
Submitted:
05 June 2023
Posted:
06 June 2023
You are already at the latest version
Abstract
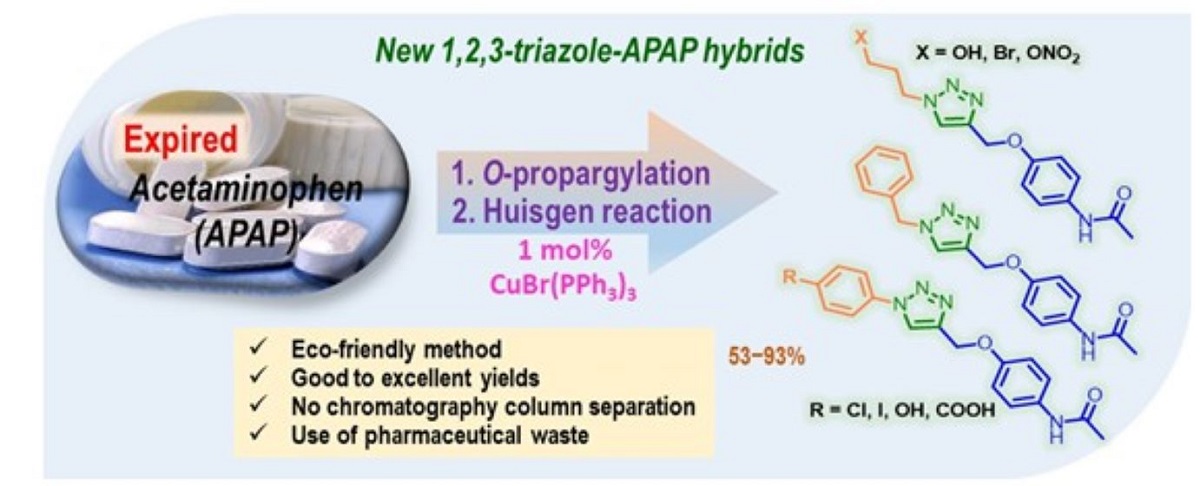
We hereby describe an efficient method for the preparation of a series of new 1-substituted 1,2,3-triazole-based acetaminophen derivatives through a clean, good-yielding, simple, and expeditious procedure based on the O-propargylation reaction of the acetaminophen (APAP) obtained from expired commercial tablets and the CuBr(PPh3)3-catalyzed Huisgen reaction be-tween O-propargylated APAP and diverse organoazides prepared from commercially available anilines as available starting reagents. An interesting nitric oxide-releasing 1,2,3-triazole hybrid of APAP was also obtained easily using the developed method. The structures of the designed hybrid compounds, which are expected to be pharmacologically active, were fully characterized by FT-IR, 1H- and 13C-NMR and are reported for the first time. According to the in-silico studies performed in this work and literature analysis, these hybrids are interesting models in search of new pharmacological nontoxic agents endowed with anti-inflammatory and anticancer properties.
Keywords:
acetaminophen
; 1
; 4-disubstituted 1
; 2
; 3-triazoles
; molecular hybrids
; azide-alkyne cycloaddition
; Huisgen reaction
; Cu(PPh3)3Br catalyst
; click chemistry
1. Introduction
Acetaminophen (APAP, paracetamol, 4-hydroxyacetanilide, or N-acetyl-p-aminophenol) is a popular medicament commonly used to treat pain and reduce high body fever. It is typically applied to relieve mild pain such as headache and toothache, and it is a preferred alternative to aspirin, particularly for patients who cannot tolerate aspirin [1,2,3,4,5]. It is safe to take, and side effects are scare in most people that consumes APAP regularly [6,7]. However, its use is one of the most common causes of poisoning worldwide, and its environmental presence has a hazardous impact on non-target organisms [8]. APAP blocks prostaglandin synthesis from arachidonic acid by inhibiting the enzymes cyclooxygenase (COX)-1 and -2 [9]. It also inhibits prostaglandin synthesis by scavenging peroxynitrite, an activator of COX [10,11]. Thus, APAP possesses antipyretic and moderate analgesic properties. However, it demonstrated a narrow therapeutic window, weak anti-inflammatory activity, and potential hepatotoxic actions [12].
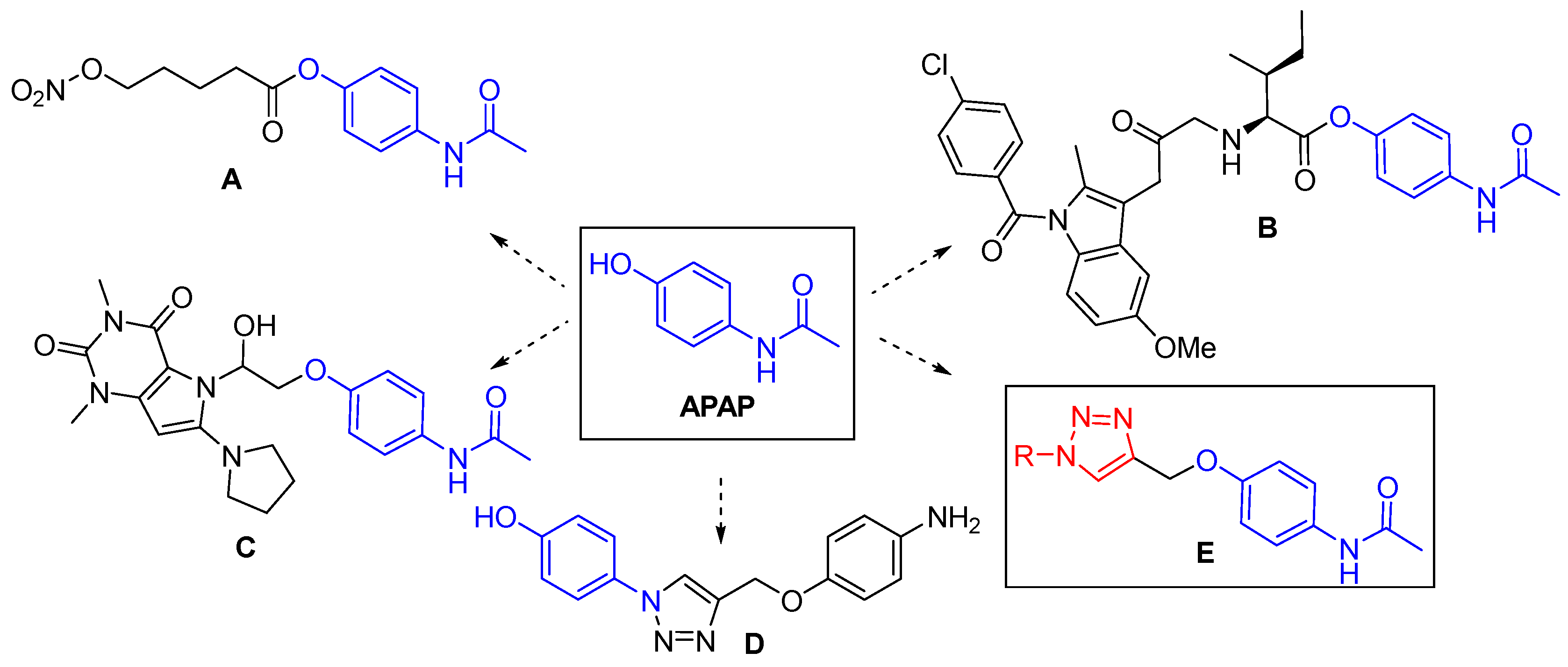
As a consequence, research on new APAP derivatives is an active area of drug development [13,14,15,16,17,18,19,20,21,22,23,24]. According to current reports, a structural modification of the N-acetyl-p-aminophenol skeleton can improve, reduce or, even change the enzymatic activities of a drug and thus, the pharmacological properties including cytotoxicity. Among these studies, APAP conjugated pharmacophores (A-D) (Figure 1) are very promising lead compounds in APAP-based drug research and development [25,26,27,28]. These so-called hybrid molecules or multi-targeted compounds that contain pharmacophore moieties of two or more bioactive molecules play an important role in the modulation of multiple biological targets exhibiting pharmacodynamic effects [29,30,31,32]. Particularly, NO-acetaminophen (nitroparacetamol, APAP-ONO2, NCX-701) (A), a nitric oxide (NO)-releasing derivative of APAP showed considerable anti-inflammatory activity and has been shown to be much more potent as an analgesic, and much less toxic to the liver, than its parent drug [33,34].
