
Submitted:
20 June 2023
Posted:
20 June 2023
You are already at the latest version
Abstract
The production and secretion of very-low density lipoproteins (VLDL) by hepatocytes has a direct impact on liver fat content as well as the concentration of cholesterol and triglycerides in the circulation and thus affects liver and cardiovascular health, respectively. Importantly, excess caloric intake and lack of physical activity are associated with overproduction of VLDL, hepatic steatosis, and increased levels of atherogenic lipoproteins. Cholesterol as well as triglycerides in remnant particles after VLDL lipolysis are risk factors for atherosclerotic cardiovascular disease (ASCVD) and have garnered increasing attention over the last few decades. To date, however, increased risk of ASCVD is not the only concern when considering today’s cardiometabolic patients, as they often also suffer from hepatic steatosis. This notion highlights the importance of understanding the molecular regulation of VLDL biogenesis. Fortunately, there has been a resurgence of interest in the intracellular assembly, trafficking, degradation, and secretion of VLDL by hepatocytes, that has led to many exciting new molecular insights, the topic of this review. We think that increasing our understanding of the biology of this pathway will help improve the health of the cardiometabolic patient in the long term.
Keywords:
Very low density lipoproteins
; biogenesis
; dyslipidemia
Introduction
After several decades of decreasing rates of atherosclerotic cardiovascular disease (ASCVD) in the world, the last two decades have seen a rise toward the very high rates of the mid-20th century. There are many potential reasons for this change in direction of the most common cause of death in most countries, but the pandemic of obesity and its sequalae, diabetes, hypertension, and dyslipidaemia, is certainly a very significant factor 1. Dyslipidaemia is not simply high levels of low-density lipoprotein (LDL) cholesterol, which in most people can be reduced significantly with statins. Rather, it is a lipid disorder driven by increased hepatic assembly and secretion of very low-density lipoproteins (VLDL), resulting in elevated levels of these triglyceride-rich lipoproteins and their remnants that are, based on genetic evidence (e.g., LPL deficiency), causal for ASCVD 2. Drugs that inhibit the assembly and secretion of VLDL are used in the clinic for patients at very high risk of ASDVD, but unfortunately this approach has led to hepatic lipid accumulation, a deleterious consequence for the liver 3. The question is whether it will be possible to reduce VLDL secretion without increasing hepatic lipids to prevent or treat ASCVD, an important unmet need in medicine.
VLDL biogenesis and secretion have been characterized extensively and based on current data, involves approximately 50 molecular entities. However, how these factors interact with apoB, or what determines how VLDL at any stage of biogenesis is targeted for either degradation or secretion remains largely unknown. It is also unclear how VLDL secretion is co-regulated with lipid synthesis and disposal to prevent hepatic steatosis. Over the last few years, however, numerous studies have considerably improved our understanding of the VLDL biogenesis pathway, and we have tried to capture the current state of the art in one scheme (Figure 1) after which each recently (±5 years) identified player is described in detail in this review.
The genesis of VLDL starts with the translation of the APOB transcript into apolipoprotein B (apoB), one of the largest proteins in human (4536 amino acids). This core protein is continuously produced and loaded with lipids in the endoplasmic reticulum (ER). It is thereafter transported to the Golgi network by dedicated VLDL transport vesicles (VTVs). Following transport through the Golgi, VLDL is subsequently secreted into the circulation to deliver energy, in the form of TG-fatty acids to the periphery. Although the liver also plays a major role in the uptake VLDL remnants or LDL, generated by lipolysis in peripheral tissues, this review only focusses on proteins required for the biogenesis of a mature VLDL particle in hepatocytes. In the legend of Figure 1, we briefly describe this process incorporating the new molecules that will be discussed along the way in this review. We have – for clarity - not included all molecular players, in the text, but for completeness, listed all factors known to regulate intracellular VLDL metabolism directly (Table 1). Not every protein listed has been studied in both cellular or animal models, or even known to carry out the same function in humans. The many different in vitro and in vivo models that are being used to characterise the function of each protein in intracellular VLDL metabolism add to the complexity of this research field. While hepatocarcinoma cell lines of human (HepG2, IHH), murine (Hepa1-6), and rat (McArdle) origin are commonly used, some investigators also use primary hepatocytes or in vivo human kinetic studies. For completeness, Table 1 gives information regarding which models were used to define roles of the numerous factors that affect hepatic VLDL production and secretion.
In the nucleus, there is increasing evidence that Apob expression is regulated at mRNA level (miRNAs, HUR). APOBEC-1, an Apob100 mRNA editing enzyme that generates apoB48, is present in the mouse but not the human hepatocyte. During translation of apoB across the ER membrane, facilitated by several chaperones*, apoB is co-translationally and co-translocationally lipidated by MTP. When proper folding of apoB is affected or when lipidation is inadequate, apoB will be ubiquitinylated followed by proteasomal degradation, facilitated by proteins involved in ER-associated degradation** (ERAD). Newly identified factors in this process include MDM2 and LZP. Next, many proteins take part in the supply of additional lipids that can derive from both lipid droplet utilization as well as de novo lipogenesis in the ER membrane (e.g., CIDEB, DGAT, PEMT) to nascent VLDL. Although MTP is essential for initial lipidation of apoB to form nascent VLDL, this figure illustrates that many other proteins are implicated in the conversion of nascent lipid poor VLDL to a more mature, well-lipidated VLDL. Newly identified players that are involved in this process are PRAP1, TORSINA (activated by LAP1), SMLR1, PLA2G12B, ERLIN1/2 and TM6SF2. Prerequisites for VLDL to leave the ER for subsequent transport to the Golgi, include phospholipid enrichment of the ER to enable vesicle budding (VMP1, TMEM41B) as well as the recruitment of transport proteins to form VLDL transport vesicles (VTV). Here, SURF4, RTN3 and RABs are added to an already long list of proteins including TANGO1, TALI, KLHL12, SAR1B, SEC13/31 and SEC23/24. After docking of VTV at the Cis-Golgi with SNAREs, VLDL is thought to be subjected to sorting, glycosylation and additional/final lipidation. SORTILIN1 is known to regulate VLDL secretion by re-directing some particles for auto-lysosomal degradation. While reviewing the literature, the scarcity of information on how VLDL leaves the Golgi and how it is transported to the cell membrane for eventual secretion into the circulation was striking.
*Chaperones = BIP, PDI, ERP57, ERP72, HSP110, Calreticulin, Calnexin, GRP94, Cyclophilin B, HSP70 and HSP90. ** ER associated degradation (ERAD) = DERLIN-1, GP78/AMFR, HRD1, HSP40, P97, P58IPK, SEC61, UBXD8. Abbreviations; CE, cholesterol esters; ER, endoplasmic reticulum; TG, triglycerides; VLDL, very low-density lipoprotein.
In the following sections, we discuss each novel regulator in detail and briefly touch upon some already well-described players.
NUCLEUS
Apob in mice or APOB in humans is generally thought to be constitutively expressed, which implies absent or minimal regulation at the transcriptional level 56,57. Recent evidence, however, suggests that this is not entirely the case. Zhang et al., showed that human antigen R (HUR) is required for splicing of Apob pre-mRNA 37. HUR, encoded by the Elavl1 gene, is an RNA-binding protein implicated in various biological processes including tumorigenesis and inflammation, lipid transport and ATP synthesis. Bioinformatic prediction models identified HUR-binding sites in intronic Apob pre-mRNA. Hepatic Elav1 knockout in mice (Elav1-LKO) was shown to increase Apob pre-mRNA levels and reduce Apob mRNA levels. Concordantly, chow fed Elav1-LKO mice have reduced plasma levels of apoB (≈43%). After a high-fat diet for four weeks, these mice show increased liver triglycerides and cholesterol and impaired liver function compared to controls. Loss of hepatic HUR, however, also increased protein levels of CYCS, NDUFB6 and UQCRB, indicating that HUR not only promotes lipid transport but also ATP synthesis.
Non-coding RNAs have also been shown to play a role in regulating the metabolism of apoB-containing lipoproteins. In a recent study, Fernández-Tussy et al. showed non-coding RNAs affecting the expression of MTTP, PCSK9 and SORT1, which can alter the assembly, secretion, and/or re-uptake of VLDL 58. Although this study does not mention miRNAs that directly affect Apob mRNA, another study indicated a role for miR-548p in regulating APOB/Apob expression in human primary hepatocytes but not mouse primary hepatocytes, respectively 59. Although more miRNAs have been linked to Apob regulation 60, studies showing causality are still lacking.
NUCLEAR MEMBRANE
A role for proteins in the nuclear envelope in the production of VLDL was established in 2019 by Shin et al 46. It was shown that hepatic deletion of lamina-associated polypeptide 1 (LAP1) results in 20% lower VLDL secretion and mild steatosis, including intranuclear lipid accumulation, in chow-fed mice. Previous research has revealed that LAP1, as well as luminal domain-like LAP1 (LULL1), binds to and activates TORSINA, an ER-resident AAA+ ATPase encoded by Tor1a 61. Whereas Tor1a-/- mice exhibit perinatal lethality, some mutations in human TOR1A are implicated in primary dystonia (OMIM#605204), a severe movement disorder. A link to lipid metabolism was implied in 2019 by genetic studies that listed regulators of lipid metabolism after mapping 107 genetically distinct mouse strains62. Hepatic loss of TORSINA in mice causes profound steatosis and marked reduction in VLDL secretion with 64% lower VLDL levels on a chow diet, a more severe phenotype compared to hepatic deletion of LAP1. It is not clear if LAP1 and LULL1 affect apoB secretion other than via TORSINA, nor is it known how and where TORSINA regulates early VLDL biogenesis.
