
Submitted:
22 June 2023
Posted:
22 June 2023
You are already at the latest version
Abstract
Metastasis is the main cause of anti-cancer therapies failure, leading to unfavorable prognosis for patients. The true challenge to increase cancer patient life expectancy by making cancer a chronic disease with periodic but manageable relapses, relies on the development of efficient therapeutic strategies specifically directed against key targets in the metastatic process.
Traditional chemotherapy with classical alkylating agents, microtubule inhibitors and antimetabolites has demonstrated its limited efficacy against metastatic cells due to their capacity to select chemo-resistant cell populations that undergo epithelial to mesenchymal transition (ETM) thus promoting the colonization of distant sites that in turn sustain the initial metastatic process. This scenario has prompted efforts aimed at discovering a wide variety of small molecules as potential anti-metastatic drugs directed against more specific targets known to be involved in the various stages of metastasis.
In this short reviews we will give an overview of the most recent advances related to three main large families of antimetastatic small molecules: intracellular tyrosine kinase inhibitors, integrin antagonists and small interfering (siRNAs). Although the majority of these small molecules are not yet approved and not available in the drug market, any information related to their stage of development could represent a precious and valuable tool to identify new targets in the endless fight against metastasis.

Keywords:
metastasis treatment
; small molecules
; tyrosine kinase inhibitors
; integrins
; siRNA
INTRODUCTION
Solid tumors are composed by different cancer cell types, generally considered to be derived from a single mutated cell clone that had undergone different mutations according to their differentiation grade. Because the degree of differentiation of these cancers is directly correlated to their sensitivity to chemotherapeutic agents [1,2] the first cycles of chemotherapy are very effective in reducing the tumor mass. This therapeutic approach, however, has an important drawback whose effects become noticeable over time: the apoptotic cell death of cancer cells confers an advantage to less differentiated or undifferentiated cancer cells who can grow in the absence of competition with other cancer cells for oxygen and nutrients. These less differentiated cancer cells, commonly referred to as cancer stem cells (CSC), can easily infiltrate healthy tissues and reach blood or lymphatic vessel being responsible for resistance to chemotherapy and metastatic spread [3,4].
However, to colonize distant sites these cells had to take a long and winding road that can be roughly divided in five steps: 1) expansion of original solid tumor mass; 2) proliferation and angiogenesis; 3) detachment and intravasation; tumor cells enter the circulation; 4) extravasation; circulating tumor cells settle at distant organ target sites; 5) local cell proliferation; change of the extracellular matrix (ECM) composition to support cell survival by shaping an appropriate tumor microenvironment. Any of the aforementioned steps implies the involvement of a plethora of different and very specialized components like adhesion proteins, soluble factors, miRNAs, extracellular vesicles (EV) and immune system cells [5].
Quite surprisingly, despite several decades of intensive and research devoted to unveil the precise mechanisms of cancer cell spreading in the body, the involvement of key players in cell spreading are still poorly understood and metastasis remains the primary cause of cancer mortality.
The (many) failures and (few) successes in the history of the search for effective and reliable antimetastatic drugs support the hypothesis that it is highly unlikely only one molecule could exert an inhibitory effect, thus making the discovery of a “one-drug fits all” antimetastatic molecule a very daunting and elusive task.
Instead, the extreme complexity of molecular mechanisms involved in the metastatic process clearly reinforces the notion that only the combined effect of several small molecules with different molecular targets, maybe supported by other tailored pharmacological therapeutic strategies, might be effective to counteract the metastatic spread [6].
Classical candidate drugs for solid tumors and with antiproliferative action are mostly screened by their ability to induce tumor shrinkage, but unfortunately this parameter does not correlate with cancer cell dissemination and therefore is not predictive of an antimetastatic effect.
The recent and spectacular rise of checkpoint inhibitors and monoclonal antibodies, that block the activation of overexpressed membrane growth factors receptors, changed the game in the treatment of solid tumors. However, the impact of checkpoint inhibitors on metastasis treatment, still in early phase of clinical testing, is uncertain and controversial thus requiring further validation and standardizations of methods dedicated to the assessment of their clinical efficacy [7].