APAP-triazole conjugated pharmacophore D also displayed more potent anti-inflammatory activity along with analgesic and antipyretic properties than APAP, while its hepatotoxic side effects were found to be less significant than its parent skeleton [25]. In general, the molecular scaffold of the 1,2,3-triazole ring displays a special and different physicochemical properties such as an amide bond, mimicking (amide bioisostere), poor basicity, metabolic stability, and ability to participate as a hydrogen-bond acceptor, which could be favorable in binding biomolecular targets as well as in increasing solubility [35,36,37,38]. Moreover, the 1,2,3-triazole ring is a good linker and valuable structural platform [39,40] possessing a broad spectrum of pharmacological activities like anti-inflammatory, anticancer, antioxidant, antitubercular, anticonvulsant, antibacterial activity, etc. [41,42,43,44,45].
Due to their marked and diverse pharmacological properties, there are several methods described in the literature for the synthesis of 1,2,3-triazoles [46,47]; however, the 1,3-dipolar cycloaddition of an azide to the triple bond of acetylenes (Huisgen reaction [48,49]) is the most important, versatile, and popular route. This is due to the use of copper catalysts in the Huisgen reaction, i.e., copper (I)-catalyzed azide−alkyne cycloaddition (CuAAC) that was discovered independently by two research groups: Sharpless and coll. [50] and Meldal and co-workers [51] in 2002.
The CuAAC reaction has become an excellent reaction for organic synthesis and represents the classic example of click chemistry [52]. Numerous Cu-catalytic systems have been reported for the CuAAC reaction but the most widely used methodology is based on a CuSO4/sodium ascorbate (NaAsC) system in a solvent mixture of water and alcohol (Sharpless−Folkin protocol) [50]. This combination of CuSO4/ NaAsC (reducing agent) is used as a precatalyst system, which generated the Cu(I) species in situ. This system is extremely efficient, but it is basically limited to water-tolerant substrates. Alternatively, copper(I) salts (iodide, bromide, chloride, acetate) and their mixed catalytic systems can be used in acetonitrile as co-solvent within the protecting ligands for the Cu(I) centers from oxidation (Meldal−Tornøe protocol) [51]. In this sense, bromotris(triphenylphosphine) copper(I) complex, CuBr(PPh3)3 was found to be an organic solvent soluble, air-stable, and efficient catalyst, whose preparation is very easy and inexpensive, for clean and quick preparation of diverse 1,4-disubstituted 1,2,3-triazoles [53,54].
From a green chemistry perspective, the use of expired drug components, i.e., pharmaceutical waste as starting materials for the organic synthesis and medicinal chemistry of new active pharmaceutical ingredients (APIs) is an important but underdeveloped principle of sustainable chemistry [55]. The generation of expired or unused drug constituents has caused the discharge of chemicals into the environment and, thereby, leads to the spreading of pollution which has already occurred on the surface, ground, and sewage. To solve these environmental and health problems, it is believed that recycling expired or unused drugs will recover the active principles of drugs in their pure forms, which can later be reformulated into new drug products [56,57,58].
Considering the above-stated features, and as a continuation of our efforts in the study on the catalyzed 1,3-dipolar cycloaddition reactions towards new potentially bioactive triazole-based heterocyclic molecules [59,60,61,62,63], we designed, synthesized, and characterized new functionalized 1,2,3-triazole-based acetaminophen derivatives using expired acetaminophen pills as starting material.
Therefore, in this work, we describe a practical method for the synthesis of the title compounds through a common strategy based on the CuAAC reaction of O-propargyl-APAP and diverse aryl- and alkyl azides using 1 mol% CuBr(PPh3)3 complex as an air-stable catalyst in a solvent mixture of tert-BuOH: H2O (1:1) at room temperature for a period of 9-13 h. In addition, the developed method allowed us to obtain a new nitric oxide-releasing 1,2,3-triazole hybrid of APAP. Finally, prepared 1,2,3-triazole-based APAP derivatives were subjected to in-silico analysis (Molinspiration online platform and OSIRIS property explorer), evaluating some physicochemical properties (Lipinski descriptors), drug-likeness and drug-score parameters, and toxicity risks.
2. Materials and Methods
2.1. Materials and Instruments
The reagents and solvents used in the synthesis of the intermediate and final compounds were of purity grade for synthesis. The composition and monitoring of the reactions, as well as the preliminary analysis of the purity of the synthesized compounds, was carried out by thin layer chromatography (TLC) on Silufol UV254 plates of 0.25 mm thickness, revealed in a UV light chamber of 254 nm.
Fisher-Johns melting point apparatus was used to measure the melting points (uncorrected). The acquisition of nuclear magnetic resonance spectra 1H and 13C was performed in a Bruker Avance–400 spectrometer (400 MHz for 1H and 100 MHz for 13C) using deuterated chloroform (CDCl3, 99.8% Merck®) as solvent. Chemical shift values (δ) are expressed in ppm. In 1H NMR spectra, the scale was adjusted from the residual chloroform signal (7.26 ppm). Similarly, the 13C NMR spectra were scaled from the signal characteristic of the solvent (CDCl3). Coupling constants (J) are described at n bonds and are given in Hz; the multiplicity of signals is expressed by the following abbreviations: (s) singlet, (d) doublet, (t) triplet, (q) quartet, and (m) multiplet.
A Hewlett Packard 5890a Series II Gas Chromatograph interfaced to an HP 5972 Mass Selective Detector (MSD) with an HP MS Chemstation Data system (PerkinElmer, Akron, OH, USA) was used for MS identification at 70 eV using a 60 m capillary column that was coated with HP-5 [5%-phenyl-poly(dimethyl-siloxane)]. Infrared spectra were taken on an FT-IR Nicolet™ iS50 spectrophotometer with attenuated reflectance (ATR) cell at 31 scans and 2 cm−1 resolution. Elemental analyses were performed on a PerkinElmer 2400 Series II analyzer and were within ± 0.4 of theoretical values.
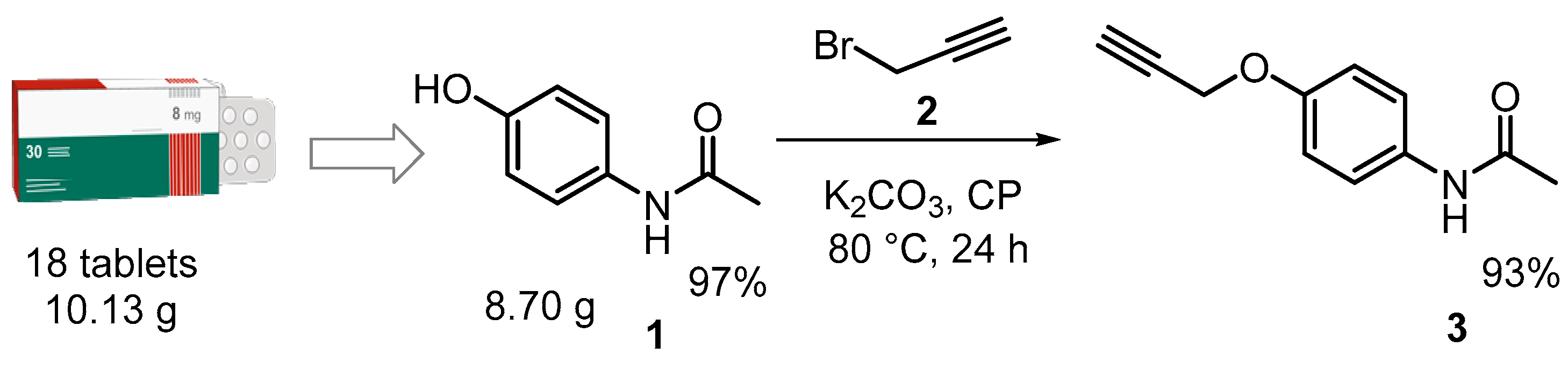
18 tablets of commercial acetaminophen brand Genfar® (10.13 g), expiration date 25 Dec. 2018 were obtained at the blue point of reception of the University Wellness department at the Industrial University of Santander and then were pulverized, treated with a CH2Cl2:MeOH (3:1) mixture, stirring vigorously at rt for 24 h, and filtered to obtain a white crystalline solid whose characteristics (Mp, Rf) correspond to the N-(4-hydroxyphenyl)acetamide (acetaminophen, paracetamol) (see, Supplementary Materials).
