
Submitted:
09 June 2023
Posted:
12 June 2023
You are already at the latest version
Abstract
Formation of the excited doublet (D) and quartet (Q) states of free radicals under their photoexcitation is discussed. Relative positions of D and Q states are compared to the positions of photoexcited states of organic molecules (Jablonsky diagram). A number of representative cases of the detected by emission or absorption excited is presented. A special case of the population lowest Q-state in some radicals of is discoursed. Spin-statistical factor in the reactions of Q and D is debated.
Keywords:
Photoexcited free radicals
; Jablonsky diagram
; reactions with dioxygen
; doublet and quartet states
1. INTRODUCTION
Organic compounds are used in chemistry and exist in everyday life. They consist of organic molecules. The vast majority of these molecules exist in the singlet state So. Upon absorption of light, a molecule reaches the excited S1 state. S1 can undergo intersystem crossing (ISC) and populate the lowest triplet state T1. These processes are presented in the Jablonsky diagram [1,2] (Figure 1). Singlet states are “close shells”; they have two electrons with opposite spins on each occupied molecular orbital (MO). Triplet state has two single-occupied MO (SOMO) with parallel spins. That way, the spin angular momentum of the singlet states is s=0; for the T-state s=1. (Spin multiplicity is 2s + 1. Spin multiplicity is presented as a superscript at a state notation.)
The Jablonsky diagram is described in detail in the text books on photochemistry [1,2]. Such books and review articles present all of the processes occurring in excited states as well as the known rare exceptions to the diagram [1,2]. Figure 1 presents only the absorption of a light quantum and the intersystem crossing (ISC) to T1 – state initiated by the spin-orbit coupling (SOC) [3]. The probabilities of the occurrence of the processes in the excited states of molecules obviously vary from 0 to 1. Say, the higher the energy of SOC, the higher the population of T1.
2. DOUBLETS AND QUARTETS
Organic free radicals (R∙ ) are species with an “open shell”. They have at least one SOMO. The spin state of R∙ is s=1/2 and such states are called doublets (D). Radicals with three one-electron orbitals exist in the quartet electronic state (Q) which has s=1½. Free radicals are important intermediates in many photoinduced reactions. There are cases when radicals absorb light and produce excited D1 or Q1-states; see below. Obviously, the chemical reactivity of the photoexcited radicals (molecules) is different from that in Do- (So-) states [4].
Synthesized stable organic p-radicals have been recently reported, which are especially interesting for application in organic light-emitting devices (OLED) due to their possibility of providing a quantum yield of luminescence of 1.0 [5]. This is one of the modern reasons for studying photoexcited free radicals.
It is logical to assume an analog of the Jablonsky scheme (Figure 1) for free radicals. SOC mixes D1 and Q; see Figure 1. All other processes (luminescence, internal conversion, and vibrational relaxation) take place in the photoexcited molecules and radicals. Luminescence from the D1- (Q1) - state is named in the literature as fluorescence (phosphorescence) apparently due to analogy with such processes in molecules.
Stable free radicals (R∙) are usually colored, whereas the parent compounds (RH) are colorless. That means that D1 lies close in energy to Do, namely 180-300 kJ/mol above Do (Figure 1). For example, colorless 2,4,6-tri-tert-butyl phenol has a longwave absorption maximum at l= 280 nm (370 kJ/mol) whereas the corresponding phenoxyl radical (blue aroxyl) has an analogous value l=626 nm (190 kJ/mol). A similar batochromic shift of the absorption maximum holds between a parent compound (say a ketone, a quinone) and its radical R∙ (a ketyl radical, a semiquinone) formed by hydrogen abstraction.
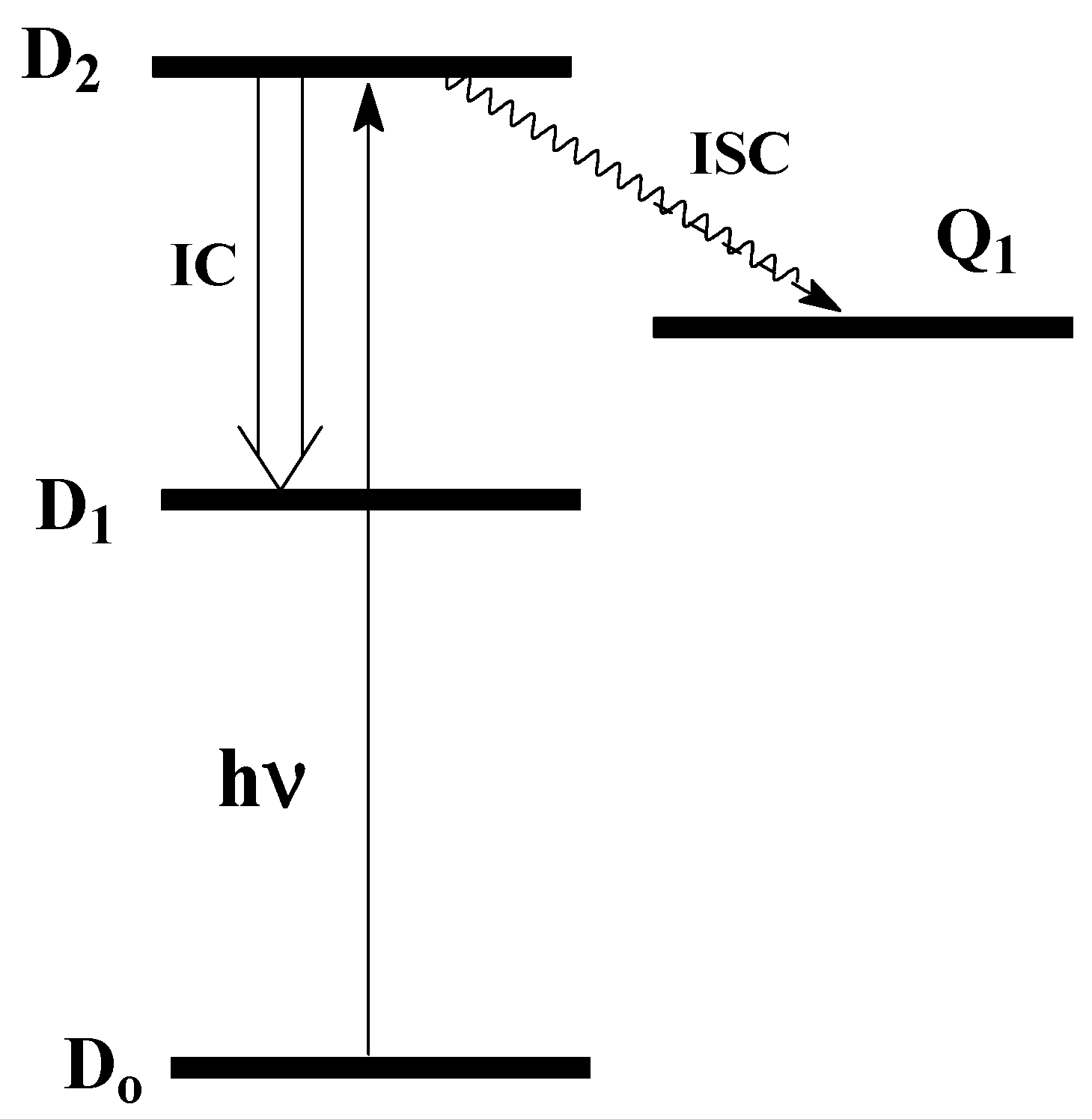
Q1 may have a higher energy than D1; see Figure 2:
The formation of Q1 requires three unpaired electrons with parallel spins, which may increase the energy of the excited R∙*compared to that of D1. (The formation of T1 in molecules requires only two unpaired electrons with parallel spins. The dominant role of the Pauli effect leads to a lower energy of T1 compared to S1 in the majority of cases [1,2].) Thus, in a number of cases, Q1 can be populated only after primary excitation into D2 or D3, see Figure 2.
