
Submitted:
19 June 2023
Posted:
20 June 2023
You are already at the latest version
Abstract
This work reports the influence of reduced graphene oxide (rGO) support on the catalytic performance of Cu@PtRu/rGO catalysts towards methanol oxidation in acidic medium. These core-shell catalysts are synthesized by the reduction method in two steps being the first using ethylene glycol to obtain Cu/rGO and the second, the reduction of copper atoms synthesized in the previous step to produce Cu@PtRu/rGO. In order to compare the catalytic performance of the prepared catalysts, Pt/C, PtRu/C, Cu@PtRu/C catalysts are also synthesized on Vulcan XC-72R carbon. The theoretical loading of Pt used in the Vulcan XC-72R carbon supported catalysts is equal to that utilized in Cu@PtRu/rGO, and higher in the other prepared catalysts. The surface composition of the nanoparticles, as well as their sizes, particle size distribution and morphology of the catalyst particles are characterized by X-ray diffraction (XRD) and high resolution transmission electron microscopy (HRTEM). Cyclic voltammetry (CV) and chronoamperometry (CA) are employed to measure the electrochemical performance of the prepared catalysts. The anodic scans show that for the region of technological interest (0.1 to 0.4 V), the Cu@PtRu/rGO-16 electrocatalyst exhibits higher current density value than the others as a result of its lower starting oxidation potential (0.250V against 0.437V for Pt/C) and its higher ratio between forward (iF) and reverse (iR) current densities (iF/iR 1.45 against 0.93 for Pt/C). Chronoamperometric tests depict that the current density value of Cu@PtRu/rGO-16 catalyst is about 4 times higher than that of Cu@PtRu/C-16 and Cu@PtRu/C-18 catalysts, ca. 4.8 times higher than that of PtRu/C-30 catalyst and around 6 times higher than that of Pt/C-30. These results suggest that Cu@PtRu/rGO-16 exhibits high bulk activity for the electrooxidation of methanol, high stability and high tolerance to CO poisoning, making it possible to reduce the platinum loading in proton exchange membrane fuel cells (PEMFCs).
Keywords:
Electrocatalysts
; direct methanol fuel cell
; reduced graphene oxide
1. Introduction
Direct methanol fuel cells (DMFCs) have storage, volumetric energy density and cost advantages over cells that use hydrogen as fuel. Thus, DMFCs are promising candidates as a source of energy for transport and electronic devices [1,2,3,4]. However, the efficiency of fuel cells depends on appropriate catalysts in the electrodes [5,6,7]. In this sense, PtRu nanoparticles based on carbonaceous materials are considered the best anodic catalyst for the electroxidation of methanol [8,9,10,11]. Two issues limit the practical applications of PtRu catalyst: the high cost of platinum and the low poison resistance caused by adsorbed intermediates [3,12].
Recent researches have sought the preparation of catalysts with low Pt loading and resistant to intermediate species that hinder the DMFCs performance. The alternative found so far to circumvent these issues is the change in the nanoparticles morphology of the catalysts; in this new arrangement, Pt is not inserted in the bulk crystal lattice, but makes an envelope on the core comprised of metal or low cost metallic alloy. These core-shell nanostructures are the most efficient manner to reduce the use of precious metals in electrocatalysts [11,12,13,14]. In literature the most common supports for these nanostructures are Vulcan Carbon XC-72R and carbon nanotubes, being graphene and its derivatives (GO and rGO) mentioned in several technological applications. However, to date, there are few papers in which graphene and its derivatives are employed as supports for catalysts used in alcohol oxidation reactions [15,16,17,18,19]. Therefore, it is proposed the utilization of rGO support to improve the performance of Cu@PtRu core-shell electrocatalysts. The reasons for choosing this catalyst in rGO are based on: (i) Cu is a low cost material, its positive potential makes it stable in acidic medium, in addition to its face-centered cubic (FCC) crystalline form, the same as Pt, favoring the epitaxial growth of platinum on the surface of its nanoparticles [14]; (ii) Pt and Ru are well known as the most promising alloys for anodic catalysis for methanol oxidation in acidic media [20,21,22]; and (iii) catalysts supported on materials derived from graphene have higher electrochemically active surface area and higher electronic conductivity than those of other carbon materials [12,17]. The prepared Cu@PtRu/rGO catalyst increases electrocatalytic activity for methanol oxidation when compared to that of Pt/C, PtRu/C, and Cu@PtRu/C, being a promising candidate material for DMFCs.
2. Experimental details
2.1. Preparation of reduced graphene oxide (rGO)
Obtaining graphene oxide was based on the modified Hummers method [15]. In a 500 mL beaker, 0.1 g of graphene and 0.075 g of NaNO3 were added, followed by 3.44 mL of concentrated sulfuric acid under slow stirring to avoid overheating. Subsequently, 0.450g of KMnO4 (KMnO4, 100% Merck) was added to the mixture, still under constant stirring, at room temperature. After all KMnO4 addition, the reagents continued under constant agitation for 24h. 10 ml H2SO4 5% m/m was then added and, after 2h, 0.9 ml H2O2 30% m/m. Due to the tendency of the stirring to stop as the viscosity of the mixture increases with time, 40 ml of 30% m/m H2SO4 + 5% m/m H2O2 was slowly added over the 5 days of synthesis.
The formation of a yellowish suspension was observed and a dark brown material was deposited at the bottom of the vessel. 30 ml of HCl (3%) was added to the suspension to remove impurities. The material was then washed extensively with ultrapure water. The obtained GO was placed to dry in an oven at a temperature of 80°C for 12 h.
The material synthesized previously was reduced to rGO in the first step of synthesis of the electrocatalysts, the step that uses ethylene glycol as a reducing agent for cupric ions in solution. The material obtained after the reduction process was Cu/rGO, which was then washed extensively with ultrapure water.
2.2. Preparation of Cu@PtRu/rGO catalysts
Core-shell Cu@PtRu/C catalysts were prepared by two-step reduction method. Two types of carbon support were used, Vulcan Carbon XC72R and GO, respectively. In the first step, initially 70 mg of carbon powder was suspended in a solution of 8 ml of ultrapure water (18.2 MΩ cm) and 2 ml of isopropyl alcohol, and stirred under sonication for 20 minutes. Then, 30 mg of Cu from a penta-hydrated copper sulfate solution (CuSO4 5.H2O) and 2 ml of ethylene glycol were added to a becker containing the previously GO suspension. The pH was adjusted to 10 with potassium hydroxide/ethylene glycol solution (5% w/w KOH/EG), and the mixture was refluxed for 6 hours at 140°C. Subsequently, the Cu/rGO system was filtered, washed excessively with ultrapure water and placed in an oven for 12 h at a temperature of 60ºC.
In the second step of the synthesis process for each catalyst, solutions with varying volumes of H2PtCl6.6H20 and RuCl3 xH2O were used. The mass proportions were 16mg of Pt and 8.3mg of Ru in Cu@PtRu/C-16 and in Cu@PtRu/rGO-16 catalysts, 18mg of Pt and 9.2mg of Ru in Cu@PtRu/C-18 , 30mg of Pt and 15.2mg of Ru in PtRu/C-30, and finally, 30mg of Pt in Pt/C-30. In each synthesis, the volumes containing these masses were added to the Cu/C suspension, this final mixture was stirred for 15 minutes under sonication. After stirring, the pH value of the solution was adjusted to 8 by adding 0.1mol.L-1 KOH. Then, the mixture was refluxed at 100°C. After reflux, it was filtered, and the Cu@PtRu/C obtained was washed excessively with ultrapure water to remove traces of the reducer or ions from the salt or acid used. Finally, it was placed in an oven for 12 h at 60°C.
2.3. Physical characterization
The catalysts were characterized by XRD and HRTEM. X-ray measurements were made on a PANalytical X-ray Diffractometer, model X'PERT PRO MPD (PW 3040/60). A copper anode monochromatic X-ray beam (Kα 1.540598 Ǻ) was used. The recordings were performed in the 2θ interval between 10 and 110º, at a scan of 2º per minute. The characteristic patterns of the samples were identified by comparison with patterns from a database that was developed by the Joint Committee on Powder Diffraction Standards (JCPDS). The HRTEM images were acquired in a transmission electron microscope, model TECNAI G2F2. Operating at 200kV beam power, in scan transmission mode (STEM) for HAADF microanalysis and imaging.
