
Submitted:
04 June 2023
Posted:
05 June 2023
You are already at the latest version
Abstract
Applying of blood biopolymers to regulate the phase composition is promising in designing the hydroxyapatite-based hybrid biomaterials with controllable resorbability. Hybrid materials based on hydroxyapatite and platelet-poor plasma (PPP) were formed in conditions of chemical precipi-tation at pH 11, [Ca2+] / [PO43–] ratio 1.67, PPP volume fraction of 6–24%, maturing time of 4–9 days. Mineral component of the materials was represented as 53% hydroxyapatite / 47% amor-phous calcium phosphate after 4 days of maturation, and 100% hydroxyapatite after 9 days of maturation. Varying PPP content from 6% to 24% provided forming of materials with rather de-fined content of amorphous calcium phosphate and biopolymer component, having desired mor-phology ranging from typical apatitic conglomerates to hybrid apatite-biopolymer fibers. Co-precipitated hybrid materials based on hydroxyapatite and PPP are promising for bone regen-eration in osteoplastic and maxillofacial applications.
Keywords:
hybrid material
; hydroxyapatite formation
; amorphous calcium phosphate
; platelet-poor plasma
; phase composition
1. Introduction
In modern medicine, repairing of damaged and degraded bone tissue is an actual problem. Low rates of bone regeneration and inability of bone itself to repair so-called “critical” defects [1,2] define ever-increasing necessity in development of novel bioactive bone implants. Resorbable materials seem to be promising due to its complete substitution by newly formed bone tissue soon after implantation [3]. The resorption of such materials is based on the action of osteoclastic cells, which are capable of creating highly acidic and enzyme-enriched areas on the material surfaces, providing its fast and complete dissolution [4]. The bioactivity of resorbable materials strongly depends on the balance between rate of its resorption and rate of new bone formation, and one of evident approaches to achieve the balance is to imitate the bone structure and / or composition.
Inorganic component of bone tissue, a bioapatite, is very similar to stoichiometric hydroxyapatite (HAp) Ca10(PO4)6(OH)2 in composition and structure [5,6]. However, the resorbability of stoichiometric HAp does not correspond to that of bioapatite, because the latter comprises additional phases, such as amorphous calcium phosphates (ACP) Ca9(PO4)6⋅nH2O, 3.0≤n≤4.5, a metastable precursor of calcium phosphate wet formation [7,8]. Correspondingly, one biomimetic approach for biomaterials design is composing a multiphasic calcium phosphates, e.g., via thermal decomposition of calcium-deficient HAp Ca10–xHx(PO4)6(OH)2–x, 0≤x≤1 [9,10].
Biopolymers perform an important role in the formation of bioapatite, and therefore fabrication of HAp / biopolymer composites in situ is a promising biomimetic approach for obtaining resorbable bone implants with excellent bioactivity. Among the most interesting biopolymer constituents in the composites are biopolymers of blood plasma, such as proteins of albumin, α/β/γ-globuline, fibrinogen etc. [11]. Blood biopolymers are capable of binding and transporting of proteins and key growth factors therefore stimulating tissue healing and remodeling [12]. Availability is another attractive feature of blood biopolymers, which provide utilization of its most bioactive autologous form. Conventionally, extracted blood plasma undergoes stabilization by citrate-containing anticoagulant, which prevents blood coagulation – a Ca2+-mediated formation of dense fibrin clot [12]. Being of autologous nature, the citrate-stabilized platelet-poor plasma (PPP) retain its bioactivity and immune safety toward the donor organism, and may be subsequently utilized for blood volume resuscitation or tissue regeneration [13,14].
Many of blood biopolymers are presented in bone tissue, e.g., osteoblastic cells express blood protein of albumin, and may adsorb on the surfaces of apatitic particles inhibiting nucleation and growth of HAp crystallites [15]. Inhibiting of apatite crystallization is essential in preventing blood vessel calcification [16], but does not interrupt normal formation of bioapatite. One explanation is that the enzyme of alkaline phosphatase, directly involved in osteoblast functioning, could abolish the inhibitory activity of biopolymers on HAp crystallization [17].
According to the study [18], additive of blood serum (blood plasma without the clotting factors) may inhibit the growth of HAp crystals, at that the predominant inhibitory activity was attributed to a high-molecular fraction of the serum, i.e. blood biopolymers. However, there are few data on the effect of blood components on the phase composition of in situ precipitated calcium phosphates, though phase composition is among the properties strongly affecting the resorbability of the material. According to our previous study [19] an effect of 6% PPP additive in precipitation medium provides formation of bioactive calcium phosphates composites instead of single-phased stoichiometric HAp. The purpose of present study was to elucidate regularities of wet precipitation of HAp-based materials in presence of 6–24% PPP additive, at pH 11 and varying maturing time from 4 to 9 days, in order to design biomimetic hybrid materials with controllable resorpability, applicable in bone replacement.
2. Materials and Methods
2.1. Reagents, Additives, and Sample Preparation
Utilized reagents for calcium phosphate preparation included soluble salts of (NH4)2HPO4 (Carl Roth, Germany) and CaCl2·2H2O (Sigma Aldrich, USA), both of analytical grade. The pH values of precipitation media were regulated by 25% water ammonia (ultrahigh purity, Baza #1 Himreaktivov, Russia).
