
Submitted:
07 June 2023
Posted:
08 June 2023
You are already at the latest version
Abstract
Hb Adana is a non-deletional α-thalassaemia variant, due to mutations in α1- or α2-globin codon 59 (αCD59) that produce unstable α-globin. Clinical appearance can range from silent carrier to blood transfusion reliance, hepatosplenomegaly, skeletal deformities, and spinal cord compression. Despite the importance of Hb Adana inheritance, this Hb variant is challenging to study since molecular tests are scarce and routine diagnosis can be missed. This study explored the distribution of Hb Adana among local high school students and evaluate the hematological parameters and haemoglobin analysis of Hb Adana in this region of Malaysia. This retrospective study included 13,721 blood samples from high school students enrolled in Malaysia's National Thalassaemia Screening Program at Hospital Raja Perempuan Zainab II (HRPZ II). Multiplex Gap PCR was used to detect deletional α-thalassaemia. Common non-deletional α-thalassaemia was detected using multiplex ARMS PCR. Evaluable data were retrieved from the HRPZ II database. Results: A total of 2327 subjects exhibited common deletional (n=1037, 44.6%) and non-deletional (n=1290, 55.4%) α-thalassaemia. Hb Constant Spring was the most frequent non-deletional α-thalassaemia. Thirty-one participants (1.33%) had αCD59α/αα and one (0.04%) had αCD59α/-α3.7. The 32 Hb Adana subjects were 87.5% Malay, and 12.5% Orang Asli. Additionally, seven cases of HbE/Hb Adana were discovered. The haemoglobin level in heterozygous Hb Adana ranged from mild anaemia to normal, 95.0 g/L to 153.0 g/L. The MCV and MCH were approximately 73 fL and 23 pg, respectively. Conclusions: We have denoted the distribution of alpha thalassaemia mutation patterns among high school students in Kelantan, Northeast Peninsular of Malaysia. Our observation showed Hb Adana was rarely uncommon in our region and the co-inheritance with an α-gene deletion generates α+-thalassaemia; with HbE, α0-thalassaemia. All heterozygous Hb Adana exhibited low MCVs and MCHs.
Keywords:
Hb Adana
; non-deletional α-thalassaemia
; Hb Constant Spring
; HbE
1. Introduction
Thalassaemia is a heterogeneous group of blood disorders involving haemoglobin synthesis inherited in an autosomal recessive pattern. Thalassaemia and related haemoglobin disorders have mutations in the globin gene that affect the globin chain production [1]. The classification is broadly divided into a quantitative reduction of the globin chain (=thalassaemia) or qualitative abnormal globin chain (=hemoglobinopathies) produced in the marrow. The two forms of thalassemia include α- and β- thalassaemias. More rare categories include γ-, δ-, and εγδβ- thalassaemias [2]. The genomic structure of α- and β- thalassaemias was illustrated in Figure 1. The population in malaria-endemic regions, such as tropical and sub-tropical areas, has a high carrier rate of alpha thalassemia, approximately 20-30% [3-6]. It is most prevalent in Southeast Asian, Mediterranean, and Middle Eastern countries. Gene selection for alpha thalassemia offers protection against malaria falciparum [6,7]. However, nowadays, population migration has increased alpha gene mutation frequency even in the malarial non-endemic region. In Malaysia, the incidence of alpha thalassemia varies between studies and ranges from 4.5-15.8% [8-12].
Alpha thalassemia is caused by either α-gene deletion or mutation, leading to the complete absence or deficient synthesis of the α-globin chain. It can be broadly categorised into deletional and non-deletional types of alpha thalassemia [13,14]. The incidence of non-deletional α-thalassaemia is comparatively lower than that of deletional α-thalassaemia, which is more common [15,16]. However, the majority of α-thalassaemia cases with severe clinical manifestations involve at least one non-deletional α-thalassaemia [17,18]. Over 70 forms of non-deletional α-thalassaemia have been documented [19], characterised by the insertion, deletion, or substitution of a single nucleotide in the α-gene, which alters the α-globin chain synthesis. Multiple processes, including a mutant RNA splice site, RNA polyadenylation, poor RNA translation, generation of extended mRNA, and termination chain alterations, are responsible for the mutation’s problems [1,20]. In Southeast Asia, the common non-deletional types are as follows; Hb Constant Spring, Hb Adana, Hb Quong Sze, and Hb Pakse. The commonly encountered deletional alpha thalassaemia are 3.7 kb deletion (-α3.7), 4.2 kb deletion (-α4.2), SEA deletion (--SEA), and THAI deletion (--THAI) [21,22].
In Malaysia, Hb Adana is the second most common non-deletional α-thalassaemia after Hb Constant Spring [15], resulting from a mutation of HBA1 or HBA2 on either the α1- or α2-globin gene at codon 59 of chromosome 16p13.3. The other functional domains of α-globins were the embryonic ζ-gene, HBZ, three pseudogenes and the θ1 gene of unknown function. The HS-40 is an alpha-globin gene cluster, situated at the very tip of the short arms of chromosome 16, which control pattern of expression according to the developmental stages [23](Figure 1). This substitution of glycine (GGC) to aspartic acid (GAC) of HBA2- globin gene leads to the formation of a larger charged aspartic acid molecule, which can compromise the stability of haemoglobin [24]. Hb Adana is characterised by a low HBA2 level, increased Hb Bart’s, elevated Zeta chain, and a little of Hb H disease. [25]
Hb Adana was first reported in 2009 in a 52 year old Malay lady with a clinical manifestation of thalassaemia intermedia and a genotype of double heterozygous Hb Adana/-α3.7 [26]. This was followed by more case series of thalassaemia intermedia or hydrops fetalis at presentation, and eventually, family screening revealed a carrier state in either of the parents [27-29]. Heterozygous Hb Adana (single codon 59 mutation) is a silent carrier. The subtle production of unstable haemoglobin in an individual remains undiagnosed unless molecular DNA analysis is done [30].
The incidence of Hb Adana varies in different regions of the world. The incidence of Hb Adana is lower in Turkey (0.5–0.6%), China (about 1%), and Iran/Iraq (1–2.5%) [16,24,31,32,33]. There is a higher prevalence of Hb Adana in the following countries: namely, Saudi Arabia (11.6%) and Indonesia (16%) [17,28,34]. In Malaysia, a few studies showed various gene frequencies of Hb Adana. The study, done by IMR (Institute for Medical Research) and UKMMC (Universiti Kebangsaan Malaysia Medical Centre), showed frequencies of Hb Adana at 2.5% and 1.0%, respectively [35,36]. Rahimah et al. showed 0.01% Hb Adana in their study on high school students involved in the National Thalassaemia Screening program in Penang, Melaka, and Sabah [37].
