Submitted:
17 June 2023
Posted:
20 June 2023
You are already at the latest version
Abstract
Brain metastasis (BrM), involving the spread of cells from a primary tumor through the blood circulation system to the brain microvasculature, eventually progresses despite multiple treatments and remains a substantial contributor to major mortality in patients with advanced- stage cancer. Molecular signatures and immune cellular components of the tumor microenvironment (TME) are emerging as essential regulators involved in establishing an organ-specific metastasis (colonization) and therapeutic response. A comprehensive understanding of detailed characterization and the immune landscape in context of process of BrM formation will greatly expand the horizon of treatments available to target these deadly diseases. In this review, we provide a comprehensive picture of the complex interactions between tumor cells and immune cellular components participated in the BrM process. Based on this knowledge, we will discuss opportunities and challenges for therapeutic strategies against BrM.
Keywords:
brain metastasis
; tumor microenvironment
; targeted therapy
; immune therapy
1. Introduction
It is estimated that 20% of patients with advanced cancer will occur brain metastases (BrM), following the spread of cancer cells from primary tumor through the blood (circulating tumor cells, CTCs) to the brain microvasculature, with the majority of BrM occurring in lung cancer, breast cancer, melanoma, colorectal cancer (CRC), and renal cell carcinoma (RCC) [1,2,3]. Upon advanced diagnosis, BrM are commonly treated with multi-therapies including surgery, radiotherapy, chemotherapy, immunotherapy and targeted therapies. However, prognosis of BrM from major cancer types is still poor, with 2-year overall survival (OS) rate less than 10% [4,5]. Underlying heterogeneity including molecular and immune cellular elements within BrM, are essential factors contributing to poor outcomes.
Elucidating some of the immunological intricacies of BrM has opened a therapeutic window to explore the potential efficacy of immune checkpoint inhibitors (ICIs) in this globally lethal disease [6,7]. Complex microenvironmental niche–tumor interactions, neovascularization and, possibly, immunological escape are involved in establishing new metastatic lesions (colonization) [7]. Nonetheless, the role of the immune checkpoint in the complex interplays between tumor and immune cells and in conferring resistance to therapy of BrM remains under investigation [8]. It is expected that newer mechanism-based therapies will play a greater part in treatment.
In this paper, we focus on the molecular elements and complex interplays between tumor and immune cells in the formative process of BrM. We conclude with a discussion of the future outlook in the field of BrM based on the molecular and immune cellular understanding of the disease, which will also drive continued development of novel targeted therapies and immunotherapies that have higher bioavailability beyond the blood–tumor barrier (BBB).
2. Elements for Cancer Cells Dissemination to the Brain
Metastasis is a multistep process characterized by tumor cells from primary tumor sites spread to distant organs or other regions within the same organ [1,9]. The trajectory of brain metastatic cells can be generally divided into five major steps (Figure 1): Invasive tumor cells that gaining stem-cell-like properties break away from the primary site and infiltrate the surrounding tissue by ECM remodeling (Proliferation and ECM remodeling) [10], then invade venules and lymphatic system (Invasion and intravasation) to become circulating cells (CTCs) with the help of platelets, macrophages and blood vessel endothelial cells (ECs) (Survival and Circulation) [11,12]. CTCs spread throughout the blood circulatory system and overcome the BBB via extravasation after adhesion with ECs and successfully infiltrate the brain (Adhesion and extravasation) [13,14]. Subsequently, most tumor cells die or enter a dormant state, while a few cells proliferate within this new microenvironment and induce formation of pre-metastatic niche (PMN) [15,16]. After micrometastases formation, cancer cells establish a more stable niche for colonization and then form brain metastases (Micro-metastases and colonization). Moreover, dormant cells would be reawakened in suitable conditions and participate in colonization and induce tumor recurrence [17,18].
However, the factors that influence the cancer cells dissemination to brain remains to be elucidated, and this will probably be addressed only by macroscopic and comprehensive certifications of a large number of original literatures. The evolving TME during all stages of cancer progression to BrM is depicted with key representative molecular signatures and immune cell types shown (Figure 2). It should be noticed that these representative elements were only partially shown and that factors will be added, removed or modified in the future.
2.1. Proliferation, ECM Remodeling, Invasion and Intravasation
In situ process, the TME conditions including angiogenesis, inflammation, hypoxia, low pH, and high stress are suitable for growth. Within the environment, mesenchymal cells, such as fibroblasts, endothelial and immune cells, are reprogrammed into tumor-associated types that provide favorable conditions for proliferation and growth, and ECM remodeling in order to enter the phase of progression: local invasion [19]. Due to host genetics (e.g., RAS mutation, loss of PTEN, inactivation of p53, EGFR-amplification) [20], environmental factors (e.g., nicotine exposure, air pollutants and angiogenesis, resulting in the preparation of the cellular proliferation and ECM remodeling for subsequent invasion [21,22]. Invasive migration is a complex multi-step process induced by activation of a series of signaling pathways, including Wnt-β-catenin [23], JNK/p38/MAPK [24], vessel abnormalization, and cytokines such as TGF-β, releasing from tumor-associated cells (TAMs) [25]. A small population of tumor cells at the primary site acquired proliferated properties before gaining stem-cell-like properties and lose cell polarity through epithelial-to-mesenchymal transition (EMT) [17], preparing for the migration and invasion.
2.2. Survival and Circulation
Intravasation of tumor cells into the circulation, either directly or via the lymphatic system, was regarded as the most essential step of all metastases [1,26]. The vast majority of CTCs will die, however, for the small proportion of CTCs that survive through the circulation system, they can evade destruction through a variety of mechanisms including CTCs clustering, which promotes stemness via induction of NANOG, SOX2, OCT4 [27,28], and HLA-E [29], association with specific immune cells of the bloodstream such as neutrophils or platelets; and, conversely, evasion of cytotoxic immune cells including NK cells [29]. Platelet, a highly abundant immune cell type in the blood, have long been recognized as key helper of CTCs survival through ‘‘CTCs-platelet’’ clustering [30,31,32]. Upon entering the circulation, CTCs derived from primary tumor are exposed to sustained physical, biochemical and congenital immune stresses (e.g., high oxygen tension, fluid shear stress and immune surveillance) which result in extreme oxidative stress characterized by increased reactive oxygen species (ROS) in CTCs, hindering the survival of the vast majority of CTCs [33,34,35]. Although current advances in enriching and analyzing connections between CTCs and the components of bloodstream were further analyzed, the mechanisms of CTCs escape immune surveillance and survive or remain dormant stage in the circulation remain largely unclear.
2.3. Adhesion and Extravasation
The CNS is protected by several functional barriers including the BBB and blood–cerebrospinal fluid (CSF) barrier. Due to the existence of these barriers, only a few CTCs can cross through the BBB via adhesion and extravasation, essentially a transition from BBB to blood-tumor barrier (BTB). CTCs enter the microvasculature and thus may initiate intraluminal growth and form an suitable niche that eventually ruptures the vessel or extravasate by breaching vascular walls [36]. For example, CTCs-associated platelets can promote extravasation by releasing TGF-β [37] or by secreting adenine nucleotides, which relax endothelial cell junctions [38]. Some factors also can promote extravasation of CTCs into brain, including cancer-cell derived sialyltransferase ST6GalNac5 [39], cathepsin S [40], and microRNAs [41,42]. In conclusion, a combination of priming signals from the tumor stroma, CTCs cluster composition and cancer cell-secrete factors determine brain metastatic infiltration of CTCs.
2.4. Formation of Pre-Metastatic Niche (PMN) in the Brain
Paget’s “seed and soil” theories were benefit for biological understanding underlying tumor metastasis including an explanation for organ-specificity in metastasis process: CTCs (the “seed”) colonize in specific organ sites (the “soil”) [43]. Studies in experimental models have provided evidence that creating ‘pre-metastatic niches’ was prior to the arrival of CTCs [44,45]. Tumor-derived secreted factors (TDSFs) and extracellular vesicle (EVs) induce the mobilization and recruitment of several cell populations to brain environment, including cancer-associated fibroblasts (CAFs), and immune cells including MDSCs, Tregs, TAMs, and tumor-associated neutrophils (TANs) [46,47]. The interaction among these TDSFs, tumor-recruited immune cells (e.g., TANs, TAMs), and local stroma including astrocytes may create a suitable growth condition (the “PMN”) for metastatic tumor cell colonization [48,49]. Thus, it’s not surprising that analyzing the molecular and cellular components of PMN in the blood may be helpful for diagnosis of cancer metastasis and for prognosis predication. Six characteristics that define the PMN including immunosuppression, inflammation, angiogenesis/vascular permeability, lymphangiogenesis, organotropism, and reprogramming [16]. These characteristics determine whether the CTCs can colonize and survive or become dormant after crossing the BBB. Monitoring the biology underlying the formation of PMN might be possible to identify new and powerful biomarkers, and thereby allowed us to early detect micrometastasis prior to macrometastatic site. However, the molecular and cellular components within PMN enhances tumor cell proliferation, and growth to macrometastasis were need to further discovered.
3. The Immune Cell Landscape of BrM
The immune cell landscapes of BrM were diverse via substantial infiltration of various immune cells such as T cells, neutrophils and macrophages. In addition, a variety of organ-specific immune cells, namely microglia, dendritic cells (DCs) and astrocytes collectively constitute the immune microenvironment of brain metastatic tumors.
