
Submitted:
12 June 2023
Posted:
13 June 2023
You are already at the latest version
Abstract
Breast cancer pathogenesis, treatment, and patient outcomes are shaped by tumor-intrinsic genomic alterations that divide breast tumors into molecular subtypes. These molecular subtypes often dictate viable therapeutic interventions, and ultimately, patient outcomes. However, heterogeneity of therapeutic response may be a result of underlying epigenetic features that may further stratify breast cancer patient outcomes. In this review we examine non-genetic mechanisms that drive functional changes to chromatin in breast cancer to contribute to cell and tumor fitness, and highlight how epigenetic activity may inform therapeutic response. We conclude by providing perspectives on the future of therapeutic targeting of epigenetic enzymes, an approach which holds untapped potential to improve breast cancer patient outcomes.
Keywords:
breast cancer
; epigenetics
; HDAC
; HAT
; histone methylation
; histone acetylation
; chromatin modification
Introduction
A high degree of breast cancer heterogeneity had led to its classification into four distinct molecular subtypes, which can vary in tumor genomics and the cell type from which tumor initiation occurs: (1) luminal A, (2) luminal B, (3) human epidermal growth factor receptor 2 (HER2)-positive, and (4) triple negative breast cancer (TNBC). Tumor heterogeneity informs patient prognosis and clinical disease management. At diagnosis, a combination of immunohistochemistry (IHC) and genomic assays define critical gene expression of three cell surface receptors: estrogen receptor (ER), progesterone receptor (PR), and HER2. Collectively, ER, PR, and HER2 expression enable breast cancer classification into one of the four established molecular subtypes, which forms the basis of clinical disease management. Approximately 60-70% of all diagnosed breast cancers fall under the luminal A subtype, which is defined as ER-positive and/or PR-positive, HER2-negative, with low proliferative capacity by Ki67 IHC[1]. These tumors often respond to hormone therapies including tamoxifen or aromatase inhibitors (AI), resulting in a favorable patient prognosis[2,3]. In contrast with luminal A, luminal B breast cancers are more aggressive with a high proliferative index via Ki67 IHC. Luminal B breast cancers represent about 10% of breast cancers; these tumors are ER-positive and exhibit either PR-positive or PR-negative staining[2]. Approximately 30% of diagnosed luminal B cancers are also HER2-positive[4]. Patients with luminal B breast cancer do not exhibit as high a response rate to hormone therapy as luminal A breast cancers, and some patients benefit from hormone therapy in conjunction with chemotherapy[5]. About 10-15% of diagnosed breast cancers are characterized by high HER2 expression in the absence of ER or PR expression[3]. HER2-positive patient prognosis has dramatically improved in the past 30 years with the development and widespread clinical application of HER2-targeted therapies, including small molecule inhibitors (e.g., lapatinib) and antibody therapies (e.g., trastuzumab) in combination with chemotherapies such as doxorubicin[6]. HER2-targeted antibody drug conjugates such as trastuzumab-emtansine (T-DM1) or trastuzumab-deruxtecan (T-Dxd) are also used clinically for HER2-positive breast cancers, as well as advanced breast cancers characterized by low HER2 levels[7,8]. Lastly, TNBCs account for 10-20% of diagnosed breast cancers and typically originate from basal cell lineage in the breast and are not characterized by ER, PR, or HER2 expression[9]. Instead, TNBC tumor genomics vary, resulting in proposed subclassifications within the TNBC subtype and can be characterized by loss of function mutations in tumor suppressors such as BRCA1/2 or TP53[10]. The heterogeneity of TNBCs and its aggressive nature render this tumor type more challenging to clinically manage and patient prognosis is usually poor.
In addition to ER, PR, and HER2 expression that stratify breast cancers into the above molecular subtypes, additional genomic alterations may be present that contribute to disease etiology, therapeutic response, and disease progression. This involves genomic alterations, including mutations and indels in BRCA1/2, inactivating mutations or deletions in tumor suppressor genes PTEN, CDH1, or TP53, and amplification or activating mutations in critical growth regulators such as PIK3CA[11–14]. While the importance of genomic alterations in breast cancer clinical management cannot be understated, in recent years it has become clear that the epigenetic landscape provides an additional and dynamic layer of insight into the dysregulation of cellular processes in breast and other cancers. In some cases, tumor genomics and epigenetics are linked; genomic alterations have been defined to modulate the activity or function of chromatin modifying enzymes, in turn play a role within epigenomic regulation[12,15]. Posttranslational epigenetic modifications that occur on histones and on DNA can alter chromatin accessibility by modifying histone-DNA interactions, recruiting transcriptional machinery to DNA, or interacting with transcription factors[16]. Delineating the specific mechanisms by which these modifications fine-tune cellular processes in normal conditions and in cancer first requires examining their environment in the structure of chromatin.
The size of the human genome in relation to the volume of the nucleus presents a need to organize the genome into a more condensed form. Nucleosome hetero-octamers containing two each of H2A, H2B, H3, and H4 organize DNA through compaction[17,18]. The nucleosome is incorporated into chromatin through the winding of DNA such that 145-147 base pairs of DNA are wrapped around each nucleosome. Histone proteins contain a globular core domain and unstructured N- and C-terminal tail domains which extend out of the globular domain on either side[17,19]. The nucleosome is assembled through interactions within the globular domains of the heterodimers, and together with nucleosome-incorporated DNA, the histone globular and tail domains support post-translational modification (PTM), which can inform transcriptional competence[20]. Covalent histone PTMs are orchestrated through the dynamic activity of chromatin modifying enzymes: writers that introduce modifications, readers that identify and bind to PTMs, and erasers that remove the modifications[21]. Histones harbor more than 20 types of unique epigenetic PTMs, including methylation, acetylation, phosphorylation, SUMOylation, ADP ribosylation, and numerous others[22,23]. Collectively, the specific combination of histone PTMs can impact chromatin accessibility, thereby regulating transcription and gene expression[24]. Beyond transcriptional regulation, the dynamic histone PTM landscape informs diverse cellular processes ranging from DNA repair to cellular differentiation[25]. The essential contributions of the epigenome and its dynamic regulation support cancer development while contributing to therapeutic response and clinical patient outcome, leading to the recognition of epigenetic reprogramming as an emerging, distinct hallmark of cancer[26].
Here, we examine the contributions of histone PTMs and the associated epigenetic modifiers that read, write, and erase these PTMs to the breast cancer landscape. We focus on preclinical mechanistic studies demonstrating the role(s) of epigenetic modifiers in essential biological processes that inform cancer initiation, development, and therapeutic response. We conclude with examining how these mechanistic studies could potentially support the clinical management of breast cancer.
Histone modifying complexes
COMPASS complex perturbation and associated histone methylation in breast cancer
The activity of multiprotein H3K4 methyltransferase-specific COMPASS (complex of proteins associated with Set1) complexes are dysregulated in breast cancers. A total of six Set1/MLL methyltransferases (Set1A-B, MLL1-4) recruit unique and shared subunits to support COMPASS complex substrate specificity across genome-wide mono-, di-, and tri-methylation of H3K4[27]. H3K4 methylation is typically associated with transcriptional activation. In HR+, PIK3CA mutant breast cancer, clinical PI3K inhibition hyperactivates ER signaling. Mechanistic studies have found that the AGC kinase AKT phosphorylates the MLL4/KMT2D methyltransferase, reducing its enzymatic activity; PI3K inhibition enhances MLL4 activity, increasing enhancer H3K4me1 and promoting an open chromatin state[28]. Functionally open chromatin supports the recruitment of the FOXA1 and PBX1 pioneer transcription factors to the genome to support ESR1 binding and subsequent transcription of ER-responsive genes. Other AGC kinases, including SGK1, mediate MLL4/KMT2D phosphorylation[29], suggesting that a wider network of growth factor-associated signal transduction pathways may integrate cellular cues with chromatin modifying enzyme function to regulate gene expression. The importance of maintaining appropriate H3K4 methylation is highlighted by reports that demonstrate the H3K4me2/3 demethylase KDM5A is also an AKT effector; AKT-mediated KDM5A phosphorylation redistributes KDM5A to the cytoplasm where it is unable to demethylate promoter H3K4me3[30]. Acute loss of promoter H3K4me3 occurs at cell cycle regulated genes and is correlated with a poor patient outcome in advanced stage breast cancers across molecular subtypes including HR+ breast cancers[30]. Targeting the MLL1 and/or the MLL4/KMT2D COMPASS complexes to reduce promoter or enhancer H3K4 methylation, respectively, impairs HR+ breast cancer cell and tumor growth[28,31].
Dysregulation of SWI/SNF in advanced stage breast cancers
The SWItch-mating type/Sucrose Non-Fermenting (SWI/SNF) family of ATP-dependent chromatin remodeling complexes regulate gene expression by supporting DNA accessibility, affecting cell proliferation, differentiation, and the cellular response to DNA damage[32]. The ATPases BRG1 (SMARCA4) and BRM1 are common to all SWI/SNF complexes (BAF, P-BAF, and ncBAF), with unique cofactors and binding partners assembling to form each of the three unique complexes. Genomic alterations in the SWI/SNF complexes are relatively rare in primary breast cancers. However, genomic alterations in the BAF complex tumor suppressor and DNA binding protein ARID1A occur in 12% of metastatic breast cancers, and its deletion or functional inactivation is associated with poor patient prognosis in metastatic luminal A and HER2-positive breast cancers, including endocrine therapy resistant, ER-positive breast cancers[33]. Recent studies using genome-wide CRISPR screens demonstrate that genes encoding the BAF complex—including ARID1A—mediate therapeutic response to ER antagonists; loss of ARID1A enhances proliferative capacity, rendering ER+ breast cancer cell lines resistant to both tamoxifen and fulvestrant[34]. ARID1A and BRG1 physically associate with ER. This interaction supports ARID1A repression of ER-dependent transcription by binding chromatin at ER-regulated enhancers using a FOXA1-dependent mechanism[34]. Complementary studies suggest that ARID1A expression supports luminal breast lineage maintenance. ARID1A loss impairs SWI/SNF recruitment to regions of the genome that support luminal lineage commitment, thereby mediating a transition to basal-like breast cancer cells that are ER-independent[35]. These studies suggest mechanisms of resistance to ER antagonists, and also additional therapeutic strategies to treat advanced ER-positive breast cancers characterized by genomic alterations in the BAF complex. Outside of ER-positive breast cancers, the SWI/SNF complexes may also be linked to other breast cancer subtypes. BRG1 increases cell proliferation via activation of cell cycle dependent genes in TNBC cell lines[36] and genetic loss of BRG1 increases HDAC1 binding to the genome[36]. The transcription factor SOX4 and BRG1 cooperate to positively regulate PI3K/AKT signaling via transcription of TGFBR2[37]; because TNBCs are characterized by high BRG1 levels[38], these reports suggest a patient cohort whose tumors may be sensitive to BRG1 and/or PI3K inhibition.