Fast internal conversion (IC) from the higher D2(3) to a lower D1 should lead to a low probability of population in Q1. This assumption allowed the authors of [2] to draw a conclusion about the insignificant role of Q1 in the photochemistry of radicals. However, there are many reports of experimental detection of Q1 (Section 3 below). Observation of a relatively long-lived R∙*leads to the assumption that R∙*is in a Q1 -state; see Section 3 below.
3. EXCITED NEUTRAL RADICALS
This brief review article does not aim to review most of the reported cases of R∙* studies. We will focus on liquid solutions of polyatomic organic radicals and radical-ions at room temperature. This section presents some instructive and exemplary results of such research. Chemical transformations of R∙ *, except a reaction with dioxygen (Section 5) are beyond the scope of this brief article.
The excited states are usually detected by their emission spectra or transient absorption spectra.
However, we will start with the seminal publication [6] that reports a Do ← Q1 phosphorescence of decacyclene radical anion in the solid state at 77 K. It is assumed that Q1 lies above D1, and luminescence is phosphorescence. The Q1 electronic state of this anion at 77 K was confirmed by transient magnetic resonance [7]. Photoexcited radical-ions are considered in Section 4.
High-spin molecular objects are of great interest due to their potential applications in molecular magnets. It was stated that some labile dehydrophenylnitrenes are in the ground Q-state [8]. Quantum-chemical calculations of the ground and excited states of several dehydrophenylnitrenes allowed the authors to get the geometry and distinguish between the Q- and D-states of these species [8]. The studied dehydrophenylnitrene system demonstrates that a fine competition between ferromagnetic Q- and antiferromagnetic D- state coupling of electron spins takes place. With a single electron orbital being orthogonal to two coplanar orbitals, the relative orientation of the latter orbitals determines which mode of electron coupling prevails; see Scheme 1:
The D1-state of benzophenone ketyl radical in a benzene solution at room temperature was observed by laser flash photolysis with time-resolved (TR) ESR and absorption spectroscopy [9]. The life-time of this D1 was ~ 2 ns [9]. It is assumed that a photoexcitation of the ketyl into D2 leads to ISC into Q1 ← D2 [9]. (Q1 is positioned above D1, see Figure 2). It seems that in the case of the studied ketyl, ISC successfully competes with the D1 ← D2. The life-time of Q1 is relatively long, namely ~ 2 ms [9]. As expected, Do ← Q1 is relatively long process compared to Do ← D1 in the same ketyl radical under the same conditions.
The ketyl radical of benzophenone and of its derivative were studied by two-color laser flash photolysis [10]. The first ns pulse (lex =266 or 355 nm) generated ketyl radicals by the usual way: hydrogen abstraction from a solvent. The second ps pulse (lex =532 nm) excited ketyls into D1. The transient absorption and fluorescence of D1 were measured. The lifetime of D1 in the studied solvents varied from 0.5 to 5 ns [10]. It was noticed that D1 had a high dipole moment of 7-10 D, which leads to a significant solvent effect on D1 [10].
