Submitted:
13 June 2023
Posted:
14 June 2023
You are already at the latest version
Abstract
The main property of tripodal ligands is the predictable type of coordination. Homo- and heteroleptic lanthanide complexes with tripodal ligands are a representative class of compounds. However, despite the fact that many of them are paramagnetic their magnetic behavior is poorly understood. This is because their photophysical and catalytic properties have been more attractive. In the present review, we are trying to summarize the available structural information and extremely few data on magnetic properties in order to draw some conclusions about the prospective of tripods using in the design of quantum molecular magnets based on Ln ions. We would like to draw the reader's attention to the fact that despite the consideration of a large part of the currently known lanthanide compounds with tripodal ligands, this review is not exhaustive. However, our goal was to draw the attention of researchers to the fact that a whole niche of air-stable Ln complexes remained outside the attention of magnetochemists and theoreticians.
Keywords:
tripodal ligands
; lanthanides
; single-molecule magnets
; single-ion magnets
; axiality
1. Introduction
Rational design of coordination compounds with specific physical properties is a challenging task for the synthetic chemists. Chemical engineering can be highly specialized (focusing on improving a specific property) or it can be aimed at preparing polyfunctional materials. The most common objective for materials scientists is to tune the electronic properties of a compound. This can be achieved by varying the ligand environment of the central atom: geometry of the polyhedron and the ligand field strength. The latter is mainly defined by the donor atoms composition, whereas the former depends not only on the geometry of the ligand, but also on the predictability of its coordination mode. The organic tripod molecules having the donor atoms on each of their three legs are the best choice for the chemical design of the different complexes with a given geometry. Moreover, the latter can be easily varied by using the methods of synthetic organic chemistry. Therefore, it is not surprising that tripodal ligands are widely used in various fields of applied coordination chemistry such as catalysis [1,2,3,4,5,6], chemo sensing [7,8,9], photo-[10,11,12] and electroluminescence [11,13,14], molecular gears and motors production [15,16], biomedical applications [17,18,19,20] and molecular magnetism [12,21,22,23,24].
Although numerous high-spin molecules based on tripodal ligands (including paramagnetic ones [25,26,27,28,29,30,31,32]) and d-metal ions have been studied to date [33,34,35,36,37,38,39,40,41,42], for 4f elements such studies are mainly focused on synthesis, the structural features and photophysical properties of compounds with diamagnetic tripods, often leaving aside the study of their magnetic behavior [10,11,14,18,43,44,45,46,47,48]. The present review is devoted to the analysis of known lanthanide complexes with tripodal ligands from the point of view of their application in the field of molecular magnetism.
2. Short theoretical background
Due to slow attenuation of magnetization and existence of magnetic hysteresis of purely molecular origin, single-molecule magnets (SMMs) could offer considerable future application in the domain of spintronics, quantum computing and, especially, for use as materials for storing information at the molecular level. There are three main characteristics of SMMs. The first is the effective energy barrier (Ueff) providing magnetic bistability due to spin reversal between the two ground states (GS). Highly efficient SMMs are very different in their behavior, since large Ueff do not ensure that magnetization is kept at the elevated temperature during a period necessary to serve as a magnetic storage. This is defined by the second characteristic called blocking temperature (TB), which is a central claim for the implementation of SMMs in practice. Usually, the spin-lattice relaxation and quantum tunneling of magnetization (QTM) lead to the magnetization disappearance in an SMM. The first associated with spin-phonon coupling, which can manifest itself through three different mechanisms: Orbach, Raman and direct. The QTM is a temperature-independent phenomenon. A thermally assisted QTM process on excited states is also possible, through which the relaxation occurs. These processes, phonon type relaxation and QTM, described in many papers [49,50,51] closely related with the third SMM characteristic – relaxation time (τ). The Ueff value is ordinarily determined from the Arrhenius dependence of relaxation time for the Orbach process. For the main SMM’s characteristic, TB, the more common definition is that the magnetization blocking temperature is a temperature below which magnetic hysteresis is opening. The stronger the SMM coercive field and the higher the residual magnetization at zero magnetic field, the better magnetic bistability the magnetic material will exhibit. In addition, higher Ueff and TB provide quantized magnetization with satisfactory suppression of spin-lattice relaxation. The TB is now much lower room temperature, limiting possible technological applications [49,50,51,52,53]. Consequently, the quest for SMMs possessing large Ueff and TB is a crucial issue in molecular magnetism. An important category of SMMs, lanthanide (Ln) single-ion magnets (or SIMs), are particularly promising due to their huge magnetic moments with immense magnetic anisotropy caused by a large spin–orbit coupling offered by the unquenched orbital momentum [54,55]. Improved (TB) and (Ueff) are indispensable to realize their practical applications.
Almost thirty years of transdisciplinary investigations have resulted in SIMs with record Ueff (up to 2000 K) and TB near liquid nitrogen temperature [56,57,58,59]. This is a result of several approaches, which have been shown that enhancing the uniaxiality of magnetic anisotropy is crucial for the design of highly performed SIMs [60,61,62,63,64]. The uniaxiality of SIMs can be provided by the corresponding symmetry of the coordination polyhedron. However, despite the similarity in chemical behavior (due to the shielded nature of the f-electrons) Ln3+ are very different in terms of electronic properties. This leads to the fact that for ions differing in electronic configurations, along with the crystal field (CF) splitting, their ligand environment must be taken into account. Among the lanthanides (Ln), the tri-valent Tb, Dy, Ho, and Er can produce large mJ ground states if the appropriate CF is provided [65]. This is due to that the electronic clouds of Ln GS have different shapes. The letter are generally subdivided into prolate and oblate spheroid types [60,66,67,68], which requires respectively different ligand field organization: axial for Pr, Tb, Dy, Nd and equatorial for Pm, Tm, Yb, and Er [60,69]. Thus, to increase the energy barrier Ueff, the symmetry of a Ln3+ ion environment, along with the ligand field approach (axial or equatorial), are required depending on the nature of the Ln+3 [70,71]. Although this provides an opportunity to design the SMM with the ground states with large magnetic moment, relaxation often occurs through excited states that have different electron densities. In addition, the energy gap separating the mJ levels (a key parameter for SMM enhancement) is difficult to forecast until a quantitative approach is applied and the mechanisms of relaxation are entirely established.
If strong intermolecular exchange interactions are absent between the mononuclear SIMs (which is precisely the case for lanthanide ions, since the magnetic electrons are on a well screened 4f shell), then the surrounding the magnetic metal ions ligand field leads to the degeneration of the 2S+1LJ multiplets resulting in 2J + 1 sublevels mJ. This generates a highly anisotropic ground spin state, which can be explained using quantum mechanics. However, we will not immerse the reader in the mathematical details for all types of magnetic anisotropy, since they can be found in the literature [72,73,74]. In regard of the fixed molecular structure, uniaxial anisotropy is important to suppress QTM and decrease thermo-assisted relaxation. In addition, a source of under barrier QTM is the transverse anisotropy caused by transverse CF that should be avoided in design of SIMs. Tong et al. [75] have studied the influence of symmetry on the transverse terms. They conclude that for the C∞v, C5h/D5h, S8/D4d, and S12/D6d symmetry vanish terms (q ≠ 0) of CF for interested Ho3+ ion, resulting in the perfect uniaxial anisotropy. Thus, by adjusting the symmetry of the coordination polyhedron and the ligand environment for a particular lanthanide center, QTM in SIM can be battled.
However, even the most accurate theoretical predictions are not easy to implement in practice, since in order to obtain a coordination polyhedron with a certain symmetry, it is necessary to use ligands with a predictable manner of coordination. Therefore, when designing any SIM, it is up to synthetic chemists to find a compromise between polyhedra with uniaxial symmetry and the choice of ligands for building magnetic molecules.
Therefore, the analysis of currently known molecular mononuclear complexes containing tripodal ter- and tetradentate ligands is aimed at drawing the attention of the scientific community to the use of such ligands in the design of Ln complexes with uniaxial anisotropy. To date, approaches have been developed to choice the most suitable Ln-ion for the appropriate ligand environment, based on the shape of 4f-electron clouds (oblate or prolate) for both the ground state and the excited levels of the central atom [60,76,77,78].
3. Lanthanide complexes with tripodal ligands
3.1. Complexes with the pyrazolyl bearing tripodal ligand
3.1.1. Complexes with tris(pyrazolyl)borates tripodal ligands
The first and most studied coordinative tripods were the scorpionate ligands hydro-tris(pyrazolyl)borates, first obtained by S. Trofimenko [33,35]. The coordination chemistry of these negatively charged ligands mainly includes d-metal complexes [37,79,80]. In the chemistry of coordination-capacious f-elements, scorpionates are often used as capping ligands that additionally compensate for the positive charge of Ln [81,82,83,84,85,86]. In the area of molecular magnetism, the bulky hydro-tris(pyrazolyl)-borate (TPzB) is commonly employed as a capping ligand to reduce intermolecular magnetic interactions and protect the air-sensitive radical cites [87]. Besides, a number of 3d ion-based SMMs and SCMs (single-chain magnets) [88,89] have been obtained by using this approach. Unfortunately, very few Ln-TPzB complexes have been magnetically studied.
Among them three Dy3+ compounds: one purely homoleptic [Dy(TpMe2)2]I (1), one heteroleptic (Me4N)[DyCl3(TpMe2)] (2) (TpMe2=tris(3,5-dimethylpyrazolyl)borate), and one mixed type complex [Dy(TpMe2)2][DyCl3(TpMe2)]·CH2Cl2 (3) are described [90]. The Dy3+ ions are 6-coordinate for all three complexes (Figure 1). The [Dy(TpMe2)2]+ cation in 3 adopts a bent sandwich-type structure with a B–Dy–B angle of 169.57(3)° [90]. In the cationic part, the Dy3+ ion is surrounded only by N-atoms of two TpMe2, while in the anionic part the coordination sphere of Dy3+ consists of three nitrogen atoms from one tripod and three chloride ions. The structural shape analysis has shown that the Dy-center in cation 3 is in an elongated trigonal antiprismatic coordination environment (Figure 1) [90]. Such a geometry has been previously found for divalent lanthanide [44,46,91].
Complex 2 contains the separate [DyCl3(TpMe2)]− anion, which is alike to that of 3 but having Me4N+ as a cation. Compound 1 is isomorphous to the Sm3+ analogue reported earlier [44]. It contains well isolated [Dy(TpMe2)2]+ and iodide ions. The cation in 1 placed in a 2/m symmetry element representing a mirror plane passing through two of the pyrazolyl rings [90]. It is noteworthy that the Dy-center in the cation 1 has a similar geometry comparatively to those of the compound 3, but it has the following key dissimilarities: the two independent Dy–N bond distances, 2.376(2) and 2.430(3) Å, are longer compared to those of 3; due to the crystallographic symmetry, the two planes defined by the nitrogen donor atoms of each tripod are parallel, whereas in 3 the angle between the planes is 10.53(2)°; both the TpMe2 –Dy–TpMe2 and B–Dy–B angles in 1 (178.64(2)° and 180.00(2)°), are broader than those of 3 (173.05(2)° and 169.57 (2)°), indicating that cation 1 reveals a more dense and linear structure than the cation in 3. In conclusion, the isolated [DyCl3(TpMe2)]− is in a alike trigonal antiprismatic coordination environment having the very similar bond distances and angles for the compounds 2 and 3 [90].
Magnetic studies have shown that complex 1 displays an energy barrier Ueff of 13.5 K with τ0 = 1.6 × 10−6 s under a 0.08 T applied field, while 3 is a SIM with Ueff = 80.7 K and τ0 = 6.2 × 10−7 s under a zero applied field [90]. The results of the first principles CASSCF + RASSI-SO calculations correspond well the experimental magnetic measurements for 3 and 1 indicating the presence of an intermolecular dipolar interaction of (zJ = −0.1 cm−1) in 3 [90]. The absence of SMM behavior in 1 and 2 under a zero dc field supports the conclusion that the slow magnetic relaxation observed for 1 is a result of minor changes in the coordination geometry of the Dy3+ ion, and/or intermolecular dipolar interactions between the anionic and cationic moieties [90]. The QTM probability of ground state for Kramers doublets is described by the crystal field (CF, Bkq) parameters. QTM is prevailing when the non-axial terms (for which q ≠ 0 and k = 2, 4, 6) are larger compared to the axial ones (for which q = 0 and k =2,4,6). For all the Dy3+ ions in 1–3, there is significant transverse anisotropy and fast QTM relaxation [90]. The cationic Dy1 units in 1 and 3, however, have relatively small temperature assisted-QTM in the first excited states. This situation can assist in magnetization relaxation via the first excited states when a dc field is applied [90].
From the point of view of the design of high-performance SIMs, it would be worth paying special attention to the recent publication by Hao Qi et all [92] devoted to the synthesis of Eu(II) compounds with bulky tris-pyrazolyl-borates. First of all, it should be noted that the obtained three complexes are stable under ordinary conditions and can be sublimated. Both of these properties are extremely important for practical applications. When extending the synthesis procedure chosen by the authors to magneto-anisotropic Ln ions, it would be interesting to test these compounds for SMM behavior. The complexes Eu(TpPh,Me)2, Eu(TpPh)2 and Eu(TpPh2)2 are all hexa-coordinated, with six N atoms from pyrazole heterocycles. Eu(TpPh)2 is isomorphous to the known compounds Sm(TpPh)2 and Yb(TpPh)2 [44]. The molecular structure of Eu(TpPh2)2 differs from those of Eu(TpPh,Me)2, Eu(TpPh)2. Due to the substituents change, the structure symmetry of the compounds rises in the following order Eu(TpPh,Me)2 → Eu(TpPh)2 → Eu(TpPh2)2, the latter having a higher symmetry of D3d, while the others possess a “bent sandwich-like” structure known for other Ln(II) ions [91,93,94,95,96,97,98]. The complex Eu(TpPh)2 (4) adopts an ideal trigonal antiprismatic (D3d symmetry) molecular structure with a linear B–Eu–B fragment (Figure 1).
The D3d single point group having C3 axis is a subgroup of D6h, which is enough effective to induce strong magnetic axiality in Ln complexes [99,100,101,102]. However, trigonal antiprismatic complexes of underexplored in the field of molecular magnetism. This is especially true for stable Ln2+ complexes of type 4. However, one must take into account the grand state electronic configuration of the central ion when choosing a lanthanide for such studies. This is because the Ln2+ ions can have two electronic configurations: 4fn+15d06s0 (for Nd, Sm, Eu, Dy, Tm, Yb) and 4fn5d16s0 (for La, Ce, Pr, Gd) [103]. The letters should be avoided in the design of new performant SIMs, due to the presence of an unpaired electron at the 5d level.
3.1.2. Complexes of tris(3,5-dimethylpyrazolyl)methane
The next group of tripods is a family of trispyrazolylmethane. In contrast to this boron congener tripod, tris(3,5-dimethylpyrazolyl)borate-anion, trispyrazolylmethane terdentate ligands are neutral in a majority known to date [Ln(TPM)(anion)3] (TPM = tris(3,5-dimethylpyrazolyl)methane) compounds. For the first time, air-stable complexes of rare earths with TPM were obtained in 2007 by Sella et al [45]. The six-coordinated complexes [Ln(TPM)Cl3](CH3CN)2 were obtained in acetonitrile solution for Ln3+ = Y, Ce, Nd, Sm, Gd, and Yb. Authors have reported the crystal structure only for Y3+. Later, Long et al [24] have studied isostructural complexes for Tb3+, Dy3+ and Er3+ with the same composition. These compounds crystallize in the space group P21/n with two solvent molecules. The molecular structure of [Y(TPM)Cl3](CH3CN)2 (5) is presented in Figure 2. The SHAPE [104] analysis reveals that the coordination environment of Ln3+-ion is a distorted octahedron in [Ln(TPM)Cl3](CH3CN)2.
The isomorphous [Ln(TPM)(OArMe2)3](thf)3 (6) (Ln = Y, Nd, Sm, ArMe2 = Ph-2,6-Me2) were obtained in a concentrated THF solution at −30 °C. These complexes crystallizes in the space group P1̄ with three solvent molecules. The molecular structure of the [Sm(TPM)(OArMe2)3](thf)3 (6) is shown in Figure 2. In all three compounds, each central ion is six coordinated with C3 symmetry having the trigonal antiprismatic coordination environment of the metal center, with one smaller triangle composed of N-atoms and a larger one having OArMe2 groups in its vertices. This is seen clearly by comparing the ∠(N−Ln−N) and ∠(X−Ln−X) angles (∼70 vs 100−108°). It should be noted, that in [Ln(TPM)(OArMe2)3], the Ln−N distances (2.565(5) Å) are longer than those in 5 (2.459(5) Å), suggesting that the binding of the TPM tripod is subtle to modifications in steric demand of the adjacent ligands in the Ln coordination sphere. Compared with six-coordinate compounds comprising the isosteric but anionic TpMe2 tripods, the average M−N distance is also noticeably longer (2.44(1) and 2.443(7) Å) in [Sm(TpMe2)]2BPh4 and [Sm(TpMe2)2]I [44], being consistent with the weaker Ln interaction of with neutral ligand.
The molecular structure of seven-coordinate [Sm(TPM)Cl3(thf)]·thf (7) is presented in Figure 2. By contrast, with the unsolvated six-coordinate complex 5, 7 contains one thf ligand, which can probably be accommodated due to larger coordination sphere of Sm than that of Y. The two studied [Ln(TPM)(OTf)3(thf)] complexes are isomorphous due to the almost the same ionic radii of Y3+ and Ho3+. The molecular structure of for the latter (8) is shown in Figure 2. The coordination sphere of the triflate or chloride complexes [Ln(TPM)(X)3(thf)] may be described differently. Firstly, as a trigonal antiprism defined by one tripod and three anionic ligands (X = Cl, OTf), with a thf-ligand lying on the C3 axis passing through the top of the tripod, and the triangular face defined by the anions. Secondly, as a tricapped trigonal pyramid, with the three N atoms of the TPM in the triangular base and with the thf oxygen defining the apex [45]. As in the case of the six-coordinate compounds, the triangles defined by the nitrogens are smaller than those defined by three anionic ligands.
Much later were synthesized and studied the three isomorphous complexes [Ln(TPM)(NO3)3]·MeCN (Ln3+ = Tb, Dy, Er), which crystallize in the P21/n space group with one heteroleptic [Ln(TPM)(NO3)3] complex in the asymmetric unit [24]. The molecular structure of [Ln(TPM)(NO3)3]·MeCN (9) is presented in Figure 2. The coordination sphere of 9 is composed of three nitrogens from the tripodal ligand and six oxygens from bidentate nitrate anions. The Tb–N distances ranging from 2.482(2) to 2.530(2) Å are slightly longer than the Tb–O ones (2.416(2)–2.458(2) Å). The analysis of the 9-coordinate polyhedron by the SHAPE software indicates that the geometry of the lanthanide site could be best described as a spherical tricapped trigonal prism [24].
Magnetic studies for TPM coordination compounds were performed only for the two type of the anionic co-ligands (chloride and triflate) for the three Ln3+ ions - Tb, Dy, Er, congeners of the compounds 5 and 9. For these complexes, the room temperature χT values correspond well to the theoretical ones predicted for single Ln ions consuming the free ion approximation. A temperature decrease induces a negative deviation of χT for all compounds, reflecting the depopulation of the mJ states [24]. Besides, the magnetization for the both series does not saturate even at a field of 7 T witnessing the pronounced magnetic anisotropy for all six complexes. Field induced slow magnetic relaxation was observed for all nitrate compounds and only for the Er congener of 5. Based on these studies, the authors argue that the manifestation of a field-induced slow magnetization relaxation is greatly reliant on the anion’s nature. While nitrate moieties appear to be suitable to stabilize the oblate electronic density of Dy3+, chloride ions generate an equatorial crystal-field allowing the slow relaxation of the prolate Er3+ ions [24].
