
Submitted:
01 June 2023
Posted:
08 June 2023
You are already at the latest version
Abstract
Middle East region experiences a high prevalence of hypovitaminosis D, yet most genetic studies on Vitamin D have focused on European populations. Furthermore, there is a lack of research on the genomic risk factors affecting the elderly population, who are more susceptible to health burden. We investigated the genetic determinants of 25-hydroxyvitamin D levels in elderly Lebanese individuals (n=199) through a whole exome-based genome-wide association study. We identified new loci with suggestive evidence of an association with Vitamin D levels, including rs141064014 in the MGAM gene (P-value of 4.40 × 10−06) and rs7036592 in PHF2 (P-value of 8.43 × 10−06). A meta-analysis of the Lebanese data and the largest European genome-wide association study confirmed consistence replication of numerous variants, including rs2725405 in SLC38A10 (P-value of 3.73 x 10-08). Despite the lower performance of European-derived polygenic risk scores compared to the European estimations, it still effectively predicted Vitamin D deficiency among elderly Lebanese individuals. Our findings provide novel insights into the genetic mechanisms of Vitamin D deficiency in Middle Eastern elderly populations, facilitating the development of personalized approaches for more effective management of hypovitaminosis D. Additionally, we demonstrated that whole exome-based genome-wide association study is an effective method for identifying genetic components associated with phenotypes.
Keywords:
Exome-wide association stud
; Vitamin D deficiency
; genetic determinants
; polygenic risk score
; Middle Eastern population.
1. Introduction
Vitamin D is a crucial micronutrient that plays a vital role in maintaining bone health. Deficient levels of Vitamin D, indicated by a serum 25-hydroxy-vitamin D (25(OH)D) level below 20 ng/mL (50 nmol/L), are prevalent worldwide. This deficiency has been associated with musculoskeletal disorders, like osteoporosis and rickets in children, as well as cardiovascular diseases, and cancer [1]. The elderly population is particularly susceptible, owing to factors such as decreased Vitamin D synthesis in the skin, inadequate consumption of Vitamin D-rich products, and intestinal malabsorption [2].
Genetic predisposition can also impact Vitamin D levels in addition to the conventional risk factors [3]. Studies have reported heritability estimates of genetic variations in 25(OH)D levels around 80%, underscoring the substantial influence of genetic factors on individual differences [4]. Genome-wide association studies (GWAS) have successfully identified specific genetic variants associated with 25(OH)D levels in genes involved in the synthesis and transport of Vitamin D. Notable genes include CYP2R1 (Cytochrome P450 Family 2R1), CYP24A1 (Cytochrome P450 Family 24A1), DHCR7 (7-Dehydrocholesterol Reductase), and GC (Vitamin D Binding Protein) [5,6,7,8]. Furthermore, studies have also determined a contribution of genetic variants, including CYP2R1 and DHCR7, to the variability in response to Vitamin D supplements [9].
Surprisingly, despite the abundant sunshine in the Middle East, there is a significant prevalence of Vitamin D deficiency, with rates reaching up to 74% [10,11] and approximately 63% in Lebanon [12]. However, most of the genetic studies on Vitamin D have predominantly focused on individuals of European ancestry, leaving a knowledge gap in the impact of genetic variables in Middle Eastern populations. We performed the first whole exome-based GWAS (ExWAS) of Vitamin D in an elderly Middle Eastern population. The ExWAS approach provides an enhanced ability to detect rare variants within protein-coding genes, thus enabling a more effective analysis of the genetic architecture underlying Vitamin D deficiency in this population [13]. To identify more novel and common genetic determinants of Vitamin D, we conducted meta-analysis of our cohort with the largest European GWAS (n = 417,580 individuals) [6]. Additionally, we assessed the effectiveness of European-derived polygenic risk scores (PRS) along with the correlation of genetic markers and Vitamin D deficiency in the elderly Lebanese individuals.
2. Materials and Methods
2.1. Study Participants
Data used in the present ExWAS study were obtained from three Lebanese centers data (the American University of Beirut – Medical Center, Hotel Dieu de France, and Rafic Hariri University Hospital). The cross-sectional study enrolled 199 participants aged 65 years or older with a body mass index (BMI) between 25 to 29 kg/m2 (overweight) or 30 kg/m2 and above. Participants with 25(OH)D concentrations between the validated value (10-30 ng/mL) were included. This study, conducted in Beirut, Lebanon from 2011 to 2013, was a randomized controlled trial that evaluated the effects of two different Vitamin D doses on indices of bone mineral density (NCT01315366) [2]. The study was approved by the Institutional Review Board (IM-GEHF-20), and all participants provided written informed consent prior to their participation.
2.2. Serum 25(OH)D Measurement and Related Covariates
Physical measurements, including anthropometric measurements such as body weight and height, were taken for all participants. Each participant completed a standardized questionnaire reporting lifestyle and medical history information. Serum 25(OH)D levels were measured using Liquid Chromatography Mass Spectroscopy at the Mayo Clinic Laboratories (Rochester, Minnesota, USA). Prior to the statistical analyses, the serum 25(OH)D levels for all individuals were normalized using rank-based inverse normal transformation by R version 4.1.3. The anthropometric measurements, such as body weight and height, were performed by the Seca 284 stadiometer and balance. BMI was calculated by dividing the weight (kg) by the square of height (m2).
2.3. Whole-Exome Sequencing and Bioinformatics
Peripheral blood cells were collected from participants through venipuncture during similar season of the year and processed for DNA extraction using a DNA extraction kit (the Qiagen QIAamp blood midi kit, catalog number: 51185) as per the manufacturer's recommendations. The resulting samples were assessed for quantity using a NanoDrop (Thermo) at the molecular core facilities located at the American University of Beirut, and subsequently stored at a temperature of -80 degrees Celsius. Subsequently, genomic DNA of the 199 samples were transferred to the Macrogen Inc. (Seoul, Korea) to create DNA sequencing libraries using 101-base-pair paired-end reads on the SureSelectXT Library Prep Kit on Illumina HiSeq 4000 platform. Each sample yielded paired-end reads with a range of 68,807,342-91,844,864 reads and a total of 6.9-9.2G base-pair reads. Of these, 95.92-96.67% of the reads passed Q30 (a phred quality score of over 30).
Sample-specific FASTQ files, representing the HiSeq reads for that sample, were aligned with BWA MEM to the GRCh37 reference genome [14]. Metric statistics were captured for all samples to evaluate genome capture, variants alignment, and calling quality using SnpEff version 4.2 [15], bcftools [16] with dbsnp version 138 [17], and ClinVar as databases [18]. SNV genotypes with read depth less than 20 and mapping quality less than 40 were excluded. After the DP genotype filtering, duplicate reads followed by SNVs and InDels genotypes were removed using SureCall 2.0 SNPPET algorithm.
To record variant calling for Lebanese samples, we utilized the HaplotypeCaller provided by Genome Analysis Toolkit (GATK version 3.4, https://software.broadinstitute.org/gatk/documentation/article?id=3238). All the individual intermediate genomic variant call files (gVCF) were used in a joint calling process to create a joint multi-samples VCF file for all the samples. This process had two steps. First, the regions for all the samples were combined using GenomicsDB. Then, GenotypeGVCFs was used to merge all regions, while also applying SNP/Indel recalibration. Only variants that passed the filter were included for downstream analysis after undergoing the GATK VQSR filtering steps.