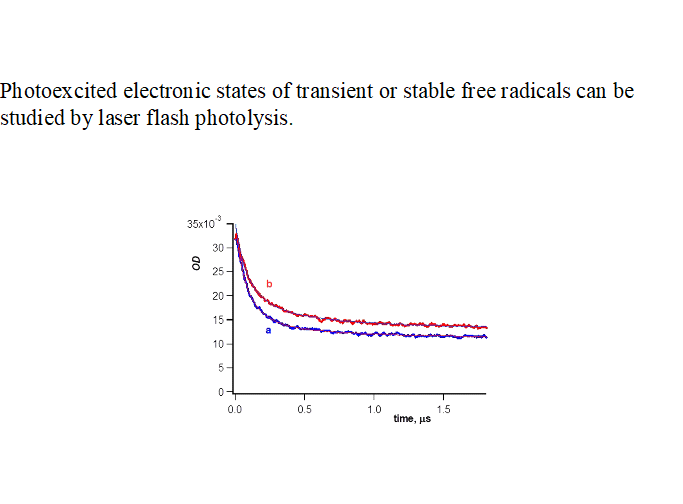
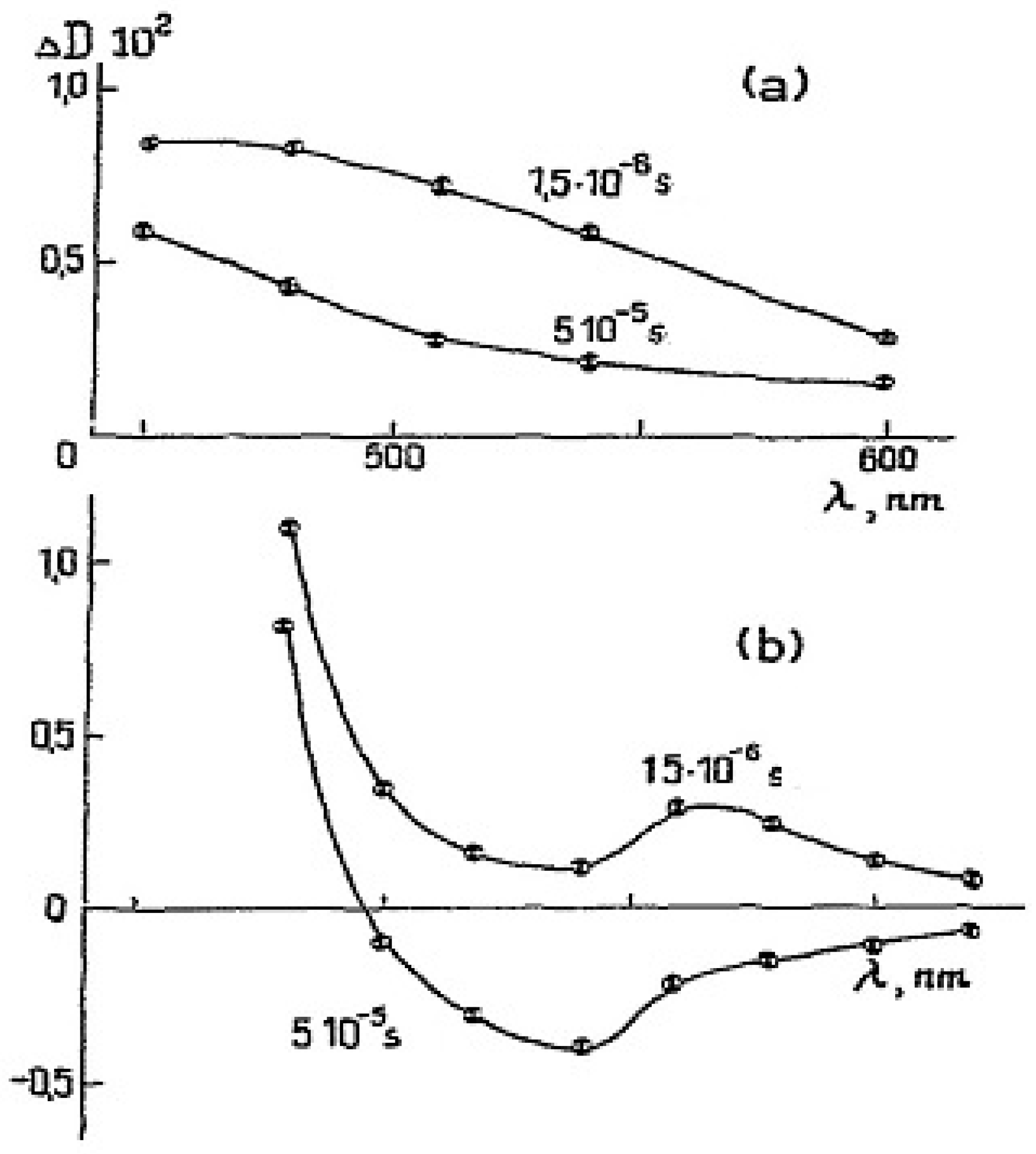
Many of the compounds reported in [5] are large, stable organic radicals that emit fluorescence. Stable aroxyl (substituted phenoxyl) radicals demonstrate transient absorption in liquid solvents in flash photolysis experiments [11]; see Figure 3. However, the fluorescence of galvinoxyl and other aroxyls has not been observed [12].
The photoexcitation was performed by UV-light (lex=260-380 nm) [11]. Quite similar spectra were observed in propan-1-ol. The spectra (Figure 3) were tentatively ascribed to Q1- state of the aroxyls due to their relatively long life-time (~ 10 ms) [11].
The D9,10 ← Do transitions were observed under further study of the photochemistry of galvinoxyl with fs flash photolysis (lex= 400 nm) [12]. However, a long-lived absorption very similar to that presented in Figure 3a was ascribed to the anion of glavinoxyl, galvinoxylate. The formation of anion in a non-polar solvent (Figure 3) is doubtful. The photochemistry of galvinoxyl deserves further study.
Phosphoresce Do ← Q1 was observed for phosphaethynyl radical in the solid argon [13]. The scheme of electronic levels for this radical follows Figure 1, and the lifetime of the emission was ~0.1 s [13].
Electron spin polarization and magnetic field effects have been investigated in photoexcited organic molecules that are chemically tethered to radical fragments (nitroxyls, others) [14,15]. With TR ESR electron spin polarization was observed in D1 and Q1 – states after photoexcitation of a large stable radical [15].
Scaiano, Johnson et al. performed a cycle of investigations on photoexcited transient radicals such as diphenylmethyl and others [16,17,18]. The radicals were generated by a laser pulse; after a short delay, the second laser pulse excited the produced radicals [16,17]. A fluorescence from D1 was apparently observed in these experiments. The lifetime of fluorescence varies from 5 to 400 ns for polyatomic radicals in liquid non-viscous solvents at room temperature [16]. The constant kq of quenching an excited state by dioxygen (Section 5) obtained for several R∙*[16,17,19] testifies to the occurrence of reactions (1) or (2) in the D1-state; see Section 5. A lifetime of photoexcited diphenylmethyl radicals in common non-viscous solvents is 250 ns at 300 K [16,17]. Once more, this R∙*is in the D1- state [20].
The excitation of perchlorodiphenylmethyl radical (PDM∙) at lex=530 nm leads to the population of D1 (lifetime t = 31 ns), from which emission is observed (ffl = 1.0 x 10-3) [20]. This state is quenched by electron donors and acceptors at a rate at or close to diffusion control and at much slower rates by dioxygen and hydrogen atom donors. Fragmentation of PDM∙ (fdec = 0.06) occurs from a higher excited doublet (Dn, n ≥2) producing intermediates that are trapped [32]. See [20] for details of the chemical reactions.