Blood components were extracted from donor blood (20–40 years old males, B+ and B– types) collected with consent of the donor in accordance with art. 44 of the Health Law of Republic of Belarus. According to technique of PPP isolation, 18 ml of donor blood was mixed with 2 ml of 3.8% sodium citrate solution (BelVitunifarm, Belarus) at 9:1 volume ratio, followed by centrifugation (3000 rpm, 15 min) and collection of upper fraction.
The HAp-based samples were prepared in conformity to existing technique for precipitation of stoichiometric HAp [10], by dropwise addition of solution I (0.30–0.34 mol/l HPO42–) to solution II (0.40–0.52 mol/l Ca2+, 0.24–1.40 mol/l OH–, PPP) at pH 11, [Ca2+]/[HPO42–] ratio of 1.67, and room temperature. In HAp-PPP samples, amounts of introduced PPP corresponded to 6–24% volume fraction of precipitation medium. Obtained calcium phosphate colloids were matured for up to 9 days, then separated from mother solution by filtering, repeatedly washed with distilled water and decanted until a supernatant pH of 7.0–7.5.
In order to investigate dynamics of HAp maturation we withdrew 4 ml aliqot of HAp colloid after its maturing under mother solution for a period of up to 7 days, then centrifuged and separated from supernatant, washed with distilled water, then dehydrated with ehanol and dried at 400°С.
2.2. Characterization
In order to determine Ca2+ concentration in supernatant, we utilized complexonometric titrimetry using ethylenediaminetetraacetic acid as titrant, eriochrome black T as indicator and 5 mmol/l MgSO4 solution as a standart, and NH4OH / NH4Cl as a buffer solution to keep pH at level of 9.
For characterization purposes, washed colloids were dried at 60°C until a constant mass of xerogels, then powdered and calcined at 800°C for 5 h.
The phase composition of the samples was determined by X-ray diffraction (XRD) using Advance D8 diffractometer (Bruker, Germany) equipped with CuKα radiation (λ=0.15406 nm), at 20°≤2θ≤45°. The XRD profiles were compared to standards compiled by the Crystallography Open Database (COD), which involved cards for α-polymorph #04-010-4348 and β-polymorph #04-008-8714 of crystalline tricalcium phosphate (TCP) Ca3(PO4)2, for hexagonal HAp #01-074-0565, using Profex software [20].
For evaluating the content of ACP in the samples, we used an extended variant of existing technique for determination of the Ca/P ratio in calcium phosphates by XRD analysis [21]. The original technique relies on thermally induced crystallization of HAp following by analyzing the ratio of formed crystalline HAp and β-TCP. In present study, key objects of investigation were biphasic calcium phosphate mixtures of HAp and ACP, which after heat treatment at 800°C for 5 h transform into mixtures of crystalline HAp and TCP. Following equations describe calculating the data on the calcium phosphate composition before heat treatment:
where χi is molar fraction, and Ca/P denotes Ca to P molar ratio of the solid mixture. The content of the phases in the article was represented as weight fractions.
χACP = (3χTCP) / (1 + 2χTCP), Ca/P = (10 – 7χTCP) / (6 – 4χTCP),
The morphological characteristics of calcium phosphate xerogel surfaces were obtained using scanning electron microscope LEO 1420 (Carl Zeiss, Germany), with gold sputtercoating of the surfaces using a K550X sputter coater (Emitech, England).
The functional groups and structural changes of the powdered samples were evaluated by Fourier Transform Infrared (FTIR) spectroscopy using Tensor-27 FTIR-spectrometer (Bruker, Germany) at ambient temperature in the range of 1850–450 cm–1, by mixing 2 mg of a sample with 800 mg of spectroscopic grade KBr. We used Origin 2018 software to integrate the spectra region at 1850–1360 cm–1, representing integrated area in a.u.
All the XRD patterns and FTIR spectra were normalized. Interpreting the XRD and FTIR spectroscopy data of HAp-based samples, we refer to our previous study [10].
The thermogravimetric analysis was performed on STA 409 PC LUXX thermal analyzer (NETZSCH, Germany), in air atmosphere at heating rate of 10°C/min, with sample masses of 30–40 mg.
3. Results
In terms of the study we considered a HAp to be monophasic and stoichiometric with Ca/P 1.67, if it crystallizes at 800°C without a detectable forming of non-apatitic phases. Certain stoichiometry variations because of CO32–, HPO42– incorporation into apatitic lattice was not taken into account.
According to XRD analysis, heat-treatment at 800°C induced transformation of the samples with varying maturing periods, yielding β-TCP (no maturation, Figure 1 a, curve 1), 50% HAp / 50% β-TCP mixture (30 min, Figure 1 a, curve 2), monophasic HAp (1–7 days, Figure 1 a, curves 3–5) in accordance to reaction:
where 0≤x≤1 denotes calcium-deficiency of the apatite.
Ca10–xHx(PO4)6(OH)2–x → (1–x) Ca10(PO4)6(OH)2 + 3x β-Ca3(PO4)2,
At the beginning of maturation, the samples represent apatitic TCP Ca9H(PO4)6OH, Ca/P 1.50, x = 1, which decomposes at 800°C into β-modification of crystalline TCP, and during maturing gradually converts into stoichiometric HAp, Ca/P 1.67, x = 0 that crystallizes at 800°C without a decomposition.