3.1.3. Complexes of anionic tripodal ligand − tris(3,5-dimethylpyrazolyl)-methanide
The electronic analog of tris(3,5-dimethylpyrazolyl)borate-anion is a deprotonated TPM − tris(3,5-dimethylpyrazolyl)-methanide (TPM*). Only a few Ln complexes are known to date [105]. Among them the three complexes with diamagnetic central Ln3+ ions [Ln(TPM*)Cl3][Li(thf)4] (Ln = Sc, Y (10), Lu) were synthesized and structurally characterized. The molecular structure of [Y(TPM*)Cl3][Li(thf)4] (10) is shown in Figure 2. All complex anions have C3 symmetry, each metal ion being surrounded by three N atoms of the TPM* ligand and three chlorides, which form a distorted-octahedron polyhedron also found for 5, despite the different space groups: P21/n and R3 for 5 and 10 respectively. It should be emphasized that the Y−N bond lengths of 2.425(5) Å for [Y(TPM*)Cl3]− (in 10) are somewhat shorter comparatively to those in the anions [Y(TPM)Cl3]− and [Tb(TPM)Cl3]− 2.459(2) Å [45] and 2.472(3)–2.490(3) Å [24]), but are closed to those of 2.425(6) Å for DyCl3(TpMe2)]− [90]. This makes sense, since the tripodal ligand in both the latter and 10 has a negative charge, while the tripod in complexes with longer Ln-N bonds is neutral.
To our great regret, we could not find a single publication describing the magnetic properties of methanide-containing complexes of paramagnetic lanthanides.
3.2. Complexes with the pyridyl bearing tripodal ligand
Pyridine containing tripodal ligands have earlier been described [1]. Moreover, a diversity of bridgehead atoms such as nitrogen, carbon, and phosphorus [106], as well as other elements: Si [107], As [108], Al and In [109], Sn [110,111], Pb [112] and B [113] have been included to alter the properties of the coordination metal complexes both in terms of polyhedral geometry and to tune the electronic structure.
Strangely enough, structurally characterized complexes of the tris(2-pyridyl)methane class for lanthanides have not yet been obtained. Only a small group of exotic organometallic complexes based on tris(2-pyridyl)metallates that are unstable under normal conditions has been obtained and structurally characterized. This family of Ln-complexes will be briefly discussed in the next subparagraph of the review.
3.2.1. Complexes of tris(2-pyridyl)metallates
The most numerous group of Ln-complexes including tris(2-pyridyl)metallates are the coordination compounds of anionic tris(pyridyl)stannate (TPS) [114,115,116,117].
Interaction of [Ln(η5-C5Me5)2(OEt2)] with the lithium [LiSn(2-Py-5-Me)3(thf)] in THF solution using a 1:2 ratio gives the sandwich like [Ln{Sn(2-Py-5-Me)3}2] (11) complexes for divalent Ln = Eu and Yb [114]. The formation of 11 displays a more pronounced steric demand of the tripodal stannate ligand than C5Me5. This is comparable with similar reactions of tris(pyrazolyl)hydroborates [HB(R2pz)3]− (R = H, Me; pz = pyrazolyl) with [Ln(η5-C5Me5)2] species giving the analogous LnII-complexes [Ln(HB(R2pz)3)2] [114].
Both compounds 11 crystallize in the same space group P212121 and have very close cell parameters. The molecular structure for europium complex is shown in Figure 3(11).
The Ln atoms of 11 are in a distorted octahedral coordination environment formed by six pyridyl nitrogens with a staggered organization of the pyridine rings around the Ln center. The coordination sphere around Ln is strongly distorted and the large size of the Ln atom affords a slight twisting of one of the pyridine rings of each Sn(2-Py-5-Me)3 ligand unit around its Sn—Cpy bond. Besides, the non-central location of the Ln atom in the cage is reflected by a bending of the Sn⋅⋅⋅Ln⋅⋅⋅Sn axis with 165.50(1)° and 167.28(13)° for Yb and Eu ions respectively [114]. The Sn bridgehead atom of the tripod reveals a trigonal pyramidal coordination mode (Figure 3(11), left).
The reaction of [LiSn(2-Py-5-Me)3(thf)] with [Ln(η5-C5Me5)2] precursors resulted in formation of the first LnII sandwich complexes 12 involving the anionic TPS in a κ3N-coordinating manner and featuring “naked” SnII centers, which can be used for following κ1Sn-metal coordination (Eu complex is shown as example in Figure 3(12)) [114].
Compound 12 crystallizes in the cubic space group Pa with the Eu2+ lying on a special crystallographic position and four molecules in the unit cell. The longer Eu—N, (2.611(3) Å) and Eu⋅⋅⋅Sn (3.8874(4) Å) distances of are consistent with the larger size of the Eu2+ vs Yb2+ cation in 11. The most pertinent feature is the Sn—Li contact of 2.792(12) Å in 12 is shorter than the covalent Sn—Li bond lengths (2.831–2.897 Å) [118]. Encapsulation of the large Eu2+ ion and additional κ1Sn coordination of the two inner TPS ligands originates an opening of the tripodal structure (C-Sn-C = 99.6(1)°) compared to both outer TPS frameworks with “naked” Sn centers (C-Sn-C = 93.6(1)°) [114]. Furthermore, the κ1Sn binding gives a practically ideal octahedral environment of the Eu2+ ion with a linear Sn⋅⋅⋅Eu⋅⋅⋅Sn axis (180.0(1)°).
Figure 3.
Molecular structures (left) and octahedral coordination polyhedra Ln-N6, with colored faces forming the tripod bases (right) for the compounds 11-16. Hydrogen atoms are omitted. The picture was made using open crystallographic data.
Figure 3.
Molecular structures (left) and octahedral coordination polyhedra Ln-N6, with colored faces forming the tripod bases (right) for the compounds 11-16. Hydrogen atoms are omitted. The picture was made using open crystallographic data.
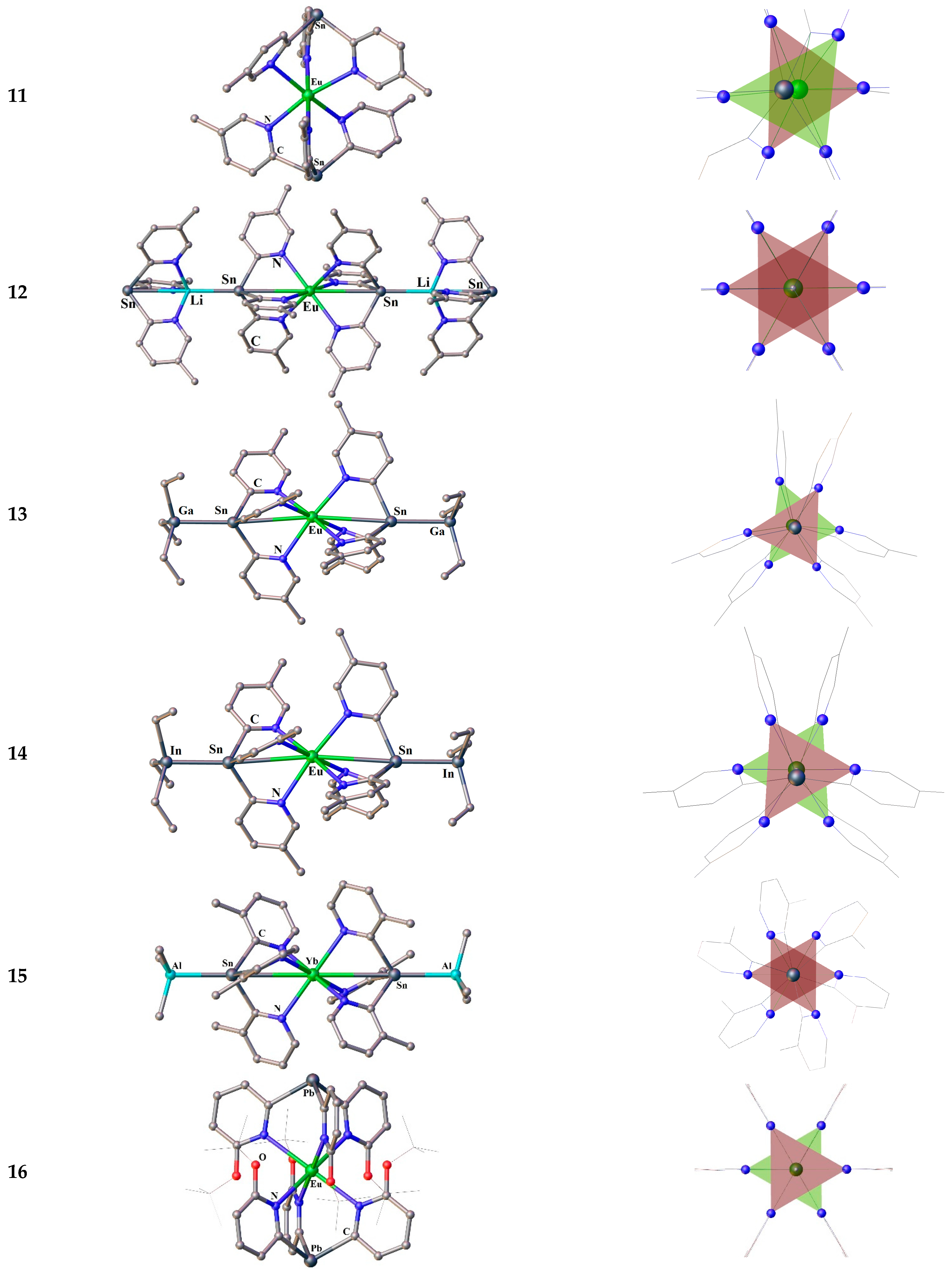
The room temperature reactions of a trialkylmetallates MAlk3 (Alk = Me for Al, and Et for Ga and In) with complex [Ln{Sn(2-Py-n-Me)3}2] gave the following organometallic species: [Eu{Sn(2-Py-4-Me)3}2{GaEt3}2] (13), Eu{Sn(2-Py-4-Me)3}2{InEt3}2] (14) and [Yb{Sn(2-Py-3-Me)3}2{AlMe3}2] (15) [115].
The compounds 13 and 14 (Figure 3) have the similar crystal structures crystallizing in the same space group C2/c [115], the Eu ions being in a distorted octahedral geometry, characteristic for the compounds 11 described above. In contrast with the complexes 11, the non-central location of the Ln ion in the coordination octahedron 13 and 14 is much less expressed in the bending of the Sn⋅⋅⋅Eu⋅⋅⋅Sn axis by 176.40(5)° and 172.05(8)° for 13 and 14, respectively. In the solid state, complex 15 (Figure 3) reveals bonding characteristics similar to those for 13 and 14, but differs in that the Yb ion possesses almost perfect octahedral environment with a strictly linear Sn⋯Yb⋯Sn axis (180.0°) [115].
A few words about the only sandwich compound of a lanthanide complex comprising tris(2-pyridyl)plumbate. The compound [Eu{Pb(2-Py-6-OtBu)3}2] (16) have been isolated from a concentrated THF solution [117]. Like 12, the complex 16 (Figure 3) crystallized in Pa space group. The Eu2+ ion is coordinated by six nitrogen donor atoms of two tripodal with Eu—N bond length of 2.699(2) Å Eu⋅⋅⋅O distance of 4.0215(1) Å, which is greater than those of 2.611(3) and 3.501(3) Å in 12. This is because of a larger radius of bridgehead ion in Pb(2-Py-6-OtBu)3 tripod. Although the cavity of the tripodal ligand offers an almost ideal octahedral coordination sphere for the Eu2+ cation, which is well encapsulated and clearly separated from the formally negatively charged lead bridgehead atoms, compound 16 is not stable in THF solution [117].
The EPR solution studies at ambient conditions for 16 in presence of nitrosobenzene (NOB) suggested the generation of a radical entity. The X-band EPR spectrum exhibits one broad signal corresponding to a EuII 4f7 spin system with the value g0=1.989±0.001 and a line width of ΔBPP=(31.0±0.1) mT. An identical EPR spectrum was obtained and without spin trap. In addition to a broad signal, the spectrum shows a triplet of multiplets. The well-resolved hyperfine structure (hfs) is due to the coupling of the unpaired electron with the 14N (I=1) nucleus and three groups of nonequivalent protons of NOB. The spectrum has been well simulated giving the values g0=2.0057±0.0005 and a0N=(1.097±0.005) mT, as well as a0H=(0.249±0.005) mT, a0H=(0.235±0.005) mT, and a0H=(0.089±0.005) mT, respectively. The trapped radical was stable over a period of few hours. However, no 207Pb hfs coupling could have been observed, indicating radical-adduct formation.
The complexes comprising tris(pyridyl)aluminate (TPAl) tripods are completed this section. As was shown by R. García-Rodríguez et al. [119,120], complexes of Ln2+ with TPAl are air unstable and can only be obtained employing sterically hindered tripods - [(Alkyl)Al(2-Py-6-R)3]−, where Alkyl = Et and R = Me or Br. It means that the coordination ability of TPAl can be adjusted by the steric and electronic character of substituents at a 6-position of the pyridyls. While [EtAl(2-py-6-Me)3]− (17) coordinates strongly to Ln2+ ions, [EtAl(2-py-6-Br)3]− (18) forms much weaker complexes, and [EtAl(2-py-6-CF3)3]− does not coordinate at all [120]. Synthetic and structural investigations were devoted essentially to the complexes of two anions [EtAl(2-Py-6-Me)3]− and [EtAl(2-Py-6-Br)3]−. The 2 : 1 ratio reactions of the 17 with LnI2 (Ln = Eu, Yb and Sm) in thf at room temperature gives deep-orange (Eu) or purple (Yb and Sm) solutions after 24 h, indicating of Ln coordination [119,120]. SC-XRD study confirmed a formation of the sandwich complexes: [{Ln(EtAl(2-py-6-Me)3}2] (Ln = Sm 18, Eu 19, Yb 20) and [{Eu(EtAl(2-py-6-Br)3}2] (21), Figure 4. The complexes 18 and 21 crystallizes in P Ī space group. The first contains one CH3C6H5 molecule as a solvate since it was crystalized from toluene. The coordination environment formed about the Sm and Eu are slightly elongated octahedron and trigonal antiprism respectively, although the both compounds have strait line passing through the bridgehead atoms of the tripods (Al-Ln-Al the angle is 180°). The compounds 19 and 20 crystallize in P 43212 and P 21/n space groups correspondingly. Molecules of 19 and 20 feature six-coordinate, distorted-octahedral lanthanide metal ions with the N–Ln–N angles being approximately 90° and slightly bent Al–Ln–Al axis (177.17° (19) and 178.23° (20))[120].
Figure 3.
Molecular structures (left) and coordination polyhedra Ln-N6, with colored faces forming the tripod bases (right) for the compounds 18-21. Hydrogen atoms are omitted. The picture was made using open crystallographic data.
Figure 3.
Molecular structures (left) and coordination polyhedra Ln-N6, with colored faces forming the tripod bases (right) for the compounds 18-21. Hydrogen atoms are omitted. The picture was made using open crystallographic data.
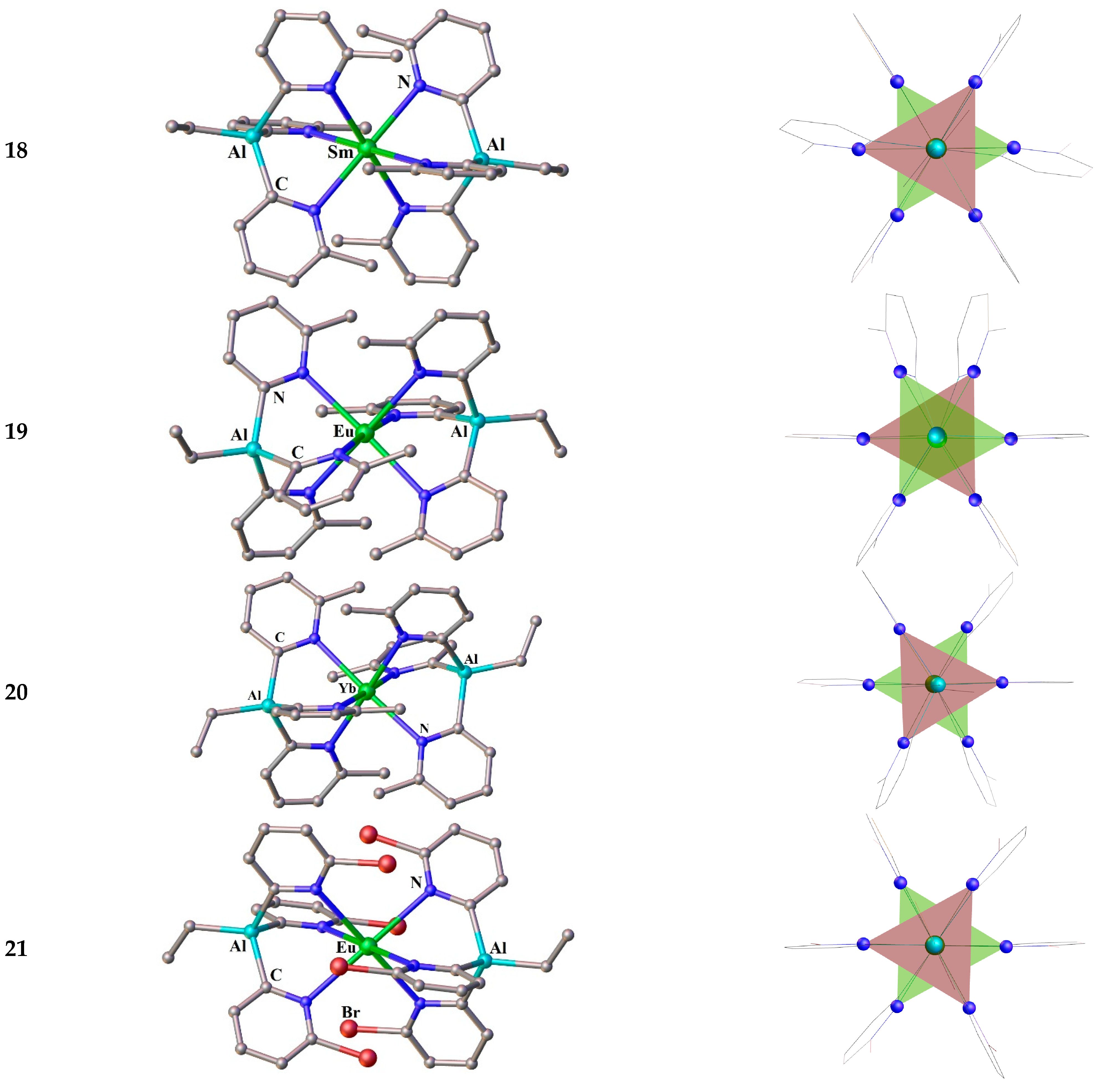
None of the Ln compounds comprising tris(2-pyridyl)metallates have been reported on their magnetic behavior. The next fairly representative group of lanthanide compounds with tripodal ligands are complexes with tris(2-pyridyl)amines.