The excitation of perchlorotriphenylmethyl radical (PTM∙) at lex =532 nm leads to an excited doublet, D1 – state (lifetime t = 7 ns) [21]. The lifetime of this state is insensitive to dioxygen, electron acceptors, and hydrogen-donating solvents. Electron donors triphenylamine, N,N-dimethylaniline, and thianthrene quench PTM∙* at diffusion-controlled rates. Cyclization of PTM∙ to form perchloro-9-phenylfluorenyl radical occurs with a quantum efficiency of fcyc=0.3 under lex=365 nm irradiation [21].
ns, ps Laser flash photolysis allowed to observe Q1 ← D1 ISC of flavin radical in DNA photolyase [22]. Thus, electronic levels in this radical follow Figure 1; see [22] for details. The lifetime of the studied D1 (Q1) is 100 ps (1 ms) in aqueous solution at room temperature [22].
Paramagnetic free radicals are efficient quenchers of the excited states of molecules [1,2]. It was observed in experiments with the stable aroxyl radicals that self-quenching occurs in solutions [11]:
R∙ * + R∙ → 2 R∙ (1)
4. EXCITED RADICAL-IONS
Electronically excited radical-cations and radical-anions were the subject of a number of experimental and theoretical investigations.
Stilbene (Sb) radical-anions and radical cations were studied by pulse radiolysis and laser flash photolysis [23]. There were four species under investigation, namely cis- and trans-Sb(+/-)∙*. It was stated that D2 of these species was observed [23]. Table 1 below summarizes most of the data obtained in this work:
Radical-cations of polycyclic aromatic hydrocarbons (PAH) play an important role in different processes and in the environment, in particular in astronomy [24]. PAH are easily ionized, producing radical-cations PAH∙+. As expected, the absorption spectra of PAH∙+ are red-shifted towards the spectra of parent PAH [24]. Excited states of 51 different PAH∙+ were studied with time-dependent density functional theory [24]. The vertical excitation energies and oscillating strengths of PAH∙+and even of PAH∙ - were obtained. p*←p transitions between individual p-orbitals in radical-ions of PAH were considered [24].
Biphotonic laser photoexcitation (lex =248 nm) of different aromatic compounds (biphenyl, naphthalene, perylene, and others) and several amines in oxygenated polar solvents led to the corresponding radical-cation R∙+ [25]. R∙+have obviously red-shifted spectra compared to the parent compounds and absorb in the visible and NIR regions. A prompt irradiation of R∙+with the second harmonic YAG:Nd laser (lex= 532 nm) led to the excited R∙+*[25]. The studied R∙+* has a lifetime ranging from hundreds of fs to tens of ps at room temperature. The nature of R∙+* is not discussed [25]; it is probably D1(2) –states.
In general, excited states of multiple radicals of radical ions in the Dn - state have very short lifetimes (see Sections 2,3) as a result of their low-lying excited state energies (energy gap law) [25,26].
It is well known that molecules in excited states essentially change their redox properties [1,2]. Not surprisingly, the same holds true for radical-ions. Wasielewski et al. studied the redox properties of radical-anions in excited doublet states [24,25]. The radical-anions of aromatic diimides have been assumed recently to be participants in different photoinduced electron transfers. Photoexcitation of these radical-anions produces powerfully reducing agents [24]. However, the properties of the π* - excited D- states of these radical-anions remain obscure. The radical-anions of three complex aromatic imides with increasingly larger π systems, namely N-(2,5-di-tert-butylphenyl)phthalimide, N-(2,5-di-tert-butylphenyl)-1,8-naphthalimide, and N-(2,5-di-tert-butylphenyl)perylene-3,4-dicarboximide, as well as the three corresponding aromatic diimides [24], were produced by electrochemical reduction of the neutral molecules. The radical-anions of these imides and diimides demonstrate intense visible and weaker NIR absorption bands corresponding to their Dn ← Do transitions [24]. Excited states of the radical anions were generated by fs excitation (lex=840 nm) into these absorption bands. Excitation of the first two radical-anions mentioned above led to their decomposition, whereas excitation of the third imide and three diimides yielded transient spectra of their Dn ← D1 transitions. The lifetimes of the observed D1-state of the radical-anions are all less than 600 ps and increase as the D0 - D1 energy gap increases [24]. These results impose constraints on the use of these excited radical-anions as electron donors in electron-transfer systems targeted at molecular electronics and solar energy utilization [24].
It was presented that the 10-phenyl-10H-phenothiazine radical cation (PTZ+•) has a manifold of excited D- states accessible using visible NIR light [25]. PTZ+• can serve as super-photooxidants with excited state potentials in excess of +2.1 V vs. SCE [25]. Photoexcitation of PTZ+• in acetonitrile with lex=517 nm laser pulse populates a Dn excited D- state that decays first to the unrelaxed lowest electronic excited state, D1' (τ < 0.3 ps), followed by relaxation to D1 (τ = 10.9 ± 0.4 ps) [25]. D1 finally decays to Do (τ = 32.3 ± 0.8 ps) [25]. To probe the oxidative power of PTZ+•* Dn - states, PTZ+• was covalently linked to each of three hole acceptors, perylene, 9,10-diphenylanthracene, and 10-phenyl-9-anthracenecarbonitrile, which have oxidation potentials of 1.04, 1.27, and 1.6 V vs. SCE, respectively [25]. In all three cases, photoexcitation wavelength dependent ultrafast hole transfer occurs from Dn, D1', or D1 of PTZ+•* to the three acceptors. The high oxidative power of the Dn -state of PTZ+•* will enable applications using this chromophore as a superoxidant for energy-demanding reactions [25].
The quantum-chemical calculations (restricted coupled cluster with perturbative triples method) were used to calculate equilibrium geometries and excitation energies of the hitherto unknown Q - states in diacetylene-, triacetylene-, and benzene radical-cations [32]. Spectroscopic data for the D - states obtained with the same approach are found to be in good agreement with the experiment [32]. In diacetylene- and triacetylene-cation there are Q-states that lie close to the minimum energies of the corresponding D1-states [32].