3.1. Tumor-Associated Macrophages and Microglia
Tumor-associated macrophages and microglia (TAMs) exhibit one of the most diverse nonmalignant cell types within BrM and have been proved to participate in metastatic colonization [40]. TAMs including organ-specific microglia and bone marrow-derived macrophages (BMDMs) within the brain tend to be pro-tumorigenic and associated with poor prognosis [50,51] (Figure 3A). However, whether microglia and BMDMs have distinct surface markers and functions in the brain TME has been controversial, and is an academic topic of active investigation. Gao et al. reported that TMEM119 expression levels may distinguish microglia (TMEM119+) from BMDMs (TMEM119-F4/80+) [52]. Until recently, however, there is still no satisfactory strategy to definitively distinguish between macrophage populations and microglia, thus determining the relative contributions of BMDMs versus microglia to brain metastases of virous cancers. Due to increasingly academic advances, the majority of literature focus on the functional contributions of TAMs types to BrM, and the principle that simple division of M1 and M2 phenotype of macrophages has been disputed [53]. Many groups are interestingly focused on defining organ-specific macrophage activation and phenotype as a measure of functional diversity [53,54]. In terms of their plasticity, some promising strategies that re-educate macrophages to specifically obtain anti-tumor phenotypes in cancer, including brain tumors. Analysis of murine breast cancer brain metastasis (BCBM) models indicates that targeting TAMs at distinct stages of the metastatic cascade using BLZ945 agents, an inhibitor of colony-stimulating factor 1 receptor (CSF1R), leads to antitumor responses in BCBM preclinical models [55]. Furthermore, TAM depletion strategies in several types of cancer can provide a survival advantage, the effects of targeting TAMs in brain tumors seem to be more environmental conditions dependent.
3.2. Dendritic Cells (DCs)
Uncontestably, DCs are critical for regulating adaptive immune responses as antigen-presenting cells (APCs) [56,57]. By integrating messages within the TME and delivering it to other immune cells, especially T cells, DCs have the potential to shape anti-tumor immunity. However, cancer cells can also use various strategies to limit and manipulate DCs activity to escape immune surveillance (Figure 3B). Various mechanisms within TME harass DCs functions, such as inhibition of differentiation [58], metabolic stress [59] and impaired handling of tumor-associated antigens (TAAs) [60,61], resulting in insufficient T cell activation and, potentially, the induction of T cell tolerance to TAAs that causing immune escape. Engineering DCs vaccines to improve immunotherapy response and the development of DC-based vaccines is an active field of cancer research [62]. Taken together, a better understanding of the diversity and functions of DCs subsets and of how these are shaped by the TME could lead to improved therapies for BrM in various cancers.
3.3. Neutrophil
In the TME, neutrophils may inhibit tumor progression by releasing anti-tumor factors, such as nitric oxide, H2O2, and TSP-1 [63,64,65], but emerging evidence indicate that neutrophils are a heterogeneous population with plasticity, and subpopulation of neutrophils including tumor-associated neutrophils (TANs) are actively involved in tumor survival, immune response, angiogenesis and metastasis [66,67,68,69]. Recruitment of neutrophils to the brain TME is regulated by various cytokines (e.g., IL-6), chemokines (e.g., G-CSF, CXCL1) and secret protein (e.g., CTSC) released by metastatic cells within brain environment (Figure 3C). Although neutrophils have the ability to inhibit tumors growth in some preclinical models [70], this property has not been well applied in clinical trials. In recent years, a close correlation between TANs in the TME and formation of neutrophil extracellular traps (NETs), which was net-like structures composed of DNA-histone complexes and proteins released by activated neutrophils, has been also observed either in primary tumors and metastatic sites [71,72,73]. TANs stimulate the proangiogenic activity of tumor cells at adjacent invading edges by inducing formation of NETs [74], and then activate tumor cells to induce formation of PMN favoring the brain colonization [74,75]. Moreover, NETs can also catch CTCs and promote metastasis. In addition, it has been reported that wake dormant cancer cells can cause tumor relapse and metastasis. These results indicated the pro-metastatic activity of NETs and highlighted the potential targets for cancer therapy. However, some urgent scientific problems need to be further discussed: (i) whether there are different phenotypes (e.g., N1, N2, N(m), N(m+1)) of neutrophils in the BrM; (ii) Is the distinguish effect (anti-tumour or pro-tumour) of neutrophils within BrM associated with different cancer phenotypes? (iii) Which factors result in different functions on tumor control of neutrophils in the context of immunotherapy? (iv) Whether the anti-tumor neutrophils can be effectively expanded by scientific intervention, such as antagonist against special receptor discovered by single-cell sequencing (scRNA-seq)?
3.4. Astrocyte
After adhesion and extravasation, CTCs are commonly surrounded by reactive astrocytes [76,77] likely influenced by damage-associated molecular patterns (DAMPs) [78]. Astrocytes play an essential role in central nervous system (CNS) in terms of the inflammation and tumor [76,79,80] (Figure 3D). Astrocytes constitute approximately ~30% of the cells in the malignant CNS disease [81]. In the context of BrM, they reduce the numbers of potential metastatic cells by activating plasmin [4]. Classically, the astrocyte has been characterized by binary polarization states in terms of functional phenotype: the neuroinflammatory and anti-tumour “A1” type and the neuroprotective and pro-tumour “A2” type [82]. A subpopulation named signal transducer and activator of transcription 3-positive (STAT3+)–reactive astrocytes that surrounding BrM and demonstrated that not only was helpful for formation of BrM, but these cells can influence the innate and adaptive immunity toward favoring tumour growth and survival [83]. Surviving cancer cells continue to interact with reactive astrocytes during brain colonization, establishing gap junctions with reactive astrocytes. Metastatic cells employ these Cx43 gap junctions to send calcium and cGAMP to astrocytes [84]. Within astrocytes, cGAMP activates signaling pathway leading to secretion of TNF and IFN-a, thus supporting promising role for cancer immunotherapy [85]. Within BrM environment, loss of PTEN increases cancer cell proliferation and induces secretion of CCL2, that attracting pro-metastatic myeloid cells to favor metastatic brain colonization [86]. In addition, astrocytes can interact with immune cells in brain metastatic tumor. For example, TANs promote STAT3 upregulation by secreting LCN2 to bind to the LCN2-specific receptor (SLC22A17) on astrocytes, thereby triggering the release of pro-inflammatory signals, leading to recruitment of LCN2-producing granulocytes within the BrM, that supporting brain colonization [87]. However, the large number of astrocytes participated in inflammatory responses in malignant CNS disease, as well as the complex astrocyte-immune cells interactions and other neural cell types, has hampered mechanistic understanding of astrocyte reactivity. Several key questions about the reactive-astrocyte remain to be addressed, including that (i) whether there are different types of reactive astrocytes (e.g., A1, A2, A(n), A(n+1)) and how they are induced? (ii) Are reactive astrocytes helpful or harmful within BrM among different cancer types, and how their effects are mediated?
3.5. T Lymphocyte Cells
In the initial stage of tumor growth, cells instinctively exhibit pMHC-I peptide complexes on their surface, usually presenting tumor-associated antigens (TAAs) to CD8+ T cells [88]. This recognition contributes to immune-mediated tumor elimination and control and is supported by the anti-tumorigenic properties of the surrounding stroma [89] (Figure 3E). Over time, however, components of the antigen presentation pathway change, resulting in the loss of pMHC-I displayed on tumor cells [90]. In addition, the stroma and other immune cells (e.g., TAMs and TANs) surrounding the tumor cells display pro-tumorigenic properties, supporting tumor immune escape and leading to tumor growth and invasion [91,92,93]. Tumor cells evade immunotoxic killing by upregulating PD-L1 binding to the PD-1 receptor on CD8+ T cells, inducing T cell dysfunction, and is one of the main mechanisms responsible for immune escape [94]. Furthermore, the characteristics of infiltrating immune cells in brain metastases were predominantly T cells and macrophages, with T cells exhibiting higher heterogeneity and consisting of five main clusters: CD8+ effector memory T cells (T: CD8+: EM cluster), central memory T cells (T: CD4+: CM1 cluster, T: CD4+: CM2 cluster and T: CM cluster) and regulatory T cells (Tregs cluster) by single-cell sequencing (scRNA-seq) of human brain metastasis tissues combined with mass spectrometry flow (CyTOF). This phenotypic shift from activated to non-functional T cells coincides with a shift in cellular metabolism from glycolysis and the tricarboxylic acid cycle to lipid metabolism, and the above results reveal a functional state of T cells (from activated/exhausted to non-functional) that is associated with concomitant metabolic and microenvironmental reprogramming [95].
4. Targeted Therapies
Despite the currently poor survival outcomes of BrM patients among mostly various cancers, our fundamental understanding of the biological principles underlying the BrM formation process is rapidly increasing. As these discoveries are shift from the field of basic science to clinical applications (Table 1), survival benefit for patients harboring BrM are almost certain to improve in future.
4.1. Targeting Molecular Signatures for Personalized Treatment of BrM
Next generation sequencing (NGS) database indicated that alterations in molecules and signal pathways can contribute to tumor growth and survival, and found that brain metastases may not share the driver mutations of the primary tumor, such as BRAF-mutation, PIK3CA-mutation, loss of PTEN, EGFR- amplifications and KRAS mutations [96,97]. These findings suggest the importance of brain metastasis specific therapies that address this unique disease. Furthermore, recent studies have demonstrated that metabolic processes as key regulators of BrM and metastatic cancer cells have distinct metabolic features within brain microenvironment differing from the primary tumor [76,98,99]. These results highlight the importance of understanding underlying mechanism of metabolic reprogramming contributing to BrM as well as essential molecular elements between BrM and primary tumors, which could provide novel therapeutic windows to improve prognosis in patients with BrM.
4.2. Principle of Targeting the “CTCs Stage” for Prevention of BrM
Circulating tumor cells (CTCs), shed by primary malignancies, function as ‘‘seeds’’ for distant metastasis [26,100]. The investigation of the interaction between tumor cells and immune cells in the bloodstream may provide a enlighten of potentiality in intervening the metastasis by escaping the immune elimination [101]. However, some scientific problems need be further addressed: (i) whether CTCs in bloodstream will conventionally face with immune surveillance by immunocytes; (ii) which subpopulation of immunocytes exhibit the function of immune elimination of CTCs; and (iii) which molecular mechanisms underlying CTCs escape this immune surveillance. Recently, platelets and neutrophils cells were reported as CTC partners (“CTCs-platelets” cluster and “CTCs-neutrophil” cluster, respectively) facilitating BrM by promoting CTCs survival and providing a regional immune suppressive microenvironment [11,102,103]. For example, CTCs escape immunosurveillance from NK cells by engaging the immune checkpoint HLA-E:CD94-NKG2A. Interrupting the checkpoint efficiently prevents BrM via the blood circulation [29]. The above results suggest that targeting the interaction of CTCs with peripheral blood immune cells will be a favorable therapeutic strategy against metastasis in the future.