We performed a comprehensive quality control assessment using PLINK (version 2.0) [19]. We excluded SNPs that had a genotyping call rate <90%, MAF < 1%, or Hardy-Weinberg equilibrium P < 1 × 10−6. We also excluded samples that had a call rate < 95% (N = 5) and checked duplicates, excess heterozygosity, and gender ambiguity. Multidimensional scaling analysis was also performed using PLINK to identify population ancestry outliers. We used a set of pruned independent autosomal SNPs (N = 14,893) to determine the pairwise identity-by-state (IBS) matrix through a window size of 200 SNPs and LD threshold of r2 = 0.05. Population outliers were identified and excluded if they deviated from the mean of the first two mds components by four standard deviation units or more (±4 SD). The genome-wide association analysis was conducted on 481,395 genetic variants obtained from 194 participants.
2.4. Exome-Wide Association Analysis
We conducted an ExWAS to investigate the relationship between each genetic variant and 25(OH)D levels. We used the generalized mixed model in SAIGE [20], which is a variance component-based linear method that correct for relatedness and genetic substructure using the genomic kinship matrix. To adjust for the mixture of populations, we performed population principal component (PC) analysis using PLINK software [19]. The first four PCs were used as covariates in the association model. We also adjusted the regression model for age and gender. The finding of interest cutoff was set at P-value < 5 × 10−5 and the nominal significance level was set at P-value < 0.05. The quantile-quantile plot, Manhattan plot, and genomic inflation factor were generated using R. The heritability was estimated using the polygenic risk model in SAIGE to determine the degree of variation in 25(OH)D levels due to inter-individual genetic variation in the population.
The pairwise linkage disequilibrium (LD) among top-associated SNPs was examined using the LD clumping analysis of PLINK software (version 1.9) [19]. We applied an r2 threshold of 0.2 within a window of 250 kb to identify SNPs exhibiting strong LD through the results of our ExWAS analysis of the Lebanese data. We then utilized LocusZoom software [21] to visualize the LD patterns, which generated regional plots based on the SAIGE summary statistics. These plots effectively highlighted clusters of SNPs demonstrating high LD.
2.5. Meta-Analysis
A meta-analysis for the suggestively associated loci was performed by combining the results of our Lebanese ExWAS and a recent large GWAS (GCST90000616) by Revez JA et al from the UK Biobank (n= 417,580 European ancestry individuals) [7]. The GCST90000616 GWAS models were characterized using the same phenotype and methods with correcting relevant covariates, including age, sex, and genotype PCs [7]. The NHGRI-EBI GWAS Catalog [22] provided summary statistics for study GCST90000616 taken in December 2022. We confirmed matching A1 and A2 alleles with the alternate and reference alleles in the GCST90000616 study. Our association statistics were canonicalized based on the alternate allele in the reference genome. Additionally, both studies used rank-based transformation to inversely normalize 25(OH)D measurements. We performed an inverse-variance weighted meta-analysis and estimated heterogeneity of effects analysis using PLINK (version 1.9) [19].
2.6. Validation of Replicated Loci Previously Associated with 25(OH)D
Our ExWAS results were compared to the European GWAS study for Vitamin D levels (GCST90000616) [7], to assess the replication and correlation of our findings in terms of effect size and allele frequency for common signals. To check if any of the SNPs we found in our meta-analysis were previously reported in the UK Biobank at a significance level of P-value < 5.0 × 10−8, we searched for each SNP in the GWAS catalog (EFO_0004631) of Vitamin D levels from the November 2022 data release [22]. We accessed this catalog in December 2022. Initially, we looked for the genetic locus associated with Vitamin D levels in our cohort and compared it to those present in the UK Biobank dataset. We then examined markers within a 250-kb region upstream and downstream of the GWAS catalog signals, to identify novel and replicated variants associated with Vitamin D.
2.7. Analysis of Polygenic Risk Scores
We evaluated the ability of a PRS derived from a European population to estimate genetic liability for Vitamin D levels in Lebanese population by PLINK (version 1.9) [19]. The PRS is an aggregation of SNPs by the sum of risk allele frequency, weighted by their corresponding effect sizes predicted by GWAS. We obtained the PRS files from one of the largest Vitamin D GWAS studies in Europeans (PGS000882: n = 417,580 individuals and 1,094,650 variants) [7], which were accessed through the Polygenic Score Catalog (https://www.PGSCatalog.org) [23] on December 2022.
We then calculated Pearson's correlation (R) between the baseline Vitamin D levels and the European-derived PRS, adjusted for age, gender, and the first four principal components using R software. We also used the area under the receiver operating characteristic (ROC) curve (AUC) to evaluate the performance of the derived PRSs in identifying individuals with Vitamin D deficiency, defined as serum 25(OH)D levels below 20 ng/mL and Vitamin D deficiency. The AUC ranges from 0.5 (no distinction) to 1 (complete distinction), indicating the effectiveness of the PRSs in identifying those at risk for Vitamin D deficiency.
2.8. SNP Annotation and Functional Analysis
The variants associated with Vitamin D levels that were found in the ExWAS and meta-analysis were annotated using the Ensembl Variant Effect Predictor release 108 (VEP, https://grch37.ensembl.org/index.html) [24]. We compared the frequencies of the identified Vitamin D variants with those in global populations using the Genome Aggregation Database (gnomAD, https://gnomad.broadinstitute.org) and the Allele Frequency Aggregator (ALFA, www.ncbi.nlm.nih.gov/snp/docs/gsr/alfa/). Expressions quantitative trait loci (eQTLs) of novel genes in human tissues were analyzed by genotype-tissue expression (GTEx, http://commonfund.nih.gov/GTEx/) and RNA-seq and ChIP-seq sample and signature search (ARCHS4) database [25].
3. Results
3.1. Study Description
This study utilizes whole exome sequence data from elderly Lebanese participants, with an average age of 71 (±4.8) years (interquartile range: 65 to 91 years) and 46.4% female participants (n = 104). Notably, the mean of 25(OH)D baseline levels was 20.12 (± 7.2) ng/mL (interquartile range: 6 to 44 ng/mL). The mean BMI (in kg/m2) was almost similar between genders, with a value of 30.2 ±4.6 (Table 1). There were no significant correlations between serum 25(OH)D levels and related covariates, such as age, gender, or BMI. Participant enrolment was distributed across all seasons. At enrolment, 19% of participants were on calcium supplementation, and only 0.5% were receiving Vitamin D supplements.
3.2. Exome-Wide Association Study on 25(OH)D
We used a generalized linear mixed model to identify the genetic basis and potential causal genes of Vitamin D levels in samples of the elderly Lebanese population who passed quality control criteria (n = 194 participants). We adjusted age, gender, and population PCs in the model to account for any population stratification and relatedness, focusing on common frequency risk alleles (MAF > 1%; N = 481,395). The Manhattan plot of the ExWAS showed top SNP signals on chromosome 7 and chromosome 9 as potential risk loci for the circulating 25(OH)D levels at a P-value ≤ 1.0 × 10-05 (Figure 1A). These variants map to novel loci not previously associated with Vitamin D levels. Our results showed no evidence of widespread inflation or significant population stratification (λGC = 1.002, stander error (SE) = 4.9 × 10-06), as shown in the Q-Q plot (Figure 1B).
The strongest association signal is at SNP rs141064014 (chromosome 7q34: 141736273: G>A) at a P-value of 4.40 × 10−06 (Table 2), in the intronic region of the maltase-glucoamylase (MGAM) gene. The pairwise LD assessment defined that rs141064014 is not in LD (r2 < 0.2) with any other variant within a window of 10 Mb (Figure 2A). Another strong association is SNP rs7036592 (chromosome 9: 96425777: C>T) at a P-value of 8.43 × 10−06 in the plant homeodomain (PHD) finger 2 (PHF2) gene (Table 2), which is in LD with several other nearby SNPs in the same locus (Figure 2B). Variants suggestively linked to 25(OH)D levels with a P-value of less than 5 × 10-04 are shown in Table S1. Additionally, we characterized the attribution of ExWAS SNPs to 25(OH)D variation in the Lebanese population, estimating the heritability of 25(OH)D using all filtered SNPs as 29%.