2.4. Electrochemical analyses
The electrochemical measurements were carried out in a glass electrochemical cell with no separation among the compartments, with five openings at the top for inlets of the working electrode. The counter electrode consisted of a platinum wire with a Pt plate in contact with the solution and the reference electrode was Ag/AgCl/KCl(3M). The working electrode was a glassy carbon disc (0.196 cm2). The thin film electrode was prepared as follows: 5 mg of catalyst was dispersed in 1 ml of isopropanol and 1 ml of water. 60 µL of Nafion (0.25% Nafion) was added and then the mixture was subjected to sonication for 15 min. 60µL of the dispersion was transferred to the glassy carbon disk using a pipette and finally, the electrode was placed in an oven until complete drying. The catalysts were characterized by cyclic voltammetry (CV) and chronoamperometry (CA) tests at room temperature. Current densities were measured in terms of A cm-2 by normalizing the electrochemical surface area of the electrode. Oxidation reactions (OR) were studied in 0.5 M H2SO4 solution saturated with N2 for 10 minutes.
3. Results and discussion
3.1. Characterization
3.1.1. X-ray analyses
The Cu@PtRu/C structure is confirmed by XRD which shows partial core-shell formation for Cu@PtRu/C-16 and Cu@PtRu/C-18, and full core-shell formation for Cu@PtRu/rGO-16 crystallites. These structures are a consequence of the substitution of atoms on the surface of Cu nanoparticles by Pt and Ru atoms. Figure 1 shows the XRD pattern for Pt/C-30, PtRu/C-30, Cu@PtRu/C-16, Cu@PtRu/C-18, and Cu@PtRu/rGO-16. For all PtRu-containing catalysts there is a slight shift in relation to the Pt/C-30 diffraction peak in the (111) plane. These shifts to values higher than 2Ɵ are indicative of the formation of PtRu alloy, either directly on the support, or in the Cu crystallites. The formation of PtRu can also be inferred from the values calculated for the lattice parameters of the PtRu catalysts that present lower values in relation to the monatomic Pt alloy. The first peak located at about 2Ɵ = 25º for Pt/C-30, PtRu/C-30, Cu@PtRu/C-16, and Cu@PtRu/C-18 is associated with the face (002) of the crystal structure of the carbon. The absence of this peak is noticed in Cu/rGO and Cu@PtRu/rGO-16 catalysts, and is associated with the efficiency in the process of exfoliation of the graphite sheets by chemical routes. Exfoliation increases the distance among the graphite planes as the oxidation process proceeds, which according to Scherrer equation (d=kλ/βcosƟ) implies in smaller angles. The other three peaks at 40°, 47° and 68° are associated with the (111), (200) and (220) planes, characteristic of face-centered cubic crystal alloys (fcc) of Pt [23,24,25,26].
As can be seen in Figure 1, the crystallinity of Cu@PtRu/C is high and the presence of peaks characteristic of the Pt structure suggests that the layer covering the core is formed, or specifically, in this case, implies that the average size of the PtRu alloy on the surface of Cu nanoparticles is higher than 1 nm [27,28,29,30,31,32,33]. The reason for the statement that the Cu@PtRu/rGO-16 nanoparticles (NPs) forms a perfect core-shell system is the absence of the standard diffraction peaks characteristic of the indication of Cu atoms and their oxides at 2Ɵ equal to 36º and 43º. The presence of these peaks for the Cu@PtRu/C-16 and Cu@PtRu/C-18 alloys suggests that the coating of Cu NPs in these alloys is only partial. The average size of the crystallites is 3.6 nm (Cu/rGO), 3.8 nm (Pt/C -30), 3.4 nm (PtRu/C-30), 4.9 nm (Cu@PtRu/C-16), 4.4 nm (Cu@PtRu/C-18) and 4.5 nm (Cu@PtRu/rGO-16)
3.1.2. HRTEM Results
The core-shell structures have been also characterized by HRTEM in order to compare with the XRD results and visualize changes in the distances between the crystallographic planes due to the substitution of atoms on the NPs surfaces that form the nuclei. Figure 2A–E display representative images obtained using HRTEM. From these images, the spherical shape is assumed for the Cu/rGO and Cu@PtRu/rGO-16 NPs and a regular dispersion of these NPs on the rGO support is observed. The average diameter measured for the Cu/rGO NPs is 3.7 nm and for the Cu@PtRu/rGO-16 NPs is 4.9 nm, suggesting that a layer with a size above 1nm of PtRu is deposited on the surface of the Cu particles. The growth of Pt alloys on the surface of copper NPs can be understood as an expansion of the copper crystal lattice, where in general the outer layer is favored by two factors: the higher oxidation potential of constituent metals of the core in relation to that of the metals concerning the covering layer, and because this layer has the same crystalline arrangement of the core atoms [34,35,36,37,38].
The PtRu formation on Cu NPs is the result of charge transfer at the Cu/solution interface, and the increase in size observed by HRTEM of Cu/rGO NPs is attributed to the reduction of PtCl42- and Ru+4 species. This reduction is favored over the Cu core because the interaction between the metal ions and the copper atoms is stronger than the interaction between them and the carbon support. Another factor favorable to the growth of the PtRu layer on the copper core is the crystalline arrangement of Pt and Cu, both FCC. From Figure 2A,B, it is possible to observe, in darker contrast, the formation of agglomerates of Cu/rGO particles resulting from the employed synthesis method. Regularly distributed regions are represented by NPs in less dark contrast. Figure 2D depicts a better distribution in the support for the Cu@PtRu/rGO-16 NPs, suggesting that the second step of the synthesis is a stage of reconfiguration of the particles distribution on the support.
The brightness contrast between the core and shell regions observed in some Cu@PtRu/rGO NPs in Figure 2D is commonly attributed to the separation between the Cu core, the darkest region, and the PtRu shell, the brighter region. However, the boundary between the core and the shell is not well defined in the images and the separation between the phases is better defined by measuring the interplanar distance of the crystal constituting the core and subsequently the formed core-shell. The lattice planes of the Cu/rGO-30 NPs in Figure 2C exhibit an interplanar spacing of 0.2206 nm, corresponding to the (111) face of the copper alloy. In this case, a single particle is chosen arbitrarily. In Figure 2E, the distance measured between the two closest rows of atoms for the Cu@PtRu/rGO-16 NPs is 0.2317 nm, referring to the orientation of the (111) crystalline plane of the FCC structure of platinum. According to the XRD results that show the absence of standard peaks characteristic of the copper crystal structure, it can be concluded that the distance between the planes in Figure 2E refers to the PtRu alloy.
Eighty particles are randomly measured to obtain the average particle size distribution of Cu@PtRu/rGO-16. The HRTEM results suggest that, in some cases, discrete Pt and PtRu particles are formed, however, this statement refers to a few measurements of particles having sizes below the measured values for most Cu@PtRu/rGO-16 NPs obtained either by HRTEM or by the Scherer equation.
3.1.3. Cyclic voltammetry
The electrochemical performance of Pt/C-30, PtRu/C-30 Cu@PtRu/C-16, Cu@PtRu/C-18 and Cu@PtRu/rGO-16 catalysts have been investigated by cyclic voltammetry in sulfuric acid, 0.5 mol.L-1H2SO4, at 20 mV/s. The cyclic voltammograms of the catalysts are shown in Figure 3. Each voltammogram is obtained after 22 cycles under N2 atmosphere, in order to obtain a more stable response from the electrode surface.
The currents of the cyclic voltammetry and chronoamperometry curves of all electrocatalysts have been normalized using the oxidation method of a monolayer of CO adsorbed on the electrodes surface. This method is used to compare the electrocatalytic activity of different electrodes, taking into account the number of surface active sites, i.e., it is an evaluation of the electrocatalytic activity intrinsic to the real area of these electrodes. The basis of this method is the fact that carbon monoxide molecules occupy, in principle, all active sites available for electrocatalytic reactions. The charge value of the conversion of COads to CO2(g) [QCO(C)] in microColoumbs (µC) divided by 420 µC is numerically equal to the electrochemically active area (EAA), defined by equation (I). The EAA values obtained for each electrode are used as normalization factors for the currents that are now expressed in A.cm-2 units. The CO amount is obtained by integrating the voltammetric peak (shaded area), as shown in Figure 4 [39].
The adsorption and oxidation of methanol occurs in few materials, in acidic medium, only in platinum and platinum alloys. In electrochemical systems, interest is the oxidation of methanol on the anodic substrate. This process goes through different stages depending on the applied potential values [40,41,42].
Despite containing a single carbon atom in its molecule, methanol has electro-oxidation in the presence of PtRu electrocatalysts in a series of steps which chemical equations below show only the additional reactions suggested by Hamnett [43]. Other similar proposals can be seen in other works dealing with the methanol oxidation mechanism [44,45].