Likewise, the limited efficacy of monoclonal antibodies in metastatic colorectal cancer, together with the finding that the pharmacological treatment with these drugs is considered to be not cost-effective, make their use as antimetastatic agents very problematic and uncertain in the future [8].
These considerations prompted projects in drug discovery to focus special attention to the wide variety of small molecules acting on different steps of the metastatic process. [9,10,11,12].
In the pretentious attempt to partially help clinical oncologists and pharmacologists researchers untangling this complex picture, in this paper we describe some recent members of three main classes of antimetastatic small molecules: a) intracellular tyrosine kinase inhibitors (TKIs); b) integrin antagonists and c) siRNAs. Despite this classification, there is an overlapping in the cellular effects modulated by these drugs: tyrosine kinase inhibitors probably show the widest spectrum of action in blocking the activation of several growth factors receptors whilst integrins antagonists mainly regulates detachment and attachment of circulating cancer cells. Even from the point of view of classical medicinal chemistry siRNA cannot be defined as small molecules (MW≤500Da), these derivatives, with a mean MW of approximately 14kDa, can be considered “small” in the increasing emerging field of biologics. Therefore, due their putative and unique ability to downregulates the levels of selected proteins involved in metastatic machinery, we felt it was important to discuss their main features. In this review we collected and critically surveyed the main features and their supposed mechanisms of action, of the most promising drugs belonging to the previous mentioned classes, focusing our attention on those currently in early phases of preclinical development for metastatic cancers or recently marketed as anticancer drugs.
We do believe that lessons learned from failures and successes gained from the study of these agents might give new insights into the molecular machinery of metastasis thus improving the development of next-generation reliable and effective antimetastatic agents [13].
Tyrosine kinase inhibitors
The common pharmacological action exerted by the majority of TKIs relies on the blocking of aberrant and dysregulated signaling pathways found in cancer cells [14]. For some selected TKIs, this blocking could be further dissected in two intracellular targets: a) catalytic pocket of tyrosine kinase receptors (TKRs) and b) different downstream kinases activated by same or different TKRs [15]. Compared to other small molecules, TKIs offer some advantages including the possibility of performing synthetic chemical modifications to optimize solubility and lipophilicity, the implementation of in vitro screening of lead compounds towards several oncogenic kinases and, last but not least, the advantage of development of dedicated formulations for oral administration.
The issue of the ability of TKIs to diffuse into and be retained by distant and difficult-to-access compartments or tissues is very critical when dealing with inhibition of metastatic processes. One example related to this problem deals with the clinical management of secondary brain metastasis treatment because drugs must cross the blood-brain barrier (BBB) to block prooncogenic kinases or aberrant signaling pathways found in metastatic cancer cells migrated into the brain.
Several recent strategies have been implemented to facilitate TKIs penetration into selected body districts [16]; 1) nanoparticles (NPs) formulations such polymeric NPs, gold NPs or liposomes, 2) the concomitant administration of efflux transporter inhibitors (ETIs) to increase the bioavailability of drugs into the brain and finally 3) refinement of chemical design to enhance drug lipophilicity by in silico quantitative structure–activity relationship (QSAR) models.
Despite these limitations, small molecule inhibitors (SMIs) are one of the most effective and widely adopted class of drugs in cancer therapy. In addition, notwithstanding the rushing impact of biotechnology drugs, big pharma’s investment for these molecules is constantly on a positive trend with large numbers of approved new molecular entities [NME] each year [17].
The majority of approved SMIs have been employed in the pharmacological treatment of solid tumors and only in recent years new clinical study describing the outcome of these molecules in the metastatic cancer cells have been reported in the literature. The extreme complexity of players involved in the metastatic process, together with our still poor knowledge of the plethora of premetastatic endogenous kinases and dysregulated signaling pathways, make the identification of suitable targets a formidable challenge. Although these considerations suggest that the use of TKIs drugs is still in its infancy, we anticipate that further clinical trials will expand their use in oncology. Here, we will briefly describe some the features and outcome of recent approved SMIs tested in clinical studies for their ability to counteract cancer cell spread in some solid metastatic tumors.