2.2. General Procedure for the Synthesis of 1,2,3-Triazole-Based APAP Derivatives 5a-f
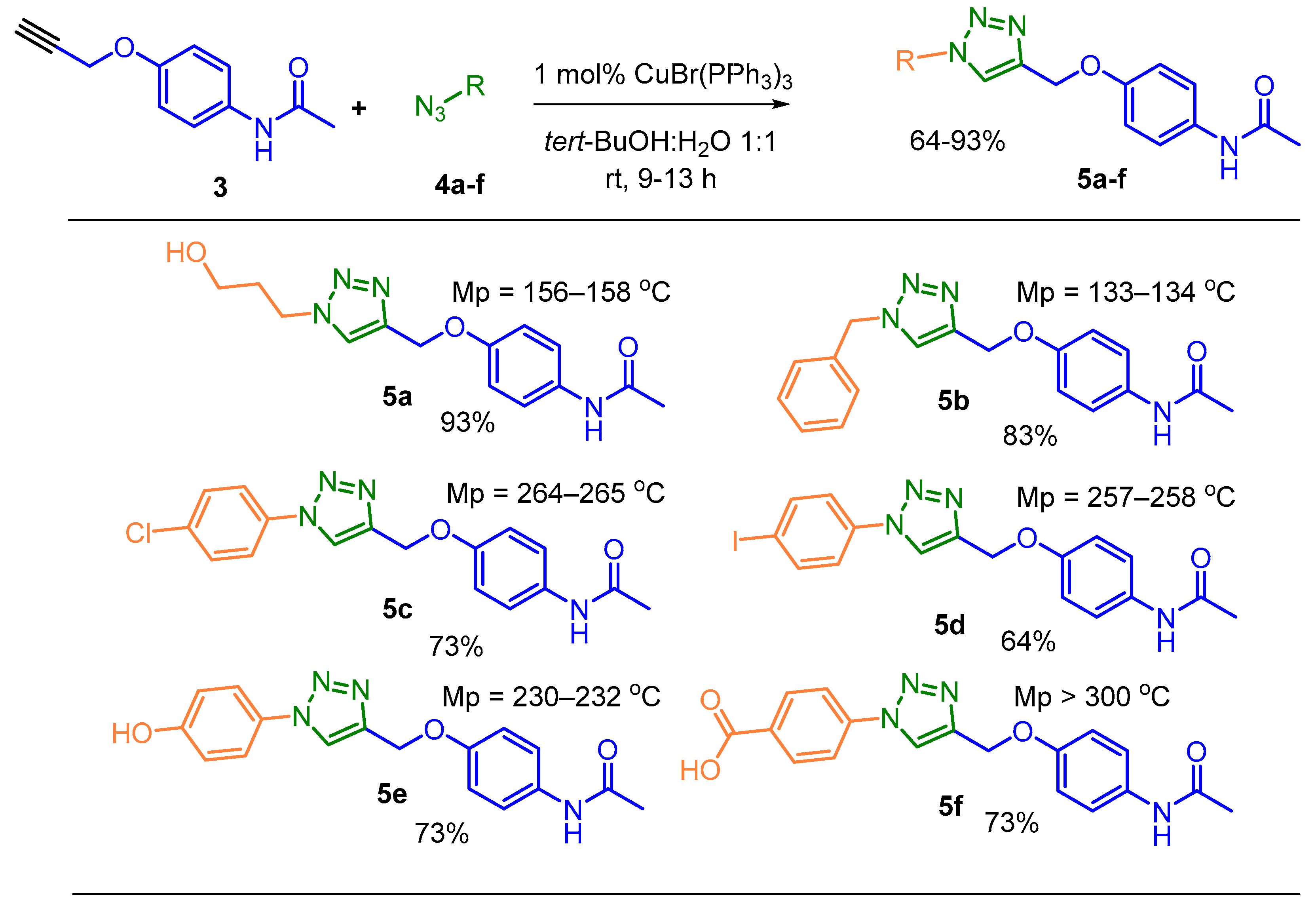
In a 20 mL flask, alkyne 3 (1mmol, 189 mg) and CuBr(PPh3)3 catalyst (1 mol%) dissolved in a 4 mL of tert-BuOH:H2O mixture (1:1) were added at room temperature and the reaction mixture was stirred for 30 min. Then, the corresponding azide 4a-f (1.3 mmol) was slowly added, and the reaction was allowed until TLC monitoring showed that the precursors had been completely consumed. The formed products 5a-f were precipitated with cold water, filtered, washed with methanol, recrystallized, and characterized as stable solid substances (Table 1).
2.3. Spectroscopic Characterization
N-(4-((1-(3-hydroxypropyl)-1H-1,2,3-triazol-4-yl)methoxy)phenyl)acetamide (5a): 270 mg (93%). Gray solid, Rf = 0.20 (ethyl acetate). Mp = 156–160 °C. IR (ATR, νmax/cm−1): 3376 (ν(N-H) amide and (O-H)), 2920 and 2842 (νAr(C-H)), 1675 (ν(C=O) amide), 1576-1393 (νAr (C=C)), 1226 (ν(C-N)), 863-759 (γAr(C-H)). 1H NMR (400 MHz, DMSO-d6) δ (ppm): 9.79 (1H, s, -NH), 8.19 (1H, s, 12-CH), 7.48 (2H, d, J = 8.2 Hz, 3- and 3’-HAr), 6.96 (2H, d, J = 8.2 Hz, 2- and 2´-HAr), 5.08 (2H, s, 7-CH2) 4.68 (1H, t, J = 5.1 Hz, 15-CH2OH) 4.42 (2H, t, J = 7.0 Hz, 13-CH2), 1.98 (7H, m, 14-CH2, 15-CH2 and 6-CH3). 13C NMR (101 MHz, DMSO-d6) δ (ppm): 167.8, 153.8, 142.7, 132.8, 124.5, 120.5 (2C), 114.7 (2C), 61.3, 57.4, 46.7, 32.9, 23.8. MS (EI, 70 eV) m/z (%): 290 (M+, 100). Anal. calcd. for C14H18N4O3 (MW = 290.32 g/mol): C, 57.92; H, 6.25; N, 19.30 %. Found: C, 68.35; H, 5.26; N, 8.15 %.
All of the spectral data of other compounds can be found in the Supplementary Materials.
3. Results and Discussion
3.1. Chemistry
Performing click chemistry and considering green chemistry principles, the synthesis of the designed 1,2,3-triazole-based APAP derivatives was planned in two synthetic steps. The initial step consisted of obtaining and functionalizing APAP 1, which was carried out by extracting the active ingredient from expired commercial with an extraction yield of 97% and its subsequent O-alkylation reaction with propargyl bromide (2) to obtain O-propargyl-APAP 3 in a basic medium (anhydrous potassium carbonate, K2CO3) using propylene carbonate (CP) as a green solvent at 80 °C for a period of 24 h (see, Supplementary Materials) (Scheme 1).
The second step was based on the CuAAC reaction of alkyne 3 and 3-azidopropan-1-ol (4a) selected for the optimization of cycloaddition reaction conditions for the synthesis of hybrid 5a as model substrates (Table 1). Azide 4a was obtained following a general protocol for azide preparation from 3-chloro-1-propanol and sodium azide in DMF at 60 oC for a period of 16 h [64]. Initially, we wanted to analyze the effectiveness of the two procedures mentioned above (Sharpless−Folkin protocol vs Meldal−Tornøe protocol) in the reaction model. Thus, the first three experiments were carried out under classical conditions of Sharpless−Folkin protocol using Cu(II)-catalyst, CuSO4 in the presence of reducing additive, NaAsC in an aqueous medium (tert-BuOH:H2O, 1:1) (Table 1, entries 1-3).

Table 1.
Optimization of CuAAC reaction conditions for the synthesis of hybrid 5aa.

| Entry | Solvent | Catalyst | Additive: mol % NaAsCb |
Load catalyst (mol%) |
Time (h) | Yield (%)c |
|---|---|---|---|---|---|---|
| 1 | tert-BuOH:H2O 1:1 | CuSO4.5H2O | 15 | 10 | 18 | 83 |
| 2 | tert-BuOH:H2O 1:1 | CuSO4.5H2O | 10 | 5 | 18 | 86 |
| 3 | tert-BuOH:H2O 1:1 | CuSO4.5H2O | 1 | 0.5 | 13 | 85 |
| 4 | tert-BuOH:H2O 1:1 | [CuBr(PPh3)3] | - | 5 | 18 | 90 |
| 5 | tert-BuOH:H2O 1:1 | [CuBr(PPh3)3] | - | 1 | 18 | 92 |
| 6 | tert-BuOH:H2O 1:1 | [CuBr(PPh3)3] | - | 1 | 12 | 90 |
| 7 | tert-BuOH:H2O 1:1 | [CuBr(PPh3)3] | - | 1 | 9 | 93 |
| 8 | tert-BuOH:H2O 1:1 | [CuBr(PPh3)3] | - | 0.5 | 11 | 87 |
| 9 | CH3CN | [CuBr(PPh3)3] | - | 1 | 12 | 73 |
| 10 | CP | [CuBr(PPh3)3] | - | 1 | 18 | NRd |
a Reaction conditions: 3 (1 mmol) and 4a (1.3 mmol) at rt; b NaAsC: sodium ascorbate; c Isolated yield after recrystallization (EtOH); d NR: no reaction.
