promising outcomes as well. For instance, research on various chromium-based catalysts supported on lamellar phosphates have revealed that CrPO4 exhibits ethylene selectivity ranging from 60% to 50% and conversion range from 30 to 20%. This result highlights the role that phosphorus can play as an additive in certain catalytic formulations [14]. ElIdrissi et al. [15,16] have reported that the selectivity for ethylene using a catalyst system composed of V/TiO2 modified with phosphorus at a 1.64 of P/V ratio, was close to 50% at a conversion rate of 33%. They attribute the high selectivity to the acid-base properties and the V4+/V5+ ratio and suggest that the presence of phosphorus adjusts the surface acidity, leading to an increase in the amount of isolated V4+ ions. These vanadium-based catalysts are still the subject of ongoing research and several recent articles have been published on their capacity for oxidative dehydrogenation. [17].
Cobalt-based supported catalysts have been proven to be effective in various processes such as hydro treatment and Fischer-Tropsch reactions, among others [18]. They have also been used in the coupling of methane to form longer-chain hydrocarbons [19]. The incorporation of cobalt in perovskite- like materials as LnCoO3, leads to catalysts with an effective performance in total oxidation and pollution control reactions [20]. The cobalt’s ability to act as an oxidant during the oxidation process is due to the presence of oxygen on the surface of the catalyst when oxygen is present in the process [21]. In ODH of ethane, the cobalt has been used either as a supported catalyst to increase stability and selectivity, incorporated as a promoter to improve catalytic performance, or explored in new materials [22,23,24,25,26,27,28,29].
Despite the promising conversion and selectivity rates reported in literature, the industrial application of oxidative dehydrogenation (ODH) is still limited. Current literature reports an ethylene yield of approximately 20%, whereas industrial applications necessitate a productivity exceeding 1kg C2H4 .kg cat-1 h-1 [30]. Previous research on cobalt supported on titania has shown promising results for oxidative dehydrogenation of ethane [12,31]. Furthermore, XRD analysis of the catalyst at the stationary state revealed the presence of the defined phases such as ilmenite CoTiO3 and spinel Co2TiO4. The best ODH performance of ethane was obtained at 550°C with a catalyst containing 7.5% wt of Co. Notably, the reaction starts with a conversion of 33% and a selectivity around 75%, and then it decreases to reach a stationary state of 22% conversion and 60% selectivity after 150 minutes of running. [12,31]. In view of climate change and the need to update industrial processes, research is focused on developing catalytic materials applied specially in the recovering and valorization of petrol-derived compounds. The purpose of this work is to investigate Co/TiO2 systems at different cobalt loadings for providing information on the formation of the defined phases, CoTiO3 and Co2TiO4, and their catalytic properties.
With this aim we have synthesized and characterized the ilmenite CoTiO3 and spinel Co2TiO4 phases using different spectroscopic techniques. Then, we have investigated their behavior in the oxidative dehydrogenation (ODH) reaction of ethane in order to correlate their activity and selectivity with the environment and the oxidation state of the active sites.
2. Materials and Methods
2.1. Chemical Products
The TiO2 anatase used as support in this work was supplied by Rhone-Poulenc and is characterized by a specific surface area of 89 m2 g-1.
Other reagents, including Co(NO3)2.6H2O, (Analytical Reagent, Sinopharm Chemical Reagent Co., Ltd.), were used without any further purification.
2.2. Preparation of Catalysts
Co/TiO2 catalysts with cobalt loadings equal to 3.8, 7.5, and 15.0 wt% were prepared according to a previously described method [12,31]. The TiO2 support was put in contact with a Co(NO3)2.6H2O solution at suitable concentration in order to obtain the desired cobalt content. The suspension was kept under stirring at 80° C until complete evaporation of the water. The recovered solid was dried at 120° C, then calcined at 550° C or at 1150°C for 4 hours under air. The CoTiO3 and Co2TiO4 phases were synthesized as previously described by using TiO2 and Co(NO3)2 .6H2O solution in stoichiometric proportions in order to form the desired phases composition. Calcination at 550 °C and or 1150 °C was performed as above reported.
2.3. Characterization Techniques
Specific surface areas of the samples were measured at −196 °C with a nitrogen sorption technique using the ASAP 2020 equipment (Micromeritics, Norcross, Catalysts 2023, 13, 606 15 of 18 GA, USA). Before the measurements, the powder samples were degassed at 250 °C for 2 h. The specific surface area was calculated via the Brunauer-Emmett-Teller (BET) method in the standard pressure range 0.05–0.3 P/P0.
The X-ray diffraction patterns were used to identify the phases present in the samples and to determine their crystalline structure. They were collected using a Siemens D500 high resolution spectrometer with Cu Kα radiation (Kα = 1.540598 A) at room temperature. The data were recorded with a step of 0.05° (2θ) and a counting time of 10 seconds per step. The assignment of the crystalline phases was based on the JCPDS powder diffraction file cards.
Diffuse reflection UV-visible-NIR spectra were recorded in the wavelength range of 190 to 2500 nm using a Varian Cary 5E double monochromator spectrometer, which was equipped with an integrating sphere. To establish a reference, polytetrafluoroethylene was utilized.
Laser Raman (LR) spectra were obtained using a Dilor XY spectrometer in backscatter geometry with a 514.5 nm excitation line from a Spectra Physics Model 165 coherent argon laser, with power kept below 20 mW. The spectrometer was equipped with a monochromator for selecting specific spectral ranges and a Jobin-Yvon 1024*256 CCD matrix thermo-electrically cooled as a multi-channel detector.
XPS spectra were recorded on a Vacuum Generators MK I spectrometer with an un-monochromated Al Kα (1486.6 eV, 200 watts) under about 10−9 Torr. The spectra were digitized, summed, smoothed, and reconstructed using Gauss–Lorentzian components. The measurements were carried out on powdered samples dispersed on an indium plaque, using the C1s peak at 285 eV as a reference.
2.4. Catalytic Tests
The oxidative dehydrogenation of ethane (ODH) was carried out in a continuous flow microreactor made of quartz, operating at atmospheric pressure, in the range of temperature 450-550°C. The catalyst, which was sieved to a particle size between 125 and 180 μm, was placed in the reactor between two plugs of quartz wool and was purged with pure nitrogen before the reaction. The reaction mixture consisted of 6 vol.% ethane, 3 vol.% O2, and 91 vol.% N2. The effluent gases were analyzed using two on-line chromatographs, one (FID) equipped with a Porapak Q column for hydrocarbons and the other (TCD) with a silica gel column for oxygenated products. At the experimental conditions chosen, the reaction light off occurred at around 300°C and produced only ethylene and COx.
3. Results and Discussion
3.1. Characterization
The activity study of the defined CoTiO3 and Co2TiO4 phases is imposed after their formation in the stationary state during a study of the x%/TiO2 catalysts in ODH of ethane, hence the importance of associating the latter to this study. The X-ray diffraction patterns of the prepared x% Co/TiO2 samples are displayed in Figure 1. An examination of the obtained results reveals that the sample diffractogram with a low concentration of cobalt 3.8 wt % (Figure 1, curve b), is characterized by peaks of the anatase titania (JCPDS file #84-1286). For the 7.5 and 15.0 wt% cobalt samples, in addition to anatase, a few peaks were observed (Figure 1, c, d), denoting Co3O4 formation (JCPDS file no. 78-1970). The intensity of the system exhibits a direct correlation with the concentration of cobalt, whereby higher cobalt concentrations lead to increased intensity levels. Additionally, when the Co content increases from 7.5% to 15%, the average particle size ranges from 17 to 19 nm, which is in agreement with the findings reported by Ho et al. [32]. The study revealed the presence of Co3O4 crystallites in samples that contain as little as 1.5% of Co within the anatase and rutile mixture, which also has a surface area of 50 m2 g-1. The findings of this work are different from those reported previously and by other researchers, which could be a result of the greater surface area of anatase present in our samples. This leads to improved dispersion of Co3O4 and potentially the formation of some amorphous species that are not identifiable by X-ray diffraction.
The XRD patterns of CoTiO3 and Co2TiO4 samples calcined at different temperatures are shown in Figure 2A and Figure 2B, respectively. For both samples, it can be observed that the calcination at 550°C leads mainly to a mixture of phases: CoTiO3, Co2TiO4 and Co3O4. The Co2TiO4 is minority compared to the other phases. Increasing temperature to 750°C for the CoTiO3 sample decreases the intensity of the Co2TiO4 lines. Similarly, a significant decrease in the Co3O4 lines can be observed. Calcination at higher temperature (1150°C) leads to single-phase systems: the ilmenite CoTiO3 and spinel Co2TiO4 phases.
On the other hand, the study of ilmenite CoTiO3 phase by XRD showed that the Ti4+ and Co2+ ions occupy slightly distorted octahedral sites. The ilmenite structure is found in oxides with the formula ABO3 when the A and B ions have similar sizes and a total charge of +6. The name ilmenite comes from the mineral FeIITiIVO3, which has a structure similar to corundum. It consists of a compact hexagonal arrangement of oxygen ions and cations occupying two thirds of the octahedral sites. Divalent and tetravalent ions form alternating layers. On the other hand, Co2TiO4 has an inverse spinel structure which can be represented by the formula Co2+(Co2+Ti4+)O4, where half of the Co2+ ions occupy the tetrahedral sites, the other half of the Co2+ ions and Ti4+ ions occupy the octahedral sites.