LAPATINIB (Tykerb®, Tyverb®)
Lapatinib is an oral TKI whose mechanism of action include blocking phosphorylation of the human epidermal growth factor receptor (EGFR) 1, HER2 and HER4, extracellular signal-regulated kinase 1 and 2 (ERK-1, 2), and protein kinase B (PKB/AKT).
Lapatinib is currently employed in combination with capecitabine [18] to treat patients with advanced HER2-positive breast cancer following standard therapy with anthracyclines, taxanes or in combination with trastuzumab for patients with metastatic HER2-positive on prior chemotherapy [19]. Due to its lipophilic properties, lapatinib has been tested in animal models of brain metastasis. Indeed, in a mice model of brain metastases induced by injection of breast of HER2 cancer cell lines, selective accumulation of lapatinib in brain metastatic cells decreased HER2 phosphorylation and the extent of brain metastasis [16]. However, despite these encouraging results, brain bioavailability of lapatinib should be enhanced and improved because it is quickly extruded from the brain to blood via P-glycoprotein and breast cancer resistance protein (BCRP) transporters thus limiting its efficacy in brain metastasis. A review aimed at identifying prognostic factors and survival parameters after developing bone radioiodine-resistant metastases (BM), together with the effects of combined therapies in patients with HER2+ breast cancer (BC), found that a combination of trastuzumab and lapatinib therapy or trastuzumab and pertuzumab therapy had the longest median survival compared with other therapies. [20]. Quite notably, the efficacy of combined therapies using lapatinib could be extended to other tissues colonized by metastatic breast cancer cells, because dual HER2 blockade with trastuzumab and lapatinib inhibited tumor growth in patient-derived xenografts of HER2-amplified metastatic colorectal cancer [21]. Among approaches exploited to increase TKIs penetration of the BBB, nanoparticles (NPs) systems seem the most promising and performant; a)NPs cross easily BBB and display enhanced permeability and retention in the tumor site; b) NPs can be used as a shuttle system for many drugs and finally c) biodegradability of NPs limit their toxicity. In this context, a nanomedicine system co-loaded with lapatinib/doxorubicine and stabilized with glycol chitosan showed a potent therapeutic effect toward triple-negative breast cancer cells in comparison to a mixture of free drugs [22].
In cancer cells, characterized by an extreme complexity and interactions among dysregulated convergent and divergent cell signaling transduction pathways, the resistance mechanism of redundancy may represents the main cause of failure or lack of efficacy of SMIs treatment [23]. In redundancy, different elements of the signaling transduction pathways may act on different elements in the same biological or dynamic manner to converge on a common target and therefore the inhibition of one of these elements, by a compensatory mechanism, does not affect the whole biological outcome in terms of combined responses of the cell. Redundant elements compensate for the blocking of targeted genes by recruiting and leveraging the ability of crosstalk among other pathways to overcome the effects of SMIs treatment in cancer cells. In the attempt to bypass this serious obstacle, the oncology community devoted considerable efforts to chemical synthesis of SMIs less specific and with a broader range of inhibition towards driver TKRs kinases involved in redundancy [24]. Among these SMIs, regorafenib and lenvatinib have recently shown promising results in clinical studies designed to test their efficacy in different types of metastatic cancers.
REGORAFENIB
Regorafenib (BAY 73-4506, Stivarga®) is a novel oral multikinase inhibitor approved for the treatment metastatic colon rectal cancer (CRC) and found to improve progression-free survival (PFS) in patients with metastatic gastrointestinal stromal tumors (GIST) [25]. Regorafenib, developed in a screening project aimed at enhancing the potency of sorafenib, differs from the parent drug for the presence of a fluorine atom onto the central aromatic ring (figure X). The main mechanism of action of regorafenib relies on the inhibition of various RTKs including c-KIT, RET, BRAF, VEGFR1–3, TIE2, PDGFR-β, and fibroblast growth factor receptor (FGF-1). Other cellular effects of regorafenib include a decrease of tumor growth and lymph node metastasis, interrupted tumor cell–MSC interaction, and modified tumor-supporting stroma [25]. However, some reports have investigated the possibility that the real mechanism of regorafenib could be broader than previously thought being not limited to inhibition of TKIs. For example, it was found that regorafenib treatment upregulates the level of PUMA, a p53 target and a critical mediator of apoptosis, thus promoting apoptosis induction in different colorectal cancer cell lines [26]. In another study, regorafenib treatment enhanced the progression-free survival and metabolic responses via downregulation of the AKT/mTOR/S6 ribosomal protein axis in refractory colorectal carcinoma [27]. The limited knowledge of the widespread mechanisms and cellular targets of regorafenib prompted the oncology community to set up several clinical trials to test the efficacy of regorafenib in other metastatic tumors such as soft tissue sarcoma (STS), renal cell carcinoma (RCC) and hepatocellular carcinoma (HCC) [28,29].