With catalyst and additive loads of 15 and 10 mol%, respectively, the desired triazole 5a was obtained after 18 h of stirring at room temperature (TLC monitoring) in 83% yield (Entry 1). When the respective catalytic and additive loads were reduced to 10 and 5 mol% and then to 5 and 0.1 mol%, yields of 5a and its reaction times practically did not change (Entries 2 and 3). Thus, this protocol works well producing the needed triazole molecule in good yields (83–86%).
Evaluating the second protocol, our attention was drawn to the influence of the nature of the solvent and the charge of the catalyst. Performing the model reaction of 3 and 4a with 5 mol% CuBr(PPh3)3 complex in the same reaction medium, we noted the increase in yield product 5a (90%) in the same reaction time (Entry 4 vs entries 2,3). The reduction of catalytic load allowed to shorten reaction time maintaining good yields of the desired triazole (90–93%) (Entries 5-7). Reduction of the charge up to 0.5 mol% led to a decrease in the product yield and a lengthening of the reaction time (Entry 8 vs Entry 7). We also tested the organic solvents (MeCN and CP) but in the case of MeCN, the cycloaddition of 3 and 4a occurred worse than the experiments carried out before (Entry 9), and in the case of CP, there not product formation formed (Entry 10).
Analyzing all the pros and cons of the results obtained, we have chosen the most effective 1 mol% CuBr(PPh3)3 / tert-BuOH:H2O 1:1 system to generate a small series of functionalized 1,2,3-triazole-based APAP derivatives 5a-f from alkyne 3 and freshly-prepared diverse organoazides R-N3 4a-f carried out by utilizing Filimonov’s procedure [65] (Scheme 2).
The 1,2,3-triazole-based APAP hybrids obtained are not only molecule targets but are also a valuable platform for generating new, more complex compounds. Considering this, hybrid 5a was transformed into nitrate propyl-triazole-APAP hybrid 7a, as a stable, yellow solid, via the two steps using the consecutive SN2 reactions (Scheme 3). Noteworthy that the latter hybrid belongs to nitric oxide-releasing agents (i.e., “hybrid nitrates”) which are explored in clinical trials in a range of indications, including cancer chemoprevention [66,67,68].
The yields for these click reactions were in the range of 53−93%. The final products were stable, colorless powders with well-defined melting points (Scheme 2 and Scheme 3). The structures of the designed hybrid compounds were fully characterized by FT-IR, 1H- and 13C-NMR spectroscopy. Whereas Infrared spectroscopy was used in the first instance for the identification of characteristic absorption bands of the functional groups present in the different synthesized hybrids, the NMR experiments strongly confirmed their structures. The proton at the C-5 position of the triazole ring of these hybrids appeared as a singlet at δ 7.62−9.02 ppm indicating that the 1,4-regioisomers were obtained (see, Supplementary Materials). Taking derivative 6a as an example, its 1H-NMR spectrum amplified in the aromatic region (Figure 2) is discussed below.
A singlet centered at 7.41 ppm corresponding to the hydrogen of the amide function corroborates the IR analysis for this compound (3278 cm−1, ν(N-H) amide). On the other hand, the signal in the form of a singlet at 7.67 ppm corresponds to the proton at the C-5 position of the triazole ring. In addition, in this region, we found two signals as doublets (d) at 7.41 ppm and 6.94 ppm with vicinal proton-proton coupling constants 3JHH (3J = 8.6 Hz) that are consistent with the hydrogens of the benzene ring as APAP skeleton. Another singlet corresponding to the hydrogens of the linker 3-Ctriazole-CH2-O- fragment is found at 5.20 ppm. Next, at 4.47 ppm and 3.38 ppm are two triplets (3J = 6.6 and 6.2 Hz, respectively) that correspond to the hydrogens of the -CH2-CH2-Ntriazole fragment. At 2.49 ppm there is a quartet (3J = 6.4 Hz) that corresponds to the Br-CH2- group and finally, at 2.17 ppm there is a singlet that belongs to the methyl group present in the acetaminophen fragment.
The analysis of its 13C NMR spectrum also indicated the nature of the diverse carbons present in the molecule through their location in the different regions of the spectrum and, in a general way, confirmed its condensed formula, finding all the carbons (see, Supplementary Materials).
3.2. In-Silico Prediction of Physicochemical Properties (Lipinski Descriptors) of Prepared Triazole-APAP Hybrids
To estimate the druggability of the APAP hybrids obtained, we complemented the present work by evaluating the potential therapeutic properties of these candidates using different in-silico tools. In particular, their physicochemical properties so-called Lipinski descriptors and toxicity risks were predicted.
First, by using the Molinspiration online platform [69], we calculated the physicochemical properties like lipophilicity (i.e., the logarithm of the partition coefficient between n-octanol and water), molecular weight, hydrogen bond donors and acceptors, rotatable bonds, and topological polar surface area according to Lipinski’s rule of five (RO5) [70] (Table 2). These properties affect the pharmacokinetics and drug-likeliness of small molecules.
The data obtained showed that all the compounds 5a-f, 6a,7a and the reference molecules (APAP and APAP-ONO2) (Figure 1) contain high bioavailability properties, and fulfill all the parameters established by this rule. It was observed that the hybrids 5b-f, 6a, 7a, and APAP-ONO2 are more lipophilic molecules having cLogP values with a short range (1.76-3.32) than compound 5a and APAP which showed similar, small lipophilicity character (cLogP = 0.60 and 0.68, respectively). Prediction results for the compounds 5a-f, 6a, and 7a molecular properties (Table 2) showed TPSA values between 69 and 124 Å confirming their drug-relevant properties. It is interesting to note that the reference molecules, APAP and APAP-ONO2, exhibited very different lipophilicity and polar surface parameters (cLogP = 0.68 vs cLogP = 2.17; TPSA = 49.33 vs TPSA = 110.46, respectively). This difference is due to the presence of the O-pentanoylnitrate fragment (NO-acetaminophen) which improved considerably the bioavailability properties of the acetaminophen drug. Noteworthy that both “nitrate hybrids” 7a and NO-acetaminophen displayed comparable Lipinski descriptors.
Then, in order to assess the possible secondary biological properties of the hybrids prepared, a toxicity profile evaluation was performed using the free software OSIRIS property explorer [71], which computes toxicity risks and drug-relevant properties of compounds and provides results as color-coded features. Thus, the APAP-triazole hybrids were predicted to have no risk of mutagenic (Mut), tumorigenic (Tum), irritant (Irr), or reproductive (Rep) effects (Table 3). The only exception was the N-ethylbromide-triazole-APAP derivative 6a, which presented a high predisposition to be tumorigenic and to have reproductive effects. By contrasting the toxicity risks obtained for molecules 5a, 6a, and 7a, it is concluded that the inclusion of bromine in this series of analogs considerably increased toxicity. Noteworthy that the OSIRIS prediction of APAP indicates that it is mutagenic, tumorigenic, and reproductively harmful, although, in practice, side effects of acetaminophen are rare if taken at appropriate doses.
Moreover, we used the OSIRIS program to predict the compounds’ drug-likeness (DL) and drug-score (DS) properties. The first is related to the similarity to drugs (with a score of -10 to 10), the qualitative calculation of the chance for a compound to become an oral drug with respect to bioavailability. Its positive value indicates that the compound comprises fragments that are frequently present in commercial drugs. The second (with a numerical value between 0 and 1) combines DL, Log P, MW, and toxicity risks in one convenient value that can be used to judge the compound’s overall potential to qualify for a drug. Compounds without predicted toxicity risks and with drug scores of DS 0.5 and above were considered promising. Table 3 includes these data for synthesized compounds and APAP and APAP-ONO2 as reference molecules.
The OSIRIS Property Explore software indicated that almost all the molecular hybrids presented high DS values ranging from 0.59 to 0.78 with the exception of compound 6a. By contrasting the DS values obtained for molecules 5a, 6a, and 7a, it is concluded that the inclusion of bromine in this series of analogs not only considerably increased toxicity but also decreased the DS property (0.12), so it could be inferred that the inclusion of this atom in the aliphatic fragment of the triazole-APAP hybrid 6a decreased also the similarity of the molecule with known drugs.