Histone Acetyltransferases
Histone acetylation is a reversible and dynamic PTM catalyzed by histone acetyltransferases (HATs) and removed by histone deacetylases (HDACs)[39]. HATs catalyze the covalent transfer of the acetyl group from acetyl-CoA to lysine groups in proteins including histones (Figure 1). While the activity of many HATs was originally described using histones as substrates, HATs have since been shown to acetylate a number of non-histone targets including p53, YY1, and STAT3[40–42]. Human HAT enzymes are divided into three major categories based on their sequence homology and mechanism of catalysis: the GNAT, MYST, and p300/CBP families (Figure 1)[43]. Functionally, HAT enzyme-mediated histone tail acetylation neutralizes the attraction between nucleosome-incorporated histone proteins and DNA, supporting a euchromatin state and contributing to enhanced transcription[44,45]. A diverse array of acetylation events have been defined on histone H3 and H4 (including but not limited to H3K4[46,47], H3K9[48], H3K18[49], H3K23[50], H3K27[51–53], H3K56[54], H3K122[55], H4K5[56], H4K8[57], H4K12[57], and H4K16[57])[58], but their biological functions have not been well-characterized. For example, H3K27 is widely-recognized as a marker for active enhancers[59], but its specific mechanistic role in transcription is not completely understood. One method to contextualize these marks within the grander scheme of breast cancer is to examine the enzymes that write or erase them and their catalytic functions within oncogenic mechanisms.
Histone acetylation in breast cancer pathogenesis
While histone acetylation is critical for normal cellular maintenance, it is also subject to dysregulation in human cancers. Aberrant histone acetylation has been closely associated with cancer progression and therapeutic response[60–63]. This is highlighted by a clinical study examining a panel of histone acetylation marks in 880 human breast cancer carcinomas. While hypoacetylation of H4K16 was observed in 78.9% breast cancer cases, its high-level expression was correlated with longer disease-free survival (DFS), thus leading authors to postulate that loss of H3K16ac is an early event in breast cancer invasion pathogenesis[60]. The presence of these acetylation marks is also associated with different subtype categories. The overall detection of high levels of global histone acetylation was significantly associated with luminal-like breast tumors, whereas moderate to low global lysine acetylation was linked to basal carcinoma and HER2-positive tumors[60]. More specifically, an evaluation of several histone PTMs including acetylation marks from patient tumors suggested that H3K9 acetylation is associated with TNBC and HER2-positive breast tumors, whereas H3K27me3 correlates with the luminal A and B molecular subtypes[64]. Furthermore, acetylation via HAT activity has been shown to impact tumor phenotypes directly. MYST3 binds to the proximal promoter region of the ESR1 gene, increasing ERα expression in a HAT-dependent manner; mutations to the HAT domain of MYST3 fail to upregulate ERα expression to the same extent as wild-type MYST3[65]. Knockdown of MYST3 in HR-positive breast cancer xenografts leads to significant tumor regression and increased progression free survival, corroborating in vitro studies that found that the genetic ablation of MYST3 significantly reduces cell growth in HR+-positive breast cancer cell lines[65]. These data suggest that further exploration of MYST3-targeting for the treatment of MYST3-high ER-positive/HER2-negative breast cancers is warranted and may benefit endocrine therapy-resistant patients. While HATs catalyse the addition of acetyl groups to histone residues, the overall maintenance of histone acetylation consists of a dynamic interplay between HATs and HDACs, both of which can be dysregulated in breast and other cancers.
HATs promote the transcription of EMT-specific markers in breast cancer
HATs have been documented to function in the epithelial-to-mesenchymal transition (EMT). While EMT occurs normally during embryonic development, it has been implicated in the acquisition of stem-cell-like qualities that accentuate tumor growth and metastasis in cancer[66]. In one study, DOT1L, a methyltransferase targeting the active transcription mark H3K79[67,68], was shown to enhance the expression of EMT transcription factors in a p300-dependent manner in noncancerous epithelial breast cells. Specifically, the HAT p300 is recruited by DOT1L to the promoters of SNAI1, ZEB1, and ZEB2, mesenchymal regulator genes, as part of a transcriptionally active complex containing DOT1L, c-MYC, and p300[69]. This is concurrent with H3 acetylation enrichment at the SNAI1, ZEB1, and ZEB2 promoters, alongside H3K79 methylation enrichment[69]. Additionally, in the TNBC MDA-MB-231 cells, pharmacologic HDAC inhibition with trichostatin A induced re-expression of Snail, ZEB1, and ZEB2 transcription factors in DOT1L-knockdown cells, reinforcing the EMT and cancer stem cell-promoting function of p300 in the presence of DOT1L[69]. This study highlights how histone acetylation, together with cross-talk with histone methylation marks, can dictate tumor differentiation and subsequent aggressiveness. Separately, elevated activity of the GNAT family HAT GCN5 supports EMT by inducing expression of Snail and Slug upon TGF-β1 treatment in TNBC cell lines including MDA-MB-231[70]. Functionally, this results in enhanced cell migration and invasion, which could facilitate cancer metastasis. Notably, genetic knockdown of GCN5 reversed the metastatic phenotypes and prevented EMT[70]. Thus, the epigenome and its HAT-mediated regulation may contribute to phenotypes that support breast cancer metastasis (Figure 1).
HAT-mediated regulation of the DNA damage response
The HAT p300/CREB-binding protein-associated factor (PCAF) acetylates H4K8 in BRCA-deficient TNBC, thereby promoting replication fork degradation[71]. This study suggests that PARP inhibitor resistance in BRCA-deficient breast cancer may be due in part to low PCAF levels; PCAF levels could serve as an indicator to assess for therapeutic response to PARP inhibition in BRCA-deficient breast and other cancers. These data simultaneously reinforce other studies that identified drug synergy via combined use of HDAC and PARP inhibitors in various cancer types[72,73,74,75], and predict that this combination therapy may increase fork degradation and subsequent cell death in BRCA-deficient cancers[71]. Similarly, Tip60, a HAT in the MYST family, regulates homologous recombination-directed DNA repair in both normal and tumor mouse mammary epithelial cells (MEC)[76]; genetic loss of Tip60 elevated MEC γH2AX[76], a marker for DNA damage. Surprisingly, Tip60-silenced MEC tumor cells were also markedly more resistant to the DNA-damaging agent cisplatin despite an increase in unrepaired DNA compared to vehicle-treated cells[76], suggesting a dual role of Tip60 in both DNA damage repair and DNA damage-induced cell death. As such, histone acetyltransferase inhibitors are in preclinical development, with early studies suggesting activity in preclinical breast cancer models. The Tip60 inhibitor TH1834 induced apoptosis by generating non-repairable DNA damage and elevated H4K8ac[77] in HR-positive MCF7 breast cancer cells and slow xenograft growth in vivo [78]. Another Tip60 inhibitor, garcinol, inhibits estradiol-induced cell proliferation by inducing G0/G1 cell cycle arrest and increasing apoptosis in MCF7 cells[79]. Collectively, the development and preclinical testing of HAT inhibitors that target specific HATs in human cancers is in its infancy. To date, HAT inhibition has not entered clinical trials, at least in part because HATs serve both oncogenic and tumor-suppressive roles within the cell. Comparatively, modulating histone acetylation through the inhibition of HDAC activity in breast and other cancers has gained more preclinical and clinical traction.
Histone Deacetylases
HDACs oppose HAT activity, catalyzing the removal of acetyl groups from the ε-amino group of a lysine residue, thereby restoring its attraction to DNA (Figure 1)[80]. 18 HDACs are encoded in the human genome, divided into four classes based on protein homology with budding yeast. Class I HDACs (HDACs 1, 2, 3, and 8) are homologous to the reduced potassium dependency 3 (Rpd3) protein in S. cerevisiae[81]. HDAC8 is the only class I HDAC that has been shown to function as a monomer; other class I HDACs are recruited to larger complexes including CoREST, MiDAC, NuRD, Sin3A (HDACs 1/2) and SMRT/NCoR (HDAC3)[82,83]. Class II HDACs are genetically similar to the yeast Hda1 protein[84], stratified into class IIa (HDACs 4, 5, 7, and 9) and class IIb (HDACs 6 and 10) based on domain composition[81]. HDAC11 is the sole member of the class IV HDAC family and is homologous to budding yeast Hos3.1[81]. Class III HDACs (SIRT1, SIRT2, SIRT3, SIRT4, SIRT5, SIRT6, SIRT7) are homologous to Sir2 and are NAD-dependent enzymes, differing from Class I, II, and IV HDACs, which all perform zinc-dependent catalysis[81]. HDACs are enzymes that can be found in numerous cellular contexts and compartments; further defining their mechanistic roles in cancer may support future studies to target their oncogenic activity or exploit their potential prognostic value. In general, HDACs are overexpressed in breast and other cancers[85,86,87,88,89,90]. The pro-oncogenic activities of HDACs along with structural and activity features that render HDACs viable therapeutic targets provide sufficient rationale to develop and utilize HDAC inhibition for the clinical management of human cancers.
HDACs as prognostic factors in breast cancer patients
While HDAC overexpression is common in some cancer types, the molecular mechanism(s) by which HDACs contribute to tumorigenesis is not yet fully understood. High HDAC1 expression is positively correlated with hormone receptor status in patient tumor samples[91,92,93]. HDAC1 and HDAC2 expression is significantly correlated with HER2 expression[92,93], and HDAC2 expression is associated with the presence of nodal metastasis[92]. Despite detectable HDAC overexpression in some breast cancer subtypes, the ability of HDAC overexpression to predict overall survival (OS) or disease-free survival (DFS) is less clear; conflicting results were published indicating potential prognostic value of HDAC6[93,94] and HDAC2[92,93,95] expression. These discrepancies in the potential prognostic value of HDAC expression could, in part, be due to differences in race and ethnicity in participating patient populations, therapeutic regimens, post-operative treatment protocols, or tumor staging[93]. However, these reports also reinforce the notion that HDACs have diverse, tissue-specific roles that vary between the HDAC classes described above, and those specific functions, rather than an overall characterization of HDACs, could guide clinical approaches in the therapeutic targeting of HDACs. Therefore, it is critical to consider HDAC functions with respect to the specific tumorigenic contexts they function within, which may vary widely by breast cancer subtype, tumor staging, or prior therapies.