There are two more relevant items regarding the mechanism of methanol oxidation. The first is that methanol adsorption takes place on energetically favored sites on the surface. The second is the occurrence of sequential loss of protons to give rise to a sequence of multiply bonded hydrogenated intermediates that convert to linearly adsorbed CO. This evidence comes from the results obtained by DEMS (Differential Electrochemical Mass Spectrometry) and FTIR (Fourier transform infrared) [46,47].
The adsorbed CO molecules (2) resulting from the partial oxidation of methanol are undesirable, since they adsorb on the surface of the platinum atoms where fuel molecules must oxidize; this poisoning of the catalytic surface results in a decrease in the activity of the electrocatalyst [48].
To circumvent the problem of poisoning the catalyst by CO, two effects can be considered from the formation of the metallic alloy of Pt with a second metal, in particular with Ru: the bifunctional mechanism and the electronic effect act together, leading the CO to oxidize at low potentials [13]. In addition, there is the free energy that originates at the interface between the core and the layer that covers it. The core-shell morphology presents an anisotropy of forces at its interface, this inequality of forces on the atoms at the phase boundary is caused by the different electronic arrangements of the atoms in that region, by the difference in crystalline structures and, in the case of the same structure, difference in the lattice parameter of the crystalline arrangement adopted by NP. The sum of these factors causes the chemical potentials to be different in the different phases of the core-shell structure, which will cause this system to release energy at appropriate potentials, in order to reach a lower energy configuration. The synergy of these effects are probably the main factors contributing to more CO tolerant PtRu catalysts [49,50,51,52].
In Figure 5, the curves obtained for the anodic scans referring to the oxidation of methanol on Pt/C-30, PtRu/C-30, Cu@PtRu/C-16, Cu@PtRu/C-18, and Cu@PtRu/rGO-16 electrocatalysts are superimposed. These curves have been obtained in solutions containing 0.5 M CH3OH plus 0.5 M H2SO4, at 20 mV/s, in the potential range between -0.2 and 0.8 V vs. Ag/AgCl.
The anodic scans shown in Figure 5 display that for the region of technological interest (0.1 to 0.4 V), the Cu@PtRu/rGO-16 electrocatalyst depicts higher current density value than the others. Similarly, its cyclic voltammograms in 0.5 M H2SO4 solution show higher current density values. The second best is Cu@PtRu/C-16, followed by Cu@PtRu/C-18, both with core-shell structure. Although the Cu@PtRu/C-16 and Cu@PtRu/C-18 catalysts show lower current densities than that of Cu@PtRu/rGO-16, their current densities are higher than those of Pt/C-30 and PtRu/C-30.
Starting oxidation potential (SOP) is an important criterion used to evaluate and compare the catalytic activities of electrodes. The values measured for the SOP of each catalyst are shown in Table 1. The oxidation-reduction peaks of the Cu@PtRu/rGO-16 catalyst are lower than all others. Cu@PtRu/C-16, Cu@PtRu/C-18 have lower SOP values than those of Pt/C-30 and PtRu/C-30. The PtRu/C electrocatalyst, although it has lower SOP value than that of Pt/C, has a higher value than the core-shell morphology electrocatalysts. Among the core-shells, Cu@PtRu/C-16 displays the lowest value in relation to Cu@PtRu/C-18. Among all tested electrocatalysts, Cu@PtRu/rGO-16 exhibits the lowest SOP value, 0.250 V. Thus, the order of catalytic activities, by the SOP value criterion, is: Pt/C < PtRu/C-30< Cu@PtRu/C-18 < Cu@PtRu/C-16 < Cu@PtRu/rGO-16.
The better performance of Cu@PtRu/rGO-16 electrocatalyst in relation to the others for methanol oxidation can be attributed to its high active surface, the higher number of oxygen-containing groups attached to the ruthenium atom (carboxyl, hydroxyl, etc.), the bifunctional mechanism and the electronic effect, and to its efficient exfoliation process or the higher thermal and mechanical stability of its carbon support [53,54].
The ratio between forward (iF) and reverse anodic (iR) peak current densities can be used to describe the catalyst's tolerance to catalytic poison accumulation [55,56]. A high iF/iR value indicates more effective removal of species that poison the catalyst surface. Table 1 shows the calculated values for iF, iR and iF/iR.
The Pt/C-30 (1.80 A/cm2) and PtRu/C-30 (1.96 A/cm2) electrocatalysts demonstrate the lowest iF values in relation to the Cu@PtRu/C-16 (2.79 A/ cm2) and Cu@PtRu/C-18 (2.89 A/cm2). The iF for Cu@PtRu/rGO-16 electrocatalyst (2.68 A/cm2) is lower than that of the other core-shell systems, however, it possesses the lowest iR (1.85 A/cm2) compared to the others, except for PtRu/C-30 (1.78 A/cm2). These results show that the Cu@PtRu/rGO-16 electrocatalyst has the highest iF/iR value (1.45 A/ cm2) among all the studied ones herein. In general, the iF/iR ratio is a measure of the efficiency of the bifunctional mechanism and the electronic effect on the electrode kinetics.
3.1.4. Chronoamperometry measuments for methanol electrooxidation
Chronoamperometric experiments have been carried out in solution of 0.5 M methanol plus 0.5 M H2SO4 to observe the stability of the catalysts over time at constant potential. The results are shown in Figure 6.
The results obtained by chronoamperometry demonstrate that the PtRu/C-30, Cu@PtRu/C-16, Cu@PtRu/C-18 and Cu@PtRu/rGO-16 systems exhibit higher current density values than that of the standard Pt/C, after 20 minutes of operation in a potential value of 500 mV. This fact is probably due to the bi-functional mechanism and/or the electronic effect resulting from the presence of ruthenium in the alloy. However, the core-shell electrocatalysts show higher stability and higher current density values than those of PtRu/C-30 and the conventional Pt/C-30, again indicating the beneficial effect of the core-shell structure for the electrocatalysts performance.
The current density value of Cu@PtRu/rGO-16 catalyst is about 4 times higher than that of Cu@PtRu/C-16 and Cu@PtRu/C-18 catalysts. In addition, it is about 5 times higher than that of PtRu/C-30 catalyst and around 6 times higher than that Pt/C-30. These results suggest that Cu@PtRu/rGO-16 exhibits high long-term bulk activity for methanol electrooxidation, high stability, and high tolerance to CO poisoning. The lower instability of Cu@PtRu/C-16 and Cu@PtRu/C-18 catalysts may be related to the pseudo core-shell morphology that allows the presence of copper and its oxides on the surface of the NPs, which according to the XRD results, does not take place in Cu@PtRu/rGO-16. The chronoamperometric results are further supported by the HRTEM images that display a regular distribution of their NPs over the rGO support.
In Cu@PtRu/C-16 and Cu@PtRu/C-18 electrocatalysts, the irregular formation of the layers that cover the core suggests that PtRu is reduced on the support in the form of a bimetallic alloy or in the form of monatomic Pt and Ru alloys. In the latter case, the absence of the bifunctional mechanism and the electronic effect on dispersed Pt NPs significantly affect the performance of these catalysts.
The following order of catalytic activities can be assigned approximately after 20 minutes of reaction: Pt/C < PtRu/C-30< Cu@PtRu/C-18 < Cu@PtRu/C-16 < Cu@PtRu/rGO-16. The results obtained by chronoamperometry agree with those obtained by cyclic voltammetry in 0.5 M H2SO4 solution, in the sense that the best electrocatalyst is Cu@PtRu/rGO-16 and the worst is Pt/C.
The higher current density values for Cu@PtRu/rGO-16 in relation to the other electrocatalysts, suggest that the surface tension and other characteristic properties of the interface may be the cause of its better performance. This energy originating at the core-shell interface may, as well as the electronic effect, be associated with the weakening of the Pt-COads bonds on the surfaces of the core-shell NPs. Thus, the free energy released at the phase boundary inside NPs with core-shell morphology, added to the electronic effect and the bi-functional mechanism, may be the cause of the greater tolerance to CO, and the consequent increase in the electrocatalytic activity of electrocatalysts with core-shell morphology. The preponderance of one of these effects depends on the potential range considered. In fact, the increased tolerance of the PtRu alloy to CO has the effect of the preponderant ligand at potentials below 0.3V, while the bifunctional mechanism at potentials above 0.3V [57,58,59,60].
The higher values of electrical current densities for Cu@PtRu/rGO-16 must be associated with the higher conductivity of rGO compared to Vulcan XC72R, thus, rGO support is crucial for the best performance of the Cu@PtRu/rGO-16 electrocatalyst.