Lenvatinib
Lenvatinib (lenvima 10®) is a broad-spectrum inhibitor that block the activation of several types of RTKs receptors including VEGFR-1 (FLT1), VEGFR-2(KDR), VEGFR-3 (FLT4), FGFR-1, FGFR-2, FGFR-3, FGFR-4, PDGFRa, RET, and c-KIT. The therapeutic indications of lenvatinib include metastatic thyroid cancer (MTC), advanced renal cell carcinoma (RCC) and hepatocellular carcinoma (HCC). Interestingly, the ability of lenvatinib to interfere and reduce the activity of the proangiogenic VEGFs-dependent signaling pathway, make it an ideal candidate as first line of treatment in limiting the vascularization processes recruited by tumor cells to support the metastatic spread.
In differentiated thyroid cancer (DTC), bone radioiodine-resistant metastases (BM) are the main cause of low survival and greater morbidity due to the formation of several severe damages associated with skeletal-related events (SRE). Because in this tumor classical antiresorptive therapy (AT) has limited benefits, an approach with concomitant therapy with multikinase TKIs and AT should be carefully considered. The validity of this strategy has been supported by a study showing that lenvatinib treatment induced a longer overall survival (OS) in DTC patients with lung metastases [30]. Future trials devoted to evaluate the outcomes and toxicity of lenvatinib in combination with AT are therefore warranted to determine the future possible utilization of multikinase TKIs in metastatic DTC [31].
In the three-arm Phase III trial CLEAR study, designed to compare lenvatinib plus everolimus and lenvatinib plus pembrolizumab versus sunitinib monotherapy for the treatment of RCC, lenvatinib plus pembrolizumab showed promising antitumor activity when the efficacy of this combination was assessed by progression-free survival (PFS) as primary end-point and other parameters, such as overall survival, safety, quality of life and pharmacokinetics, as secondary end-point [32]. Another phase III multi-national trial, termed REFLECT, designed to screen a possible less toxic first line treatment of patients with unresectable HCC, demonstrated that lenvatinib, although non-inferior compared to sorafenib for overall survival, showed a better and more controllable toxicity profile [33].
Finally, one report described another approach aimed at improving the bioavailability of combined therapy of multikinase TKIs with immunochemo-therapy in metastatic triple-negative breast cancer TNBC. Using an in vivo animal model, the authors found that lenvatinib- and vadimezan-loaded synthetic high-density lipoprotein (LV-sHDL) inhibited the growth of orthotopic tumors, reduced pulmonary metastasis and improved the survival of animals thus underscoring the potential use of TKIs in combined therapy to treat metastatic TNBC [34].
In recent years, the current widespread use in therapy, together with the growing number of new molecules in development, has clearly assessed the central and fundamental role of TKIs as valuable tool in the pharmacology of cancer.
This consideration is strongly supported by the large numbers of clinical studies currently ongoing. At the present time, the designing and the screening of TKIs by drug companies is a topic field in metastatic oncology. A search in the portal https://clinicaltrials.gov, updated on June 20, 2023, querying with the entries “metastatic cancers and TKIs” in US retrieved 14 entries in phase I, 49 in phase II, 12 in phase III and 1 in phase IV for clinical studies with reported results.
Due to space constraints, here we do not comment all these results, and hence readers interested in deepening their interests in in this topic are warmly invited to look at the above link.