Electron acceptor methyl viologen (N,N′-dimethyl-4,4′-bipyridine, abbreviated MV2+) plays an important role in redox reactions (MV2+ ↔ MV•+, see e.g. [34]). Femtosecond pump-probe spectroscopy was used to investigate the excited state dynamics of the electrochemically generated MV•+ in acetonitrile solution [31]. fs Excitation of the D1 ← Do transition at lex=730 nm led to rapid relaxation (700 fs), generating two intermediates in the transient absorption spectra [31]. The longer-lived intermediate, with a lifetime of 16 ps, could be assigned to a vibrationally excited ground state of MV•+. Its absorption spectrum was very similar to the ground-state spectrum of MV•+ in both shape and extinction coefficients, but red-shifted by ~ 810 cm-1 [31]. The shorter-lived transient decayed with a characteristic time of t =1.0 ±0.1 ps and was possibly also a vibrationally excited ground state. Thus, these results show that the excited D1-state of MV•+* in acetonitrile solution relaxes on the fs time scale via at least one long-lived (τ =16 ps) vibrationally excited ground state.
5. QUENCHING OF PHOTOEXCIED RADICALS BY DIOXYGEN
The majority of free radicals (especially C-centered alkyl, aromatic benzyl, and others) react with dioxygen at rates controlled by diffusion in solution. The same is expected for highly reactive R∙*. The rate of a diffusion-controlled reaction estimated by the Debye formula is kdiff ≈ 7 x109 M-1.s-1 at room temperature (T) and solvent viscosity h = 1 cP [31]. In the case of a reagent with non-zero spin, the expected rate constant of a diffusion reaction is lower: kq= s kdiff, where s is spin-statistical factor [19,31]. There are two types of quenching of R∙*- physical and chemical [19]:
nR∙ * + 3O2 → 2R∙ + 1O2 (1Dg)
nR∙* +3O2 → 2RO2∙
Photoexcited R∙*can be in the D1- state (n=2) or in the Q1-state (n=4). It is easy to get that for reactions (1,2) s = 1/3 and 1/6 for n=2 (D1) and n=4 (Q1), respectively. Thus, one can expect kq ≈ 2 x109 (1x109) M-1.s-1 for quenching of R∙*for D1 (Q1) in the reactions (1) or (2) under T and h mentioned above. (It is known that kdiff ~ h-1; the rate constant of a reaction with the fast diffusing O2 may have higher kdiff - values than those predicted by the Debye formula.)
6. CONCLUSIONS
The main elementary photophysical processes in the excited free radicals are depicted in
Figures 1,2. There is an analogy with the relevant photophysical processes in molecules (Figure 1) but the photophysics of R∙*is more complex (Figure 2). Usually, D1 or Q1 have been observed in the experiments by their emission or absorption spectra. Additional TR ESR experiments would make such assignments more reliable. Quantum chemical calculations should help in the identification of R∙*, their electronic nature and their structure as well.
It is possible to conclude that Q1-state has a much longer life than the D1-state of the same radical. It is obvious that Do ← Q1 transition occurs with a change of spin multiplicity and is analogous to So ← T1 ISC or phosphorescence in molecules.
Stable nitroxyl radicals (TEMPO and others) are widely used in the chemistry research, as antioxidants and as photostabilizers of polymers for many decades. To the best of our knowledge, there is no experimental data on the excited states of nitroxyls.
There are tens of colored stable free radicals that are commercially available or can be synthesized. The photochemistry of a small portion of them has been studied, see some examples in Sections 3,4 above. Stable radicals are valuable and convenient objects for the study of their excited states and their transformations. The first triphenylmethyl free radical discovered by Gomberg in 1900 is in equilibrium with its corresponding dimer [33]. Once more, to the best of our knowledge, there is no experimental data on the excited state of the first radical.
It would be of interest to compare ESR spectra of radicals in the ground and in excited states, to compare g-factors in particular.
The measured numerical values of the rate constant kq of quenching R∙* by O2 give a hint on the nature of a spin state of R∙*.
Funding
The research received no external funding.
Conflicts of Interest
The author declares no conflict of interest, financial or otherwise.
References
- Turro, N.J., Ramamurthy, V., Scaiano J.C. Modern Molecular Photochemistry of Organic Molecules; University Science Books: Sausalito, 2010.
- Klán, P., Wirtz, J. Photochemistry of Organic Compounds; Wiley: Wiltshire, 2009.
- Khudyakov, I.V., Serebrennikov, Yu.A., Turro, N.J. Spin-Orbit Coupling in Free-Radical Reactions: On the Way to Heavy Elements. Chem. Rev., 1993, 93, 537. [CrossRef]
- Melnikov, M.Ya., Smirnov, V.A. Handbook of Photochemistry of Organic Radicals; Begell House: New York, 1996.
- Teki, Y. Excited-State Dynamics of Non-Luminescent and Luminescent p-Radicals. Chem. Eur. J., 2019, 25, 1.
- Brugman, C.J.M., Rettschnick, R.P.H., Hoytink, G.J. Quartet-doublet phosphorescence from an aromatic radicals. The decacyclene mononegative ion. Chem. Phys. Lett., 1971, 8, 263.
- Kothe, G., Kim, S.S., Weissman, S.I. Transient magnetic resonance of a photoexcited quartet state. Chem. Phys. Lett., 1980, 71, 445. [CrossRef]
- Bettinger, H.F., Sander, W. Dehydrophenylnitrenes: Quartet versus Doublet States. J. Am. Chem. Soc., 2003, 125, 9726. [CrossRef]
- Thurnauer, M.C., Meisel, D. Time-resolved EPR studies of the benzophenone-diphenyl ketyl radicals system. Possible evidence for quartet-doublet intersystem crossing. Chem. Phys. Lett., 1982, 92, 343.