4. Conclusions
In summary, it has been demonstrated that the core-shell/rGO combination is superior in catalytic activity to the traditional Pt/C, PtRu/C and Cu@PtRu/C catalysts for the methanol oxidation reaction. The core-shell/rGO structure has been confirmed by XRD and HRTEM. The lower activity of the core-shell catalyst with incomplete layer (pseudo core-shell) and supported on Vulcan Carbon is related to this partial encapsulation and the properties of the support. In the case of partial encapsulation, the formation of Pt crystals outside the PtRu alloy made its best use impossible. It is also concluded that the catalytic activity of Cu@PtRu/C catalyst can indeed be improved by using rGO as a support instead of Carbon Vulcan XC-72. Furthermore, considering that the Cu core significantly decreases the Pt loading on the catalyst, Cu@PtRu/rGO is a promising candidate material for large-scale fabrication in electrocatalysis. Cu@PtRu/rGO nanostructures have excellent catalytic properties and their syntheses are simple, which paves the way for their utilization in fuel cell technologies.
Acknowledgments
The authors would like to thank Federal University of Pará and Capes for financial support. Also, the X-ray diffractometry and transmission electron microscope laboratories from Chemistry Institute of São Carlos (USP) are grateful acknowledged for the measurements.
References
- Cheng, Y.; Zhang, J.; Lu, S.; Jiang, S.P. Significantly enhanced performance of direct methanol fuel cells at elevated temperatures. Journal of Power Sources 2020, 450, 1–7. [Google Scholar] [CrossRef]
- Sun, X.; Yang, C.; Xia, Z.; Qi, F.; Sun, H.; Sun, G. Molecular sieve as an effective barrier for methanol crossover in direct methanol fuel cells. International Journal of Hydrogen Energy 2020, xx, 1–10. [Google Scholar] [CrossRef]
- Guo, J.; Zhang, M.; Xu, J.; Fang, J.; Luo, S.; Yang, C. ; Core–shell Pd–P@Pt–Ni nanoparticles with enhanced activity and durability as anode electrocatalyst for methanol oxidation reaction. RSC advances 2022, 12, 2246–2252. [Google Scholar] [CrossRef] [PubMed]
- Pereira, P. A.; Andrade, J. B. Fontes, reatividade e quantificação de metanol e etanol na atmosfera. Química Nova 1998, 21, 744–754. [Google Scholar] [CrossRef]
- Liu, M.; Zhao, Z.; Duan, X.; Huang, Y. Nanoscale Structure Design for High-Performance Pt-Based ORR Catalysts. Advanced Materials 2018, 31, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Abdelkareem, M. A.; Sayed, E. T.; Nakagawa, N. Significance of diffusion layers on the performance of liquid and vapor feed passive direct methanol fuel cells. Energy 2020, 209, 1–11. [Google Scholar] [CrossRef]
- Sekar, A.; Metzger, N.; Rajendran, S.; Elangovan, A.; Cao, Y.; Peng, F.; Li, X.; Li, J. PtRu Catalysts on Nitrogen-Doped Carbon Nanotubes with Conformal Hydrogenated TiO2 Shells for Methanol Oxidation. ACS Applied Nano Materials 2022, 5, 3275–3288. [Google Scholar] [CrossRef]
- Xu, H.; Shang, H.; Wang, C.; Du, Y. Recent Progress of Ultrathin 2D Pd-Based Nanomaterials for Fuel Cell Electrocatalysis. Small 2021, 17, 1–25. [Google Scholar] [CrossRef]
- Mohanapriya, S.; Gopi, D. Microwave assisted synthesis of core-shell Ni-Co/graphene nanosheets and their catalytic activity for methanol electro-oxidation. Materials Today 2022, 51, 1797–1881. [Google Scholar] [CrossRef]
- Wang, X.; Li, H.; Feng, H.; Wang, H.; Lei, Z. Preparation of carbon-supported core@shell PdCu@PtRu nanoparticles for methanol oxidation. Journal Power Sources 2010, 195, 1099–1102. [Google Scholar] [CrossRef]
- Lee, D.; Gok, S.; Kim, Y.; Sung, Y. E.; Lee, E.; Jang, J. H.; Lim, T. Methanol Tolerant Pt–C Core–Shell Cathode Catalyst for Direct Methanol Fuel Cells. ACS Applied Materials & Interfaces 2020, xx, 4-10. [CrossRef]
- Salgado, J. R. C.; Alcaide, F.; Álvarez, G.; Calvillo, L.; Lázaro, M. J.; Pastor, E. Pt-Ru electrocatalysts supported on ordered mesoporous carbon for direct methanol fuel cell. Journal power Sources 2010, 195, 4022–4029. [Google Scholar] [CrossRef]
- Christoffersen, E.; Liu, P.; Ruban, A.; Skriver, H.; Norskov, J. Anode Materials for Low-Temperature Fuel Cells: A Density Functional Theory Study. Journal of Catalysis 2000, 199, 123–131. [Google Scholar] [CrossRef]
- Sang, Y.; Zhang, R.; Xu, B.; Yang, J.; Zhao, C.; Xu, H. Ultrafine and Highly Dispersed PtRu Alloy on Polyacrylic Acid-Grafted Carbon Nanotube@Tin Oxide Core/Shell Composites for Direct Methanol Fuel Cells. ACS Applied Energy Materials 2022, xx, 1–9. [Google Scholar] [CrossRef]
- Yola, M. L.; Eren, T.; Atar, N.; Saral, H.; Ermiş, İ. Direct-methanol Fuel Cell Based on Functionalized Graphene Oxide with Mono-metallic and Bi-metallic Nanoparticles: Electrochemical 2015, xx, 1-9. [CrossRef]
- Li, Y.; Gao, W.; Ci, L.; Wang, C.; Ajayan, P. M. Catalytic performance of Pt nanoparticles on reduced graphene oxide for methanol electro-oxidation. Carbon 2010, 48, 1124–1130. [Google Scholar] [CrossRef]
- Lee, S. H.; Kakati, N.; Jee, S. H.; Maiti, J.; Yoon, Y. S. Hydrothermal synthesis of PtRu nanoparticles supported on graphene sheets for methanol oxidation in direct methanol fuel cell. Materials Letters 2011, 65, 3281–3284. [Google Scholar] [CrossRef]
- Zhang, Y.; Guo, Z.; Zhu, H.; Xing, W.; Tao, P.; Shang, W.; Fu, B.; Song, C.; Hong, Y.; Dickei, M. D. Synthesis of Liquid Gallium@Reduced Graphene Oxide Core–Shell Nanoparticles with Enhanced Photoacoustic and Photothermal Performance. Journal of the American Chemical Society 2022, 144, 6779–6790. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.; Li, S.; Zhang, L.; Yu, T.; Li, F.; Zhang, W.; Wang, J.; Zhang, D.; Yu, Y.; Li, X. Reduced Graphene Oxide/Ni Foam Supported ZIF-67 Derived CuCo2S4@CoS2 Core-Shell Heterostructure for Boosted Electrochemical Energy Storage. Journal of Energy Storage 2022, 47, 103–112. [Google Scholar] [CrossRef]
- Roh, G.; Lee, H.; Jeong, Y.; Kim, J. H. Preparation of Carbon-Supported PtRu Core-Shell Nanoparticles Using Carbonized Polydopamine and Ozone for a CO Tolerant Electrocatalyst. International Journal of Hydrogen Energy 2019, 44, 21588–21596. [Google Scholar] [CrossRef]
- Wu, Y. N.; Liao, S. J.; Liang, Z. X.; Yang, L. J.; Wang, R. F. High-Performance Core-Shell PdPt@Pt/C Catalysts via Decorating PdPt Alloy Cores with Pt. Journal of Power Sources 2009, 194, 805–810. [Google Scholar] [CrossRef]
- Patra, S.; Munichandraiah, N. Electrooxidation of Methanol on Pt-Modified Conductive Polymer PEDOT. Langmuir 2009, 25, 1732–1738. [Google Scholar] [CrossRef] [PubMed]
- Mcbreen, J.; Mukerjee, S. In situ X-ray Absorption Studies of a Pt-Ru Electrocatalyst. Journal of the Electrochemical Society, 1995, 142, 3399-3404. [CrossRef]
- Mukerjee, S. In situ X-ray Absorption Studies of Carbon-Supported Pt and Alloy Nanoparticles. Journal of Electroanalytical Chemistry 2003, 151, 151,501–509. [Google Scholar] [CrossRef]
- Rigsby, M. A.; Zhou, W. P.; Lewera, A.; Duong, H. T.; Bagus, O. S.; Jaegermann, W.; Hunger, R.; Wieckowski, A. Experiment and Theory of Fuel Cell Catalysis: Methanol and Formic Acid Decomposition on Nanoparticle Pt/Ru. Journal of Physical Chemistry 2008, 112, 15595–15601. [Google Scholar] [CrossRef]
- Neto, A. O.; Linardi, M.; Gonzales, E. R. Oxidação eletroquímica do metanol sobre partículas de PtRu e PtMo suportadas em carbono de alta área superficial. Eclética Química 2003, 28, 55-62. [CrossRef]
- Minghang, J.; Xiao, L.; Wenjun, H.; Mengyu, G.; Liangqing, H.; Hongmei, H.; Huanhuan, Z.; Fei, X.; Li, M. Fe2O3@FeP core-shell nanocubes/C composites supported irregular PtP nanocrystals for enhanced catalytic methanol oxidation. Electrochimica Acta 2019, 323, 1–11. [Google Scholar] [CrossRef]