Despite these enormous efforts in discovery of novel reliable and effective TKIs, unfortunately there is a very arduous issue related to the intrinsic mechanism of action of this category of drugs. After some beneficial initial cycles of treatments with TKIs, a large percentage of patients do not respond anymore to the therapy because their cancer cells develop acquired drug resistance [35,36]. Point mutation within their catalytic kinase domain, decreasing the affinity of TKIs to their specific binding site by steric hindrance, accounts for the most common drug resistance mechanism. However, other concomitant additional mechanisms have been documented such as gene amplification or overexpression, alternative splicing of RTKs, variations of elements regulating signaling pathways functionality, overexpression or mutations of drug transporters and epigenetic modifications including changes in microRNA formation and turnover [37].
Strategies to overcome this heavy limitations of TKIs include either experimental in vitro/in vivo and in silico approaches. The first approach relies on the development of organoids and lab-on-a-chip systems to screen toxicity and efficacy of new compounds. The second approach will greatly exploit the extraordinary potentiality of protein folding computational algorithms, such as RoseTTA fold [38] and Alphafold2 [39], that allows the identification of possible RTKs pockets domains, at a previously unattainable 3D spatial resolution, suitable to be targeted by new molecules. A better understanding of the TKIs resistance is the necessary prerequisite and a potent stimulus to develop next generations of TKIs as starting point to concretize the promises and claims of personalized medicine in cancer therapy [40].
INTEGRINS ANTAGONISTS
Integrins are cell adhesion proteins involved in the interaction with the extracellular matrix (ECM), in the transmission of biochemical and mechanical signals between cells and their environment, and in a wide range of biological functions. In particular, αvβ3 and ανβ5 integrin subtypes turned out to be overexpressed in different cancer types such as colon cancer, melanoma, breast cancer, and glioblastoma [41]. Therefore, in the last decades, these receptors have been widely explored as therapeutic targets in anticancer therapy [42,43,44]
Since αvβ3 and αvβ5 can be expressed by both tumor cells and tumor endothelial cells, it has been speculated that drugs capable of inhibiting the adhesive function of these integrins can hamper tumor growth in at least two ways: by directly targeting the tumor and by inhibiting angiogenesis. Cyclic peptides carrying the Arg-Gly-Asp (RGD) sequence are potent antagonist of αvβ3 and ανβ5 integrin subtypes. Cilengitide (EMD121974, Scheme 1) is undoubtedly the most known integrin antagonist characterized by a cyclic peptidomimetic structure, and it has been evaluated in almost 30 different clinical trials for cancer [45]. In Cilengitide, or [c(RGDfNMeV)], the presence of dipeptide D-phenylalanyl–N-methyl valine (fNMeV) allowed to fix the RGD sequence into the correct bioactive conformation to bind in nanomolar concentrations αvβ3 and αvβ5 integrin receptors.
Despite promising preliminary data, the use of this compound as an anticancer drug against glioblastoma has been discontinued due to failure in phase-III clinical trials, where it showed no improvement in overall patients’ survival [46]. Moreover, in a paper published in 2009, Reynolds and co-workers reported that Cilengitide had the unfavorable property of paradoxically increasing angiogenesis at concentrations below its IC50 [47].
Indeed, the binding of Cilengitide generates major conformational change in αvβ3 receptor that induces it to adopt a high-affinity ligand-binding state which is associated with enhancement of tumor growth in vivo. To circumvent this drawback, the design of pure small-molecule antagonists such as TDI-4161 (Scheme 2), which do not induce conformational changes in the receptor, while maintaining high affinity and potent antitumor activity, have emerged as an area of intense interest [48]. Interestingly, the pure antagonist TDI-4161 was synthesized through rational modification of highly active αvβ3 ligand MK-0429 (vide infra) guided by a three-dimensional molecular model of the interaction between MK-0429 and the αVβ3 binding pocket, refined by molecular dynamics (MD) simulations.
The partial agonist effect of RGD-based small molecules derivatives, albeit somewhat debated, has led for some years to putting aside the use of these peptidomimetic antagonists alone as anticancer agents [49]. Nevertheless, given their intrinsic very low toxicity towards healthy cells, and their high affinity and selectivity for integrin overexpressed in various tumor forms, RGD-based antagonist found a second life as drug delivery systems [50,51,52].