4.3. Astrocytes as a Potential Promising Therapeutic Target of BrM
Given more rapidly research evidences focusing on the effect of some astrocyte subpopulations participating in BrM progression, these cells naturally become attractive targets for treatment [104,105]. Soto et al. demonstrated that STAT3+ reactive astrocytes can induce vascular dysfunction within brain environment, which was reversible by using STAT3 signaling pathway inhibitor agents, such as WP1066 [106]. In addition, reeducation of astrocytes by tumor cells into tumor-supportive phenotypes via established gap junctions has led to interest in targeting these cell-to-cell signaling structures for preventing BrM [107,108,109]. However, some key questions need to further addressed: (i) Identification of specific markers of reactive astrocytes and (ii) whether they can provide potential therapeutic targets of BrM via astrocyte-specific manner.
4.4. Targeting the PMN for Brain Metastatic Therapeutics
Targeting the molecular and cellular components facilitating the PMN may be a promisingly therapeutic targets for suppressing PMN formation and consequently preventing BrM [15,16]. These targets including that reducing the levels of molecular components promoting the PMN formation, suppressing the recruitment of immunosuppressive cells, interfering crosstalk between cancer-associated fibroblasts (CAFs) and immune cells within distant PMN such as tumor-associated macrophages (TAMs), TANs, and Tregs. Reshaping the immunosuppressive niche, as an essential immune feature of PMN, and reactivating anti-tumor immune response may be potential strategy for BrM treatment. In conclusion, a deeply understanding of the mechanisms driving PMN formation, and identification of their characteristics and effects on tumor metastasis will provide novel insights and promisingly therapeutic windows for the prevention and treatment of BrM.
5. Conclusions and Future Perspectives
BrM is a problematic and increasingly scientific topic in patients with cancer. Although ICIs have revolutionized the treatment pattern of BrM within various cancers such as melanoma, breast cancer and lung cancer, the determinants of anti-tumour response remain incompletely clarified. Increased understanding of the unique molecular, immune cellular characteristics of the BrM is paramount in developing novel BrM-directed targeted strategies. Mechanistic insights into the process of individual components for BrM have led to the identification of multiple potential therapeutic targets in context of molecular and immune cellular elements, several of which are now under clinical evaluation. Improved understanding of immunobiological principles unique to the BrM and dissection of those that govern the activity of ICIs are promising toward unlocking BrM-specific antitumor immunity. However, until recently, translation of promising therapeutic strategies based on preclinical in vitro and in syngeneic murine models to clinical applications remains a major challenge. Organoid platform or patient-derived xenograft (PDX) models may be promising tools currently or future in use [110,111]. Another experimental model that may be exciting for accurately targeting therapy applied into BrM is immunodeficient mice engrafted with human immune cells or tissues, named “human immune system (HIS)” mice [112]. Additionally, ongoing appearances of increasingly available tools such as scRNA-seq, spatial transcriptome (ST) to combat tissue heterogeneity, and the future is optimistic for therapies that will effectively unlock molecular targeting or antitumor immunity in the BrM. However, we are also looking forward to an exciting event ahead for basic research and clinical translation in brain metastatic biology.
Author Contributions
A.Z. and S.L conceived the manuscript. S.L., Y.S., C.D., and S.Y. prepared the draft manuscript. S.L., A.Z., and D.Z. revised the manuscript. A.Z. supervised the study. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by National Natural Science Foundation of China (Grants 31871406), Natural Science Foundation of Guangdong Province (Grants 2021A1515011067 and 2023A1515010419). Funds for Construction of High-Level Universities in Guangdong Province.
Acknowledgments
We thank members of our laboratories for helpful discussions.
Conflicts of interest
The authors declare no conflict of interest.
References
- Achrol, A.S.; Rennert, R.C.; Anders, C.; Soffietti, R.; Ahluwalia, M.S.; Nayak, L.; Peters, S.; Arvold, N.D.; Harsh, G.R.; Steeg, P.S.; et al. Brain metastases. Nat Rev Dis Primers 2019, 5, 5. [Google Scholar] [PubMed]
- Berghoff, A.S.; Schur, S.; Füreder, L.M.; Gatterbauer, B.; Dieckmann, K.; Widhalm, G.; Hainfellner, J.; Zielinski, C.C.; Birner, P.; Bartsch, R.; et al. Descriptive statistical analysis of a real life cohort of 2419 patients with brain metastases of solid cancers. ESMO open 2016, 1, e000024. [Google Scholar] [CrossRef]
- Sacks, P.; Rahman, M. Epidemiology of Brain Metastases. Neurosurgery clinics of North America 2020, 31, 481–488. [Google Scholar] [CrossRef] [PubMed]
- Valiente, M.; Ahluwalia, M.S.; Boire, A.; Brastianos, P.K.; Goldberg, S.B.; Lee, E.Q.; Le Rhun, E.; Preusser, M.; Winkler, F.; Soffietti, R. The Evolving Landscape of Brain Metastasis. Trends Cancer 2018, 4, 176–196. [Google Scholar]
- Hall, W.A.; Djalilian, H.R.; Nussbaum, E.S.; Cho, K.H. Long-term survival with metastatic cancer to the brain. Medical oncology (Northwood, London, England) 2000, 17, 279–286. [Google Scholar] [CrossRef] [PubMed]
- Galldiks, N.; Kocher, M.; Ceccon, G.; Werner, J.M.; Brunn, A.; Deckert, M.; Pope, W.B.; Soffietti, R.; Le Rhun, E.; Weller, M.; et al. Imaging challenges of immunotherapy and targeted therapy in patients with brain metastases: response, progression, and pseudoprogression. Neuro Oncol 2020, 22, 17–30. [Google Scholar]
- Fares, J.; Ulasov, I.; Timashev, P.; Lesniak, M.S. Emerging principles of brain immunology and immune checkpoint blockade in brain metastases. Brain 2021, 144, 1046–1066. [Google Scholar] [CrossRef]
- Strickland, M.R.; Alvarez-Breckenridge, C.; Gainor, J.F.; Brastianos, P.K. Tumor Immune Microenvironment of Brain Metastases: Toward Unlocking Antitumor Immunity. Cancer Discov 2022, 12, 1199–1216. [Google Scholar]
- El-Kenawi, A.; Hanggi, K.; Ruffell, B. The Immune Microenvironment and Cancer Metastasis. Cold Spring Harb Perspect Med 2020, 10. [Google Scholar] [CrossRef]
- Novikov, N.M.; Zolotaryova, S.Y.; Gautreau, A.M.; Denisov, E.V. Mutational drivers of cancer cell migration and invasion. Br J Cancer 2021, 124, 102–114. [Google Scholar]
- de Visser, K.E.; Joyce, J.A. The evolving tumor microenvironment: From cancer initiation to metastatic outgrowth. Cancer Cell 2023, 41, 374–403. [Google Scholar] [PubMed]
- Chhichholiya, Y.; Ruthuparna, M.; Velagaleti, H.; Munshi, A. Brain metastasis in breast cancer: focus on genes and signaling pathways involved, blood-brain barrier and treatment strategies. Clin Transl Oncol 2023. [Google Scholar] [CrossRef] [PubMed]
- Tobar, L.E.; Farnsworth, R.H.; Stacker, S.A. Brain Vascular Microenvironments in Cancer Metastasis. Biomolecules 2022, 12. [Google Scholar] [CrossRef]
- Massague, J.; Obenauf, A.C. Metastatic colonization by circulating tumour cells. Nature 2016, 529, 298–306. [Google Scholar] [PubMed]
- Patras, L.; Shaashua, L.; Matei, I.; Lyden, D. Immune determinants of the pre-metastatic niche. Cancer Cell 2023, 41, 546–572. [Google Scholar] [PubMed]
- Liu, Y.; Cao, X. Characteristics and Significance of the Pre-metastatic Niche. Cancer Cell 2016, 30, 668–681. [Google Scholar] [CrossRef]
- Wang, Y.; Ye, F.; Liang, Y.; Yang, Q. Breast cancer brain metastasis: insight into molecular mechanisms and therapeutic strategies. Br J Cancer 2021, 125, 1056–1067. [Google Scholar]