- Mekazni, S. D.; Arán-Ais, R. M.; Ferre-Vilaplana, A.; Herrero, E. Why Methanol Electro-oxidation on Platinum in Water Takes Place Only in the Presence of Adsorbed OH. ACS Catalysis 2022, 12, 1965–1970. [Google Scholar] [CrossRef]
- Liu, H.; Sun, F.; Chen, M.; Wang, H. Reconciling the experimental and computational methanol electro-oxidation activity via potential-dependent kinetic mechanism analysis. Journal of Materials Chemistry A 2022, 10, 23551–23561. [Google Scholar] [CrossRef]
- Antolini, E.; Cardellini, F. Formation of carbon supported alloy: an XRD analysis. Journal of Alloy and Compounds 2001, 315, 118–127. [Google Scholar] [CrossRef]
- Perrozzi, F. ; Prezioso, S. ; Ottaviano, L. Graphene oxide: from fundamentals to applications to cite this article: Journal of Physics Condensed Matter 2015, 27, 1-21. [CrossRef]
- Hsin, Y. L.; Hwang, K. C.; Yeh, C. T. Poly(vinylpyrrolidone)-Modified Graphite Carbon Nanofibers as Promising Supports for PtRu Catalysts in Direct Methanol Fuel Cells. Journal of the American Chemical Society 2007, 129, 9999–10010. [Google Scholar] [CrossRef]
- Abdolhosseinzadeh, S.; Asgharzadeh, H.; Seop Kim, H. Fast and fully-scalable synthesis of reduced graphene oxide. Scientific Reports 2015, 5, 1–7. [Google Scholar] [CrossRef]
- Hummers, W. S.; Offeman, R. E. Preparation of Graphitic Oxide. Journal of the American Chemical Society 1958, 80, 1339. [Google Scholar] [CrossRef]
- Mohanaprya, S.; Gopi, D. Microwave assisted synthesis of core-shell Ni-Co/graphene nanosheets and their catalytic activity for methanol electro-oxidation. Materials today 2022, 51, 1797–1881. [Google Scholar] [CrossRef]
- Wu, Q.; Huang, X.; Wang, T.; Xiang, D.; Li, X.; Wang, K.; Yuan, X.; Li, P.; Zhu, M. Enhancing Electrocatalytic Methanol Oxidation on PtCuNi Core–Shell Alloy Structures in Acid Electrolytes. Inorganic Chemistry 2022, 61, 2612–2618. [Google Scholar] [CrossRef] [PubMed]
- Ferrarelli, G.; Giordano, G.; Migliori, M. ZSM-5@Sil-1 core shell: Effect of synthesis method over textural and catalytic properties. Catalysis Today 2022, 390, 176–184. [Google Scholar] [CrossRef]
- Zhu, H.; Li, X.; Wang, F. Synthesis and characterization of Cu@Pt/C core-shell structured catalysts for Exchange membrane fuel cell. Journal of Hydrogen Energy 2011, 36, 9151–9154. [Google Scholar] [CrossRef]
- Frelink, T.; Visscher, W.; Cox, A. P.; Van Veen, J. A. R. Ellipsometry and dems study of the electrooxidation of methanol at Pt and Ru- and Sn- promoted Pt. Electrochimica Acta 1995, 40, 1537–1543. [Google Scholar] [CrossRef]
- Szabo, T.; Berkesi, O.; Forgo, P.; Josepovits, K.; Sanakis, Y.; Petridis, X. D.; De’ka’ny, I. Evolution of Surface Functional Groups in a Series of Progressively Oxidized Graphite Oxides. Chemical Materials 2006, 18, 2740–2749. [Google Scholar] [CrossRef]
- Dohyeon, L.; Sujin, G.; Younkwang, K.; Yung, E.; Eunjik, L.; Ji-Hoon, J.; Jee Youn, H.; Oh Jooh, K.; Taeho, L. Methanol Tolerant Pt-C Core-Shell Cathode Catalyst for Direct Methanol Fuel Cells. ACS Applied Materials & Interfaces 2020, 15, 1-10. [CrossRef]
- Wang, Z.; Hu, S.; Ali, A.; Chen, H.; Shen, P. K. Facile One-Pot Synthesis of a PtRh Alloy Decorated on Ag Nanocubes as a Trimetallic Core−Shell Catalyst for Boosting Methanol Oxidation Reaction. ACS Applied Energy Materials 2021, 4, 1085–1092. [Google Scholar] [CrossRef]
- Hamnett, A.; Mechanism and electrocatalysis in the direct methanol fuel cell; Catalysis Today 1997; 38, 445-457. [CrossRef]
- Yang, S.; Yang, X.; Tong, X.; Yang, N. A. Highly Active Rh@Pd Nanocube Catalyst for Methanol Electrooxidation. Advanced Energy and Sustainability Research 2021, 20, 1–9. [Google Scholar] [CrossRef]
- Li, Z.; Gao, F.; Zou, B.; Wu, Z.; Zhang, Y.; Du, Y. Core@shell PtAuAg@PtAg Hollow Nanodendrites as Effective Electrocatalysts for Methanol and Ethylene Glycol Oxidation. Inorganic Chemistry 2021, 60, 9977–9986. [Google Scholar] [CrossRef]
- Souza, J. P. I.; Queiroz, S. L.; Nart, F. C. Uso de espectrometria de massas em medidas eletroquímicas - A técnica de DEMS. Química Nova 2000, 23, 384-391. [CrossRef]
- Iwasita, T.; Nart, F. C.; Vielstich, W. An FTIR Study of the Catalytic Activity of a 85:15 Pt:Ru Alloy for Methanol Oxidation. Berichte Der Bunsengesellschaft Für Physikalische Chemie 1990, 94, 1030-1034. [CrossRef]
- Babu, P. K.; Kim, H. S.; Oldfield, E.; Wieckowski, A. Electronic Alterations Caused by Ruthenium in Pt-Ru Alloy Nanoparticles as Revealed by Electrochemical NMR; Electro-oxidation of small organic molecules. Plenum 1992, 22, 97–104. [Google Scholar] [CrossRef]
- Hoster, H.; Iwasita, T.; Baumgartner, H.; Vielstich, W. ; Pt-Ru model catalysts for anodic methanol oxidation: Influence of structure and composition on the reactivity. Physical Chemistry 2001, 3, 337–346. [Google Scholar] [CrossRef]
- Mukerjee, S.; Urian, R. C. Bifunctionality in Pt alloy nanocluster electrocatalysts for enhanced methanol oxidation and CO tolerance in PEM fuel cell: electrochemical and in situ synchrotron spectroscopy. Electrochimica Acta 2002, 47, 3219–3231. [Google Scholar] [CrossRef]
- Rhee, C. K.; Kim, B. J.; Ham, C.; Kim, Y. J.; Song, K.; Kwon, K. Size Effect of Pt Nanoparticle on Catalytic Activity in Oxidation of Methanol and Formic Acid: Comparison to Pt(111), Pt(100), and Polycrystalline Pt Electrodes; Langmuir 2009, 25, 7140-7147. [CrossRef]
- Marković, N. M.; Gasteiger, H. A.; Ross, P. N.; Jiang, X.; Villegas, I.; Weaver, M. J. ; Electro-oxidation mechanisms of methanol and formic acid on Pt-Ru alloy surfaces. Electrochimica Acta 1995, 40, 91–98. [Google Scholar] [CrossRef]
- Smith, A.S.; Lachance, A.M.; Zeng, S.; Liu, B.; Sun, L. ; Synthesis, properties, and applications of graphene oxide/reduced graphene oxide and their nanocomposites. Nano Materials Science 2019, 1, 31–37. [Google Scholar] [CrossRef]
- Zhou, X.; Liu, Z. ; A scalable, solution-phase processing route to graphene oxide and graphene ultralarge sheets. Chemical Communications 2010, 46, 2611–2611. [Google Scholar] [CrossRef]
- Wang, H. ; Wang, R. ; Li, H. ; Wang, Q. ; Kang, J. ; Lei, Z.; Facile synthesis of carbon-supported pseudo-core@shell PdCu@Pt nanoparticles for direct methanol fuel cells; International Journal of Hydrogen Energy 2011, 36, 839-848. [CrossRef]
- Muthuswamy, N.; La Fuente, J.L.G.; Dung, T.; Walmsley, J.; Tsypkin, M.; Raaen, S.; Sunde, S.; Ronning, M.; Chen, D.; Ru@Pt core-shell nanoparticles for methanol fuel cell catalyst: Control and effects of shell composition. International Journal of Hydrogen Energy 2013; 38, 16631-16641. [CrossRef]
- Muthuswamy, N.; La Fuente, J. L. G.; Dung, T.; Walmsley, J.; Tsypkin, M.; Raaen, S.; Sunde, S.; Ronning, M.; Chen, D. Ru@Pt core-shell nanoparticles for methanol fuel cell catalyst: Control and effects of shell composition. International Journal of Hydrogen Energy 2013, 38, 16631–16641. [Google Scholar] [CrossRef]
- Baomin, L.; Qiang, Z.; Yezhen, Z.; Lijuan, W.; Fenfen, L.; Haiquan, X. Core-shell Ag nanowires@Pt nanorods catalyst: Synthesis and application in direct methanol fuel cells. Materials Letters 2018, 233, 138–141. [Google Scholar] [CrossRef]
- Watanabe, M.; Motoo, S. Electrocatalysis by ad-atoms – part II: Enhancement of the oxidation of carbon monoxide on platinum by ruthenium ad-atoms. Journal of Electroanalytical Chemistry and Interfacial Electrochemistry 1975, 60, 267–273. [Google Scholar] [CrossRef]
- Watanabe, M.; Motoo, S. Electrocatalysis by ad-atoms – part III: Enhancement of the oxidation of carbon monoxide on platinum by ruthenium adatoms. Journal of Electroanalytical Chemistry and Interfacial Electrochemistry 1975, 60, 275–283. [Google Scholar] [CrossRef]
Figure 1.