3.2.2. Complexes of tris(2-pyridyl)amines
The interaction of tris(2-pyridylmethyl)amine (TPA) with the precursors LnHal3(thf)4 and Ln(OTf)3 was investigated in anhydrous environment and in the presence of water [3,7]. In the absence of water, the succeeding formation of mono- and bis-(TPA) complexes were detected using Ln/ligand ratio of 1 and 2, respectively. The mono-TPA complexes [Ce(TPA)I3] (22) [3], [Ln(TPA)Cl3] (Ln(III) = Eu, Tb, Lu ([7]), and the bis(tpa) complexes [Ln(TPA)2]X3 (X = I, Ln3+ = La, Ce, Nd, Lu [3] and Sm [6]; X = OTf, bis(tpa) complexes [Ln(TPA)2]X3 (X = I, Ln3+ = La, Ce, Nd, Lu [3] and Sm [6]; X = OTf, Eu [3]) were obtained under anhydrous conditions and their crystal structures were determined.
Figure 4.
Molecular structures (left) and coordination polyhedra Ln-N6, with colored faces forming the tripod bases (right) for the compounds 22-25. Hydrogen atoms are omitted. The picture was made using open crystallographic data.
Figure 4.
Molecular structures (left) and coordination polyhedra Ln-N6, with colored faces forming the tripod bases (right) for the compounds 22-25. Hydrogen atoms are omitted. The picture was made using open crystallographic data.
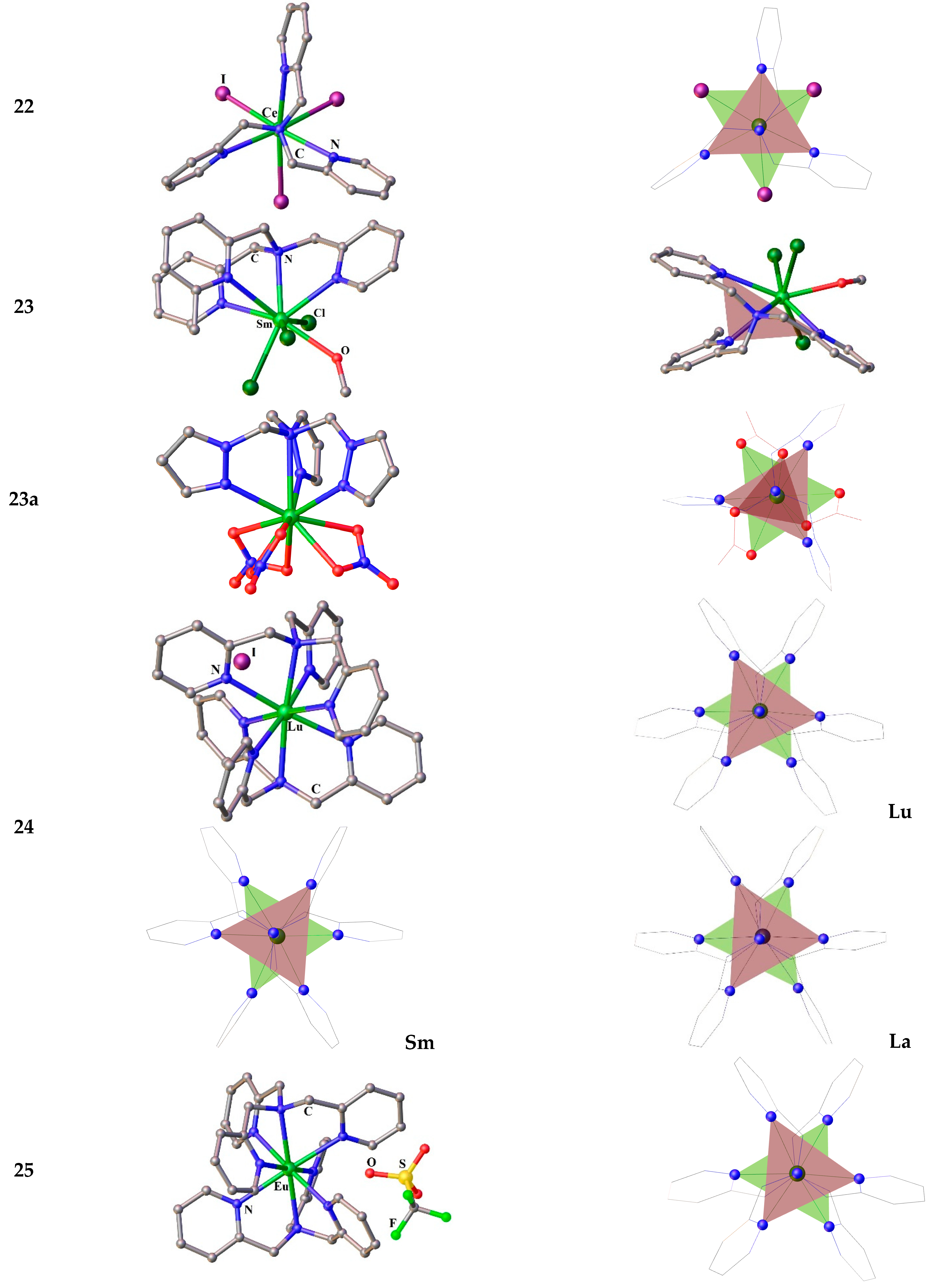
The complexes [Ln(TPA)Hal3] (see as example Figure 4(22)) are isomorphous and crystalize in the space group P21/c without any solvent molecules per a crystal cell. Note that the tripod base and a plane passing through the halide ions are not parallel.The mono-tripod complexes [Ln(TPA)Cl3(CH3OH)]·CH3OH (23) (Ln3+ = La, Nd. Sm) (Figure 4) were also obtained starting from the corresponding hydrated lanthanide halide [LnCl3(H2O)6] [121]. Nonetheless, the compounds 23 are not of interest as high performance SMMs design because they do not have monoaxiality.
In contrast, a small group of heteroleptic complexes containing three nitrates and one tripodal tetradentate ligand tris((1H-pyrazol-1-yl)methyl)amine) (TPzMA) are enough axial, Figure 4(23a) [12]. The authors present the synthesis, structures, and photophysical and magnetic properties of a sequence complexes [Ln(TPzMA)(NO3)3]·nMeCN (Ln = Eu, Tb, Dy, Er, n = 0.5; Yb, n = 0). The SCXRD analysis reveals that, among the investigated compounds, the compounds of Eu, Tb, Dy, Er are isomorphous and crystallize in the triclinic P1̅ space group with a single complex molecule and 1/2 of an CH3CN molecule in the asymmetric unit. Their structures includes a mononuclear complex in which the Ln3+ ion coordinated by four N-atoms from TPzMA and six oxygens atoms belonging to three bidentate nitrate anions, Figure 4(23a). The tripodal ligand coordinates to the central ion in a symmetrical manner frowning together with nitrates the Ln3+ coordination polyhedron, which is better described as a distorted spherocorona [12]. Contrary to their congeners, the Yb complex crystallizes in the monoclinic P21/c space group without solvates. Probably, due to the Ln contraction and the fall in intramolecular distances resulting in steric repulsions in the polyhedron, only two nitrate moieties are bidentate and the other one is monodentate, leading to a muffin geometry of the Yb site with Cs symmetry [12].
The europium, terbium, and dysprosium analogues exhibit a lanthanide-based luminescence, while the dysprosium, erbium, and ytterbium compounds show a field-induced slow relaxation of their magnetization involving Raman and direct processes.
The complexes [Ln(TPA)2]I3·CH3CN (Ln = La, Ce, Nd, Sm) are isostructural to each other (see Figure 4(24)) [3,6]. The metal ions are eight coordinate by the two-tetradentate ligands that wrap around the metal in a pseudo-D3-symmetric arrangement [3]. All the complexes crystallize as a single enantiomer in the non-centrosymmetric space group P212121 [3]. The coordination polyhedra are best defined as distorted cubes with a less distorted geometry for Lu [3].
It was not possible to obtain the crystal structure of [Eu(TPA)2]·3OTf under water-free conditions. The single crystals of [Eu(TPA)2]·3OTf·CH3CN·0.3H2O (25) were obtained in the presence of ~0.5 equivalent of H2O. The molecular structure of 25 (Figure 4(25)) is very close to those of the Ln-bis(tpa) iodides. Contrarily to the complexes 24, complex 25 crystallizes in a centro-symmetric space group P 21/c with two complex molecules per asymmetric unit and 0.15 molecule of H2O, which apparently helps the crystallization [3].
Along with to the Ln3+ TPA complexes, their congeners for Ln2+ were also isolated. Starting from LnI2 the stable mono- and bis-TPA complexes: [Yb(TPA)I2(MeCN)]·MeCN (26, Figure 5(26)) and [Ln(TPA)I2] (Ln2+ = Sm, Eu) (27), [Ln(TPA)2]·2I·0.5MeCM (Ln2+ = Sm, Yb) (28, Figure 5(28)) were prepared and characterized [6]. When a bulk anion NaBPh4 is added to the previous reaction mixture, a number of compounds with TPA and its analogs methylated, a family of Ln2+ complexes was also obtained in inert conditions [122]. The compounds [Ln(MenTPA)2](BPh4)2 (n = 0–3 reliant to methylation degree of the 6-position of the pyridyl rings of MenTPA, when n = 0 — Ln2+ = Eu, Yb 29 (Figure 5(29)), n = 2 — Ln2+ = Eu, Yb (30) and n = 3 — Ln2+ = Eu (31, Figure 5(31))) have been synthesized and their structural, electrochemical and photophysical properties studied [122].
The complexes 27, [Ln(TPA)2]·2I, crystallize in the monoclinic centrosymmetric P21/c space group while the complexes 28, [Ln(TPA)2]·2I·0.5CH3CN crystallize in the non-centrosymmetric monoclinic Cc space group [6].
Complexes 29 crystallize in the monoclinic P21/n space group, contrary to 30 and 31, which crystallize in the triclinic P1ˉ space group, with the lowering of symmetry attributed to the presence of the methyl substituents that result in crystallographic disorder [122]. The Ln2+ ion is 8-coordinate in all sandwich compounds. Continuous shape measures using the software SHAPE 2.1 were employed to determine the geometry of the Ln centers and suggest that the coordination geometry is closest to cubic in all cases [122]. The Shape distortion parameters are in the range 0.69–1.53 for the cubic geometry, the further the value being from zero, the greater the distortion from ideal geometry [122].
Unfortunately, for all the above compounds comprising TPA (both for sandwich and semisandwich), the magnetic properties have not been studied. Only two [Ln(TPA)(Anion)3] compounds and four [Ln(TPzMA)(NO3)3]·complexes have been magnetically characterized [123]. The description of their magnetic behavior we conclude the current section of the review.
Two mononuclear seven-coordinate Dy3+ complexes [Dy(TPA)Cl3] (32) and [Dy(TPA)(OPhCl2NO2)3]·0.5CH2Cl2 (33) have been synthesized based on the neutral ligand TPA and either the strong strength ligand 2,6-dichloro-4-nitrophenol (Cl2NO2PhOH) or the weak ligand field donor Cl− [123]. As in the tri-iodine compounds of type 22, mentioned above, the Dy3+ ions in complexes 32 and 33 have seven-coordinated capped octahedral and capped trigonal prismatic coordination geometries, respectively [123]. Magnetic studies showed that both Dy compounds possess field-induced slow magnetic relaxation. The energy barrier for 33 is higher than that of 32, which is due to the strong ligand field of Cl2NO2PhO−versus Cl−, resulting in a larger magnetic anisotropy of 33 as compared to 32 [123]. The direction of the magnetic anisotropy axes in both complexes deviate remarkably from the symmetry axis of the capped octahedron (C3v) and the capped trigonal prismatic (C2v), which explains the poor performance of SIM behavior for both complexes [123].
The room-temperature χT values of 12.97, 13.92, 11.54, and 3.20 emu/mol for 23a, respectively for [Ln(TPzMA)(NO3)3]·(Ln = Tb, Dy, Er, Yb), are in quite good accordance with the theoretical values of 11.82, 14.17, 11.48, and 2.57 emu/mol estimated for a unique Ln3+ ion [12]. Upon cooling, all compounds exhibit the typical decrease in χT caused by the thermal depopulation of the mJ levels reaching values at 1.8 K of 5.25, 7.35 [12]. For all complexes, the absence of saturation for the magnetization curves testifies to the existence of magnetic anisotropy, as expected for such lanthanide ions [12]. Under a zero-dc field, no strong out-of-phase susceptibility (χ″) components was observed for any of the samples pointing out the occurrence of fast QTM.
Excluding the Tb complex, for which the out-of-phase component remains weak, all other complexes show a strong χ″ component upon applying dc field. For the Er and Yb compounds, the appearance of a second low-frequency peak was detected at high magnetic fields. It might originate from different relaxation mechanisms actuated by large magnetic fields [12]. Appears therefore that the neutral tetradentate TPzMA ligand in association with nitrates and the resulting low-symmetry of the lanthanide site do not provide the requirements to maximize the anisotropy for either oblate or prolate lanthanide ions in zero dc field [12].
4.1. Complexes of the tripodal nitroxyl radicals
Metal-radical systems are recognized to display better SMM characteristics [124]. For example, an extra electron transforms (n-Bu)4N[TbPc2] (Pc is a dianion of phthalocyanine) into [TbPc2] (Kramers system), guaranteeing twofold degeneracy of all electronic levels enforced by time reversal symmetry, irrespective to ligand field symmetry[125]. Park et al. [126] stated that the higher energy levels sturdily depend on the ligand type, molecular symmetry, and overall charge of the molecules. Additionally, ligand distortion and molecular symmetry play a key role in transverse CF parameters leading to tunnel splitting. The latter induces QTM. Compared to the anionic complex [TbPc2]–, for the neutral complex, no tunnel splitting was observed for the ground and excited state quasi-doublets [126]. For the anionic complex, the tunnel splittings of 0.007, 0.090, and 7.969 cm–1 have been observed for the ground, first and second excited state quasi doublets. The perceived dissimilarity in the magnetic behavior was attributed to the significant an exchange interaction, Jex of ~ 7 cm–1, existing in the compound [TbPc2] compared to the anionic one [126]. Moreover, the axial CF parameter (B20 = –5.93 vs –5.05 cm–1) is larger, and the transverse CF parameter (B22 = 0.3 to 0.4 cm–1 vs. 0.8 to 3.6 cm–1) is small for the latter [126].
Real influence of exchange coupling on magnetic relaxation dynamics was reported by Long et al., firstly reported very large TB values [127]. They have obtained the binuclear Tb3+ complex [{[(Me3Si)2N]2(THF)Tb}2(µ-η2:η2-N2)–] (TbN2Tb), in which the large magnetic interaction between Tb3+ and radical established to quench QTM in the absence of magnetic field. This SMM relaxes in the thermally activated direct process having (QTM probability B~10–6) an Ueff=227 cm–1 and TB~14 K [127]. Later, the strong coupling of –27 cm–1 was communicated for the Gd congener of TbN2Tb [128]. For the GdN2Gd the electronic structure calculations have been performed, which clearly evidence about the direct overlap between the Gd3+ 4f-orbitals and the π* orbital of the radical [129].
4.1.1. The functionalized by 2-pyridyl groups paramagnetic tripods and their complexes
Nitroxyl stable radicals a widely used in the field of molecular magnetism [130,131,132,133,134,135]. However, till now only three tripodal nitroxyl radical functionalized by pyridyl groups (Figure 6) are known [25,136]. The complexes of the 3d metal ions with the radical tripods 34-36 are most of a sandwich type [25,26,27,28,29,30,31,32,137]), while a number of semi-sandwich Ln-complexes were recently synthesized [138,139] (Figure 7(37)). In contrast to the tetradentate tripods of TPA described above, the tripodal oxazolidine radical (Rad) 35 is sterically rigid, forming a pyramid upon coordination with an almost right triangle at the face for both monoradical (37) and biradical complexes (38) (Figure 7) [138,140].
4.1.2. Structural features of the paramagnetic tripod complexes
According to PRXD study, the monoradical complexes [LnRad(NO3)3] (37) are isostructural. The crystal structures for the 37 complexes of Ln3+ = Dy, Tm, Y, Eu and Lu ascertained by SC-RXD experiments. Like their closest relatives, the nitrate complexes of Ln with diamagnetic tripod TPM (9), the compounds 37 crystallize in the non-centro symmetric P21/n space group [138]. In this complexes, Rad coordinates the Ln ion in a tridentate manner via two N atoms of the pyridyl substituents and an O atom of an NO group. The coordination sphere is further complemented by three bidentate nitrates to give an LnO7N2 polyhedron [138]. Consistent with the continuous symmetry measures method [104], the polyhedron is best described as a spherical tricapped trigonal prism. The radical donor atoms compose a triangular face of the prism. In addition, the coordinated oxygen atoms of the nitrate ligands, they also form triangular planes parallel to that of Rad (Figure 7(37), right). Moreover, the centers of all triangles lie on the same axis passing through the central Ln atom, which bears witness to with S3 symmetry.
Figure 7.
Molecular structures (left) and coordination Ln polyhedra with colored triangles forming the tripod bases (right) for the compounds 37-39. Hydrogen atoms are omitted. The picture was made using open crystallographic data.
Figure 7.
Molecular structures (left) and coordination Ln polyhedra with colored triangles forming the tripod bases (right) for the compounds 37-39. Hydrogen atoms are omitted. The picture was made using open crystallographic data.