The N-benzyl-triazole-APAP hybrid 5b showed the best DS parameter (0.78), acceptable to become a potential drug, while the acetaminophen DS displayed a low value of 0.20, comparable with that of hybrid 6a. This difference suggests that its combination with the N-substituted triazole fragment in the same molecule could contribute to improving the properties that influence the possibility of being potential drugs. Moreover, all the hybrids synthesized 5a-f including the N-propylnitrate-triazole-APAP hybrid 7a exhibited DS values (0.59-0.78) very close to, or higher, than that of NO-acetaminophen (0.45), confirming the potential of these compounds to become potential drugs and, therefore, other types of substitutions in these molecules should be evaluated in order to increase their values.
4. Conclusions
In conclusion, we have successfully applied an eco-friendly method for obtaining new 1,2,3-triazole-based acetaminophen derivatives through the 1 mol% CuBr(PPh3)3 / tert-BuOH:H2O 1:1 system from easily accessible starting materials. These drug hybrids were obtained with good to excellent yields (64-93%) without chromatography column separation using the expired acetaminophen drug, i.e., pharmaceutical waste as raw material. Taking advantage of the chemical reactivity of hybrid 5a, a new nitric oxide-releasing N-propylnitrate-triazole-APAP hybrid 7a was synthesized in two easy steps with good performance (61%) for the first time. Based on our observations, most of the new molecules appeared to be stable solids under laboratory conditions. Furthermore, the in-silico studies indicated the potential of the hybrid compounds due to their interesting physicochemical properties containing high bioavailability levels and fulfilling all parameters established by Lipinski´s rule. According to the Osiris platform, almost all the molecular hybrids designed and obtained did not show any risk of toxicity and presented higher Drug-Score values ranging from 0.59 to 0.78 than those of reference drugs acetaminophen and NO-acetaminophen. Finally, our study demonstrated that the hybridization of acetaminophen by a 1-substituted 1,2,3-triazole ring is a valuable approach for the research and development of new agents based on acetaminophen
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, synthetic procedures FT-IR and NMR.
Author Contributions
Conceptualization, V.V.K.; methodology, D.C.L.; formal analysis, C.E.G.P.; writing—original draft preparation, D.C.L. and V.V.K; writing—review and editing V.V.K. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Data Availability Statement
Synthetic procedures and FT-IR and NMR data are reported in Supplementary Materials.
Acknowledgments
We are grateful to Escuela de Química of the Universidad Industrial de Santander for financial support.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Anderson, B.J. Paracetamol (Acetaminophen): mechanisms of action. Paediatr. Anaesth. 2008, 18, 915–921. [Google Scholar] [CrossRef] [PubMed]
- Ayoub, S.S. Paracetamol (acetaminophen): A familiar drug with an unexplained mechanism of action. Temp. 2021, 8, 351–371. [Google Scholar] [CrossRef]
- Klotz, U. Paracetamol (acetaminophen)–a popular and widely used nonopioid analgesic. Arzneimittelforschung 2012, 62, 355–359. [Google Scholar] [CrossRef]
- Brune, K.; Renner, B.; Tiegs, G. Acetaminophen/paracetamol: a history of errors, failures and false decisions. Eur. J. Pain 2015, 19, 953–965. [Google Scholar] [CrossRef]
- Freo, U.; Ruocco, C.; Valerio, A.; Scagnol, I.; Nisoli, E. Paracetamol: a review of guideline recommendations. J. Clin. Med. 2021, 10, 3420. [Google Scholar] [CrossRef]
- FDA drug safety communication: prescription acetaminophen products are to be limited to 325 mg per dosage unit; a boxed warning will highlight the potential for severe liver failure. Available online: https://www.fda.gov/drugs/drug-safety-and-availability/fda-drug-safety-communication-prescription-acetaminophen-products-be-limited-325-mg-dosage-unit.
- Aminoshariae, A.; Khan, A. Acetaminophen: old drug, new issues. J. End. 2015, 41, 588–593. [Google Scholar] [CrossRef] [PubMed]
- Bunchorntavakul, C.; Reddy, K.R. Acetaminophen (APAP or N-acetyl-p-aminophenol) and acute liver failure. Clin. Liver Dis. 2018, 22, 325–346. [Google Scholar] [CrossRef] [PubMed]
- Hanel, A.M.; Lands, W.E. Modification of anti-inflammatory drug effectiveness by ambient lipid peroxides. Biochem. Pharmacol. 1982, 31, 3307–3311. [Google Scholar] [CrossRef]
- Boutaud, O.; Aronoff, D.M.; Richardson, J.H.; Marnett, L.J.; Oates, J.A. Determinants of the cellular specificity of acetaminophen as an inhibitor of prostaglandin H2 synthases. Proc. Natl. Acad. Sci. USA 2002, 99, 7130–7135. [Google Scholar] [CrossRef]
- Schildknecht, S.; Daiber, A.; Ghisla, S.; Cohen, R.A.; Bachschmid, M.M. Acetaminophen inhibits prostanoid synthesis by scavenging the PGHS-activator peroxynitrite. FASEB J. 2008, 22, 215–224. [Google Scholar] [CrossRef]
- Graham, G.G.; Davies, M.J.; Day, R.O.; Mohamudally, A.; Scott, K.F. The modern pharmacology of paracetamol: therapeutic actions, mechanism of action, metabolism, toxicity and recent pharmacological findings. Inflammopharmacol. 2013, 21, 201–232. [Google Scholar] [CrossRef] [PubMed]
- Bessems, J.G.; Gaisser, H.D.; Te Koppele, J.M.; Van Bennekom, W.P.; Commandeur, J.N.; Vermeulen, N.P. 3, 5-Disubstituted analogues of paracetamol. Synthesis, analgesic activity and cytotoxicity. Chem. Biol. Interact. 1995, 98, 237–250. [Google Scholar] [CrossRef] [PubMed]
- Tavakolinejad-Kermani, E.; Saidi, K.; Islami, M.R. New synthesis of acetaminophen derivatives containing a phosphorus atom. Phosphorus Sulfur Silicon Relat. Elem. 2005, 180, 1879–1884. [Google Scholar] [CrossRef]
- Santos, C.; Mateus, M.L.; dos Santos, A.P.; Moreira, R.; de Oliveira, E.; Gomes, P. Cyclization-activated prodrugs. Synthesis, reactivity and toxicity of dipeptide esters of paracetamol. Bioorg. Med. Chem. Lett. 2005, 15, 1595–1598. [Google Scholar] [CrossRef] [PubMed]
- Jung, S.; Tsukuda, Y.; Kawashima, R.; Ishiki, T.; Matsumoto, A.; Nakaniwa, A.; Takagi, M.; Noguchi, T.; Imai, N. Convenient synthesis of acetaminophen analogues containing α-amino acids and fatty acids via their mixed carbonic carboxylic anhydrides in aqueous organic solvent. Tetrahedron Lett. 2013, 54, 5718–5720. [Google Scholar] [CrossRef]