Along the years, different groups have worked on the synthesis of Cilengitide analogues by replacing the D-Phe–N-MeVal dipeptide unit with various molecular scaffolds [53]. In this context, constrained dipeptide mimics such as azabicycloalkanone amino acids (ABA, Scheme 1) [Khashper-54] proved to be useful intermediates for the obtainment of cRGD antagonists such as 1a-RGD, a nanomolar inhibitor of αvβ3 and αvβ5 integrin subtypes [55]. Analogously, synthesis of “easy-to-functionalize” azabicycloalkanone amino acids (f-ABA, Scheme 1) [56,57,58] allowed the design of modified cRGD derivatives like BIO1 and BIO2, in which a side chain installed onto the azabicycloalkanone core has been exploited for the conjugation of bioactive compounds [57]. This strategy resulted into the preparation of cRGD-cytotoxic bioconjugates [60], cRGD-based imaging probes [61,62] and cRGD-functionalized nanosystems [63,64].
The high selectivity of cRGD-functionalized nanoparticles towards cancer cell lines overexpressing αvβ3 and αvβ5 integrin receptors [65] could pave the way to the use of highly active anticancer agents such as naphthalene diimide derivative NDI-1 (Scheme 1). The reverse transcriptase enzyme telomerase, overexpressed in many cancer cells, plays a key role in maintaining the telomere length for the immortal division of malignant cells. Naphthalene diimide derivatives are potent anticancer agents that act by inducing/stabilizing G-quadruplex DNA structures, thus sequestering the enzyme substrate in the single-stranded telomeric DNA [66,67,68]. As a consequence, the telomere length can gradually return to shorten, leading, as in the case of non-malignant cells, to senescence and apoptosis. Although some derivatives have reached clinical trials in humans [69], owing to their indiscriminate cell entry and very tight therapeutic window, naphthalene diimides can cause severe side effects which strongly limits their applications in anticancer therapy. By loading NDI-1 into silk fibroin nanoparticles endowed with cRGD derivatives, our group observed a significantly higher cytotoxic effect on human glioma cell lines U373, which overexpress αvβ3 and αvβ5 integrin subtypes, than on D384 cell lines, characterized by a lower expression of the same integrins [70]. Moreover, the encapsulation of the active inside the nanoparticles considerably reduces its distribution in the body and therefore its toxicity towards healthy cells/organs.
As previously discussed in the case of Cilengitide, despite the good premises of being able to synthesize novel anticancer agents by exploiting the selective binding of small molecule integrin antagonists (SMIA) to specific integrin receptors, this approach has not led to the expected clinical success. Indeed, the high adaptive capacity of cancer cells probably demands that additional biological mechanisms need to be targeted in the tumor microenvironment for SMIA to be effective.
Despite above considerations, to conclude this section we think it could be useful to trace a brief overview of the SMIA that, in the last years, have reached clinical trials in cancer therapy [71]. The broad-spectrum (or pan) αv antagonist GLPG0187 (Scheme 2) was tested in phase Ib trial in patients with solid tumors [72]. In preclinical models and in mouse cancer models GLPG0187 showed to be very active, inhibiting the formation and progression of bone and visceral metastases in prostate cancer and breast cancer. Although in humans turned out to be well tolerated, GLPG0187 was dismissed due to its low efficacy because continuous infusion of this compound failed to show signs of monotherapy efficacy. The previously cited MK-0429 is a nonpeptide pan-αv integrin inhibitor that was tested in a phase I randomized double-blind study on men with hormone refractory prostate cancer and metastatic bone disease (MDB) [73]. MK-0429 was generally well tolerated and displayed a potential for clinical use in MDB but, unexpectedly, an increasing in serum prostate specific antigen (SPA, a marker for disease activity) took place during the experimentation. While since 2012 no clinical activity has been reported for MK-0429 as anticancer drug, in 2019, the interesting preclinical in vivo data on the obese ZSF1 (Zucker fatty and spontaneously hypertensive) rat model of diabetic nephropathy [74], prompted Merck to patent MK-0429 for chronic kidney disease [75].