Results of investigation of the dynamics of calcium phosphate maturing under mother solution (Figure 1 b) showed that:
1) the Ca/P molar ratio of the precipitate increased from initial value of 1.50 to equilibrium value of 1.67 over 1 day of maturation;
2) the concentration of Ca2+ ions decreased from 33.4 mmol/l at 30 min of maturing, to ~8.6 mmol/l over 4 days.
Both phenomena correspond to the known model of HAp formation [10], including crystallization of ACP to apatitic TCP and following reprecipitation of stoichiometric HAp, which is described by following scheme:
Ca9(PO4)6⸱nH2O + H2O → Ca9H(PO4)6OH + Ca2+ + 2OH– → Ca10(PO4)6(OH)2,
Basing on obtained data, we pointed out that a period of 4 days corresponds to complete formation of stoichiometric HAp.
The influence of PPP additive on the precipitate phase composition were studied on the example of the samples, matured for 4 days (complete formation of individual HAp) and 9 days (extra-maturing for transformation of PPP-stabilized intermediates). The XRD pattern of individual HAp, matured for 4 days, (Figure 2, curve 1) after 800°C showed sharp peaks of well-crystalline apatite phase, and that of HAp / PPP additionally exhibited formation of up to 45% α-TCP phase (Figure 2, curves 2, 3). The latter is associated with crystallization of ~47% of ACP at 650°C, according to reaction (4):
Ca9(PO4)6⸱nH2O → 3α-Ca3(PO4)2 + nH2O,
In contrast, all the XRD patterns of the samples, matured for 9 days and heat-treated at 800°C (Figure 2, curves 4–6), contained only the peaks of HAp phase. The Table 1 summarizes obtained XRD data.
The observed stabilizing of ACP in HAp / PPP samples, matured for 4 days, presumably, occurs due to the surface adsorption of biopolymer macromolecules on the calcium phosphate particles, which is a common mechanism for a polyelectrolyte-mediated preventing of ACP crystallization [22]. At that increasing of PPP concentration from 6% to 24% negligibly affected amount of ACP stabilized, and the total Ca/P ratio remained to be 1.59 (table 1). We may suppose that the stabilizing effect has already reached its maximum at PPP concentration of 6%. After continuous maturation for 9 days the effect of ACP stabilization is no longer observed implying only a partial inhibition of ACP crystallization into HAp rather than its complete prevention. We associate the latter with permeability of adsorbed layer of hydrophilic biopolymers toward water molecules, participating in conversion of ACP into apatite [8].
As is shown, in conditions of HAp synthesis at initial [Ca2+]/[HPO42–] ratio of 1.67, precipitation medium contains Ca2+ ions in concentrations of 8–33 mmol/l, which is induced PPP coagulation and formation of fibrin network throughout the volume of maturation medium. For that reason, formed colloid precipitates of HAp / PPP had reduced flowability comparing to that obtained without PPP. During maturation for 4 days, the HAp / PPP precipitates gradually recovered its flowability due to hydrolytic disruption of fibrin network at pH 11. For comparison, individual dense fibrin clot completely dissolves in alkaline solutions for 1 day at pH 11 [19].
According to the SEM images, xerogels of HAp / 6% PPP (Figure 3 a, b) represent typical apatite appearance [23] being composed of irregular agglomerates with rough surface. For the sample, we observe no evidence of biopolymer structures on the xerogel surface that, possibly, owes to inclusion of the biopolymer macromolecules into the bulk of HAp xerogel. The SEM images of the HAp / 24% PPP xerogels (Figure 3 c, d) exhibited dense biopolymer fibers up to 30 μm in diameter with rough surface, unlike the fibers of an individual fibrin that has a smooth surface and discernible transparence to an electronic beam [24]. Moreover, the fibers of the HAp / 24% PPP samples retain its structural integrity under maturation at pH 11 for 4 days in contrast to an individual fibrin that completely dissolves in the same conditions. The reason of these differences possibly lies in fine mixing of the HAp particles and fibrin fibers on the submicron scale. Another prove to above-mentioned presumption one observes after mechanical disruption of the fibers, which result in the formation of fragments (Figure 3 d) with a typical apatitic appearance. Correspondingly, the obtained data imply formation of a hybrid structure under conditions of simultaneous HAp precipitation and PPP coagulation.
In terms on the FTIR spectroscopy, we studied the spectra region at 1850–450 cm–1 that represents set of bands sufficient to detect both the HAp and blood biopolymers, and also contain no extra-widened bands, e.g. stretching vibrational modes of adsorbed water. Due to predominantly apatitic nature of the mineral constituent of the samples, all the FTIR spectra after drying at 60°C (Figure 4 a) showed characteristic bands for apatite at 1090, 1040, 956, 603, 566, 472 cm–1 due to O–P–O vibrations and at 633 cm–1 due to O–H libration. The PPP-free samples (Figure 4 a, curves 1, 4) has bands at 1650 cm–1 due to H–O–H bending vibration of adsorbed water as well as bands at 1500–1350 cm–1 due to O–C–O stretching vibrations of CO32– ions in apatite lattice. The integrated intensity of these bands of 13±5 a.u. mostly arises from vibrations of apatitic CO32– ions that incorporate in the precipitate structure during maturing in alkaline solution. For HAp precipitation in highly alkaline, CO32–-poor medium, the incorporation proceeds via substitution of PO43–for CO32– (B-type), for which one of the most intense band locates at ~1480 cm–1 [25].