HDACs support the epithelial-to-mesenchymal transition (EMT) in breast cancer
The Nucleosome Remodeling and Deacetylation (NuRD) complex is a repressor complex that reduces DNA accessibility within chromatin[96]. The complex contains class I HDAC1 and HDAC2, an ATP-dependent chromatin remodeling protein (CHD3/4/5), methyl-CpG-binding domain protein (MBD2/3), nuclear zinc finger protein (Gata2a/2b), SANT and GATA DNA-binding domain (MTA1/2/3), as well as additional scaffolding proteins (Rbbp4, Rbbp7)[96]. Prior studies identified NuRD interacting with the EMT-related transcription factor ZEB1 to contribute to non-small cell lung cancer metastasis[97]. In TNBC, the transcription factor RUNX2 has been shown to recruit NuRD to repress expression of tumor suppressor genes, thereby promoting EMT. RUNX2 binds with the NuRD complex member MTA1, and either RUNX2 or MTA1 overexpression augments fibronectin, N-cadherin, and vimentin mesenchymal markers while depleting E-cadherin, α-catenin, and γ-catenin epithelial markers[98]. The HDAC class I/II inhibitor vorinostat upregulates E-cadherin, enhancing differentiation in TNBC cells[99]. The authors found that the use of either class I and II HDAC-targeting inhibitor vorinostat or entinostat decreases protein expression of TCF4, a major Wnt signaling effector. In this context, HDAC inhibition reprograms the epigenome to repress Wnt signaling[99], thereby reducing EMT[100,101,102]. These data suggests that HDAC inhibition can support cancer cell differentiation, reducing tumorigenesis while improving treatment efficacy. In a murine mammary carcinoma model, HDAC8 siRNA knockdown reverses the upregulation of EMT-driver genes VIM, CDH2, WNT5A, and ZEB1 in surviving cells following treatment with cisplatin and/or paclitaxel[103]. Conversely, HDAC8 knockdown rescued the chemotherapy-induced downregulation of genes associated with mesenchymal to epithelial transition (e.g., ELF, GATA3, RORA, GRHL2) that support the epithelial phenotype by reversing EMT[103]. These same genes exhibited a significant decrease of promoter-proximal H3K27ac occupancy following chemotherapy[103], suggesting HDAC8-mediated EMT antagonizes chemotherapy efficacy. HDAC inhibition via entinostat hinders EMT, reducing the expression of the EMT transcription factors twist and snail, as well as N-cadherin[104]. Chromatin immunoprecipitation (ChIP) demonstrates that twist and snail binding to the E-cadherin-encoding CDH1 promoter decreases with entinostat treatment, leading to an increase in CDH1 mRNA expression[104]. This coincides with earlier studies that have found that snail represses CDH1 by recruiting the Sin3A-HDAC1/2 complex to the CDH1 promoter[105]. Cytokeratin 8/18, filaments lost during EMT[106], were upregulated via entinostat treatment[104]. Numerous HDACs have been implicated in the EMT process, thus inhibiting HDACs that regulate gene expression associated with EMT may enhance the anti-tumorigenic effects of other therapies including DNA damaging agents commonly used in breast cancer treatment.
HDACs modulate ER expression and signaling
The NuRD complex may also repress ER expression in breast cancer. In advanced stage TNBC, the NuRD complex interacts with the ESR1 promoter, which reduces promoter H3K27ac via an HDAC1-dependent mechanism, reducing ERα expression[107]. Further, when NuRD is suppressed by knocking down the NuRD-recruiting protein MUC1, an increase in ESR1 promoter H3K27ac is observed, along with induction of ERα expression at both mRNA and protein levels[107]. Complementary studies in melanoma found that the NuRD complex is recruited to the twist-binding chromatin region of the ESR1 gene by twist to decrease H3K9ac and increase H3K9me1 in melanoma, consistent with the repressive role of NuRD in ERα expression.[108] These results suggest HDACs are involved in epigenetic changes that enable the transition of luminal breast cancer into more aggressive basal breast cancer. Pharmacologically targeting HDACs that contribute to NuRD complex function may reverse the de-differentiation of TNBC and enhance ER expression, which could potentially define new patient populations who may benefit from hormone therapy.
NuRD complex constituents also play a role in the downstream ER pathway. Within the normal estrogen pathway, estrogen binds to estrogen-responsive elements (EREs) to transactivate downstream genes[109]. The NuRD complex member MTA1 suppresses ERE-mediated transcription by recruiting HDACs and interacting with the activation domain of ERα in HR-positive breast cancer[110]. Class I HDAC inhibition via trichostatin A mediates a reversal of the inhibition of ERE transcription by MTA1[110]. HDAC2 localizes with MTA1 to the PS2 promoter, which is an ER-responsive gene and a tumor suppressor[110,111]. Other studies in HR-positive breast cancer demonstrate that HDAC1 and HDAC3 are recruited to the PS2 and c-MYC promoters in a tamoxifen-dependent manner as part of chromatin-remodeling complexes: HDAC3-containing NCoR and HDAC1-containing NuRD[112]. The introduction of tamoxifen reverses estrogen-induced H3 and H4 acetylation, leading to global H3 hypoacetylation and an increase in euchromatin, as well as a reduction in RNA pol II occupancy at PS2 and c-MYC genes[112]. The ER antagonist tamoxifen is one of the most common first-line treatment drugs for patients with ER-positive breast cancer[113,114]. However, tamoxifen resistance occurs in 50% of these tumors[114]. Targeting HDACs may reduce tamoxifen resistance, which could restore hormone therapy benefit in some patients with HR-positive breast cancers.
HDACs as a therapeutic target to overcome treatment resistance
Tamoxifen resistance is also caused by upregulation of the embryonic transcription factor SOX9, which localizes to the nucleus in ER-positive breast cancer to support tamoxifen resistance[115]. HDAC5-mediated SOX9 deacetylation induces SOX9 nuclear localization in HR-positive breast cancer, and HDAC5 overexpression promotes cell growth under tamoxifen exposure[116]. Proto-oncogene c-MYC encodes c-MYC, which regulates HDAC5 gene expression[116]. In turn, c-MYC also maintains the nuclear localization of SOX9 in an HDAC5-dependent manner; HDAC5 rescues a reduced growth rate in tamoxifen-resistant, MCF7 cells that have been treated with shRNAs targeting c-MYC[116].
Radiotherapy remains a treatment mainstay for many breast cancer patients. However, resistance occurs from radiation most commonly due to intratumoral heterogeneity or activation of downstream signaling pathways[117], including ERK1/2 and PI3K/AKT[118]. Several studies suggest that HDACs may also contribute radioresistance in other cancers[119,120,121,122]. HDAC inhibition in breast cancer may enhance radiotherapy efficacy, improving patient outcome. To investigate the mechanisms of radioresistance, radioresistant HR-positive and TNBC cell lines have been engineered and are characterized by reduced H3K9ac and H3K27ac and an increase in HDAC activity compared to radiosensitive parental cell lines[123]. In radioresistant HR-positive breast cancer cell lines, the loss of H3K9 and H3K27 acetylation occurred independently of the cell cycle, suggesting that these events are connected to the acquisition of radio-resistance[123]. Treatment with the class I HDAC inhibitor valproic acid increases H3K9ac and H3K56ac while also increasing γH2AX retention, signaling sustained DNA damage, in both acquired and intrinsically radio-resistant cells[123]. These studies suggest that HDAC inhibition may sensitize some breast cancer subtypes to DNA damaging agents including radiotherapy, although a more thorough investigation is necessary to determine the broader implications of these studies.
Clinical HDAC inhibition
To date, four HDAC inhibitors have achieved US FDA-approval to treat cancer (Table 1). In 2006, the class I/II HDAC inhibitor vorinostat was approved to treat advanced cutaneous T-cell lymphoma (CTCL), which was followed by the 2009 approval of romidepsin for the same indication[124,125]. Romidepsin achieved accelerated approval as a class I HDAC inhibitor for peripheral T-cell lymphoma (PTCL) initially in 2011; however, its PTCL approval was withdrawn in 2021 due to its failure to reach the endpoint of PFS in combination with the chemotherapy CHOP[126]. The pan HDAC inhibitor belinostat was FDA-approved for the treatment of PTCL in 2014[127], and the pan HDAC inhibitor panobinostat gained accelerated approval in 2015 to treat multiple myeloma in combination with dexamethasone and the proteosome inhibitor bortezomib[128]. Currently, the clinical utility of HDAC inhibition is restricted to the hematological malignancies described above. For solid tumors including breast cancer, while HDAC inhibition is under limited clinical investigation, including four open U.S. clinical trials (Table 1). Given the importance of HDACs and histone acetylation in breast cancer development and the metastatic cascade, rationale remains for the continued exploration of clinical HDAC inhibition as part of a combination treatment regimen.
Currently, preclinical mechanistic data support the utility of HDAC inhibition in multiple breast cancer subtypes via unique mechanisms described above and elsewhere, HDAC inhibition is not a currently approved therapeutic modality for the treatment of breast cancer. Preclinical data demonstrated that the class I and II HDAC inhibitor entinostat significantly reduces the growth of letrozole-resistant tumors in the presence of the aromatase inhibitor (AI) letrozole or exemestane, overcoming AI resistance[129]. These data led to a US multi-center phase III clinical trial in patients with advanced HR+ breast cancer, but the trial did not meet the endpoint in AI-resistant HR-positive/HER2-negative breast cancer when treated with exemestane with entinostat[130]. Patients treated with entinostat with exemestane showed a median overall survival of 23.4 months (95% CI, 21.2 to 25.6) compared to 21.7 months (95% CI, 19.3 to 27.1) for patients treated with a placebo with exemestane (p = 0.94)[130]. Similarly, median progression free survival (PFS) was 3.3 months (95% CI, 3.1 to 5.3) in patients treated with entinostat with exemestane and 3.1 months (95% CI, 3.0 to 3.3) in the placebo/exemestane group (p = 0.30)[130]. This contrasts a similar international study based in China that examined the use of the class I/II HDAC inhibitor tucidinostat with exemestane in the treatment of advanced, HR-positive breast cancer. Patients who received tucidinostat with exemestane experienced 9.2 months of PFS compared to the placebo group of 3.8 months (p = 0.024)[131]. This study was limited to a single racial patient cohort, and quality of life metrics were not reported. Furthermore, patients in this study were less likely to have received prior endocrine therapy with advanced disease, which all could account for differences in efficacy between these two reported HDAC inhibitor trials[130,131]. Given the importance of HDACs in mediating multiple cancer hallmarks and the promising results of the above tucindostat trial, preclinical and clinical HDAC inhibition should continue to be explored. Moving forward, rational combination therapies may enhance the on-target activity of HDAC inhibition.
Perspectives on HDAC inhibition in breast cancer
As descbied above and elsewhere, class I HDACs including HDAC1/2/3 participate in repressive multi-unit complexes that have been shown to decrease ER expression and estrogen signaling, such as reducing pS2 and c-MYC expression. Moving forward, it could be valuable to investigate the action of class I-specific HDAC inhibitors for the treatment of advanced stage breast cancer. Because of the role of class I HDACs in repressing ER expression and promoting EMT, there may also be value in the preclinical investigation of sequential class I HDAC inhibition followed by aromatase inhibition or SERM treatment in TNBC or other HR- breast cancers. This approach may support tumor de-differentiation of TNBC into an ER-dependent tumor, especially in advanced-stage tumors that rely on de-differentiation in order to invade other cell types[132]. These proposed studies could expand on the clinical trial that utilized entinostat with exemestane, which focused only on overcoming aromatase inhibitor resistance in HR+ cancers. Because HDAC5 has been shown to mediate tamoxifen resistance in HR-positive breast cancer[116], it may worth developing and testing class II HDAC inhibitors for overcoming resistance in advanced stage HR-positive breast cancer. Moreover, the class I/II HDAC inhibitors vorinostat and entinostat demonstrate preclinical efficacy in repressing EMT in TNBC cell culture models; testing vorinostat or entinostat in in vivo models of TNBC in combination with chemotherapy may demonstrate greater treatment efficacy, as it has been shown in other cancers that chemo-sensitivity significantly increases with tumor differentiation[133,134]. Lastly, the relationship and functional overlap between epigenetic and genetic mechanisms of tumor development and metastasis should be considered for maximizing clinical benefit for breast cancer patients. Inhibitors such as the dual PI3K/HDAC inhibitor fimepinostat may overcome PI3K inhibitor resistance, given the established antagonism between PI3K and ER signaling. PI3K inhibition is associated with a compensatory increase in ER-dependent transcription[29,135] and HDAC-inhibition has been shown to decrease ER-dependent transcription[110].