Table 1 shows the Specific Surface Areas (m2/g) (SSA) of Co(x%)/TiO2 and CoTiO3 and Co2TiO4 defined phases. It has been noticed that the SSA of the CoTiO3 and Co2TiO4 phases are lower than the Co(x)/TiO2 samples and these values decrease when calcination temperature of CoTiO3 and Co2TiO4 increase, which could be due to sintering and reduction in the number of active sites available for catalysis.
Diffuse reflection UV-visible-NIR spectroscopy offers valuable insights into the oxidation state of transition metals, their coordination, and the symmetry of their surrounding environment. Co/TiO2 absorption spectra in the UV-visible-NIR are displayed in Figure 3. It should be noted that distinguishing between different species is challenging for two distinct considerations: (i) in the 1100 - 1500 nm wavelength range, the v1 transition of octahedral ions partially overlaps with the v2 transition of tetrahedral species. In addition, the high coefficient of absorption of the tetrahedral species makes it difficult to detect the first transition; (ii) in TiO2, a Co2+ → Ti4+ charge transfer can take place. Indeed, an absorption of Co2+ modified MgTi2O5 in the 400-500 nm wavelength region was assigned to this transition mode [12,33]. It is important to consider Co2+ to Ti4+ charge transfers since cations of active phase and the support have variable valence. The UV spectrum of TiO2 shows strong absorption related to the interband transition and maxima around 320 and 220 nm are attributed to Co2+ to Ti4+ charge transfers. The 3.8 wt% Co sample has slight absorption in the visible range (Figure 3), however for 7.5 and 15 wt% Co samples, new bands appear and intensify with increasing Co load. The near-infrared (NIR) region has a broad band in the 1200 and 1600 nm interval, as well as the band of 2"(OH) harmonics of residual hydroxyl groups, which is located close to 1380 nm. The visible range has another band around 700 nm, and a shoulder with a maximum at 420 nm in the range of 400 and 600 nm observed only in the loaded 15 wt% Co sample (Figure 3, curve d). These results indicate the presence of different cobalt species [12].
The UV-visible-NIR spectra of the CoTiO3 and Co2TiO4 samples calcined at different temperatures (550, 750 and 1150°C) are shown in Figure 4A andFigure 4B. In general, the calcination temperature changes drastically the position, shape and relative intensities of the bands.
In the case of the CoTiO3 sample calcined at 550° C, two broad bands A and B appear at 596 and 760 nm respectively. The calcination temperature has no effect on the position of these bands, however, a variation in their relative intensities is observed after calcination. In the NIR part, a notable modification of the maxima of the bands was observed. Indeed, the sample calcined at 550°C is characterized by the appearance of the bands at 1250, 1350 and 1530 nm and a shoulder at 1750 nm (Figure 4A; spectrum a) which are already attributed to the transition 4A2(W)→4 T1 (F) divalent cobalt in tetrahedral site. The treatment at 1150°C leads to the appearance of an intense and broad band centered at 1530 nm (Figure 4A, spectrum c). Also note that in the ilmenite CoTiO3 structure, the Co2+ ions are in octahedral symmetry. In the olivine Co2SiO4 structure taken as reference, the Co2+ occupy the octahedral interstices. The UV-visible spectrum of this compound (data not shown) has a similar appearance with that of the ilmenite CoTiO3 phase (Figure 4, spectrum c). The bands at 600, 760 and 1530 nm are assigned to the three 4 T 1g → 4 T 1g (P); 4T 1g →_ 4 A 2g (F) and 4 T 1g→ 4 T 2g (F) divalent Cobalt in octahedral sites.
The fundamental 4F level of the Co2+ ion in the tetrahedral field breaks down into the lowest ,4A2 4 T1 and 4 T2. The transition 4A2 (F) →4T2 (F) is forbidden to the electric dipole approximation and allowed to the magnetic dipole approximation. It is indeed very weak and often appears in the infrared region. The two transitions 4A2 (F) →4T1 (F) and 4A2 (F) →4T1 (P) give very high intense absorption bands, often in three components because of the spin-orbit coupling.
The spectrum of the Co2TiO4 sample calcined at 550°C is similar to that of CoTiO3 calcined at the same temperature of 550°C (Figure 4B, curve a). After treatment at 750°C (Figure 4B, curve b), its appearance changes radically in the visible part. Indeed, it is noticed the appearance of several absorptions: 588,638 and 688 nm and a shoulder around 750 nm.
At higher calcination temperature, a broad absorption spreads between 1300 nm up to 1820 nm which has already been observed in the case of soda-lime glasses where divalent cobalt and in tetrahedral symmetry [12]. The two shoulders around 535 and 720 nm are respectively attributed to the 4T1g →4T1g (F) and 4T1g→4T1g (F) of Co2+ in octahedral sites. The band at 720 nm has already been observed in the case of CoNaY zeolites [34].
Laser Raman spectroscopy is a widely used tool in the field of supported and unsupported catalysis. The obtained data for the three Co/TiO2 catalysts are depicted in Figure 5A. The band observed at 397, 514 and 640 cm-1 are characteristic of TiO2 anatase structure, (curve a). When 3.8% wt of cobalt was added, the spectrum was not impacted (spectrum b). In contrast, higher cobalt levels spectra show bands close to 692 cm-1 and two shoulders at 487 and 528 cm-1, suggesting the formation of Co3O4 [12,35].
The Raman spectra of the two CoTiO3 and Co2TiO4 phases calcined at 1150°C are shown in Figure 5B. As far as the spinel is concerned, no Raman vibration mode has been observed in the frequency range between 200 and 1000 cm-1 (Figure 5B-a). On the other hand, the Raman spectrum of the ilmenite CoTiO3 phase (Figure 5B, spectrum b) shows several bands respectively at 688, 450, 379, 332, 263 and 233 cm-1 and two shoulders at 602 and 204 cm-1. The mode of vibration at 688 cm-1 is the most intense. Several isostructural compounds such as (Ni; Fe; Mg)TiO3 exhibit the same Raman line around 700 cm-1 . It corresponds to the mode of vibration A1g of symmetrical elongation of octahedron MO6 [36].
The XPS spectra of the CoTiO3 and Co2TiO4 phases are illustrated in Figure 6A and Figure 6B, respectively. The spectral lines of the Co2p of these compounds such as the Co2p3/2 binding energies, the spin-orbit coupling ΔE (Co2p1/2 -Co2p3/2) as well as the S/M ratios (satellite intensity/main peak intensity ratio) are reported in Table 2.
The Co2p3/2 binding energies (CoTiO3 : 781.2 eV; Co2TiO4 : 781 eV) as well as the ratio (S/M) are in good agreement with the presence of paramagnetic ions (Co2+). The energy difference ΔE (Co2p1/2 -Co2p3/2) is 16 eV in the case of CoO [12]. It is close to the CoTiO3 phase ΔE value, which contains only Co2+ in octahedral sites. For the spinel phase, a ΔE value of 15.8 eV is observed. It should be noted that this difference in energy of the 2p1/2 and 2p3/2 electrons usually allows the distinction of the tetrahedral sites, the octahedral sites, however, in the case of the mixed spinel, this discrimination is practically impossible.
3.2. Catalytic Results and Discussion
In order to find a Co/TiO2 compound adequate to compare the studied CoTiO3 and Co2TiO4 phases, the experiments were conducted at temperature of 550°C. Firstly, the conversion on pure TiO2 as support has been less than 5%. At steady state, for some Co/TiO2 catalysts, the ethane conversion increase with reaction temperature in the range 450-550°C (Figure 7). The conversion rate increases as the cobalt amount increases, reaching a plateau from 7.5 wt% Co. The catalyst loaded 7.5wt% of Co shows a conversion, at 550°C, around 22.2% with an ethylene yield close to 13.3% (60% selectivity).
This result allows us to select the sample containing 7.5 wt % Co as a reference to compare the catalytic performances of the CoTiO3 and Co2TiO4 phases. Figure 8 shows the variation of ethane conversion as a function of time on Co(7.5)/TiO2 at 550°C, compared to CoTiO3 and Co2TiO4 catalytic activities. For the Co (7.5)/TiO2, the conversion decreases from 33% to around 22.2% when it reaches steady state after 3 h of reaction. During this time, the specific surface has been significantly reduced and the color of the catalyst changes from light gray to green, probably due to the change in the coordination and/or the oxidation state of the cobalt cations. Figure 8 displays also the conversion of ethane with time over the defined phases CoTiO3 and Co2TiO4 calcined at 550 and 1150 °C, respectively. The evolution of the ethane conversion shows that these solids are stable over time regardless of the temperature of their calcination. At 550°C the ethane conversion is of the same order of magnitude as that measured with the Co (7.5)/TiO2 sample. Therefore, it can be surmised that the octahedral Co2+ ions, present in these phases, are responsible of the activity [12,31]. This result is in agreement with the behavior observed in catalysts modified by phosphorus [12] and confirms the role played by the Co3+ ions in the orientation of the reaction.
On the other hand, the catalytic results obtained for the CoTiO3 and Co2TiO4 samples calcined at different temperatures (550°C and 1150°C) are reported in Table 3. By comparing these performances, the following observations can be made:
• The evolution of overall conversion (Figure 8) shows that these materials are stable over time regardless of their calcination temperature. At 550°C, the overall conversion of ethane is of the same order of magnitude as that measured with the Co(7.5)/TiO2 sample (αg= 22.2%).