In case of the PPP-containing samples (Figure 4 a, curves 2, 3, 5, 6) the spectra region at 1850–1350 cm–1 is mainly composed of bands assigned to vibrations of biopolymer functional groups [26]: amide I is at 1660, 1640 cm–1; amide II is at 1540 cm–1; bands at 1450, 1415 cm–1 due to vibrations of hydrophobic side groups of amino acids. We did not considered low-intensity amide III bands at 1330–1190 cm–1 due to its overlapping with intense apatitic O–P–O bands. One may notice the gap on the spectra at 1480 cm–1 (Figure 4 a, denoted by dash line), where previously mentioned B-type carbonate band are expected to overlap with the biopolymer bands. Integrated area of the biopolymer bands at 1850–1350 cm–1 is related to total biopolymer content in the samples. As is seen from obtained values, the four-fold increasing of introduced PPP concentration from 6% to 24% provides comparably lesser increasing of total biopolymer amount, entrapped by solid precipitate, from 24±3 a.u. to 38±3 a.u. One explanation of the observed refers to partial removal of biopolymers during washing-decantation, due to which HAp / 6% PPP sample (apatitic morphology) and HAp / 24% PPP sample (biopolymer-like morphology) comprises comparable total amount of biopolymers.
In order to test the validity of the latter regularities revealed by FTIR spectroscopy, we performed the thermogravimetric analysis of the samples. According to data obtained (Figure 4 b), HAp-based samples undergone three-staged thermal transformations:
- I.
- A stage of dehydration is observed at 25–250°C, accompanied by mass loss of 5.4–9.8%, and corresponds to elimination of absorbed water and decomposition of HAp crystallohydrate [27].
- II.
- Further heating of PPP-based samples induced biopolymer decomposition at 250–750°C. For individual HAp (Figure 4 b, curve 1), mass decreasing of 2.7% is due to removal of residual water, but in case of HAp / PPP samples (Figure 4 b, curves 2, 3) the mass loss values correspond to weight content of biopolymer and organic substances in the samples. At that increasing of introduced PPP amount from 6% to 24% provides increasing of content of decomposing substances from 18.6% only to 22.5%, that is in accordance with FTIR data (Figure 4 a).
- III.
- Crystallization of calcium phosphates occurred at 750–1000°C. Corresponding mass loss is maximal (0.6%) for individual HAp (Figure 4 b, curve 1), and is attributed to gradual decomposition of lattice CO32– ions with removal of CO2 [25]. For HAp / PPP samples (Figure 4 b, curves 2, 3), one observes abrupt mass loss of 0.2% (pointed by arrows) at 750–780°C due to crystallization of ACP according to reaction (4). After 780°C, the mass of the HAp / PPP samples remained virtually unchanged, implying insignificant decomposition of CO32– ions. As is seen, the degree of carbonation in HAp / PPP is much lesser than in individual HAp, that gives a reason to absence of carbonate bands on the FTIR spectra of HAp / PPP. We associate the preventing CO32– incorporation in the apatitic lattice with barrier effect of the adsorbed biopolymers of PPP.
4. Conclusions
Formation of HAp in presence of PPP was studied in conditions of co-precipitation at pH 11, [Ca2+] / [PO43–] 1.67, PPP volume fraction of 6–24%. The effect of PPP on HAp formation partially inhibited crystallization of ACP and CO32–-incorporation into the apatite structure. Therefore, composites of HAp with up to 47% ACP were formed after 4 day of maturing. At that, increasing the introduced PPP content from 6 to 24% insignificantly affected the amount of stabilized ACP, whereas continuous maturation for 9 days led to formation of monophasic HAp. Varying PPP content in range of 6%–24% provided forming of materials with rather predictable biopolymer content of 18.6–22.5% and desired morphology ranging from typical apatitic conglomerates to hybrid apatite-biopolymer fibers. Co-precipitated hybrid materials based on HAp and PPP are promising for bone regeneration in osteoplastic and maxillofacial applications.
Author Contributions
Conceptualization, IG and VK; methodology, IG and VK; validation, VK and TS; formal analysis, NS; investigation, IG, NK, RV, OM and AK; resources, RV; writing—original draft preparation, IG and NS; writing—review and editing, TS, NK and OM; supervision, VK; project administration, VK; funding acquisition, AK. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by the State Research Program “Chemical Processes, Reagents, and Technologies, Bioregulators and Bioorganic Chemistry” according to assignment 2.1.04.7 for 2021–2025 and by Belarusian Republican Foundation for Fundamental Research (grant no. X22M-043).