3.3. Evaluation of Known Loci Replication
To evaluate the extent of replication, we compared our findings to the largest and most comprehensive GWAS on Vitamin D from the UK Biobank (GCST90000616). The GCST90000616 study, similar to our study, used an association analysis approach and rank-based inverse normalization to analyse Vitamin D levels [7], facilitating the comparison of effect sizes for the loci determined in both cohorts. In the Lebanese cohort, we observed replication of 7,151 loci that showed significant association in the GCST90000616 study. Specifically, we replicated 58 variants that exhibited suggestive significance in the GCST90000616 study (Table S2).
A significant association was observed between the allele frequencies of the replicated variants, which displayed a Pearson's coefficient (R) of 0.62 (95% CI = 0.26 to 0.83) at a P-value of 0.0025. In addition, the correlation analysis of effect directions and sizes for the common SNPs showed consistent directionality, with larger effect sizes than the previous findings of the GCST90000616 (n = 12, R = 0.92, 95% CI = 0.74 to 0.98, P-value < 0.0001). The other SNPs (n = 46 variants) exhibited an opposite association direction as compared to GCST90000616, which could be influenced by genetic diversity within the populations, study design, and environmental factors that contribute to Vitamin D levels.
3.4. GWAS Meta-Analysis for Vitamin D
We then conducted a meta-analysis to uncover potential novel variants that demonstrate significant genome-wide associations in the Lebanese dataset. This analysis involved combining our ExWAS results with summary statistics obtained from the largest European GWAS conducted by Revez JA et al. for Vitamin D, consisting of 417,580 individuals. The replication GCST90000616 study has been previously described in detail [7]. Through our meta-analysis, we validated the replication of a missense variant rs2725405 (chromosome 17:79220224 C>G) at a P-value of 3.73 x 10-08, Beta = 0.0109. This variant is in the solute carrier family 38A10 (SLC38A10) gene that had been previously reported in the GWAS Catalog (P-value = 2 x 10-8, Beta = 0.0114) [7]. We further identified several variants in the same loci of known variants (n = 70 variants), as presented in Table S3.
3.5. Analysis of Functional Variant Expression and Frequency
The frequency of the most significant Vitamin D variants in the Lebanese cohort were compared with control populations from the gnomAD and ALFA browsers. The frequency of rs141064014 was higher, but rs7036592 and rs2725405 were lower in the Lebanese elderly population compared to the European population in gnomAD and ALFA (Table 3).
We performed a targeted tissue enrichment analysis using the GTEx map tool to gain insights into the potential functions of the top linked SNPs involved in Vitamin D. We discovered notable correlations between particular alleles and decreased gene expression in the digestive tract and skin tissues when compared to other tissues. The homozygous risk allele “CC” and heterozygous “TC” of rs141064014 were significantly associated with reduced MGAM gene expression in the small intestine, with a P-value of 1.9 x 10-21 (Figure 3A). The presence of the "CC" risk allele of rs2725405 was associated with a significant reduction of the SLC38A10 gene expression in the colon (Figure 3C) and small intestine (Figure 3D), with P-values of 1.8 x 10-48 and 1.5 x 10-23, respectively. Finally, an enriched expression of the PHF2 gene was detected in multiple tissues through the RNA-seq public resource ARCHS4, including chondrocyte of bone marrow and osteoblast in bone tissues. The normalized gene expression, measured as transcript per million (nTPM), was approximately 9.7 (Figure S1).
3.6. Analysis of Polygenic Risk Score
We then assessed the performance of a European-derived PRS from panel PGS000882 [7]. Out of the 1,094,650 variants in this panel, we detected 41,736 in our whole exome sequence data. The performance of these polygenic risk scores in predicting Vitamin D levels in the Lebanese population is shown in Figure 4.
Our results indicate that the European-derived PRS has a lower predictive performance on the Lebanese cohort (R = 0.2033, 95% CI = 0.0643 to 0.3346, P-value = 0.0044) compared to the previously reported R values of 0.31 in the PGS000882 study. Despite this, the PRS was able to efficiently predict the risk of Vitamin D deficiency in Lebanese individuals, with an AUC of 0.644 (P-value = 0.0192, Odds Ratio = 1.6, 95% CI = 1.3 to 13.7), as shown in Figure 5.
4. Discussion
Our study represents the first known attempt to identify common and new genetic variants that are linked to Vitamin D levels in elderly individuals from the Middle East. Through a whole exome analysis of 194 elderly Lebanese individuals, we discovered essential loci with suggestive evidence of an association with Vitamin D levels. The main findings of our study deviate from previous GWAS, where the most significant associated loci are located within four major genes, namely GC, CYP2R1, CYP24A1, and DHCR7 [26]. Our research sheds light on previously unidentified genes that potentially contribute to the process of intestinal absorption of Vitamin D. Importantly, this observation may be influenced by the specific characteristics of our cohort, which primarily consists of elderly individuals. Further validation of our results in independent populations, with an expanded sample size and utilizing a GWAS approach, would be advantageous. Indeed, such replication is the most reliable method to verify the validity of our findings.
In our study, the top genetic predisposition was the novel MGAM locus, rs141064014, with no indication of LD. The MGAM encodes the maltase-glucoamylase protein belonging to the glycoside hydrolase family 31 that breaks down complex carbohydrates in the small intestine [27]. In elderly individuals, reduced MGAM activity may exacerbate age-related changes in the digestive system, leading to reduced intestinal and hepatic enzymes, intestinal motility, and malabsorption of nutrients [28]. Our functional analyses showed that the "C" risk allele significantly reduces the intestinal expression of MGAM, which is highly elevated in our regional populations compared to other populations. These observations suggest a potential regulatory role in the intestinal absorption of Vitamin D in Middle Easterners, which requires further investigation.
We also identified a novel suggestive SNP, rs7036592, in the PHF2 gene belonging to the Jumonji C family. It plays a role in various physiological processes, including the regulation of gene expression, different tissue functions, metabolism, and adipogenesis [29]. Interestingly, there is evidence that PHF2 might be involved in regulating Vitamin D metabolism and signalling [30]. PHF2 has been shown to physically interact with and regulate the activity of CYP27B1, thereby affecting the production of 1,25(OH)2D [31]. On the other hand, PHF2 was found to interact with and enhance the transcriptional activity of the Vitamin D receptor in osteoblasts, suggesting a potential role in regulating Vitamin D-dependent gene expression [32]. Recent studies indicate that PHF2 plays a crucial role in controlling the methylation pattern and subsequent expression of genes responsible for osteoblast differentiation in mice. Moreover, deleting PHF2 in mice results in inadequate bone formation [33]. The high expression of PHF2 in bone tissues suggests direct links with Vitamin D in regulating osteoblast differentiation that requires further investigation.
The heritability of Vitamin D in Middle Eastern populations remains unknown and requires further investigation. Previous GWAS has approximated the heritability of 25(OH)D to be between 7.5% to 16% among Europeans [5,8]. We found that the SNP-based heritability of 25(OH)D in the Lebanese group surpassed the estimation observed in UK Biobank participants, with a higher estimate of approximately 29%. This observation may be due to several factors, including study design, genetic diversity, and environmental exposures, which lead to increased heritability estimates [8]. Further research is needed to understand the underlying mechanisms driving these differences.
To ensure the validity and reliability of our findings, we examined data from the UK Biobank since the frequency and impact of alleles may differ across populations. Our analysis of this dataset enabled us to confirm several Vitamin D-related variants identified in the GCST90000616 study, indicating the consistency of our results. Nonetheless, we acknowledge that differences in the genetic backgrounds of study populations and environmental exposures may have led to variants with opposing effect sizes. In order to better understand the reasons for opposing effect sizes and insufficient statistical power, further investigation may be needed, including studies in larger and more diverse populations.