ENDOPLASMIC RETICULUM AND PROTEIN DEGRADATION
Translation of the 16kb transcript of Apob100 into an appropriately folded full-length protein requires multiple chaperone (see Table 1). Misfolding and/or inadequate lipidation of apoB result in its co-translational ubiquitinylation and subsequent degradation by the proteasome during translocation. ApoB100 degradation is a type of ER-associated degradation (ERAD). The ERAD of apoB requires several proteins noted in Table 1 and can be regulated by microsomal triglyceride transfer protein (MTP) and sterol regulatory element-binding protein 1c (SREBP1c, regulates lipogenesis), but also cytosolic chaperones like HSP70 and HSP90 (for a recent review see 57). In addition, post-ER, pre-secretory proteolysis (PERPP) also affects intracellular apoB100 degradation 63.
Liver-specific Zona Pellucida domain-containing protein (LZP), encoded by the OIT3 gene, can be regarded as a new member of this group of proteins that mediate apoB100 degradation. LZP was initially studied in the context of hepatocarcinogenesis 64, but Oit3-/- mice were also found to exhibit hepatic lipid accumulation and reduced plasma triglycerides on chow and HFD, associated with a hepatic apoB levels and a 70% decrease in VLDL secretion compared to controls (diet unknown) 38. It was furthermore shown that LZP interacts with apoB in immunoprecipitation experiments, while proteasome inhibition restores intracellular apoB levels in primary hepatocytes of Oit3-/- mice. The investigators show that LZP is in the ER and Golgi and that it stabilizes apoB in the ER by preventing ubiquitinylation of the nascent protein by the ER transmembrane E3 ubiquitin ligase, GP78/autocrine motility factor receptor (AMFR)10.
Despite the high energy demand to generate the very large apoB protein, there is always production and degradation of apoB. Inhibition of proteins facilitating this breakdown might lead to increased apoB secretion. This was recently shown after hepatic deletion of murine double minute 2 (MDM2), a protein that acts as an E3 ubiquitin ligase and targets apoB for proteasomal degradation 39. Other groups have shown a role for MDM2 in the nuclear export and activation of p53 (tumor suppressor gene) but whether it regulates apoB100 is unknown. Liver-specific deletion of MDM2 in mice on chow did not alter hepatic triglyceride levels but on high fat high cholesterol diet prevented hepatic steatosis by a 50% reduction of hepatic triglycerides. This was in line with increased circulating apoB and plasma triglycerides in a context of a 25% elevation of TG-VLDL secretion upon inhibition of VLDL lipolysis (by Tyloxapol injection). The potential clinical significance of this finding was provided through pharmacological inhibition of MDM2 which alleviated non-alcoholic steatohepatitis in choline-deficient amino acid-defined high-fat diet fed mice. The authors also showed that the opposite is true; overexpressing MDM2, but not an inactive MDM2 mutant, increased ubiquitinylation of apoB in HepG2 cells. Whether this leads to excess hepatic lipids in mice was not investigated. Taken together, blocking MDM2 may be tool to fight hepatic lipid accumulation but this will be associated with increased levels of atherogenic plasma lipids.
ENDOPLASMIC RETICULUM AND LIPID LOADING
As mentioned in the introduction, other ER-resident proteins including MTP, are essential to stabilize apoB. MTP was identified as a microsomal lipid transfer protein in 1985 65. It was only after the identification of abetalipoproteinemia patients (#OMIM 200100) with loss-of-function mutations in the encoding gene MTTP, that MTP was recognized for its essential role in the assembly of apoB-containing lipoproteins. MTP exists in a heterodimeric complex with protein disulfide isomerase (PDI) 66. MTP transfers phospholipids, triglycerides, and cholesterol esters from the ER membrane into the apoB lipid-binding pocket, i.e., the N-terminal domain of approximately 1000 amino acids. Initial lipidation comes from the ER bilayer lipids. MTP also mediates the translocation of apoB across the ER membrane 20 (detailed information about the interactions between apoB and MTP can be found in studies by Hussain or Ginsberg and co-workers; 21,67).
The transcription factor hepatocyte nuclear factor 4α (HNF-4α) governs the expression of numerous genes with roles in glucose and lipid homeostasis including e.g., Mttp, Apob as well as Pla2g12b which encodes for phospholipase A2, group XIIB 68. PLA2G12B was also identified when searching the transcriptome for genes that are co-expressed with MTTP and APOB in humans 69. Livers from chow fed whole body Pla2g12b-/- mice have a 2- and 3-fold increase in liver cholesterol and triglycerides respectively, which was associated with an approximate 50% reduction in VLDL-TG secretion as determined by Tyloxapol injection. Interestingly, plasma cholesterol and triglyceride levels showed a marked reduction of 92% and 79%, respectively 69. Phospholipase A2 (PLA2) catalyses the lipolysis of glycerophospholipids to lysophospholipids and fatty acids but PLA2G12B type lacks this catalytic phospholipase activity 70, leaving the mechanisms by which PLA2G12B affects lipoprotein metabolism unclear. In 2012, Aljakna et al. already linked a mutation in Pla2g12b with a 58% reduction in plasma triglyceride levels in mice 71. In 2022, complete loss of Pla2g12b in zebrafish larvae was associated with large lipid droplets (LD) in the lumen of the ER and secretion of abnormally small lipoproteins suggesting a defect in the formation of large apoB-containing lipoproteins 72. Additional studies in HepG2 and Caco-2 (intestinal) cells implicated a role for PLA2G12B in calcium and MTP recruitment to the ER membrane to enhance lipid transfer to VLDL and chylomicrons 72. The utilization of cytosolic LD to generate luminal LD, which are subsequently used to lipidate VLDL, has already been proposed by Lehner et al. in 2012 73. Thierer and colleges propose PLA2G12B as the first regulator of moving triglycerides from a luminal LD to nascent VLDL.
Another protein also closely linked to MTP is proline-rich acidic protein 1 (PRAP1). PRAP1 was recently identified to facilitate MTP-mediated lipid transfer in the intestine 40. Wolfarth et al. performed transcriptomic analysis on murine intestine to identify novel cytoprotective genes induced by probiotics. Prap1 was a top candidate gene which protected the barrier from oxidative insult 74. To delineate its function, they used pull-down assays and found MTP as main interacting partner of PRAP1, which changed their attention towards lipid metabolism 40. Isolated intestinal epithelial cells from Prap1-/- mice displayed almost 50% lower triglyceride and phospholipid transfer activities, and near absent apoB48 secretion in pulse chase experiments. Oil-Red-O staining of enterocytes revealed marked lipid accumulation in the Prap1-/- mice compared to controls 2 hours after receiving an intragastric bolus of lipids. While plasma triglycerides were lower in Prap1-/- mice (on both chow and a high fat diet), total cholesterol levels were unaffected. This phenotype resulted mainly from a defect in lipid absorption noted by increased faecal lipid content with similar food intake in the Prap1-/- mice compared to controls. Only on a high-fat diet, Prap1-/- mice appeared leaner with reduced body weight increase compared to controls. [73]. PRAP1 levels are very low in the liver and fractionation of plasma by density gradient ultracentrifugation revealed only a slight reduction in VLDL levels, suggesting that most of the phenotype stems from the loss of PRAP1 in the intestine. Interestingly, PRAP1 is also secreted in the circulation, but whether this affects lipoprotein metabolism has not been studied.
Important to lipid loading of apoB, there are several steps leading up to the generation of lipids in the ER. These lipids are mostly triglycerides, which are synthesized from diacylglycerides by two acyl-CoA: diacylglycerol acyltransferase enzymes named DGAT1 and DGAT2. These triglycerides are used for cytosolic LD biogenesis but, in hepatocytes and enterocytes, are also critical for the production and secretion of apoB-containing lipoproteins. Although both DGATs are key in regulating triglyceride synthesis, DGAT1 differs from DGAT2 in several ways. DGAT1 is most highly expressed in the small intestine and mainly utilizes exogenous (dietary) fatty acids for triglyceride synthesis whereas DGAT2 is primarily expressed in the liver and utilizes fatty acids which are newly generated (de novo lipogenesis) 75. More details and a working model of how these proteins synthesize and incorporate triglycerides in the ER can be found in the study from Li et al., 2015 76. When it comes to their role in VLDL biology, it has been shown that liver specific Dgat1 KO mice produce smaller VLDL (55% decrease in TG/apoB ratio) without changing apoB particle number 23. DGAT2 inhibition in mice resulted in effective reduction of triglyceride secretion into blood (>33%) but no such effect was observed in Rhesus primates 22.