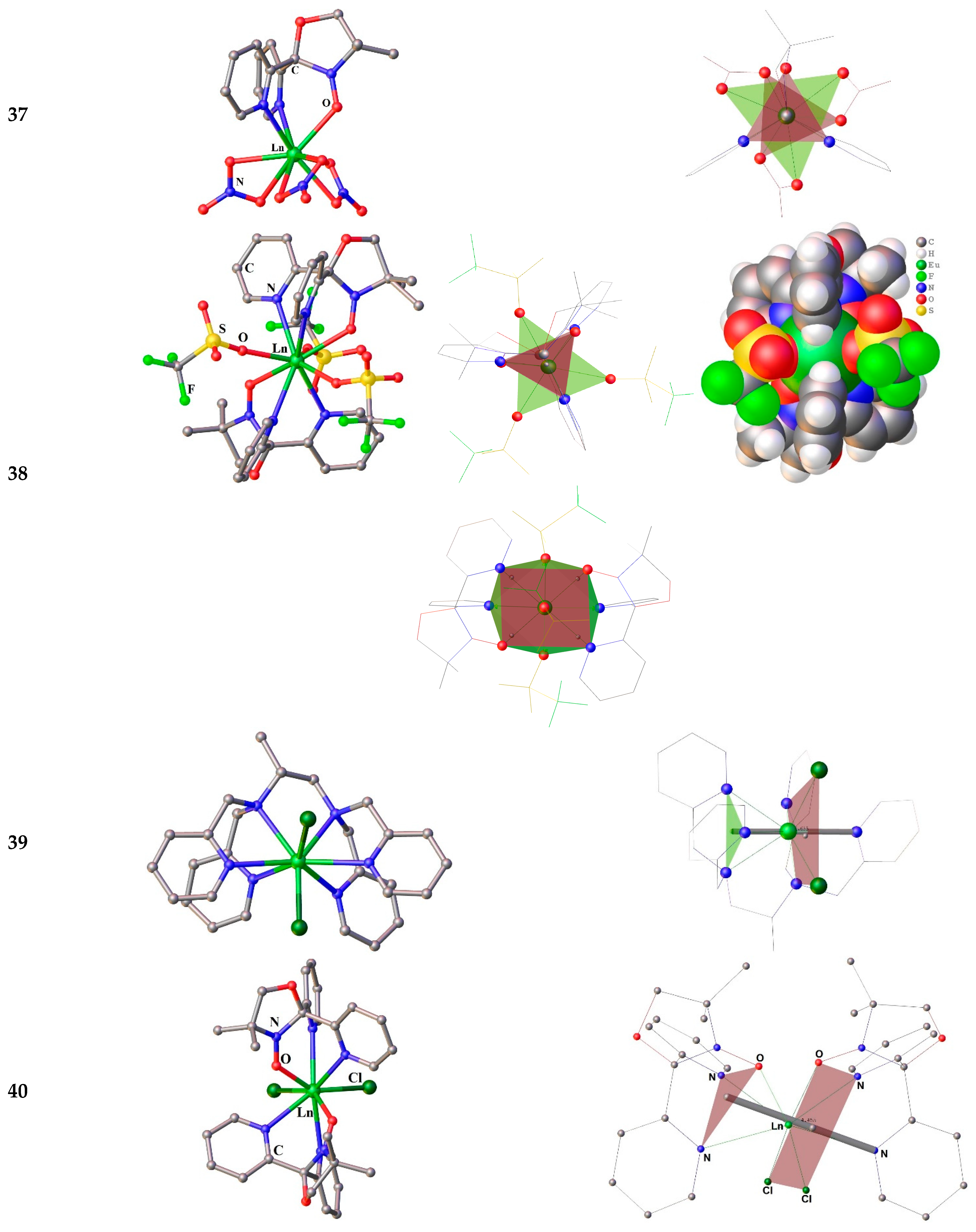
The Ln–O bond distances related to the NO moiety vary in region of 2.34 – 2.38 Å. N – O bonds length of the nitroxyl group fluctuate in the 1.25–1.28 Å interval. Markedly, all bond lengths and angles of the complexes are in the expected range [138]. In biradical complexes 38, the mode of ligand coordination does not differ from that of monoradical complexes 37. The Ln environment is composed of three monodentate anions to give LnO5N4 polyhedron. Unlike nitrate ligands, triflates are bound to Ln by only one donor O atom and are located in the equatorial plane of the compounds 38. At first glance, the latter have D3h pseudosymmetry, since both tripod bases are placed one above the other, and the donor atoms of the OTf ligands form a triangle (Figure 7(38)). However, due to the “sandwich” bending, all triangles are highly distorted. Though, the SHAPE analysis [104] gave the polyhedron geometry as a spherical capped square antiprism with the point symmetry close to C4v [140]. The radical donor atoms and two OTf compose faces of the antiprism while oxygen of the third triflate arranges above the rectangular face [140].
Only two related compounds for the sandwich complex 38 are Ln3+ complexes with a quasi tripodal ligand 1,3,5-trimethyl-triazacyclohexane (Me3tach) – [Pr(Me3tach)2(OTf)3] [141] and HMe3tach[La(Me3tach)2(OTf)4] [142], Figure 8. The first is so bent that the three triflates are not evenly distributed in the equatorial plane, while the latter is capable to house even more triflates. Such a geometrical organization of the Ln coordination sphere is favorable to stabilize the highest mJ ground states for the prolate tripositive lanthanides ions (Pm, Sm, Er, Tm, and Yb). That is why the 38-Dy complex studied by us earlier does not exhibit slow magnetic relaxation. The most attractive geometry (for prolate type Ln3+) among compounds with this pseudotripodal ligand has the recently studied the only non-bent sandwich complex [La(Me3tach)2Cl3] [142] having three chlorine anionic ligands in equatorial plane, Figure 9. However, chloride anions are bulky enough to enter threesome the inner sphere of smaller Ln3+ ion beginning from samarium. Nevertheless, two chlorides can fit quite well in the equatorial plane of the Ln coordination sphere. We did not find an example of such a specie for any [Ln(tripod)2Cl2]+. According to the CCDC, there are only a few examples of such compounds with bis-tripodal ligands. One of which is the Eu3+ complex [EuIIICl2{R)-tppn}]ClO4 (39) [143], Figure 7. The coordination geometry of 39 is best described as a distorted dodecahedron. The authors point out that the four coordinated pyridyl nitrogens and one from the two Cl-atoms are approximately on a plane and form a pentagon, the other Cl being coordinated almost perpendicular to this plane. However, from our point of view, it is better to choose a different description of the geometry of the compound 39 in order to predict the magnetic behavior. Both Cl anions are best placed in the same plane, as shown in Figure 8. In this case, the molecule have an axis, which could serve as an axis of anisotropy. Our preliminary studies have shown that sandwich-type complexes containing two chloride ligands in the equatorial plane of the cationic complex 40 (Figure 7) can also be obtained for the paramagnetic tripod 35 [144].
4.1.3. Magnetic properties of the paramagnetic tripod complexes
At 300 K, the χT product values are 7.814, 12.210, 14.366, 7.376 emu K/mol for the 37 type compounds of Gd, Tb, Dy, and Tm, respectively. These values are well consistent with the expected ones for the appropriate LnIII ion plus a radical (8.255, 12.195, 14.545, 7.525 emu K/mol) [138]. With the temperature lowering, the χT value for all derivatives drops considerably, which can be ascribed to CFS and/or the antiferromagnetic metal-radical interaction. It could be assumed a dominance of the CFS for all Ln except Gd, which is magnetically isotropic at first order. It should be emphasized, that the 37-Tm complex, for which the nonmagnetic mJ = 0 ground state is stabilized (χT is equal to 0.375 emu K/mol corresponding to the value of a radical tripod). Furthermore, the important decrease in χT for 37-Gd bears witness to a strong antiferromagnetic (AFM) coupling between the GdIII ion and the radical. The plot M(H) for the 37-Gd complex is very informative. Considering the low crystal field splitting typical of Gd3+, the saturation value of 6 μB is consistent with an AFM coupling between a spin S = 7/2 and a radical spin S = ½ (Stot = 7/2-1/2 = 3) [138]. X band EPR has been performed on [GdRad(NO3)3] in order to obtain precise information about the electronic structure. The spectrum of Gd exhibits several broad transitions.
A satisfactory fitting of both the EPR and the static magnetic measurements can be obtained using the following Hamiltonian:
where the first three terms parametrize the CFS of the GdIII ion, the fourth term is the isotropic magnetic coupling between Gd and the radical and the last two terms are the Zeeman splitting for Gd and the radical, respectively. The finest simulation result was achieved with the values b20 = 5.2 10-2 cm-1, b22 = 1.1 10-2 cm-1, b40 = 3.8 10-4 cm-1, j = +23 cm-1, gGd = 2.0023 and grad = 1.998. Notably, the value of +23 cm-1 of the isotropic coupling constant is unusually large for gadolinium complexes of organic radicals. For example, for Gd3+ hexafluoroacetylacetonate complexes containing one acyclic nitroxyl radical [145,146,147] the |j| value of fluctuates between 9.6 and 12.5 cm-1. For the similar compounds including one nitronyl nitroxyl radical [148,149,150,151,152,153], exchange coupling is also inferior (|j| = 0.77 ÷ 8.35 cm-1). For the Gd-complex comprising congener of Rad TEMPO (TEMPO = 2,2,6,6-tetramethylpiperidin-1-oxyl) metal-radical interaction is very small (j = 2.43 cm-1) [154]. For the mono-semiquinone complex [Gd(HBTp3)2SQ] j = 11.4 cm-1 [155]. Moreover, the exchange interaction strength for Gd-radical in the [GdRad(NO3)3] species is comparable with for the binuclear Gd complex of the purely inorganic single radical N23- obtained in the group of Long [128]. The origin of such a strong magnetic exchange interaction in 37 must be ascribed to a favorable metal-to-ligand orbital superposition. In contrast to the most studied complexes with nitronyl and imino-nitroxyl radicals, in which the spin density is mainly delocalized over four or three centers (O - N ... N – O [156], N ... N - O), in 37, an unpaired electron is shared by only two cites. Consequently, the spin density on the donor oxygen atom in Rad is two times higher than that in the nitronyl nitroxyl radical. Notably, the exchange interaction is directly related to magnetic orbital overlapping, which depends on the symmetry and donor strength of all ligands [138].
Dynamic magnetic studies were to get information on the magnetization dynamics at low temperatures. A frequency scan at various applied fields for 37 was performed at the temperature of 2 K. The only compound to display a relevant nonzero out of phase magnetic susceptibilities was 37-Tb. The latter displays slow relaxation both with and without an external applied field. In zero applied field the relaxation of 37-Tb at 2 K is at the upper edge of accessible frequencies with relaxation time ca. 16 μs, for this reason a temperature study was impossible. When an external magnetic field was applied, the slow relaxation of 37-Tb slows by several orders of magnitude [138]. The relaxation time best fit was obtained using a combination of quantum tunnelling and Orbach processes, giving τQT=12(1) ms, τ0 = 0.9(3) ns and ΔE = 57 cm-1 [138].
The magnetic studies on powder samples of the complexes 38 were also fulfilled. The χmT-value at 300 K for the 38-Dy compound (14.735 emu K/mol) is slightly smaller than theoretical one (14.92 emu K/mol) for the non-interacting two radicals and one Dy3+ ion (S = 1/2, g = 2.0023, 0.375 emu K/mol) (4f9, S = 5/2, L = 5, J = 15/2, gJ = 4/3, 6H15/2, 14.17 emu K/mol). However, this value corresponds well to those of 14.365 emu K /mol, previously obtained for the monoradical complex 37-Dy [138] taking into account the fact that the experimental values for Dy3+ in a related coordination environment are somewhat lower than those for a free ion: 13.92–14.00 [12,157]. On cooling, the χmT value remains almost unchanged up to 90 K, and then steadily falls down to 10 emu K mol−1 at 20 K and finely drops to 4.803 emu K mol−1 at 2 K. The latter is considerably lower than that for the monoradical analogue (6.50 emu K/mol). Since significant intermolecular exchange interactions in 38-Dy can be excluded, the cause of such behavior could be attributed to the radical-radical coupling, which can reach high values (JTEMPO1-TEMPO2/kB = −24.9 K) in [Y(hfac)3(TEMPO)2]) [158]. Moreover, the metal-to-radical coupling in the [Gd(hfac)3(TEMPO)2] has been reported to be either positive or negative for the different radicals of the same molecule (JGd–TEMPO/kB = −6.45 and +4.0 K, obscuring the picture [158]. Therefore, for the very anisotropic Dy, it is difficult to obtain more information also due to the high mixing of the |J,mj> levels favored by the relatively low-symmetry ligand field. In order to elucidate the nature of exchange interactions in 38-Dy [158].
The temperature dependence of χmT for Eu3+ compounds is determined by the thermal population of the 7F1 level nearest to the ground nonmagnetic level 7F0. The excited 7F1 state, closely sited to the ground one, is partly populated already at room temperature. At 300 K, χmT for Eu3+ complexes with diamagnetic organic ligands can vary in the range of 1.032–1.386 emu K/mol1 depending on the ligand field parameters [159]. Therefore, for the complex 38-Eu, the 300 K χmT value of 1.778 emu K/mol is reasonable for the two uncoupled radicals and one Eu3+ ion, but lower than the value of 1.93 emu K/mol found for the bis-nitronylnitroxide (NN) complex [Eu(NN)2(NO3)3] with a different coordination environment (N10 instead of N4O5) [160]. As the temperature pulls down, the χmT decreases gradually, and, starting from 9 K, drops to 0.348 emu K/mol, which is somewhat higher than that for [Eu(NN)2(NO3)3] compound (0.24 emu K/mol). No ac signal was observed in either sample in the measurable interval of frequencies (0.1–10,000 Hz).
4.1.4. Rational design of the paramagnetic tripods and their complexes
As mentioned above, in the presence of anionic ligands in the equatorial plane, it is better to use prolate Ln ions: Pm, Sm, Er, Tm, and Yb. While for more "magnetic" dysprosium and terbium, the presence of anionic ligands at the equator is strictly contraindicated. Therefore, it is necessary to change the structure of the paramagnetic tripod so that there is no space for anionic ligands or solvent molecules in the Ln coordination sphere. An analysis of the molecular structure of sandwich complexes with diamagnetic tripods has showed that low-coordination compounds without anionic ligands are formed in the presence of steric hindrance in a position adjacent to donor tripod atoms giving propeller-like structures with ideal axiality (see compounds 1, 4, 16, 18, 21, 31). It should be noted that in most of the bis-tripodal complexes considered in this review, the ligands are symmetrical tripods, in contrast to the radical 35, which consists of two six- and one five-membered heterocycles. Moreover, the latter comprises a bridgehead carbon atom in ligand. While, other described tripods are more spatial ligands. In complexes with the radical 35, the average value for the side length of the triangular base is about 2.8 Å, while for other bridgeheads it is much larger: 2.9-3.1 for boron, 3.8-4.0 for nitrogen and metallic bridgehead. The larger dimensions allow to the tripods with hetero bridgehead atom “to more deeply set on the head” of the central Ln ion.
On the space filling image of the "burger" 38 (Figure 7), it is clearly seen that two pyridyl substituents of the different oxazolidine radicals 35 are in close proximity, and the shortest contact H…H between them is equal to 2.41 Å. In complex 40, there are also sufficiently close distances between the pyridine rings and the methyl groups of the radical (2.613 and 3.007 Å, Figure 10). Therefore, if an additional methyl group is located in the fourth position of the pyridyl rings (just near donor nitrogen atom), then most likely this will be enough for the paramagnetic tripods to turn relative to each other with the formation of a propeller-like structure in which there will be no place for additional molecules. In addition to increasing the steric load of the pyridyl substituents, it is necessary to make the paramagnetic tripod more symmetrical about the bridgehead carbon atom.
This can be done by going from a five-membered oxazolidine ring to a six-membered oxazinane (41), Figure 11a. The synthesis of such a tripodal radical becomes rather complicated due to commercially unavailable bis(6-methyl-2-pyridyl)methanone and 3-amino-3-methyl-butan-1-ol, the condensation of which yields its diamagnetic precursor.
Other way is a choice of 1-imidazole as a functional group (instead of pyridine) for oxazolidine nitroxide, Figure 11b. This is slightly easier root because the starting aminoalcohol is cheap, but corresponding bis-ketone must be prepared.
If the use of the hindered tripod 41 is intended to prevent coordination of hetero-ligands in the equatorial plane, which is favorable for complexes with oblate Ln-ions, then its unhindered congener is better to be used for heteroleptic complexes like 39 and 40 for the prolate series. The latter can also be chosen for heteroleptic complexes with tripod 42.
5. Conclusions
Despite numerous publications on the coordination chemistry of lanthanides with tripodal ligands containing detailed crystallographic information, their magnetic behavior has not been practically studied.
In contrast to the complexes of dysprosium and terbium, the coordination compounds of the prolate Pm, Sm, Er, Tm, and Yb are much less studied. For the latter, unhindered tripods with a shallow fit are more suitable, which makes it possible to ensure equatorial coordination of anionic hetero-ligands. Whereas sterically demanding tripods of the 4 type are brilliantly suited for obtaining stable divalent Ln complexes [44,92], which is especially promising for the design of magneto-luminescent materials based on Eu(II) complexes, because their tricationic congener Eu3+ has nonmagnetic ground state.
The review of complexes with diamagnetic tripodal ligands was made with the hope that physicochemists will pay attention to this family of compounds, and make more precisely magneto-structural predictions based on CF parameters for the lanthanide ions in the respective ligand environments.
In magnetic terms, mono- and biradical Ln-complexes are very interesting, but difficult for theoretical calculations. An important feature of potential materials is stability under normal conditions. Complexes with tripodal radicals possess this property. In addition, tripods have a predictable type of coordination, which is extremely important in the rational design of compounds with desired properties.
The strong magnetic interaction between the radical center and the metal ion has a number of advantages for the design of advanced SIMs. For example, an additional unpaired electron of an organic radical may transform the resulting cationic complex into the Kramers system, while free Ln has not this property. In addition, the direct interaction of the unpaired 2p electron of the radical with the 4f shell also removes the degeneracy of the energy levels, which can reduce QTM.
Without theoretical calculations, it is difficult to give precise prescriptions about the structure of the paramagnetic tripod for a particular lanthanide ion. However, we have tried to guess the general trend for cyclic nitroxyl radicals functionalized by aromatic nitrogen-containing heterocycles: pyridine and imidazoline.
Author Contributions
Not applicable.
Funding
This research received no external funding” or “This research was funded by funded by Russian Science Foundation, Grant No. 23-23-00437 and partially supported by Ministry of Science and Higher Education of the Russian Federation (crystal structure determination for the complexes with nitroxide radicals, 121031700313-8)
Data Availability Statement
Not applicable
Conflicts of Interest
The author declares no conflict of interest.
References
- Szczepura, L.F.; Witham, L.M.; Takeuchi, K.J. Tris(2-Pyridyl) Tripod Ligands. Coord. Chem. Rev. 1998, 174, 5–32. [CrossRef]
- Bellemin-Laponnaz, S.; Gade, L.H. A Modular Approach to C1 and C3 Chiral N-Tripodal Ligands for Asymmetric Catalysis. Angew. Chemie Int. Ed. 2002, 41, 3473–3475. [CrossRef]
- Natrajan, L.; Pécaut, J.; Mazzanti, M.; LeBrun, C. Controlled Hydrolysis of Lanthanide Complexes of the N-Donor Tripod Tris(2-Pyridylmethyl)Amine versus Bisligand Complex Formation. Inorg. Chem. 2005, 44, 4756–4765. [CrossRef]
- Chen, Y.-J.; Chen, H.-H. 1,1,1-Tris(Hydroxymethyl)Ethane as a New, Efficient, and Versatile Tripod Ligand for Copper-Catalyzed Cross-Coupling Reactions of Aryl Iodides with Amides, Thiols, and Phenols. Org. Lett. 2006, 8, 5609–5612. [CrossRef]
- Eckert, M.; Brethon, A.; Li, Y.-X.; Sheldon, R.A.; Arends, I.W.C.E. Study of the Efficiency of Amino-Functionalized Ruthenium and Ruthenacycle Complexes as Racemization Catalysts in the Dynamic Kinetic Resolution of 1-Phenylethanol. Adv. Synth. Catal. 2007, 349, 2603–2609. [CrossRef]
- Andrez, J.; Bozoklu, G.; Nocton, G.; Pécaut, J.; Scopelliti, R.; Dubois, L.; Mazzanti, M. Lanthanide(II) Complexes Supported by N,O-Donor Tripodal Ligands: Synthesis, Structure, and Ligand-Dependent Redox Behavior. Chem. – A Eur. J. 2015, 21, 15188–15200. [CrossRef]
- Wietzke, R.; Mazzanti, M.; Latour, J.-M.; Pécaut, J.; Cordier, P.-Y.; Madic, C. Lanthanide(III) Complexes of Tripodal N-Donor Ligands: Structural Models for the Species Involved in Solvent Extraction of Actinides(III). Inorg. Chem. 1998, 37, 6690–6697. [CrossRef]
- Kuswandi, B.; N/a, N.; Verboom, W.; Reinhoudt, D.N. Tripodal Receptors for Cation and Anion Sensors. Sensors 2006, 6, 978–1017.