To enhance the statistical significance of our observations, we merged our findings from Lebanese samples with data from the largest European GWAS [7]. Our meta-analysis has revealed several SNPs associated with Vitamin D levels that were not previously considered in the GWAS catalog (Table S2). Furthermore, we have confirmed the replication of multiple SNPs from the GCST90000616 study (Table S3), including a missense variant, rs2725405, located in the SLC38A10 gene. This gene is responsible for regulating protein transportation, synthesis, and cellular stress responses. In some cases, SLC38A10 protein can also mediates the intestinal transportation of some Vitamins into the blood and lipid metabolism [34]. Notably, the expression of SLC38A10 decreases in the presence of “C” risk allele in the intestinal and skin. The enrichment of SLC38A10 in intestinal and skin tissues and regional populations, suggesting possible mechanisms in Vitamin D absorption and metabolism. However, more research is needed to understand how it may impact Vitamin D status.
The comprehensiveness of the UK Biobank cohort provided the possibility of deriving PRS for Lebanese individuals from European populations. The effectiveness of European-derived PRS in the Lebanese population was lower than that of estimated in Europeans. This variation in performance might be due to various factors, such as differences in variant effect sizes and frequencies of causal alleles across ethnicities as well as the number of genetic variants and participants utilized in the study [35]. This emphasizes the need for a larger genome-wide association study tailored specifically for the Middle Eastern population to improve the performance of PRS estimation. Nevertheless, our polygenic risk score model for Vitamin D was able to predict Vitamin D deficiency efficiently in the Lebanese cohort. The modest performance of the PRS in predicting Vitamin D levels in individuals of European and Lebanese descent, with R values of 0.31 and 0.2033, respectively, may suggest the influence of non-genetic factors associated with Vitamin D deficiency, such as inadequate sunlight exposure and lifestyle/environmental factors. Therefore, the development of a more effective risk score tool for Vitamin D levels may require the incorporation of such factors.
5. Conclusions
Overall, we explored the first suggestive evidence of an association between several loci and Vitamin D levels in an elderly Middle Eastern population through our ExWAS. Our study showed that ExWAS can more easily identify the genes and biological pathways associated with Mendelian phenotypes. The results of our PRS model may provide a new avenue for guiding the personalized treatment of Vitamin D deficiency. However, further replications with larger sample sizes are necessary to confirm the potential of these findings and advance their application in the development of personalized medicine.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Supplementary Information Table S1 Table S2 Table S3 Figure S1.
Author Contributions
GN: GEF, and MC conceived the work and secured funding. DB, MC, and GEF carried out the clinical analysis. NNH carried out the ExWAS analysis, meta-analysis, and PRS analysis, generated all the results, and wrote the first draft of the manuscript. NNH, YA, and OA wrote the codes for the computational analyses. All authors participate in analyzing the final data and the write up of the current version of the manuscript.
Informed Consent Statement
All individuals who participated in the study provided informed consent.
Data Availability Statement
All the data produced in this study have been examined and incorporated into this published article or documented in the data repositories mentioned in the References section. Publicly available software tools were used for all analyses, as indicated in both the main text and the Methods section.
Acknowledgments
N.N.H is supported by a Ph.D. scholarship from Hamad Bin Khalifa University (HBKU) funded by the Qatar Foundation.
Conflicts of Interest
The authors declare no competing interests.
References
- Holick, M.F. , Vitamin D deficiency. N Engl J Med, 2007. 357(3): p. 266-81.
- Rahme, M. , et al., Impact of Calcium and Two Doses of Vitamin D on Bone Metabolism in the Elderly: A Randomized Controlled Trial. J Bone Miner Res, 2017. 32(7): p. 1486-1495.
- Mitchell, B.L. , et al., Half the Genetic Variance in Vitamin D Concentration is Shared with Skin Colour and Sun Exposure Genes. Behav Genet, 2019. 49(4): p. 386-398.
- Wjst, M. , et al., A genome-wide linkage scan for 25-OH-D(3) and 1,25-(OH)2-D3 serum levels in asthma families. J Steroid Biochem Mol Biol, 2007. 103(3-5): p. 799-802.
- Manousaki, D. , et al., Genome-wide Association Study for Vitamin D Levels Reveals 69 Independent Loci. Am J Hum Genet, 2020. 106(3): p. 327-337.
- Sinnott-Armstrong, N. , et al., Author Correction: Genetics of 35 blood and urine biomarkers in the UK Biobank. Nat Genet, 2021. 53(11): p. 1622.
- Revez, J.A. , et al., Genome-wide association study identifies 143 loci associated with 25 hydroxyvitamin D concentration. Nat Commun, 2020. 11(1): p. 1647.
- Jiang, X. , et al., Genome-wide association study in 79,366 European-ancestry individuals informs the genetic architecture of 25-hydroxyvitamin D levels. Nat Commun, 2018. 9(1): p. 260.
- Autier, P. , et al., Effect of vitamin D supplementation on non-skeletal disorders: a systematic review of meta-analyses and randomised trials. Lancet Diabetes Endocrinol, 2017. 5(12): p. 986-1004.
- Lips, P. , et al., Current vitamin D status in European and Middle East countries and strategies to prevent vitamin D deficiency: a position statement of the European Calcified Tissue Society. Eur J Endocrinol, 2019. 180(4): p. P23-P54.
- Chakhtoura, M. , et al., Vitamin D in the Middle East and North Africa. Bone Rep, 2018. 8: p. 135-146.
- Salman, S. , et al., Prevalence and Predictors of Vitamin D Inadequacy: A Sample of 2,547 Patients in a Mediterranean Country. Cureus, 2021. 13(5): p. e14881.
- McMahon, A. , et al., Sequencing-based genome-wide association studies reporting standards. Cell Genom, 2021. 1(1).
- Li, H. and R. Durbin, Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics, 2009. 25(14): p. 1754-60.
- Cingolani, P. , et al., A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin), 2012. 6(2): p. 80-92.
- Li, H. , et al., The Sequence Alignment/Map format and SAMtools. Bioinformatics, 2009. 25(16): p. 2078-9.
- Sherry, S.T., M. Ward, and K. Sirotkin, dbSNP-database for single nucleotide polymorphisms and other classes of minor genetic variation. Genome Res, 1999. 9(8): p. 677-9.
- Landrum, M.J. , et al., ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res, 2018. 46(D1): p. D1062-D1067.
- Chang, C.C. , et al., Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience, 2015. 4: p. 7.
- Zhou, W. , et al., Efficiently controlling for case-control imbalance and sample relatedness in large-scale genetic association studies. Nat Genet, 2018. 50(9): p. 1335-1341.
- Pruim, R.J. , et al., LocusZoom: regional visualization of genome-wide association scan results. Bioinformatics, 2010. 26(18): p. 2336-7.
- Buniello, A. , et al., The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res, 2019. 47(D1): p. D1005-D1012.
- Lambert, S.A. , et al., The Polygenic Score Catalog as an open database for reproducibility and systematic evaluation. Nat Genet, 2021. 53(4): p. 420-425.
- Hunt, S.E. , et al., Annotating and prioritizing genomic variants using the Ensembl Variant Effect Predictor-A tutorial. Hum Mutat, 2022. 43(8): p. 986-997.
- Lachmann, A. , et al., Massive mining of publicly available RNA-seq data from human and mouse. Nat Commun, 2018. 9(1): p. 1366.
- Wang, T.J. , et al., Common genetic determinants of vitamin D insufficiency: a genome-wide association study. Lancet, 2010. 376(9736): p. 180-8.
- Zhang, E. , et al., Identification of subgroups along the glycolysis-cholesterol synthesis axis and the development of an associated prognostic risk model. Hum Genomics, 2021. 15(1): p. 53.