• For a calcination temperature of 550°C, the selectivity towards ethylene (Table 3) is slightly higher in the case of CoTiO3.
• No significant modification of catalytic activity was observed after calcination at 1150°C, but the production of COx becomes higher.
Apparent activation energy of C2H6 oxydehydrogenation on the defined phases catalysts was calculated using the Arrhenius equation (Eq. 1). The relationship between the reaction rate r and the reaction rate constant k was obtained using the theory of reaction series. The expression for the catalytic reaction rate r is identical to that used by Wang et al. [37].
k is the reaction rate constant, A is the pre-exponential factor, E is the apparent activation energy, and R is the molar gas constant. In Eq. 2, 𝛼, 𝛽, and 𝛾 are reaction orders; C1, C2, and Cn are reactant concentrations. In Eq. 4, α is the conversion of reactants, F is the gas flow rate, and M is the mass of the catalyst.
By combining (1), (2) and (3) equations, if the linear relationship between ln(r) and 1/T can be fitted, then the slope of the line is determined. After further calculation, the apparent activation energy E can be obtained (Table 3).
The obtained values of E for each of the defined phases calcined at 550-1150°C are represented in Table 3. The values are quite low and the phases calcined at higher temperatures are higher. The order of magnitude of the values suggests a certain influence of the homogeneous phase reaction.
The performance of the catalysts has been compared, determining their selectivity for the same conversion at the identical experimental conditions. At temperature reaction of 550°C, all the catalytic results obtained for the CoTiO3 and Co2TiO4 samples calcined at different temperatures (550°C and 1150°C) are reported in Figure 9. The evolution of the total conversion shows a notable modification of the catalytic activity after calcination at 1150°C, however the production of COx becomes higher. It has been observed that both catalysts have comparable performances when calcined at 550°C. After calcination at 1150°C, the CoTiO3 phase became more efficient.
Impact of Water Vapor in ODH reaction
In general, water increases the surface acidity and, in this sense, facilitates desorption. Table 4 summarizes the results obtained when steam was added to the reaction mixture at different percentages. It was observed that both phases CoTiO3 and Co2TiO4 show the same catalytic behavior in the presence of water, whatever their calcination temperature.
It can be pointed out that there is a decrease in the overall ethane conversion and an increase in the ethylene selectivity when the percentage of water increases (Table 4). On the other hand, it is also noticeable that the yield of ethylene increases slightly with a reaction mixture containing 1.7% of H2O.
The comparison of the selectivity, obtained for different catalysts, can be appreciated from the plots shown in Figure 9 and Figure 10. It is possible to classify the samples in two categories, the first one, the less selective in ethylene, includes the samples tested with the dry mixture. The second category, more selective, is obtained in the presence of steam.
From these obtained results, it can be considered that the limiting step is the desorption of the ethylene molecule from the catalyst surface. The presence of water vapor in the reaction mixture promotes this desorption through competition for site occupation. However, at high percentages of water vapor, a significant portion of the active sites are occupied, leading to a significant decrease in overall conversion. The increase in C2H4 selectivity is therefore due to an increase in the OH* species at the catalyst surface [38] relative to the O* species, which favors total oxidation.
4. Conclusion
This work focuses on catalysts with high cobalt contents supported on anatase titanium oxide and analyzes their steady-state composition. The samples characterization with cobalt contents of 7.5% and 15wt% demonstrated highly satisfactory catalytic performance at 550°C. Furthermore, post-catalytic testing revealed that the attainment of a stationary state is dependent on the formation of two phases: ilmenite CoTiO3 and spinel Co2TiO4. Therefore, these defined phases have been further studied in the ODH.
The catalytic results obtained on these phases show that they are active but less selective for ethylene. It therefore seems that the divalent cobalt ions present in these phases are responsible for their activity. Ethylene selectivity could be related to the presence of Co3+ ions.
The presence of water in the reaction mixture was examined revealing a slightly increased yield of ethylene, but also a lower catalytic activity. The increased surface acidity due to the higher surface concentration of hydroxyl species (OH) is likely the cause of this behavior.
In the overall it has also been demonstrated that there is a correlation between the catalytic activity, surface acidity and the chemical environment of the active species.
Further investigations by employing new synthesis protocols will be carried out in order to optimize the [Co3+]/ [Co+2 oct] ratio. Moreover, the addition to the active phase of new elements, such as alkali metals, could be favorable for the selective production of ethylene.
Author Contributions
H.M. and Y.B; preparation and characterization of catalysts performing experiments and original draft preparation; E.L.G. and L.C. characterization of catalysts; A. B. and A. E.H. supervised experiments and finalized the writing of the manuscript; M.K. designed the catalyst; J.T.; L.F.L. contributed to complete the writing of the manuscript in its final form. All authors have read, agreed and contributed to the achievement of this work.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Blay, V.; Epelde, E.; Miravalles, R.; Perea, L.A. Converting olefins to propene: Ethene to propene and olefin cracking. Catalysis Reviews 2018, 60, 278–335. [Google Scholar] [CrossRef]
- Cnudde, P.; De Wispelaere, K.; Vanduyfhuys, L.; Demuynck, R.; Van der Mynsbrugge, J.; Waroquier, M.; Van Speybroeck, V. How Chain Length and Branching Influence the Alkene Cracking Reactivity on H-ZSM-5. ACS Catalysis 2018, 8, 9579–9595. [Google Scholar] [CrossRef] [PubMed]
- Gholami, Z.; Gholami, F.; Tišler, Z.; Tomas, M.; Vakili, M. A Review on Production of Light Olefins via Fluid Catalytic Cracking. Energies 2021, 14. [Google Scholar] [CrossRef]
- Usman, A.; Siddiqui, M.A.B.; Hussain, A.; Aitani, A.; Al-Khattaf, S. Catalytic cracking of crude oil to light olefins and naphtha: Experimental and kinetic modeling. Chemical Engineering Research and Design 2017, 120, 121–137. [Google Scholar] [CrossRef]
- Sun, M.; Zhang, J.; Putaj, P.; Caps, V.; Lefebvre, F.; Pelletier, J.; Basset, J.-M. Catalytic Oxidation of Light Alkanes (C1–C4) by Heteropoly Compounds. Chemical Reviews 2014, 114, 981–1019. [Google Scholar] [CrossRef] [PubMed]
- Owusu, P.A.; Asumadu-Sarkodie, S. A review of renewable energy sources, sustainability issues and climate change mitigation. Cogent Engineering 2016, 3, 1167990. [Google Scholar] [CrossRef]
- Wu, X.; Tian, Z.; Guo, J. A review of the theoretical research and practical progress of carbon neutrality. Sustainable Operations and Computers 2022, 3, 54–66. [Google Scholar] [CrossRef]
- Sattler, J.J.H.B.; Ruiz-Martinez, J.; Santillan-Jimenez, E.; Weckhuysen, B.M. Catalytic Dehydrogenation of Light Alkanes on Metals and Metal Oxides. Chemical Reviews 2014, 114, 10613–10653. [Google Scholar] [CrossRef]
- Monai, M.; Gambino, M.; Wannakao, S.; Weckhuysen, B.M.J.C.S.R. Propane to olefins tandem catalysis: a selective route towards light olefins production. 2021, 50, 11503–11529. [Google Scholar] [CrossRef]
- Melnikov, D.P.; Novikov, A.A.; Glotov, A.P.; Reshetina, M.V.; Smirnova, E.M.; Wang, H.Q.; Vinokurov, V.A. Dehydrogenation of Light Alkanes (A Review). Petroleum Chemistry 2022, 62, 1027–1046. [Google Scholar] [CrossRef]
- Ai-Zegbayer, Y.S.; Ai- Mayman, S.I.; Ai-Smarei, T.A. Oxidative Dehydrogenation of Ethane to Ethylene Over Mo-V-Nb Catalysts: Effect of Calcination Temperature and Type of Support. Journal of King Saud University - Engineering Sciences 2010, 22, 21–27. [Google Scholar] [CrossRef]
- Brik, Y.; Kacimi, M.; Ziyad, M.; Bozon-Verduraz, F. Titania-Supported Cobalt and Cobalt–Phosphorus Catalysts: Characterization and Performances in Ethane Oxidative Dehydrogenation. Journal of Catalysis 2001, 202, 118–128. [Google Scholar] [CrossRef]
- Zhang, Z.; Ding, J.; Chai, R.; Zhao, G.; Liu, Y.; Lu, Y. Oxidative dehydrogenation of ethane to ethylene: A promising CeO2-ZrO2-modified NiO-Al2O3/Ni-foam catalyst. Applied Catalysis A: General 2018, 550, 151–159. [Google Scholar] [CrossRef]
- Loukah, M.; Coudurier, G.; Vedrine, J.C.; Ziyad, M. Oxidative dehydrogenation of ethane on V- and Cr-based phosphate catalysts. Microporous Materials 1995, 4, 345–358. [Google Scholar] [CrossRef]
- El-Drissi, J.; Kacimi, M.; Loukah, M.; Ziyad, M.J.J.C.P. Activité de V2O5/TiO2 modifié par le phosphore dans la réaction de déshydrogénation oxydante de l’éthane en éthylène. 1997, 94, 1984-1992.