The inheritance of non-deletional α-thalassaemia provides diverse clinical manifestation that span from the asymptomatic silent carrier to dependency on blood transfusion, hepatosplenomegaly, skeletal anomalies, and spinal cord compression as sequelae of extramedullary haematopoiesis [30,38,39]. Homozygous Hb Adana (αCD59α/αCD59α) and compound heterozygous Hb Adana with Southeast Asian (SEA) deletion (--SEA/ααCD59) manifest as hydrops fetalis, where the foetus is not compatible with life [38,39]. Interactions of deletional with non-deletional α-thalassaemia mutations produce HbH disease with moderate to severe anaemia and some significant hepatosplenomegaly [18,39]. Heterozygous Hb Adana carrier status is generally asymptomatic except in pregnancy, where it may present with severe anaemia [38].
Hb Adana showed subtle changes in haematological profiles due to the unstable Hb variant and decreased expression of α-globin genes, which may associate with the cellular processing of an unstable mRNA. This is characterised by a reduced lifespan, and red cell precipitation causing haemolysis [19]. The full blood count indices may show mildly hypochromic microcytic indices with normal haemoglobin levels. The haemoglobin analysis is usually unremarkable [30]. Hence, the final diagnosis of Hb Adana requires confirmation by DNA studies with multiplex ARMS PCR [39].
Hb Adana inheritance may have a negative influence on health, yet the study of the disease is limited as the diagnosis may be missed with the routine method and the availability of molecular tests is sparse, making a correct diagnosis difficult. The possibility of utilising haematology parameters and haemoglobin analysis studies to understand Hb Adana’s disease behaviour needs to be explored. Thus, determining certain haematological features could be helpful to identify Hb Adana carriers from other more common deletional type α-thalassaemia.
2. Materials and Methods
2.1. Study site
This retrospective study was conducted in the Haematology Unit, Pathology Department, Hospital Raja Perempuan Zainab II (HRPZ II), Kota Bharu, Kelantan, Malaysia. The study obtained ethical approval from the NMRR Medical Research & Ethics Committee of the Ministry of Health (Research Code: NMRR-19-3700-52411 (IIR)) and Human Research Ethics Committee USM (HREC) (JEPeM Code: USM/JEPeM/20010012).
2.2. Study population
A total of 13,721 blood samples were screened for α-thalassaemia based on the HRPZ II registry under the National Thalassaemia Screening for high school students (year 16) for three years, dated June 2017 till June 2020. This program was carried out at the school level by respective district clinics in charge, where medical officers and their team were assigned to carry out screening procedures at school. The informed consent was obtained from the parents. The samples were sent to HRPZ II for laboratory testing. Based on the National Thalassaemia Screening Programme guidelines, samples with MCH <27 and iron deficiency state were excluded while the rest of the samples were screened for thalassaemia carrier through Hb analysis and molecular testing to detect common deletional and non-deletional α-thalassaemia. For molecular study, the samples were outsourced to Molecular Laboratory, Hospital Kuala Lumpur (HKL). The study sampling was illustrated in a flowchart (Figure 2).
2.3. Sampling
There were 2327 subjects recruited from the convenience sampling of 13,721 subjects screened for α-thalassaemia, with confirmed molecular diagnosis of α-thalassaemia. Based on the molecular results, samples with heterozygous Hb Adana, other non-deletional α-thalassaemia, common deletional α-thalassaemia, and compound heterozygous were included in this study. For the molecular study (outsourced), as for the record, multiplex Gap PCR was utilised to detect single α-gene deletions (-α3.7 and -α4.2) and two α-gene deletions (--SEA, --THAI, --FIL, --MED and --(α)20.5) using an optimised method and primer sets published by Chong et al. [40]. The second phase single-tube multiplex amplification refractory mutation system-PCR (ARMS PCR) method was used to detect common non-deletional α-thalassaemia; initiation codon (ATG>A–G), codon 30 (ΔGAG), codon 35 (TCC>CCC), codon 59 (GGC>GAC) Hb Adana, codon 125 (CTG>CCG) or Hb Quong Sze [Hb QS, HBA2: c.377 T>C (or HBA1)] and termination codon TAA>CAA or Hb CS. The method was based on the standardised protocol published by Eng et al. [41]. Secondary data consisting of demographic data, haematological parameters (i.e., Hb, RBC, MCV, MCH, and MCHC), and haemoglobin analysis (i.e., HbA, HbA2, and HbF) were obtained from the HRPZ II database. Subjects with incomplete documentation of haematological parameters (such as any single missed-out haematology parameter for the study, including Hb, RBC, MCV, MCH, MCHC, HbA, HbA2, or HbF) were excluded from this study.
3. Results
3.1. Detection of Common deletional and non-deletional α-thalassaemia
Our analysis detected 2327 out of 13,721 subjects screened for α-thalassaemia as confirmed α-thalassaemia carriers by molecular analysis. They consist of common deletional (n = 1037, 44.6%) and non-deletional α-thalassaemia (n = 1290, 55.4%). We reported 31 (1.33%) subjects with heterozygous Hb Adana and 1 (0.04%) compound heterozygous Hb Adana with a single α-gene (3.7kb) deletion. They were the second commonest non-deletional α-thalassaemia after Hb Constant Spring (n = 1234, 53.0%). Six subtypes under common deletional α-thalassaemia were observed, namely heterozygous α+-thalassaemia 3.7 kb deletion (αα/-α3.7), heterozygous α+-thalassaemia 4.2 kb deletion (αα/-α4.2), homozygous α0-thalassaemia 3.7 kb deletion (-α3.7/-α3.7), compound heterozygous α0-thalassaemia with 3.7 kb and 4.2 kb deletion (-α3.7/-α4.2), heterozygous α0-thalassaemia SEA deletion (--SEA) and heterozygous α0-thalassaemia THAI deletion (--THAI). The distribution of α-thalassaemia among high school students in Kelantan is tabulated in Table 1.