The contribution of human genetic studies as a starting point to identify novel VLDL genes is highlighted by the discovery of TM6SF2, encoding for transmembrane 6 superfamily member 2, which was found to affect the development of non-alcoholic fatty liver disease (NAFLD) in humans 77. TM6SF2 is a transmembrane protein of the ER, the ER-Golgi intermediate compartment (ERGIC) and the Golgi. A recent study shows that TM6SF2 stabilizes apoB through complex formation with ER lipid raft protein (ERLIN)-1 and ERLIN242. These proteins were found in pull-down assays of TM6SF2 in a rat hepatoma cell line. The ERLINs were formerly known as KE04p and C8orf2 and were later defined as lipid-raft like domains in the ER 78. Short hairpin RNA-mediated silencing of Tm6sf2 or both Erlins in mouse liver has been shown to reduce plasma cholesterol (≈80%, ≈70%) and triglyceride (≈50%, ≈35%) levels, but resulted in a 3-fold or 1.4-fold increase in liver triglycerides in mice on chow diet, respectively. There is evidence showing that TM6SF2 is involved in VLDL lipidation and maybe its apoB secretion 43,79, but the molecular mechanisms underlying these observations are still unclear.
VLDL VESICLE FORMATION
When lipoproteins exit the ER through vesicle budding, it is important that ER membrane integrity is maintained, which is controlled by various proteins including transmembrane protein 41B (TMEM41B) and vacuole membrane protein 1 (VMP1). TMEM41B is an integral ER membrane protein with phospholipid scramblase activity. The equilibration of phospholipids between the inner and outer leaflet of the membranes is also necessary to meet the high demand of phospholipids at the luminal side of the ER for lipoprotein assembly. TMEM41B was initially linked to autophagosome formation 80 but has also been described to play a role in LD formation. Loss of TMEM41B in Hela or HuH7 cells revealed impaired cellular distribution of phosphatidylserine and cholesterol in the ER and giant LDs 81. Others showed that liver specific ablation of Tmem41b in mice on chow led to depletion of plasma triglycerides due to reduced lipidation of apoB and lipoprotein trafficking [37], which was seen in the context of depleted intracellular apoB and rapid development of NASH. VMP1 also has phospholipid scramblase activity and is associated with TMEM41B to mediate autophagy 83, which initiated a study on VMP1 in lipoprotein biogenesis. Liver-specific VMP1 ablation in mice results in similar outcomes as seen in TMEM41B KO mice 81. Like MDM2 39 as well as MTP 84, overexpression of VMP1 in mice results in increased VLDL secretion and reduced hepatic steatosis.
VLDL VESICLE TRANSPORT
For the canonical transport of cargos from the ER to the Golgi, cells use Coat Protein Complex II (COPII) vesicles with an estimated size of 50-90nm in yeast and around 50nm in HepG2 cells 85,86. The formation of these COPII vesicles is initiated by the activation of GTPase-SAR1a by Sec12, the concurrent recruitment of Sec23/24 and subsequently Sec13/31 to form an inner and outer coat respectively 87 (see Figure 1). The transport of larger protein cargos like procollagen XII (300-400nm 88) requires larger vesicles and calls for the assisting proteins; transport and golgi organization protein 1 (TANGO1) 89, TANGO1-like (TALI) 90 and kelch-like protein 12 (KLHL12) 88.
VLDLs typically have diameters between 30-70 nm but can have diameters up to 120nm 91, which also require the assistance of TANGO and KELCH proteins 27,28. The authors suggest that these proteins induce VTV formation via the recruitment of Sec12 for more efficient activation of SAR1b or ubiquitinylation of Sec31. Whereas SAR1a has been implicated in COPII vesicle formation for protein transport, SAR1b has instead been shown to be key in the formation of transport vesicles for apoB-containing lipoproteins 92. This is clearly demonstrated by chylomicron retention disease in subjects suffering from loss-of-function mutation in SAR1B.
There are more proteins known to affect apoB/VLDL transport via the recruitment of motor proteins or vesicle tethers including some members of RAB family of small GTPases 51. In addition, a role for RAB GTPase-activating protein GP73 in VLDL secretion was first established in 2021 50. Because GP73 expression is low in normal liver tissues but increases upon ER stress or liver damage, the investigators overexpressed GP73 in mouse livers. They found increased intracellular apoB levels together with a 50% reduction in VLDL-TG secretion three hours after Tyloxapol injection. Although GP73 is a Golgi-resident protein which can traffic to the cell surface, the authors suggest that it acts via RAB23, a protein that regulates ER to Golgi transport and possibly secretion of lipoproteins. Whether these RABs are in direct contact with apoB and to what degree each RAB contributes to VLDL secretion is unclear.
To identify proteins associated with VTVs, proteomics analysis of isolated ER-derived vesicles revealed cell death-inducing DFFA-like effector b (CIDEB) and small VCP interacting protein (SVIP) 93, which were later confirmed to be important for intracellular VLDL transport 26,29. Reticulin 3 (RTN3) was also listed and more recently studied by Siddiqi et al. They showed that RTN3 is localized to VTVs through using immunogold labelling and co-localization and co-immunoprecipitation with apoB100 in hepatocytes 52. Blocking RTN3 in an in vitro VTV budding assay resulted in decreased triacylglycerol in cytosol which was used as read-out for the generation of VTVs. To attain the impact of RTN3 silencing on VLDL secretion, secreted triglyceride levels were measured in media of cultured cells. Although it is unclear whether they used HepG2s or primary hepatocytes, triglyceride levels were reduced by ≈30% compared to controls. How RTN3 controls the genesis of vesicles still is unknown.
To identify novel receptors that can bind cargos and recruit them to nascent vesicles, like the cargo receptors TANGO1 or SAR1b, Wang et al. (2021) performed a proximity-dependent proteomics approach with SAR1b 53. This study led to the identification of surfeit locus protein 4 (SURF4), the mammalian homologue of SFT-4, which was previously linked to the exit of vitellogenin 2 (VIT-2) (also synthesised as a lipoprotein complex) from the ER in yeast 94. They showed that apoB interacts with SURF4 in HepG2 cells 94, and subsequently studied its role in mice 53. Like Sar1b, liver-specific loss of Surf4 results in a drastic reduction of VLDL secretion (>85%) and 90% lower plasma lipids in mice. This was accompanied by increased hepatic lipid accumulation as assessed by oil-red-O staining. In contrast to the results from this acute CRISPR-mediated KO model, other studies using Surf4fl/flAlb-Cre+ mice or a liver specific knockdown of Surf4 in Ldrl-/- or ApoE-/- mice describe no changes in liver lipid levels compared to controls 54. These contrasting results require additional studies looking into the possibility of reducing hepatic and plasma lipids simultaneously. SURF4 however does not only regulate VLDL trafficking but also regulates PCSK9 transport 55, erythropoietin 95 and growth hormones 96. It was also found to play a role in maintaining the architecture of the ERGIC and the Golgi 97.
One of the latest novel players in intracellular VLDL metabolism is small leucine rich protein 1 (SMLR1). In contrast to abovementioned proteins, there was no literature in the public domain on SMLR1. This gene was identified upon contextual co-expression analysis with MTTP/Mttp in human and murine transcriptome datasets 47. Liver-specific ablation of SMLR1 in mice has been shown to reduce VLDL secretion by 45% while increasing hepatic triglycerides 7-fold on a chow diet compared to controls. SMLR1 was found to be located in the ER membrane and Cis-Golgi, and a role for VLDL transport was implicated but the underlying molecular mechanisms remain to be elucidated.
Altogether, these studies emphasize the need for a unique combination of numerous proteins to form and transport VTVs from the ER to the Golgi.
GOLGI APPARATUS
Thus far, proteins involved in docking of VTVs at the Cis-Golgi are poorly defined. A role for a unique set of SNARE proteins has been suggested for fusion-complex formation and includes Sec22b, GOS28, Syntaxin5 and rBet1 33. These studies have been exclusively conducted in cultured cells with little or no evidence of their roles in rodents or humans. In stark contrast to the ER, very little is known about which proteins are needed for the subsequent transport of VLDL into the Golgi apparatus. Studies in McA-RH7777 cells show that glycosylation and phosphorylation of apoB occurs in the Golgi 98,99 but it remains to be shown whether this is required for eventual VLDL secretion. At this point it is also not clear whether VLDL is or can be fully lipidated in the ER or that further lipidation of VLDL occurs in the Golgi 100–102. Evidence for specific cellular VLDL secretion routes comes from human kinetic studies in which smaller triglyceride-poor VLDL2 particles and larger triglyceride rich-VLDL1 are studied 100,103. In such studies, however, it cannot be excluded that these VLDL subpopulations are a product of peripheral lipolysis.
GWAS studies identified SORT1, encoding for SORTILIN1, as a regulator of apoB levels. 104. One of the proposed functions of this multi-ligand receptor includes protein transport from the Golgi to lysosomes or to the cell surface. Although SORTILIN1 is also recognized as plasma membrane receptor for VLDL, LDLR and PCSK9, the role of SORTILIN1 in apoB secretion remains incompletely understood, also due to conflicting results on apoB secretion in mice 105. In 2022, Mitok et al. summarized evidence on the role of SORTILIN1 in cardiovascular and metabolic diseases, and how it regulates LDL levels 36. This review did not include the latest insights provided by Conlon et al., (2022) who showed that loss-of-function of SORTILIN1 under basal non-stressed conditions has little effect on apoB secretion whereas, under ER stress or a lipid loading conditions, absence of SORTILIN1 leads to increased apoB secretion 35. This suggests that hepatic SORTILIN1, under stress conditions, may direct apoB towards lysosomal degradation, whereas it targets apoB for secretion in the absence of stress. The dual function of SORTILIN1 is supported by a recent report which suggests that mutating two different binding sites on SORTILIN1 either increase or decrease VLDL secretion in McA-RH7777 cells 106, suggesting allosteric conformational changes depending on the activated binding site.