Conclusion and Perspectives
Despite the categorization of existing breast cancers into the luminal A, luminal B, HER2, and triple negative subtypes, intratumoral epigenetic heterogeneity and epigenetic plasticity can impact breast cancer aggressiveness and response to existing therapeutics. Targeting the enzymes and enzyme complexes responsible for these epigenetic changes may reduce breast cancer growth and proliferation. This epigenetics-forward approach may address some of the mechanisms of resistance to standard-of-care therapies commonly employed for breast cancer clinical management, and identify mechanisms to promote tumor differentiation which can open avenues for enhanced efficacy in later-line treatments. As more information is obtained about how the cellular roles of histone methyltransferases, HATs, HDACs, and chromatin-remodeling complexes are perturbed in cancer initiation, development, and therapeutic response, it remains critical to investigate their therapeutic targeting to advance current personalized treatment strategies in breast cancer.
Author Contributions
Conceptualization, R.S.L, K.S., and J.M.S.; Writing – Original Draft Preparation, R.S.L, K.S., D.V.F., and J.M.S.; Writing – Review and Editing, R.S.L, K.S., and J.M.S; Visualization, R.S.L, K.S., D.V.F., and J.M.S; Supervision, J.M.S,; Funding Acquisition, J.M.S.
Funding
This research was funded by the National Institutes of Health, grant number R00CA204601, to J.M.S.
Acknowledgements
We thank Drs. Anita Corbett, Andrew Hong, and David Frank for helpful discussions centered around HDACs, histone methyltransferases, and SWI/SNF. Figure 1 created with BioRender.com.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Acheampong, T.; Kehm, R.D.; Terry, M.B.; Argov, E.L.; Tehranifar, P. Incidence Trends of Breast Cancer Molecular Subtypes by Age and Race/Ethnicity in the US From 2010 to 2016. JAMA Netw Open 2020, 3, e2013226. [Google Scholar] [CrossRef]
- Yersal, O.; Barutca, S. Biological subtypes of breast cancer: Prognostic and therapeutic implications. World J Clin Oncol 2014, 5, 412–424. [Google Scholar] [CrossRef]
- Orrantia-Borunda, E.; Anchondo-Nunez, P.; Acuna-Aguilar, L.E.; Gomez-Valles, F.O.; Ramirez-Valdespino, C.A. Subtypes of Breast Cancer. In Breast Cancer, Mayrovitz, H.N., Ed.; Brisbane (AU), 2022.
- Li, Z.H.; Hu, P.H.; Tu, J.H.; Yu, N.S. Luminal B breast cancer: patterns of recurrence and clinical outcome. Oncotarget 2016, 7, 65024–65033. [Google Scholar] [CrossRef] [PubMed]
- Ahn, H.J.; Jung, S.J.; Kim, T.H.; Oh, M.K.; Yoon, H.K. Differences in Clinical Outcomes between Luminal A and B Type Breast Cancers according to the St. Gallen Consensus 2013. J Breast Cancer 2015, 18, 149–159. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.; Unni, N.; Peng, Y. The Changing Paradigm for the Treatment of HER2-Positive Breast Cancer. Cancers (Basel) 2020, 12. [Google Scholar] [CrossRef] [PubMed]
- von Minckwitz, G.; Huang, C.S.; Mano, M.S.; Loibl, S.; Mamounas, E.P.; Untch, M.; Wolmark, N.; Rastogi, P.; Schneeweiss, A.; Redondo, A.; et al. Trastuzumab Emtansine for Residual Invasive HER2-Positive Breast Cancer. N Engl J Med 2019, 380, 617–628. [Google Scholar] [CrossRef] [PubMed]
- Modi, S.; Jacot, W.; Yamashita, T.; Sohn, J.; Vidal, M.; Tokunaga, E.; Tsurutani, J.; Ueno, N.T.; Prat, A.; Chae, Y.S.; et al. Trastuzumab Deruxtecan in Previously Treated HER2-Low Advanced Breast Cancer. N Engl J Med 2022, 387, 9–20. [Google Scholar] [CrossRef]
- Yin, L.; Duan, J.J.; Bian, X.W.; Yu, S.C. Triple-negative breast cancer molecular subtyping and treatment progress. Breast Cancer Res 2020, 22, 61. [Google Scholar] [CrossRef]
- Shi, Y.; Jin, J.; Ji, W.; Guan, X. Therapeutic landscape in mutational triple negative breast cancer. Mol Cancer 2018, 17, 99. [Google Scholar] [CrossRef]
- Nik-Zainal, S.; Davies, H.; Staaf, J.; Ramakrishna, M.; Glodzik, D.; Zou, X.; Martincorena, I.; Alexandrov, L.B.; Martin, S.; Wedge, D.C.; et al. Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature 2016, 534, 47–54. [Google Scholar] [CrossRef]
- Cancer Genome Atlas, N. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar] [CrossRef]
- FitzGerald, M.G.; Marsh, D.J.; Wahrer, D.; Bell, D.; Caron, S.; Shannon, K.E.; Ishioka, C.; Isselbacher, K.J.; Garber, J.E.; Eng, C.; et al. Germline mutations in PTEN are an infrequent cause of genetic predisposition to breast cancer. Oncogene 1998, 17, 727–731. [Google Scholar] [CrossRef] [PubMed]
- Pharoah, P.D.; Guilford, P.; Caldas, C.; International Gastric Cancer Linkage, C. Incidence of gastric cancer and breast cancer in CDH1 (E-cadherin) mutation carriers from hereditary diffuse gastric cancer families. Gastroenterology 2001, 121, 1348–1353. [Google Scholar] [CrossRef] [PubMed]
- Ciriello, G.; Gatza, M.L.; Beck, A.H.; Wilkerson, M.D.; Rhie, S.K.; Pastore, A.; Zhang, H.; McLellan, M.; Yau, C.; Kandoth, C.; et al. Comprehensive Molecular Portraits of Invasive Lobular Breast Cancer. Cell 2015, 163, 506–519. [Google Scholar] [CrossRef] [PubMed]
- Gibney, E.R.; Nolan, C.M. Epigenetics and gene expression. Heredity (Edinb) 2010, 105, 4–13. [Google Scholar] [CrossRef] [PubMed]
- Fyodorov, D.V.; Zhou, B.R.; Skoultchi, A.I.; Bai, Y. Emerging roles of linker histones in regulating chromatin structure and function. Nat Rev Mol Cell Biol 2018, 19, 192–206. [Google Scholar] [CrossRef] [PubMed]
- Simpson, B.; Tupper, C.; Al Aboud, N.M. Genetics, DNA Packaging. In StatPearls; Treasure Island (FL), 2023.
- Luger, K.; Mader, A.W.; Richmond, R.K.; Sargent, D.F.; Richmond, T.J. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature 1997, 389, 251–260. [Google Scholar] [CrossRef] [PubMed]
- Mersfelder, E.L.; Parthun, M.R. The tale beyond the tail: histone core domain modifications and the regulation of chromatin structure. Nucleic Acids Res 2006, 34, 2653–2662. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.; Rao, C.M. Epigenetic tools (The Writers, The Readers and The Erasers) and their implications in cancer therapy. Eur J Pharmacol 2018, 837, 8–24. [Google Scholar] [CrossRef]
- Ramazi, S.; Allahverdi, A.; Zahiri, J. Evaluation of post-translational modifications in histone proteins: A review on histone modification defects in developmental and neurological disorders. J Biosci 2020, 45. [Google Scholar] [CrossRef]
- Cavalieri, V. The Expanding Constellation of Histone Post-Translational Modifications in the Epigenetic Landscape. Genes (Basel) 2021, 12. [Google Scholar] [CrossRef]
- Millan-Zambrano, G.; Burton, A.; Bannister, A.J.; Schneider, R. Histone post-translational modifications - cause and consequence of genome function. Nat Rev Genet 2022, 23, 563–580. [Google Scholar] [CrossRef]
- Tyagi, M.; Imam, N.; Verma, K.; Patel, A.K. Chromatin remodelers: We are the drivers!! Nucleus 2016, 7, 388–404. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov 2022, 12, 31–46. [Google Scholar] [CrossRef]
- Cenik, B.K.; Shilatifard, A. COMPASS and SWI/SNF complexes in development and disease. Nat Rev Genet 2021, 22, 38–58. [Google Scholar] [CrossRef] [PubMed]
- Erratum for the Report "PI3K pathway regulates ER-dependent transcription in breast cancer through the epigenetic regulator KMT2D" by E. Toska, H. U. Osmanbeyoglu, P. Castel, C. Chan, R. C. Hendrickson, M. Elkabets, M. N. Dickler, M. Scaltriti, C. S. Leslie, S. A. Armstrong, J. Baselga. Science 2019, 363. [CrossRef]
- Toska, E.; Castel, P.; Chhangawala, S.; Arruabarrena-Aristorena, A.; Chan, C.; Hristidis, V.C.; Cocco, E.; Sallaku, M.; Xu, G.; Park, J.; et al. PI3K Inhibition Activates SGK1 via a Feedback Loop to Promote Chromatin-Based Regulation of ER-Dependent Gene Expression. Cell Rep 2019, 27, 294–306 e295. [Google Scholar] [CrossRef] [PubMed]
- Spangle, J.M.; Dreijerink, K.M.; Groner, A.C.; Cheng, H.; Ohlson, C.E.; Reyes, J.; Lin, C.Y.; Bradner, J.; Zhao, J.J.; Roberts, T.M.; et al. PI3K/AKT Signaling Regulates H3K4 Methylation in Breast Cancer. Cell Rep 2016, 15, 2692–2704. [Google Scholar] [CrossRef]
- Jones, R.B.; Farhi, J.; Adams, M.; Parwani, K.K.; Cooper, G.W.; Zecevic, M.; Lee, R.S.; Hong, A.L.; Spangle, J.M. Targeting MLL Methyltransferases Enhances the Antitumor Effects of PI3K Inhibition in Hormone Receptor-positive Breast Cancer. Cancer Res Commun 2022, 2, 1569–1578. [Google Scholar] [CrossRef]
- Centore, R.C.; Sandoval, G.J.; Soares, L.M.M.; Kadoch, C.; Chan, H.M. Mammalian SWI/SNF Chromatin Remodeling Complexes: Emerging Mechanisms and Therapeutic Strategies. Trends Genet 2020, 36, 936–950. [Google Scholar] [CrossRef]
- Razavi, P.; Chang, M.T.; Xu, G.; Bandlamudi, C.; Ross, D.S.; Vasan, N.; Cai, Y.; Bielski, C.M.; Donoghue, M.T.A.; Jonsson, P.; et al. The Genomic Landscape of Endocrine-Resistant Advanced Breast Cancers. Cancer Cell 2018, 34, 427–438 e426. [Google Scholar] [CrossRef]