Of the 32 Hb Adana subjects, 28 (87.5%) were of Malay ethnicity, and the remaining 4 (12.5%) were Orang Asli (aboriginal people). The gender distribution of Hb Adana showed a slight female predominance among 16 female and 15 male subjects. Overall, we noted a female thalassaemia carrier preponderance among males by a ratio of 1.36:1 (female to male ratio) in our study population. The ethnic distribution showed a mainly Malay population with a minority of Chinese, Siamese, and Orang Asli. There were no Indian ethnic subjects found in this study. The ethnic and gender distribution of the study population was tabulated in Table 2 and Table 3.
Seven cases of double heterozygous HbE/Hb Adana were discovered by chance when tracing DNA analysis results for subjects suspected of α-thalassaemia. The haemoglobin level for heterozygous Hb Adana varied from mild anaemia to a normal level, where the lowest value was 95.0 g/L and the highest was 153.0 g/L. Their MCV was about 73 fL (mean 72.93 ± 3.57 SD), and their MCH was less than 24 pg (mean 23.39 ± 1.36 SD). Heterozygous Hb Adana, when co-inherited with a 3.7kb single gene deletion (αCd59α/-α3.7) exhibited a normal Hb level (124.0 g/L) but lower MCV (71.1 fL) and MCH (21.5 pg) compared to heterozygous Hb Adana alone. The RBC count for both Hb Adana and when co-inherited with a single α-gene was approximately 5.7 x 109/L. For haemoglobin analysis, heterozygous Hb Adana showed normal HbA2 compared with a mildly reduced HbA2 percentage in compound heterozygous Hb Adana with the 3.7kb single gene deletion (αCd59α/-α3.7) (Table 4).
All the cases of double heterozygous HbE/Hb Adana showed normal Hb levels with mild hypochromic microcytosis. Their MCV was consistently less than 73 fL, which corresponds with the heterozygous Hb Adana. The lowest MCH of HbE/Hb Adana was 21.1 pg, which was higher than the minimum value of MCH of heterozygous Hb Adana in this current study. The HbA2 percentage of double heterozygous HbE/Hb Adana was borderline raised, ranging from 3.4% to 3.9%. The HbE percentage was, on average, less than 23%. There was no remarkable rise in HbF level (Table 5).
4. Discussion
This study was conducted in Kelantan, the 6th largest state in Malaysia, among the 13 states (Negeri) and 3 federal territories (Wilayah Persekutuan). Kelantan is on the northeast coast of Peninsular Malaysia. Thailand bounds it in the north, Pahang in the south, Terengganu in the east, and Perak in the west, with approximately 15,040 km2. There are ten districts in Kelantan, as follows: Kota Bharu, Bachok, Tumpat, Pasir Mas, Tanah Merah, Kuala Krai, Machang, Pasir Putih, Jeli, and Gua Musang [42]. Kota Bharu is the capital city of Kelantan, and Hospital Raja Perempuan Zainab II (HRPZ II), the only Ministry of Health (MOH) tertiary hospital in the state. All samples from the districts, inclusive of those from the National Thalassaemia Screening Programme for high school students, were catered to by this hospital.
Our study revealed that Hb Adana inheritance was a prevalent non-deletional form of α-thalassaemia discovered in Kelantan, which was in agreement with the Hockham et al. comprehensive review on the prevalence of α-thalassaemia in Southeast Asia [15]. Our study’s distribution of Hb Adana superseded the other similar study conducted across three states (Penang, Melaka, and Sabah) in Malaysia by Rahimah et al. [37]. From their research, non-deletional α-thalassaemia consisted of only one Hb Adana case, twenty Hb Constant Spring cases and two Hb Quong Sze cases [37]. Jameela et al. described a single case of Hb Quong Sze with no reported Hb Adana or Hb Constant Spring in a secondary school in Ampang, Selangor [10].
This current study found that 87.5% of Hb Adana were Malay and 12.5% were Orang Asli. This finding agreed with a previous studyin our neighbouring country, Singapore, that reported 93% of the Hb Adana cases (12% from 83 cases) were Malay ethnicity among their patient registries [18]. The rest of the α-thalassaemia carriers in the study also mainly affected the Malay population, with the predominant Malay (97.9%) ethnicity followed by Chinese (1.2%), Siamese (0.5%), and Orang Asli (0.4%). It was an expected finding in accordance with Kelantan’s population distribution. According to the Department of Statistics Malaysia Official Portal, the ethnic distribution in Kelantan consists of Malay and Bumiputera (95.7%), Chinese (3.4%), Indian (0.3%), and other minorities (0.6%) [43]. However, the data on α-thalassaemia carriers among Orang Asli in this study may not be representative. Orang Asli are the aboriginal people who populate the heart of the deepest jungle and rarely venture out of their comfort zones unless in exceptional circumstances [44]. The National Thalassaemia Screening Programme was conducted at the school level by district clinics and might omit Orang Asli. Most of the Orang Asli had financial and cultural restrictions, which underprivileged their chances of attending school, thus missing out on their opportunities for screening [45].
There was a slight female predominance with a ratio of 1.36 to 1 (female to male) among our study population, which may explain the higher female Hb Adana than male Hb Adana inheritance. The study source population was obtained from the National Thalassaemia Screening Programme among high school students, whereby the sample collection by MOH was done at the school. According to data from the United Nations Educational, Scientific, and Cultural Organisation (UNESCO), the rates of completion of secondary-level schooling were consistently higher among girls than boys [46]. It may explain the higher female population in this study, possibly due to boys’ higher school dropout rate.
A previous study by Bozdogan et al. [24] showed haematology parameters of heterozygous Hb Adana with mutated codon 59 at α1 globin gene identified in Adana Province, Turkey, which were similar to our study. The results manifested a normal Hb level (mean 134 g/L ± 10.0 SD), range 12.8–14.4), a low MCV value (mean 75.5 fL ± 2.8 SD, range 72.6–78.2) and a low MCH value (mean 25.5 pg ± 1.0 SD, range 24.4–26.3) [24]. However, the mean RBC (mean 5.2 x 109/L ± 0.2 SD, 5.0–5.5) was lower. A single case of compound heterozygous Hb Adana with a 3.7kb single gene deletion (αCD59α/-α3.7) was also detected, presented with mild anaemia and more marked hypochromic microcytosis. The study shows that heterozygous Hb Adana in the Turkey population, had haematological parameters that resembled α+-thalassaemia phenotypes and α0-thalassaemia when compounded with a single α-gene deletion. However, in the Malaysian population, heterozygous Hb Adana had haematological parameters that only resembled α+-thalassaemia phenotypes. Despite Hb Adana being compounded with a single α-gene deletion, it manifests α+-thalassaemia phenotypes by haematological parameters. Another study by Singh et al. [17] in the US population reported that compound heterozygous Hb Adana with 3.7kb single gene deletion (αCD59α/-α3.7) showed dissimilarly by low Hb but equally moderately low MCV and MCH [Hb mean value (91.0 g/L ± 14.0 SD), MCV (mean 72.5 fL ± 5.8 SD, range 59.5-83.8), and MCH (mean 23.3 pg ± 1.7 SD, range 19.0–25.9]. Separately, a novel compound heterozygous Hb Adana that consists of two-point mutations (HBA1: c.179G>A) and codon 127 (A>T) (HBA2: c.382A>T) was reported in a Kurdish family in Iran lead to a severe form of α+-thalassaemia. Results of haematological parameters showed abnormal indices, with the blood smear conferring hypochromia, anisocytosis, poikilocytosis, tear-drops and fragmented cells [47].