- Chiruvella, V. , et al., Sucrase-Isomaltase Deficiency Causing Persistent Bloating and Diarrhea in an Adult Female. Cureus, 2021. 13(4): p. e14349.
- Okuno, Y. , et al., Epigenetic regulation of adipogenesis by PHF2 histone demethylase. Diabetes, 2013. 62(5): p. 1426-34.
- Luo, P. , et al., HIF-1alpha-mediated augmentation of miRNA-18b-5p facilitates proliferation and metastasis in osteosarcoma through attenuation PHF2. Sci Rep, 2022. 12(1): p. 10398.
- Pereira, F. , et al., Vitamin D has wide regulatory effects on histone demethylase genes. Cell Cycle, 2012. 11(6): p. 1081-9.
- Sawatsubashi, S. , et al., The Function of the Vitamin D Receptor and a Possible Role of Enhancer RNA in Epigenomic Regulation of Target Genes: Implications for Bone Metabolism. J Bone Metab, 2019. 26(1): p. 3-12.
- Kim, H.J. , et al., Plant homeodomain finger protein 2 promotes bone formation by demethylating and activating Runx2 for osteoblast differentiation. Cell Res, 2014. 24(10): p. 1231-49.
- Tripathi, R. , et al., SLC38A10 Regulate Glutamate Homeostasis and Modulate the AKT/TSC2/mTOR Pathway in Mouse Primary Cortex Cells. Front Cell Dev Biol, 2022. 10: p. 854397.
- Patel, R.A. , et al., Genetic interactions drive heterogeneity in causal variant effect sizes for gene expression and complex traits. Am J Hum Genet, 2022. 109(7): p. 1286-1297.
Figure 1.
Manhattan plot and Q-Q plot of the ExWAS results for Vitamin D levels. A, Manhattan plot displays chromosomal positions of genetic variants, and the –log10 P-value with a horizontal grey line represents the top signals (P-value < 1 x 10-5). The plot shows novel genomic regions on chromosome 7 and 9 that exceeds the significance threshold of Vitamin D ExWAS (n = 481,395 variants). B, Q-Q plot displays a good fit between the observed –log10 P-values and the expected –log10 P-values, indicating that the ExWAS results are not biased and are consistent with the null hypothesis.
Figure 1.