- Nagarajan, S.; Rao, S.V.; Sutton, J.; Cheeseman, D.; Dunn, S.; Papachristou, E.K.; Prada, J.G.; Couturier, D.L.; Kumar, S.; Kishore, K.; et al. ARID1A influences HDAC1/BRD4 activity, intrinsic proliferative capacity and breast cancer treatment response. Nat Genet 2020, 52, 187–197. [Google Scholar] [CrossRef]
- Xu, G.; Chhangawala, S.; Cocco, E.; Razavi, P.; Cai, Y.; Otto, J.E.; Ferrando, L.; Selenica, P.; Ladewig, E.; Chan, C.; et al. ARID1A determines luminal identity and therapeutic response in estrogen-receptor-positive breast cancer. Nat Genet 2020, 52, 198–207. [Google Scholar] [CrossRef]
- Sobczak, M.; Pietrzak, J.; Ploszaj, T.; Robaszkiewicz, A. BRG1 Activates Proliferation and Transcription of Cell Cycle-Dependent Genes in Breast Cancer Cells. Cancers (Basel) 2020, 12. [Google Scholar] [CrossRef]
- Mehta, G.A.; Angus, S.P.; Khella, C.A.; Tong, K.; Khanna, P.; Dixon, S.A.H.; Verzi, M.P.; Johnson, G.L.; Gatza, M.L. SOX4 and SMARCA4 cooperatively regulate PI3k signaling through transcriptional activation of TGFBR2. NPJ Breast Cancer 2021, 7, 40. [Google Scholar] [CrossRef] [PubMed]
- Do, S.I.; Yoon, G.; Kim, H.S.; Kim, K.; Lee, H.; Do, I.G.; Kim, D.H.; Chae, S.W.; Sohn, J.H. Increased Brahma-related Gene 1 Expression Predicts Distant Metastasis and Shorter Survival in Patients with Invasive Ductal Carcinoma of the Breast. Anticancer Res 2016, 36, 4873–4882. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zang, C.; Cui, K.; Schones, D.E.; Barski, A.; Peng, W.; Zhao, K. Genome-wide mapping of HATs and HDACs reveals distinct functions in active and inactive genes. Cell 2009, 138, 1019–1031. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.L.; Yang, W.M.; Seto, E. Regulation of transcription factor YY1 by acetylation and deacetylation. Mol Cell Biol 2001, 21, 5979–5991. [Google Scholar] [CrossRef]
- Wang, R.; Cherukuri, P.; Luo, J. Activation of Stat3 sequence-specific DNA binding and transcription by p300/CREB-binding protein-mediated acetylation. J Biol Chem 2005, 280, 11528–11534. [Google Scholar] [CrossRef] [PubMed]
- Sakaguchi, K.; Herrera, J.E.; Saito, S.; Miki, T.; Bustin, M.; Vassilev, A.; Anderson, C.W.; Appella, E. DNA damage activates p53 through a phosphorylation-acetylation cascade. Genes Dev 1998, 12, 2831–2841. [Google Scholar] [CrossRef]
- Marmorstein, R.; Zhou, M.M. Writers and readers of histone acetylation: structure, mechanism, and inhibition. Cold Spring Harb Perspect Biol 2014, 6, a018762. [Google Scholar] [CrossRef] [PubMed]
- Sheikh, B.N.; Akhtar, A. The many lives of KATs - detectors, integrators and modulators of the cellular environment. Nat Rev Genet 2019, 20, 7–23. [Google Scholar] [CrossRef] [PubMed]
- Kouzarides, T. Chromatin modifications and their function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef] [PubMed]
- Messier, T.L.; Gordon, J.A.; Boyd, J.R.; Tye, C.E.; Browne, G.; Stein, J.L.; Lian, J.B.; Stein, G.S. Histone H3 lysine 4 acetylation and methylation dynamics define breast cancer subtypes. Oncotarget 2016, 7, 5094–5109. [Google Scholar] [CrossRef] [PubMed]
- Guillemette, B.; Drogaris, P.; Lin, H.H.; Armstrong, H.; Hiragami-Hamada, K.; Imhof, A.; Bonneil, E.; Thibault, P.; Verreault, A.; Festenstein, R.J. H3 lysine 4 is acetylated at active gene promoters and is regulated by H3 lysine 4 methylation. PLoS Genet 2011, 7, e1001354. [Google Scholar] [CrossRef]
- Gates, L.A.; Shi, J.; Rohira, A.D.; Feng, Q.; Zhu, B.; Bedford, M.T.; Sagum, C.A.; Jung, S.Y.; Qin, J.; Tsai, M.J.; et al. Acetylation on histone H3 lysine 9 mediates a switch from transcription initiation to elongation. J Biol Chem 2017, 292, 14456–14472. [Google Scholar] [CrossRef]
- Halasa, M.; Wawruszak, A.; Przybyszewska, A.; Jaruga, A.; Guz, M.; Kalafut, J.; Stepulak, A.; Cybulski, M. H3K18Ac as a Marker of Cancer Progression and Potential Target of Anti-Cancer Therapy. Cells 2019, 8. [Google Scholar] [CrossRef]
- Ma, L.; Yuan, L.; An, J.; Barton, M.C.; Zhang, Q.; Liu, Z. Histone H3 lysine 23 acetylation is associated with oncogene TRIM24 expression and a poor prognosis in breast cancer. Tumour Biol 2016, 37, 14803–14812. [Google Scholar] [CrossRef]
- Huang, H.; Hu, J.; Maryam, A.; Huang, Q.; Zhang, Y.; Ramakrishnan, S.; Li, J.; Ma, H.; Ma, V.W.S.; Cheuk, W.; et al. Defining super-enhancer landscape in triple-negative breast cancer by multiomic profiling. Nat Commun 2021, 12, 2242. [Google Scholar] [CrossRef]
- Li, Q.L.; Wang, D.Y.; Ju, L.G.; Yao, J.; Gao, C.; Lei, P.J.; Li, L.Y.; Zhao, X.L.; Wu, M. The hyper-activation of transcriptional enhancers in breast cancer. Clin Epigenetics 2019, 11, 48. [Google Scholar] [CrossRef] [PubMed]
- Bao, C.; Duan, J.; Xie, Y.; Liu, Y.; Li, P.; Li, J.; Zhao, H.; Guo, H.; Men, Y.; Ren, Y.; et al. A novel oncogenic enhancer of estrogen receptor-positive breast cancer. Mol Ther Nucleic Acids 2022, 29, 836–851. [Google Scholar] [CrossRef] [PubMed]
- Stejskal, S.; Stepka, K.; Tesarova, L.; Stejskal, K.; Matejkova, M.; Simara, P.; Zdrahal, Z.; Koutna, I. Cell cycle-dependent changes in H3K56ac in human cells. Cell Cycle 2015, 14, 3851–3863. [Google Scholar] [CrossRef] [PubMed]
- Tropberger, P.; Pott, S.; Keller, C.; Kamieniarz-Gdula, K.; Caron, M.; Richter, F.; Li, G.; Mittler, G.; Liu, E.T.; Buhler, M.; et al. Regulation of transcription through acetylation of H3K122 on the lateral surface of the histone octamer. Cell 2013, 152, 859–872. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Wang, J.; Rousseaux, S.; Tan, M.; Pan, L.; Peng, L.; Wang, S.; Xu, W.; Ren, J.; Liu, Y.; et al. Metabolically controlled histone H4K5 acylation/acetylation ratio drives BRD4 genomic distribution. Cell Rep 2021, 36, 109460. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zang, C.; Rosenfeld, J.A.; Schones, D.E.; Barski, A.; Cuddapah, S.; Cui, K.; Roh, T.Y.; Peng, W.; Zhang, M.Q.; et al. Combinatorial patterns of histone acetylations and methylations in the human genome. Nat Genet 2008, 40, 897–903. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; He, C.; Wang, M.; Ma, X.; Mo, F.; Yang, S.; Han, J.; Wei, X. Targeting epigenetic regulators for cancer therapy: mechanisms and advances in clinical trials. Signal Transduct Target Ther 2019, 4, 62. [Google Scholar] [CrossRef] [PubMed]
- Rada-Iglesias, A.; Bajpai, R.; Swigut, T.; Brugmann, S.A.; Flynn, R.A.; Wysocka, J. A unique chromatin signature uncovers early developmental enhancers in humans. Nature 2011, 470, 279–283. [Google Scholar] [CrossRef] [PubMed]
- Elsheikh, S.E.; Green, A.R.; Rakha, E.A.; Powe, D.G.; Ahmed, R.A.; Collins, H.M.; Soria, D.; Garibaldi, J.M.; Paish, C.E.; Ammar, A.A.; et al. Global histone modifications in breast cancer correlate with tumor phenotypes, prognostic factors, and patient outcome. Cancer Res 2009, 69, 3802–3809. [Google Scholar] [CrossRef]
- Mungamuri, S.K.; Murk, W.; Grumolato, L.; Bernstein, E.; Aaronson, S.A. Chromatin modifications sequentially enhance ErbB2 expression in ErbB2-positive breast cancers. Cell Rep 2013, 5, 302–313. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, Z.; Yang, X.; Lu, W.; Chen, Y.; Lin, Y.; Wang, J.; Lin, S.; Yun, J.P. H3K27 acetylation activated-COL6A1 promotes osteosarcoma lung metastasis by repressing STAT1 and activating pulmonary cancer-associated fibroblasts. Theranostics 2021, 11, 1473–1492. [Google Scholar] [CrossRef]
- Wang, F.; Zhang, W.; Song, Z.; Wang, M.; Wu, H.; Yang, Y.; Chen, R. A novel miRNA inhibits metastasis of prostate cancer via decreasing CREBBP-mediated histone acetylation. J Cancer Res Clin Oncol 2021, 147, 469–480. [Google Scholar] [CrossRef]
- Karsli-Ceppioglu, S.; Dagdemir, A.; Judes, G.; Lebert, A.; Penault-Llorca, F.; Bignon, Y.J.; Bernard-Gallon, D. The Epigenetic Landscape of Promoter Genome-wide Analysis in Breast Cancer. Sci Rep 2017, 7, 6597. [Google Scholar] [CrossRef]
- Yu, L.; Liang, Y.; Cao, X.; Wang, X.; Gao, H.; Lin, S.Y.; Schiff, R.; Wang, X.S.; Li, K. Identification of MYST3 as a novel epigenetic activator of ERalpha frequently amplified in breast cancer. Oncogene 2017, 36, 2910–2918. [Google Scholar] [CrossRef]
- Roche, J. The Epithelial-to-Mesenchymal Transition in Cancer. Cancers (Basel) 2018, 10. [Google Scholar] [CrossRef]
- Frederiks, F.; Tzouros, M.; Oudgenoeg, G.; van Welsem, T.; Fornerod, M.; Krijgsveld, J.; van Leeuwen, F. Nonprocessive methylation by Dot1 leads to functional redundancy of histone H3K79 methylation states. Nat Struct Mol Biol 2008, 15, 550–557. [Google Scholar] [CrossRef] [PubMed]
- Min, J.; Feng, Q.; Li, Z.; Zhang, Y.; Xu, R.M. Structure of the catalytic domain of human DOT1L, a non-SET domain nucleosomal histone methyltransferase. Cell 2003, 112, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Cho, M.H.; Park, J.H.; Choi, H.J.; Park, M.K.; Won, H.Y.; Park, Y.J.; Lee, C.H.; Oh, S.H.; Song, Y.S.; Kim, H.S.; et al. DOT1L cooperates with the c-Myc-p300 complex to epigenetically derepress CDH1 transcription factors in breast cancer progression. Nat Commun 2015, 6, 7821. [Google Scholar] [CrossRef]