The linear pentapeptide ATN-161 (Ac-PHSCN-NH2) is a non-competitive inhibitor of the fibronectin “synergy region” (PHSRN sequence) [76]. This region enhances the affinity and specificity of the RGD-mediated binding, thus increasing the interaction between fibronectin and the integrin receptor. ATN-161 binds to beta subunits of α5β1 and αvβ3 integrins, outside the RGD-binding site, displaying antitumorigenic and antimetastatic activities in various cancers [77,78]. The peptide has been designed by introducing a cysteine residue in place of arginine of the original fibronectin PHSRN sequence, along with acetylation of the N-terminal proline and amidation of the C-terminal asparagine. The said chemical modifications were introduced to improve the pharmaceutical properties of ATN-161, which reached phase II clinical trial in patient with solid tumors such as renal cell cancer and brain and central nervous system tumors. Since 2012 no clinical activity has been reported for ATN-161 as antineoplastic agent, but recent in vitro experiments highlighted the effectiveness of this compound in reducing the entity of SARS-CoV-2 infection [79]. These findings, which are in agreement with the hypothesis that RGD-binding integrins are co-receptors for angiotensin-converting enzyme 2 (ACE2), used by SARS-CoV-2 to entry into the host cells [80], open the door at the possible future use of integrin inhibitors in viral infections [81]. Alintegimod (7HP349) is a clinical-stage orally active immunostimulant which acts as an allosteric activator of T cells’leukocyte-specific integrins αLβ2 and α4β1. The interaction with these receptors enhances T cell activation and adhesion, thus increasing the penetration of T cells into tumors in mouse model of melanoma and colon carcinoma [82]. Following a successful first-in-human Phase I clinical trial completed at the end of 2021, Alintegimod will be entering Phase Ib/IIa trials in programmed cell death 1 (PD-1) refractory solid tumors and influenza vaccination of the elderly. Moreover, in 2022 the FDA has granted fast track designation to 7HP349, in combination with a cytotoxic T-lymphocyte antigen 4 (CTLA-4) inhibitor, for the treatment of patients with unresectable or metastatic malignant melanoma [83].

siRNA
siRNAs, also known as short interfering RNAs, are a type of non-coding double-stranded RNAs of 20-23 nucleotide base pairs in length that act by interfering with the expression of the specific gene having short stretches complementary to sequences to target mRNA [84]. The siRNAs are similar to endogenous microRNAs (miRNA) in terms of functions, except that the microRNAs can regulate the expression of hundreds of genes via imperfect base pairing whilst siRNAs bind more specifically to the single mRNA at a particular location. The significant and peculiar difference between these two non-coding RNAs is that the miRNA has multiple targets, whereas siRNA has only one specific mRNA target.
The great advantage of siRNA as drug therapy platform, compared to “conventional” drugs such as small molecule or antibodies, relies on their capability to be taken up by target cells, processed by the RNA-induced silencing complex (RISC) to release single strand RNA (ssRNA) that in turn degrades its complementary mRNA thus blocking the translation of the target protein [85,86].
The intrinsic structure of non-modified naked siRNA therapeutics, together with the hurdle to overcome systemic delivery issues, poses some critical and important challenges regarding their bioavailability (such as serum stability and potency), off-target effects and ability to trigger an immunogenic unwanted response [87].
Efforts to cope with these challenges identified two founding pillars required for a successful siRNA drug therapy. The first pillar relies on modification of the structure of siRNAs to limit their degradation, increasing their stability and at the same time improving their binding affinity toward target mRNAs [88].
The second pillar relies on the design of delivery systems to enhance the amount of siRNA that enters the target cell [89]. Indeed, naked siRNAs are polar and poor lipophilic compounds that do not cross the cell membranes, thus requiring a lipophilic “shuttle” system to enhance their entrance and subsequent bioavailability inside the cells.
siRNA modifications
1. Termini Modification
The addition of a phosphate groups at 5’ end of the siRNA antisense strand is necessary for the recognition by Argonaute 2 (Ago2), a component of the RISC (RNA-induced silencing complex) machinery that process double strand siRNA thus allowing its pairing with target mRNA. However, caution is required when using heavily modified siRNAs because these chemical modifications could decrease siRNAs binding capacity for the RISC complex; this problem can be bypassed by using non-hydrolyzable 5’-phosphate analogs such as 5’-(E)-vinyl-phosphonate [90] (Figure 1).