X-ray patterns of Pt/C-30, PtRu/C-30, Cu@PtRu/rGO-16, Cu@PtRu/C-16, Cu@PtRu/C-18, and Cu/rGO-30 catalysts.
Figure 1.
X-ray patterns of Pt/C-30, PtRu/C-30, Cu@PtRu/rGO-16, Cu@PtRu/C-16, Cu@PtRu/C-18, and Cu/rGO-30 catalysts.
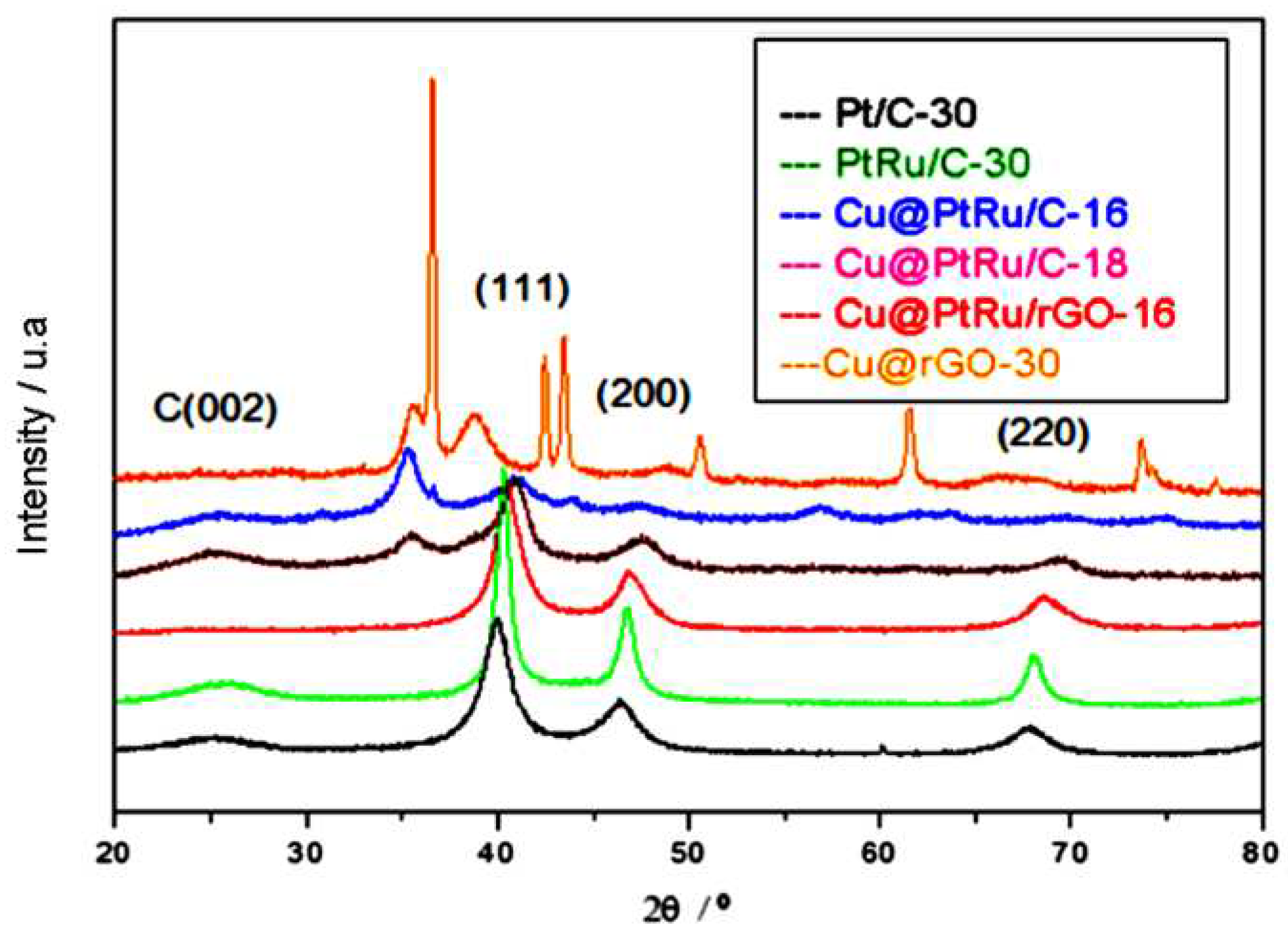
Figure 2.
HRTEM images of Cu/rGO-30 (A), (B) and (C). (D), and (E) images of catalysts Cu@PtRu/rGO-16.
Figure 2.
HRTEM images of Cu/rGO-30 (A), (B) and (C). (D), and (E) images of catalysts Cu@PtRu/rGO-16.