Manhattan plot and Q-Q plot of the ExWAS results for Vitamin D levels. A, Manhattan plot displays chromosomal positions of genetic variants, and the –log10 P-value with a horizontal grey line represents the top signals (P-value < 1 x 10-5). The plot shows novel genomic regions on chromosome 7 and 9 that exceeds the significance threshold of Vitamin D ExWAS (n = 481,395 variants). B, Q-Q plot displays a good fit between the observed –log10 P-values and the expected –log10 P-values, indicating that the ExWAS results are not biased and are consistent with the null hypothesis.
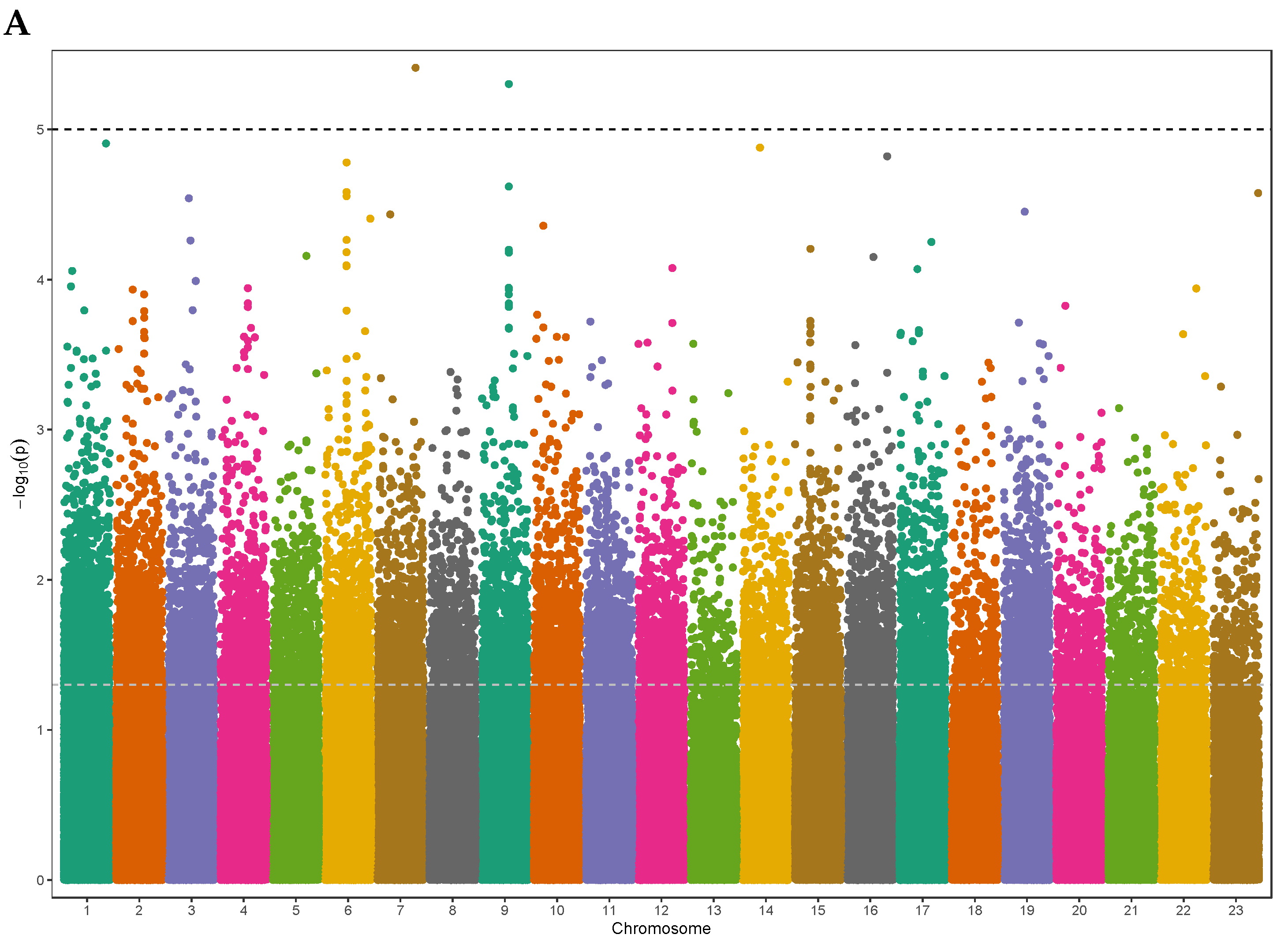
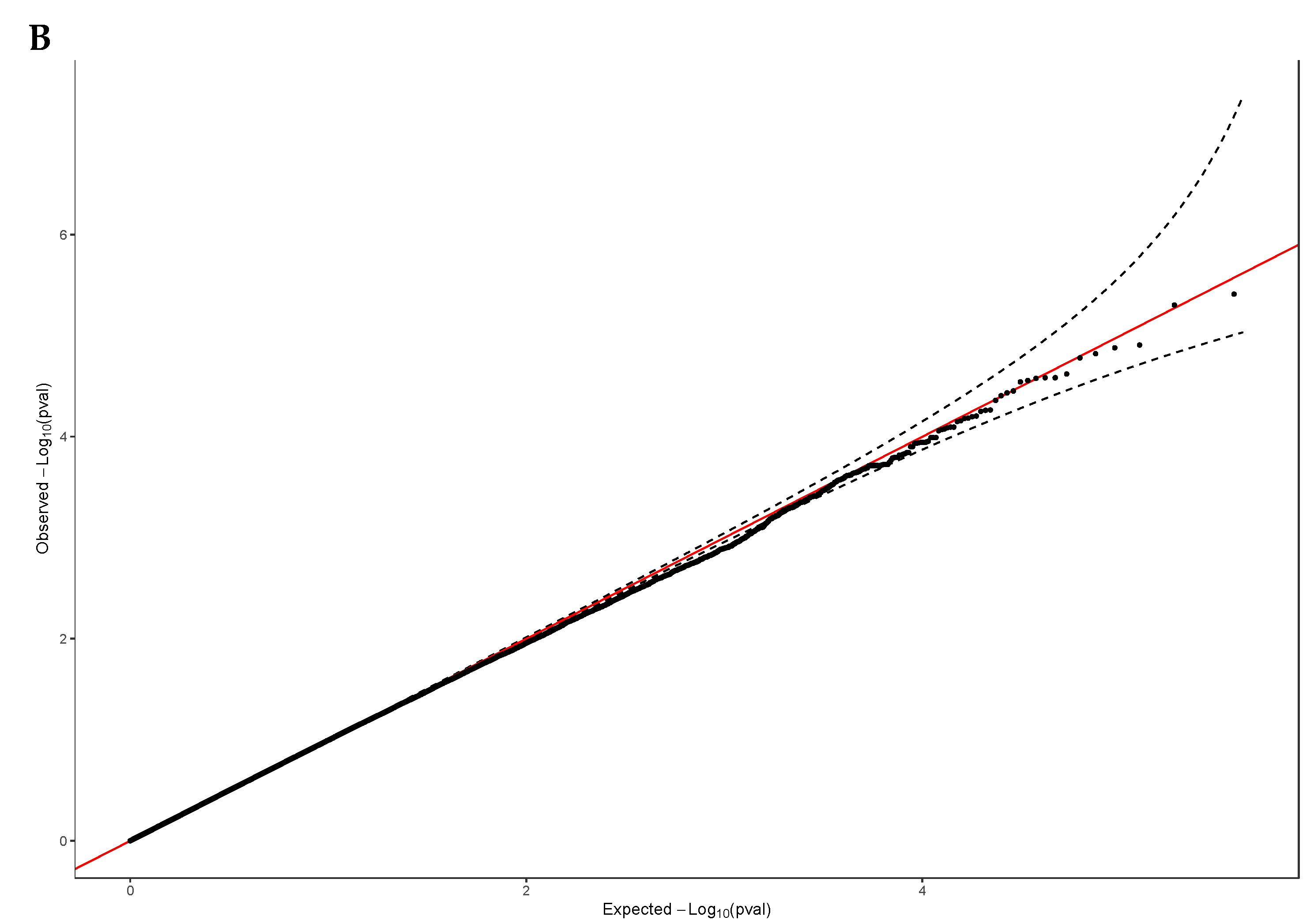
Figure 2.
Regional Plot for the top novel regions. LocusZoom plots of strongest correlated SNPs to Vitamin D, A, rs141064014 on chromosome 9, and B, rs7036592 on chromosome 7 (lead SNP–shown in purple diamonds). P-values in-log 10 scale, as in the Manhattan plot, are shown on the left vertical axis, the recombination rates are on the right vertical axis as a blue line, and the chromosomal positions are on the horizontal axis. The bottom panel shows the name and location of genes. Genes within the region are annotated and shown as arrows, and r2 of linkage disequilibrium relationships of each SNP with lead SNP are colored as indicated.
Figure 2.
Regional Plot for the top novel regions. LocusZoom plots of strongest correlated SNPs to Vitamin D, A, rs141064014 on chromosome 9, and B, rs7036592 on chromosome 7 (lead SNP–shown in purple diamonds). P-values in-log 10 scale, as in the Manhattan plot, are shown on the left vertical axis, the recombination rates are on the right vertical axis as a blue line, and the chromosomal positions are on the horizontal axis. The bottom panel shows the name and location of genes. Genes within the region are annotated and shown as arrows, and r2 of linkage disequilibrium relationships of each SNP with lead SNP are colored as indicated.
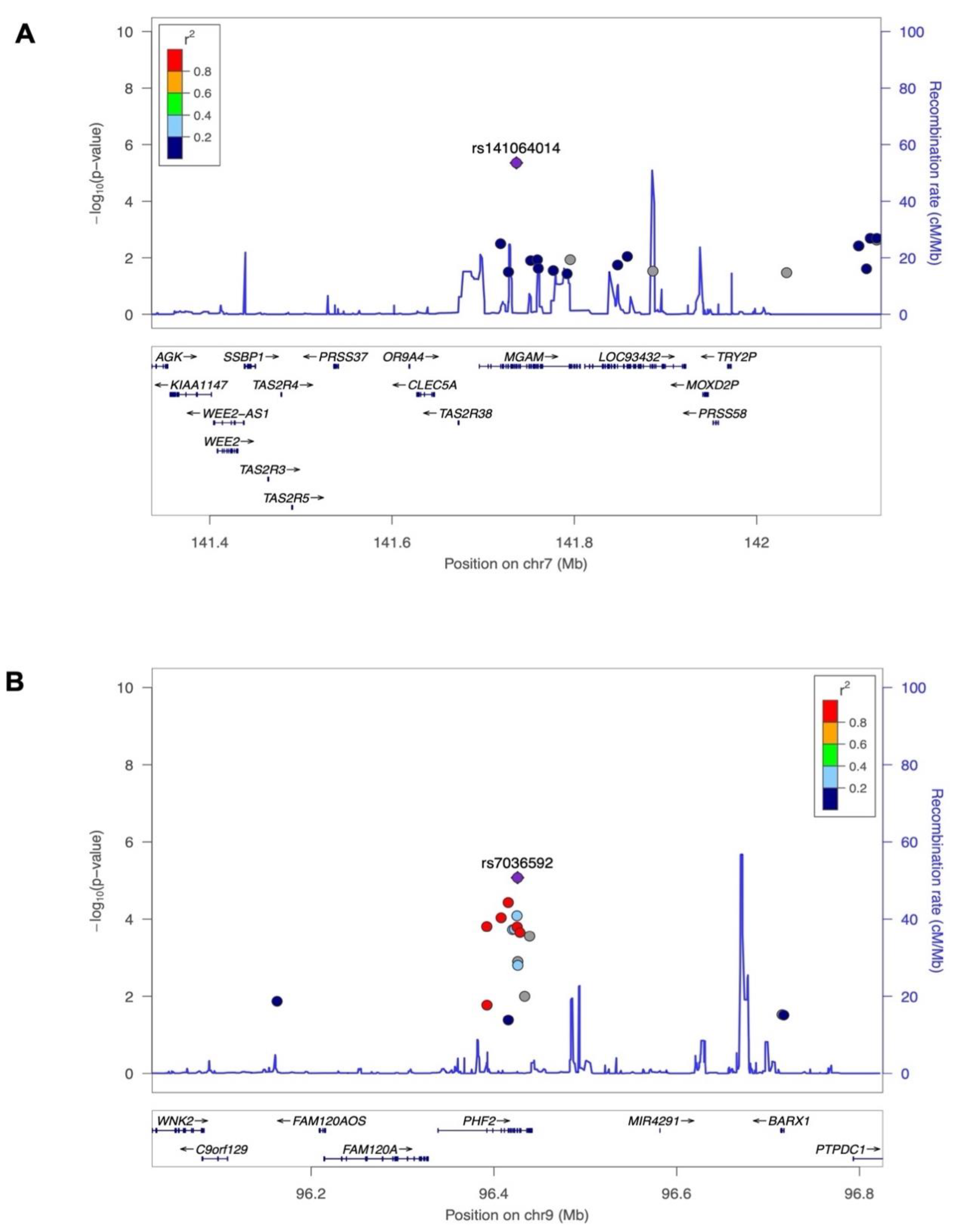
Figure 3.