- Tiwari, A.D.; Panda, S.S.; Girgis, A.S.; Sahu, S.; George, R.F.; Srour, A.M.; La Starza, B.; Hall, C.D.; Asiri, A.M.; Katritzky, A.R. Microwave assisted synthesis and QSAR study of novel NSAID acetaminophen conjugates with amino acid linkers. Org. Biomol. Chem. 2014, 12, 7238–7249. [Google Scholar] [CrossRef] [PubMed]
- Ahmadi, A.; Khalili, M.; Sadeghi, S.; Soleimani, N.; Nahri-Niknafs, B. Synthesis of New Acetaminophen Analogs and Their Ibuprofen Conjugates as Novel Analgesic Drugs. Pharm. Chem. J. 2016, 50, 369–376. [Google Scholar] [CrossRef]
- Vaccarino, A.L.; Paul, D.; Mukherjee, P.K.; de Turco, E.B.R.; Marcheselli, V.L.; Xu, L.; Trudell, M.L.; Minguez, J.M.; Matía, M.P.; Sunkel, C.; Alvarez-Builla, J.; Bazan, N.G. Synthesis and in vivo evaluation of non-hepatotoxic acetaminophen analogs. Bioorg. Med. Chem. 2007, 15, 2206–2215. [Google Scholar] [CrossRef]
- Queiroz, L.M.; Rocha, J.R.; Leitão, A.; Montanari, C.A.; da Silva, A.B.; Sousa, P.J.; Borges, R.S. A Combined Study Using Ligand-Based Design, Synthesis, and Pharmacological Evaluation of Analogues of the Acetaminophen Ortho-Regioisomer with Potent Analgesic Activity. Chem. Biol. Drug Des. 2012, 80, 99–105. [Google Scholar] [CrossRef]
- Alisi, M.A.; Brufani, M.; Cazzolla, N.; Ceccacci, F.; Dragone, P.; Felici, M.; Furlotti, G.; Garofalo, B.; La Bella, A.; Dragone, P.; Lanzalung, O.; Leonelli, F.; Bettolo, R.M.; Maugeri, C.; Maria Migneco, L.M.; Russo, V. DPPH radical scavenging activity of paracetamol analogues. Tetrahedron 2012, 68, 10180–10187. [Google Scholar] [CrossRef]
- Reddy, Y.D.; Kumari, Y.B.; Dubey, P.K. Synthesis of a novel water soluble phthalimide derivative of acetaminophen as potential analgesic and antipyretic agent. Indian J. Chem. 2013, 52B, 691–693. [Google Scholar] [CrossRef]
- Raj, N.U.I. Synthesis, single crystal XRD and CT DNA/BSA binding studies of new paracetamol derivatives. J. Mol. Struct. 2020, 1208, 127911. [Google Scholar]
- Asghari, A.; Ameri, M.; Taghipour, S.; Ghaderi, O. Facile and clean electrochemical synthesis of new acetaminophen derivatives through electrochemical oxidation of acetaminophen in the presence of thiouracil derivatives. J. Sulphur Chem. 2017, 38, 163–172. [Google Scholar] [CrossRef]
- Profire, L.; Sunel, V.; Lupascu, D.; Baican, M.C.; Bibire, N.; Vasile, C. New theophylline derivatives with potential pharmacological activity. Farmacia 2010, 58, 170–176. [Google Scholar]
- Das, M.; Bhattacharjee, S.; Fronczek, F.R.; Bazan, N.G.; Trudell, M.L. Synthesis, hepatotoxic evaluation and antipyretic activity of nitrate ester analogs of the acetaminophen derivative SCP-1. Bioorg. Med. Chem. Lett. 2018, 28, 3798–3801. [Google Scholar] [CrossRef] [PubMed]
- Sahu, A.; Das, D.; Sahu, P.; Mishra, S.; Sakthivel, A.; Gajbhiye, A.; Agrawal, R. Bioisosteric replacement of amide group with 1, 2, 3-triazoles in acetaminophen addresses reactive oxygen species-mediated hepatotoxic insult in Wistar albino rats. Chem. Res. Toxicol. 2020, 33, 522–535. [Google Scholar] [CrossRef] [PubMed]
- Al-Swayeh, O.A.; Futter, L.E.; Clifford, R.H.; Moore, P.K. Nitroparacetamol exhibits anti-inflammatory and anti-nociceptive activity. Br. J. Pharmacol. 2000, 130, 1453–1456. [Google Scholar] [CrossRef]
- Proschak, E.; Stark, H.; Merk, D. Polypharmacology by design: a medicinal chemist’s perspective on multitargeting compounds. J. Med. Chem. 2018, 62, 420–444. [Google Scholar] [CrossRef]
- Bansal, Y.; Silakari, O. Multifunctional compounds: Smart molecules for multifactorial diseases. Eur. J. Med. Chem. 2014, 76, 31–42. [Google Scholar] [CrossRef]
- Bérubé, G. An overview of molecular hybrids in drug discovery. Expert Opin. Drug Discov. 2016, 11, 281–305. [Google Scholar] [CrossRef]
- Abdolmaleki, A.; Ghasemi, J.B. Dual-acting of hybrid compounds-a new dawn in the discovery of multi-target drugs: lead generation approaches. Curr. Top. Med. Chem. 2017, 17, 1096–1114. [Google Scholar] [CrossRef] [PubMed]
- Joshi, G.P. NCX-701. NCX-701. NicOx. Curr. Opin. Investig. 2004, 5, 755–759. [Google Scholar]
- Romero-Sandoval, E.A.; Curros-Criado, M.M.; Gaitan, G.; Molina, C.; Herrero, J.F. Nitroparacetamol (NCX-701) and Pain: First in a Series of Novel Analgesics. CNS Drug Reviews, 2007, 13, 279–295. [Google Scholar] [CrossRef] [PubMed]
- Massarotti, A.; Aprile, S.; Mercalli, V.; Del Grosso, E.; Grosa, G.; Sorba, G.; Tron, G.C. Are 1, 4-and 1, 5-Disubstituted 1, 2, 3-Triazoles Good Pharmacophoric Groups? ChemMedChem 2014, 9, 2497–2508. [Google Scholar] [CrossRef] [PubMed]
- Lengerli, D.; Ibis, K.; Nural, Y.; Banoglu, E. The 1,2,3-triazole ‘all-in-one’ring system in drug discovery: a good bioisostere, a good pharmacophore, a good linker, and a versatile synthetic tool. Expert Opin. Drug Discov. 2022, 17, 1209–1236. [Google Scholar] [CrossRef]
- Bonandi, E.; Christodoulou, M.S.; Fumagalli, G.; Perdicchia, D.; Rastelli, G.; Passarella, D. The 1, 2, 3-triazole ring as a bioisostere in medicinal chemistry. Drug Discov. Today 2017, 22, 1572–1581. [Google Scholar] [CrossRef] [PubMed]
- Rani, A.; Singh, G.; Singh, A.; Maqbool, U.; Kaur, G.; Singh, J. CuAAC-ensembled 1, 2, 3-triazole-linked isosteres as pharmacophores in drug discovery. RSC Adv. 2020, 10, 5610–5635. [Google Scholar] [CrossRef]
- Sahu, A.; Sahu, P.; Agrawal, R. A recent review on drug modification using 1, 2, 3-triazole. Curr. Chem. Biol. 2020, 14, 71–87. [Google Scholar] [CrossRef]
- Bozorov, K.; Zhao, J.; Aisa, H.A. 1, 2, 3-Triazole-containing hybrids as leads in medicinal chemistry: A recent overview. Bioorg. Med. Chem. 2019, 27, 3511–3531. [Google Scholar] [CrossRef]
- Keri, R.S.; Patil, S.A.; Budagumpi, S.; Nagaraja, B.M. Triazole: a promising antitubercular agent. Chem. Biol. Drug. Des. 2015, 86, 410–423. [Google Scholar] [CrossRef]
- Song, M.X.; Deng, X.Q. Recent developments on triazole nucleus in anticonvulsant compounds: a review. J. Enzyme Inhib. Med. Chem. 2018, 33, 453–478. [Google Scholar] [CrossRef] [PubMed]
- Lal, K.; Yadav, P. Recent advancements in 1, 4-disubstituted 1H-1, 2, 3-triazoles as potential anticancer agents. Anti-Cancer Agents Med. Chem. 2018, 18, 21–37. [Google Scholar] [CrossRef]
- Alam, M.M. 1,2,3-Triazole hybrids as anticancer agents: A review. Arch. Pharm. 2022, 355, 2100158. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B. Comprehensive review on the anti-bacterial activity of 1, 2, 3-triazole hybrids. Eur. J. Med. Chem. 2019, 168, 357–372. [Google Scholar] [CrossRef] [PubMed]