- Dai, Z.; Canary, J.W. Tailoring Tripodal Ligands for Zinc Sensing. New J. Chem. 2007, 31, 1708–1718. [CrossRef]
- Machado, K.; Mukhopadhyay, S.; Videira, R.A.; Mishra, J.; Mobin, S.M.; Mishra, G.S. Polymer Encapsulated Scorpionate Eu3+ Complexes as Novel Hybrid Materials for High Performance Luminescence Applications. RSC Adv. 2015, 5, 35675–35682. [CrossRef]
- Yu, T.; Zhao, Y.; Fan, D.; Hong, Z.; Su, W. Preparation, Photo- and Electro-Luminescent Properties of a Novel Complex of Tb (III) with a Tripod Ligand. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2008, 69, 654–658. [CrossRef]
- Long, J.; Lyubov, D.M.; Mahrova, T. V.; Lyssenko, K.A.; Korlyukov, A.A.; Fedorov, Y. V.; Chernikova, E.Y.; Guari, Y.; Larionova, J.; Trifonov, A.A. Heteroleptic Lanthanide Complexes Coordinated by Tripodal Tetradentate Ligand: Synthesis, Structure, and Magnetic and Photoluminescent Properties. Cryst. Growth Des. 2020, 20, 5184–5192. [CrossRef]
- Zhu, L.; Tang, H.; Harima, Y.; Yamashita, K.; Hirayama, D.; Aso, Y.; Otsubo, T. Electrochemical Properties of Self-Assembled Monolayers of Tripod-Shaped Molecules and Their Applications to Organic Light-Emitting Diodes. Chem. Commun. 2001, 1830–1831. [CrossRef]
- Zheng, X.-L.; Liu, Y.; Pan, M.; Lü, X.-Q.; Zhang, J.-Y.; Zhao, C.-Y.; Tong, Y.-X.; Su, C.-Y. Bright Blue-Emitting Ce3+ Complexes with Encapsulating Polybenzimidazole Tripodal Ligands as Potential Electroluminescent Devices. Angew. Chemie Int. Ed. 2007, 46, 7399–7403. [CrossRef]
- Chen, K.-Y.; Ivashenko, O.; Carroll, G.T.; Robertus, J.; Kistemaker, J.C.M.; London, G.; Browne, W.R.; Rudolf, P.; Feringa, B.L. Control of Surface Wettability Using Tripodal Light-Activated Molecular Motors. J. Am. Chem. Soc. 2014, 136, 3219–3224. [CrossRef]
- Kammerer, C.; Rapenne, G. Scorpionate Hydrotris(Indazolyl)Borate Ligands as Tripodal Platforms for Surface-Mounted Molecular Gears and Motors. Eur. J. Inorg. Chem. 2016, 2016, 2214–2226. [CrossRef]
- Rabinovich, D. Synthetic Bioinorganic Chemistry: Scorpionates Turn 50 BT - 50 Years of Structure and Bonding – The Anniversary Volume. In; Mingos, D.M.P., Ed.; Springer International Publishing: Cham, 2017; pp. 139–157 ISBN 978-3-319-35138-4.
- Eliseeva, S. V.; Bünzli, J.-C.G. Lanthanide Luminescence for Functional Materials and Bio-Sciences. Chem. Soc. Rev. 2010, 39, 189–227. [CrossRef]
- Klitsche, F.; Ramcke, J.; Migenda, J.; Hensel, A.; Vossmeyer, T.; Weller, H.; Gross, S.; Maison, W. Synthesis of Tripodal Catecholates and Their Immobilization on Zinc Oxide Nanoparticles. Beilstein J. Org. Chem. 2015, 11, 678–686. [CrossRef]
- A Fluorescent Sensor-Based Tripodal-Bodipy for Cu (II) Ions: Bio-Imaging on Cells. TURKISH J. Chem. 2021, 45. [CrossRef]
- Brechin, E.K. Using Tripodal Alcohols to Build High-Spin Molecules and Single-Molecule Magnets. Chem. Commun. 2005, 5141–5153. [CrossRef]
- Milios, C.J.; Manoli, M.; Rajaraman, G.; Mishra, A.; Budd, L.E.; White, F.; Parsons, S.; Wernsdorfer, W.; Christou, G.; Brechin, E.K. A Family of [Mn6] Complexes Featuring Tripodal Ligands. Inorg. Chem. 2006, 45, 6782–6793. [CrossRef]
- Domingo, N.; Bellido, E.; Ruiz-Molina, D. Advances on Structuring, Integration and Magnetic Characterization of Molecular Nanomagnets on Surfaces and Devices. Chem. Soc. Rev. 2012, 41, 258–302. [CrossRef]
- Long, J.; Lyubov, D.M.; Mahrova, T. V.; Cherkasov, A. V.; Fukin, G.K.; Guari, Y.; Larionova, J.; Trifonov, A.A. Synthesis, Structure and Magnetic Properties of Tris(Pyrazolyl)Methane Lanthanide Complexes: Effect of the Anion on the Slow Relaxation of Magnetization. Dalt. Trans. 2018, 47, 5153–5156. [CrossRef]
- Ito, A.; Nakano, Y.; Urabe, M.; Tanaka, K.; Shiro, M. Structural and Magnetic Studies of Copper(II) and Zinc(II) Coordination Complexes Containing Nitroxide Radicals as Chelating Ligands. Eur. J. Inorg. Chem. 2006, 2006, 3359–3368. [CrossRef]
- Gass, I.A.; Gartshore, C.J.; Lupton, D.W.; Moubaraki, B.; Nafady, A.; Bond, A.M.; Boas, J.F.; Cashion, J.D.; Milsmann, C.; Wieghardt, K.; et al. Anion Dependent Redox Changes in Iron Bis-Terdentate Nitroxide {NNO} Chelates. Inorg. Chem. 2011, 50, 3052–3064. [CrossRef]
- Gass, I.A.; Tewary, S.; Nafady, A.; Chilton, N.F.; Gartshore, C.J.; Asadi, M.; Lupton, D.W.; Moubaraki, B.; Bond, A.M.; Boas, J.F.; et al. Observation of Ferromagnetic Exchange, Spin Crossover, Reductively Induced Oxidation, and Field-Induced Slow Magnetic Relaxation in Monomeric Cobalt Nitroxides. Inorg. Chem. 2013, 52, 7557–7572. [CrossRef]
- Gass, I.A.; Tewary, S.; Rajaraman, G.; Asadi, M.; Lupton, D.W.; Moubaraki, B.; Chastanet, G.; Létard, J.-F.; Murray, K.S. Solvate-Dependent Spin Crossover and Exchange in Cobalt(II) Oxazolidine Nitroxide Chelates. Inorg. Chem. 2014, 53, 5055–5066. [CrossRef]
- Gass, I.A.; Asadi, M.; Lupton, D.W.; Moubaraki, B.; Bond, A.M.; Guo, S.-X.; Murray, K.S. Manganese(II) Oxazolidine Nitroxide Chelates: Structure, Magnetism, and Redox Properties. Aust. J. Chem. 2014, 67, 1618. [CrossRef]
- Pedersen, A.H.; Geoghegan, B.L.; Nichol, G.S.; Lupton, D.W.; Murray, K.S.; Martínez-Lillo, J.; Gass, I.A.; Brechin, E.K. Hexahalorhenate(IV) Salts of Metal Oxazolidine Nitroxides. Dalt. Trans. 2017, 46, 5250–5259. [CrossRef]
- Gass, I.A.; Lu, J.; Asadi, M.; Lupton, D.W.; Forsyth, C.M.; Geoghegan, B.L.; Moubaraki, B.; Cashion, J.D.; Martin, L.L.; Bond, A.M.; et al. Use of the TCNQF 4 2− Dianion in the Spontaneous Redox Formation of [Fe III (L − ) 2 ][TCNQF 4 ⋅− ]. Chempluschem 2018, 83, 658–668. [CrossRef]
- Gass, I.A.; Lu, J.; Ojha, R.; Asadi, M.; Lupton, D.W.; Geoghegan, B.L.; Moubaraki, B.; Martin, L.L.; Bond, A.M.; Murray, K.S. [FeII(L•)2][TCNQF4•−]2: A Redox-Active Double Radical Salt. Aust. J. Chem. 2019, 72, 769–777. [CrossRef]
- Trofimenko, S. Boron-Pyrazole Chemistry. J. Am. Chem. Soc. 1966, 88, 1842–1844. [CrossRef]
- Oliver, J.D.; Mullica, D.F.; Hutchinson, B.B.; Milligan, W.O. Iron-Nitrogen Bond Lengths in Low-Spin and High-Spin Iron(II) Complexes with Poly(Pyrazolyl)Borate Ligands. Inorg. Chem. 1980, 19, 165–169. [CrossRef]
- Trofimenko, S. Recent Advances in Poly(Pyrazolyl)Borate (Scorpionate) Chemistry. Chem. Rev. 1993, 93, 943–980. [CrossRef]
- Wu, L.P.; Yamagiwa, Y.; Ino, I.; Sugimoto, K.; Kuroda-Sowa, T.; Kamikawa, T.; Munakata, M. Unique Tetranuclear Copper(II) Cluster and Monomeric Iron(II), (III) Complexes with a Tris(Imidazolyl) Chelating Ligand. Polyhedron 1999, 18, 2047–2053. [CrossRef]
- SCORPIONATES II: CHELATING BORATE LIGANDS - Dedicated To Swiatoslaw Trofimenko; Pettinari, C., Ed.; IMPERIAL COLLEGE PRESS and distribiuted by World Scientific: London, 2008; ISBN 978-1-86094-876-3.
- Dougherty, W.G.; Kassel, W.S. Synthesis of a Series of First-Row Tris-Imidazolylphosphine Sandwich Complexes and Their Potential for Formation of Polymetallic Species. Inorganica Chim. Acta 2010, 364, 120–124. [CrossRef]
- Saouma, C.T.; Peters, J.C. ME and ME Complexes of Iron and Cobalt That Emphasize Three-Fold Symmetry (E=O, N, NR). Coord. Chem. Rev. 2011, 255, 920–937. [CrossRef]
- Lavrenova, L.G.; Strekalova, A.D.; Smolentsev, A.I.; Naumov, D.Y.; Bogomyakov, A.S.; Sheludyakova, L.A.; Vasilevskii, S.F. Mono- and Heteroligand Iron(II) Complexes with Tris(3,5-Dimethylpyrazol-1-Yl)Methane. Russ. J. Coord. Chem. 2016, 42, 711–718. [CrossRef]
- Feng, M.; Tong, M.-L. Single Ion Magnets from 3d to 5f: Developments and Strategies. Chem. – A Eur. J. 2018, 24, 7574–7594. [CrossRef]
- Landart-Gereka, A.; Quesada-Moreno, M.M.; Palacios, M.A.; Díaz-Ortega, I.F.; Nojiri, H.; Ozerov, M.; Krzystek, J.; Colacio, E. Pushing up the Easy-Axis Magnetic Anisotropy and Relaxation Times in Trigonal Prismatic CoII Mononuclear SMMs by Molecular Structure Design. Chem. Commun. 2023, 59, 952–955. [CrossRef]
- Apostolidis, C.; Carvalho, A.; Domingos, A.; Kanellakopulos, B.; Maier, R.; Marques, N.; Matos, A.P. de; Rebizant, J. Chloro-Lanthanide, and Plutonium Complexes Containing the Hydrotris (3,5-Dimethylpyrazol-1-Yl)Borate Ligand: The Crystal and Molecular Structures of [PrCl(μ-Cl)TpMe2(3,5-Me2pzH)]2 and YbCl2TpMe2 (THF). Polyhedron 1998, 18, 263–272. [CrossRef]
- Hillier, A.C.; Zhang, X.W.; Maunder, G.H.; Liu, S.Y.; Eberspacher, T.A.; Metz, M. V.; McDonald, R.; Domingos, A.; Marques, N.; Day, V.W.; et al. Synthesis and Structural Comparison of a Series of Divalent Ln(TpR,R)2 (Ln = Sm, Eu, Yb) and Trivalent Sm(TpMe2)2X (X = F, Cl, I, BPh4) Complexes. Inorg. Chem. 2001, 40, 5106–5116. [CrossRef]
- Sella, A.; Brown, S.E.; Steed, J.W.; Tocher, D.A. Synthesis and Solid-State Structures of Pyrazolylmethane Complexes of the Rare Earths. Inorg. Chem. 2007, 46, 1856–1864. [CrossRef]
- Liu, S.-Y.; Maunder, G.H.; Sella, A.; Stevenson, M.; Tocher, D.A. Synthesis and Molecular Structures of Hydrotris(Dimethylpyrazolyl)Borate Complexes of the Lanthanides. Inorg. Chem. 1996, 35, 76–81. [CrossRef]
- Werner, E.J.; Biros, S.M. Supramolecular Ligands for the Extraction of Lanthanide and Actinide Ions. Org. Chem. Front. 2019, 6, 2067–2094. [CrossRef]
- Barraza, R.; Sertage, A.G.; Kajjam, A.B.; Ward, C.L.; Lutter, J.C.; Schlegel, H.B.; Allen, M.J. Properties of Amine-Containing Ligands That Are Necessary for Visible-Light-Promoted Catalysis with Divalent Europium. Inorg. Chem. 2022, 61, 19649–19657. [CrossRef]
- Frost, J.M.; Harriman, K.L.M.; Murugesu, M. The Rise of 3-d Single-Ion Magnets in Molecular Magnetism: Towards Materials from Molecules? Chem. Sci. 2016, 7, 2470–2491. [CrossRef]
- Zhu, Z.; Guo, M.; Li, X.-L.; Tang, J. Molecular Magnetism of Lanthanide: Advances and Perspectives. Coord. Chem. Rev. 2019, 378, 350–364. [CrossRef]
- Zabala-Lekuona, A.; Seco, J.M.; Colacio, E. Single-Molecule Magnets: From Mn12-Ac to Dysprosium Metallocenes, a Travel in Time. Coord. Chem. Rev. 2021, 441, 213984. [CrossRef]
- Gatteschi, D.; Sessoli, R. Quantum Tunneling of Magnetization and Related Phenomena in Molecular Materials. Angew. Chemie Int. Ed. 2003, 42, 268–297. [CrossRef]
- Goodwin, C.A.P.; Ortu, F.; Reta, D. Strangely Attractive: Collaboration and Feedback in the Field of Molecular Magnetism. Int. J. Quantum Chem. 2020, 120, e26248. [CrossRef]
- Tang, J.; Zhang, P. Lanthanide Single Molecule Magnets; Springer Berlin Heidelberg: Berlin, Heidelberg, 2015; ISBN 978-3-662-46998-9.
- Sorace, L.; Gatteschi, D. Electronic Structure and Magnetic Properties of Lanthanide Molecular Complexes. In Lanthanides and Actinides in Molecular Magnetism; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2015; pp. 1–26.
- Goodwin, C.A.P.; Ortu, F.; Reta, D.; Chilton, N.F.; Mills, D.P. Molecular Magnetic Hysteresis at 60 Kelvin in Dysprosocenium. Nature 2017, 548, 439–442. [CrossRef]
- Guo, F.-S.; Day, B.M.; Chen, Y.-C.; Tong, M.-L.; Mansikkamäki, A.; Layfield, R.A. A Dysprosium Metallocene Single-Molecule Magnet Functioning at the Axial Limit. Angew. Chemie Int. Ed. 2017, 56, 11445–11449. [CrossRef]
- Guo, F.-S.; Day, B.M.; Chen, Y.-C.; Tong, M.-L.; Mansikkamäki, A.; Layfield, R.A. Magnetic Hysteresis up to 80 Kelvin in a Dysprosium Metallocene Single-Molecule Magnet. Science (80-. ). 2018, 362, 1400–1403. [CrossRef]
- Vincent, A.H.; Whyatt, Y.L.; Chilton, N.F.; Long, J.R. Strong Axiality in a Dysprosium(III) Bis(Borolide) Complex Leads to Magnetic Blocking at 65 K. J. Am. Chem. Soc. 2023, 145, 1572–1579. [CrossRef]
- Rinehart, J.D.; Long, J.R. Exploiting Single-Ion Anisotropy in the Design of f-Element Single-Molecule Magnets. Chem. Sci. 2011, 2, 2078–2085. [CrossRef]
- Chilton, N.F. Design Criteria for High-Temperature Single-Molecule Magnets. Inorg. Chem. 2015, 54, 2097–2099. [CrossRef]
- Harriman, K.L.M.; Errulat, D.; Murugesu, M. Magnetic Axiality: Design Principles from Molecules to Materials. Trends Chem. 2019, 1, 425–439. [CrossRef]
- Guo, F.-S.; Bar, A.K.; Layfield, R.A. Main Group Chemistry at the Interface with Molecular Magnetism. Chem. Rev. 2019, 119, 8479–8505. [CrossRef]
- Chiesa, A.; Cugini, F.; Hussain, R.; Macaluso, E.; Allodi, G.; Garlatti, E.; Giansiracusa, M.; Goodwin, C.A.P.; Ortu, F.; Reta, D.; et al. Understanding Magnetic Relaxation in Single-Ion Magnets with High Blocking Temperature. Phys. Rev. B 2020, 101, 174402. [CrossRef]
- Woodruff, D.N.; Winpenny, R.E.P.; Layfield, R.A. Lanthanide Single-Molecule Magnets. Chem. Rev. 2013, 113, 5110–5148. [CrossRef]
- Jiang, S.-D.; Wang, B.-W.; Gao, S. Advances in Lanthanide Single-Ion Magnets BT - Molecular Nanomagnets and Related Phenomena. In; Gao, S., Ed.; Springer Berlin Heidelberg: Berlin, Heidelberg, 2015; pp. 111–141 ISBN 978-3-662-45723-8.
- Gupta, S.K.; Murugavel, R. Enriching Lanthanide Single-Ion Magnetism through Symmetry and Axiality. Chem. Commun. 2018, 54, 3685–3696. [CrossRef]
- Vogel, R.; Müntener, T.; Häussinger, D. Intrinsic Anisotropy Parameters of a Series of Lanthanoid Complexes Deliver New Insights into the Structure-Magnetism Relationship. Chem 2021, 7, 3144–3156. [CrossRef]
- Jiang, S.-D.; Qin, S.-X. Prediction of the Quantized Axis of Rare-Earth Ions: The Electrostatic Model with Displaced Point Charges. Inorg. Chem. Front. 2015, 2, 613–619. [CrossRef]
- Liu, J.-L.; Chen, Y.-C.; Tong, M.-L. Symmetry Strategies for High Performance Lanthanide-Based Single-Molecule Magnets. Chem. Soc. Rev. 2018, 47, 2431–2453. [CrossRef]
- Chen, Y.-C.; Tong, M.-L. Single-Molecule Magnets beyond a Single Lanthanide Ion: The Art of Coupling. Chem. Sci. 2022. [CrossRef]
- Molecular Nanomagnets and Related Phenomena —Structure and Bonding; Gao, S., Ed.; Springer Berlin: Heidelberg, 2015; ISBN 978-3-662-45722-1.
- Kahn, O. Molecular Magnetism; VCH: New York, NY, USA, 1993; ISBN 978-1-56081-566-2.
- Series Page. In Theoretical Foundations of Molecular Magnetism; Boča, R.B.T.-C.M. in I.C., Ed.; Elsevier, 1999; Vol. 1, p. ii ISBN 1873-0418.