Relationship between the genotypes of Vitamin D-associated SNPs and the gene expression enrichment. The bean plots display the normalized intron-excision ratio and their median (indicated by a white horizontal line) and interquartile range (represented by a black box) for A, MGAM rs141064014 expression in small intestine (P-value = 1.9 x 10-21), SLC38A10 rs2725405 expression in B, colon (P-value = 1.8 x 10-48), and C, small intestine (P-value = 1.5 x 10-23). The data presented is derived from the GTEx database.
Figure 3.
Relationship between the genotypes of Vitamin D-associated SNPs and the gene expression enrichment. The bean plots display the normalized intron-excision ratio and their median (indicated by a white horizontal line) and interquartile range (represented by a black box) for A, MGAM rs141064014 expression in small intestine (P-value = 1.9 x 10-21), SLC38A10 rs2725405 expression in B, colon (P-value = 1.8 x 10-48), and C, small intestine (P-value = 1.5 x 10-23). The data presented is derived from the GTEx database.
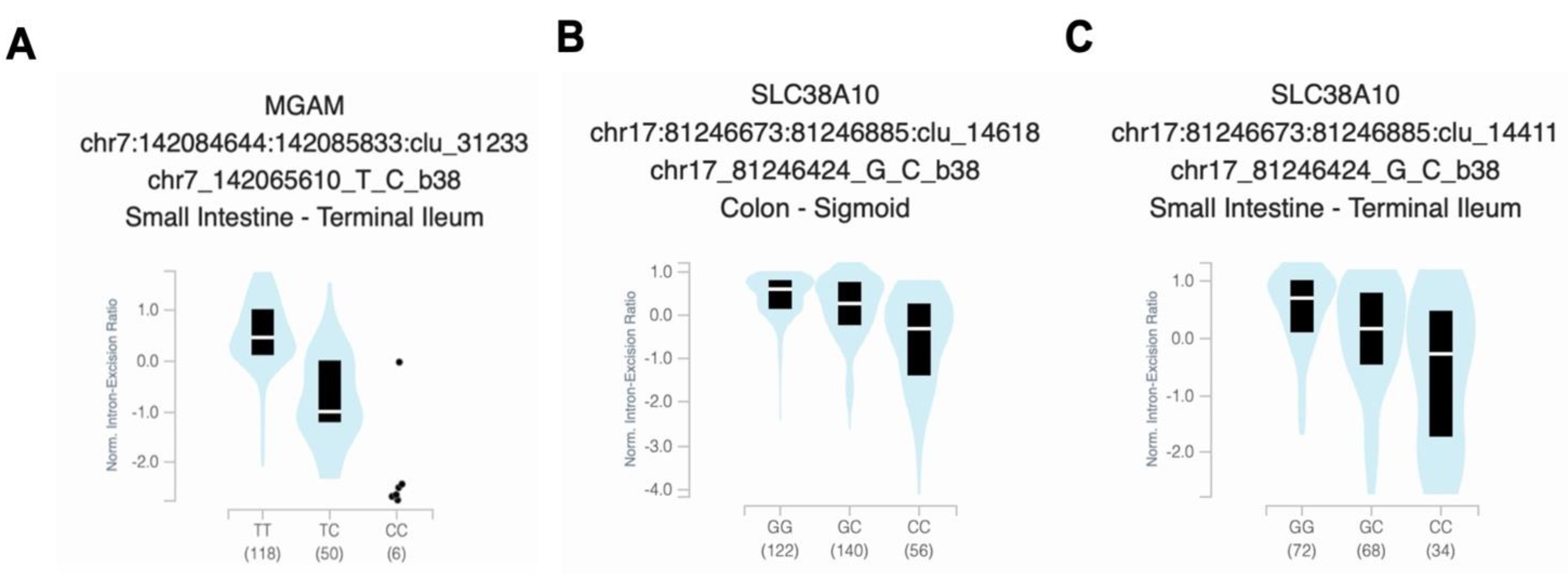
Figure 4.
European-derived polygenic risk score performance on the Lebanese population. Linear regression of baseline Vitamin D levels and polygenic risk scores (PRS) derived from a large European dataset (PGS000882: R= 0.2033, P-value = 0.0044). The blue line represents the best fit of linear regression analysis.
Figure 4.
European-derived polygenic risk score performance on the Lebanese population. Linear regression of baseline Vitamin D levels and polygenic risk scores (PRS) derived from a large European dataset (PGS000882: R= 0.2033, P-value = 0.0044). The blue line represents the best fit of linear regression analysis.
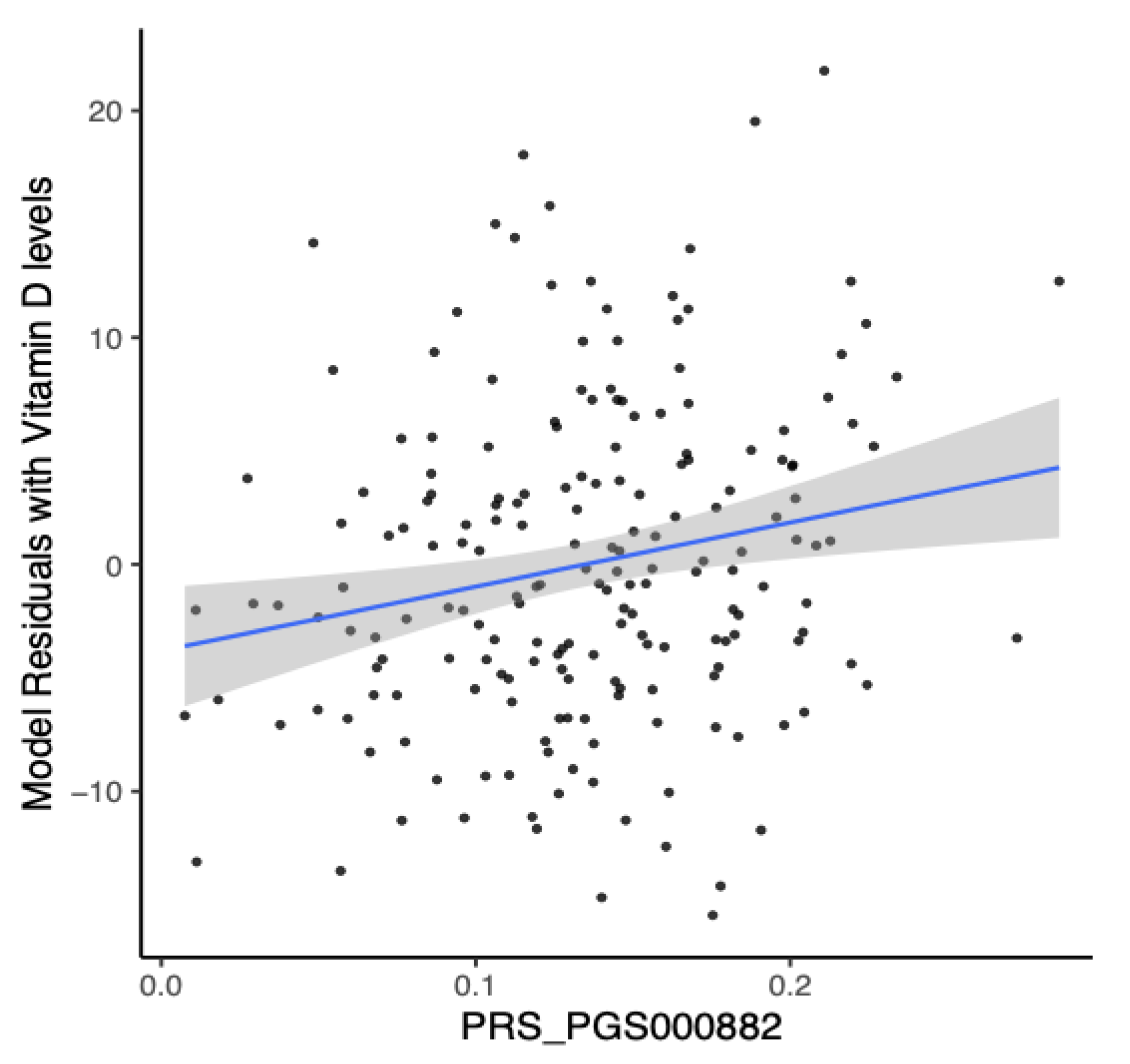
Figure 5.