- Nemallapudi, B.R.; Guda, D.R.; Ummadi, N.; Avula, B.; Zyryanov, G.V.; Reddy, C.S.; Gundala, S. New methods for synthesis of 1,2,3-triazoles: A review. Polycycl. Aromat. Compd. 2022, 42, 3874–3892. [Google Scholar] [CrossRef]
- Vala, D.P.; Vala, R.M.; Patel, H.M. Versatile Synthetic Platform for 1,2,3-Triazole Chemistry. ACS Omega 2022, 7, 36945–36987. [Google Scholar] [CrossRef] [PubMed]
- Huisgen, R. Cycloadditions—definition, classification, and characterization. Angew. Chem. Int. Ed. 1968, 7, 321–328. [Google Scholar] [CrossRef]
- Breugst, M.; Reissig, H.U. The Huisgen Reaction: Milestones of the 1, 3-Dipolar Cycloaddition. Angew. Chem. Int. Ed. 2020, 59, 12293–12307. [Google Scholar] [CrossRef] [PubMed]
- Rostovtsev, V.V.; Green, L.G.; Fokin, V.V.; Sharpless, K.B. A Stepwise Huisgen Cycloaddition Process: Copper(I)-Catalyzed Regioselective “Ligation” of Azides and Terminal Alkynes. Angew. Chem. Int. Ed. 2002, 41, 2596–2599. [Google Scholar] [CrossRef]
- Tornøe, C.W.; Christensen, C.; Meldal, M. Peptidotriazoles on Solid Phase: [1,2,3]-Triazoles by Regiospecific Copper(I)-Catalyzed 1,3-Dipolar Cycloadditions of Terminal Alkynes to Azides. J. Org. Chem. 2002, 67, 3057–3064. [Google Scholar] [CrossRef]
- Haldón, E.; Nicasio, M.C.; Pérez, P.J. Copper-catalysed azide−alkyne cycloadditions (CuAAC): an update. Org. Biomol. Chem. 2015, 13, 9528–9550. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Balderas, F.; Ortega-Munoz, M.; Morales-Sanfrutos, J.; Hernández-Mateo, F.; Calvo-Flores, F.G.; Calvo-Asín, J.A.; Isac-García, J.; Santoyo-González, F. Multivalent neoglycoconjugates by regiospecific cycloaddition of alkynes and azides using organic-soluble copper catalysts. Org. Lett. 2003, 5, 1951–1954. [Google Scholar] [CrossRef] [PubMed]
- Lal, S.; Diez-Gonzalez, S. [CuBr (PPh3)3] for azide− alkyne cycloaddition reactions under strict click conditions. J. Org. Chem. 2011, 76, 2367–2373. [Google Scholar] [CrossRef]
- Han, J. Barcoding drug information to recycle unwanted household pharmaceuticals: a review. Environ. Chem. Lett. 2022, 20, 2989–3003. [Google Scholar] [CrossRef]
- Pratama, D.E.; Hsieh, W.C.; Elmaamoun, A.; Lee, H.L.; Lee, T. Recovery of active pharmaceutical ingredients from unused solid dosage-form drugs. ACS Omega 2020, 5, 29147–29157. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, D.S.; Lindrud, M.; Lu, X.; Zordan, C.; Tang, L.; Davies, M. A process for active pharmaceutical ingredient recovery from tablets using green engineering technology. Org. Process Res. Dev. 2017, 21, 1272–1285. [Google Scholar] [CrossRef]
- Messina, L.C.; de Espindola, C.S.; Omori, A.T. Direct One-Pot Synthesis of Propofol from Paracetamol Tablets. ACS Sustainable Chem. Eng. 2023, 11, 1638–1642. [Google Scholar] [CrossRef]
- Kouznetsov, V.V.; Vargas-Méndez, L.Y.; Zubkov, F.I. Recent Advances in Synthesis of Bioactive Quinoline-based 1,2,3-Triazoles via Cu-catalyzed Huisgen 1,3-Dipolar Cycloaddition (“Click reaction”). Mini-Rev. Org. Chem. 2016, 13, 488–503. [Google Scholar] [CrossRef]
- Luna-Parada, L.K.; Vargas-Méndez, L.Y.; Kouznetsov, V.V. Quinoline-Substituted 1,2,3-Triazole-Based Molecules, As Promising Conjugated Hybrids in Biomedical Research. Organic Medicinal Chem. IJ. 2018, 7, 555708. [Google Scholar] [CrossRef]
- Luna-Parada, L.K.; Kouznetsov, V.V. 5-Chloro-8-{[1-(2-chlorobenzyl)-1H-1,2,3-triazol-4-yl] methoxy}quinoline. Molbank 2019, 2019, M1038. [Google Scholar] [CrossRef]
- Rosado-Solano, D.N.; Barón-Rodríguez, M.A.; Sanabria-Florez, P.L.; Luna-Parada, L.K.; Puerto-Galvis, C.E.; Zorro-González, A.F.; Kouznetsov, V.V.; Vargas-Méndez, L.Y. Synthesis, Biological Evaluation and in silico Computational Studies of 7-Chloro-4-(1H-1,2,3-triazol-1-yl)quinoline Derivatives. Search for new controlling agents against Spodoptera frugiperda (Lepidoptera: Noctuidae) larvae. J. Agric. Food Chem. 2019, 67, 9210–9219. [Google Scholar] [CrossRef]
- Calderón Lamus, D.; Puerto Galvis, C.E.; Kouznetsov, V.V. Cu(PPh3)3Br-catalyzed synthesis of new paracetamol-1,2,3-triazole molecular hybrids from expired commercial tablets and their in silico assessment to study their pharmacological properties. Med. Sci. Forum 2022, 14, 85, (8th International Electronic Conference on Medicinal Chemistry Session Small molecules as drug candidates, 01-30 November 2022, MDPI). [Google Scholar]
- Pak, J.K.; Hesse, M. Synthesis of penta-N-protected homocaldopentamine and its selective acylation. J. Org. Chem. 1998, 63, 8200–8204. [Google Scholar] [CrossRef]
- Kutonova, K.V.; Trusova, M.E.; Postnikov, P.S.; Filimonov, V.D.; Parello, J. A simple and effective synthesis of aryl azides via arenediazonium tosylates. Synthesis 2013, 45, 2706–2710. [Google Scholar]
- Vong, L.B.; Nagasaki, Y. Nitric oxide nano-delivery systems for cancer therapeutics: Advances and challenges. Antioxidants 2020, 9, 791. [Google Scholar] [CrossRef]
- Serafim, R.A.; Pernichelle, F.G.; Ferreira, E.I. The latest advances in the discovery of nitric oxide hybrid drug compounds. Expert Opin. Drug Discov. 2017, 12, 941–953. [Google Scholar] [CrossRef]
- Kumar, S.; Singh, R.K.; Bhardwaj, T.R. Therapeutic role of nitric oxide as emerging molecule. Biomed. Pharmacother. 2017, 85, 182–201. [Google Scholar] [CrossRef]
- Molinspiration Cheminformatics. Available online: http://www.molinspiration.com (accessed on 15 January 2023).
- Lipinski, C.A. Lead-and drug-like compounds: the rule-of-five revolution. Drug Discov. Today Technol. 2004, 1, 337–341. [Google Scholar] [CrossRef]
- Organic Chemistry Portal. The OSIRIS Property Explorer open-source program. Available online: http://www.organic-chemistry.org/prog/peo/ (accessed on 15 January 2023).
Figure 1.
Structure of acetaminophen (APAP), its hybrid compounds A-D exhibiting more anti-inflammatory activity than the parent APAP (A-C) and bronchodilator effect than theophylline drug (D), and the designed 1,2,3-triazole-based acetaminophen derivatives E.
Figure 1.
Structure of acetaminophen (APAP), its hybrid compounds A-D exhibiting more anti-inflammatory activity than the parent APAP (A-C) and bronchodilator effect than theophylline drug (D), and the designed 1,2,3-triazole-based acetaminophen derivatives E.
Scheme 1.
Preparation of O-propargyl-APAP 3.
Scheme 2.
Preparation of functionalized 1,2,3-triazole-based APAP derivatives 5a-f via the CuAAC reaction.
Scheme 2.
Preparation of functionalized 1,2,3-triazole-based APAP derivatives 5a-f via the CuAAC reaction.
Scheme 3.
Synthesis of nitrate propyl-triazole-APAP hybrid 7a through simple transformations.