- El-Idrissi, J.; Kacimi, M.; Bozon-Verduraz, F.; Ziyad, M. Oxidative dehydrogenation of ethane over Cr/TiO2 modified by phosphorus. Catalysis Letters 1998, 56, 221–225. [Google Scholar] [CrossRef]
- Benzaouak, A.; Mahir, H.; El Hamidi, A.; Kacimi, M.; Liotta, L.F. Investigation of Phosphorus Loaded V2O5/ZrO2 Catalysts for the Oxidative Dehydrogenation of Propane (ODH). Catalysts 2022, 12. [Google Scholar] [CrossRef]
- Munirathinam, R.; Pham Minh, D.; Nzihou, A. Effect of the Support and Its Surface Modifications in Cobalt-Based Fischer–Tropsch Synthesis. Industrial & Engineering Chemistry Research 2018, 57, 16137–16161. [Google Scholar] [CrossRef]
- Lögdberg, S.; Yang, J.; Lualdi, M.; Walmsley, J.C.; Järås, S.; Boutonnet, M.; Blekkan, E.A.; Rytter, E.; Holmen, A. Further insights into methane and higher hydrocarbons formation over cobalt-based catalysts with γ-Al2O3, α-Al2O3 and TiO2 as support materials. Journal of Catalysis 2017, 352, 515–531. [Google Scholar] [CrossRef]
- Dreyer, M.; Krebs, M.; Najafishirtari, S.; Rabe, A.; Friedel Ortega, K.; Behrens, M. The Effect of Co Incorporation on the CO Oxidation Activity of LaFe1−xCoxO3 Perovskites. Catalysts 2021, 11. [Google Scholar] [CrossRef]
- Dreyer, M.; Rabe, A.; Budiyanto, E.; Friedel Ortega, K.; Najafishirtari, S.; Tüysüz, H.; Behrens, M. Dynamics of Reactive Oxygen Species on Cobalt-Containing Spinel Oxides in Cyclic CO Oxidation. Catalysts 2021, 11. [Google Scholar] [CrossRef]
- Aaddane, A.; Kacimi, M.; Ziyad, M. Oxidative dehydrogenation of ethane and propane over magnesium-cobalt phosphates CoxMg3-x(PO4)2. Catalysis Letters 2001, 73, 47–53. [Google Scholar] [CrossRef]
- Aaddane, A.; Kacimi, M.; Ziyad, M. Oxidative dehydrogenation of ethane and propane over Ca-Co-P catalysts. In Proceedings of the Journal De Physique. IV : JP; 2004; pp. 151–154. [Google Scholar]
- Concepción, P.; Blasco, T.; López Nieto, J.M.; Vidal-Moya, A.; Martínez-Arias, A. Preparation, characterization and reactivity of V- and/or Co-containing AlPO-18 materials (VCoAPO-18) in the oxidative dehydrogenation of ethane. Microporous and Mesoporous Materials 2004, 67, 215–227. [Google Scholar] [CrossRef]
- Čapek, L.; Vaněk, L.; Adam, J.; Smoláková, L. Dehydrogenation of ethane over vanadium, cobalt and nickel based catalysts. Studies in Surface Science and Catalysis 2008, 174, 1175–1178. [Google Scholar] [CrossRef]
- Alifanti, M.; Bueno, G.; Parvulescu, V.; Parvulescu, V.I.; Cortés Corberán, V. Oxidation of ethane on high specific surface SmCoO3 and PrCoO3 perovskites. Catalysis Today 2009, 143, 309–314. [Google Scholar] [CrossRef]
- Tian, X.; Zheng, C.; Li, F.; Zhao, H. Co and Mo Co-doped Fe2O3for Selective Ethylene Production via Chemical Looping Oxidative Dehydrogenation. ACS Sustainable Chemistry and Engineering 2021, 9, 8002–8011. [Google Scholar] [CrossRef]
- Xiao, F.; Guo, D.; Zhao, F.; Zhao, Y.; Wang, S.; Ma, X. Catalytic oxidative dehydrogenation of ethane using carbon dioxide as a soft oxidant over Co-HMS catalysts to ethylene. Asia-Pacific Journal of Chemical Engineering 2022, 17. [Google Scholar] [CrossRef]
- Povari, S.; Alam, S.; Somannagari, S.; Nakka, L.; Chenna, S. Oxidative Dehydrogenation of Ethane with CO2 over the Fe-Co/Al2O3 Catalyst: Experimental Data Assisted AI Models for Prediction of Ethylene Yield. Industrial and Engineering Chemistry Research 2023, 62, 2573–2582. [Google Scholar] [CrossRef]
- Koirala, R.; Safonova, O.V.; Pratsinis, S.E.; Baiker, A. Effect of cobalt loading on structure and catalytic behavior of CoOx/SiO2 in CO2-assisted dehydrogenation of ethane. Applied Catalysis A: General 2018, 552, 77–85. [Google Scholar] [CrossRef]
- Brik, Y.; Kacimi, M.; Bozon-Verduraz, F.; Ziyad, M. Characterization and Comparison of the Activity of Boron-Modified Co/TiO2 Catalysts in Butan-2-ol Conversion and Oxidative Dehydrogenation of Ethane. Journal of Catalysis 2002, 211, 470–481. [Google Scholar] [CrossRef]
- Advanced materials in catalysis / edited by James, J. Burton and Robert L. Garten; Academic Press: New York, 1977. [Google Scholar]
- Blasse, G. Optical electron transfer between metal ions and its consequences. In Proceedings of the Complex Chemistry, Berlin, Heidelberg, 1991; 1991//; pp. 153–187. [Google Scholar]
- Verberckmoes, A.A.; Weckhuysen, B.M.; Schoonheydt, R.A. Spectroscopy and coordination chemistry of cobalt in molecular sieves1Dedicated to Professor Lovat V.C. Rees in recognition and appreciation of his lifelong devotion to zeolite science and his outstanding achievements in this field.1. Microporous and Mesoporous Materials 1998, 22, 165–178. [Google Scholar] [CrossRef]
- Farhadi, S.; Javanmard, M.; Nadri, G. Characterization of Cobalt Oxide Nanoparticles Prepared by the Thermal Decomposition. Acta Chimica Slovenica 2016, 63. [Google Scholar] [CrossRef]
- Vennari, C.E.; Williams, Q. A high-pressure Raman study of FeTiO3 ilmenite: Fermi resonance as a manifestation of Fe-Ti charge transfer. Physics and Chemistry of Minerals 2021, 48, 34. [Google Scholar] [CrossRef]
- Peng, W.; Ziqi, W.; Zhongqing, Y.; Zhilei, L.; Jingyu, R.; Mingnv, G. Selective catalytic and kinetic studies on oxydehydrogenation of ethane with CO2 over lanthanide metal catalysts. Comptes Rendus. Chimie 2020, 23, 33–46. [Google Scholar] [CrossRef]
- Zeng, T.; Sun, G.; Miao, C.; Yan, G.; Ye, Y.; Yang, W.; Sautet, P. Stabilizing Oxidative Dehydrogenation Active Sites at High Temperature with Steam: ZnFe2O4-Catalyzed Oxidative Dehydrogenation of 1-Butene to 1,3-Butadiene. ACS Catalysis 2020, 10, 12888–12897. [Google Scholar] [CrossRef]
Figure 1.
XRD patterns of Co(x)/TiO2 samples, TiO2 :(a), Co(3.8) : (b), Co(7.5) :(c) and Co(15:(d).
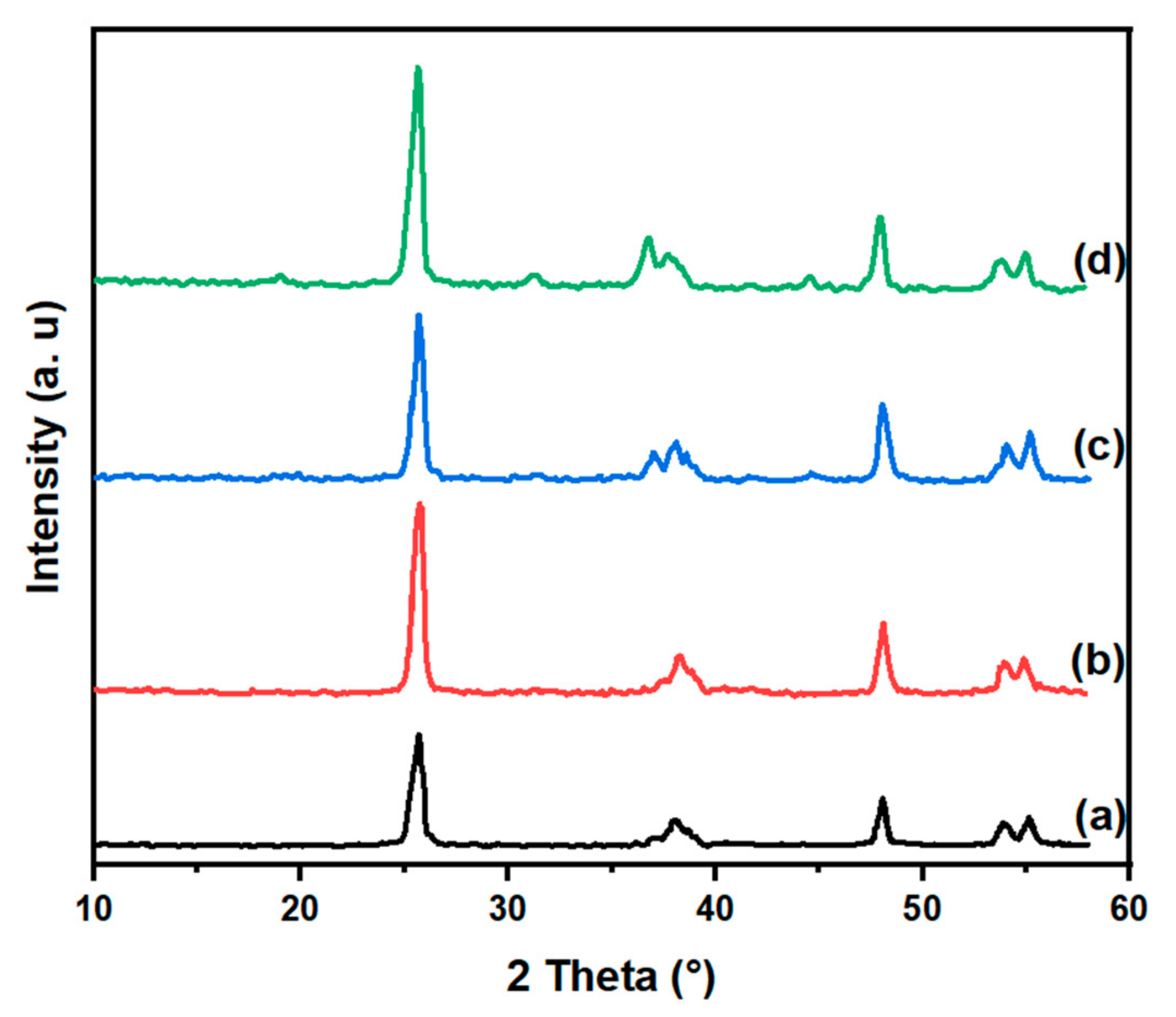
Figure 2.