Prediction of Vitamin D deficiency using European-derived PRS. Receiver Operating Characteristic (ROC) curve of the European-derived PRS on the Lebanese cohort for the prediction of Vitamin D deficiency (25(OH)D < 20 ng/mL). Area under the ROC curve (AUC) is reported in the figure.
Figure 5.
Prediction of Vitamin D deficiency using European-derived PRS. Receiver Operating Characteristic (ROC) curve of the European-derived PRS on the Lebanese cohort for the prediction of Vitamin D deficiency (25(OH)D < 20 ng/mL). Area under the ROC curve (AUC) is reported in the figure.
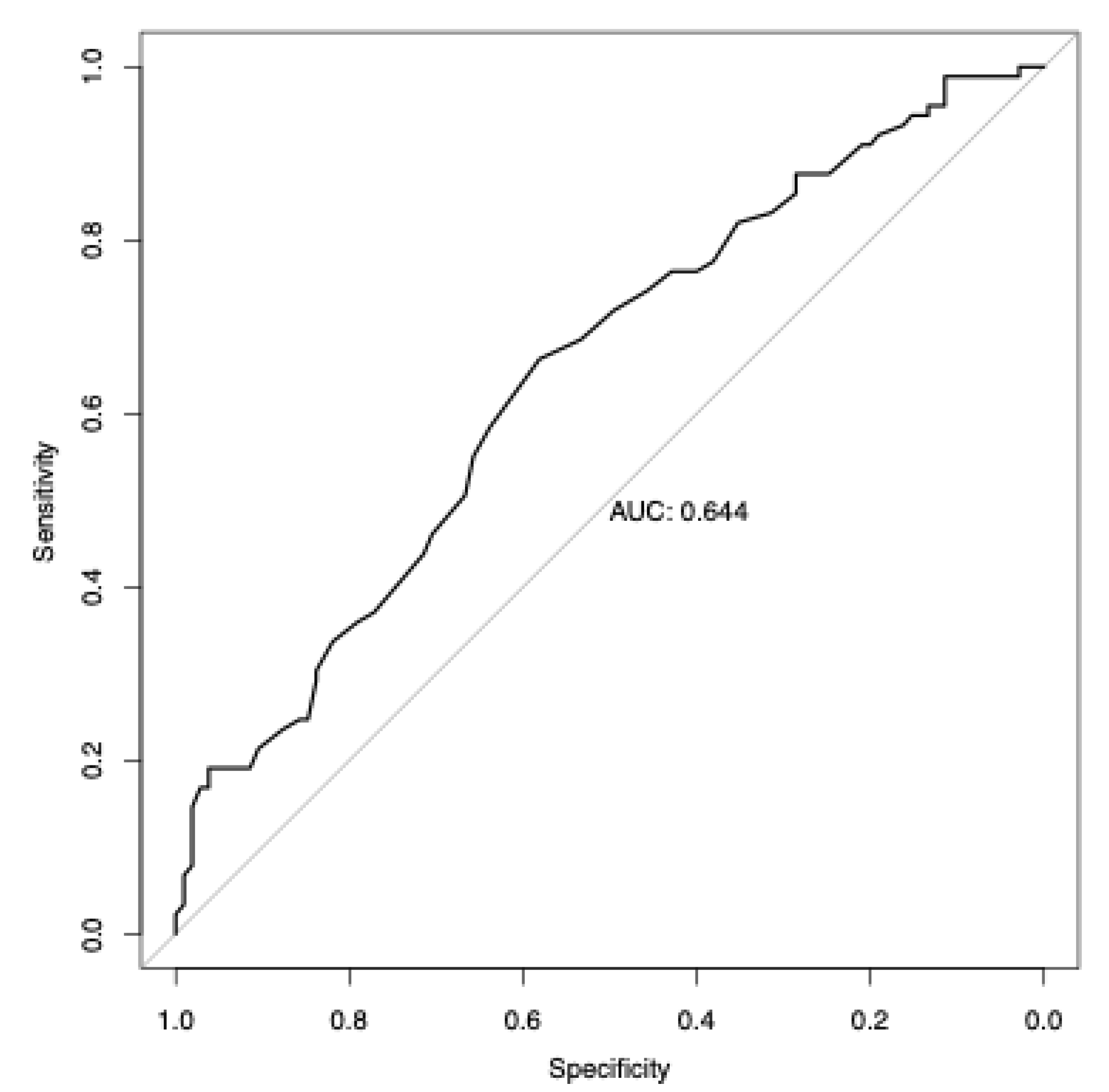

Table 1.
Baseline characteristics of the enrolled sample population.
| Characteristic | Male | Female | Total |
| Age (years) | 72.7 (±5.50) | 69.77 (±3.51) | 71.13 (±4.82) |
| BMI (kg/m2) | 28.69 (±3.35) | 31.60 (±5.10) | 30.24 (±4.60) |
| Serum 25(OH)D (ng/ml) | 19.36 (±6.25) | 20.83 (±7.93) | 20.12 (±7.22) |
| Sample Size | 90 (46.4) | 104 (53.6) | 194 |
| Abbreviations: BMI, body mass index; SD, standard deviation. | |||
Table 2.
Variants identified in exome-wide association analysis for 25-hydroxyVitamin D levels.
| SNP | Gene | HGVS ID | CHR | Position | A1 | A2 | Beta | SE (Beta) | P-value |
|---|---|---|---|---|---|---|---|---|---|
| rs141064014 | MGAM | NC_000007.13:g.141736273G>A | 7 | 141736273 | G | A | -2.38 | 0.52 | 4.4E-06 |
| rs7036592 | PHF2 | NC_000009.11:g.96425777C>T | 9 | 96425777 | C | T | -0.54 | 0.12 | 8.4E-06 |
Statistical summary of the Lebanese ExWAS analysis using linear mixed models adjusting for age, sex, principal population components, and relatedness with a P-value < 1 x 10-5. We used GRCh37/hg19 genome reference. Columns are SNP, single nucleotide polymorphism rs ID; Gene, mapped genes affected by a variant from ANNOVAR; HGVS ID, Human Genome Variation Society sequence variant nomenclature descriptions from Ensembl; CHR, chromosome; BP, Base pair; A1, reference allele; A2, alternative allele; MAF, minor allele frequency the A1; Beta, variants effect size for A1; SE, the standard error for Beta; P-value, P-value of ExWAS analysis. Abbreviations: MGAM, Maltase-glucoamylase; PHF2, PHD finger 2.
Table 3.
Allele frequency of the top Vitamin D-variants across different populations.
| Populations | Frequency for rs141064014 in MGAM | Frequency for rs7036592 in PHF2 | Frequency for rs2725405 in SLC38A10 |
| Lebanese elderly population | 0.0103 | 0.2408 | 0.4845 |
| European population of ALFA | 0.00794 | 0.39533 | 0.5729 |
| Controls of gnomAD populations | |||
| European | 0.00634 | 0.3829 | 0.5468 |
| East Asian | 0.001 | 0.2136 | 0.3467 |
| African | 0.001 | 0.2783 | 0.9234 |
| All populations | 0.00553 | 0.3216 | 0.5084 |
| Abbreviations: gnomAD, Genome Aggregation Database; ALFA, Allele Frequency Aggregator. | |||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