HbE is a common structural hemoglobinopathy in Asia involving a mutation of the β globin gene that substitutes glutamic acid for lysine at codon 26 (GAG→AAG). The incidence of double heterozygous HbE/Hb Adana was unusually high based on the figure obtained, in contrast to the rarity mentioned by Achour et al., who reported the first case combination of HbE with Hb Adana in the year 2018 [48]. The prevalence of HbE varies between studies, ranging from 11.25 -19.3% in Malaysia and 12.9% in southern Thailand [9,49,50]. Thus, we postulate there is a higher chance of different inheritance combinations of thalassaemia in the Malaysia region involving α-thalassaemia with these HbE. The nature of the disease for HbE showed similarities with β-thalassaemia due to a mutation that activates the cryptic splice site. It is now well known that compound heterozygosity of β-thalassaemia with α-thalassaemia reduces the imbalance in the globin chain and ameliorates the clinical phenotypes. Thus, inheritance of α-thalassaemia with HbE can decrease the globin chain imbalance as well [51,52]. The haematological parameters had been reported as normal Hb levels with lower MCV, MCH, and HbE percentages compared with the inheritance of the HbE trait [1,53].
Based on the seven cases of double heterozygous HbE/Hb Adana detected in our study, we observed pretty uniform result parameters similar to HbE interaction with deletional type α0-thalassaemia (two α-gene deletions) as discussed by Fucharoen et al. in their previous study [51]. The study exhibited slightly lower mean Hb (mean 12.5 ± 1.4 SD), MCV level (mean 77 ± 5 SD) and HbE percentage (20.7 ± 1.2 SD) for double heterozygous HbE with αo-thalassaemia [54]. It was reported that double heterozygous HbE with α+-thalassaemia showed near normal FBC indices and a higher HbE percentage, with Hb (mean 13.1 ± 1.4 SD), MCV (mean 88 ± 4 SD) and HbE percentage (mean 28.5 ± 1.5 SD) [51,55]. Our study reported normal Hb for double heterozygous HbE/Hb Adana, which complied with the Achour et al. study, but our result showed more marked microcytosis than their study. Achour et al. study showed normal Hb of 14.1 g/dL, MCV of 83.0 fL, and mild hypochromia of MCH 25.1 pg. Their percentage of HbE was 20.2%, which was also very similar to our findings [48]. Thus, heterozygous Hb Adana in our population had haematological parameters that mimic α0-thalassaemia phenotypes when co-inheriting with the HbE trait.
As for other types of non-deletional α-thalassaemia, double heterozygous HbE/Hb Constant Spring showed lower Hb, normal MCV, and mildly low MCH compared to HbE/Hb Adana in our study. Their result parameters were as follows; Hb (mean 114.0 g/L ± 14.0 SD), MCV (mean 80.2 fL ± 4.2 SD) and MCH (mean 24.6 pg ± 1.8 SD) [56]. We noticed that Hb Adana behaviour was quite similar to Hb Constant Spring when inherited with the HbE trait, as supported by previous studies [56].
5. Conclusions
To summarise, we noted heterozygous Hb Adana was rarely uncommon in our region, and the haematological features of the disease change depending on the types of thalassaemia it co-inherits. It is concluded that heterozygous Hb Adana, when compounded with single α-gene deletion is α+-thalassaemia and co-inheritance of HbE would change the disease nature to α0-thalassaemia. The MCV for all types of heterozygous Hb Adana was consistently less than 80 fL, and the MCH was less than 24 pg. Thus, we can assure that most heterozygous Hb Adana can be detected by Malaysia’s National Thalassaemia Screening Programme. Emphasising on the importance of adherence to the screening programme is highly important to ensure early detection of carrier status, which will be beneficial for genetic counselling and future spouse selection. A new generation without deleterious outcomes from the complications of thalassaemia is crucial to reducing the country’s health burden.
Several limitations were noted in this study. The number of tested samples was slightly lower than expected from March 2020 until June 2020 due to pandemic COVID-19 resulting in school closures, bringing a halt to screening work, wherein we foresee higher student enrolment, which may improve data analysis. Some of the DNA analyses (n=101) sent during the study period had pending results possibly related to delay testing whereby Covid-19 pandemic had affected much of laboratories services. The review of secondary data made it impossible for Hb Adana cases to be further molecularly tested to identify the affected α1 or α2 globin genes. The postulation of α+-thalassaemia or α0-thalassaemia phenotypes was based on the haematological features from the laboratory testing. We could not obtain documentation on the clinical phenotypes of the subjects enrolled in this study to better characterise the disease, as this study was done based on convenience sampling of secondary data.
Author Contributions
Conceptualization, R.B and M.R; Data curation, N.A.Z.; Formal analysis, L.M.P. and S.H.; Project administration, M.R.; Resources, M.N.H., Z.Z., S.I., M.A., N.H.M.N. and S.M.Y.; Supervision, R.B. and M.R.; Validation, M.N.H., Z.Z., N.H.M.N., and S.M.Y.; Writing – original draft, L.M.P.; Writing – review & editing, M.J.S.A. All authors have read and agreed to the published version of the manuscript.
Funding
No funding.
Institutional Review Board Statement
The study was conducted in accordance with the Declaration of Helsinki, and ethical approved by NMRR Medical Research & Ethics Committee of the Ministry of Health (Research Code: NMRR-19-3700-52411 (IIR)) and Human Research Ethics Committee USM (HREC) (JEPeM Code: USM/JEPeM/20010012).