- Zang, Y.W.; Gu, X.D.; Xiang, J.B.; Chen, Z.Y. Brain metastases from colorectal cancer: microenvironment and molecular mechanisms. Int J Mol Sci 2012, 13, 15784–15800. [Google Scholar] [CrossRef]
- Liu, W.; Powell, C.A.; Wang, Q. Tumor microenvironment in lung cancer-derived brain metastasis. Chin Med J (Engl) 2022, 135, 1781–1791. [Google Scholar] [CrossRef]
- Shih, D.J.H.; Nayyar, N.; Bihun, I.; Dagogo-Jack, I.; Gill, C.M.; Aquilanti, E.; Bertalan, M.; Kaplan, A.; D’Andrea, M.R.; Chukwueke, U.; et al. Genomic characterization of human brain metastases identifies drivers of metastatic lung adenocarcinoma. Nature genetics 2020, 52, 371–377. [Google Scholar] [CrossRef]
- Egleton, R.D.; Brown, K.C.; Dasgupta, P. Nicotinic acetylcholine receptors in cancer: multiple roles in proliferation and inhibition of apoptosis. Trends in pharmacological sciences 2008, 29, 151–158. [Google Scholar] [CrossRef] [PubMed]
- Hill, W.; Lim, E.L.; Weeden, C.E.; Lee, C.; Augustine, M.; Chen, K.; Kuan, F.C.; Marongiu, F.; Evans, E.J., Jr.; Moore, D.A.; et al. Lung adenocarcinoma promotion by air pollutants. Nature 2023, 616, 159–167. [Google Scholar] [PubMed]
- Liu, J.; Xiao, Q.; Xiao, J.; Niu, C.; Li, Y.; Zhang, X.; Zhou, Z.; Shu, G.; Yin, G. Wnt/β-catenin signalling: function, biological mechanisms, and therapeutic opportunities. Signal transduction and targeted therapy 2022, 7, 3. [Google Scholar] [PubMed]
- Wagner, E.F.; Nebreda, A.R. Signal integration by JNK and p38 MAPK pathways in cancer development. Nat Rev Cancer 2009, 9, 537–549. [Google Scholar]
- Chen, P.; Bonaldo, P. Role of macrophage polarization in tumor angiogenesis and vessel normalization: implications for new anticancer therapies. International review of cell and molecular biology 2013, 301, 1–35. [Google Scholar]
- Green, T.L.; Cruse, J.M.; Lewis, R.E.; Craft, B.S. Circulating tumor cells (CTCs) from metastatic breast cancer patients linked to decreased immune function and response to treatment. Exp Mol Pathol 2013, 95, 174–179. [Google Scholar] [CrossRef]
- Gkountela, S.; Castro-Giner, F.; Szczerba, B.M.; Vetter, M.; Landin, J.; Scherrer, R.; Krol, I.; Scheidmann, M.C.; Beisel, C.; Stirnimann, C.U.; et al. Circulating Tumor Cell Clustering Shapes DNA Methylation to Enable Metastasis Seeding. Cell 2019, 176, 98–112. [Google Scholar] [CrossRef]
- Pan, G.; Thomson, J.A. Nanog and transcriptional networks in embryonic stem cell pluripotency. Cell Res 2007, 17, 42–49. [Google Scholar] [CrossRef]
- Liu, X.; Song, J.; Zhang, H.; Liu, X.; Zuo, F.; Zhao, Y.; Zhao, Y.; Yin, X.; Guo, X.; Wu, X. Immune checkpoint HLA-E:CD94-NKG2A mediates evasion of circulating tumor cells from NK cell surveillance. Cancer Cell 2023, 41, 272–287. [Google Scholar] [CrossRef]
- Placke, T.; Örgel, M.; Schaller, M.; Jung, G.; Rammensee, H.G.; Kopp, H.G.; Salih, H.R. Platelet-derived MHC class I confers a pseudonormal phenotype to cancer cells that subverts the antitumor reactivity of natural killer immune cells. Cancer Res 2012, 72, 440–448. [Google Scholar] [CrossRef]
- Ward, M.P.; E. K. L; A. N. L; Mohamed, B.M.; Kelly, T.; Bates, M.; Clarke, A.; Brady, N.; Martin, C.M.; Brooks, R.D.; et al. Platelets, immune cells and the coagulation cascade, friend or foe of the circulating tumour cell? Mol Cancer 2021, 20, 59. [Google Scholar] [CrossRef]
- Kanikarla-Marie, P.; Lam, M.; Menter, D.G.; Kopetz, S. Platelets, circulating tumor cells, and the circulome. Cancer metastasis reviews 2017, 36, 235–248. [Google Scholar] [CrossRef]
- Ruan, H.; Zhou, Y.; Shen, J.; Zhai, Y.; Xu, Y.; Pi, L.; Huang, R.; Chen, K.; Li, X.; Ma, W.; et al. Circulating tumor cell characterization of lung cancer brain metastases in the cerebrospinal fluid through single-cell transcriptome analysis. Clinical and translational medicine 2020, 10, e246. [Google Scholar] [CrossRef] [PubMed]
- Castro-Giner, F.; Aceto, N. Tracking cancer progression: from circulating tumor cells to metastasis. Genome medicine 2020, 12, 31. [Google Scholar] [PubMed]
- Williams, S.C. Circulating tumor cells. Proc Natl Acad Sci U S A 2013, 110, 4861. [Google Scholar] [CrossRef]
- Chambers, A.F.; Groom, A.C.; MacDonald, I.C. Dissemination and growth of cancer cells in metastatic sites. Nat Rev Cancer 2002, 2, 563–572. [Google Scholar] [CrossRef]
- Labelle, M.; Begum, S.; Hynes, R.O. Direct signaling between platelets and cancer cells induces an epithelial-mesenchymal-like transition and promotes metastasis. Cancer Cell 2011, 20, 576–590. [Google Scholar] [CrossRef]
- Schumacher, D.; Strilic, B.; Sivaraj, K.K.; Wettschureck, N.; Offermanns, S. Platelet-derived nucleotides promote tumor-cell transendothelial migration and metastasis via P2Y2 receptor. Cancer Cell 2013, 24, 130–137. [Google Scholar] [CrossRef]
- Bos, P.D.; Zhang, X.H.; Nadal, C.; Shu, W.; Gomis, R.R.; Nguyen, D.X.; Minn, A.J.; van de Vijver, M.J.; Gerald, W.L.; Foekens, J.A.; et al. Genes that mediate breast cancer metastasis to the brain. Nature 2009, 459, 1005–1009. [Google Scholar] [CrossRef]
- Sevenich, L.; Bowman, R.L.; Mason, S.D.; Quail, D.F.; Rapaport, F.; Elie, B.T.; Brogi, E.; Brastianos, P.K.; Hahn, W.C.; Holsinger, L.J.; et al. Analysis of tumour- and stroma-supplied proteolytic networks reveals a brain-metastasis-promoting role for cathepsin S. Nat Cell Biol 2014, 16, 876–888. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Fong, M.Y.; Min, Y.; Somlo, G.; Liu, L.; Palomares, M.R.; Yu, Y.; Chow, A.; O’Connor, S.T.; Chin, A.R.; et al. Cancer-secreted miR-105 destroys vascular endothelial barriers to promote metastasis. Cancer Cell 2014, 25, 501–515. [Google Scholar]
- Tominaga, N.; Kosaka, N.; Ono, M.; Katsuda, T.; Yoshioka, Y.; Tamura, K.; Lötvall, J.; Nakagama, H.; Ochiya, T. Brain metastatic cancer cells release microRNA-181c-containing extracellular vesicles capable of destructing blood-brain barrier. Nat Commun 2015, 6, 6716. [Google Scholar] [CrossRef] [PubMed]
- Akhtar, M.; Haider, A.; Rashid, S.; Al-Nabet, A. Paget’s “Seed and Soil” Theory of Cancer Metastasis: An Idea Whose Time has Come. Advances in anatomic pathology 2019, 26, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, R.N.; Riba, R.D.; Zacharoulis, S.; Bramley, A.H.; Vincent, L.; Costa, C.; MacDonald, D.D.; Jin, D.K.; Shido, K.; Kerns, S.A.; et al. VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature 2005, 438, 820–827. [Google Scholar] [CrossRef] [PubMed]
- McAllister, S.S.; Weinberg, R.A. The tumour-induced systemic environment as a critical regulator of cancer progression and metastasis. Nat Cell Biol 2014, 16, 717–727. [Google Scholar] [CrossRef]
- Dong, Q.; Liu, X.; Cheng, K.; Sheng, J.; Kong, J.; Liu, T. Pre-metastatic Niche Formation in Different Organs Induced by Tumor Extracellular Vesicles. Front Cell Dev Biol 2021, 9, 733627. [Google Scholar] [CrossRef]
- Wang, H.; Pan, J.; Barsky, L.; Jacob, J.C.; Zheng, Y.; Gao, C.; Wang, S.; Zhu, W.; Sun, H.; Lu, L.; et al. Characteristics of pre-metastatic niche: the landscape of molecular and cellular pathways. Molecular biomedicine 2021, 2, 3. [Google Scholar] [PubMed]
- Schulz, M.; Sevenich, L. TAMs in Brain Metastasis: Molecular Signatures in Mouse and Man. Front Immunol 2021, 12, 716504. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.J.; Wei, K.C.; Chen, P.Y.; Lim, M.; Hwang, T.L. Roles of Neutrophils in Glioma and Brain Metastases. Front Immunol 2021, 12, 701383. [Google Scholar]
- Andersen, B.M.; Akl, C.F.; Wheeler, M.A.; Chiocca, E.A.; Reardon, D.A.; Quintana, F.J. Glial and myeloid heterogeneity in the brain tumour microenvironment. Nat Rev Cancer 2021, 21, 786–802. [Google Scholar]
- Bowman, R.L.; Klemm, F.; Akkari, L.; Pyonteck, S.M.; Sevenich, L.; Quail, D.F.; Dhara, S.; Simpson, K.; Gardner, E.E.; Iacobuzio-Donahue, C.A.; et al. Macrophage Ontogeny Underlies Differences in Tumor-Specific Education in Brain Malignancies. Cell Rep 2016, 17, 2445–2459. [Google Scholar] [CrossRef]