- Chen, Y.-C.; Liu, J.-L.; Wernsdorfer, W.; Liu, D.; Chibotaru, L.F.; Chen, X.-M.; Tong, M.-L. Hyperfine-Interaction-Driven Suppression of Quantum Tunneling at Zero Field in a Holmium(III) Single-Ion Magnet. Angew. Chemie Int. Ed. 2017, 56, 4996–5000. [CrossRef]
- Skomski, R.; Sellmyer, D.J. Anisotropy of Rare-Earth Magnets. J. Rare Earths 2009, 27, 675–679. [CrossRef]
- Zhu, Z.; Tang, J. Lanthanide Single-Molecule Magnets with High Anisotropy Barrier: Where to from Here? Natl. Sci. Rev. 2022, 9, nwac194. [CrossRef]
- Wang, J.; Sun, C.; Zheng, Q.; Wang, D.; Chen, Y.; Ju, J.; Sun, T.; Cui, Y.; Ding, Y.; Tang, Y. Lanthanide Single-molecule Magnets: Synthetic Strategy, Structures, Properties and Recent Advances. Chem. – An Asian J. 2023, 18. [CrossRef]
- Trofimenko, S. The Coordination Chemistry of Polypyrazolylborate Ligand; IMPERIAL COLLEGE PRESS, 1999; ISBN 978-1-86094-172-6.
- Cheng, J.; Takats, J.; Ferguson, M.J.; McDonald, R. Heteroleptic Tm(II) Complexes: One More Success for Trofimenko’s Scorpionates. J. Am. Chem. Soc. 2008, 130, 1544–1545. [CrossRef]
- Dei, A.; Gatteschi, D.; Pécaut, J.; Poussereau, S.; Sorace, L.; Vostrikova, K. Crystal Field and Exchange Effects in Rare Earth Semiquinone Complexes. Comptes Rendus l’Academie des Sci. - Ser. IIC Chem. 2001, 4. [CrossRef]
- Zhang, P.; Perfetti, M.; Kern, M.; Hallmen, P.P.; Ungur, L.; Lenz, S.; Ringenberg, M.R.; Frey, W.; Stoll, H.; Rauhut, G.; et al. Exchange Coupling and Single Molecule Magnetism in Redox-Active Tetraoxolene-Bridged Dilanthanide Complexes. Chem. Sci. 2018, 9, 1221–1230. [CrossRef]
- Xu, G.-F.; Gamez, P.; Tang, J.; Clérac, R.; Guo, Y.-N.; Guo, Y. MIIIDyIII3 (M = FeIII, CoIII) Complexes: Three-Blade Propellers Exhibiting Slow Relaxation of Magnetization. Inorg. Chem. 2012, 51, 5693–5698. [CrossRef]
- Xu, G.-F.; Wang, Q.-L.; Gamez, P.; Ma, Y.; Clérac, R.; Tang, J.; Yan, S.-P.; Cheng, P.; Liao, D.-Z. A Promising New Route towards Single-Molecule Magnets Based on the Oxalate Ligand. Chem. Commun. 2010, 46, 1506–1508. [CrossRef]
- Mikhalyova, E.A.; Zeller, M.; Jasinski, J.P.; Butcher, R.J.; Carrella, L.M.; Sedykh, A.E.; Gavrilenko, K.S.; Smola, S.S.; Frasso, M.; Cazorla, S.C.; et al. Combination of Single-Molecule Magnet Behaviour and Luminescence Properties in a New Series of Lanthanide Complexes with Tris(Pyrazolyl)Borate and Oligo(β-Diketonate) Ligands. Dalt. Trans. 2020, 49, 7774–7789. [CrossRef]
- Kandel, A. V.; Mikhalyova, E.A.; Zeller, M.; Addison, A.W.; Pavlishchuk, V. V. Influence of the Structure of 3-Arylacetylacetonate Ligands on the Luminescence Properties of Eu3+ and Tb3+ Complexes. Theor. Exp. Chem. 2017, 53, 180–186. [CrossRef]
- Depperman, E.C.; Bodnar, S.H.; Vostrikova, K.E.; Shultz, D.A.; Kirk, M.L. Spin Robustness of a New Hybrid Inorganic−Organic High-Spin Molecule. J. Am. Chem. Soc. 2001, 123, 3133–3134. [CrossRef]
- Wang, S.; Zuo, J.-L.; Zhou, H.-C.; Choi, H.J.; Ke, Y.; Long, J.R.; You, X.-Z. [(Tp)8(H2O)6CuII6FeIII8(CN)24]4+: A Cyanide-Bridged Face-Centered-Cubic Cluster with Single-Molecule-Magnet Behavior. Angew. Chemie Int. Ed. 2004, 43, 5940–5943. [CrossRef]
- Wang, S.; Zuo, J.-L.; Gao, S.; Song, Y.; Zhou, H.-C.; Zhang, Y.-Z.; You, X.-Z. The Observation of Superparamagnetic Behavior in Molecular Nanowires. J. Am. Chem. Soc. 2004, 126, 8900–8901. [CrossRef]
- Alexandropoulos, D.I.; Vignesh, K.R.; Xie, H.; Dunbar, K.R. Switching on Single-Molecule Magnet Properties of Homoleptic Sandwich Tris(Pyrazolyl)Borate Dysprosium(III) Cations via Intermolecular Dipolar Coupling. Dalt. Trans. 2019, 48, 10610–10618. [CrossRef]
- Kühling, M.; Wickleder, C.; Ferguson, M.J.; Hrib, C.G.; McDonald, R.; Suta, M.; Hilfert, L.; Takats, J.; Edelmann, F.T. Investigation of the “Bent Sandwich-like” Divalent Lanthanide Hydro-Tris(Pyrazolyl)Borates Ln(TpIPr2)2 (Ln = Sm, Eu, Tm, Yb). New J. Chem. 2015, 39, 7617–7625. [CrossRef]
- Qi, H.; Zhao, Z.; Zhan, G.; Sun, B.; Yan, W.; Wang, C.; Wang, L.; Liu, Z.; Bian, Z.; Huang, C. Air Stable and Efficient Rare Earth Eu(II) Hydro-Tris(Pyrazolyl)Borate Complexes with Tunable Emission Colors. Inorg. Chem. Front. 2020, 7, 4593–4599. [CrossRef]
- Takats, J.; Zhang, X.W.; Day, V.W.; Eberspacher, T.A. Synthesis and Structure of Bis[Hydrotris(3,5-Dimethylpyrazolyl)Borato]Samarium(II), Sm[HB(3,5-Me2pz)3]2, and the Product of Its Reaction with Azobenzene. Organometallics 1993, 12, 4286–4288. [CrossRef]
- Maunder, G.H.; Sella, A.; Tocher, D.A. Synthesis and Molecular Structures of a Redox-Related Pair of Lanthanide Complexes. J. Chem. Soc. Chem. Commun. 1994, 885. [CrossRef]
- Momin, A.; Carter, L.; Yang, Y.; McDonald, R.; Essafi (née Labouille), S.; Nief, F.; Del Rosal, I.; Sella, A.; Maron, L.; Takats, J. To Bend or Not To Bend: Experimental and Computational Studies of Structural Preference in Ln(TpiPr2)2 (Ln = Sm, Tm). Inorg. Chem. 2014, 53, 12066–12075. [CrossRef]
- Saliu, K.O.; Takats, J.; Ferguson, M.J. Bis[Tris(3-Tert-Butyl-5-Methylpyrazol-1-Yl)Hydridoborato]Ytterbium(II) Toluene Solvate. Acta Crystallogr. Sect. E Struct. Reports Online 2009, 65, m643–m644. [CrossRef]
- Suta, M.; Kühling, M.; Liebing, P.; Edelmann, F.T.; Wickleder, C. Photoluminescence Properties of the “Bent Sandwich-like” Compounds [Eu(TpIPr2)2] and [Yb(TpIPr2)2]– Intermediates between Nitride-Based Phosphors and Metallocenes. J. Lumin. 2017, 187, 62–68. [CrossRef]
- Arikawa, Y.; Inada, K.; Onishi, M. Side-on Coordination Mode of a Pyrazolyl Group in the Structure of a Divalent [Sm{B(3-Mepz)4}2] Complex (3-Mepz Is 3-Methylpyrazol-1-Yl). Acta Crystallogr. Sect. C Struct. Chem. 2016, 72, 838–841. [CrossRef]
- Li, Z.-H.; Zhai, Y.-Q.; Chen, W.-P.; Ding, Y.-S.; Zheng, Y.-Z. Air-Stable Hexagonal Bipyramidal Dysprosium(III) Single-Ion Magnets with Nearly Perfect D6h Local Symmetry. Chem. – A Eur. J. 2019, 25, 16219–16224. [CrossRef]
- Canaj, A.B.; Dey, S.; Martí, E.R.; Wilson, C.; Rajaraman, G.; Murrie, M. Insight into D 6 h Symmetry: Targeting Strong Axiality in Stable Dysprosium(III) Hexagonal Bipyramidal Single-Ion Magnets. Angew. Chemie Int. Ed. 2019, 58, 14146–14151. [CrossRef]
- Li, Q.-W.; Wan, R.-C.; Chen, Y.-C.; Liu, J.-L.; Wang, L.-F.; Jia, J.-H.; Chilton, N.F.; Tong, M.-L. Unprecedented Hexagonal Bipyramidal Single-Ion Magnets Based on Metallacrowns. Chem. Commun. 2016, 52, 13365–13368. [CrossRef]
- Gil, Y.; Castro-Alvarez, A.; Fuentealba, P.; Spodine, E.; Aravena, D. Lanthanide SMMs Based on Belt Macrocycles: Recent Advances and General Trends. Chem. – A Eur. J. 2022, 28, e202200336. [CrossRef]
- Meyer, G. All the Lanthanides Do It and Even Uranium Does Oxidation State +2. Angew. Chemie Int. Ed. 2014, 53, 3550–3551. [CrossRef]
- Llunell, M.; Casanova, D.; Cirera, J.; Alemany, P.; Alvarez, S. SHAPE, Version 2.1, Program for the Stereochemical Analysis of Molecular Fragments by Means of Continuous Shape Measures and Associated Tools. Univ. Barcelona, Barcelona, Spain 2013, 2103.
- Li, T.; Zhang, G.; Guo, J.; Wang, S.; Leng, X.; Chen, Y. Tris(Pyrazolyl)Methanide Complexes of Trivalent Rare-Earth Metals. Organometallics 2016, 35, 1565–1572. [CrossRef]
- Keene, F.R.; Snow, M.R.; Stephenson, P.J.; Tiekink, E.R.T. Ruthenium(II) Complexes of the C3v Ligands Tris(2-Pyridyl)Amine, Tris(2-Pyridyl)Methane, and Tris(2-Pyridyl)Phosphine. 1. Synthesis and x-Ray Structural Studies of the Bis(Ligand) Complexes. Inorg. Chem. 1988, 27, 2040–2045. [CrossRef]
- García, F.; Hopkins, A.D.; Humphrey, S.M.; McPartlin, M.; Rogers, M.C.; Wright, D.S. The First Example of a Si-Bridged Tris(Pyridyl) Ligand; Synthesis and Structure of [MeSi(2-C5H4N)3LiX] (X = 0.2Br, 0.8Cl). Dalt. Trans. 2004, 361–362. [CrossRef]
- MANN, F.G.; WATSON, J. Conditions of Salt Formation in Polyamines And Kindred Compounds. Salt Formation in the Tertiary 2-Pyridylamines, Phosphines and Arsines. J. Org. Chem. 1948, 13, 502–531. [CrossRef]
- García, F.; Hopkins, A.D.; Kowenicki, R.A.; McPartlin, M.; Rogers, M.C.; Silvia, J.S.; Wright, D.S. Syntheses and Structure of Heterometallic Complexes Containing Tripodal Group 13 Ligands [RE(2-Py)3]- (E = Al, In). Organometallics 2006, 25, 2561–2568. [CrossRef]
- Beswick, M.A.; Belle, C.J.; Davies, M.K.; Halcrow, M.A.; Raithby, P.R.; Steiner, A.; Wright, D.S. One-Pot Synthesis of a Novel Tridentate Tin(IV) Ligand; Syntheses and Structures of [BunSn(NC5H4-C,N)3MBr](M = Li, Cu). Chem. Commun. 1996, 2619. [CrossRef]
- Zeckert, K.; Zahn, S.; Kirchner, B. Tin–Lanthanoid Donor–Acceptor Bonds. Chem. Commun. 2010, 46, 2638. [CrossRef]
- Beswick, M.A.; Davies, M.K.; Raithby, P.R.; Steiner, A.; Wright, D.S. Synthesis and Structure of [Pb(2-Py)3Li·THF], Containing a Low-Valent Group 14 Tris(Pyridyl) Ligand (2-Py = 2-Pyridyl). Organometallics 1997, 16, 1109–1110. [CrossRef]
- Goura, J.; McQuade, J.; Shimoyama, D.; Lalancette, R.A.; Sheridan, J.B.; Jäkle, F. Electrophilic and Nucleophilic Displacement Reactions at the Bridgehead Borons of Tris(Pyridyl)Borate Scorpionate Complexes. Chem. Commun. 2022, 58, 977–980. [CrossRef]
- Reichart, F.; Kischel, M.; Zeckert, K. Lanthanide(II) Complexes of a Dual Functional Tris(2-Pyridyl)Stannate Derivative. Chem. – A Eur. J. 2009, 15, 10018–10020. [CrossRef]
- Zeckert, K. Syntheses and Structures of Lanthanoid(Ii) Complexes Featuring Sn–M (M = Al, Ga, In) Bonds. Dalt. Trans. 2012, 41, 14101–14106. [CrossRef]
- Zeckert, K. Pyridyl Compounds of Heavier Group 13 and 14 Elements as Ligands for Lanthanide Metals. Organometallics 2013, 32, 1387–1393. [CrossRef]
- Zeckert, K.; Griebel, J.; Kirmse, R.; Weiß, M.; Denecke, R. Versatile Reactivity of a Lithium Tris(Aryl)Plumbate(II) Towards Organolanthanoid Compounds: Stable Lead-Lanthanoid-Metal Bonds or Redox Processes. Chem. - A Eur. J. 2013, 19, 7718–7722. [CrossRef]
- Hellmann, K.W.; Gade, L.H.; Gevert, O.; Steinert, P.; Lauher, J.W. Tripodal Triamidostannates and -Plumbates. Inorg. Chem. 1995, 34, 4069–4078. [CrossRef]
- García-Rodríguez, R.; Simmonds, H.R.; Wright, D.S. Formation of a Heterometallic AlIII/SmIII Complex Involving a Novel [EtAl(2-Py)2O]2– Ligand (2-Py = 2-Pyridyl). Organometallics 2014, 33, 7113–7117. [CrossRef]
- García-Rodríguez, R.; Kopf, S.; Wright, D.S. Modifying the Donor Properties of Tris(Pyridyl)Aluminates in Lanthanide(Ii) Sandwich Compounds. Dalt. Trans. 2018, 47, 2232–2239. [CrossRef]
- Hajiashrafi, T.; Nemati Kharat, A.; Love, J.A.; Patrick, B.O. Synthesis, Characterization and Crystal Structure of Three New Lanthanide (III) Complexes with the [(6-Methyl-2-Pyridyl)Methyl]Bis(2-Pyridylmethyl)Amine (MeTPA) Ligand; New Precursors for Lanthanide (III) Oxide Nano-Particles. Polyhedron 2013, 60, 30–38. [CrossRef]
- Hay, M.A.; Gable, R.W.; Boskovic, C. Modulating the Electronic Properties of Divalent Lanthanoid Complexes with Subtle Ligand Tuning. Dalt. Trans. 2023, 52, 3315–3324. [CrossRef]
- Zhang, C.; Cheng, Z.; Tan, P.; Lv, W.; Cui, H.; Chen, L.; Cai, X.; Zhao, Y.; Yuan, A. Tuning the Ligand Field in Seven-Coordinate Dy(III) Complexes to Perturb Single-Ion Magnet Behavior. New J. Chem. 2021, 45, 8591–8596. [CrossRef]
- Demir, S.; Jeon, I.-R.; Long, J.R.; Harris, T.D. Radical Ligand-Containing Single-Molecule Magnets. Coord. Chem. Rev. 2015, 289–290, 149–176. [CrossRef]
- Ishikawa, N.; Sugita, M.; Tanaka, N.; Ishikawa, T.; Koshihara, S.; Kaizu, Y. Upward Temperature Shift of the Intrinsic Phase Lag of the Magnetization of Bis(Phthalocyaninato)Terbium by Ligand Oxidation Creating an S = 1 / 2 Spin. Inorg. Chem. 2004, 43, 5498–5500. [CrossRef]
- Pederson, R.; Wysocki, A.L.; Mayhall, N.; Park, K. Multireference Ab Initio Studies of Magnetic Properties of Terbium-Based Single-Molecule Magnets. J. Phys. Chem. A 2019, 123, 6996–7006. [CrossRef]
- Rinehart, J.D.; Fang, M.; Evans, W.J.; Long, J.R. A N 2 3– Radical-Bridged Terbium Complex Exhibiting Magnetic Hysteresis at 14 K. J. Am. Chem. Soc. 2011, 133, 14236–14239. [CrossRef]
- Rinehart, J.D.; Fang, M.; Evans, W.J.; Long, J.R. Strong Exchange and Magnetic Blocking in N23−-Radical-Bridged Lanthanide Complexes. Nat. Chem. 2011, 3, 538–542. [CrossRef]
- Vieru, V.; Iwahara, N.; Ungur, L.; Chibotaru, L.F. Giant Exchange Interaction in Mixed Lanthanides. Sci. Rep. 2016, 6, 24046. [CrossRef]
- Eugene V Tretyakov; Victor I Ovcharenko The Chemistry of Nitroxide Radicals in the Molecular Design of Magnets. Russ. Chem. Rev. 2009, 78, 971. [CrossRef]
- Vostrikova, K.E. High-Spin Molecules Based on Metal Complexes of Organic Free Radicals. Coord. Chem. Rev. 2008, 252, 1409–1419. [CrossRef]
- Ovcharenko, V.I.; Vostrikova, K.E.; Romanenko, G. V; Ikorski, V.N.; Podberezskaya, N. V; Larionov, S. V Synthesis, Crystal Structure and Magnetic Properties of Di(Methanol) and Di(Ethanol)-Bis 2,2,5,5-Tetramethyl-1-Oxyl-3-Imidazoline-4-(3’,3’,3’-Trifluoromethyl-1-Propenyl-2’-Oxyato Nickel(II) - a New Type of Low Temperature Ferromagnetics. Dokl. Akad. Nauk SSSR 1989, 306, 115–118, doi:WOS:A1989U689100027.
- Meng, X.; Shi, W.; Cheng, P. Magnetism in One-Dimensional Metal–Nitronyl Nitroxide Radical System. Coord. Chem. Rev. 2019, 378, 134–150. [CrossRef]
- Fegy, K.; Vostrikova, K.E.; Luneau, D.; Rey, P. New Nitroxide Based Molecular Magnetic Materials. Mol. Cryst. Liq. Cryst. Sci. Technol. Sect. A. Mol. Cryst. Liq. Cryst. 1997, 305, 69–80. [CrossRef]
- Ovcharenko, V.I.; Vostrikova, K.E.; Ikorskii, V.N.; Larionov, S. V; Sagdeev, R.Z. The Low Temperature Ferromagnet Dimethanol-Bis-[2,2,5,5-Tetramethyl-1-Oxyl-3-Imidazoline-4-(3’,3’,3’-Tri Fluoromethyl-1’-Propenyl-2’-Oxyato)] Cobalt(II), CoL2(CH3OH)2. Dokl. Akad. Nauk SSSR 1989, 306, 660–662, doi:WOS:A1989AC74400036.