Informed Consent Statement
Informed consent was obtained from the parents of the subjects involved in the study.
Data Availability Statement
No supplementary data.
Acknowledgments
The specialists, scientific officers, and laboratory technologists at Haematology Unit, Pathology Department of Hospital Raja Perempuan Zainab II, Kelantan, Malaysia and Molecular laboratory, Haematology Unit, Department of Pathology of Hospital Kuala Lumpur, Kuala Lumpur, Malaysia who had involved in the processing of patient samples.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Bain, B.J. Haemoglobinopathy diagnosis; Blackwell Publising: 2006.
- Hoffbrand, A.V.; Keeling, D.M.; Mehta, A.B.; Higgs, D.R. Haemoglobin and The Inherited Disorders of Globin Synthesis. 2016; pp. 72-97.
- Anders Enevold; Michael Alifrangis; Juan J. Sanchez; Ilona Carneiro; Cally Roper; Claus Børsting; John Lusingu; Lasse S. Vestergaard; Martha M. Lemnge; Niels Morling; et al. Associations between α+-Thalassemia and Plasmodium falciparum Malarial Infection in Northeastern Tanzania The Journal of Infectious Diseases 2007, 196, 451-459. [CrossRef]
- Mockenhaupt, F.P.; Ehrhardt, S.; Gellert, S.; Otchwemah, R.N.; Dietz, E.; Anemana, S.D.; Bienzle, U. α+-thalassemia protects African children from severe malaria. Blood 2004, 104, 2003-2006.
- Ghartey-Kwansah, G.; Boampong, J.N.; Aboagye, B.; Afoakwah, R.; Ameyaw, E.O.; Quashie, N.B. The prevalence of α-thalassemia and its relation to plasmodium falciparum infection in patients presenting to clinics in two distinct ecological zones in Ghana. Hemoglobin 2016, 40, 32-37.
- Opoku-Okrah, C.; Gordge, M.; Kweku Nakua, E.; Abgenyega, T.; Parry, M.; Robertson, C.; Smith, C.L. An investigation of the protective effect of alpha+-thalassaemia against severe P lasmodium falciparum amongst children in K umasi, G hana. International journal of laboratory hematology 2014, 36, 62-70.
- Helena Lamptey; Michael Fokuo Ofori; Bright Adu; Kwadwo Asamoah Kusi; Emmanuel Kakra Dickson; Isabella Quakyi; Alifrangis, M. Association between alpha-thalassaemia trait, Plasmodium falciparum asexual parasites and gametocyte carriage in a malaria endemic area in Southern Ghana. BMC Res Notes 2019, 12, 134. [CrossRef]
- Rosnah, B.; Rosline, H.; Zaidah, A.W.; Noor Haslina, M.N.; Marini, R.; Shafini, M.Y. Detection of Common Deletional Alpha-Thalassemia Spectrum by Molecular Technique in Kelantan, Northeastern Malaysia. International Scholarly Research Network Hematology 2012, 2012, 3-3. [CrossRef]
- Rosline, H.; Ahmed, S.A.; Al-Joudi, F.S.; Rapiaah, M.; Naing, N.N.; Nor Atifah, M.A. Thalassemia among blood donors at the Hospital Universiti Sains Malaysia. Southeast Asian J Trop Med Public Health 2006, 37, 549-552.
- Jameela, S.; Sabirah, S.O.S.; Phan, C.L.; Visalachy, P.; Chang, K.M.; Salwana, M.A.; Zuraidah, A.; Subramanian, Y.; Rahimah, A. Thalassaemia screening among students in a secondary school in Ampang, Malaysia. Med J Malaysia 2011, 66, 522-524.
- Azma, R.Z.; Ainoon, O.; Hafiza, A.; Azlin, I.; Noor Farisah, A.R.; Nor Hidayati, S.; Noor Hamidah, H. Molecular characteristic of alpha thalassaemia among patients diagnosed in UKM Medical Centre. The Malaysian Journal of Pathology 2014, 36, 27-32.
- Rahimah, A.; Saleem, M.; Aloysious, N.S.; Yelumalai, P.; Mohamed, N.; Hassan, S. Distribution of Alpha Thalassaemia Gene variants in Diverse Ethnic Populations in Malaysia: Data from the Institute For Medical Research. International Journal of Molecular Sciences 2013, 14, 18599-18614. [CrossRef]
- Cornelis L Harteveld; Higgs, D.R. α-Thalassaemia. Orphanet Journal of Rare Diseases 2010, 5, 13.
- Farashi, S.; Harteveld, C.L. Molecular basis of alpha-thalassemia. Blood Cells Mol Dis 2018, 70, 43-53. [CrossRef]
- Hockham, C.; Ekwattanakit, S.; Bhatt, S.; Penman, B.S.; Gupta, S.; Viprakasit, V.; Piel, F.B. Estimating the burden of α-thalassaemia in Thailand using a comprehensive prevalence database for Southeast Asia. Elife 2019, e40580. [CrossRef]
- Alibakhshi, R.; Mehrabi, M.; Omidniakan, L.; Shafieenia, S. The spectrum of α-thalassemia mutations in Kermanshah Province, West Iran. Hemoglobin 2015, 39, 403-406. [CrossRef]
- Singh, S.A.; Sarangi, S.; Appiah-Kubi, A.; Hsu, P.; Smith, W.B.; Gallagher, P.G.; Glader, B.; Chui, D.H.K. Hb Adana (HBA2 or HBA1: c.179G > A) and alpha thalassemia: genotype-phenotype correlation. Pediatr Blood Cancer 2018, 65, e27220. [CrossRef]
- Lam, J.C.M.; Soh, S.Y.; Law, H.Y. Clinical and haematological features of non-deletional alpha thalassaemia mutations in Singapore. Pathology 2014, 46, S94. [CrossRef]
- Fucharoen, S.; Viprakasit, V. Hb H disease: clinical course and disease modifiers. Hematology 2009, 2009, 26-34. [CrossRef]
- Galanello, R.; Cao, A. Gene test review. Alpha-thalassemia. Genet Med 2011, 13, 83-88. [CrossRef]
- Viprakasit, V. Alpha-thalassemia syndromes: from clinical and molecular diagnosis to bedside management. Hematology Education: the education program for the annual congress of the European Hematology Association 2013, 7, 329-338.