SECRETION
To our knowledge, the export of VLDL from the Golgi and subsequent transport to plasma membrane has only been studied by Siddiqi et al. 107. His group has developed a trans-Golgi network budding assay in primary rat hepatocytes to examine post-trans Golgi VLDL transport vesicles (PG-VTV). Besides cytosol, ATP, GTP hydrolysis and incubation at 37°C, no other conditions were needed for vesicle formation from the Golgi. Electron microscopy revealed vesicle size ranging from 300-350nm. The reaction mixture of this assay was subsequently resolved on a sucrose gradient and showed apoB, apoA4, apoA1, and apoE, but not albumin or transferrin to be present on the vesicles. Whereas the acquisition of apoA1 on chylomicrons has been described 108, it is unclear whether apoA1 is secreted together with VLDL. This warrants further study of the relationship between the intracellular regulation and secretion of HDL and VLDL in hepatocytes. The same group also identified two post-Golgi SNARE proteins on the PG-VTVs named VAMP7 and SNAP23, which suggests that SNARE proteins direct the particle from the trans Golgi network to the plasma membrane 107.
Discussion
The VLDL biogenesis and secretion pathway has been studied continuously for five decades, but over the last few years numerous new insights have been generated. This increased focus on VLDL is likely related to an growing interest in the association between plasma apoB, triglycerides, and risk of ASCVD, and how VLDL secretion is related to the development of NAFLD. We have tried to capture all the latest findings as well as highlight several knowledge gaps in the understanding of VLDL biogenesis. There are a few key points that deserve special attention, which will be discussed below with referral back to Figure 1.
VLDL BIOGENESIS IN THE ER
The list of proteins required for VLDL biogenesis in the ER has recently been expanded to include PLA2G12B, ERLIN1/2, TM6SF2, LAP1, TORSINA, and SMLR1. Mechanistic studies have convincingly shown that PLA2G12B is required for the MTP-driven assembly of nascent VLDL in the ER 109. In the case of TM6SF2, however, a well-studied gene and protein with strong translational aspects, it is still not clear whether it solely affects lipidation of apoB or has additional functions. Hepatic ablation of TORSINA, as well as SMLR1, in chow-fed mice has been shown to strongly affect VLDL biogenesis/secretion accompanied by marked hepatic steatosis, but the molecular mechanisms how and where these proteins affect intracellular VLDL metabolism remains to be elucidated.
LEAVING THE ER
The formation of VTVs was originally proposed by Siddiqi et al. in 2008 87 but important additional evidence on how these VLDL containing vesicles can leave the ER came from the groups of Huang et al. and Chen et al. They showed that hepatic loss of TMEM41B or SURF4 blocks VTV budding from the ER which caused reduced VLDL secretion in mice 53,82. Interestingly, analyses of transmission electron microscopy analyses suggest that – in one of the studies - only the loss of hepatic SURF4 causes the accumulation of VLDL in the ER 48. Although TMEM41B is a scramblase and SURF4 a cargo receptor, it is surprising that blocking the exit of VLDL from the ER would only lead to VLDL accumulation in one of the cases. Although the abovementioned discrepancy may be related to the differences in the underlying biology, it emphasizes the need for improved tools to identify colocalization of apoB with lipids to confirm the presence of a VLDL particle in the ER.
GOLGI AND HEPATIC SECRETION
Only a few groups have studied the fate of VLDL after it has exited the ER. One study provides evidence that VLDL in wild-type mice docks to ERGIC and the cis-Golgi 33. VLDL trafficking through the Golgi is required to explain the extensive glycosylation of apoB (for review see 110) and it has further been suggested that VLDL undergoes additional lipidation (see review 111). However, there are almost no studies how apoB/VLDL finds its way through the Golgi. Little if anything is known regarding the steps needed for VLDL exciting the Golgi, how it is transported to the cell membrane and how VLDL is secreted.
LIMITATIONS
In this review, we have focused on novel players that have a physical or supposedly direct impact on the assembly of VLDL and its subsequent trafficking through the hepatocyte. We have not discussed the factors that regulate the availability of phospholipids, triglycerides, free cholesterol, and esterified cholesterol for VLDL production in the ER and possibly the Golgi. In the ER, these are prerequisites for the synthesis of nascent VLDL to prevent ubiquitinylation and proteasomal degradation of apoB/VLDL. While key enzymes in the de novo synthesis of triglycerides (DGAT1,275), and cholesterol esters (ACAT1/2, reviewed in 112) are long known to affect VLDL biogenesis, Lipin-1 has also been shown to control VLDL secretion by converting phosphatidic acid to diacylglycerol 113. The availability of triglycerides for VLDL lipidation is on the other hand determined by various factors including access to triglycerides in cytosolic LDs via membrane contact sites 114. The latter pathway involves a role for CIDEB in LD biology (apart from a role in VTV formation), that upon deletion in hepatocytes, results in steatosis and reduced VLDL secretion 25,26. This interesting example illustrates the strong interrelation between cytosolic LD, VLDL biogenesis, and hepatic steatosis. The entire hepatic VLDL secretory pathway is also regulated via feeding-axis dependent orphan GPR146 (G-coupled protein receptor 146) mediated Erk-signalling and Srebp2 gene regulation 115. Last but not least, various cellular processes such as ER stress, mitochondrial function (mitochondrial fission factor [14], RAB24 116), retrograde trafficking (ARF1, 117) and uptake of plasma free fatty acids and (remnant) lipoproteins by the liver, as well as VLDL reuptake by the liver, all affect VLDL production and secretion, but are not discussed here.
This review focuses on the hepatic production of apoB-containing lipoproteins, which is in humans limited to the production of apoB100-VLDL. Mice produce APOBEC1 in hepatocytes that edits apoB100 mRNA which leads to the production of apoB48-VLDL as well as apoB100-VLDL. It must be noted that the biogenesis of chylomicrons in the small intestine, with apoB48 as its critical structural protein, requires similar proteins as in the liver, including MTP, SAR1B and several chaperones. It is in this regard likely that APOBEC1 and apoB48 in rodent hepatocytes will affect the apoB100-VLDL production machinery 118. This may limit the translation of findings in mice that are discussed in this review to relevance in humans.
Conclusion and future perspectives
Exciting new insights, including the importance of proper lipidation of apoB/VLDL by PLA2G12B and the key role of SURF4 in VLDL trafficking, demonstrate that there are still many things to learn about the assembly and trafficking of VLDL. Intriguingly, however, almost all the knowledge gained during the past five decades involves the biogenesis of VLDL in the ER and its exit from the ER. This contrasts with the scarcity of data on VLDL transport to and in the Golgi, and how this lipoprotein leaves the Golgi for subsequent transport to the cell membrane for secretion from hepatocytes into the circulation. This prompts the question whether these later steps in the secretion pathway may not be as heavily regulated as the initial stages of VLDL assembly in the ER.
The ablation of any of the new players described in this review all result in hepatic steatosis. These finding often lead to the suggestion that these players are potential targets to alleviate NAFLD. In a few cases (MTP, VMP1, MDM2), hepatic overexpression is actually shown to reduce hepatic steatosis 39,49,84. However, increased VLDL secretion, without increasing its catabolism, will also increase plasma levels of apoB, cholesterol, triglycerides, and remnant lipoproteins and thus promote the risk of atherogenesis. Any approach to reducing VLDL secretion in humans would have to be specific and target only the pathway of interest. As seen for many proteins in this review, their function is not limited to VLDL but also autophagy or cellular trafficking of other proteins, making any such protein an unappealing target. Although the advances in gene silencing technologies in the human liver 119 have paved the road for targeting the hepatic VLDL pathway, this review highlights the need to increase our basic knowledge in VLDL biogenesis to identify targets and design combinatorial strategies that will reduce NAFLD as well as ASCVD.
Abbreviations
- ASCVD Atherosclerotic cardiovascular disease
- ERAD ER-associated degradation
- ER Endoplasmic Reticulum
- LD Lipid droplet
- LDL Low-density lipoprotein
- SNARE Soluble N-ethylmaleimide-sensitive-factor attachment protein receptor
- VLDL Very low-density lipoprotein
- VTV VLDL transport vesicle
Abbreviations proteins
- ApoB Apolipoprotein B
- APOBEC-1 Apolipoprotein B mRNA editing enzyme, catalytic polypeptide 1
- BIP/GRP78 Binding immunoglobulin protein / Glucose-regulated protein 78
- CIDEB Cell death-inducing DFFA-like effector b
- DERLIN-1 Degradation in endoplasmic reticulum protein 1
- DGAT Diglyceride acyltransferase
- ERLIN ER lipid raft-associated protein
- ERP57/PDIA3 ER resident protein 57 / Protein disulfide-isomerase A3
- ERP72/PDIA4 ER resident protein 72 / Protein disulfide-isomerase A4
- GRP94 Glucose-regulated protein 94
- GP78/AMFR Glycoprotein 78 / Autocrine motility factor receptor
- HRD1 HMG-CoA reductase degradation protein 1
- HSP Heat shock protein
- KLHL12 Kelch-like protein 12
- LAP1 Lamina-associated polypetide 1
- LULL1 Luminal domain-like LAP1
- LZP Zona pellucida domain-containing protein
- MDM2 Mouse/murine double minute 2 homolog
- MTP Microsomal triglyceride transfer protein
- PCSK9 Proprotein convertase subtilisin/kexin type 9
- PDI/PDIA1 Protein disulfide-isomerase / Protein disulfide-isomerase A1
- PEMT Phosphatidylethanolamine N-Methyltransferase
- PRAP1 Proline-rich acidic protein 1
- P97/VCP Valosin-containing protein
- SAR1 Secretion associated ras related GTPase 1
- SCREBP-1c Sterol regulatory element-binding protein 1c
- SMLR1 Small leucine rich protein 1
- SURF4 Surfeit locus protein 4
- SVIP Small VCP interacting protein
- TALI TANGO1-like
- TANGO1 Transport and golgi organization protein 1
- TMEM41B Transmembrane protein 41b
- TM6SF2 Transmembrane 6 superfamily member 2
- UBXD8 Ubiquitin regulatory x domain-containing protein
- VMP1 Vacuole membrane protein
References
- Henning, R. J. Obesity and obesity-induced inflammatory disease contribute to atherosclerosis: a review of the pathophysiology and treatment of obesity. Am J Cardiovasc Dis 11, 504–529 (2021).