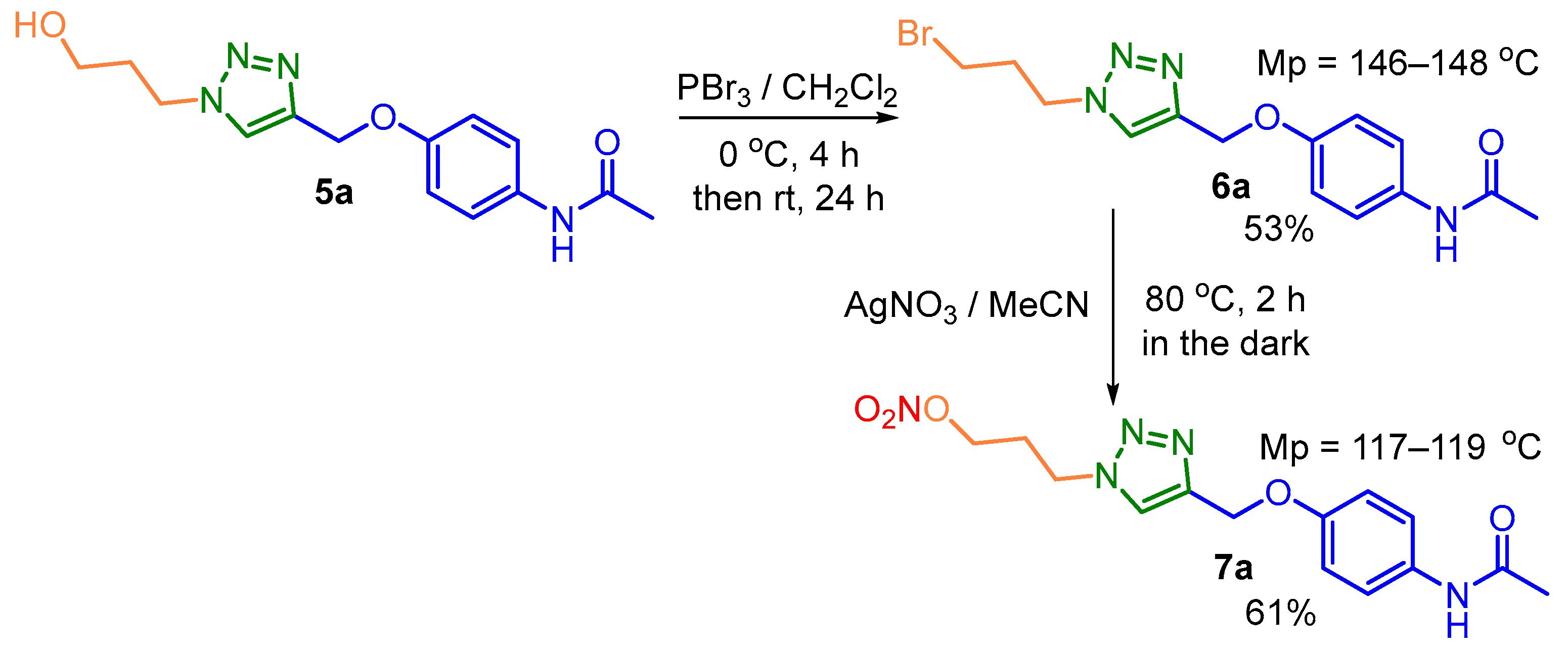
Figure 2.
1H-NMR of the hybrid 6a (400 MHz, CDCl3, δ, ppm).
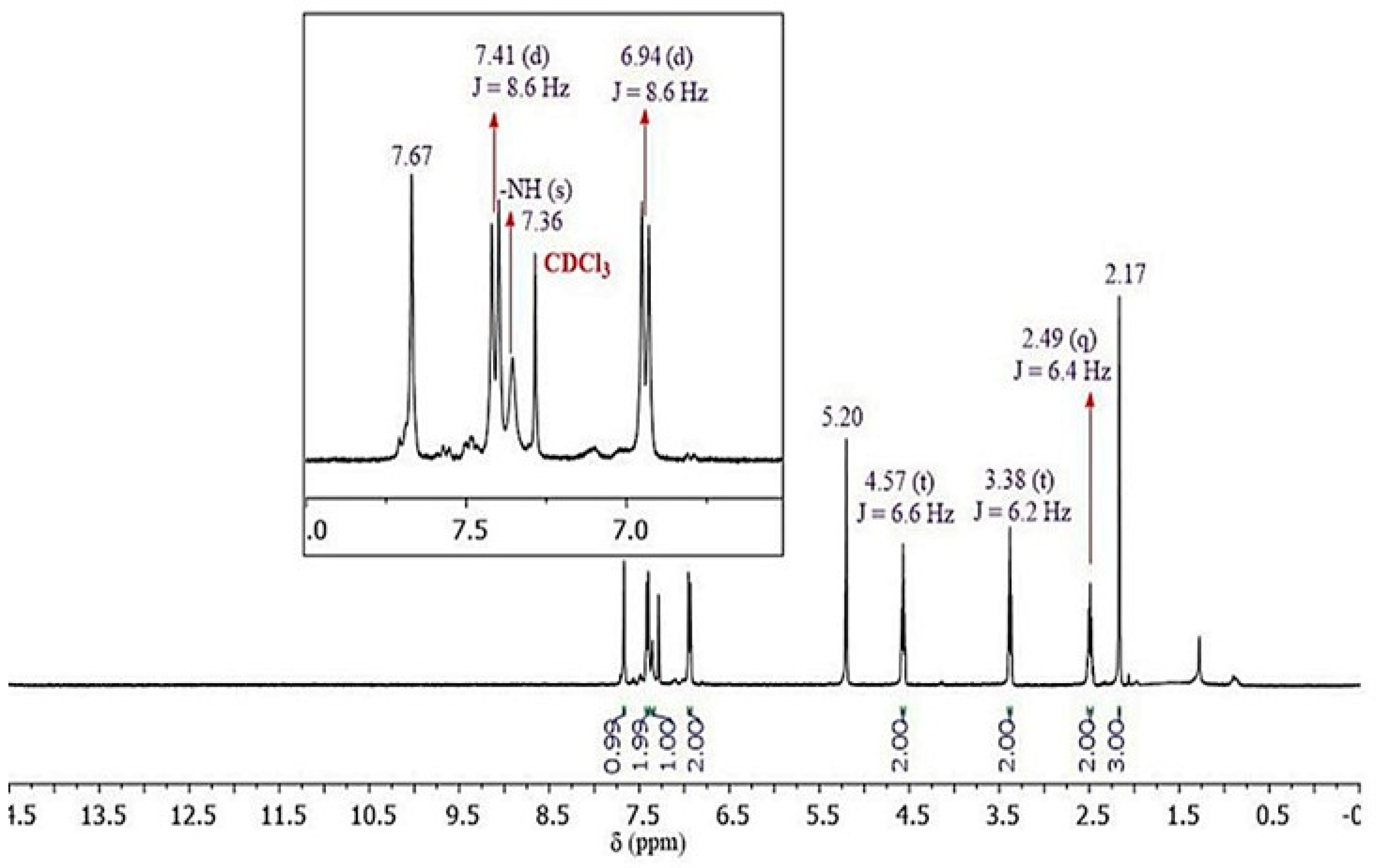
Table 2.
Calculated Lipinski’s rule of five parameters a for the synthesized molecules.
| Comp. | MWb | cLogPc | HBAd | HBDe | ROTBf | TPSAg | n violations |
|---|---|---|---|---|---|---|---|
| 5a | 290.32 | 0.60 | 7 | 2 | 7 | 89.28 | 0 |
| 5b | 322.37 | 2.56 | 6 | 1 | 6 | 69.05 | 0 |
| 5c | 342.79 | 2.92 | 6 | 1 | 5 | 69.05 | 0 |
| 5d | 434.24 | 3.32 | 6 | 1 | 5 | 69.05 | 0 |
| 5e | 324.34 | 1.76 | 7 | 2 | 5 | 89.28 | 0 |
| 5f | 352.35 | 2.15 | 8 | 2 | 6 | 106.35 | 0 |
| 6a | 353.22 | 1.97 | 6 | 1 | 7 | 69.05 | 0 |
| 7a | 335.32 | 1.86 | 10 | 1 | 9 | 124.11 | 0 |
| APAP | 151.16 | 0.68 | 3 | 2 | 1 | 49.33 | 0 |
| APAP-ONO2 | 296.28 | 2.17 | 8 | 1 | 9 | 110.46 | 0 |
a Lipinski´s rule of bioavailability: molecular weight ≤ 500 Da, lipophilicity assessed by the cLogP ≤ 5, number of hydrogen bond donors and acceptors ≤ 5 and ≤ 10 respectively, number of rotatable bonds ≤ 10, and topological polar surface area ≤ 140.; b Molecular Weight (g/mol); c Logarithm of the partition coefficient between n-octanol and water; d Number of hydrogen-bond acceptors; e Number of hydrogen-bond donors; f Number of rotatable bonds; g Polar Surface Area (Å2).
Table 3.
Toxicity risks and drug-likeliness predicted by OSIRIS Property Explorer a.
| Comp. | Mut | Tum | Irr | Rep | Drug-likeness | Drug-score |
|---|---|---|---|---|---|---|
| 5a |  |
|
|
|
-0.75 | 0.62 |
| 5b | |
|
|
|
1.72 | 0.78 |
| 5c | |
|
|
|
1.06 | 0.70 |
| 5d | |
|
|
|
0.65 | 0.59 |
| 5e | |
|
|
|
0.46 | 0.72 |
| 5f | |
|
|
|
-0.52 | 0.59 |
| 6a |  |
 |
|
|
-8.16 | 0.12 |
| 7a | |
|
|
|
-0.71 | 0.59 |
| APAP | |
|
|
|
1.93 | 0.20 |
| APAP-ONO2 | |
|
|
|
-8.25 | 0.45 |
a = No risk, = Moderated risk, = High risk
= No risk, = Moderated risk, = High risk
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