- Carinna, H.; Samit, B.; Bridget S., P.; Sunetra, G.; Vip, V.; Frederic B., P.; Supachai, E. Estimating the burden of alpha thalassaemia in Thailand using cmprehensive prevalence databae for Southeast Asia. eLife Research Communication 2019, 8. [CrossRef]
- Hua-bing, Z.; De-Pei, L.; Chih-Chuan, L. The Control of Expression of the α-Globin Gene Cluster. International Journal of Hematology 2002, 76, 420-426. [CrossRef]
- Bozdogan, S.T.; Yuregir, O.O.; Buyukkurt, N.; Aslan, H.; Ozdemir, Z.C.; Gambin, T. Alpha-thalassemia mutations in Adana Province, Southern Turkey: genotype-phenotype correlation. Indian J Hematol Blood Transfus 2015, 31, 223-228. [CrossRef]
- Aksu, T.; Yaralı, N.; Bayram, C.; Fettah, A.; Avcı, Z.; Tunç, B. Homozygosity for HBA1: c.179G > A: Hb Adana in an Infant. Hemoglobin 2014, 38, 449-450. [CrossRef]
- George, E.; Jama, T.; Azian, A.S.; Rahimah, A.; Zubaidah, Z. A rare case of alpha-thalassaemia intermedia in a Malay patient double heterozygous for alpha(+)-thalassaemia and a mutation in alpha1 globin gene CD59 (GGC --> GAC). Med J Malaysia 2009, 64, 321-322.
- Alauddin, H.; Jaapar, N.A.; Azma, R.Z.; Ithnin, A.; Razak, N.F.; Loh, C.K.; Alias, H.; Abdul-Latiff, Z.; Othman, A. A case series of α-thalassemia intermedia due to compound heterozygosity for Hb Adana [HBA2: c179G>A (or HBA1); p.Gly60Asp] with other α-thalassemias in Malay families. Hemoglobin 2014, 38, 277-281. [CrossRef]
- Nainggolan, I.M.; Harahap, A.; Setianingsih, I. Hydrops fetalis associated with homozygosity for Hb Adana [alpha59(E8)Gly-->Asp (alpha2)]. Hemoglobin 2010, 34, 394-401. [CrossRef]
- Nainggolan, I.M.; Harahap, A.; Ambarwati, D.D.; Liliani, R.V.; Megawati, D.; Swastika, M.; Setianingsih, I. Interaction of Hb Adana (HBA2: c.179G>A) with deletional and nondeletional α(+)-thalassemia mutations: diverse hematological and clinical features. Hemoglobin 2013, 37, 297-305. [CrossRef]
- Lee, T.Y.; Lai, M.I.; Ismail, P.; Ramachandran, V.; Tan, J.A.; Teh, L.K.; Othman, R.; Hussein, N.H.; George, E. Analysis of α1 and α2 globin genes among patients with hemoglobin Adana in Malaysia. Genet Mol Res 2016, 15. [CrossRef]
- Al-Allawi N.A.;Badi A.I.;Imanian H. et al. Molecular characterization of alpha-thalassemia in the Dohuk region of Iraq. Hemoglobin. 2009,33, pp. 37-44.
- Li B.;Han X.;Ma J. et al. Mutation spectrum and erythrocyte indices characterisation of α-thalassaemia and β-thalassaemia in Sichuan women in China: a thalassaemia screening survey of 42 155 women. J Clin Pathol. 2021,74, pp. 182-186.
- Chen F.E.;Ooi C.;Ha S.Y. et al. Genetic and clinical features of Hemoglobin H disease in Chinese patients. N Engl J Med. 2000,343, pp. 544-50.
- Akhtar M.S.;Qaw F.;Borgio J.F. et al. Spectrum of α-thalassemia mutations in transfusion-dependent β-thalassemia patients from the Eastern Province of Saudi Arabia. Hemoglobin. 2013,37, pp. 65-73.
- Azma, R.Z.; Ainoon, O.; Hafiza, A.; Azlin, I.; Noor Farisah, A.R.; Nor Hidayati, S.; Noor Hamidah, H. Molecular characteristic of alpha thalassaemia among patients diagnosed in UKM Medical Centre. Malay J Pathol 2014, 36, 27-32.
- Ahmad, R.; Saleem, M.; Aloysious, N.S.; Yelumalai, P.; Mohamed, N.; Hassan, S. Distribution of alpha thalassaemia gene variants in diverse ethnic populations in malaysia: data from the institute for medical research. Int J Mol Sci 2013, 14, 18599-18614. [CrossRef]
- Rahimah, A.N.; Nisha, S.; Safiah, B.; Roshida, H.; Punithawathy, Y.; Nurul, H.; Syahzuwan, H.; Zubaidah, Z. Distribution of alpha thalassaemia in 16 year old Malaysian Students in Penang, Melaka and Sabah. Med J Malaysia 2012, 67, 565-570.
- Nur Zaireena, Z.; Alauddin, H.; Ahmad, S.; Hamidah Hussin, N.; Hamidah Hussin, N. Thalassemia with Haemoglobin Adana mutation: prenatal diagnosis Malay J Pathol 2014, 36, 207-211.
- Tan, J.; Kho, S.L.; Ngim, C.F.; Chua, K.H.; Goh, A.S.; Yeoh, S.L.; George, E. DNA studies are necessary for accurate patient diagnosis in compound heterozygosity for Hb Adana (HBA2:c.179>A) with deletional or nondeletional alpha-thalassaemia. Sci Rep 2016, 6, 26994. [CrossRef]
- Chong, S.S.; Boehm, C.D.; Higgs, D.R.; Cutting, G.R. Single-tube multiplex-PCR screen for common deletional determinants of α-thalassemia. Blood 2000, 95, 360-362. [CrossRef]
- Eng, B.; Patterson, M.; Walker, L.; Chui, D.H.K.; Waye, J.S. Detection of Severe Nondeletional α-Thalassemia Mutations Using a Single-Tube Multiplex ARMS Assay. Genetic Testing 2001, 5, 327-329. [CrossRef]
- Department of Statistics, M. Population estimates based on the adjusted population and housing census of Malaysia 2010. 2021. Availabe online: https://www.dosm.gov.my/v1/index.php?r=column/cone&menu_id=RU84WGQxYkVPeVpodUZtTkpPdnBmZz09 (accessed on 18 Oct 2021).