- She, X.; Shen, S.; Chen, G.; Gao, Y.; Ma, J.; Gao, Y.; Liu, Y.; Gao, G.; Zhao, Y.; Wang, C.; et al. Immune surveillance of brain metastatic cancer cells is mediated by IFITM1. EMBO J 2023, 42, e111112. [Google Scholar] [CrossRef]
- Ginhoux, F.; Schultze, J.L.; Murray, P.J.; Ochando, J.; Biswas, S.K. New insights into the multidimensional concept of macrophage ontogeny, activation and function. Nat Immunol 2016, 17, 34–40. [Google Scholar] [CrossRef] [PubMed]
- Ginhoux, F.; Greter, M.; Leboeuf, M.; Nandi, S.; See, P.; Gokhan, S.; Mehler, M.F.; Conway, S.J.; Ng, L.G.; Stanley, E.R.; et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science (New York, N.Y.) 2010, 330, 841–845. [Google Scholar] [CrossRef] [PubMed]
- Klemm, F.; Möckl, A.; Salamero-Boix, A.; Alekseeva, T.; Schäffer, A.; Schulz, M.; Niesel, K.; Maas, R.R.; Groth, M.; Elie, B.T.; et al. Compensatory CSF2-driven macrophage activation promotes adaptive resistance to CSF1R inhibition in breast-to-brain metastasis. Nat Cancer 2021, 2, 1086–1101. [Google Scholar] [CrossRef] [PubMed]
- Wculek, S.K.; Cueto, F.J.; Mujal, A.M.; Melero, I.; Krummel, M.F.; Sancho, D. Dendritic cells in cancer immunology and immunotherapy. Nat Rev Immunol 2020, 20, 7–24. [Google Scholar] [CrossRef] [PubMed]
- Gardner, A.; Ruffell, B. Dendritic Cells and Cancer Immunity. Trends Immunol 2016, 37, 855–865. [Google Scholar] [CrossRef]
- Salmon, H.; Idoyaga, J.; Rahman, A.; Leboeuf, M.; Remark, R.; Jordan, S.; Casanova-Acebes, M.; Khudoynazarova, M.; Agudo, J.; Tung, N.; et al. Expansion and Activation of CD103(+) Dendritic Cell Progenitors at the Tumor Site Enhances Tumor Responses to Therapeutic PD-L1 and BRAF Inhibition. Immunity 2016, 44, 924–938. [Google Scholar] [CrossRef]
- Veglia, F.; Tyurin, V.A.; Mohammadyani, D.; Blasi, M.; Duperret, E.K.; Donthireddy, L.; Hashimoto, A.; Kapralov, A.; Amoscato, A.; Angelini, R.; et al. Lipid bodies containing oxidatively truncated lipids block antigen cross-presentation by dendritic cells in cancer. Nat Commun 2017, 8, 2122. [Google Scholar] [CrossRef]
- Tang, M.; Diao, J.; Gu, H.; Khatri, I.; Zhao, J.; Cattral, M.S. Toll-like Receptor 2 Activation Promotes Tumor Dendritic Cell Dysfunction by Regulating IL-6 and IL-10 Receptor Signaling. Cell Rep 2015, 13, 2851–2864. [Google Scholar] [CrossRef]
- Ruffell, B.; Chang-Strachan, D.; Chan, V.; Rosenbusch, A.; Ho, C.M.; Pryer, N.; Daniel, D.; Hwang, E.S.; Rugo, H.S.; Coussens, L.M. Macrophage IL-10 blocks CD8+ T cell-dependent responses to chemotherapy by suppressing IL-12 expression in intratumoral dendritic cells. Cancer Cell 2014, 26, 623–637. [Google Scholar] [CrossRef] [PubMed]
- Perez, C.R.; De Palma, M. Engineering dendritic cell vaccines to improve cancer immunotherapy. Nat Commun 2019, 10, 5408. [Google Scholar] [CrossRef] [PubMed]
- Catena, R.; Bhattacharya, N.; El Rayes, T.; Wang, S.; Choi, H.; Gao, D.; Ryu, S.; Joshi, N.; Bielenberg, D.; Lee, S.B.; et al. Bone marrow-derived Gr1+ cells can generate a metastasis-resistant microenvironment via induced secretion of thrombospondin-1. Cancer Discov 2013, 3, 578–589. [Google Scholar] [PubMed]
- Finisguerra, V.; Di Conza, G.; Di Matteo, M.; Serneels, J.; Costa, S.; Thompson, A.A.R.; Wauters, E.; Walmsley, S.; Prenen, H.; Granot, Z.; et al. MET is required for the recruitment of anti-tumoural neutrophils. Nature 2015, 522, 349-+. [Google Scholar] [CrossRef]
- Granot, Z.; Henke, E.; Comen, E.A.; King, T.A.; Norton, L.; Benezra, R. Tumor Entrained Neutrophils Inhibit Seeding in the Premetastatic Lung. Cancer Cell 2011, 20, 300–314. [Google Scholar] [CrossRef] [PubMed]
- Acharyya, S.; Oskarsson, T.; Vanharanta, S.; Malladi, S.; Kim, J.; Morris, P.G.; Manova-Todorova, K.; Leversha, M.; Hogg, N.; Seshan, V.E.; et al. A CXCL1 Paracrine Network Links Cancer Chemoresistance and Metastasis. Cell 2012, 150, 165–178. [Google Scholar] [CrossRef]
- Coffelt, S.B.; Kersten, K.; Doornebal, C.W.; Weiden, J.; Vrijland, K.; Hau, C.S.; Verstegen, N.J.M.; Ciampricotti, M.; Hawinkels, L.; Jonkers, J.; et al. IL-17-producing γδ T cells and neutrophils conspire to promote breast cancer metastasis. Nature 2015, 522, 345–348. [Google Scholar] [CrossRef]
- Zhuang, X.Q.; Zhang, H.; Li, X.Y.; Li, X.X.; Cong, M.; Peng, F.L.; Yu, J.Y.; Zhang, X.; Yang, Q.F.; Hu, G.H. Differential effects on lung and bone metastasis of breast cancer by Wnt signalling inhibitor DKK1. Nature Cell Biology 2017, 19, 1274-+. [Google Scholar] [CrossRef]
- Wculek, S.K.; Malanchi, I. Neutrophils support lung colonization of metastasis-initiating breast cancer cells. Nature 2015, 528, 413-+. [Google Scholar] [CrossRef]
- Linde, I.L.; Prestwood, T.R.; Qiu, J.; Pilarowski, G.; Linde, M.H.; Zhang, X.; Shen, L.; Reticker-Flynn, N.E.; Chiu, D.K.; Sheu, L.Y.; et al. Neutrophil-activating therapy for the treatment of cancer. Cancer Cell 2023, 41, 356–372.e310. [Google Scholar] [CrossRef]
- Jaillon, S.; Ponzetta, A.; Di Mitri, D.; Santoni, A.; Bonecchi, R.; Mantovani, A. Neutrophil diversity and plasticity in tumour progression and therapy. Nat Rev Cancer 2020, 20, 485–503. [Google Scholar] [CrossRef]
- Xiong, S.; Dong, L.; Cheng, L. Neutrophils in cancer carcinogenesis and metastasis. Journal of hematology & oncology 2021, 14, 173. [Google Scholar]
- Kaltenmeier, C.; Simmons, R.L.; Tohme, S.; Yazdani, H.O. Neutrophil Extracellular Traps (NETs) in Cancer Metastasis. Cancers (Basel) 2021, 13. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Liu, Q.; Zhang, X.; Liu, X.; Zhou, B.; Chen, J.; Huang, D.; Li, J.; Li, H.; Chen, F.; et al. DNA of neutrophil extracellular traps promotes cancer metastasis via CCDC25. Nature 2020, 583, 133–138. [Google Scholar] [CrossRef]
- Xiao, Y.; Cong, M.; Li, J.; He, D.; Wu, Q.; Tian, P.; Wang, Y.; Yang, S.; Liang, C.; Liang, Y.; et al. Cathepsin C promotes breast cancer lung metastasis by modulating neutrophil infiltration and neutrophil extracellular trap formation. Cancer Cell 2021, 39, 423–437.e427. [Google Scholar] [CrossRef] [PubMed]
- Valiente, M.; Obenauf, A.C.; Jin, X.; Chen, Q.; Zhang, X.H.; Lee, D.J.; Chaft, J.E.; Kris, M.G.; Huse, J.T.; Brogi, E.; et al. Serpins promote cancer cell survival and vascular co-option in brain metastasis. Cell 2014, 156, 1002–1016. [Google Scholar] [CrossRef]
- Lorger, M.; Felding-Habermann, B. Capturing changes in the brain microenvironment during initial steps of breast cancer brain metastasis. The American journal of pathology 2010, 176, 2958–2971. [Google Scholar] [CrossRef]
- Sofroniew, M.V. Astrocyte barriers to neurotoxic inflammation. Nature reviews. Neuroscience 2015, 16, 249–263. [Google Scholar]
- Sofroniew, M.V. Astrocyte Reactivity: Subtypes, States, and Functions in CNS Innate Immunity. Trends Immunol 2020, 41, 758–770. [Google Scholar] [CrossRef]
- Linnerbauer, M.; Wheeler, M.A.; Quintana, F.J. Astrocyte Crosstalk in CNS Inflammation. Neuron 2020, 108, 608–622. [Google Scholar] [CrossRef]
- Liddelow, S.A.; Barres, B. Reactive Astrocytes: Production, Function, and Therapeutic Potential. Immunity 2017, 46, 957–967. [Google Scholar]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Münch, A.E.; Chung, W.S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Priego, N.; Zhu, L.C.; Monteiro, C.; Mulders, M.; Wasilewski, D.; Bindeman, W.; Doglio, L.; Martinez, L.; Martinez-Saez, E.; Cajal, S.R.Y.; et al. STAT3 labels a subpopulation of reactive astrocytes required for brain metastasis. Nat Med 2018, 24, 1024-+. [Google Scholar] [CrossRef] [PubMed]