Acknowledgments
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Pereira, H.F.; Cengiz, I.F.; Silva, F.S.; Reis, R.L.; Oliveira, J.M. Scaffolds and coatings for bone regeneration. J. Mater. Sci. Mater. Med. 2020, 31, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Safronova, T.V. Inorganic materials for regenerative medicine. Inorg. mater. 2021, 57, 443–474. [Google Scholar] [CrossRef]
- Schilling, A. F; Linhart, W; Filke, S.; Gebauer, M.; Schinke, T.; Rueger, J.M.; Amling, M. Resorbability of bone substitute biomaterials by human osteoclasts. Biomater. 2004, 25, 3963–3972. [Google Scholar] [CrossRef] [PubMed]
- Everts, V.; Jansen, I.D.C.; de Vries, T.J. Mechanisms of bone resorption. Bone. 2022, 163, 116499. [Google Scholar] [CrossRef] [PubMed]
- Edén, M. Structure and formation of amorphous calcium phosphate and its role as surface layer of nanocrystalline apatite: Implications for bone mineralization. Materialia. 2021, 17, 101107. [Google Scholar] [CrossRef]
- DileepKumar, V.G.; Sridhar, M.S.; Aramwit, P.; Krut’ko,V. K.; Musskaya, O.N.; Glazov, I.E.; Reddy, N. A Review on the Synthesis and Properties of Hydroxyapatite. J. Biomater. Sci. Polymer Ed. 2022, 33, 229–261. [Google Scholar] [CrossRef]
- Dorozhkin, S.V. Synthetic amorphous calcium phosphates (ACPs): Preparation, structure, properties, and biomedical applications. Biomater. Sci. 2021, 9, 7748–7798. [Google Scholar] [CrossRef] [PubMed]
- Combes, C.; Rey, C.C. Amorphous calcium phosphates: synthesis, properties and uses in biomaterials. Acta Biomater. 2010, 6, 3362–3378. [Google Scholar] [CrossRef]
- Dorozhkin, S.V. Biphasic, triphasic and multiphasic calcium orthophosphates. Acta Biomater. 2012, 8, 963–977. [Google Scholar] [CrossRef]
- Glazov, I.E.; Krut’ko, V.K.; Musskaya, O.N.; Kulak, A.I. Calcium Phosphate Apatites: Wet Formation, Thermal Transformations, Terminology, and Identification. Russ. J. Inorg. Chem. 2022, 67, 173–182. [Google Scholar] [CrossRef]
- Krebs, H.A. Chemical composition of blood plasma and serum. Ann. Rev. Biochem. 1950, 19, 409–430. [Google Scholar] [CrossRef] [PubMed]
- Noori, A.; Ashrafi, S.J.; Vaez-Ghaemi, R.; Hatamian-Zaremi, A.; Webster, T.J. A review of fibrin and fibrin composites for bone tissue engineering. Intern. J. Nanomed. 2017, 12, 4937–4961. [Google Scholar] [CrossRef] [PubMed]
- Ehrenfest, D.M.D.; Rasmusson, L.; Albrektsson, T. Classification of platelet concentrates: from pure platelet-rich plasma (P-PRP) to leucocyte- and platelet-rich fibrin (L-PRF). Trends Biotech. 2009, 27, 158–167. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, J.; Pepkowitz, S.H.; Klapper, E. Platelet-rich plasma for the replenishment of bone. Curr. Osteopor. Rep. 2011, 9, 258–263. [Google Scholar] [CrossRef] [PubMed]
- Ishida, K.; Sawada, N.; Yamaguchi, M. Expression of albumin in bone tissues and osteoblastic cells: Involvement of hormonal regulation. Int. J. Mol. Med. 2004, 14, 891–895. [Google Scholar] [CrossRef]
- Hunter, G.K. Role of osteopontin in modulation of hydroxyapatite formation. Calcif. Tiss. Int. 2013, 93, 348–354. [Google Scholar] [CrossRef]
- Vimalraj, S. Alkaline phosphatase: Structure, expression and its function in bone mineralization. Gene. 2020, 754, 144855. [Google Scholar] [CrossRef]
- Garnett, J.; Dieppe, P. The effects of serum and human albumin on calcium hydroxyapatite crystal growth. Biochem. J. 1990, 266, 863–868. [Google Scholar]
- Glazov, I.E.; Krut’ko, V.K.; Kulak, A.I.; Musskaya, O.N.; Vlasov, R.A.; Malakhovsky, P.O.; DileepKumar, V.G.; Surya, P.S. , Sridhar M. S.; Reddy N. Effect of platelet-poor plasma additive on the formation of biocompatible calcium phosphates Mater. Today Commun. 2021, 47. [Google Scholar] [CrossRef]
- Doebelin, N.; Kleeberg, R. Profex: a graphical user interface for the Rietveld refinement program BGMN. J. App. Crystallograph. 2015, 48(5), 1573–1580. [Google Scholar] [CrossRef]
- Ishikawa, K.; Ducheyne, P.; Radin, S. Determination of the Ca/P ratio in calcium-deficient hydroxyapatite using X-ray diffraction analysis. J. Mater. Sci. Mater. Med. 1993, 4(2), 165–168. [Google Scholar] [CrossRef]
- Bar-Yosef Ofir, P.; Govrin-Lippman, R.; Garti, N.; Füredi-Milhofer, H. The influence of polyelectrolytes on the formation and phase transformation of amorphous calcium phosphate Cryst. Gr. Des. 2004, 4, 177–183. [Google Scholar] [CrossRef]
- Koutsopoulos, S. Synthesis and characterization of hydroxyapatite crystals: a review study on the analytical methods. J. Biomed. Mater. Res. 2002, 62, 600–612. [Google Scholar] [CrossRef] [PubMed]
- Pretorius, E.; Oberholzer, H.M.; van der Spuy, W.J.; Swanepoel, A.C.; Soma, P. Scanning electron microscopy of fibrin networks in rheumatoid arthritis: a qualitative analysis. Rheumatolog. Int. 2012, 32, 1611–1615. [Google Scholar] [CrossRef] [PubMed]
- Glazov, I.E.; Krut’ko, V.K.; Musskaya, O.N.; Kulak, A.I. Low-temperature formation and identification of biphasic calcium carbonate-phosphates. Russ. J. Inorg. Chem. 2022, 67, 1718–1730. [Google Scholar] [CrossRef]
- Litvinov, R.I.; Faizullin, D.A.; Zuev, Y.F.; Weisel, J.W. The α-helix to β-sheet transition in stretched and compressed hydrated fibrin clots. Biophys. J. 2012, 103, 1020–1027. [Google Scholar] [CrossRef]
- Krut'ko, V.K.; Kulak, A.I.; Lesnikovich, L.A.; Trofimova, I.V.; Musskaya, O.N.; Zhavnerko, G.K.; Paribok, I.V. Influence of the Dehydration Procedure on the Physicochemical Properties of Nanocrystalline Hydroxylapatite Xerogel. Russ. J. Gen. Chem. 2007, 77(3), 336–342. [Google Scholar] [CrossRef]
Figure 1.