XRD diagrams of the (A) CoTiO3 and (B) Co2TiO4 samples as a function of the calcination temperature.
Figure 2.
XRD diagrams of the (A) CoTiO3 and (B) Co2TiO4 samples as a function of the calcination temperature.


Figure 3.
DRS spectra of Co (x)/TiO2 samples, TiO2 :(a), Co(3.8) : (b), Co(7.5) :(c) and Co(15:(d).
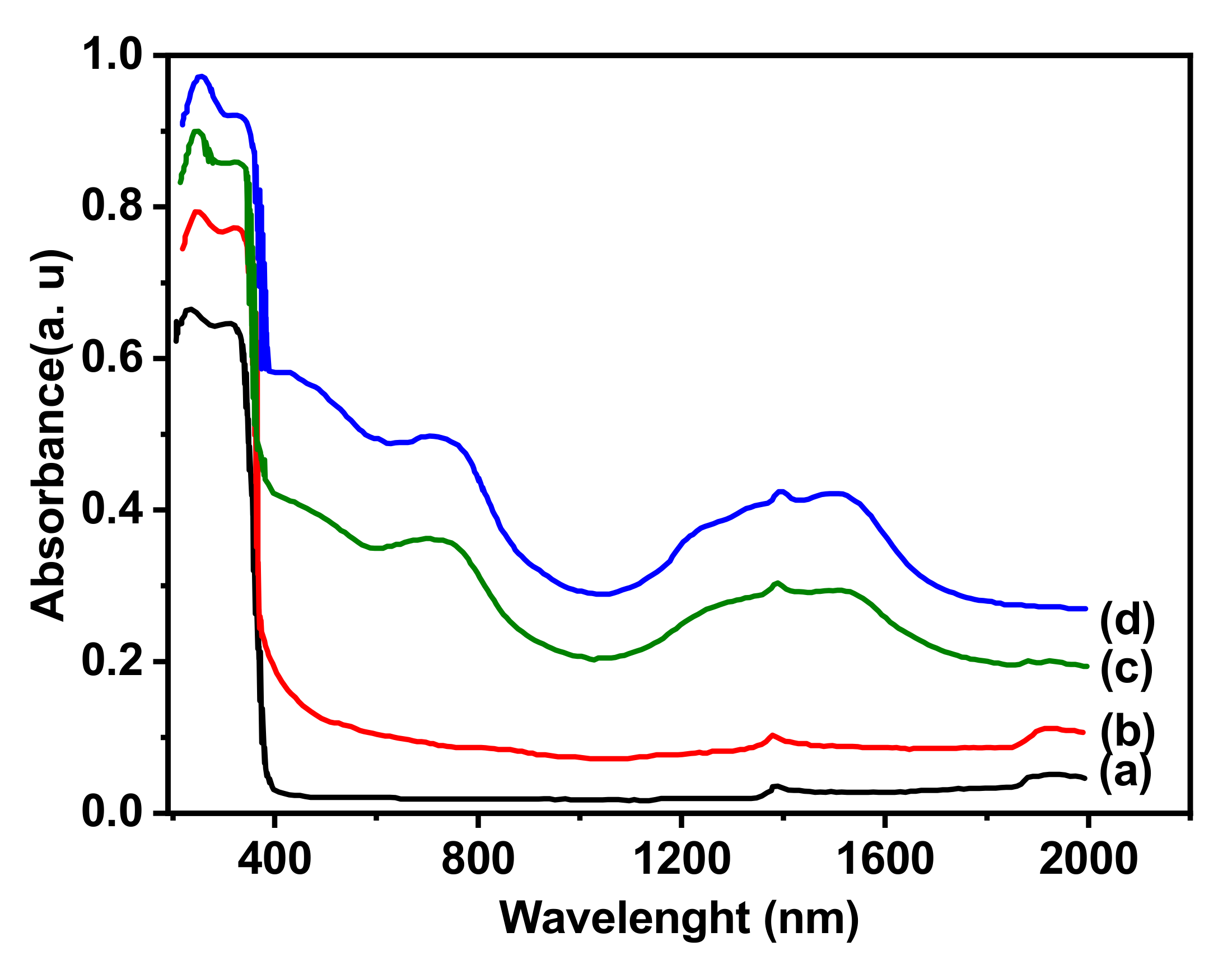
Figure 4.
DRS spectra of the CoTiO3 and Co2TiO4 phases as a function of the calcination temperature: (a) 550°C, (b) 750°C, (c) 1150°C.
Figure 4.
DRS spectra of the CoTiO3 and Co2TiO4 phases as a function of the calcination temperature: (a) 550°C, (b) 750°C, (c) 1150°C.
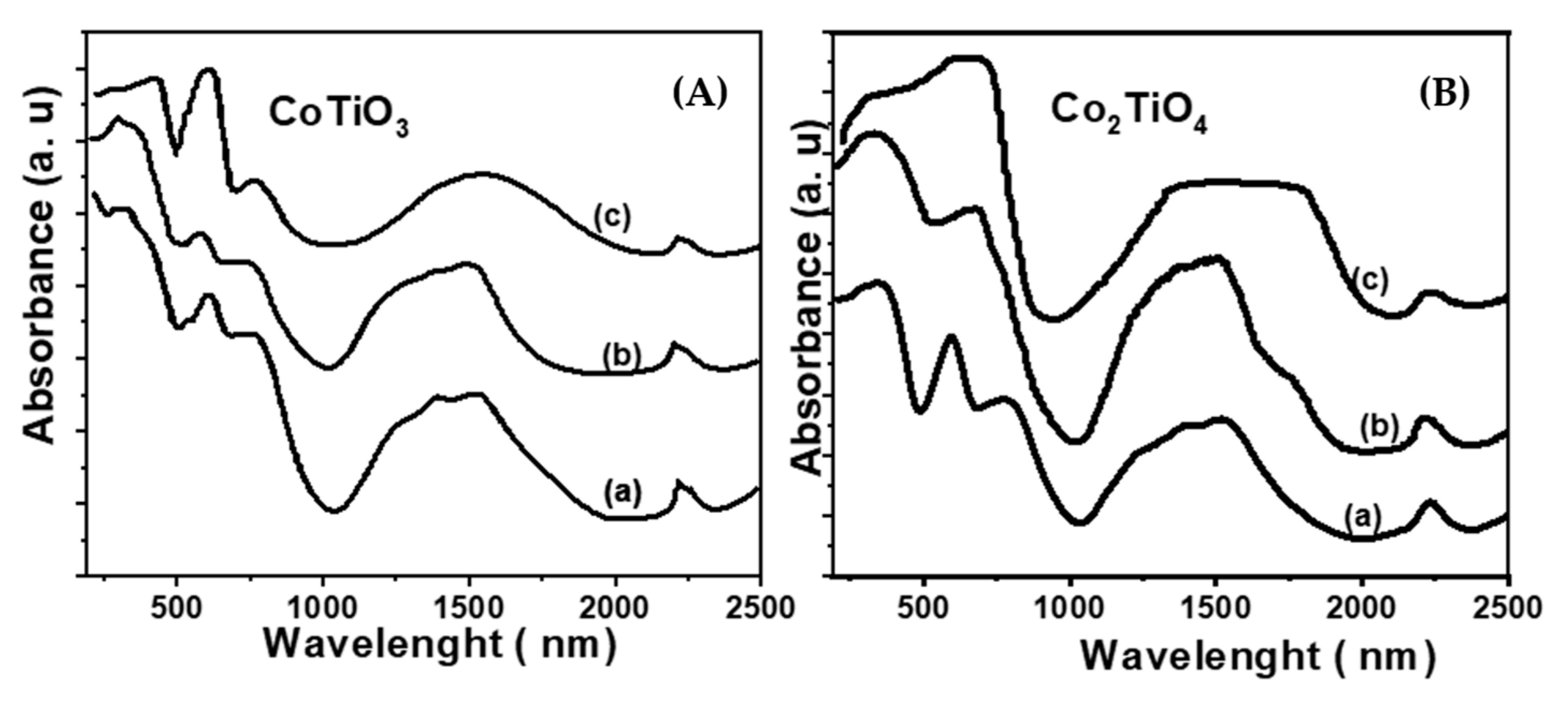
Figure 5.