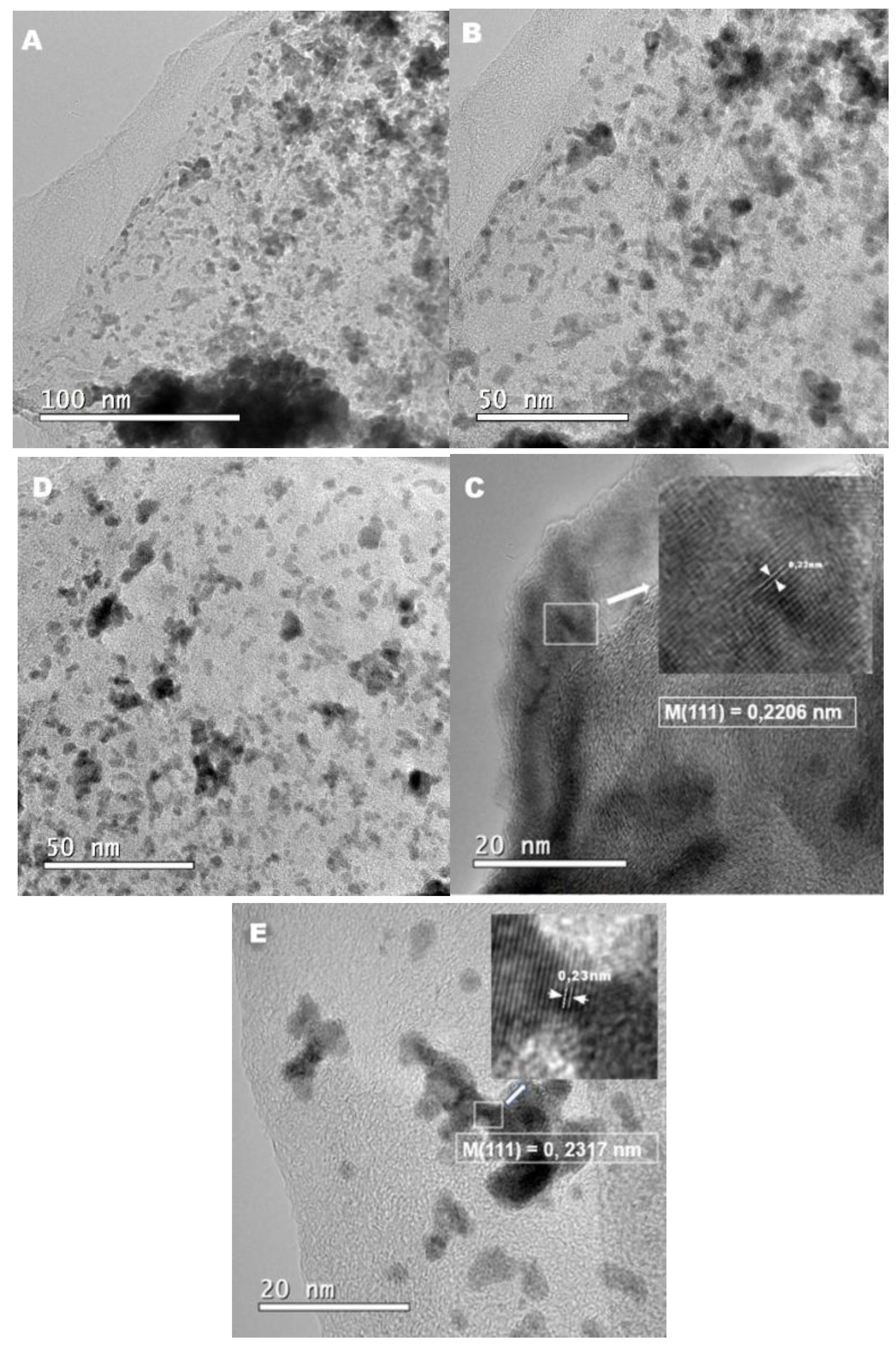
Figure 3.
Cyclic voltammograms of Pt/C-30, PtRu/C-30, Cu@PtRu/C-16, Cu@PtRu/C-18, and Cu@PtRu/rGO-16, obtained in 0.5 M H2SO4 solution, at a scan rate of 20 mV/s.
Figure 3.
Cyclic voltammograms of Pt/C-30, PtRu/C-30, Cu@PtRu/C-16, Cu@PtRu/C-18, and Cu@PtRu/rGO-16, obtained in 0.5 M H2SO4 solution, at a scan rate of 20 mV/s.
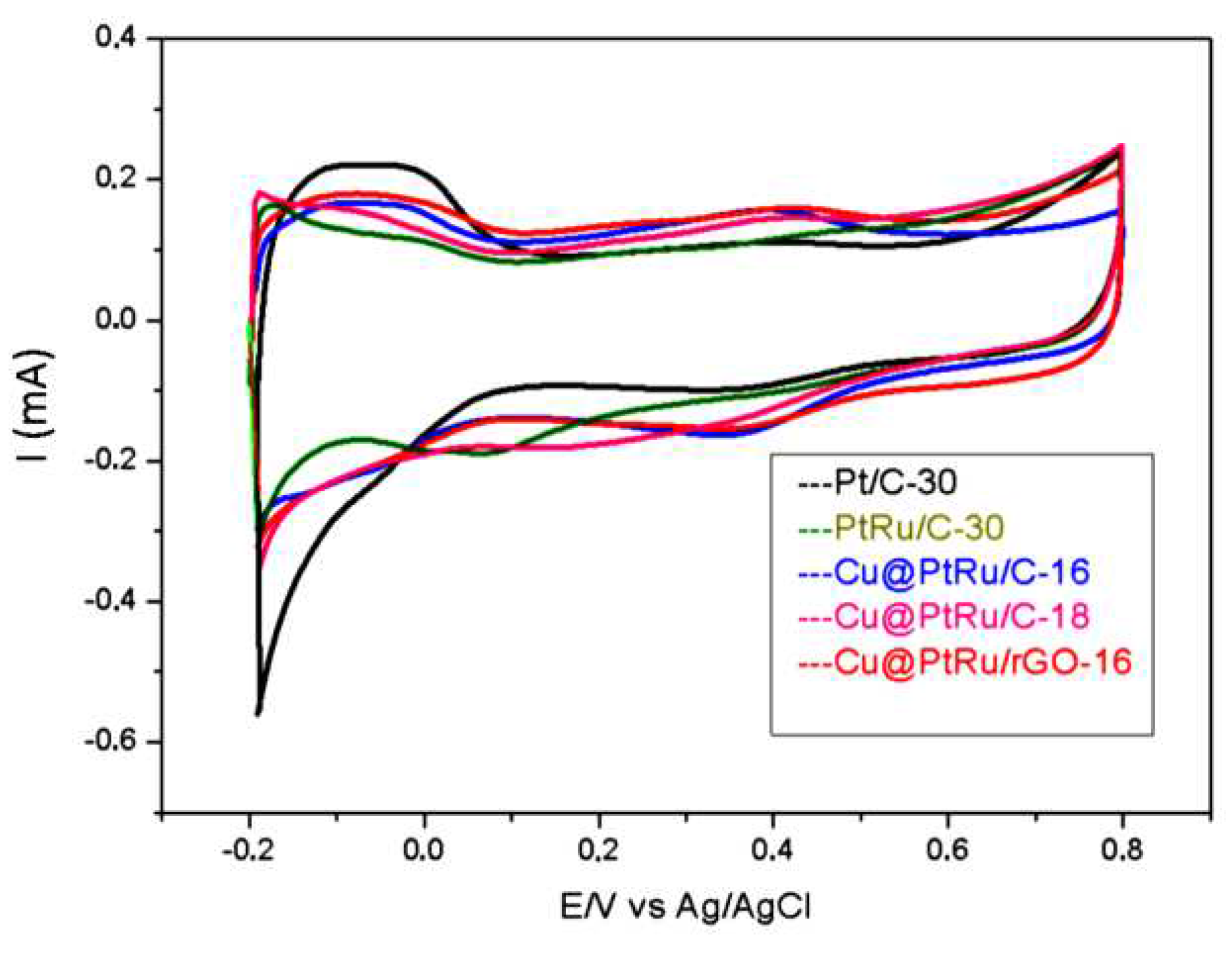
Figure 4.
Cyclic voltammogram of Pt/C-30 catalyst obtained in 0.5 M H2SO4 solution, at 20 mV/s. The shaded area represents the CO oxidation peak.
Figure 4.
Cyclic voltammogram of Pt/C-30 catalyst obtained in 0.5 M H2SO4 solution, at 20 mV/s. The shaded area represents the CO oxidation peak.
Figure 5.
Anodic scans of Pt/C-30, PtRu/C-30, Cu@PtRu/C-16, Cu@PtRu/C-18 e Cu@PtRu/rGO-16 obtained in solution containing 0.5 M CH3OH plus 0.5 M H2SO4, at 20 mV/s.
Figure 5.
Anodic scans of Pt/C-30, PtRu/C-30, Cu@PtRu/C-16, Cu@PtRu/C-18 e Cu@PtRu/rGO-16 obtained in solution containing 0.5 M CH3OH plus 0.5 M H2SO4, at 20 mV/s.