XRD patterns of the samples, matured for different periods, after heat-treating at 800°C (a): 1 – no maturation; 2 – 30 min; 3 – 1 day; 4 – 4 days; 5 – 7 days; Changing of calculated Ca/P molar ratio of the precipitate (solid) and Ca2+ concentration in precipitation medium (dash) during maturation (b).
Figure 1.
XRD patterns of the samples, matured for different periods, after heat-treating at 800°C (a): 1 – no maturation; 2 – 30 min; 3 – 1 day; 4 – 4 days; 5 – 7 days; Changing of calculated Ca/P molar ratio of the precipitate (solid) and Ca2+ concentration in precipitation medium (dash) during maturation (b).
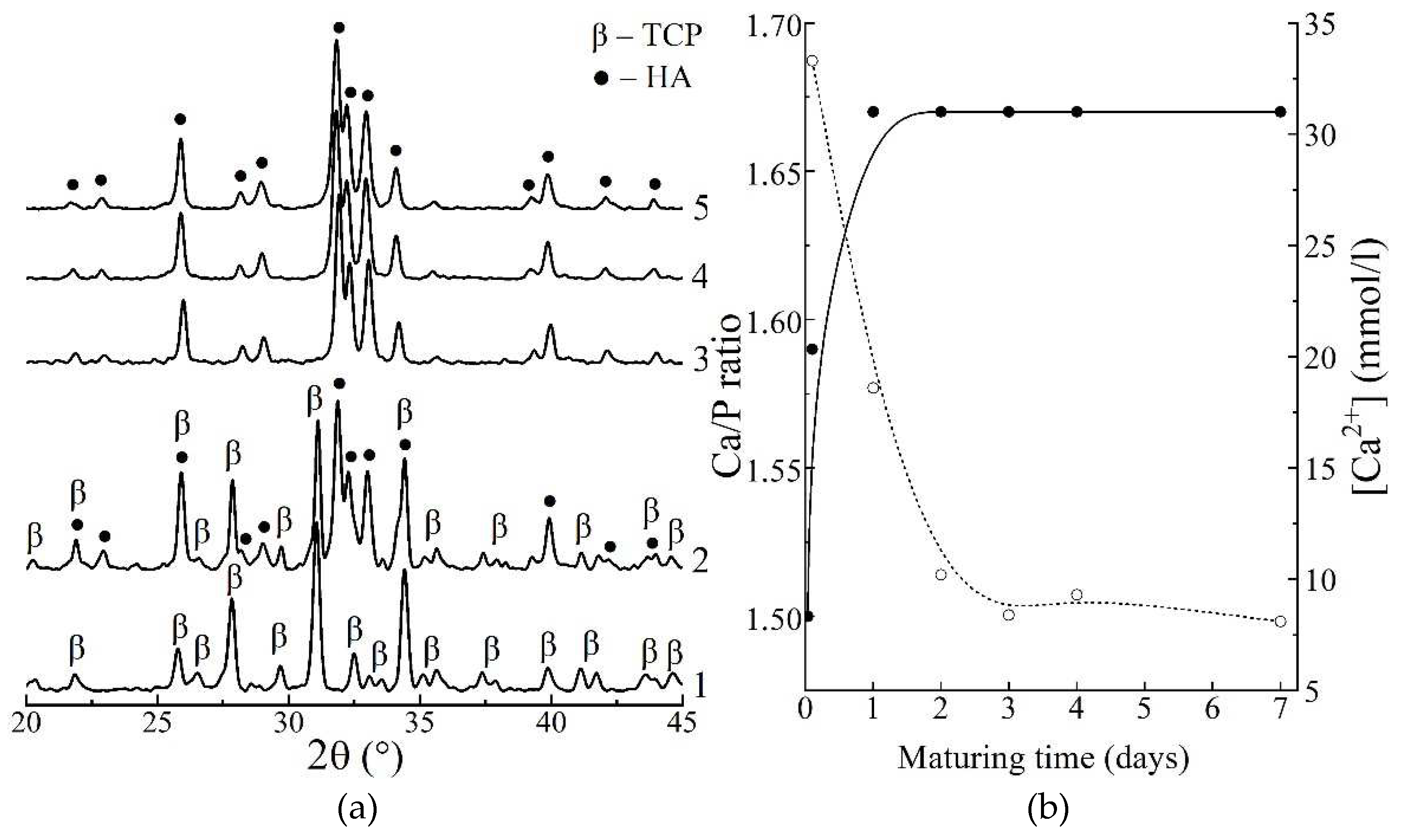
Figure 2.