Raman spectra of Co (x)/TiO2 samples (A); TiO2 :(a), Co(3.8) : (b), Co(7.5) :(c) and Co(15):(d) and (B) of CoTiO3 (b) and Co2TiO4 (a) phases.
Figure 5.
Raman spectra of Co (x)/TiO2 samples (A); TiO2 :(a), Co(3.8) : (b), Co(7.5) :(c) and Co(15):(d) and (B) of CoTiO3 (b) and Co2TiO4 (a) phases.
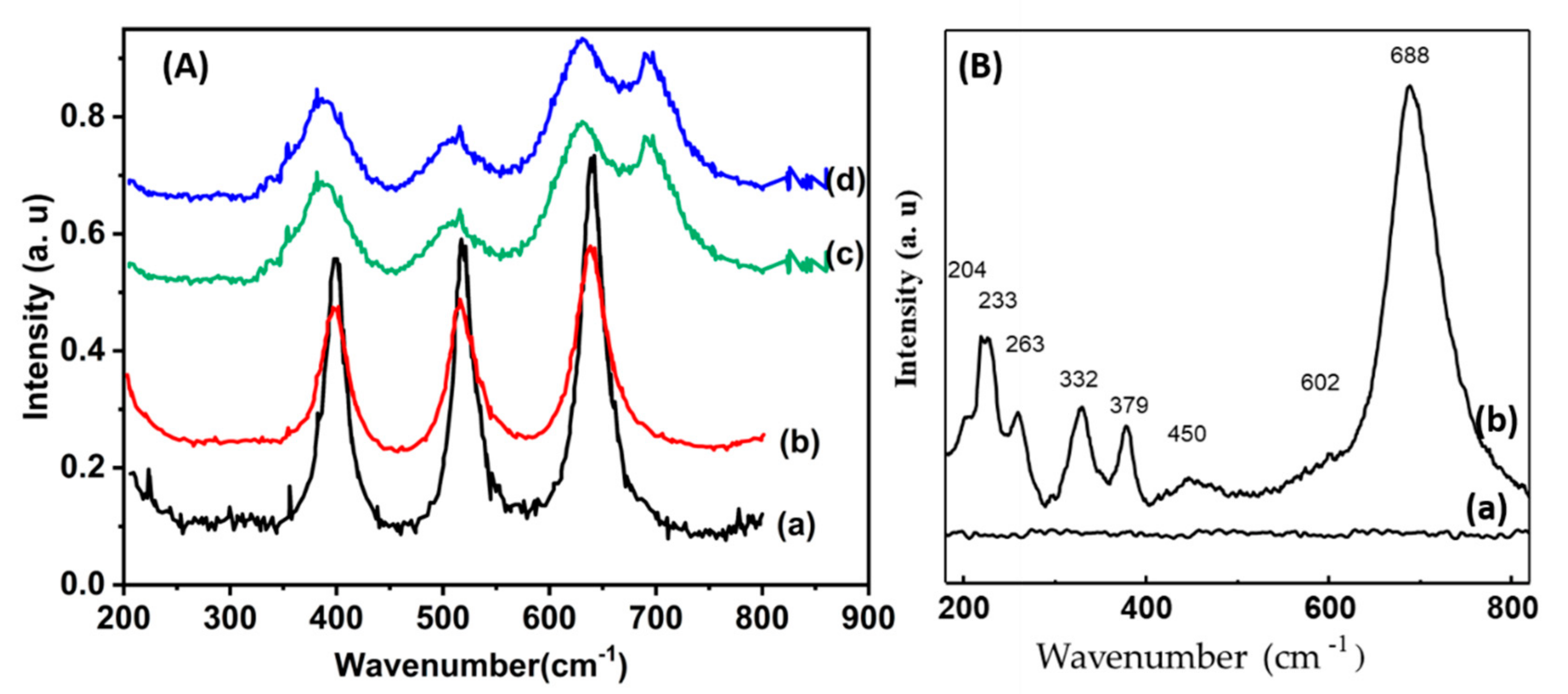
Figure 6.
XPS spectra of CoTiO3 (A) and Co2TiO4 (B) phases.
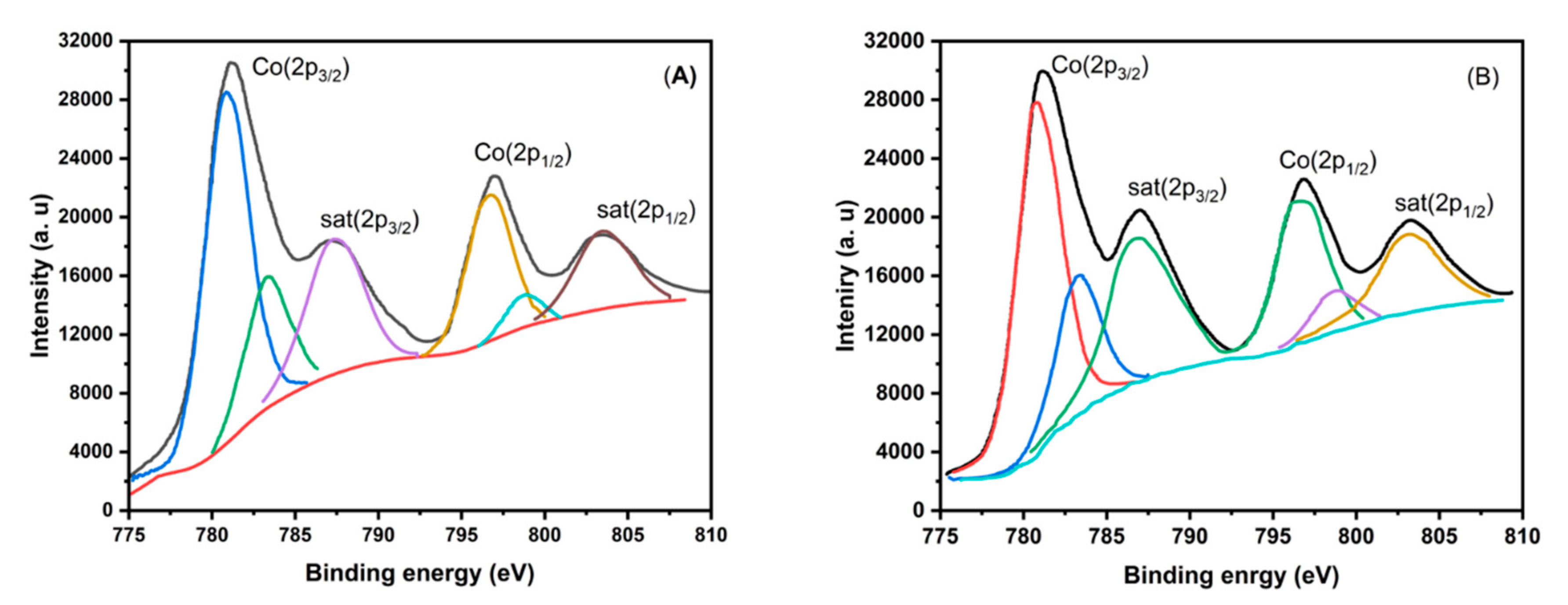
Figure 7.
Ethane conversion as a function of Cobalt content at three different temperatures: (a) 450, (b) 500 and (c) 550°C.
Figure 7.
Ethane conversion as a function of Cobalt content at three different temperatures: (a) 450, (b) 500 and (c) 550°C.