- Kelantan State, Malaysia. Population, Charts, Maps. 2010. Availabe online: https://www.citypopulation.de/en/malaysia/admin/03__kelantan/ (accessed on 5 Jan).
- Khor, G.L.; Shariff, Z.M. Do not neglect the indigenous peoples when reporting health and nutrition issues of the socio-economically disadvantaged populations in Malaysia. BMC Public Health 2019, 19, 1685. [CrossRef]
- Vengadesan, M. Bullied, separated-national education failing Orang Asli children. 2019. Availabe online: https://www.malaysiakini.com/news/472558 (accessed on 3 March).
- UNESCO. 2011. Malaysia: the millennium development goals at 2010. Availabe online: (accessed on 20 Oct 2021).
- Azimi, A.; Tahmasebi, S.; Moradi, K.; Nejati, P.; Alibakhshi, R. Severe α-Thalassemia Due to Compound Heterozygosity for Hb Adana (α59 Gly>Asp) (HBA1: c.179G > A) and Codon 127 (A > T) (HBA2: c.382A > T) in an Iranian Family. Hemoglobin 2020, 44, 139-142. [CrossRef]
- Achour, A.; de Grouw, E.; van Erp, F.; Arkesteijn, S.; Schaap, R.; Huurne, J.T.; Bisoen, S.; Verschuren, M.; Harteveld, C.L. The first report of Hemoglobin E in combination with the highly unstable alpha-globin variant Hb Adana: the importance of molecular confirmation. Int J Lab Hematol 2019, 41, e76-e78. [CrossRef]
- Chin-yuet M.;Ezalia E.;Aliza M.Y. et al. Screening for hemoglobinopathies among patients in a government hospital and health clinics in Perlis, Malaysia. J Med Sci Technol. 2014,3, pp. 82-86.
- Nuinoon M.;Kruachan K.;Sengking W. et al. Thalassemia and Hemoglobin E in Southern Thai blood donors. Adv Hematol. 2014,2014, pp. 932306.
- Fucharoen, S.; Weatherall, D.J. The Hemoglobin E thalassemias. Cold Spring Harb Perspect Med 2012, 2, a011734-a011734. [CrossRef]
- Thein, S.L.; Rees, D. Haemoglobin and the inherited disorders of globin synthesis. In Postgraduate Haematology, Hoffbrand, A.V., Catovsky, D., Tuddenham, E.G., Green, A.R., Eds. Blackwell Publishing Ltd: 2016; pp. 72-97.
- Pornprasert, S.; Tookjai, M.; Punyamung, M.; Pongpunyayuen, P. HbE level and red cell parameters in heterozygous HbE with and without α0-thalassemia trait. Indian J Hematol Blood Transfus 2018, 34, 662-665. [CrossRef]
- Vichinsky, E. Hemoglobin E syndromes. Hematology Am Soc Hematol Educ Program 2007, 79-83. [CrossRef]
- Fucharoen, G.; Srivorakun, H.; Singsanan, S.; Fucharoen, S. Presumptive diagnosis of common haemoglobinopathies in Southeast Asia using a capillary electrophoresis system. Int J Lab Hematol 2011, 33, 424-433. [CrossRef]
- Jomoui, W.; Fucharoen, G.; Sanchaisuriya, K.; Nguyen, V.H.; Fucharoen, S. Hemoglobin Constant Spring among Southeast Asian Populations: Haplotypic Heterogeneities and Phylogenetic Analysis. PLoS One 2015, 10, e0145230. [CrossRef]
Figure 1.
Genomic structure of α and β globin gene.
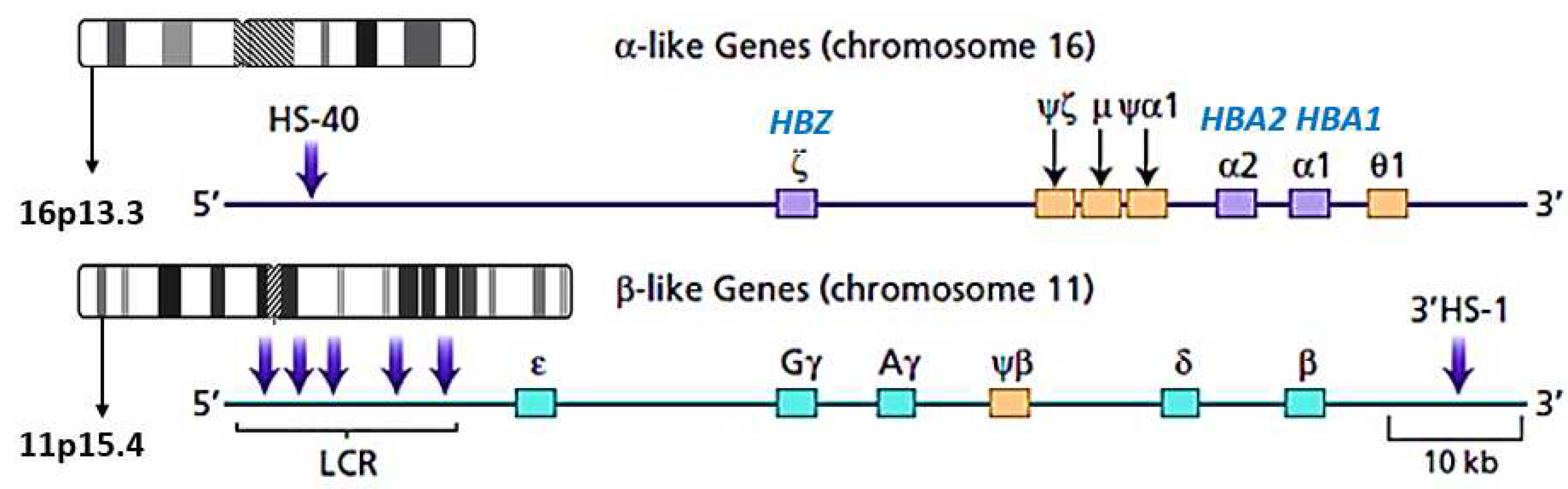
Figure 2.
Study sampling flowchart.


Table 1.
The distribution of α-thalassaemia among screened high school students in Kelantan.
| DNA Analysis Results | Frequency (n) | Percentage (%) |
|---|---|---|
| Non deletional α-thalassaemia αCD59 α / αα αQZα / αα αCSα / αα |
31 24 1234 |
1.33% 1.03% 53.03% |
| Deletional α-thalassaemia -α3.7/αα -α4.2/αα -α3.7/-α3.7 -α3.7/ -α4.2 --SEA/αα --THAI/αα |
659 43 55 11 260 9 |
28.32% 1.85% 2.36% 0.47% 11.17% 0.39% |
| Compound heterozygous αCD59α / -α3.7 |
1 |
0.04% |
| Total | 2327 | 100.00% |
Table 2.