- Lin, Q.; Balasubramanian, K.; Fan, D.; Kim, S.J.; Guo, L.; Wang, H.; Bar-Eli, M.; Aldape, K.D.; Fidler, I.J. Reactive astrocytes protect melanoma cells from chemotherapy by sequestering intracellular calcium through gap junction communication channels. Neoplasia (New York, N.Y.) 2010, 12, 748–754. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; Chen, P.; Wang, L.; Li, W.; Chen, B.; Liu, Y.; Wang, H.; Zhao, S.; Ye, L.; He, Y.; et al. cGAS-STING, an important pathway in cancer immunotherapy. Journal of hematology & oncology 2020, 13, 81. [Google Scholar]
- Zhang, L.; Zhang, S.; Yao, J.; Lowery, F.J.; Zhang, Q.; Huang, W.C.; Li, P.; Li, M.; Wang, X.; Zhang, C.; et al. Microenvironment-induced PTEN loss by exosomal microRNA primes brain metastasis outgrowth. Nature 2015, 527, 100–104. [Google Scholar] [CrossRef]
- Adler, O.; Zait, Y.; Cohen, N.; Blazquez, R.; Doron, H.; Monteran, L.; Scharff, Y.; Shami, T.; Mundhe, D.; Glehr, G.; et al. Reciprocal interactions between innate immune cells and astrocytes facilitate neuroinflammation and brain metastasis via lipocalin-2. Nat Cancer 2023, 4, 401–418. [Google Scholar] [CrossRef]
- Godfrey, D.I.; Rossjohn, J.; McCluskey, J. The fidelity, occasional promiscuity, and versatility of T cell receptor recognition. Immunity 2008, 28, 304–314. [Google Scholar]
- Thomas, C.; Tampé, R. MHC I chaperone complexes shaping immunity. Current opinion in immunology 2019, 58, 9–15. [Google Scholar] [CrossRef]
- Kass, I.; Buckle, A.M.; Borg, N.A. Understanding the structural dynamics of TCR-pMHC complex interactions. Trends Immunol 2014, 35, 604–612. [Google Scholar] [CrossRef]
- Cheng, S.; Li, Z.; Gao, R.; Xing, B.; Gao, Y.; Yang, Y.; Qin, S.; Zhang, L.; Ouyang, H.; Du, P.; et al. A pan-cancer single-cell transcriptional atlas of tumor infiltrating myeloid cells. Cell 2021, 184, 792–809. [Google Scholar] [CrossRef]
- De Jaeghere, E.A.; Denys, H.G.; De Wever, O. Fibroblasts Fuel Immune Escape in the Tumor Microenvironment. Trends Cancer 2019, 5, 704–723. [Google Scholar] [CrossRef]
- Del Alcazar, C.R.G.; Alečković, M.; Polyak, K. Immune Escape during Breast Tumor Progression. Cancer immunology research 2020, 8, 422–427. [Google Scholar] [CrossRef]
- Jiang, X.; Wang, J.; Deng, X.; Xiong, F.; Ge, J.; Xiang, B.; Wu, X.; Ma, J.; Zhou, M.; Li, X.; et al. Role of the tumor microenvironment in PD-L1/PD-1-mediated tumor immune escape. Mol Cancer 2019, 18, 10. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, H.; Mei, W.; Robles, I.; Hagerling, C.; Allen, B.M.; Okholm, T.L.H.; Nanjaraj, A.; Verbeek, T.; Kalavacherla, S.; van Gogh, M.; et al. Cellular architecture of human brain metastases. Cell 2022, 185, 729–745. [Google Scholar] [CrossRef] [PubMed]
- Brastianos, P.K.; Carter, S.L.; Santagata, S.; Cahill, D.P.; Taylor-Weiner, A.; Jones, R.T.; Van Allen, E.M.; Lawrence, M.S.; Horowitz, P.M.; Cibulskis, K.; et al. Genomic Characterization of Brain Metastases Reveals Branched Evolution and Potential Therapeutic Targets. Cancer Discov 2015, 5, 1164–1177. [Google Scholar] [CrossRef] [PubMed]
- Saunus, J.M.; Quinn, M.C.; Patch, A.M.; Pearson, J.V.; Bailey, P.J.; Nones, K.; Reed, A.E.M.; Miller, D.; Wilson, P.J.; Al-Ejeh, F.; et al. Integrated genomic and transcriptomic analysis of human brain metastases identifies alterations of potential clinical significance. The Journal of pathology 2015, 237, 363–378. [Google Scholar] [CrossRef]
- Neman, J.; Termini, J.; Wilczynski, S.; Vaidehi, N.; Choy, C.; Kowolik, C.M.; Li, H.; Hambrecht, A.C.; Roberts, E.; Jandial, R. Human breast cancer metastases to the brain display GABAergic properties in the neural niche. Proc Natl Acad Sci U S A 2014, 111, 984–989. [Google Scholar] [CrossRef]
- Tyagi, A.; Wu, S.Y.; Watabe, K. Metabolism in the progression and metastasis of brain tumors. Cancer Lett 2022, 539, 215713. [Google Scholar] [CrossRef]
- Mohme, M.; Riethdorf, S.; Pantel, K. Circulating and disseminated tumour cells - mechanisms of immune surveillance and escape. Nat Rev Clin Oncol 2017, 14, 155–167. [Google Scholar] [CrossRef]
- Lin, D.; Shen, L.; Luo, M.; Zhang, K.; Li, J.; Yang, Q.; Zhu, F.; Zhou, D.; Zheng, S.; Chen, Y.; et al. Circulating tumor cells: biology and clinical significance. Signal transduction and targeted therapy 2021, 6, 404. [Google Scholar] [PubMed]
- Sun, Y.F.; Wu, L.; Liu, S.P.; Jiang, M.M.; Hu, B.; Zhou, K.Q.; Guo, W.; Xu, Y.; Zhong, Y.; Zhou, X.R.; et al. Dissecting spatial heterogeneity and the immune-evasion mechanism of CTCs by single-cell RNA-seq in hepatocellular carcinoma. Nat Commun 2021, 12, 4091. [Google Scholar] [CrossRef]
- Szczerba, B.M.; Castro-Giner, F.; Vetter, M.; Krol, I.; Gkountela, S.; Landin, J.; Scheidmann, M.C.; Donato, C.; Scherrer, R.; Singer, J.; et al. Neutrophils escort circulating tumour cells to enable cell cycle progression. Nature 2019, 566, 553–557. [Google Scholar] [CrossRef]
- Hosonaga, M.; Saya, H.; Arima, Y. Molecular and cellular mechanisms underlying brain metastasis of breast cancer. Cancer metastasis reviews 2020, 39, 711–720. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.Y.; Huo, J. A1/A2 astrocytes in central nervous system injuries and diseases: Angels or devils? Neurochem Int 2021, 148, 105080. [Google Scholar] [PubMed]
- Soto, M.S.; Larkin, J.R.; Martin, C.; Khrapitchev, A.A.; Maczka, M.; Economopoulos, V.; Scott, H.; Escartin, C.; Bonvento, G.; Serres, S.; et al. STAT3-Mediated Astrocyte Reactivity Associated with Brain Metastasis Contributes to Neurovascular Dysfunction. Cancer Res 2020, 80, 5642–5655. [Google Scholar] [CrossRef]
- Chen, Q.; Boire, A.; Jin, X.; Valiente, M.; Er, E.E.; Lopez-Soto, A.; Jacob, L.; Patwa, R.; Shah, H.; Xu, K.; et al. Carcinoma-astrocyte gap junctions promote brain metastasis by cGAMP transfer. Nature 2016, 533, 493–498. [Google Scholar] [CrossRef]
- Abbott, N.J.; Rönnbäck, L.; Hansson, E. Astrocyte-endothelial interactions at the blood-brain barrier. Nature reviews. Neuroscience 2006, 7, 41–53. [Google Scholar] [CrossRef]
- Han, R.T.; Kim, R.D.; Molofsky, A.V.; Liddelow, S.A. Astrocyte-immune cell interactions in physiology and pathology. Immunity 2021, 54, 211–224. [Google Scholar] [CrossRef]
- LeSavage, B.L.; Suhar, R.A.; Broguiere, N.; Lutolf, M.P.; Heilshorn, S.C. Next-generation cancer organoids. Nature materials 2022, 21, 143–159. [Google Scholar] [CrossRef]
- Jenkins, R.W.; Aref, A.R.; Lizotte, P.H.; Ivanova, E.; Stinson, S.; Zhou, C.W.; Bowden, M.; Deng, J.; Liu, H.; Miao, D.; et al. Ex Vivo Profiling of PD-1 Blockade Using Organotypic Tumor Spheroids. Cancer Discov 2018, 8, 196–215. [Google Scholar] [CrossRef] [PubMed]
- Allen, T.M.; Brehm, M.A.; Bridges, S.; Ferguson, S.; Kumar, P.; Mirochnitchenko, O.; Palucka, K.; Pelanda, R.; Sanders-Beer, B.; Shultz, L.D.; et al. Humanized immune system mouse models: progress, challenges and opportunities. Nat Immunol 2019, 20, 770–774. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Cancer cell metastatic dissemination to brain. Brain metastases develop following a small population of cells from primary tumor to the brain microvasculature, with subsequent tumor growth in the pre-microenvironment niche, immune escape and colonization. TAN, Tumor-associated neutrophil; TAM, tumor-associated macrophage; NETs, neutrophil extracellular traps; MDSCs, myeloid-derived suppressor cells; BMDCs, bone-marrow-derived cells; Tregs, regulatory cells; CAF, cancer-associated fibroblasts; CTCs, circulating tumor cells. Created by Biorender.com.
Figure 1.
Cancer cell metastatic dissemination to brain. Brain metastases develop following a small population of cells from primary tumor to the brain microvasculature, with subsequent tumor growth in the pre-microenvironment niche, immune escape and colonization. TAN, Tumor-associated neutrophil; TAM, tumor-associated macrophage; NETs, neutrophil extracellular traps; MDSCs, myeloid-derived suppressor cells; BMDCs, bone-marrow-derived cells; Tregs, regulatory cells; CAF, cancer-associated fibroblasts; CTCs, circulating tumor cells. Created by Biorender.com.