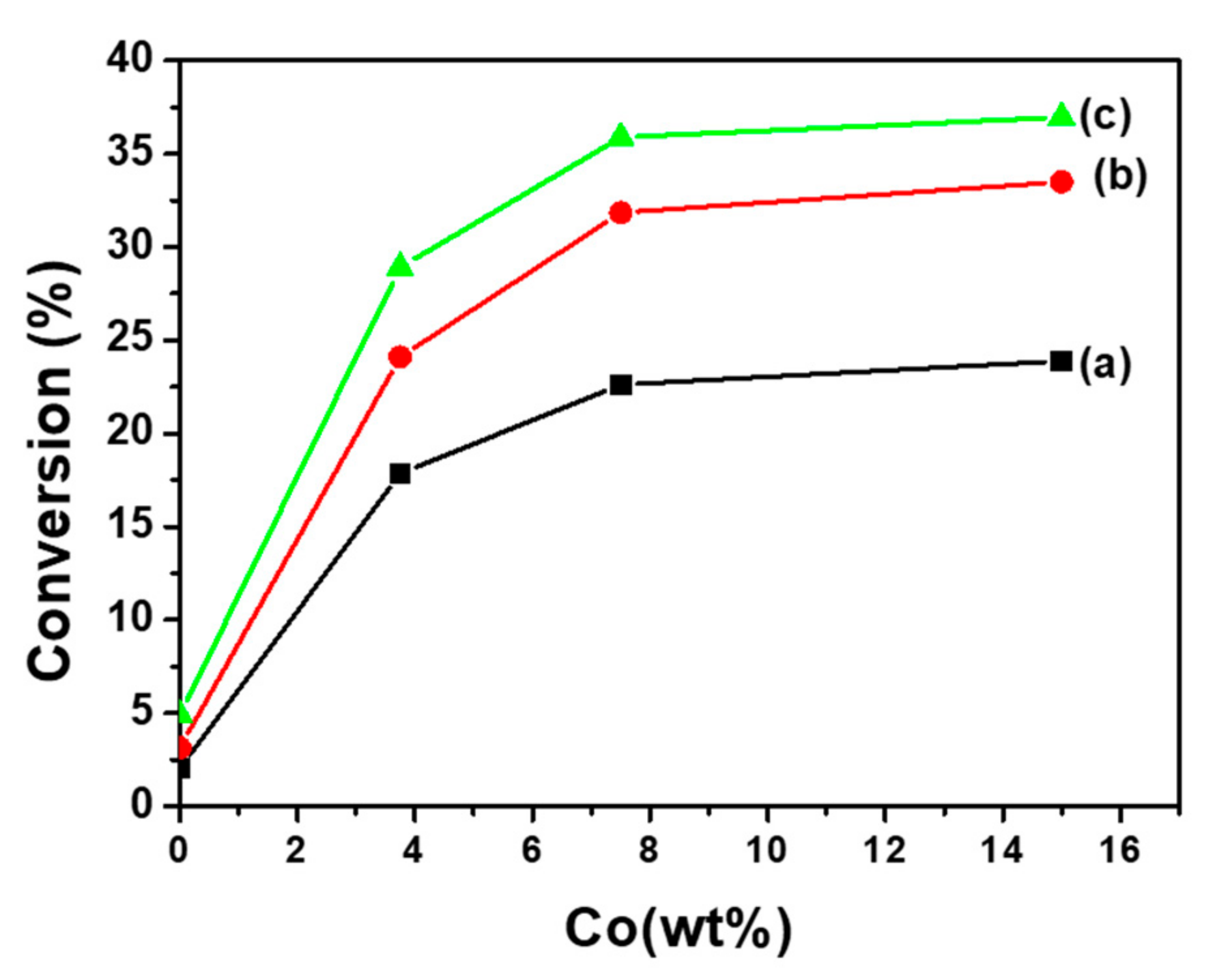
Figure 8.
Variation of conversion of ethane vs time, at 550 °C, for the bulk phases: Co2TiO4 calcined at 1150°C (a) and 550°C (c); CoTiO3 calcined at 550°C (b) and 1150°C (d) and Co(7.5)/TiO2 (e).
Figure 8.
Variation of conversion of ethane vs time, at 550 °C, for the bulk phases: Co2TiO4 calcined at 1150°C (a) and 550°C (c); CoTiO3 calcined at 550°C (b) and 1150°C (d) and Co(7.5)/TiO2 (e).
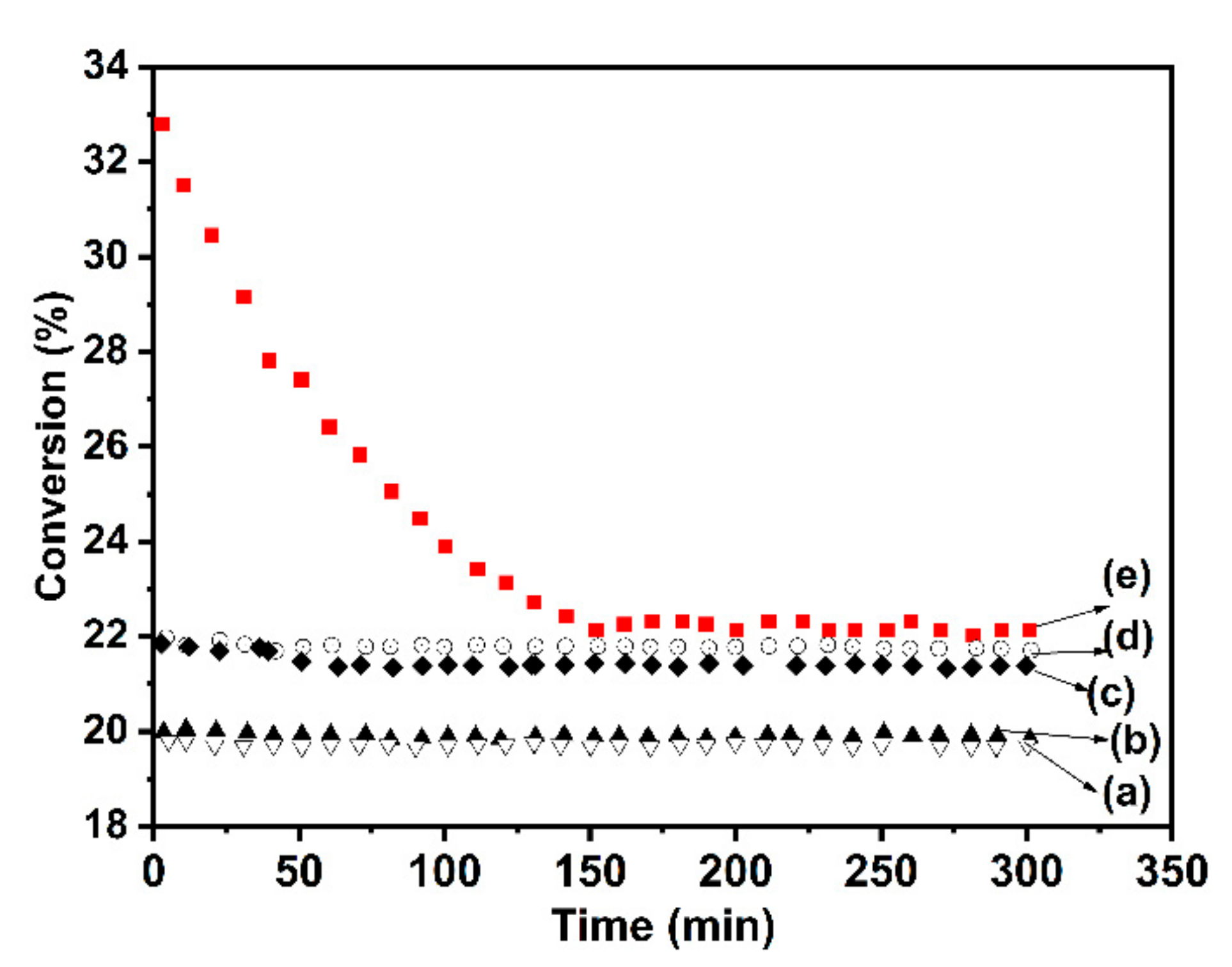
Figure 9.
Ethylene Selectivity versus Ethane Conversion on CoTiO3 calcined at 550°C (a) and 1150°C (b) and Co2TiO4 calcined at 550°C (c) and 1150°C (d).
Figure 9.
Ethylene Selectivity versus Ethane Conversion on CoTiO3 calcined at 550°C (a) and 1150°C (b) and Co2TiO4 calcined at 550°C (c) and 1150°C (d).
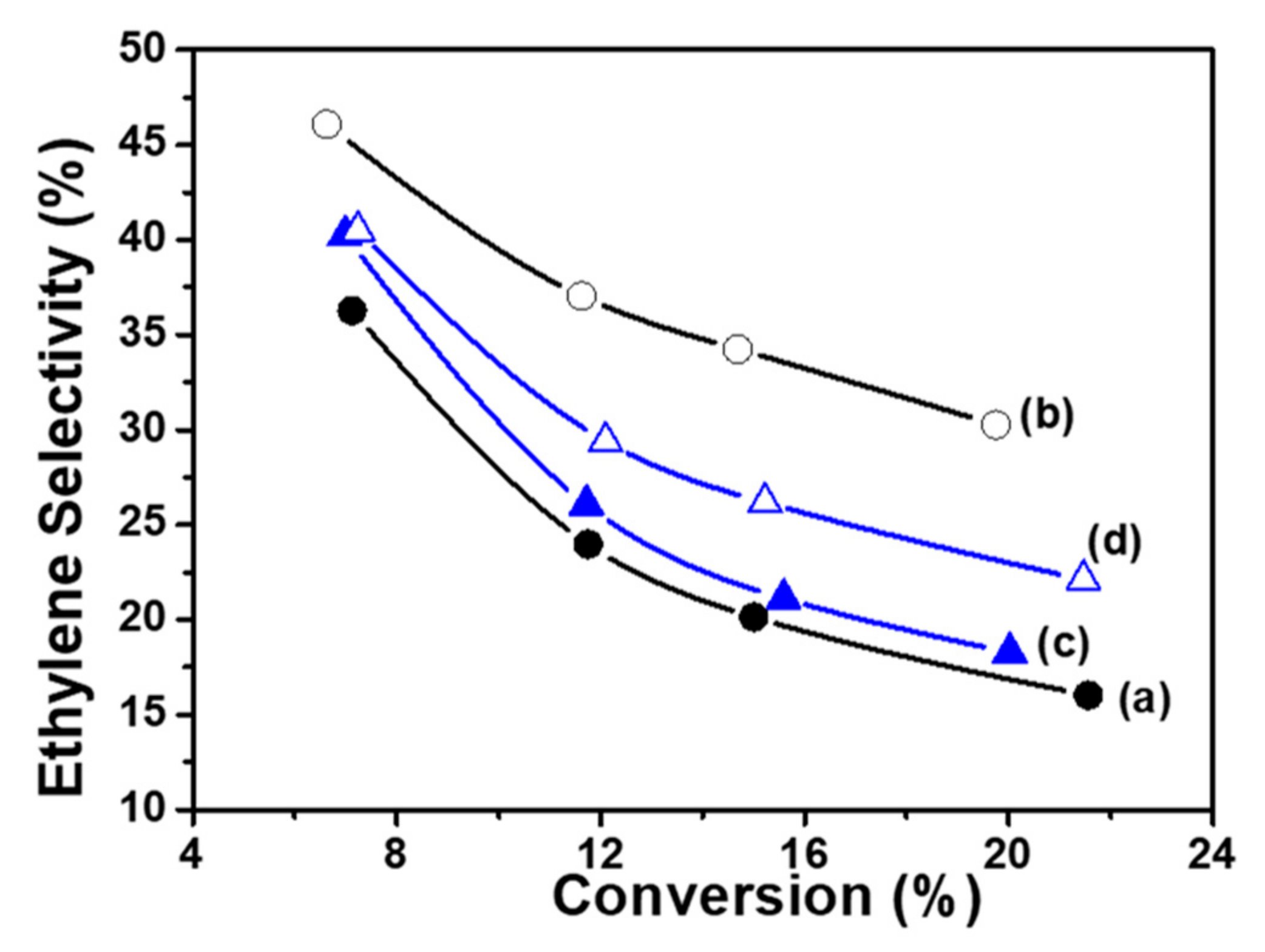
Figure 10.