XRD patterns of the powdered xerogels after heat-treating at 800°C: 1 – HAp, 4 days; 2 – HAp / 6% PPP, 4 days; 3 – HAp / 24% PPP, 4 days; 4 – HAp, 9 days; 5 – HAp / 6% PPP, 9 days; 6 – HAp / 24% PPP, 9 days.
Figure 2.
XRD patterns of the powdered xerogels after heat-treating at 800°C: 1 – HAp, 4 days; 2 – HAp / 6% PPP, 4 days; 3 – HAp / 24% PPP, 4 days; 4 – HAp, 9 days; 5 – HAp / 6% PPP, 9 days; 6 – HAp / 24% PPP, 9 days.
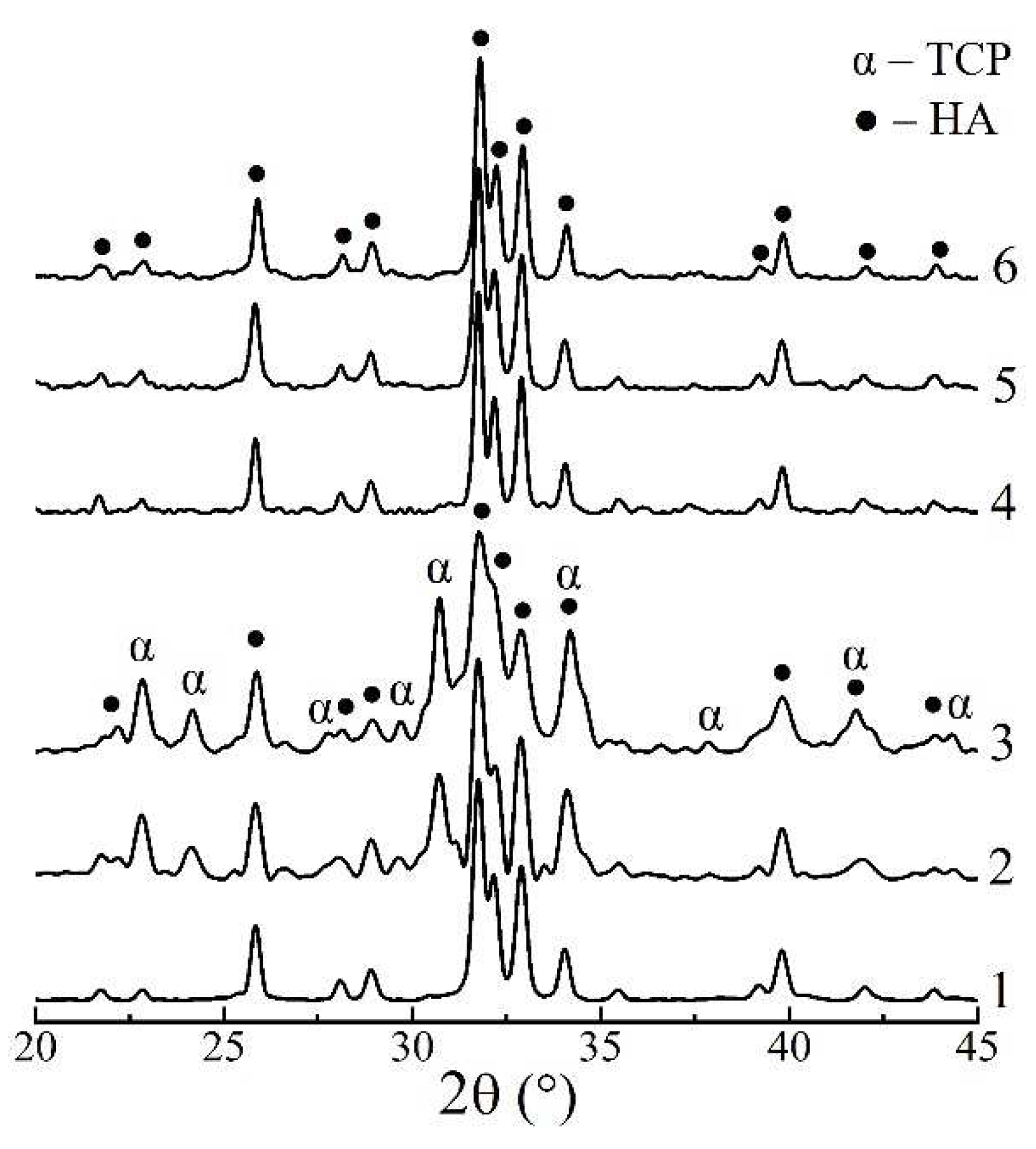
Figure 3.
SEM images of the xerogel surfaces of HAp / 6% PPP (a, b) and HAp / 24% PPP (c, d), matured for 4 days, after drying at 60°C, before (a, c) and after (b, d) mechanical disruption.
Figure 3.
SEM images of the xerogel surfaces of HAp / 6% PPP (a, b) and HAp / 24% PPP (c, d), matured for 4 days, after drying at 60°C, before (a, c) and after (b, d) mechanical disruption.

Figure 4.