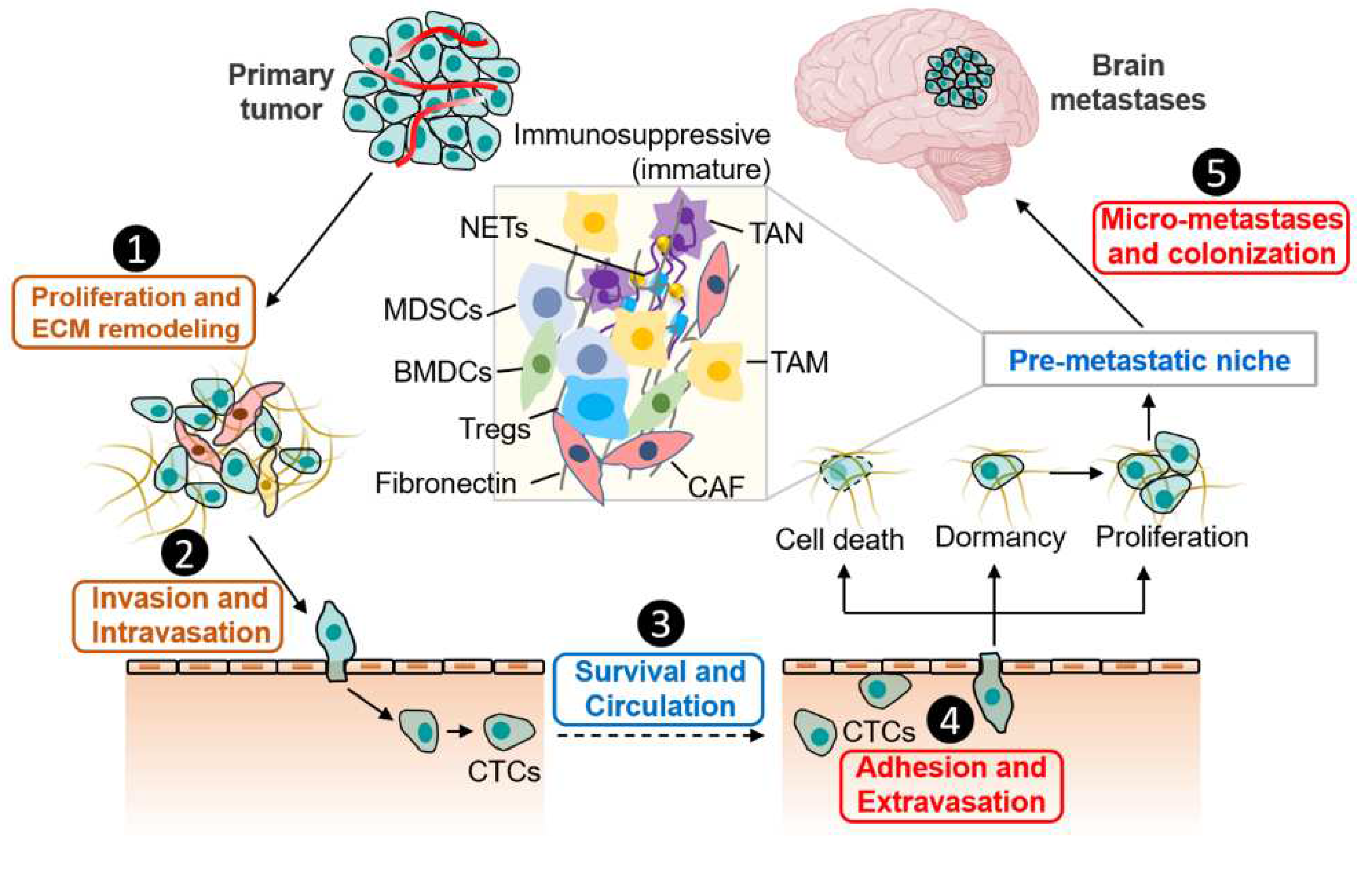
Figure 2.
Schematic representation of molecular and immune factors that influence the brain metastases. A map of molecular and immunity showing the factors that affect the brain metastases. The factors are placed on rings that denote their type, and each factor is also placed in the step of the primary-brain metastatic cycle in which they mainly act. TNF, tumor necrosis factor; TAMs, tumor-associated macrophages; MDSCs, myeloid-derived suppressor cells; TDSFs, tumor-derived secreted factors; ROS, reactive oxygen species; NF-kB, nuclear factor-k-gene binding; BTB, brain-tumor barrier; BBB, blood-brain barrier; MHC, major histocompatibility complex; DC, dendritic cell; HLA, human leukocyte antigen; TILs, Tumor Infiltrating Lymphocytes.
Figure 2.
Schematic representation of molecular and immune factors that influence the brain metastases. A map of molecular and immunity showing the factors that affect the brain metastases. The factors are placed on rings that denote their type, and each factor is also placed in the step of the primary-brain metastatic cycle in which they mainly act. TNF, tumor necrosis factor; TAMs, tumor-associated macrophages; MDSCs, myeloid-derived suppressor cells; TDSFs, tumor-derived secreted factors; ROS, reactive oxygen species; NF-kB, nuclear factor-k-gene binding; BTB, brain-tumor barrier; BBB, blood-brain barrier; MHC, major histocompatibility complex; DC, dendritic cell; HLA, human leukocyte antigen; TILs, Tumor Infiltrating Lymphocytes.
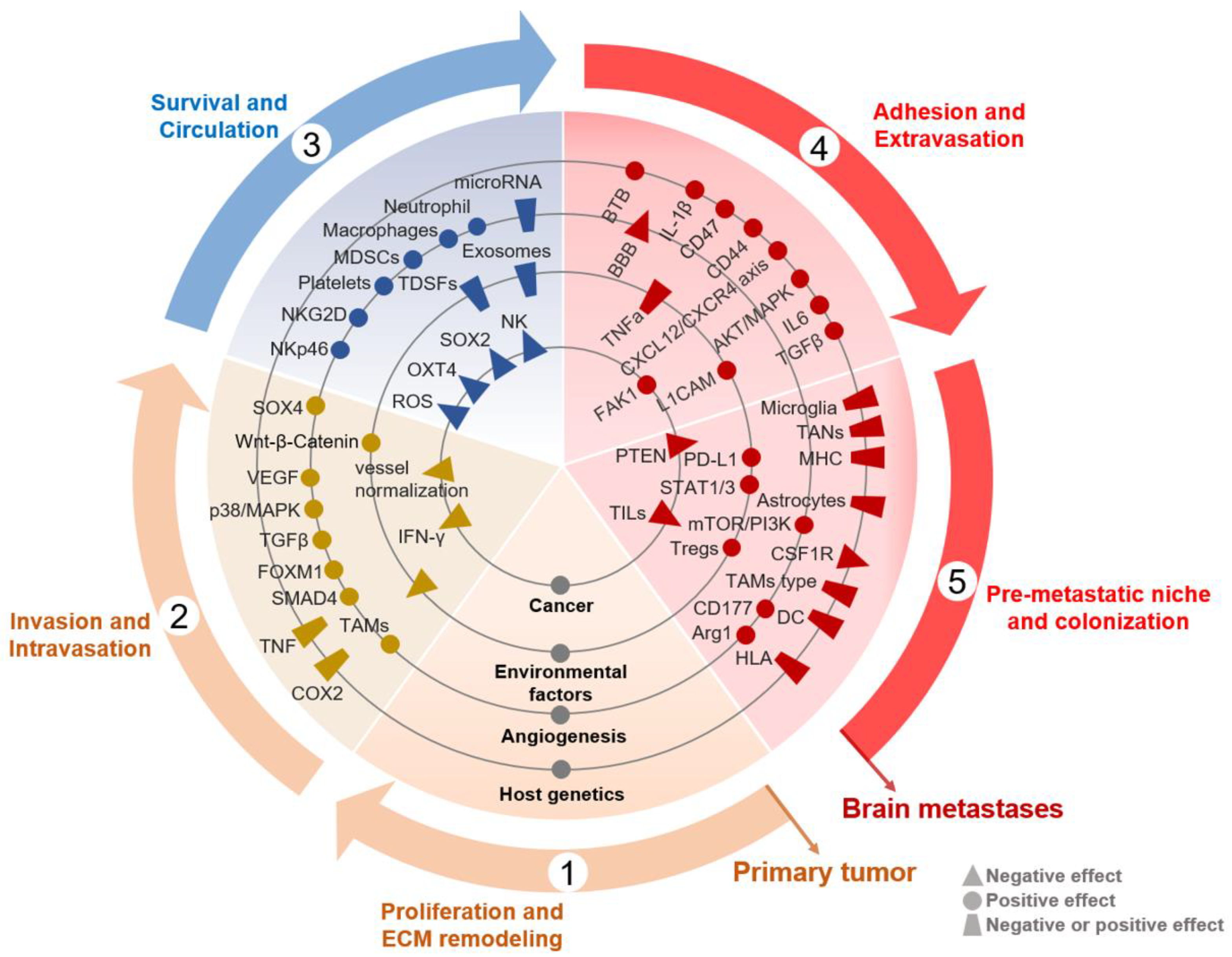
Figure 3.
The Crosstalk between Immune Cellular Components and Tumor Cells in Brain Metastatic Microenvironment. BrM are composed of diverse immune cellular components, including various specialized organ-resident cell types such as astrocyte and microglia. Each of these immune cells contributes to brain metastatic biology in unique ways. (A) Macrophages and microglia engage in essential crosstalk with tumor cells (TC) within the brain metastases, whereby brain tumor cells could release cytokines to recruit TAMs to the brain microenvironment, and TAMs in turn supply pro-tumorigenic, pro-growth factors. (B) Dendritic cells (DCs) can present tumor-associated antigen (TAAs) to T cells to promote anti-tumor immune response. However, function of DCs can be disturbed by factors which released into the TME by tumor cells, such as IL-10, TGF-β and prostaglandin E2 (PGE2). (C) Mature neutrophils are abundant in adult human peripheral blood and TME, promoting tumor growth and metastases. Tumor cells also in turn secrete chemokines (e.g., G-CSF, IL-6) to recruit TANs infiltration. NETosis act as scaffolds mediating the capture of cancer cells and providing a soil that can support protumorigenic niche to metastasis-initiating cancer cells. (D) Astrocytes are unique to the CNS and play important roles in mediating tissue-specific communication in the brain tumors including jap with TANs and TAMs. For example, LCN2 drives an inflammatory activation of astrocytes in the brain metastatic niche, via the LCN2-specific receptor, leading to recruitment of LCN2-producing granulocytes to the BrM microenvironment. Granulocyte-derived LCN2 is central to further enhancing neuroinflammation and BrM progression. (E) During effector phases of anti-tumor immunity, T lymphocytes are further educated by the BrM microenvironment to progressively extend non-function. This can occur via PD-1/PD-L1 pathway, defective antigen presentation including low MHC expression, leading to an immunosuppressive microenvironment that is permissive to tumor growth. Created by Biorender.com.
Figure 3.