Variation of C2H4 selectivity with overall conversion of CoTiO3 (A) and Co2TiO4 (B) samples depending on the percentage of steam in the reaction mixture.
Figure 10.
Variation of C2H4 selectivity with overall conversion of CoTiO3 (A) and Co2TiO4 (B) samples depending on the percentage of steam in the reaction mixture.
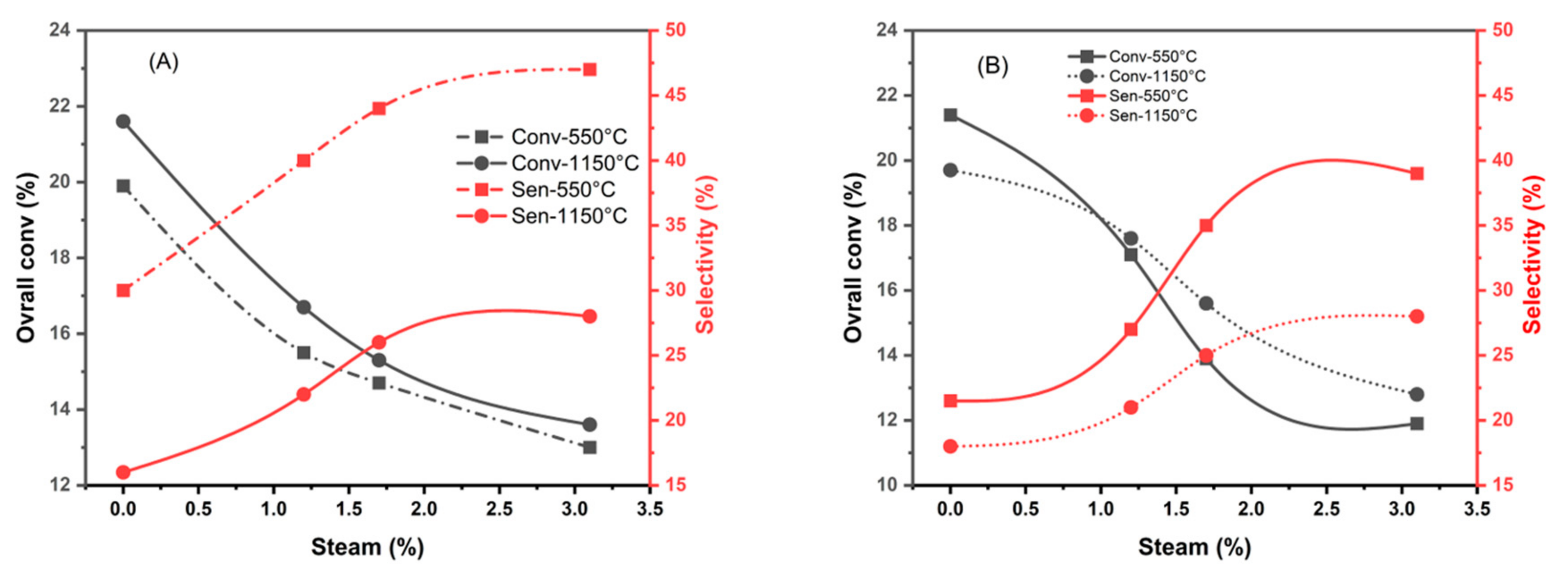

Table 1.
Specific Surface Areas (m2/g) of Co(x)/TiO2 and defined phases : CoTiO3 and Co2TiO4.
| Samples | Co(3.8)/TiO2 | Co(7.5)/TiO2 | Co(15)/TiO2 | CoTiO3 | Co2TiO4 |
| Surface area BET(m2/g) | 65 | 60 | 56 | a 46 | a 45 |
| - | - | - | b 9.4 | b10 |
a : calcined at 550 b : calcined at 1150°C.
Table 2.
XPS Characterization of the Co(x)/TiO2 and defined phases: CoTiO3 and Co2TiO4.
| samples | Co2p3/2 | ΔEc | |
| B.E. S/M* ratio | |||
| Co(3.8)/TiO2 | 781.0 | 0.42 | 15.4 |
| Co(7.5)/TiO2 | 780.2 | 0.27 | 15.2 |
| Co(15)/TiO2 | 779.9 | 0.19 | 15.1 |
| Co2TiO4 | 781 | 0.40 | 15.8 |
| CoTiO3 | 781.2 | 0.47 | 15.9 |
| Co3O4 | 779.9 | 0.18 | 15.1 |
| CoO | 780.4 | 0.4 | 16.0 |
* S/M =(satellite intensity/main peak intensity)
Table 3.
Catalytic results of CoTiO3 and Co2TiO4 at steady state and activation energy.
| Samples calcined at 550°C | ||||||||
| Reaction Temperature | CoTiO3 | Co2TiO4 | ||||||
| αg | αene | Sene | SCOx | αg | αene | Sene | SCOx | |
| 450°C | 12.9 | 4.1 | 32 | 72 | 13.8 | 3.8 | 28 | 72 |
| 500°C | 16.8 | 5.2 | 31 | 69 | 16.8 | 4.5 | 27 | 69 |
| 550°C | 19.9 | 6 | 30 | 70 | 21.4 | 4.6 | 21.5 | 78.5 |
| Activation Energy (kJ.mol-1) | 21.6 | 21.6 | ||||||
| Samples calcined at 1150°C | ||||||||
| Reaction Temperature | CoTiO3 | Co2TiO4 | ||||||
| αg | αene | Sene | SCOx | αg | αene | Sene | SCOx | |
| 450°C | 13.1 | 2.5 | 19 | 71 | 13.8 | 2.6 | 19 | 81 |
| 500°C | 17 | 3 | 18 | 72 | 17.7 | 3.3 | 19 | 81 |
| 550°C | 21.6 | 3.8 | 16 | 84 | 19.7 | 3.8 | 18 | 82 |
| Activation Energy (kJ.mol-1) | 24.5 | 24.4 | ||||||
αg: overall conversion of ethane, αene: ethylene yield, S: selectivity
Table 4.
Catalytic data from the effect study of H2O presence in reaction mixture at reaction temperature of 550°C.
Table 4.
Catalytic data from the effect study of H2O presence in reaction mixture at reaction temperature of 550°C.
| Samples calcined at 550°C | |||||||||
| %H2O | CoTiO3 | Co2TiO4 | |||||||
| αg | αene | Sene | SCOx | αg | αene | Sene | SCOx | ||
| Dry mixture | 19.9 | 6 | 30 | 70 | 21.4 | 4.6 | 21.5 | 78.5 | |
| 1.2% | 15.5 | 6.2 | 40 | 60 | 17.1 | 4.7 | 27 | 73 | |
| 1.7% | 14.7 | 6.5 | 44 | 56 | 13.9 | 4.9 | 35 | 65 | |
| 3.1% | 13 | 6.1 | 47 | 53 | 11.9 | 4.6 | 39 | 61 | |
| Samples calcined at 1150°C | |||||||||
| %H2O | CoTiO3 | Co2TiO4 | |||||||
| αg | αene | Sene | SCOx | αg | αene | Sene | SCOx | ||
| Dry mixture | 21.6 | 3.8 | 16 | 84 | 19.7 | 3.8 | 18 | 82 | |
| 1.2% | 16.7 | 3.7 | 22 | 78 | 17.6 | 3.7 | 21 | 79 | |
| 1.7% | 15.3 | 4 | 26 | 74 | 15.6 | 3.9 | 25 | 75 | |
| 3.1% | 13.6 | 3.8 | 28 | 72 | 12.8 | 3.6 | 28 | 72 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