Ethnic distribution of α-thalassaemia carrier among screened high school students in Kelantan.
Table 2.
Ethnic distribution of α-thalassaemia carrier among screened high school students in Kelantan.
| DNA Analysis Results | Ethnic, n(%) | |||
|---|---|---|---|---|
| Malay | Chinese | Orang Asli | Siamese | |
| Non deletional α-thalassaemia | ||||
| αCD59 α / αα | 27 (1.19) | 0 | 4 (44.44) | 0 |
| αQZα / αα | 23 (1.01) | 1 (3.45) | 0 (0.00) | 0 (0.00) |
| αCSα / αα | 1223 (53.69) | 6 (20.69) | 5 (55.56) | 0 (0.00) |
| Deletional α-thalassaemia | ||||
| -α3.7/αα | 645 (28.31) | 7 (24.14) | 0 (0.00) | 7 (63.64) |
| -α4.2/αα | 43 (1.89) | 0 (0.00) | 0 (0.00) | 0 (0.00) |
| -α3.7/-α3.7 | 53 (2.33) | 1 (3.45) | 0 (0.00) | 1 (9.09) |
| -α3.7/ -α4.2 | 10 (0.44) | 1 (3.45) | 0 (0.00) | 0 (0.00) |
| --SEA/αα | 244 (10.71) | 13 (44.83) | 0 (0.00) | 3 (27.27) |
| --THAI/αα | 9 (0.39) | 0 | 0 | 0 |
| Compound heterozygous | ||||
| αCD59α / -α3.7 | 1 (0.04) | 0 | 0 | 0 |
| Total | 2278 | 29 | 9 | 11 |
Table 3.
Gender distribution of α-thalassaemia carrier among screened high school students in Kelantan.
Table 3.
Gender distribution of α-thalassaemia carrier among screened high school students in Kelantan.
| DNA Analysis Results | Gender, n (%) | |
|---|---|---|
| Male | Female | |
| Non deletional α-thalassaemia | ||
| αCD59 α / αα | 15 (1.52) | 16 (1.19) |
| αQZα / αα | 11 (1.11) | 13 (0..97) |
| αCSα / αα | 583 (59.01) | 651 (48.62) |
| Deletional α-thalassaemia | ||
| -α3.7/αα | 204 (20.65) | 455 (33.98) |
| -α4.2/αα | 20 (2.02) | 23 (1.72) |
| -α3.7/-α3.7 | 22 (2.23) | 33 (2.46) |
| -α3.7/ -α4.2 | 6 (0.61) | 5 (0.37) |
| --SEA/αα | 123 (12.45) | 137 (10.23) |
| --THAI/αα | 4 (0.40) | 5 (0.37) |
| Compound heterozygous | ||
| αCD59α / -α3.7 | 0 (0.00) | 1 (0.07) |
| Total | 988 | 1339 |
Table 4.
Haematological parameters (RBC, Hb, MCV, MCH, MCHC, HbA2, HbE, and HbF) of Heterozygous Hb Adana (n=31) and Compound heterozygous Hb Adana/ -α3.7 (n=1).
Table 4.
Haematological parameters (RBC, Hb, MCV, MCH, MCHC, HbA2, HbE, and HbF) of Heterozygous Hb Adana (n=31) and Compound heterozygous Hb Adana/ -α3.7 (n=1).
| Parameters | Heterozygous Hb Adana αCD59 α / αα (n=31) Range |
Mean (SD) | Compound heterozygous αCD59α / -α3.7 (n=1) |
|---|---|---|---|
| Hb (g/l) | 95.00-153.00 | 133.61 (±12.31) | 124.0 |
| RBC (x109/L) | 4.82-6.50 | 5.71 (±0.42) | 5.78 |
| MCV (fL) | 58.90-79.60 | 72.93 (±3.57) | 71.1 |
| MCH (pg) | 17.50-24.90 | 23.39 (±1.36) | 21.5 |
| MCHC (g/dl) | 29.80- 34.20 | 32.06 (±0.99) | 30.2 |
| Hb A (%) | 96.70- 98.10 | 97.46 (±0.25) | 97.5 |
| Hb A2 (%) | 1.90-3.00 | 2.51 (±0.20) | 2.1 |
| Hb F (%) | 0.00-0.50 | 0.03 (±0.10) | 0.4 |
Table 5.
Haematological parameters (RBC, Hb, MCV, MCH, and MCHC) and haemoglobin analysis (HbA, HbA2, HbE, and HbF) of double heterozygous Hb E/ Hb Adana (n=7).
Table 5.
Haematological parameters (RBC, Hb, MCV, MCH, and MCHC) and haemoglobin analysis (HbA, HbA2, HbE, and HbF) of double heterozygous Hb E/ Hb Adana (n=7).
| Double heterozygous Hb E/ Hb Adana (n = 7) | |||||||
|---|---|---|---|---|---|---|---|
| Parameters | 1 | 2 | 3 | 4 | 5 | 6 | 7 |
| Hb (g/l) | 122.0 | 124.0 | 131.0 | 132.0 | 133.0 | 136.0 | 141 |
| RBC (x109/L) | 4.94 | 5.12 | 5.45 | 5.42 | 6.31 | 5.91 | 5.83 |
| MCV (fL) | 73.50 | 70.30 | 72.70 | 72.90 | 64.8 | 69.5 | 71.0 |
| MCH (pg) | 24.70 | 24.20 | 24.0 | 24.40 | 21.1 | 23.0 | 24.2 |
| MCHC (g/dl) | 33.60 | 34.50 | 33.10 | 33.40 | 32.5 | 33.10 | 34.1 |
| Hb A (%) | 74.4 | 72.60 | 75.6 | 75.6 | 73.50 | 74.9 | 74.4 |
| Hb A2 (%) | 3.5 | 3.6 | 3.4 | 3.6 | 4.1 | 3.7 | 3.9 |
| Hb E (%) | 21.9 | 22.4 | 21.0 | 20.8 | 22.4 | 21.4 | 21.7 |
| Hb F (%) | 0.2 | 1.4 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