- Sakamoto, M., Cai, X., Fujitsuka, M., Majima, T. Solvent Effect on the Deactivation Processes of Benzophenone Ketyl Radicals in the Excited State. J. Phys. Chem. A, 2006, 110, 11800. [CrossRef]
- Kuzmin, V.A., Khudyakov, I.V., Tatikolov, A.S. Electronically-Excited States of Phenoxy Radicals. Chem. Phys. Lett., 1977, 49, 495. [CrossRef]
- Grilj, J., Zonca, C., Max, L., Daku, L., Vauthey, E. Photophysics of the galvinoxyl free radical revisited. Phys. Chem. Chem. Phys., 2012, 14, 6352. [CrossRef]
- Ganesan, E., Custer, A.-L., Guillemin, J.-C., Kołos, R. Unusual Quartet-Doublet Phosphorescence from the Phosphaethynyl Radical, CP. Angew. Chem. Intern. Edn. 2022, 134, e202210521. [CrossRef]
- Turro, N.J., Khudyakov, I.V. Application of chemically induced dynamic electron polarization to mechanistic photochemistry. Res. Chem. Interm., 1999, 25, 505. [CrossRef]
- Giacobbe, E.M., Mi, Q., Colvin, M.T., Cohen, B., Ramanan, C., Scott, A.M., Yeganeh, S., Marks, T,J., Ratner, M.A., Wasielewski, M.R. Ultrafast Intersystem Crossing and Spin Dynamics of Photoexcited Perylene-3,4:9,10-bis(dicarboximide) Covalently Linked to a Nitroxide Radical at Fixed Distances. J. Amer. Chem. Soc., 2009, 131, 3700. [CrossRef]
- Scaiano, J.C., Tanner, M., Weir, D. Exploratory Study of the Intermolecular Reactivity of Excited Diphenylmethyl Radicals. J. Am. Chem. Soc., 1985, 107, 4396. [CrossRef]
- Scaiano, J.C., Johnson, L.J., McGimsey, W.G., Wier, D. Photochemistry of Organic Reaction Intermediates: Novel Reaction Paths Induced by Two-Photon Laser Excitation. Acc. Chem. Res., 1988, 21, 22. [CrossRef]
- Johnston, L.J. Photochemistry of Radicals and Biradicals. Chem. Rev., 1993, 93, 251. [CrossRef]
- Darmanyan, A.P., Gregory, D.D., Guo, Y., Jenks, W.S. Generation and Decay of Aryl Sulfinyl and Sulfenyl Radicals: A Transient Absorption and Computational Study. J. Phys. Chem. A, 1997, 101, 6855. [CrossRef]
- Ruberu, S.R., Fox, M.A. Photochemical Behavior of Stable Free Radicals: The Photochemistry of Perchlorodiphenylmethyl Radical J. Phys. Chem., 1993, 97, 143.
- Fox, M.A., Gaillard, E., Chen, C. Photochemistry of Stable Free Radicals: The Photolysis of Perchlorotriphenylmethyl Radicals. J. Am. Chem. Soc., 1987, 109, 708. [CrossRef]
- Okamura, T., Sancar, A., Heelis, P F., Hirata, Y., Mataga, N. Doublet-Quartet Intersystem Crossing of Flavin Radical in DNA Photolyase. J. Am. Chem. Soc., 1989, 111, 5961. [CrossRef]
- Majima, T., Fukui, M., Ishida, A., Takamuku, S. Stilbene Radical Anions in the Excited Doublet State. J. Phys. Chem., 1996, 100, 8913. [CrossRef]
- Hirata, S., Head-Gordon, M., Szczepanski, J., Vala, M. Time-Dependent Density Functional Study of the Electronic Excited States of Polycyclic Aromatic Hydrocarbon Radical Ions. J. Phys. Chem. A, 2003, 107, 4940. [CrossRef]
- Shkrob, I.A., Sauer, M.C., ALiu, A.D., Crowell, R.A., Trifunac, A.D. Reactions of Photoexcited Aromatic Radical Cations with Polar Solvents. J. Phys. Chem. A, 1998, 102, 4976. [CrossRef]
- Gosztola, D., Niemczyk, M.P., Svec, W., Lukas, A.S., Wasielewski. M.R. Excited Doublet States of Electrochemically Generated Aromatic Imide and Diimide Radical Anions. J. Phys. Chem. A, 2000, 104, 6545. [CrossRef]
- Christensen, J.A., Phelan, B.T., Chaudhuri, S., Acharya, A., Batista, V.S., Wasielewski. M.R. Phenothiazine Radical Cation Excited States as Super-oxidants for Energy Demanding Reactions. J. Am. Chem. Soc. 2018, 140, 5290. [CrossRef]
- Englman, R.; Jortner, J. Energy Gap Law for Radiationless Transitions in Large Molecules. Mol. Phys., 1970, 18, 145. [CrossRef]
- Häupl, M., Lomoth, R., Hammarström, L. Femtosecond Dynamics of the Photoexcited Methyl Viologen Radical Cation. J. Phys. Chem. A, 2003, 107, 435. [CrossRef]
- Turro, N.J., Khudyakov, I.V., Gopidas, K.R. A laser flash photolysis study of magnetic field effects in photoinduced electron transfer between Ru (bpy )32+ and N,N ̛ -dimethylviologen in micellar solution. Chem. Phys., 1992, 162, 131.
- Khudyakov, I.V. Transient free radicals in viscous solvents. Res. Chem. Interm., 2013, 39, 781. [CrossRef]
- Komiha, N., Rosmus, P, Maier, J.P. Low lying quartet states in diacetylene, triacetylene and benzene radical cations. Mol. Phys., 2007, 105, 893. [CrossRef]
- Khudyakov, I.V., Levin, P.P., Kuzmin, V.A. Reversible recombination of radicals. Russ. Chem. Rev., 1980, 49, 1990. [CrossRef]
Figure 1.
Jablonsky diagram for a molecule. Abbreviations of the electronic states of a molecule are presented below the horizontal lines reflecting the position of a state. An analog of the Jablonsky diagram for a radical is presented on the same diagram, with notions of the states placed above the horizontal lines.
Figure 1.
Jablonsky diagram for a molecule. Abbreviations of the electronic states of a molecule are presented below the horizontal lines reflecting the position of a state. An analog of the Jablonsky diagram for a radical is presented on the same diagram, with notions of the states placed above the horizontal lines.