- Ginsberg, H. N. et al. Triglyceride-rich lipoproteins and their remnants: metabolic insights, role in atherosclerotic cardiovascular disease, and emerging therapeutic strategies—a consensus statement from the European Atherosclerosis Society. Eur Heart J 42, 4791–4806 (2021).
- Rader, D. J. & Kastelein, J. J. P. Lomitapide and mipomersen: Two first-in-class drugs for reducing low-density lipoprotein cholesterol in patients with homozygous familial hypercholesterolemia. Circulation 129, 1022–1032 (2014).
- Zhang, J. & Herscovitz, H. Nascent lipidated apolipoprotein B is transported to the Golgi as an incompletely folded intermediate as probed by its association with network of endoplasmic reticulum molecular chaperones, GRP94, ERp72, BiP, calreticulin, and cyclophilin B. Journal of Biological Chemistry (2003). [CrossRef]
- Rutledge, A. C., Su, Q. & Adeli, K. Apolipoprotein B100 biogenesis: A complex array of intracellular mechanisms regulating folding, stability, and lipoprotein assembly. in Biochemistry and Cell Biology (2010). [CrossRef]
- Hrizo, S. L. et al. The Hsp110 Molecular Chaperone Stabilizes Apolipoprotein B from Endoplasmic Reticulum-associated Degradation (ERAD). Journal of Biological Chemistry 282, 32665–32675 (2007). [CrossRef]
- Linnik, K. M. & Herscovitz, H. Multiple Molecular Chaperones Interact with Apolipoprotein B during Its Maturation. Journal of Biological Chemistry 273, 21368–21373 (1998). [CrossRef]
- Gusarova, V., Caplan, A. J., Brodsky, J. L. & Fisher, E. A. Apoprotein B Degradation Is Promoted by the Molecular Chaperones hsp90 and hsp70. Journal of Biological Chemistry 276, 24891–24900 (2001). [CrossRef]
- Zhou, M., Wu, X., Huang, L.-S. & Ginsberg, H. N. Apoprotein B100, an Inefficiently Translocated Secretory Protein, Is Bound to the Cytosolic Chaperone, Heat Shock Protein 70. Journal of Biological Chemistry 270, 25220–25224 (1995). [CrossRef]
- Liang, J. et al. Overexpression of the Tumor Autocrine Motility Factor Receptor Gp78, a Ubiquitin Protein Ligase, Results in Increased Ubiquitinylation and Decreased Secretion of Apolipoprotein B100 in HepG2 Cells. Journal of Biological Chemistry 278, 23984–23988 (2003). [CrossRef]
- Fisher, E. A., Khanna, N. A. & McLeod, R. S. Ubiquitination regulates the assembly of VLDL in HepG2 cells and is the committing step of the apoB-100 ERAD pathway. J Lipid Res 52, 1170–1180 (2011). [CrossRef]
- Fisher, E. A., Lapierre, L. R., Junkins, R. D. & McLeod, R. S. The AAA-ATPase p97 facilitates degradation of apolipoprotein B by the ubiquitin-proteasome pathway. J Lipid Res 49, 2149–2160 (2008). [CrossRef]
- Kumari, D., Fisher, E. A. & Brodsky, J. L. Hsp40s play distinct roles during the initial stages of apolipoprotein B biogenesis. Mol Biol Cell 1–45 (2021). [CrossRef]
- Oyadomari, S. et al. Cotranslocational Degradation Protects the Stressed Endoplasmic Reticulum from Protein Overload. Cell 126, 727–739 (2006). [CrossRef]
- Suzuki, M. et al. Derlin-1 and UBXD8 are engaged in dislocation and degradation of lipidated ApoB-100 at lipid droplets. Mol Biol Cell 23, 800–810 (2012). [CrossRef]
- Rutledge, A. C. et al. Mechanisms Targeting Apolipoprotein B100 to Proteasomal Degradation. Arterioscler Thromb Vasc Biol 29, 579–585 (2009). [CrossRef]
- Chen, Y., Le Cahérec, F. & Chuck, S. L. Calnexin and Other Factors That Alter Translocation Affect the Rapid Binding of Ubiquitin to ApoB in the Sec61 Complex. Journal of Biological Chemistry 273, 11887–11894 (1998). [CrossRef]
- Wetterau, J. R., Combs, K. A., McLean, L. R., Spinner, S. N. & Aggerbeck, L. P. Protein disulfide isomerase appears necessary to maintain the catalytically active structure of the microsomal triglyceride transfer protein. Biochemistry 30, 9728–9735 (1991). [CrossRef]
- Wang, L., Fast, D. G. & Attie, A. D. The Enzymatic and Non-enzymatic Roles of Protein-disulfide Isomerase in Apolipoprotein B Secretion. Journal of Biological Chemistry 272, 27644–27651 (1997). [CrossRef]
- Pan, M., Liang, J. S., Fisher, E. A. & Ginsberg, H. N. Inhibition of translocation of nascent apolipoprotein B across the endoplasmic reticulum membrane is associated with selective inhibition of the synthesis of apolipoprotein B. Journal of Biological Chemistry 275, 27399–27405 (2000). [CrossRef]
- Hussain, M. M., Shi, J. & Dreizen, P. Microsomal triglyceride transfer protein and its role in apoB-lipoprotein assembly. J Lipid Res 44, 22–32 (2003).
- McLaren, D. G. et al. DGAT2 Inhibition Alters Aspects of Triglyceride Metabolism in Rodents but Not in Non-human Primates. Cell Metab 27, 1236-1248.e6 (2018). [CrossRef]
- Irshad, Z., Chmel, N., Adya, R. & Zammit, V. A. Hepatic VLDL secretion: DGAT1 determines particle size but not particle number, which can be supported entirely by DGAT2. J Lipid Res 60, 111–120 (2019). [CrossRef]
- Amin, N. B. et al. Efficacy and safety of an orally administered DGAT2 inhibitor alone or coadministered with a liver-targeted ACC inhibitor in adults with non-alcoholic steatohepatitis (NASH): rationale and design of the phase II, dose-ranging, dose-finding, randomised, placebo-controlled MIRNA (Metabolic Interventions to Resolve NASH with fibrosis) study. BMJ Open 12, e056159 (2022). [CrossRef]
- Ye, J. et al. Cideb, an ER- and Lipid Droplet-Associated Protein, Mediates VLDL Lipidation and Maturation by Interacting with Apolipoprotein B. Cell Metab 9, 177–190 (2009). [CrossRef]
- Tiwari, S., Siddiqi, S. & Siddiqi, S. A. CideB Protein Is Required for the Biogenesis of Very Low Density Lipoprotein (VLDL) Transport Vesicle. Journal of Biological Chemistry 288, 5157–5165 (2013).
- Butkinaree, C. et al. A Regulator of Secretory Vesicle Size, Kelch-Like Protein 12, Facilitates the Secretion of Apolipoprotein B100 and Very-Low-Density Lipoproteins—Brief Report. Arterioscler Thromb Vasc Biol 34, 251–254 (2014).
- Santos, A. J. M., Nogueira, C., Ortega-Bellido, M. & Malhotra, V. TANGO1 and Mia2/cTAGE5 (TALI) cooperate to export bulky pre-chylomicrons/VLDLs from the endoplasmic reticulum. Journal of Cell Biology 213, 343–354 (2016).
- Tiwari, S., Siddiqi, S., Zhelyabovska, O. & Siddiqi, S. A. Silencing of Small Valosin-containing Protein-interacting Protein (SVIP) Reduces Very Low Density Lipoprotein (VLDL) Secretion from Rat Hepatocytes by Disrupting Its Endoplasmic Reticulum (ER)-to-Golgi Trafficking. Journal of Biological Chemistry 291, 12514–12526 (2016).