- Zhao, L.; Pang, A.; Li, Y. Function of GCN5 in the TGF-beta1-induced epithelial-to-mesenchymal transition in breast cancer. Oncol Lett 2018, 16, 3955–3963. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.J.; Lee, S.Y.; Choi, J.H.; Woo, H.G.; Xhemalce, B.; Miller, K.M. PCAF-Mediated Histone Acetylation Promotes Replication Fork Degradation by MRE11 and EXO1 in BRCA-Deficient Cells. Mol Cell 2020, 80, 327–344 e328. [Google Scholar] [CrossRef] [PubMed]
- Ha, K.; Fiskus, W.; Choi, D.S.; Bhaskara, S.; Cerchietti, L.; Devaraj, S.G.; Shah, B.; Sharma, S.; Chang, J.C.; Melnick, A.M.; et al. Histone deacetylase inhibitor treatment induces 'BRCAness' and synergistic lethality with PARP inhibitor and cisplatin against human triple negative breast cancer cells. Oncotarget 2014, 5, 5637–5650. [Google Scholar] [CrossRef] [PubMed]
- Marijon, H.; Lee, D.H.; Ding, L.; Sun, H.; Gery, S.; de Gramont, A.; Koeffler, H.P. Co-targeting poly(ADP-ribose) polymerase (PARP) and histone deacetylase (HDAC) in triple-negative breast cancer: Higher synergism in BRCA mutated cells. Biomed Pharmacother 2018, 99, 543–551. [Google Scholar] [CrossRef]
- Rasmussen, R.D.; Gajjar, M.K.; Jensen, K.E.; Hamerlik, P. Enhanced efficacy of combined HDAC and PARP targeting in glioblastoma. Mol Oncol 2016, 10, 751–763. [Google Scholar] [CrossRef]
- Yin, L.; Liu, Y.; Peng, Y.; Peng, Y.; Yu, X.; Gao, Y.; Yuan, B.; Zhu, Q.; Cao, T.; He, L.; et al. PARP inhibitor veliparib and HDAC inhibitor SAHA synergistically co-target the UHRF1/BRCA1 DNA damage repair complex in prostate cancer cells. J Exp Clin Cancer Res 2018, 37, 153. [Google Scholar] [CrossRef] [PubMed]
- Bassi, C.; Li, Y.T.; Khu, K.; Mateo, F.; Baniasadi, P.S.; Elia, A.; Mason, J.; Stambolic, V.; Pujana, M.A.; Mak, T.W.; et al. The acetyltransferase Tip60 contributes to mammary tumorigenesis by modulating DNA repair. Cell Death Differ 2016, 23, 1198–1208. [Google Scholar] [CrossRef] [PubMed]
- Gao, C.; Bourke, E.; Scobie, M.; Famme, M.A.; Koolmeister, T.; Helleday, T.; Eriksson, L.A.; Lowndes, N.F.; Brown, J.A. Rational design and validation of a Tip60 histone acetyltransferase inhibitor. Sci Rep 2014, 4, 5372. [Google Scholar] [CrossRef] [PubMed]
- Idrissou, M.; Judes, G.; Daures, M.; Sanchez, A.; El Ouardi, D.; Besse, S.; Degoul, F.; Penault-Llorca, F.; Bignon, Y.J.; Bernard-Gallon, D. TIP60 Inhibitor TH1834 Reduces Breast Cancer Progression in Xenografts in Mice. OMICS 2019, 23, 457–459. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.; Yuan, L.; Zhang, L.; Zhao, J.; Zhang, C.M.; Deng, H.Y. Garcinol, an acetyltransferase inhibitor, suppresses proliferation of breast cancer cell line MCF-7 promoted by 17beta-estradiol. Asian Pac J Cancer Prev 2014, 15, 5001–5007. [Google Scholar] [CrossRef] [PubMed]
- Chessum, N.; Jones, K.; Pasqua, E.; Tucker, M. Recent advances in cancer therapeutics. Prog Med Chem 2015, 54, 1–63. [Google Scholar] [CrossRef]
- Park, S.Y.; Kim, J.S. A short guide to histone deacetylases including recent progress on class II enzymes. Exp Mol Med 2020, 52, 204–212. [Google Scholar] [CrossRef]
- Luo, Y.; Li, H. Structure-Based Inhibitor Discovery of Class I Histone Deacetylases (HDACs). Int J Mol Sci 2020, 21. [Google Scholar] [CrossRef]
- Watson, P.J.; Millard, C.J.; Riley, A.M.; Robertson, N.S.; Wright, L.C.; Godage, H.Y.; Cowley, S.M.; Jamieson, A.G.; Potter, B.V.; Schwabe, J.W. Insights into the activation mechanism of class I HDAC complexes by inositol phosphates. Nat Commun 2016, 7, 11262. [Google Scholar] [CrossRef]
- Clocchiatti, A.; Florean, C.; Brancolini, C. Class IIa HDACs: from important roles in differentiation to possible implications in tumourigenesis. J Cell Mol Med 2011, 15, 1833–1846. [Google Scholar] [CrossRef] [PubMed]
- Wilson, A.J.; Byun, D.S.; Popova, N.; Murray, L.B.; L'Italien, K.; Sowa, Y.; Arango, D.; Velcich, A.; Augenlicht, L.H.; Mariadason, J.M. Histone deacetylase 3 (HDAC3) and other class I HDACs regulate colon cell maturation and p21 expression and are deregulated in human colon cancer. J Biol Chem 2006, 281, 13548–13558. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.H.; Laban, M.; Leung, C.H.; Lee, L.; Lee, C.K.; Salto-Tellez, M.; Raju, G.C.; Hooi, S.C. Inhibition of histone deacetylase 2 increases apoptosis and p21Cip1/WAF1 expression, independent of histone deacetylase 1. Cell Death Differ 2005, 12, 395–404. [Google Scholar] [CrossRef] [PubMed]
- Zhu, P.; Huber, E.; Kiefer, F.; Gottlicher, M. Specific and redundant functions of histone deacetylases in regulation of cell cycle and apoptosis. Cell Cycle 2004, 3, 1240–1242. [Google Scholar] [CrossRef] [PubMed]
- Ishihama, K.; Yamakawa, M.; Semba, S.; Takeda, H.; Kawata, S.; Kimura, S.; Kimura, W. Expression of HDAC1 and CBP/p300 in human colorectal carcinomas. J Clin Pathol 2007, 60, 1205–1210. [Google Scholar] [CrossRef]
- Choi, J.H.; Kwon, H.J.; Yoon, B.I.; Kim, J.H.; Han, S.U.; Joo, H.J.; Kim, D.Y. Expression profile of histone deacetylase 1 in gastric cancer tissues. Jpn J Cancer Res 2001, 92, 1300–1304. [Google Scholar] [CrossRef]
- Shan, W.; Jiang, Y.; Yu, H.; Huang, Q.; Liu, L.; Guo, X.; Li, L.; Mi, Q.; Zhang, K.; Yang, Z. HDAC2 overexpression correlates with aggressive clinicopathological features and DNA-damage response pathway of breast cancer. Am J Cancer Res 2017, 7, 1213–1226. [Google Scholar]
- Krusche, C.A.; Wulfing, P.; Kersting, C.; Vloet, A.; Bocker, W.; Kiesel, L.; Beier, H.M.; Alfer, J. Histone deacetylase-1 and -3 protein expression in human breast cancer: a tissue microarray analysis. Breast Cancer Res Treat 2005, 90, 15–23. [Google Scholar] [CrossRef]
- Muller, B.M.; Jana, L.; Kasajima, A.; Lehmann, A.; Prinzler, J.; Budczies, J.; Winzer, K.J.; Dietel, M.; Weichert, W.; Denkert, C. Differential expression of histone deacetylases HDAC1, 2 and 3 in human breast cancer--overexpression of HDAC2 and HDAC3 is associated with clinicopathological indicators of disease progression. BMC Cancer 2013, 13, 215. [Google Scholar] [CrossRef]
- Seo, J.; Min, S.K.; Park, H.R.; Kim, D.H.; Kwon, M.J.; Kim, L.S.; Ju, Y.S. Expression of Histone Deacetylases HDAC1, HDAC2, HDAC3, and HDAC6 in Invasive Ductal Carcinomas of the Breast. J Breast Cancer 2014, 17, 323–331. [Google Scholar] [CrossRef]
- Zhang, Z.; Yamashita, H.; Toyama, T.; Sugiura, H.; Omoto, Y.; Ando, Y.; Mita, K.; Hamaguchi, M.; Hayashi, S.; Iwase, H. HDAC6 expression is correlated with better survival in breast cancer. Clin Cancer Res 2004, 10, 6962–6968. [Google Scholar] [CrossRef]
- Garmpis, N.; Damaskos, C.; Dimitroulis, D.; Kouraklis, G.; Garmpi, A.; Sarantis, P.; Koustas, E.; Patsouras, A.; Psilopatis, I.; Antoniou, E.A.; et al. Clinical Significance of the Histone Deacetylase 2 (HDAC-2) Expression in Human Breast Cancer. J Pers Med 2022, 12. [Google Scholar] [CrossRef] [PubMed]
- Basta, J.; Rauchman, M. The nucleosome remodeling and deacetylase complex in development and disease. Transl Res 2015, 165, 36–47. [Google Scholar] [CrossRef] [PubMed]
- Manshouri, R.; Coyaud, E.; Kundu, S.T.; Peng, D.H.; Stratton, S.A.; Alton, K.; Bajaj, R.; Fradette, J.J.; Minelli, R.; Peoples, M.D.; et al. ZEB1/NuRD complex suppresses TBC1D2b to stimulate E-cadherin internalization and promote metastasis in lung cancer. Nat Commun 2019, 10, 5125. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.; Teng, X.; Ma, T.; Yang, T.; Zhang, J.; Huo, M.; Liu, W.; Yang, Y.; Yuan, B.; Yu, H.; et al. RUNX2 recruits the NuRD(MTA1)/CRL4B complex to promote breast cancer progression and bone metastasis. Cell Death Differ 2022, 29, 2203–2217. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.; Hopfinger, N.R.; Nguyen, T.D.; Pogash, T.J.; Santucci-Pereira, J.; Russo, J. Epigenetic reprogramming of epithelial mesenchymal transition in triple negative breast cancer cells with DNA methyltransferase and histone deacetylase inhibitors. J Exp Clin Cancer Res 2018, 37, 314. [Google Scholar] [CrossRef]
- Jiang, Y.G.; Luo, Y.; He, D.L.; Li, X.; Zhang, L.L.; Peng, T.; Li, M.C.; Lin, Y.H. Role of Wnt/beta-catenin signaling pathway in epithelial-mesenchymal transition of human prostate cancer induced by hypoxia-inducible factor-1alpha. Int J Urol 2007, 14, 1034–1039. [Google Scholar] [CrossRef] [PubMed]
- Coelho, B.P.; Fernandes, C.F.L.; Boccacino, J.M.; Souza, M.; Melo-Escobar, M.I.; Alves, R.N.; Prado, M.B.; Iglesia, R.P.; Cangiano, G.; Mazzaro, G.R.; et al. Multifaceted WNT Signaling at the Crossroads Between Epithelial-Mesenchymal Transition and Autophagy in Glioblastoma. Front Oncol 2020, 10, 597743. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.C.; Zhu, Y.T.; Chen, S.Y.; Tseng, S.C. Wnt signaling induces epithelial-mesenchymal transition with proliferation in ARPE-19 cells upon loss of contact inhibition. Lab Invest 2012, 92, 676–687. [Google Scholar] [CrossRef]