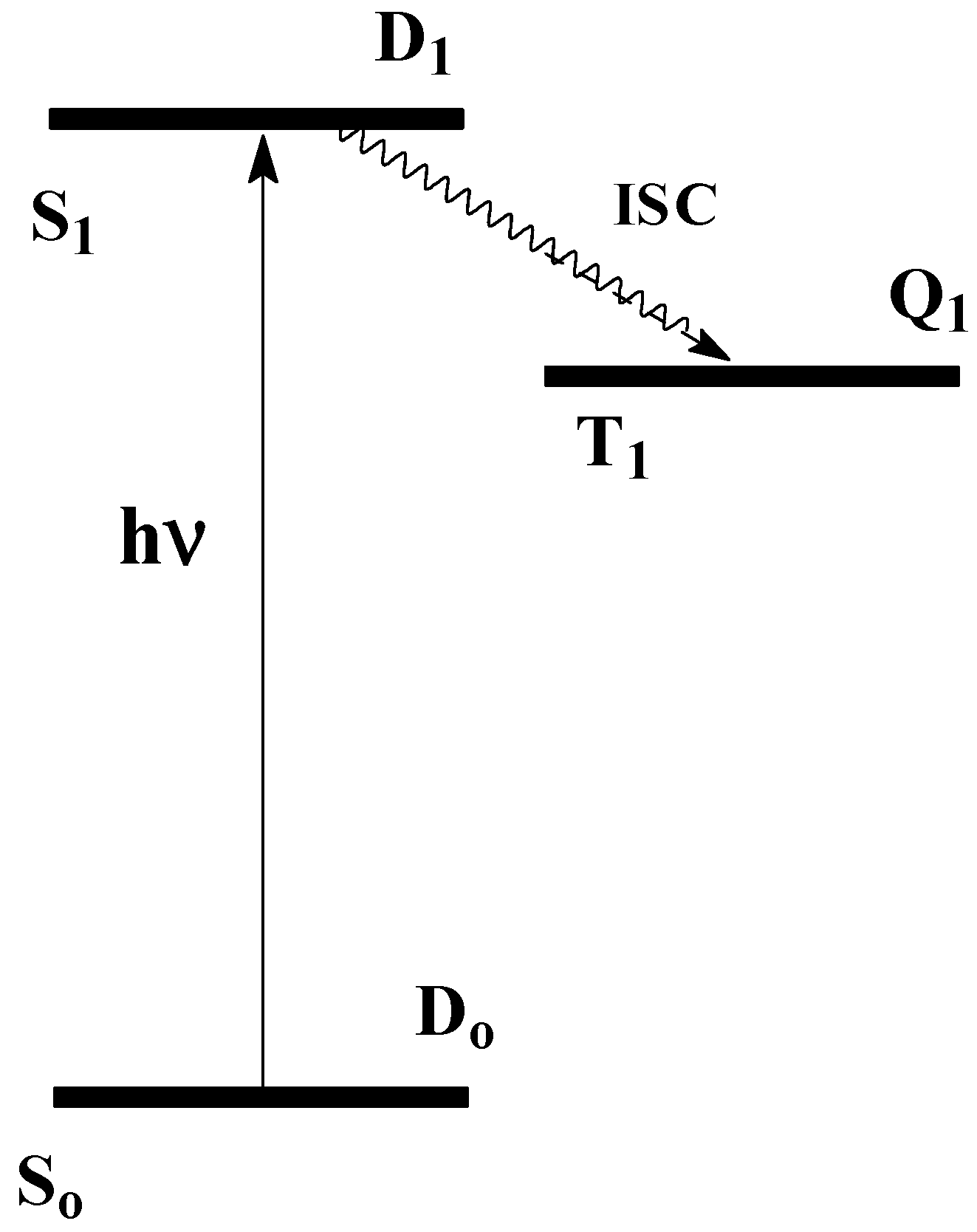
Figure 2.
Possible positions of the excited states of a radical where the Q1-state has a higher energy than the D1-state. IC stands for the internal conversion.
Figure 2.
Possible positions of the excited states of a radical where the Q1-state has a higher energy than the D1-state. IC stands for the internal conversion.
Scheme 1.
Resonance Structures of 4-dehydro-2,3,5,6-tetrafluorophenylnitrene. Hint to its phenylnitrene/phenyl radical (left) and carbene/iminyl radical (right) [8].
Scheme 1.
Resonance Structures of 4-dehydro-2,3,5,6-tetrafluorophenylnitrene. Hint to its phenylnitrene/phenyl radical (left) and carbene/iminyl radical (right) [8].
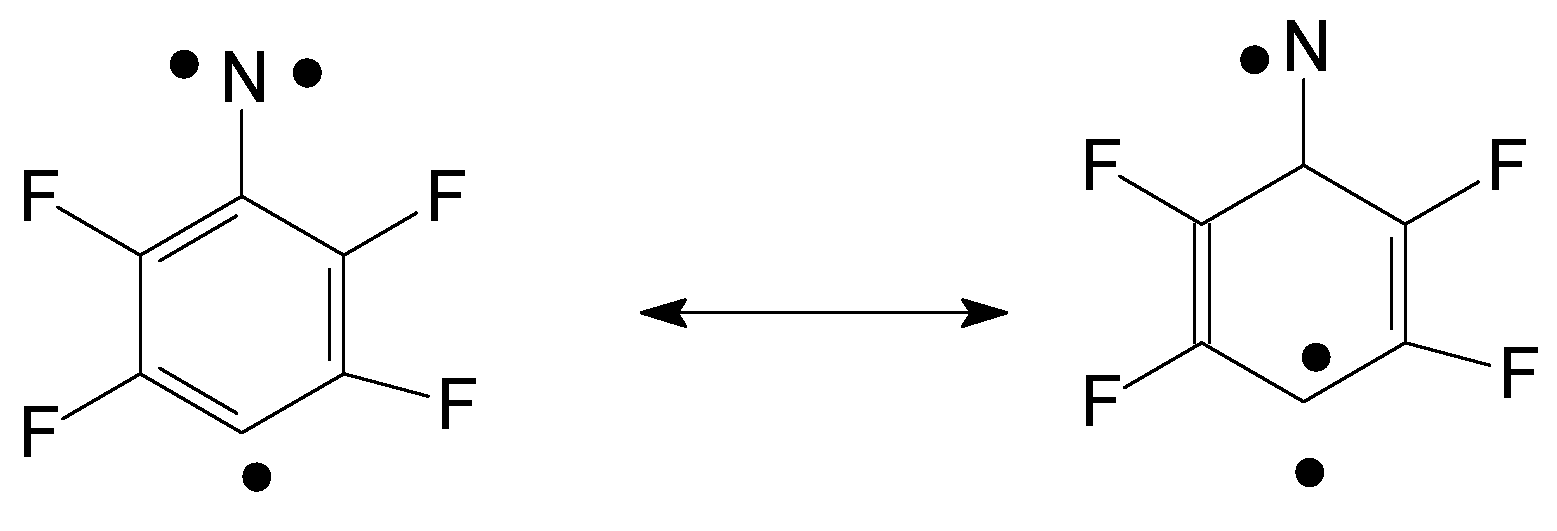
Figure 3.