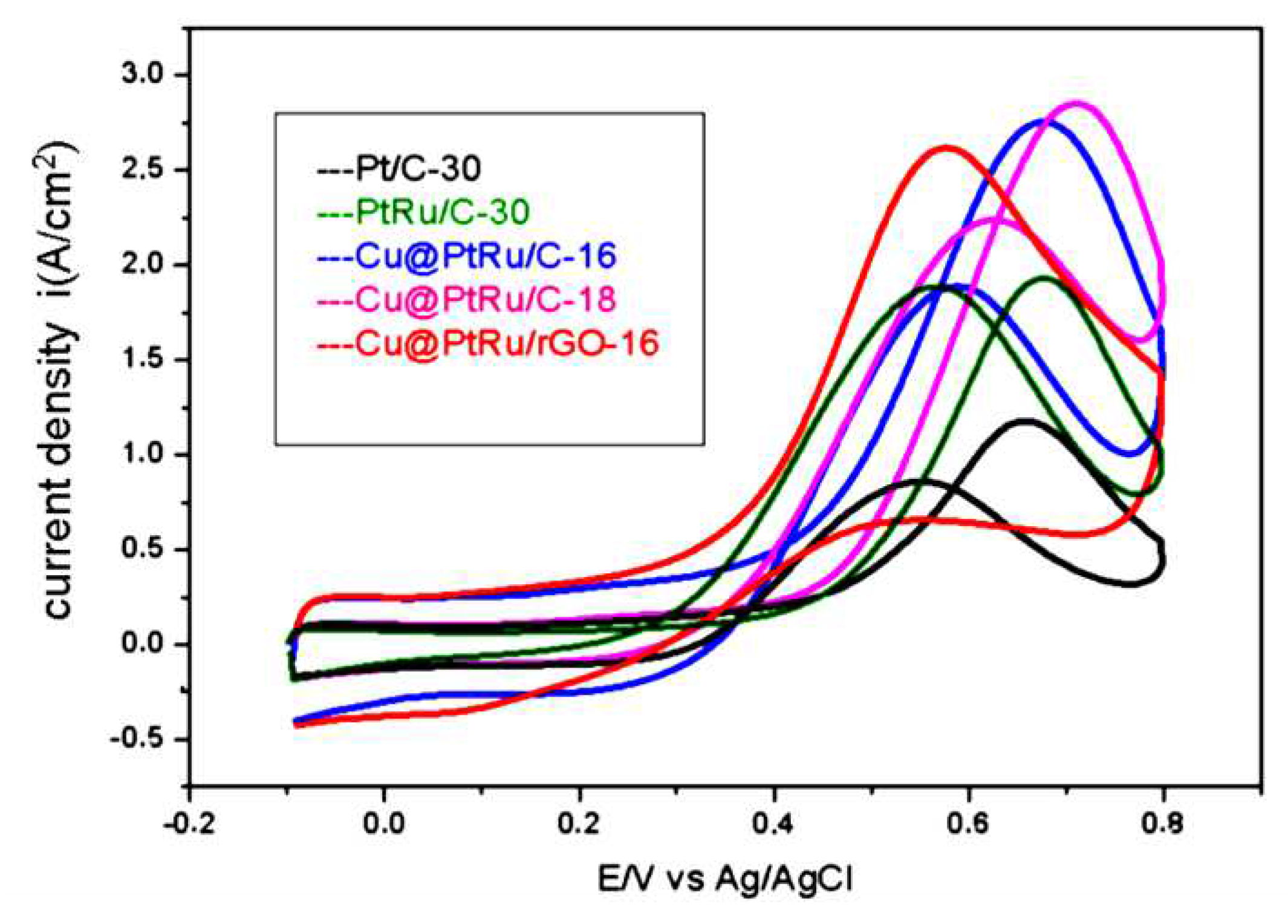
Figure 6.
Chronoamperometric curves for metanol electrooxidation on Pt/C-30, PtRu/C-30, Cu@PtRu/C-16, Cu@PtRu/C-18 and Cu@PtRu/rGO-16 in 0.5 M methanol plus 0.5 M H2SO4 solution.
Figure 6.
Chronoamperometric curves for metanol electrooxidation on Pt/C-30, PtRu/C-30, Cu@PtRu/C-16, Cu@PtRu/C-18 and Cu@PtRu/rGO-16 in 0.5 M methanol plus 0.5 M H2SO4 solution.
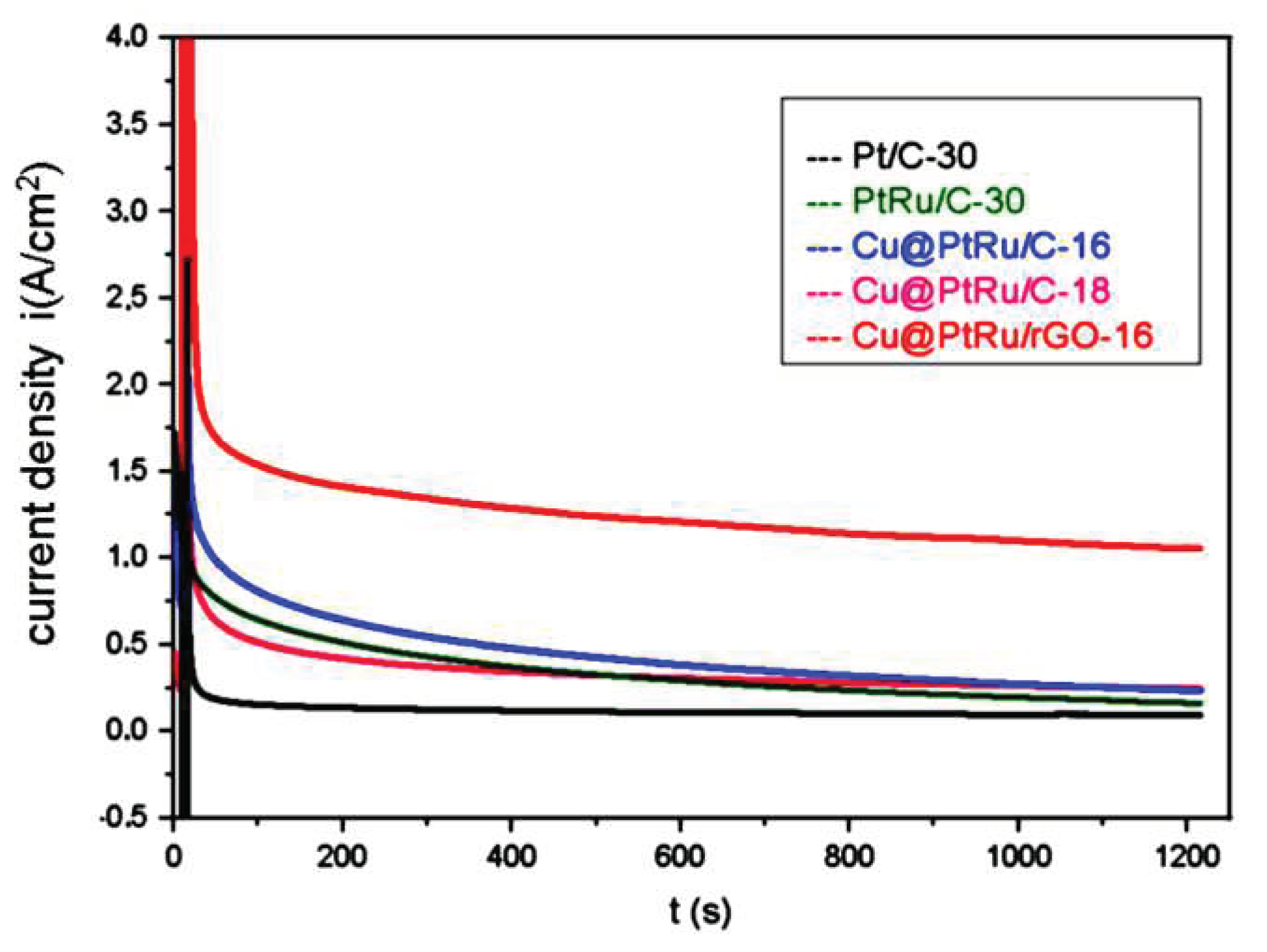

Table 1.
Starting oxidation potential (SPO) in volts, forward peak current density (iF) in Ampere-Coulomb, reverse peak current density (iR) and iF/IR ratio for Pt/C-30, PtRu/C-30, Cu@PtRu/C-16, Cu@PtRu/C-18 and Cu@PtRu/rGO-16 electrocatalysts obtained by reduction method.
Table 1.
Starting oxidation potential (SPO) in volts, forward peak current density (iF) in Ampere-Coulomb, reverse peak current density (iR) and iF/IR ratio for Pt/C-30, PtRu/C-30, Cu@PtRu/C-16, Cu@PtRu/C-18 and Cu@PtRu/rGO-16 electrocatalysts obtained by reduction method.
| Electrocatalyst SOP IF IR (V) (A/cm2) (A/cm2) |
IF/IR | |||
| Pt/C-30 | 0.437 | 1.80 | 1.92 | 0.93 |
| PtRu/C-30 | 0.425 | 1.96 | 1.78 | 1.10 |
| Cu@PtRu/C-16 | 0.300 | 2.79 | 2.02 | 1.38 |
| Cu@PtRu/C-18 | 0.391 | 2.89 | 2.17 | 1.33 |
| Cu@PtRu/rGO-16 | 0.250 | 2.68 | 1.85 | 1.45 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