- Tiwari, S. & Siddiqi, S. A. Intracellular trafficking and secretion of VLDL. Arterioscler Thromb Vasc Biol 32, 1079–1086 (2012). [CrossRef]
- Auclair, N. et al. Sar1b mutant mice recapitulate gastrointestinal abnormalities associated with chylomicron retention disease. J Lipid Res 62, 100085 (2021). [CrossRef]
- Fryer, L. G. D. et al. The Endoplasmic Reticulum Coat Protein II Transport Machinery Coordinates Cellular Lipid Secretion and Cholesterol Biosynthesis. Journal of Biological Chemistry 289, 4244–4261 (2014). [CrossRef]
- Siddiqi, S., Mani, A. M. & Siddiqi, S. A. The identification of the SNARE complex required for the fusion of VLDL-transport vesicle with hepatic cis-Golgi. Biochemical Journal (2010). [CrossRef]
- Amengual, J. et al. Autophagy Is Required for Sortilin-Mediated Degradation of Apolipoprotein B100. Circ Res 122, 568–582 (2018). [CrossRef]
- Conlon, D. M. et al. Sortilin restricts secretion of apolipoprotein B-100 by hepatocytes under stressed but not basal conditions. Journal of Clinical Investigation 132, (2022). [CrossRef]
- Mitok, K. A., Keller, M. P. & Attie, A. D. Sorting through the extensive and confusing roles of sortilin in metabolic disease. J Lipid Res 63, 100243 (2022). [CrossRef]
- Zhang, Z. et al. Hepatic HuR modulates lipid homeostasis in response to high-fat diet. Nat Commun 11, 3067 (2020). [CrossRef]
- Wu, J.-X. et al. LZP is required for hepatic triacylglycerol transportation through maintaining apolipoprotein B stability. PLoS Genet 17, e1009357 (2021). [CrossRef]
- Lin, H. et al. Hepatic MDM2 Causes Metabolic Associated Fatty Liver Disease by Blocking Triglyceride-VLDL Secretion via ApoB Degradation. Advanced Science 9, 2200742 (2022). [CrossRef]
- Peng, H. et al. PRAP1 is a novel lipid-binding protein that promotes lipid absorption by facilitating MTTP-mediated lipid transport. Journal of Biological Chemistry 296, 100052 (2021). [CrossRef]
- Thierer, J. H. et al. Pla2g12b is Essential for Expansion of Nascent Lipoprotein Particles. [CrossRef]
- Li, B. T. et al. Disruption of the ERLIN–TM6SF2–APOB complex destabilizes APOB and contributes to non-alcoholic fatty liver disease. PLoS Genet 16, 1–20 (2020). [CrossRef]
- Smagris, E., Gilyard, S., BasuRay, S., Cohen, J. C. & Hobbs, H. H. Inactivation of Tm6sf2, a gene defective in fatty liver disease, impairs lipidation but not secretion of very low density lipoproteins. Journal of Biological Chemistry 291, 10659–10676 (2016). [CrossRef]
- Ehrhardt, N. et al. Hepatic Tm6sf2 overexpression affects cellular ApoB-trafficking, plasma lipid levels, hepatic steatosis and atherosclerosis. Hum Mol Genet 26, 2719–2731 (2017).
- Luo, F. et al. Hepatic TM6SF2 Is Required for Lipidation of VLDL in a Pre-Golgi Compartment in Mice and Rats. Cell Mol Gastroenterol Hepatol 13, 879–899 (2022). [CrossRef]
- Shin, J.-Y. et al. Nuclear envelope–localized torsinA-LAP1 complex regulates hepatic VLDL secretion and steatosis. Journal of Clinical Investigation 129, 4885–4900 (2019).
- van Zwol, W. et al. Loss of hepatic SMLR1 causes hepatosteatosis and protects against atherosclerosis due to decreased hepatic VLDL secretion. Hepatology (2022). [CrossRef]
- Huang, D. et al. TMEM41B acts as an ER scramblase required for lipoprotein biogenesis and lipid homeostasis. Cell Metab 33, 1655-1670.e8 (2021). [CrossRef]
- Jiang, X. et al. Lack of VMP1 Impairs Hepatic Lipoprotein Secretion and Promotes Nonalcoholic Steatohepatitis. J Hepatol (2022). [CrossRef]
- Peng, Y. et al. GP73 is a TBC-domain Rab GTPase-activating protein contributing to the pathogenesis of non-alcoholic fatty liver disease without obesity. Nat Commun 12, 7004 (2021). [CrossRef]
- Takacs, C. N. et al. Differential Regulation of Lipoprotein and Hepatitis C Virus Secretion by Rab1b. Cell Rep 21, 431–441 (2017). [CrossRef]
- Siddiqi, S., Zhelyabovska, O. & Siddiqi, S. A. Reticulon 3 regulates very low density lipoprotein secretion by controlling very low density lipoprotein transport vesicle biogenesis. Can J Physiol Pharmacol 96, 668–675 (2018).
- Wang, X. et al. Receptor-Mediated ER Export of Lipoproteins Controls Lipid Homeostasis in Mice and Humans. Cell Metab 33, 350-366.e7 (2021). [CrossRef]
- Tang, V. T. et al. Hepatic inactivation of murine Surf4 results in marked reduction in plasma cholesterol. BioRxiv (2022). [CrossRef]
- Wang, B. et al. Atherosclerosis-associated hepatic secretion of VLDL but not PCSK9 is dependent on cargo receptor protein Surf4. J Lipid Res 62, 100091 (2021). [CrossRef]
- Pullinger, C. R. et al. The apolipoprotein B gene is constitutively expressed in HepG2 cells: regulation of secretion by oleic acid, albumin, and insulin, and measurement of the mRNA half-life. J Lipid Res 30, 1065–1077 (1989).
- Fisher, E. A. & Ginsberg, H. N. Complexity in the secretory pathway: The assembly and secretion of apolipoprotein B-containing lipoproteins. Journal of Biological Chemistry Preprint at (2002). [CrossRef]
- Fernández-Tussy, P., Ruz-Maldonado, I. & Fernández-Hernando, C. MicroRNAs and Circular RNAs in Lipoprotein Metabolism. Current Atherosclerosis Reports Preprint at (2021). [CrossRef]
- Zhou, L. & Hussain, M. M. Human MicroRNA-548p Decreases Hepatic Apolipoprotein B Secretion and Lipid Synthesis. Arterioscler Thromb Vasc Biol 37, 786–793 (2017). [CrossRef]
- Khalifeh, M. et al. A novel regulatory facet for hypertriglyceridemia: The role of microRNAs in the regulation of triglyceride-rich lipoprotein biosynthesis. Prog Lipid Res 89, 101197 (2023). [CrossRef]
- Goodchild, R. E. & Dauer, W. T. The AAA+ protein torsinA interacts with a conserved domain present in LAP1 and a novel ER protein. J Cell Biol 168, 855–62 (2005).
- Parker, B. L. et al. An integrative systems genetic analysis of mammalian lipid metabolism. Nature 567, 187–193 (2019).
- Doonan, L. M., Fisher, E. A. & Brodsky, J. L. Can modulators of apolipoproteinB biogenesis serve as an alternate target for cholesterol-lowering drugs? Biochimica et Biophysica Acta (BBA) - Molecular and Cell Biology of Lipids 1863, 762–771 (2018).
- Xu, Z. G. et al. Identification of LZP gene from Mus musculus and Rattus norvegicus coding for a novel liver-specific ZP domain-containing secretory protein. DNA Sequence - Journal of DNA Sequencing and Mapping (2004). [CrossRef]
- Wettereau, J. R. & Zilversmit, D. B. Purification and characterization of microsomal triglyceride and cholesteryl ester transfer protein from bovine liver microsomes. Chem Phys Lipids 38, 205–222 (1985). [CrossRef]
- Sirwi, A. & Hussain, M. M. Lipid transfer proteins in the assembly of apoB-containing lipoproteins. J Lipid Res 59, 1094–1102 (2018).
- Liang, J. & Ginsberg, H. N. Microsomal Triglyceride Transfer Protein Binding and Lipid Transfer Activities Are Independent of Each Other, but Both Are Required for Secretion of Apolipoprotein B Lipoproteins from Liver Cells. Journal of Biological Chemistry 276, 28606–28612 (2001). [CrossRef]
- Zhang, M. et al. Andrographolide modulates HNF4α activity imparting on hepatic metabolism. Mol Cell Endocrinol 513, 110867 (2020). [CrossRef]
- Guan, M., Qu, L., Tan, W., Chen, L. & Wong, C.-W. Hepatocyte nuclear factor-4 alpha regulates liver triglyceride metabolism in part through secreted phospholipase A2 GXIIB. Hepatology 53, 458–466 (2011). [CrossRef]
- Murakami, M., Taketomi, Y., Sato, H. & Yamamoto, K. Secreted phospholipase A2 revisited. J Biochem 150, 233–55 (2011). [CrossRef]
- Aljakna, A. et al. Pla2g12b and Hpn Are Genes Identified by Mouse ENU Mutagenesis That Affect HDL Cholesterol. PLoS One 7, e43139 (2012). [CrossRef]
- Thierer, J. H. et al. Pla2g12b is Essential for Expansion of Nascent Lipoprotein Particles. bioRxiv 2022.08.02.502564 (2022). [CrossRef]
- Lehner, R., Lian, J. & Quiroga, A. D. Lumenal Lipid Metabolism. Arterioscler Thromb Vasc Biol 32, 1087–1093 (2012).