FTIR spectra (a) and thermogravimetric curves (b) of the powdered xerogels after heat-treating at 60°C: 1 – HAp, 4 days; 2 – HAp / 6% PPP, 4 days; 3 – HAp / 24% PPP, 4 days; 4 – HAp, 9 days; 5 – HAp / 6% PPP, 9 days; 6 – HAp / 24% PPP, 9 days. Arrows denote abrupt mass decrease due to ACP crystallization.
Figure 4.
FTIR spectra (a) and thermogravimetric curves (b) of the powdered xerogels after heat-treating at 60°C: 1 – HAp, 4 days; 2 – HAp / 6% PPP, 4 days; 3 – HAp / 24% PPP, 4 days; 4 – HAp, 9 days; 5 – HAp / 6% PPP, 9 days; 6 – HAp / 24% PPP, 9 days. Arrows denote abrupt mass decrease due to ACP crystallization.
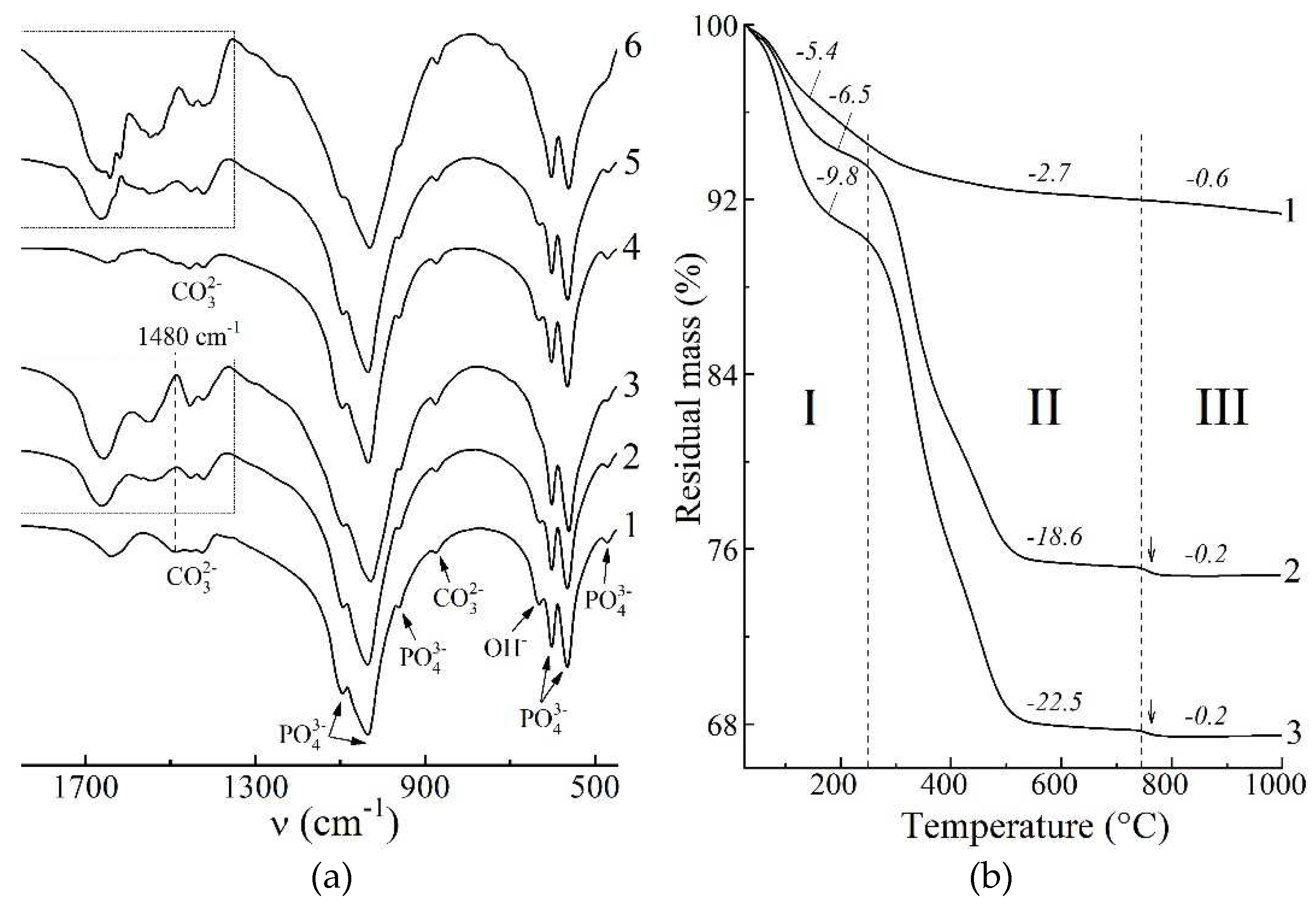

Table 1.
Results of the XRD phase analysis of the HAp / PPP samples.
| Sample | Maturingtime, days | Content of phases, % | Ca/P | |
|---|---|---|---|---|
| ACP (60°C) 1 | α-TCP (800°C) | |||
| HAp | 4 | – | – | 1.67 |
| HAp / 6% PPP | 46 | 44 | 1.59 | |
| HAp / 24% PPP | 47 | 45 | 1.59 | |
| HAp | 9 | – | – | 1.67 |
| HAp / 6% PPP | – | – | 1.67 | |
| HAp / 24% PPP | – | – | 1.67 | |
1 Calculated in accordance to Section 2.2; “–“ denotes absence of the phase in the sample.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