The Crosstalk between Immune Cellular Components and Tumor Cells in Brain Metastatic Microenvironment. BrM are composed of diverse immune cellular components, including various specialized organ-resident cell types such as astrocyte and microglia. Each of these immune cells contributes to brain metastatic biology in unique ways. (A) Macrophages and microglia engage in essential crosstalk with tumor cells (TC) within the brain metastases, whereby brain tumor cells could release cytokines to recruit TAMs to the brain microenvironment, and TAMs in turn supply pro-tumorigenic, pro-growth factors. (B) Dendritic cells (DCs) can present tumor-associated antigen (TAAs) to T cells to promote anti-tumor immune response. However, function of DCs can be disturbed by factors which released into the TME by tumor cells, such as IL-10, TGF-β and prostaglandin E2 (PGE2). (C) Mature neutrophils are abundant in adult human peripheral blood and TME, promoting tumor growth and metastases. Tumor cells also in turn secrete chemokines (e.g., G-CSF, IL-6) to recruit TANs infiltration. NETosis act as scaffolds mediating the capture of cancer cells and providing a soil that can support protumorigenic niche to metastasis-initiating cancer cells. (D) Astrocytes are unique to the CNS and play important roles in mediating tissue-specific communication in the brain tumors including jap with TANs and TAMs. For example, LCN2 drives an inflammatory activation of astrocytes in the brain metastatic niche, via the LCN2-specific receptor, leading to recruitment of LCN2-producing granulocytes to the BrM microenvironment. Granulocyte-derived LCN2 is central to further enhancing neuroinflammation and BrM progression. (E) During effector phases of anti-tumor immunity, T lymphocytes are further educated by the BrM microenvironment to progressively extend non-function. This can occur via PD-1/PD-L1 pathway, defective antigen presentation including low MHC expression, leading to an immunosuppressive microenvironment that is permissive to tumor growth. Created by Biorender.com.
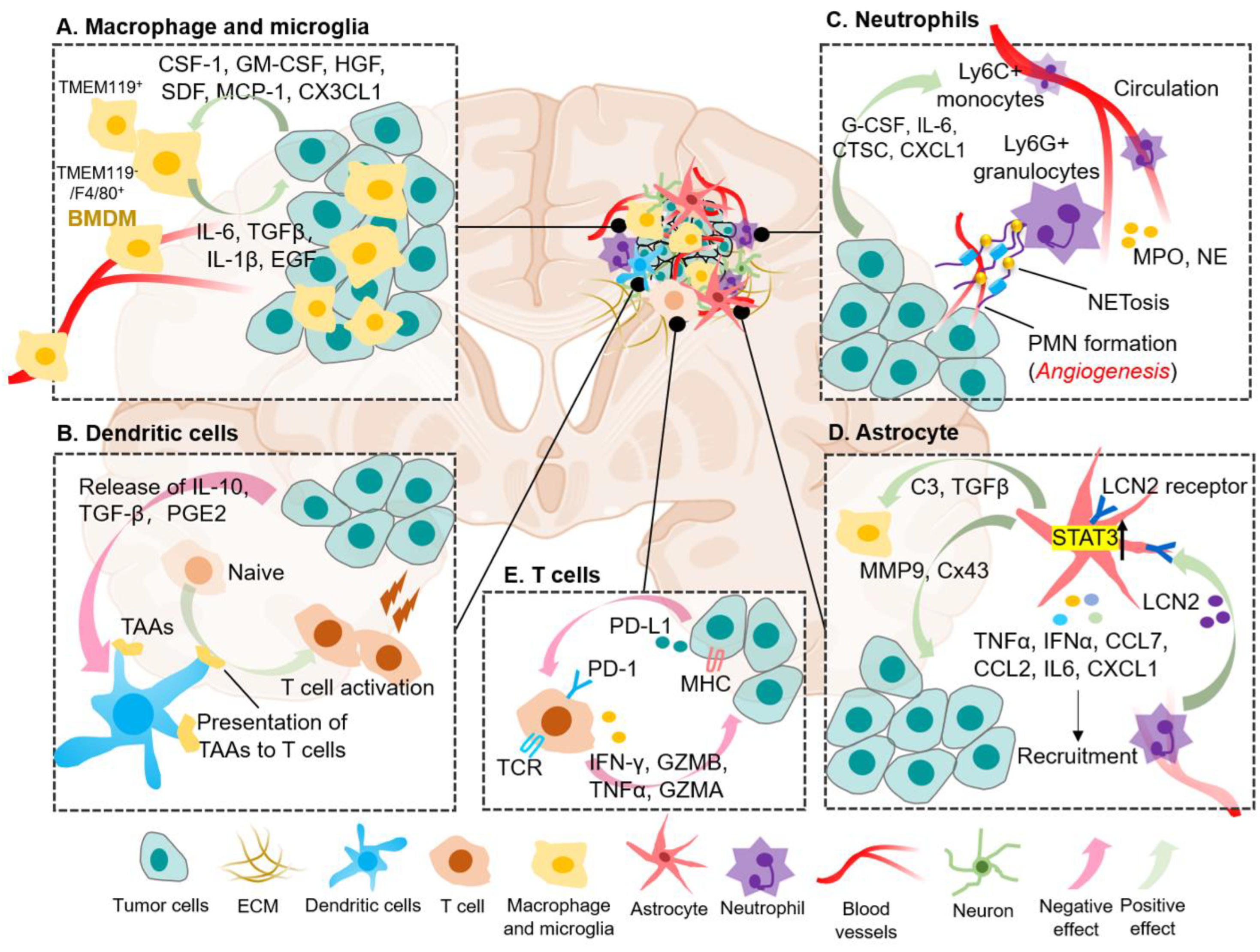

Table 1.
Ongoing key clinical trials of targeted therapies in brain metastasis in various cancers.
| Targeting molecular signatures for personalized treatment of BrM | ||||||
|---|---|---|---|---|---|---|
| Study design | Primary histology | Total cohort size | Molecular signatures | Treatment regimen | Primary endpoints | ClinicalTrial (No.)/ Status |
| Phase I | Solid tumor | 36 | PIK3CA-mut | GDC-0084 plus WBRT | MTD | NCT04192981/ R |
| Phase II | NSCLC | 30 | EGFR-mut | Keynatinib | ORR | NCT04824079/ R |
| Phase II | NSCLC | 47 | EGFR-mut | Almonertinib | CNS-DOR | NCT04643847/ A |
| Phase II | NSCLC | 54 | EGFR-mut | Anlotinib plus Almonertinib | iPFS | NCT04978753/ R |
| Phase II | NSCLC | 40 | EGFR-mut | AZD9291 | iORR | NCT02736513/ A |
| Phase I/II | NSCLC | 43 | KRAS G12C-mut | AMG 510 | Dose exploration and expansion | NCT05180422/ R |
| Phase III | NSCLC | 232 | EGFR-mut | Almonertinib plus brain RT |
OS | NCT05768490/ R |
| Phase II | Solid tumor | 30 | HER2-mut | Tucatinib plus T-DM1 | Intracranial antitumor activity | NCT05673928/ N |
| Phase I | Breast cancer | 10 | HER2 (+) | ExAblate BBBD | Adverse events | NCT03714243/ R |
| Phase II | Breast cancer | 30 | HER2 (+) | T-DM1 | Intracranial antitumor activity | NCT05673928/ R |
| Phase II | Breast cancer | 120 | HER2 (+) | Pertuzumab plus Taxane | ORR | NCT04760431/ N |
| Phase II | Breast cancer | 130 | HER2 (+) | Afatinib plus T-DM1 | Safety and tolerability | NCT04158947/ R |
| Phase II | Melanoma | 150 | BRAFV600-mut | Encorafenib plus Pembrolizumab | iPFS | NCT04074096/ R |
| Phase II | Melanoma | 20 | BRAF-mut | Vemurafenib plus Cobimetinib | ORR | NCT03430947/ A |
| Phase I | Melanoma, NSCLC | 140 | BRAF and RAS/MAPK-mut | BDTX-4933 | Dose Escalation | NCT05786924/ N |
| Phase II | Melanoma | 150 | BRAF-mut | Triple therapy +/- SRS | iPFS | NCT04074096/ R |
| Targeting TME or immune cellular elements for BrM treatment | ||||||
| Study design | Primary histology | Total cohort size | Targeting Elements | Treatment regimen | Primary endpoints |
ClinicalTrial (No.)/ Status |
| Phase II | Renal cell carcinoma | 40 | CD8+T, APCs | Nivolumab plus Ipilimumab | iPFS | NCT05048212/ R |
| Observational | NSCLC | 50 | ctDNA, TCR | WBRT | OS | NCT05737589/ R |
| Phase II | NSCLC | 35 | MET-Amp on cfDNA | Capmatinib | CNS-ORR | NCT05567055/ N |
| Phase II | Melanoma | 76 | CD8+T | Nivolumab plus Ipilimumab | Intracranial response rate | NCT02374242/ A |
| Phase II | Melanoma | 53 | CD8+T, VEGF | Pembrolizumab plus Bevacizumab | BMRR | NCT02681549/ R |
| Phase II | Breast cancer | 100 | VEGF | Utidelone Plus Bevacizumab | CNS-ORR | NCT05357417/ R |
| Phase I/II | Melanoma | 29 | CAFs | CA4948* plus Pembrolizumab | iORR | NCT05669352/ N |
| Phase I | Melanoma | 10 | TILs | Lifileucel (LN-144) | Feasible effect | NCT05640193/ R |
| Phase I/II | Melanoma | 30 | NK | UD TGFβ NK plus Temozolomide | Safety and tolerability | NCT05588453/ R |
| Phase III | NSCLC | 20 | BBB | Exablate plus Pembrolizumab | AEs | NCT05317858/ R |
Index: TIME, tumor immune microenvironment; BrM, brain metastasis; MTD, maximum tolerated dose; WBRT, whole-brain radiotherapy; NSCLC, non-small cell lung cancer; CNS-DOR, central nerve system duration of response (RECIST 1.1); iPFS, intracranial progression-free survival; BBBD, blood-brain barrier disruption; BCBM, breast cancer brain metastasis; T-DM1, tucatinib and Ado-trastuzumab emtansine; ORR, objective response rate; SRS, stereodirectional radiotherapy; Triple therapy, binimetinib plus encorafenib plus pembrolizumab; APCs, antigen-presenting cell; ctDNA, circulating tumor DNA; TCR, T cell receptor; OS, overall survival; BMRR, brain metastasis response rate (mRECIST); CA4948*, IRAK4 inhibitor; VEGF, vascular endothelial growth factor; CAFs, cancer-associated fibroblast; TILs, Tumor-infiltrating lymphocytes; cfDNA, circulating free DNA; CNS-ORR, central nerve system overall response rate; NK, natural killer cell; BBB, blood-brain barrier; AEs, adverse events. R: Recruiting; A: Active, not recruiting; N: Not yet recruiting.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