- Wolfarth, A. A. et al. Proline-Rich Acidic Protein 1 (PRAP1) Protects the Gastrointestinal Epithelium From Irradiation-Induced Apoptosis. CMGH (2020). [CrossRef]
- Yen, C.-L. E., Stone, S. J., Koliwad, S., Harris, C. & Farese, R. V. Thematic review series: glycerolipids. DGAT enzymes and triacylglycerol biosynthesis. J Lipid Res 49, 2283–301 (2008). [CrossRef]
- Li, C. et al. Roles of Acyl-CoA:Diacylglycerol Acyltransferases 1 and 2 in Triacylglycerol Synthesis and Secretion in Primary Hepatocytes. Arterioscler Thromb Vasc Biol 35, 1080–1091 (2015).
- Li, X.-Y., Liu, Z., Li, L., Wang, H.-J. & Wang, H. TM6SF2 rs58542926 is related to hepatic steatosis, fibrosis and serum lipids both in adults and children: A meta-analysis. Front Endocrinol (Lausanne) 13, (2022). [CrossRef]
- Browman, D. T., Resek, M. E., Zajchowski, L. D. & Robbins, S. M. Erlin-1 and erlin-2 are novel members of the prohibitin family of proteins that define lipid-raft-like domains of the ER. J Cell Sci 119, 3149–3160 (2006). [CrossRef]
- Newberry, E. P. et al. Liver-Specific Deletion of Mouse Tm6sf2 Promotes Steatosis, Fibrosis, and Hepatocellular Cancer. Hepatology 74, 1203–1219 (2021).
- Morita, K. et al. Genome-wide CRISPR screen identifies TMEM41B as a gene required for autophagosome formation. Journal of Cell Biology 217, 3817–3828 (2018). [CrossRef]
- Li, Y. E. et al. TMEM41B and VMP1 are scramblases and regulate the distribution of cholesterol and phosphatidylserine. Journal of Cell Biology 220, (2021). [CrossRef]
- Huang, D. et al. TMEM41B acts as an ER scramblase required for lipoprotein biogenesis and lipid homeostasis. Cell Metab (2021). [CrossRef]
- Morita, K., Hama, Y. & Mizushima, N. TMEM41B functions with VMP1 in autophagosome formation. Autophagy 15, 922–923 (2019). [CrossRef]
- Tietge, U. J. F. et al. Hepatic overexpression of microsomal triglyceride transfer protein (MTP) results in increased in vivo secretion of VLDL triglycerides and apolipoprotein B. J Lipid Res 40, 2134–2139 (1999). [CrossRef]
- Matsuoka, K. et al. COPII-Coated Vesicle Formation Reconstituted with Purified Coat Proteins and Chemically Defined Liposomes. Cell 93, 263–275 (1998). [CrossRef]
- Zeuschner, D. et al. Immuno-electron tomography of ER exit sites reveals the existence of free COPII-coated transport carriers. Nat Cell Biol 8, 377–383 (2006). [CrossRef]
- Siddiqi, S. A. VLDL exits from the endoplasmic reticulum in a specialized vesicle, the VLDL transport vesicle, in rat primary hepatocytes. Biochemical Journal (2008). [CrossRef]
- Jin, L. et al. Ubiquitin-dependent regulation of COPII coat size and function. Nature 482, 495–500 (2012). [CrossRef]
- Saito, K. et al. TANGO1 Facilitates Cargo Loading at Endoplasmic Reticulum Exit Sites. Cell 136, 891–902 (2009). [CrossRef]
- Saito, K. et al. cTAGE5 mediates collagen secretion through interaction with TANGO1 at endoplasmic reticulum exit sites. Mol Biol Cell 22, 2301–2308 (2011). [CrossRef]
- Siddiqi, S. A. VLDL exits from the endoplasmic reticulum in a specialized vesicle, the VLDL transport vesicle, in rat primary hepatocytes. Biochemical Journal 413, 333–342 (2008). [CrossRef]
- Ginsberg, H. N. ApoB SURFs a Ride from the ER to the Golgi. Cell Metab 33, 231–233 (2021). [CrossRef]
- Rahim, A. et al. Proteomic Analysis of the Very Low Density Lipoprotein (VLDL) transport vesicles. J Proteomics 75, 2225–2235 (2012). [CrossRef]
- Saegusa, K., Sato, M., Morooka, N., Hara, T. & Sato, K. SFT-4/Surf4 control ER export of soluble cargo proteins and participate in ER exit site organization. Journal of Cell Biology 217, 2073–2085 (2018).
- Lin, Z. et al. The Endoplasmic Reticulum Cargo Receptor SURF4 Facilitates Efficient Erythropoietin Secretion. Mol Cell Biol 40, (2020). [CrossRef]
- Yin, Y. et al. Surf4 (Erv29p) binds amino-terminal tripeptide motifs of soluble cargo proteins with different affinities, enabling prioritization of their exit from the endoplasmic reticulum. PLoS Biol 16, e2005140 (2018). [CrossRef]
- Mitrovic, S., Ben-Tekaya, H., Koegler, E., Gruenberg, J. & Hauri, H.-P. The Cargo Receptors Surf4, Endoplasmic Reticulum-Golgi Intermediate Compartment (ERGIC)-53, and p25 Are Required to Maintain the Architecture of ERGIC and Golgi. Mol Biol Cell 19, 1976–1990 (2008). [CrossRef]
- Tran, K. et al. Intracellular assembly of very low density lipoproteins containing apolipoprotein B100 in rat hepatoma McA-RH7777 cells. Journal of Biological Chemistry (2002). [CrossRef]
- Swift, L. L. Role of the Golgi apparatus in the phosphorylation of apolipoprotein B. Journal of Biological Chemistry (1996). 1996. [CrossRef]
- Stillemark-Billton, P., Beck, C., Borén, J. & Olofsson, S. O. Relation of the size and intracellular sorting of apoB to the formation of VLDL 1 and VLDL 2. J Lipid Res (2005). [CrossRef]
- Gusarova, V., Brodsky, J. L. & Fisher, E. A. Apolipoprotein B100 Exit from the Endoplasmic Reticulum (ER) Is COPII-dependent, and Its Lipidation to Very Low Density Lipoprotein Occurs Post-ER. Journal of Biological Chemistry (2003). [CrossRef]
- Yamaguchi, J., Gamble, M. V., Conlon, D., Liang, J. & Ginsberg, H. N. The Conversion of ApoB100 Low Density Lipoprotein/High Density Lipoprotein Particles to ApoB100 Very Low Density Lipoproteins in Response to Oleic Acid Occurs in the Endoplasmic Reticulum and Not in the Golgi in McA RH7777 Cells. Journal of Biological Chemistry 278, 42643–42651 (2003). [CrossRef]
- Zhao Shui-Ping et al. Separation of VLDL subfractions by density gradient ultracentrifugation. Journal of Laboratory and Clinical Medicine (1995).
- Strong, A., Patel, K. & Rader, D. J. Sortilin and lipoprotein metabolism: Making sense out of complexity. Current Opinion in Lipidology Preprint at (2014). [CrossRef]
- Su, X. & Peng, D. New insight into sortilin in controlling lipid metabolism and the risk of atherogenesis. Biological Reviews (2020). [CrossRef]
- Sparks, R. P. et al. An Allosteric Binding Site on Sortilin Regulates the Trafficking of VLDL, PCSK9, and LDLR in Hepatocytes. Biochemistry (2020). [CrossRef]
- Hossain, T., Riad, A., Siddiqi, S., Parthasarathy, S. & Siddiqi, S. A. Mature VLDL triggers the biogenesis of a distinct vesicle from the trans -Golgi network for its export to the plasma membrane. Biochemical Journal 459, 47–58 (2014). [CrossRef]
- Glickman, R. M. & Green, P. H. The intestine as a source of apolipoprotein A1. Proceedings of the National Academy of Sciences 74, 2569–2573 (1977).
- Thierer, J. H. et al. Pla2g12b is Essential for Expansion of Nascent Lipoprotein Particles. (2022). 2022. [CrossRef]
- Subramanian, S. P. & Gundry, R. L. The known unknowns of apolipoprotein glycosylation in health and disease. iScience 25, 105031 (2022). [CrossRef]
- Borén, J., Taskinen, M.-R., Björnson, E. & Packard, C. J. Metabolism of triglyceride-rich lipoproteins in health and dyslipidaemia. Nat Rev Cardiol 19, 577–592 (2022). [CrossRef]
- Buhman, K. Mammalian acyl-CoA:cholesterol acyltransferases. Biochimica et Biophysica Acta (BBA) - Molecular and Cell Biology of Lipids 1529, 142–154 (2000).