- Hintermaier, F.; Volodarsky, L.B.; Polborn, K.; Beck, W. New 2,5-Dihydroimidazole-1-Oxyls with Functional Side Groups (N, O, S Donors). Liebigs Ann. 1995, 1995, 2189–2194. [CrossRef]
- Hintermaier, F.; Sünkel, K.; Volodarsky, L.B.; Beck, W. Synthesis, Structure, and Magnetic Properties of Transition Metal Complexes of the Nitroxide 2,5-Dihydro-4,5,5-Trimethyl-2,2-Bis(2-Pyridyl)Imidazole-1-Oxyl. Inorg. Chem. 1996, 35, 5500–5503. [CrossRef]
- Perfetti, M.; Caneschi, A.; Sukhikh, T.S.; Vostrikova, K.E. Lanthanide Complexes with a Tripodal Nitroxyl Radical Showing Strong Magnetic Coupling. Inorg. Chem. 2020, 59, 16591–16598. [CrossRef]
- Rey, P.; Smolentsev, A.I.; Vostrikova, K.E. Oxazolidine Nitroxide Transformation in a Coordination Sphere of the Ln3+ Ions. Molecules 2022, 27, 1626. [CrossRef]
- Rey, P.; Caneschi, A.; Sukhikh, T.S.; Vostrikova, K.E. Tripodal Oxazolidine-N-Oxyl Diradical Complexes of Dy3+ and Eu3+. Inorganics 2021, 9, 91. [CrossRef]
- Köhn, R.D.; Pan, Z.; Kociok-Köhn, G.; Mahon, M.F. New Sandwich Complexes of Praseodymium(Iii) Containing Triazacyclohexane Ligands. J. Chem. Soc. Dalt. Trans. 2002, 2344–2347. [CrossRef]
- Wedal, J.C.; Ziller, J.W.; Evans, W.J. Trimethyltriazacyclohexane Coordination Chemistry of Simple Rare-Earth Metal Salts. Dalt. Trans. 2023, 52, 4787–4795. [CrossRef]
- Hazama, R.; Umakoshi, K.; Kabuto, C.; Kabuto, K.; Sasaki, Y. A Europium(III)-N,N,N′,N′-Tetrakis(2-Pyridylmethyl)-(R)-Propylenediamine Complex as a New Chiral Lanthanide NMR Shift Reagent for Aqueous Neutral Solution. Chem. Commun. 1996, 15–16. [CrossRef]
- Kira E. Vostrikova, Denis G. Samsonenko, CCDC 2268033: Experimental Crystal Structure Determination, DOI: 10.5517/ccdc.csd.cc2g42c2.
- Ishida, T.; Murakami, R.; Kanetomo, T.; Nojiri, H. Magnetic Study on Radical-Gadolinium(III) Complexes. Relationship between the Exchange Coupling and Coordination Structure. Polyhedron 2013, 66, 183–187. [CrossRef]
- Kanetomo, T.; Ishida, T. Strongest Exchange Coupling in Gadolinium(III) and Nitroxide Coordination Compounds. Inorg. Chem. 2014, 53, 10794–10796. [CrossRef]
- Kanetomo, T.; Yoshitake, T.; Ishida, T. Strongest Ferromagnetic Coupling in Designed Gadolinium(III)–Nitroxide Coordination Compounds. Inorg. Chem. 2016, 55, 8140–8146. [CrossRef]
- Hu, P.; Zhu, M.; Mei, X.; Tian, H.; Ma, Y.; Li, L.; Liao, D. Single-Molecule Magnets Based on Rare Earth Complexes with Chelating Benzimidazole-Substituted Nitronyl Nitroxide Radicals. Dalt. Trans. 2012, 41, 14651. [CrossRef]
- Wang, X.; Zhu, M.; Wang, J.; Li, L. Unusual Gd–Nitronyl Nitroxide Antiferromagnetic Coupling and Slow Magnetic Relaxation in the Corresponding Tb Analogue. Dalt. Trans. 2015, 44, 13890–13896. [CrossRef]
- Wang, J.; Miao, H.; Xiao, Z.-X.; Zhou, Y.; Deng, L.-D.; Zhang, Y.-Q.; Wang, X.-Y. Syntheses, Structures and Magnetic Properties of the Lanthanide Complexes of the Pyrimidyl-Substituted Nitronyl Nitroxide Radical. Dalt. Trans. 2017, 46, 10452–10461. [CrossRef]
- Benelli, C.; Caneschi, A.; Gatteschi, D.; Pardi, L. Gadolinium(III) Complexes with Pyridine-Substituted Nitronyl Nitroxide Radicals. Inorg. Chem. 1992, 31, 741–746. [CrossRef]
- Hu, P.; Sun, Z.; Wang, X.; Li, L.; Liao, D.; Luneau, D. Magnetic Relaxation in Mononuclear Tb Complex Involving a Nitronyl Nitroxide Ligand. New J. Chem. 2014, 38, 4716–4721. [CrossRef]
- Chen, P.Y.; Wu, M.Z.; Shi, X.J.; Tian, L. A Family of Multi-Spin Rare-Earth Complexes Based on a Triazole Nitronyl Nitroxide Radical: Synthesis, Structure and Magnetic Properties. RSC Adv. 2018, 8, 15480–15486. [CrossRef]
- Nakamura, T.; Ishida, T. Magnetic Exchange Interaction in Gadolinium(III) Complex Having Aliphatic Nitroxide Radical TEMPO.; 2016; p. 020016.
- Caneschi, A.; Dei, A.; Gatteschi, D.; Sorace, L.; Vostrikova, K. Antiferromagnetic Coupling in a Gadolinium(III) Semiquinonato Complex. Angew. Chemie - Int. Ed. 2000, 39. [CrossRef]
- Zheludev, A.; Barone, V.; Bonnet, M.; Delley, B.; Grand, A.; Ressouche, E.; Rey, P.; Subra, R.; Schweizer, J. Spin Density in a Nitronyl Nitroxide Free Radical. Polarized Neutron Diffraction Investigation and Ab Initio Calculations. J. Am. Chem. Soc. 1994, 116, 2019–2027. [CrossRef]
- Hamada, D.; Fujinami, T.; Yamauchi, S.; Matsumoto, N.; Mochida, N.; Ishida, T.; Sunatsuki, Y.; Tsuchimoto, M.; Coletti, C.; Re, N. Luminescent DyIII Single Ion Magnets with Same N6O3 Donor Atoms but Different Donor Atom Arrangements, ‘Fac’-[DyIII(HLDL-Ala)3]·8H2O and ‘Mer’-[DyIII(HLDL-Phe)3]·7H2O. Polyhedron 2016, 109, 120–128. [CrossRef]
- Murakami, R.; Nakamura, T.; Ishida, T. Doubly TEMPO-Coordinated Gadolinium(III), Lanthanum(III), and Yttrium(III) Complexes. Strong Superexchange Coupling across Rare Earth Ions. Dalt. Trans. 2014, 43, 5893–5898. [CrossRef]
- Tkachenko, I.A.; Petrochenkova, N. V.; Mirochnik, A.G.; Karasev, V.E.; Kavun, V.Y. Carboxylato-Bis-Dibenzoylmethanates of Europium(III): Luminescence and Magnetic Properties. Russ. J. Phys. Chem. A 2012, 86, 681–684. [CrossRef]
- Kahn, M.L.; Sutter, J.-P.; Golhen, S.; Guionneau, P.; Ouahab, L.; Kahn, O.; Chasseau, D. Systematic Investigation of the Nature of the Coupling between a Ln(III) Ion (Ln = Ce(III) to Dy(III)) and Its Aminoxyl Radical Ligands. Structural and Magnetic Characteristics of a Series of {Ln(Radical)2} Compounds and the Related {Ln(Nitrone)2} Derivat. J. Am. Chem. Soc. 2000, 122, 3413–3421. [CrossRef]
Figure 1.
Molecular structures (left) and trigonal antiprismatic coordination polyhedra (right) for the compounds 1-4. Hydrogen atoms are omitted. The picture was made using open crystallographic data.
Figure 1.
Molecular structures (left) and trigonal antiprismatic coordination polyhedra (right) for the compounds 1-4. Hydrogen atoms are omitted. The picture was made using open crystallographic data.
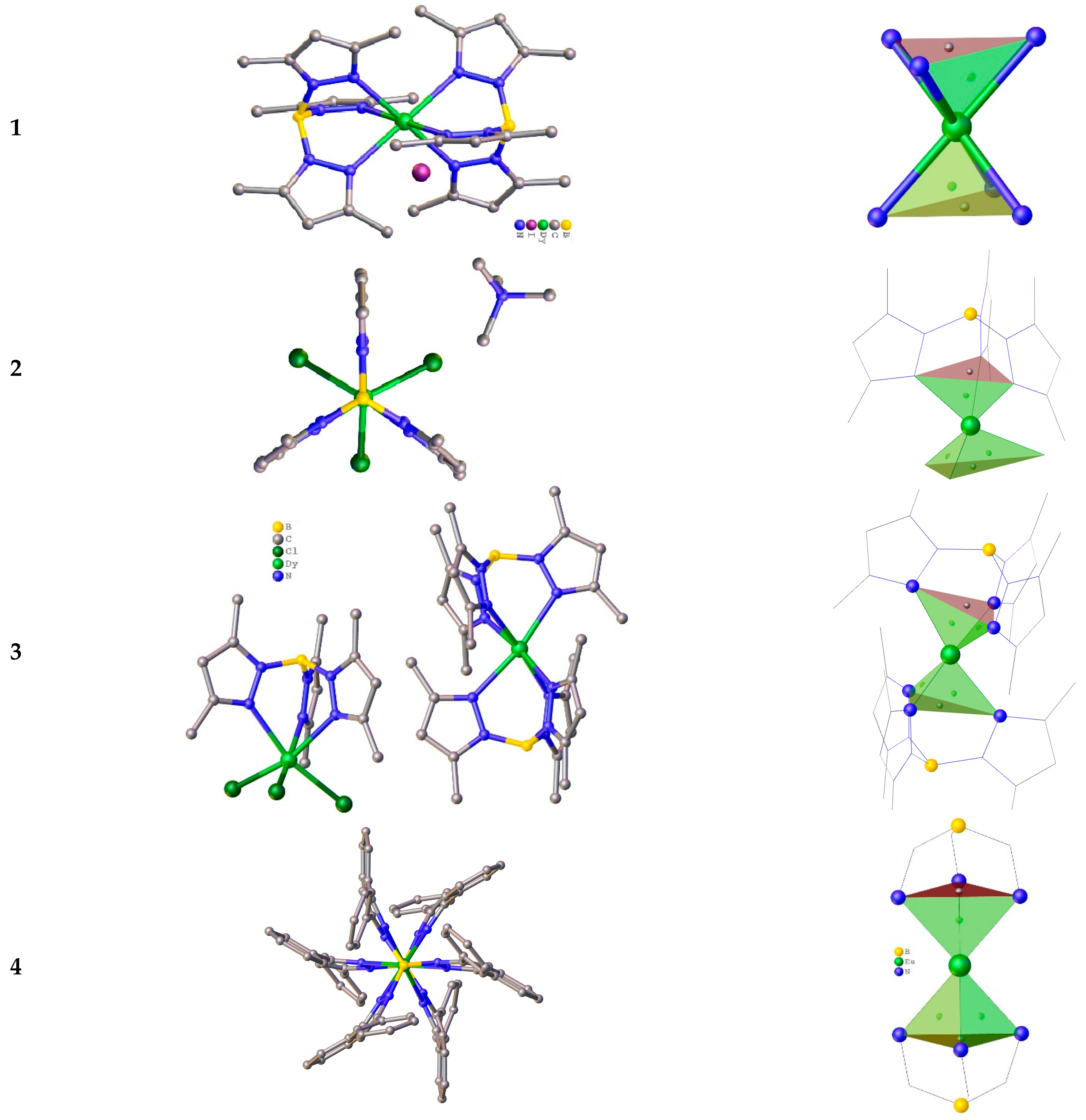
Figure 2.
Molecular structures (left) and trigonal antiprismatic coordination polyhedra (right) for the compounds 5-10. Hydrogen atoms are omitted. The picture was made using open crystallographic data.
Figure 2.
Molecular structures (left) and trigonal antiprismatic coordination polyhedra (right) for the compounds 5-10. Hydrogen atoms are omitted. The picture was made using open crystallographic data.
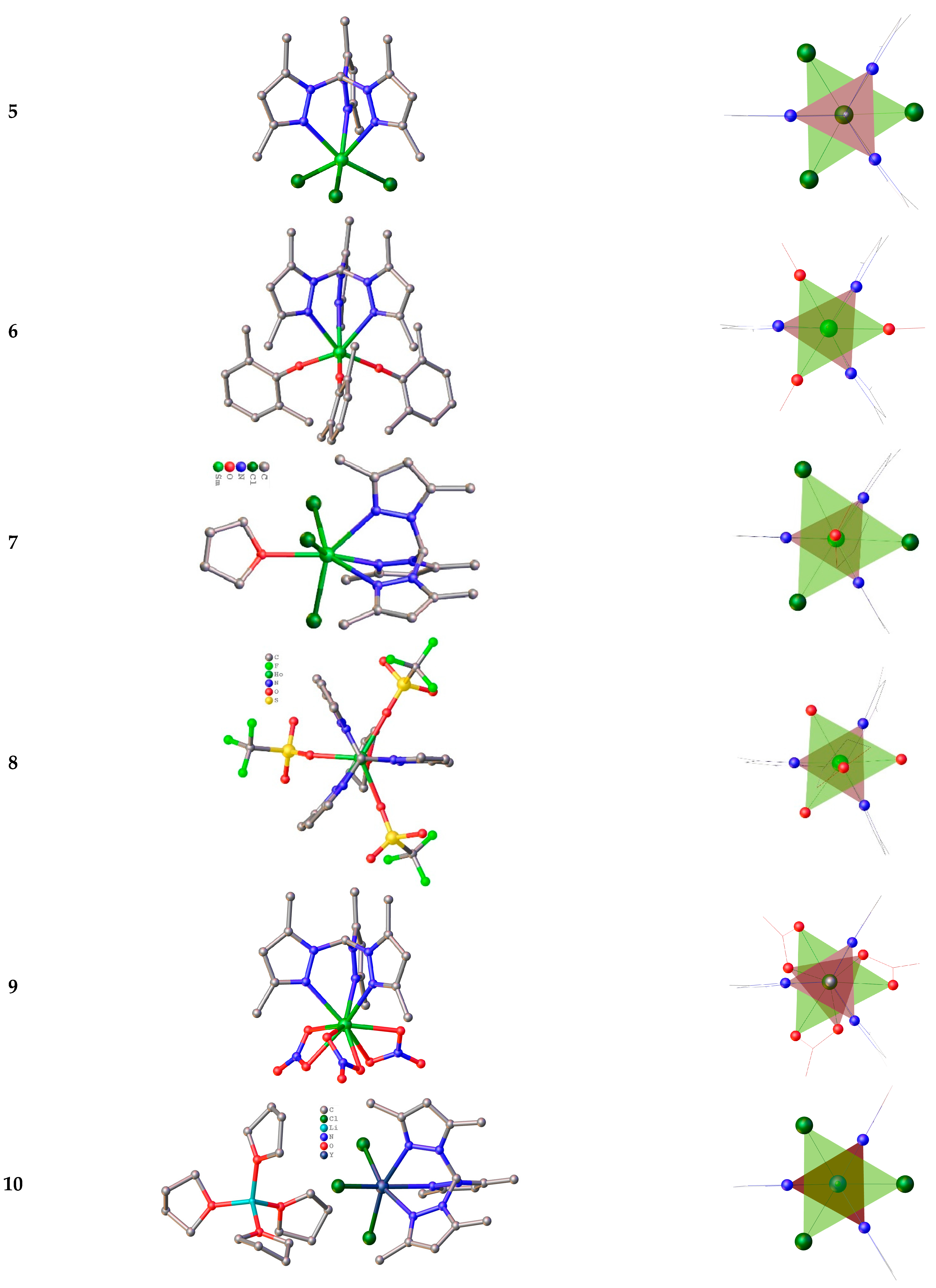
Figure 5.
Molecular structures (left) and coordination polyhedra Ln-N6, with colored faces forming the tripod bases (right) for the compounds 26-33. Hydrogen atoms are omitted. The picture was made using open crystallographic data.
Figure 5.
Molecular structures (left) and coordination polyhedra Ln-N6, with colored faces forming the tripod bases (right) for the compounds 26-33. Hydrogen atoms are omitted. The picture was made using open crystallographic data.
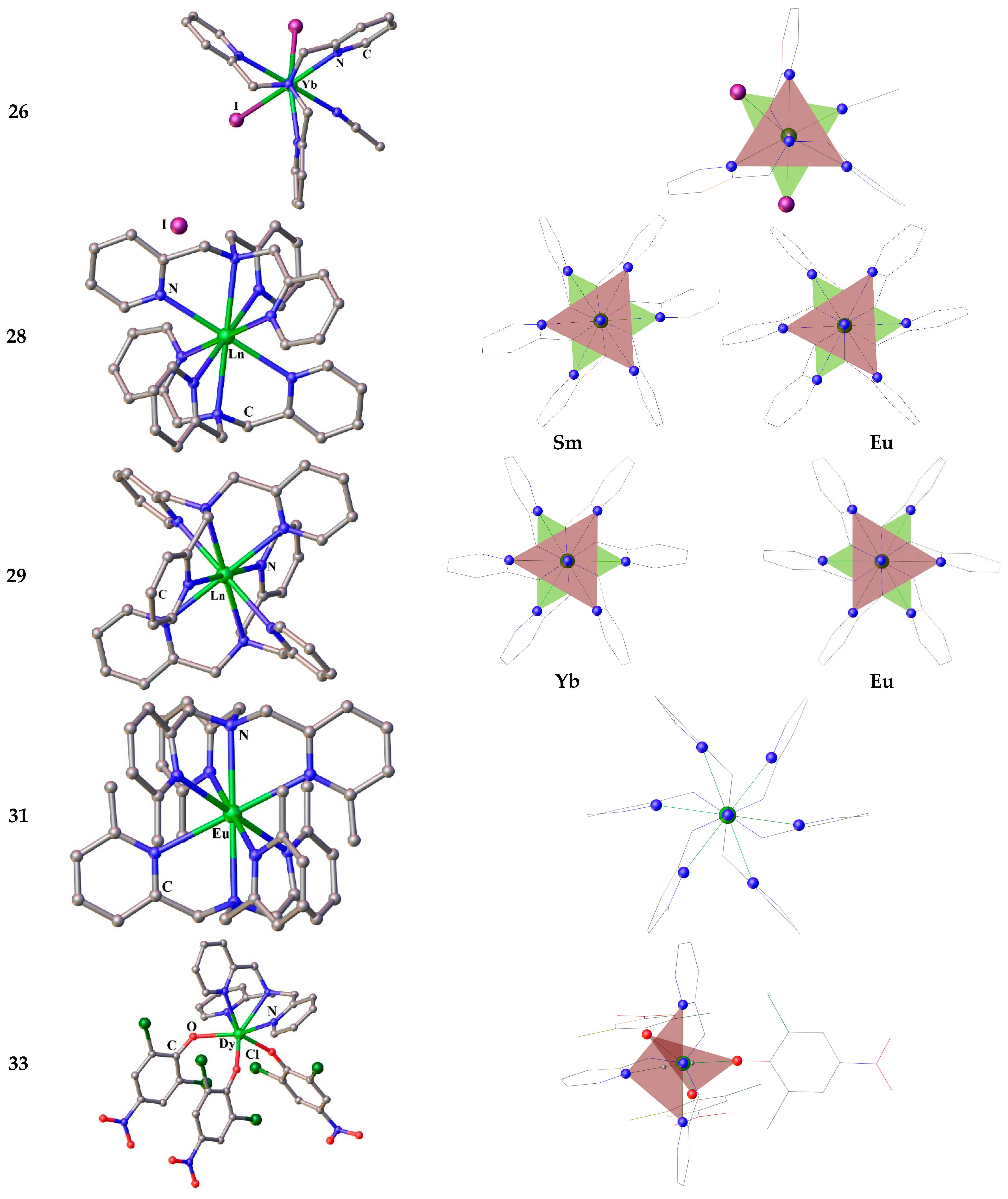
Figure 6.