- Pantelaiou-Prokaki, G.; Mieczkowska, I.; Schmidt, G.E.; Fritzsche, S.; Prokakis, E.; Gallwas, J.; Wegwitz, F. HDAC8 suppresses the epithelial phenotype and promotes EMT in chemotherapy-treated basal-like breast cancer. Clin Epigenetics 2022, 14, 7. [Google Scholar] [CrossRef]
- Shah, P.; Gau, Y.; Sabnis, G. Histone deacetylase inhibitor entinostat reverses epithelial to mesenchymal transition of breast cancer cells by reversing the repression of E-cadherin. Breast Cancer Res Treat 2014, 143, 99–111. [Google Scholar] [CrossRef]
- Peinado, H.; Ballestar, E.; Esteller, M.; Cano, A. Snail mediates E-cadherin repression by the recruitment of the Sin3A/histone deacetylase 1 (HDAC1)/HDAC2 complex. Mol Cell Biol 2004, 24, 306–319. [Google Scholar] [CrossRef]
- Fortier, A.M.; Asselin, E.; Cadrin, M. Keratin 8 and 18 loss in epithelial cancer cells increases collective cell migration and cisplatin sensitivity through claudin1 up-regulation. J Biol Chem 2013, 288, 11555–11571. [Google Scholar] [CrossRef]
- Hata, T.; Rajabi, H.; Takahashi, H.; Yasumizu, Y.; Li, W.; Jin, C.; Long, M.D.; Hu, Q.; Liu, S.; Fushimi, A.; et al. MUC1-C Activates the NuRD Complex to Drive Dedifferentiation of Triple-Negative Breast Cancer Cells. Cancer Res 2019, 79, 5711–5722. [Google Scholar] [CrossRef]
- Fu, J.; Zhang, L.; He, T.; Xiao, X.; Liu, X.; Wang, L.; Yang, L.; Yang, M.; Zhang, T.; Chen, R.; et al. TWIST represses estrogen receptor-alpha expression by recruiting the NuRD protein complex in breast cancer cells. Int J Biol Sci 2012, 8, 522–532. [Google Scholar] [CrossRef]
- Klinge, C.M. Estrogen receptor interaction with estrogen response elements. Nucleic Acids Res 2001, 29, 2905–2919. [Google Scholar] [CrossRef] [PubMed]
- Mazumdar, A.; Wang, R.A.; Mishra, S.K.; Adam, L.; Bagheri-Yarmand, R.; Mandal, M.; Vadlamudi, R.K.; Kumar, R. Transcriptional repression of oestrogen receptor by metastasis-associated protein 1 corepressor. Nat Cell Biol 2001, 3, 30–37. [Google Scholar] [CrossRef]
- Heuer, J.; Heuer, F.; Sturmer, R.; Harder, S.; Schluter, H.; Braga Emidio, N.; Muttenthaler, M.; Jechorek, D.; Meyer, F.; Hoffmann, W. The Tumor Suppressor TFF1 Occurs in Different Forms and Interacts with Multiple Partners in the Human Gastric Mucus Barrier: Indications for Diverse Protective Functions. Int J Mol Sci 2020, 21. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.F.; Bagchi, M.K. Recruitment of distinct chromatin-modifying complexes by tamoxifen-complexed estrogen receptor at natural target gene promoters in vivo. J Biol Chem 2004, 279, 15050–15058. [Google Scholar] [CrossRef]
- Criscitiello, C.; Fumagalli, D.; Saini, K.S.; Loi, S. Tamoxifen in early-stage estrogen receptor-positive breast cancer: overview of clinical use and molecular biomarkers for patient selection. Onco Targets Ther 2010, 4, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Ali, S.; Rasool, M.; Chaoudhry, H.; P, N.P.; Jha, P.; Hafiz, A.; Mahfooz, M.; Abdus Sami, G.; Azhar Kamal, M.; Bashir, S.; et al. Molecular mechanisms and mode of tamoxifen resistance in breast cancer. Bioinformation 2016, 12, 135–139. [Google Scholar] [CrossRef]
- Jeselsohn, R.; Cornwell, M.; Pun, M.; Buchwalter, G.; Nguyen, M.; Bango, C.; Huang, Y.; Kuang, Y.; Paweletz, C.; Fu, X.; et al. Embryonic transcription factor SOX9 drives breast cancer endocrine resistance. Proc Natl Acad Sci U S A 2017, 114, E4482–E4491. [Google Scholar] [CrossRef] [PubMed]
- Xue, Y.; Lian, W.; Zhi, J.; Yang, W.; Li, Q.; Guo, X.; Gao, J.; Qu, H.; Lin, W.; Li, Z.; et al. HDAC5-mediated deacetylation and nuclear localisation of SOX9 is critical for tamoxifen resistance in breast cancer. Br J Cancer 2019, 121, 1039–1049. [Google Scholar] [CrossRef] [PubMed]
- Yard, B.; Chie, E.K.; Adams, D.J.; Peacock, C.; Abazeed, M.E. Radiotherapy in the Era of Precision Medicine. Semin Radiat Oncol 2015, 25, 227–236. [Google Scholar] [CrossRef] [PubMed]
- Ouellette, M.M.; Zhou, S.; Yan, Y. Cell Signaling Pathways That Promote Radioresistance of Cancer Cells. Diagnostics (Basel) 2022, 12. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.L.; Liu, W.L.; Hsu, F.M.; Yang, P.S.; Yen, R.F.; Tzen, K.Y.; Cheng, A.L.; Chen, P.J.; Cheng, J.C. Targeting histone deacetylase 4/ubiquitin-conjugating enzyme 9 impairs DNA repair for radiosensitization of hepatocellular carcinoma cells in mice. Hepatology 2018, 67, 586–599. [Google Scholar] [CrossRef] [PubMed]
- Chiu, H.W.; Yeh, Y.L.; Wang, Y.C.; Huang, W.J.; Chen, Y.A.; Chiou, Y.S.; Ho, S.Y.; Lin, P.; Wang, Y.J. Suberoylanilide hydroxamic acid, an inhibitor of histone deacetylase, enhances radiosensitivity and suppresses lung metastasis in breast cancer in vitro and in vivo. PLoS One 2013, 8, e76340. [Google Scholar] [CrossRef] [PubMed]
- Baschnagel, A.; Russo, A.; Burgan, W.E.; Carter, D.; Beam, K.; Palmieri, D.; Steeg, P.S.; Tofilon, P.; Camphausen, K. Vorinostat enhances the radiosensitivity of a breast cancer brain metastatic cell line grown in vitro and as intracranial xenografts. Mol Cancer Ther 2009, 8, 1589–1595. [Google Scholar] [CrossRef]
- Wang, S.; Song, M.; Zhang, B. Trichostatin A enhances radiosensitivity and radiation-induced DNA damage of esophageal cancer cells. J Gastrointest Oncol 2021, 12, 1985–1995. [Google Scholar] [CrossRef]
- Sharda, A.; Rashid, M.; Shah, S.G.; Sharma, A.K.; Singh, S.R.; Gera, P.; Chilkapati, M.K.; Gupta, S. Elevated HDAC activity and altered histone phospho-acetylation confer acquired radio-resistant phenotype to breast cancer cells. Clin Epigenetics 2020, 12, 4. [Google Scholar] [CrossRef]
- Grant, S.; Easley, C.; Kirkpatrick, P. Vorinostat. Nat Rev Drug Discov 2007, 6, 21–22. [Google Scholar] [CrossRef]
- Grant, C.; Rahman, F.; Piekarz, R.; Peer, C.; Frye, R.; Robey, R.W.; Gardner, E.R.; Figg, W.D.; Bates, S.E. Romidepsin: a new therapy for cutaneous T-cell lymphoma and a potential therapy for solid tumors. Expert Rev Anticancer Ther 2010, 10, 997–1008. [Google Scholar] [CrossRef]
- Braunstein, Z.; Ruiz, M.; Hanel, W.; Shindiapina, P.; Reneau, J.C.; Brammer, J.E. Recent Advances in the Management of Relapsed and Refractory Peripheral T-Cell Lymphomas. J Pers Med 2022, 12. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.Z.; Kwitkowski, V.E.; Del Valle, P.L.; Ricci, M.S.; Saber, H.; Habtemariam, B.A.; Bullock, J.; Bloomquist, E.; Li Shen, Y.; Chen, X.H.; et al. FDA Approval: Belinostat for the Treatment of Patients with Relapsed or Refractory Peripheral T-cell Lymphoma. Clin Cancer Res 2015, 21, 2666–2670. [Google Scholar] [CrossRef]
- Raedler, L.A. Farydak (Panobinostat): First HDAC Inhibitor Approved for Patients with Relapsed Multiple Myeloma. Am Health Drug Benefits 2016, 9, 84–87. [Google Scholar]
- Sabnis, G.J.; Goloubeva, O.G.; Kazi, A.A.; Shah, P.; Brodie, A.H. HDAC inhibitor entinostat restores responsiveness of letrozole-resistant MCF-7Ca xenografts to aromatase inhibitors through modulation of Her-2. Mol Cancer Ther 2013, 12, 2804–2816. [Google Scholar] [CrossRef]
- Connolly, R.M.; Zhao, F.; Miller, K.D.; Lee, M.J.; Piekarz, R.L.; Smith, K.L.; Brown-Glaberman, U.A.; Winn, J.S.; Faller, B.A.; Onitilo, A.A.; et al. E2112: Randomized Phase III Trial of Endocrine Therapy Plus Entinostat or Placebo in Hormone Receptor-Positive Advanced Breast Cancer. A Trial of the ECOG-ACRIN Cancer Research Group. J Clin Oncol 2021, 39, 3171–3181. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Li, W.; Hu, X.; Zhang, Q.; Sun, T.; Cui, S.; Wang, S.; Ouyang, Q.; Yin, Y.; Geng, C.; et al. Tucidinostat plus exemestane for postmenopausal patients with advanced, hormone receptor-positive breast cancer (ACE): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol 2019, 20, 806–815. [Google Scholar] [CrossRef]
- Heerboth, S.; Housman, G.; Leary, M.; Longacre, M.; Byler, S.; Lapinska, K.; Willbanks, A.; Sarkar, S. EMT and tumor metastasis. Clin Transl Med 2015, 4, 6. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Ma, Z.; Ye, X.; Kang, W.; Yu, J. Clinicopathological factors affecting the effect of neoadjuvant chemotherapy in patients with gastric cancer. World J Surg Oncol 2021, 19, 44. [Google Scholar] [CrossRef]
- Sun, L.B.; Zhao, G.J.; Ding, D.Y.; Song, B.; Hou, R.Z.; Li, Y.C. Comparison between better and poorly differentiated locally advanced gastric cancer in preoperative chemotherapy: a retrospective, comparative study at a single tertiary care institute. World J Surg Oncol 2014, 12, 280. [Google Scholar] [CrossRef]
- Bosch, A.; Li, Z.; Bergamaschi, A.; Ellis, H.; Toska, E.; Prat, A.; Tao, J.J.; Spratt, D.E.; Viola-Villegas, N.T.; Castel, P.; et al. PI3K inhibition results in enhanced estrogen receptor function and dependence in hormone receptor-positive breast cancer. Sci Transl Med 2015, 7, 283ra251. [Google Scholar] [CrossRef]
- Cacabelos, R.T., C. Chapter 32 – Pharmacoepigenomics; Tollefsbol, T.O., Ed.; Academic Press: 2016.