- Khalil, M. B. et al. The level and compartmentalization of phosphatidate phosphatase-1 (lipin-1) control the assembly and secretion of hepatic VLDL. J Lipid Res 50, 47–58 (2009). [CrossRef]
- Herker, E., Vieyres, G., Beller, M., Krahmer, N. & Bohnert, M. Lipid Droplet Contact Sites in Health and Disease. Trends Cell Biol 31, 345–358 (2021). [CrossRef]
- Yu, H. et al. GPR146 Deficiency Protects against Hypercholesterolemia and Atherosclerosis. Cell 179, 1276-1288.e14 (2019). [CrossRef]
- Seitz, S. et al. Hepatic Rab24 controls blood glucose homeostasis via improving mitochondrial plasticity. Nat Metab 1, 1009–1026 (2019). [CrossRef]
- Asp, L. et al. Role of ADP Ribosylation Factor 1 in the Assembly and Secretion of ApoB-100–Containing Lipoproteins. Arterioscler Thromb Vasc Biol 25, 566–570 (2005). [CrossRef]
- Galloway, C. A., Ashton, J., Sparks, J. D., Mooney, R. A. & Smith, H. C. Metabolic regulation of APOBEC-1 Complementation Factor trafficking in mouse models of obesity and its positive correlation with the expression of ApoB protein in hepatocytes. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease 1802, 976–985 (2010). [CrossRef]
- Nordestgaard, B. G., Nicholls, S. J., Langsted, A., Ray, K. K. & Tybjærg-Hansen, A. Advances in lipid-lowering therapy through gene-silencing technologies. Nature Reviews Cardiology Preprint at (2018). [CrossRef]
Figure 1.
A cellular model of the hepatic VLDL assembly and secretion pathway, with a focus on recently discovered key players. For clarity, the proteins are classified into sections where in the cell they affect apoB/VLDL, although this may not be the only subcellular location of the protein.
Figure 1.
A cellular model of the hepatic VLDL assembly and secretion pathway, with a focus on recently discovered key players. For clarity, the proteins are classified into sections where in the cell they affect apoB/VLDL, although this may not be the only subcellular location of the protein.
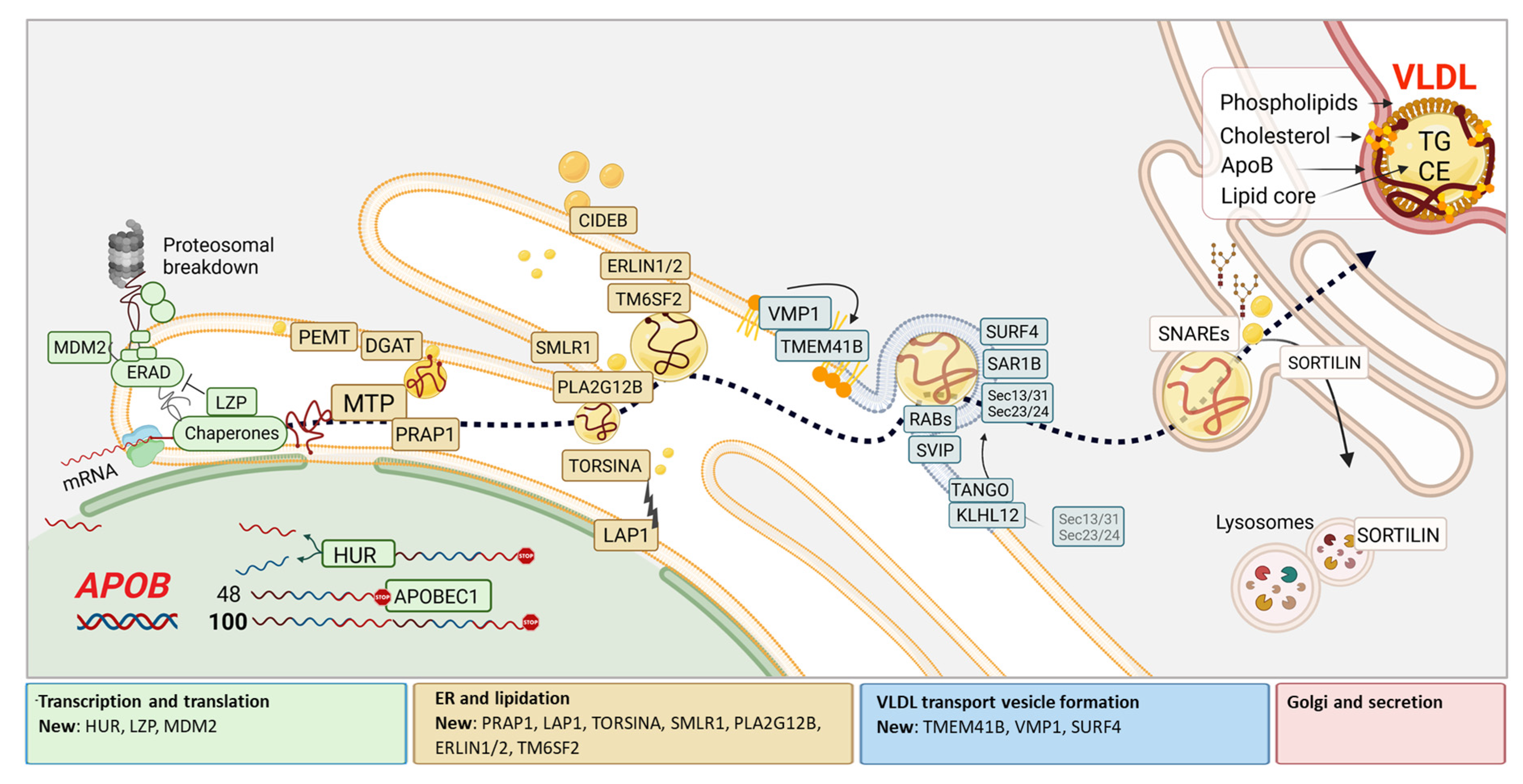

Table 1.
Established and new players in the VLDL assembly, trafficking, and secretion pathway.
| Protein name | Role in VLDL biogenesis | Studies performed in | Ref. |
|---|---|---|---|
| Previously described players in VLDL biogenesis | |||
| BIP, HSP110, Calreticulin, Calnexin, ERP57, ERP72, GRP94, Cyclophilin B. | ER-chaperones, mediating correct folding of apoB. | HepG2 and McArdle RH-7777 cells, yeast. | 4–7 |
| HSP70, HSP90 | Cytosolic chaperones, participate in proteasomal targeting of apoB. | HepG2 and McArdle RH-7777 cells | 8,9 |
| GP78 (=AMFR) | E3 ligase that targets apoB for proteasomal degradation | HepG2 cells | 10,11 |
| HSP40, P58IPK, P97, GP78, HRD1, UBXD8, BIP, DERLIN-1, SEC61 | Involved in proteasomal degradation of apoB | Yeast, HepG2 and HuH7 cells, whole body KO mice (P58) | 12–17 |
| PDI | Subunit of MTP, ER-chaperone | Bovine liver, Sf21 cells. | 18,19 |
| MTP | Lipidation and translocation of apoB across the ER membrane | HepG2 and McArdle-RH7777 cells, rodent models, human genetics/drug |
20,21 |
| DGAT1 and DGAT2 | Lipid supply for VLDL via de novo TG synthesis in the ER | Rodents, primates, and Phase 2 trials in humans | 22–24 |
| CIDEB | Lipidation and VTV transport | Whole body KO mice and both mouse and rat primary hepatocytes. | 25,26 |
| KLHL12 | VTV transport | McArdle-RH7777 cells | 27 |
| TANGO1, TALI | VTV transport | Caco-2 and HepG2 cells | 28 |
| SVIP | VTV transport | siRNAs for rat primary hepatocytes or McArdle-RH7777 cells. | 29 |
| SEC13/31, SEC23/24, | VTV transport | See review | 30 |
| SAR1B | VTV transport | Intestinal and liver-specific KO mice, CMRD patients, McArdle-RH7777 cells | 31,32 |
| SNARES; SEC22b, GOS28, Syntaxin5 and RBET1 | Cis-Golgi docking of VTVs | Rat hepatocytes | 33 |
| SORTILIN1 | Deviation from VLDL secretion to lysosomal degradation | Rodent models, McArdle RH-7777 cells | 34–36 |
| New players in VLDL biogenesis | |||
| HUR | ApoB mRNA splicing | Liver-specific KO in mice, Hepa1-6 cells | 37 |
| LZP | ERAD | Whole body KO mice, HepG2 cells | 38 |
| MDM2 | E3 ligase that targets ApoB for proteasomal degradation | Liver-specific KO mice, HepG2 cells | 39 |
| PRAP1 | Facilitates apoB lipidation | Whole body KO mice | 40 |
| PLA2G12B | Facilitates apoB lipidation | Zebrafish, liver-specific KO mice, Caco-2 and HepG2 cells | 41 |
| ERLIN1/2 | Stabilizes TM6SF2 | Liver-specific KO and KD in mice, HuH7 cells | 42 |
| TM6SF2 | Lipidates and/or traffics apoB | Whole body and liver-specific KO in rodents, HuH7, HepG2, McArdle-RH7777 cells, humans | 42–45 |
| LAP1, TORSINA | Affects apoB secretion | Whole body and liver-specific KO mice | 46 |
| SMLR1 | Affects apoB secretion | Liver-specific KO mice | 47 |
| TMEM41B, VMP1 | Phospholipid scramblase activity for VTV formation in the ER | Liver-specific KO mice, HEK293 and HuH7 cells | 48,49 |
| RAB1b, GP73 | Facilitate VLDL transport | HuH7 cells and liver-specific overexpression in mice (GP73) | 50,51 |
| RTN3 | VTV transport | HepG2 cells | 52 |
| SURF4 | VTV transport | Liver-specific KO mice and HepG2 cells | 53–55 |
Proteins are ordered by the place in the VLDL biogenesis pathway. Abbreviations: KO; knockout, KD; knock down, CMRD; chylomicron retention disease, Rodents; both mouse and rat models, TG; triglycerides.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