2,5-Dihydro-4,5,5-Trimethyl-2,2-Bis(2-Pyridyl)Imidazole-1-Oxyl. 34; 4,4-dimethyl-2,2-di(2-pyridyl) oxazolidine-N-oxyl 35 and 4,4-dimethyl-2,2-bis[2-(4-methylpyridyl)]oxazolidine-N-oxyl 36 − radical tripods.
Figure 6.
2,5-Dihydro-4,5,5-Trimethyl-2,2-Bis(2-Pyridyl)Imidazole-1-Oxyl. 34; 4,4-dimethyl-2,2-di(2-pyridyl) oxazolidine-N-oxyl 35 and 4,4-dimethyl-2,2-bis[2-(4-methylpyridyl)]oxazolidine-N-oxyl 36 − radical tripods.
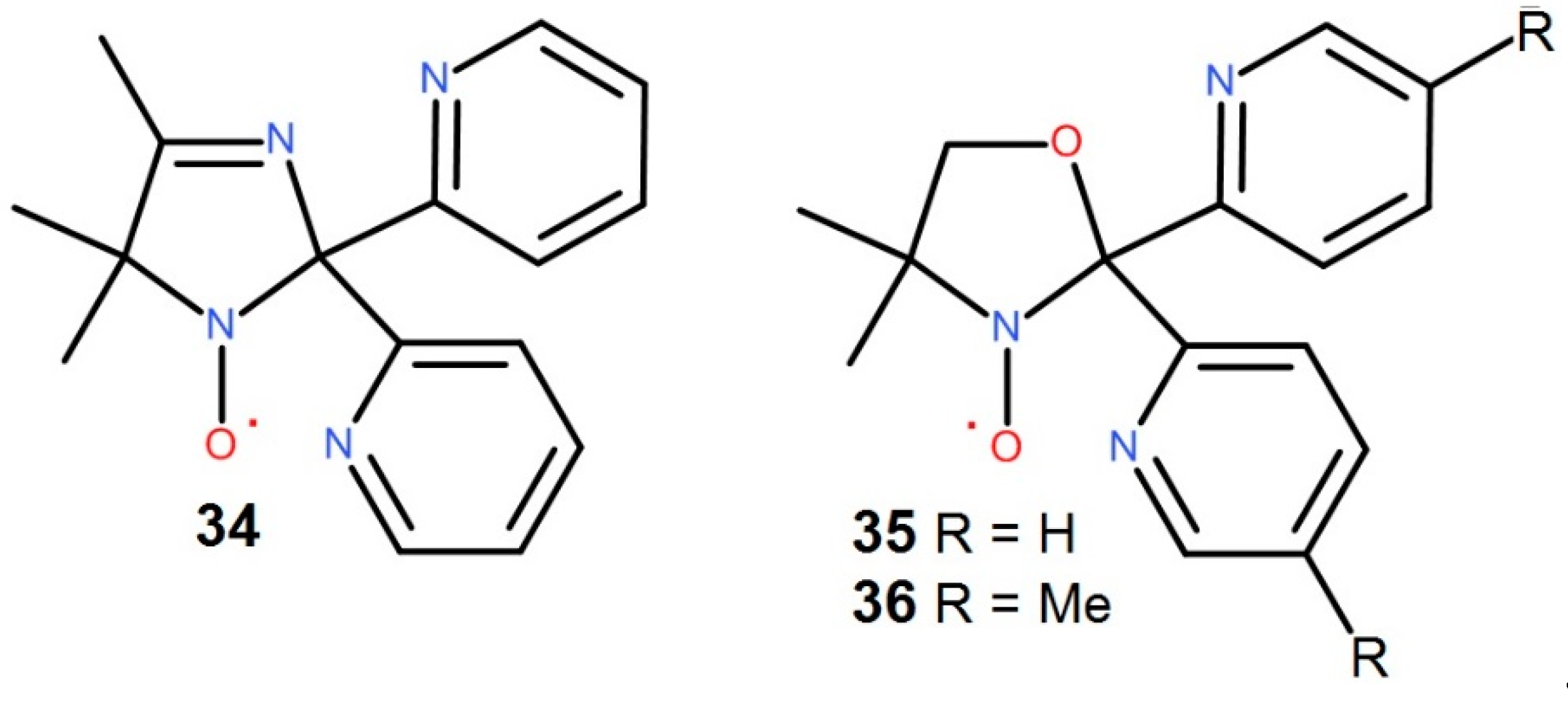
Figure 8.
(a) Tri-triflate [Pr(Me3tach)2(OTf)3] [141]; (b) Four-triflate HMe3tach[La(Me3tach)2(OTf)4] [142] complexes. The picture was made using open crystallographic data.
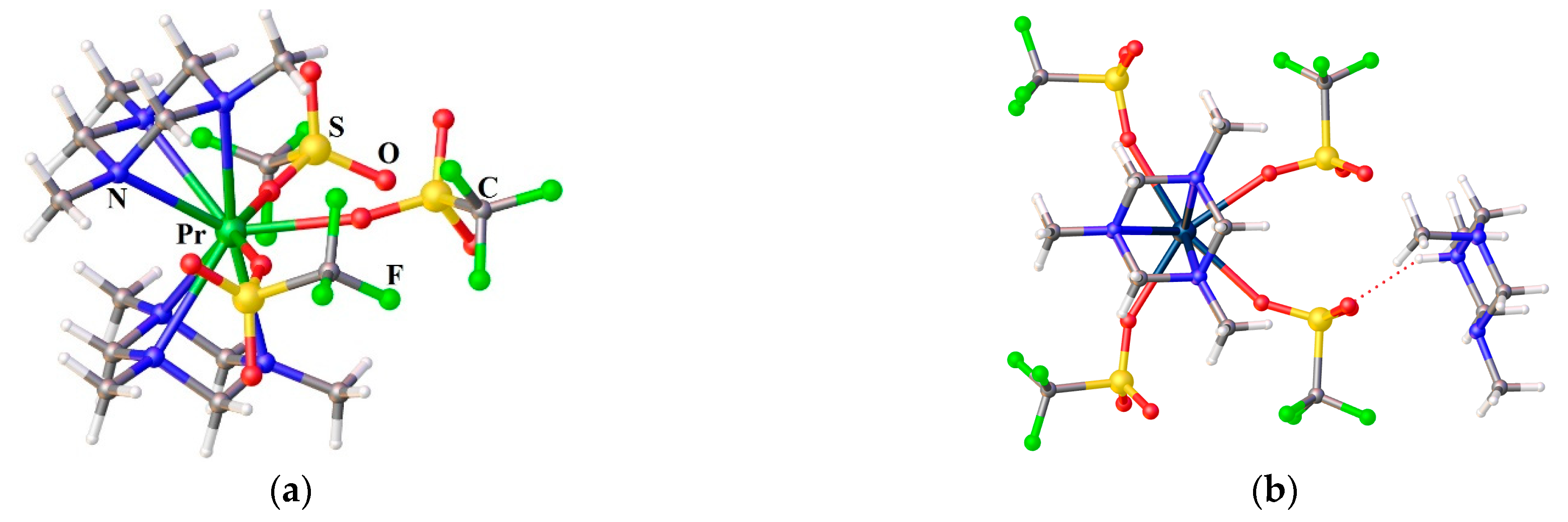
Figure 9.
The compound [La(Me3tach)2Cl3] having three chlorine anionic ligands in equatorial plane.
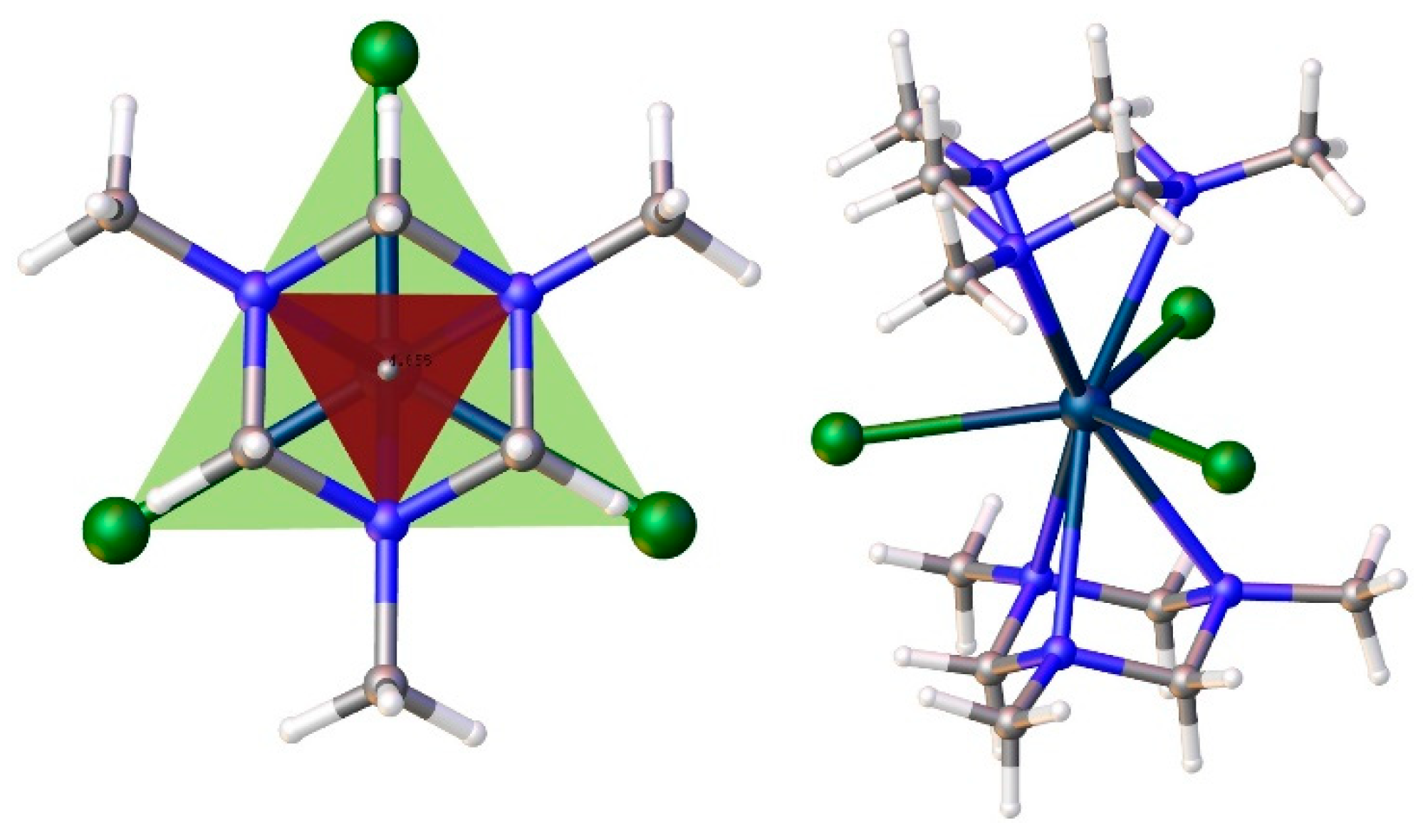
Figure 10.
(a) The closest contacts in [DyRad2Cl2]+; (b) Bis-(oxazinane-radical) Ln complex cation.
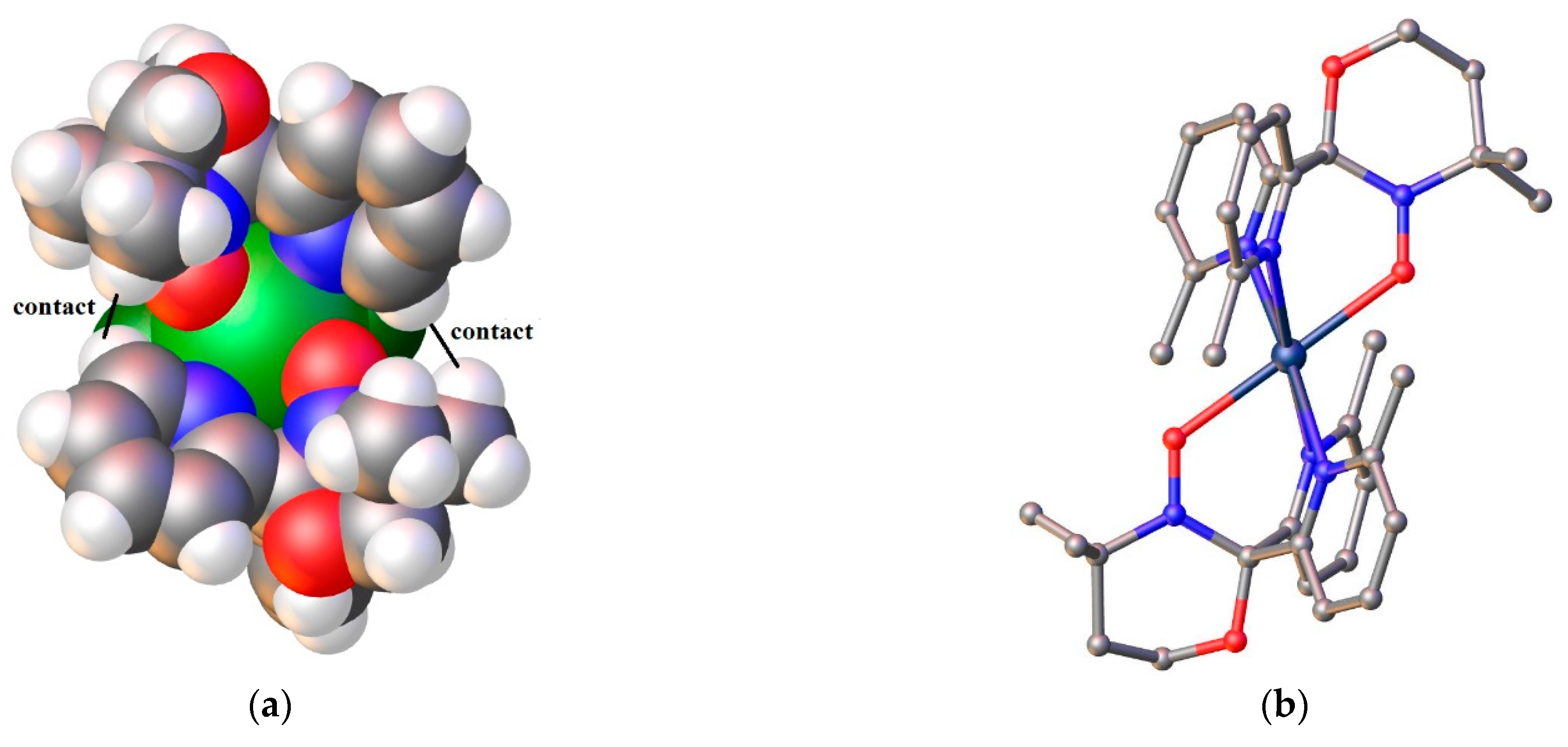
Figure 11.
More sterically demanding and more symmetrical candidates as tripodal nitroxyl radicals: (a) 4,4-dimethyl-2,2-bis[2-(3-methylpyridyl)]oxazinane-N-oxyl (41); (b) 2,2-bis[2-(1-methyl-imidazol-2-yl)]oxazolidine-N-oxyl (42).
Figure 11.
More sterically demanding and more symmetrical candidates as tripodal nitroxyl radicals: (a) 4,4-dimethyl-2,2-bis[2-(3-methylpyridyl)]oxazinane-N-oxyl (41); (b) 2,2-bis[2-(1-methyl-imidazol-2-yl)]oxazolidine-N-oxyl (42).
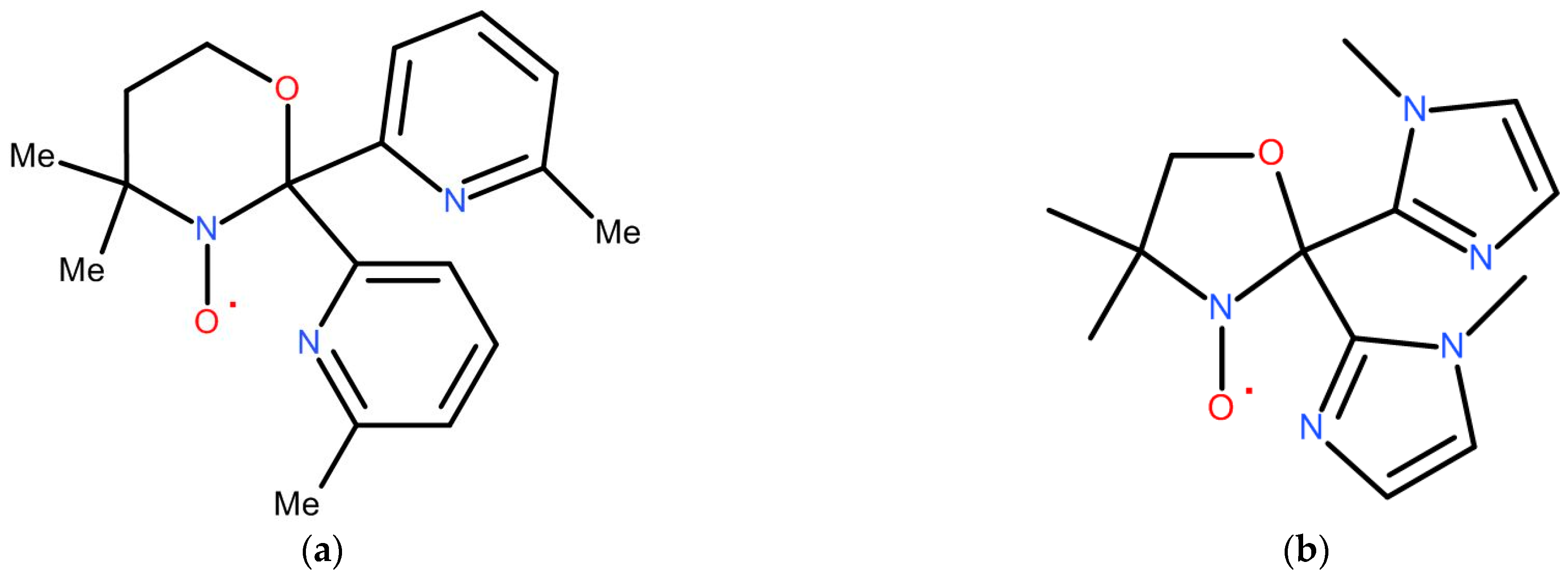
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