Transient absorption spectra obtained under ms flash photolysis of stable aroxyl radicals (a) galvinoxyl, (b) indophenoxyl in hexane at room temperature [11]. The structures of the radicals are presented in Scheme 2:.
Scheme 2.
Chemical structures of galvinoxyl (a) and indophenoxyl (b).
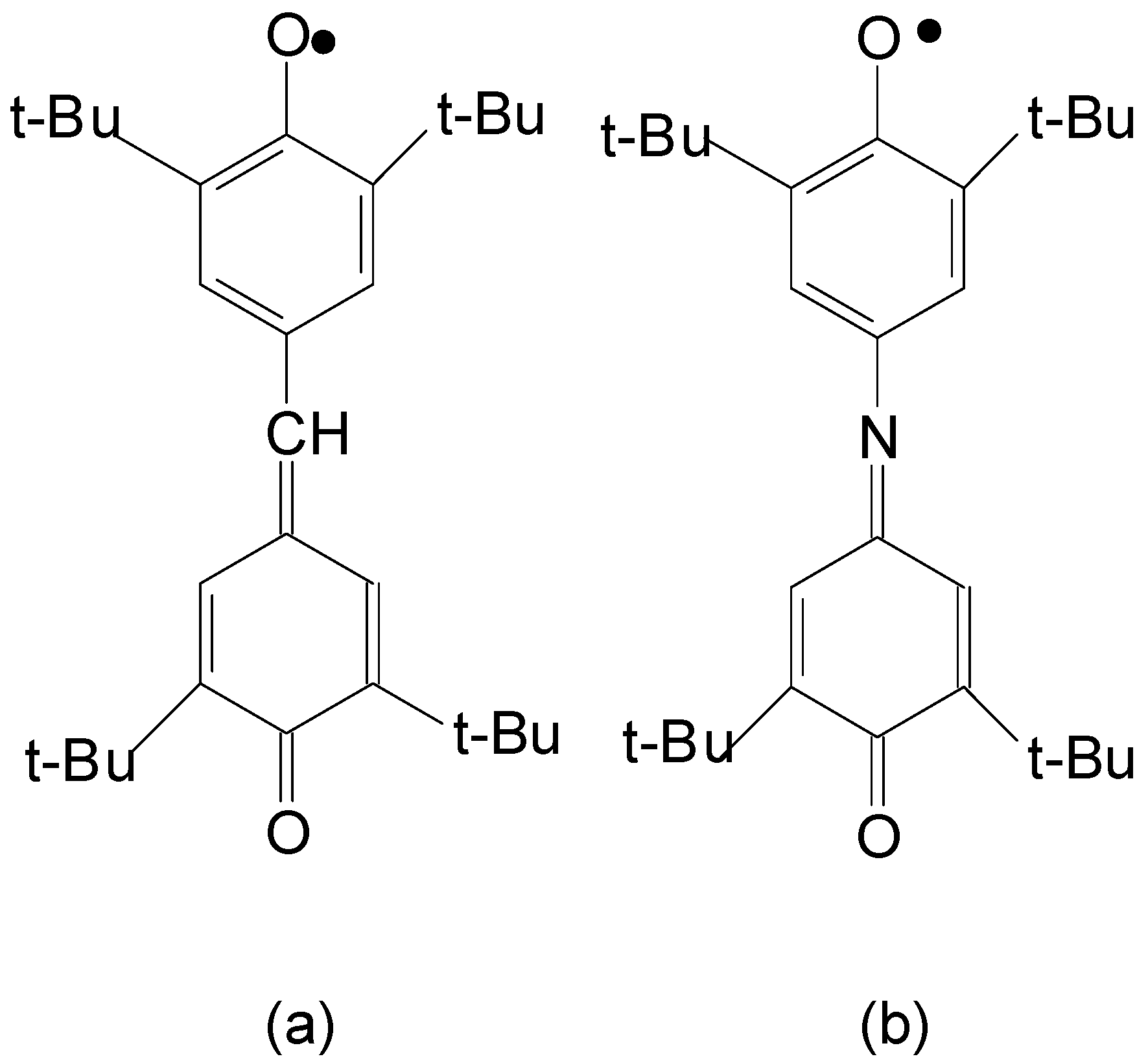

Table 1.
Properties of stilbene radical-anion and radical-cation in the D2 state: Life-time (t), rate constants of internal conversion (kic) and isomerization (ki), quantum yield of isomerization (fi), and excitation energies (E)1 [23].
Table 1.
Properties of stilbene radical-anion and radical-cation in the D2 state: Life-time (t), rate constants of internal conversion (kic) and isomerization (ki), quantum yield of isomerization (fi), and excitation energies (E)1 [23].
| Radical-ion | t, ns | kic x 10-8, s -1 | ki x 10-9, s -1 | fi | E, kJ/mol |
|---|---|---|---|---|---|
| trans-Sb -∙ * | 2.5±1.0 | 4.0±1.6 | 0 | 0 | 202 |
| cis-Sb -∙ * | 1.5±0.4 | 5.6±1.7 | 1.0±0.3 | 0.14±0.05 | 189 |
| trans-Sb +∙ * | 0.24±0.05 | 41±11 | 0 | 0 | 223 |
| cis-Sb +∙ * | 0.12±0.03 | 29±7 | 4.1±1.0 | 0.49±0.12 | 210 |
1In DMF at room temperature. See [23] for additional details.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