- Feng, X.; Han, H.; Zou, D.; Zhou, J.; Zhou, W. Suberoylanilide hydroxamic acid-induced specific epigenetic regulation controls Leptin-induced proliferation of breast cancer cell lines. Oncotarget 2017, 8, 3364–3379. [Google Scholar] [CrossRef]
- Palczewski, M.B.; Kuschman, H.P.; Bovee, R.; Hickok, J.R.; Thomas, D.D. Vorinostat exhibits anticancer effects in triple-negative breast cancer cells by preventing nitric oxide-driven histone deacetylation. Biol Chem 2021, 402, 501–512. [Google Scholar] [CrossRef]
- Mann, B.S.; Johnson, J.R.; Cohen, M.H.; Justice, R.; Pazdur, R. FDA approval summary: vorinostat for treatment of advanced primary cutaneous T-cell lymphoma. Oncologist 2007, 12, 1247–1252. [Google Scholar] [CrossRef]
- Petrich, A.; Nabhan, C. Use of class I histone deacetylase inhibitor romidepsin in combination regimens. Leuk Lymphoma 2016, 57, 1755–1765. [Google Scholar] [CrossRef] [PubMed]
- Sawas, A.; Radeski, D.; O'Connor, O.A. Belinostat in patients with refractory or relapsed peripheral T-cell lymphoma: a perspective review. Ther Adv Hematol 2015, 6, 202–208. [Google Scholar] [CrossRef]
- Tate, C.R.; Rhodes, L.V.; Segar, H.C.; Driver, J.L.; Pounder, F.N.; Burow, M.E.; Collins-Burow, B.M. Targeting triple-negative breast cancer cells with the histone deacetylase inhibitor panobinostat. Breast Cancer Res 2012, 14, R79. [Google Scholar] [CrossRef] [PubMed]
- Zucchetti, B.; Shimada, A.K.; Katz, A.; Curigliano, G. The role of histone deacetylase inhibitors in metastatic breast cancer. Breast 2019, 43, 130–134. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.; Zhao, S.; Yang, Q.; Shi, Z.; Liu, J.; Pan, T.; Zhou, D.; Zhang, J. Chidamide Combined With Doxorubicin Induced p53-Driven Cell Cycle Arrest and Cell Apoptosis Reverse Multidrug Resistance of Breast Cancer. Front Oncol 2021, 11, 614458. [Google Scholar] [CrossRef]
Figure 1.
Epigenetic regulation of histone acetylation and deacetylation. HATs and HDACs have a crucial role in gene regulation. These enzymes are responsible for the transfer and removal of acetyl group from histones on lysine residues. HDACs are classified into class I (HDAC 1, 2, 3, and 8), class II (HDAC 4, 5, 6, 7, 9, and 10), class III (SIRT1-7), and class IV (HDAC 11). HAT-directed lysine acetylation on histones promotes led an open chromatin structure that enhances transcriptional competence. HDAC-directed lysine deacetylation supports chromatin compaction, thereby reducing transcriptional activity. Dysregulation of these chromatin modifying enzymes aids in aberrant cellular proliferation, angiogenesis, EMT, as well as escape from cell cycle arrest and evasion of apoptosis. While HAT inhibition remains in preclinical development, four HDAC inhibitors are currently FDA approved in human cancers, and are denoted by *.
Figure 1.
Epigenetic regulation of histone acetylation and deacetylation. HATs and HDACs have a crucial role in gene regulation. These enzymes are responsible for the transfer and removal of acetyl group from histones on lysine residues. HDACs are classified into class I (HDAC 1, 2, 3, and 8), class II (HDAC 4, 5, 6, 7, 9, and 10), class III (SIRT1-7), and class IV (HDAC 11). HAT-directed lysine acetylation on histones promotes led an open chromatin structure that enhances transcriptional competence. HDAC-directed lysine deacetylation supports chromatin compaction, thereby reducing transcriptional activity. Dysregulation of these chromatin modifying enzymes aids in aberrant cellular proliferation, angiogenesis, EMT, as well as escape from cell cycle arrest and evasion of apoptosis. While HAT inhibition remains in preclinical development, four HDAC inhibitors are currently FDA approved in human cancers, and are denoted by *.
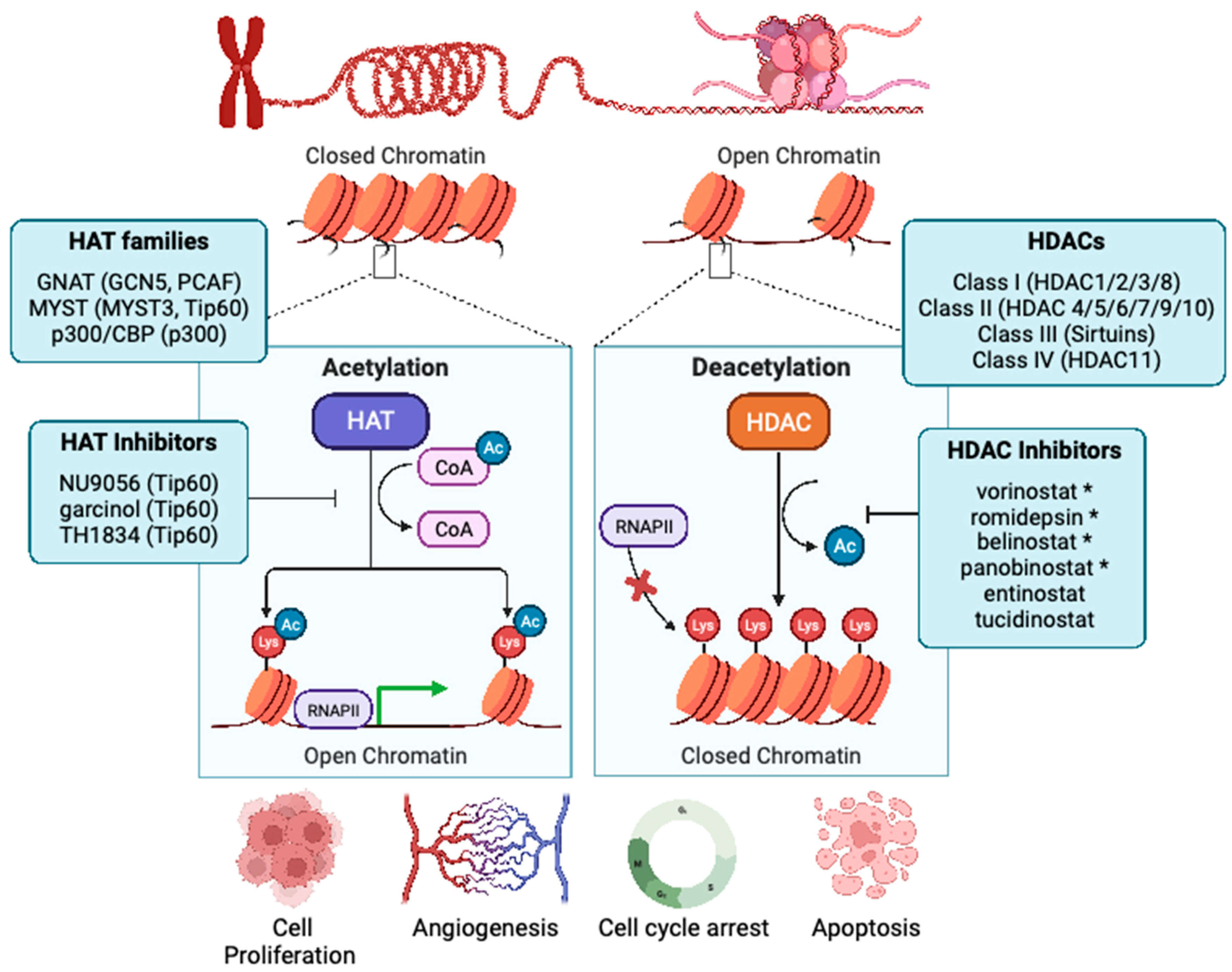

Table 1.
Clinically viable HDAC inhibitors and their targets. HDAC inhibitors that have achieved FDA-approval in non-breast cancers or have an active, ongoing clinical trial in breast cancer. The HDAC targets of each inhibitor, along with the known histone acetylation marks that are regulated via the inhibitor are indicated. When known, the context in which histone acetylation is regulated is described, along with the disease indication. While multiple HDAC inhibitors have entered US-based clinical trials for the treatment of breast cancer, 12 clinical trials are currently active.
Table 1.
Clinically viable HDAC inhibitors and their targets. HDAC inhibitors that have achieved FDA-approval in non-breast cancers or have an active, ongoing clinical trial in breast cancer. The HDAC targets of each inhibitor, along with the known histone acetylation marks that are regulated via the inhibitor are indicated. When known, the context in which histone acetylation is regulated is described, along with the disease indication. While multiple HDAC inhibitors have entered US-based clinical trials for the treatment of breast cancer, 12 clinical trials are currently active.
| Agent | Target(s) | PTM-regulation | Approval indication | Trial stage in breast cancer | Clinical trial status |
| Vorinostat (SAHA) | HDAC1, 2, 3, 8 (Class I) & HDAC6 (Class IIb)[136] | H3K14ac & H3K27ac (MCF7), H3K27ac, H3K18ac, H4K5ac (MDA-MB-231)[137], H3K9ac (TNBC)[138] | Cutaneous T-cell lymphoma (FDA)[139] |
I, II | Active; NCT03742245, NCT00616967, NCT04190056, NCT03878524 |
| Romidepsin (FK228) | HDAC1, 2, 3, 8 (Class I)[140] | Unknown | Cutaneous T-cell lymphoma (FDA)[125] | I, II | Active; NCT02393794, NCT01638533 |
| Belinostat (PXD101) | Pan-inhibitor for zinc-dependent HDAC[141] | Unknown | Relapsed/Refractory peripheral T-cell lymphoma (FDA)[127] |
I | Active; NCT04315233, NCT04703920 |
| Panobinostat (LBH-589) | Pan-inhibitor for zinc-dependent HDAC[136] | H3K9ac, H4K8ac (TNBC)[142] | Multiple myeloma (FDA)[128] | I | Active; NCT03878524 |
| Entinostat (MS-275) | HDAC1, 2, 3 (Class I), and HDAC9 (Class II)[143] | Unknown | Breakthrough designation (advanced breast cancer) | III | Active; NCT01349959, NCT02115282, NCT02453620, NCT03280563. NCT03538171 (China) |
| Tucidinostat | HDAC1, 2, 3 (Class I), HDAC10 (Class II)[143] | H3K9ac, H3K18ac (TNBC, HR+)[144] | Peripheral T-cell lymphoma (CFDA) | III (China) | Active (China only); NCT05276713, NCT05390476, NCT05632848, NCT05633914, NCT05335473, NCT05411380, NCT04192903, NCT05747313, NCT05186545, NCT05047848, NCT05890287, NCT05749575, NCT05085626, NCT05464173, NCT05400993 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