2. Backbone Modifications
The stability and in vivo bioavailability of siRNAs can be improved by replacing standard phosphodiester linkage with boron, sulfur or acetate thus forming phosphonothioate, boranophosphate, and phosphonoacetate linkages. The presence of these exogenous linkages increases siRNAs resistance to nuclease hydrolysis but these modifications could potentially reduce siRNA potency by interfering with their recognition by the RISC machinery and could also increase siRNA immunogenicity. The insertion of a phosphotriester enhances cellular uptake of siRNAs by eliminating the negative charge thus increasing siRNA half-life in circulation. These small interfering ribonucleic neutrals [siRNNs] are then transformed in siRNAs by the action of esterases inside the cells.
3. Sugar Modifications
The 2’-hydroxy group of the ribose sugar can be replaced with less nucleophilic moieties to gain 2’-O-methyl, 2’-O-methoxyethyl and 2’-fluoro derivatives. These replacements, when inserted in a siRNA in which the ribose ring is constrained by a methylene linkage between the 2′-oxygen and the 4′-carbon (locked nucleic acid), lead to an increased binding affinity of the siRNA toward its target mRNA. The main advantage of sugar modifications compared to backbone modifications are their inability to affect siRNA recognition by RISC.
4. Base Modification
Immune cells can distinguish foreign siRNAs from endogenous siRNAs because the former usually have chemical modifications that are not present in mammalian cells. These include sugar modifications such as 2’-O-methyl and base substitution with modified bases such as pseudouridine, 5’-methylcytidine, N6-methyladenosine, inosine, and N7-methylguanosine. Therefore, modifying siRNAs at these sites will allow them to escape immune detection, but can also reduce their silencing potency.
5. Lipid Conjugation
The conjugation of lipids to the 5’ or 3’ termini of siRNA increases their binding with serum proteins and their uptake, thus improving their whole in vivo bioavailability; siRNA uptake in several tissues upon intravenous injection is greatly enhanced by conjugation with cholesterol and vitamin E.
6. bioconjugations
Bioconjugation has recently emerged as one of the most innovative and promising approach to bypass hurdles and problems associated with direct chemical modifications of siRNAs [91]. The principle of this approach relies on the formation of a covalent binding between siRNA and a variety of other biogenic molecules such as antibodies, aptamers, carbohydrates, ligands, lipophilic derivatives, oligonucleotides, peptides and polymers. Bioconjugates display several advantages over classical non-conjugated nanosystems such as lipid nanoparticles, polymeric nanoparticles, metallic core nanoparticles, dendrimers and polymeric micelles. Indeed, siRNA-bioconjugates complexes could be assembled without a positive charge, they are less immunogenic due to their small size and in addition the chemical nature of the link (such as cleavable or photosensitive bonds) between siRNA and its cognate bioconjugate could be designed to modulate stability, efficiency, accumulation and resistance to RNase. The main issue associated with siRNA-bioconjugate deals with the lack of specificity of the different conjugated chemical entities that hampers the recognition of specific targets on the membranes of siRNA recipient cells. Although this drawback could be not so critical when high selectivity of delivery is not required and accumulation of the drug into non-target cells do not induce side effects, future studies aimed at understanding the specificity of the conjugates-target surface molecules interactions are mandatory to boost the application of siRNA-bioconjugates into clinical practice.
siRNA delivery
The various siRNA delivery systems could be roughly divided in two big families: a) a physiological approach based on viral vectors and small extracellular vesicles (EV) such as exosomes (EXs) and b) a synthetic approach based on a wide variety of nanoparticles (LNP) that encompass liposomes and other formulations.
Viral vectors
Viral vectors display optimal properties as siRNA delivery system; they have high transfection efficiency and could target in a very specific way sensitive cells that do not withstand harsh and stressful treatments linked to other delivery systems [89]. However, the implementation of siRNA incapsulated in viral vectors is a rather expensive and complex procedure that necessitates trained staff with good skills in molecular biology and virology.
In addition, safety concerns in term of the ability to induce dangerous mutations or unwanted incorporation of viral genes into the host DNA [92], together with the risk to trigger immunogenic and inflammatory processes, have slowed attempts to set up the production of safe vectors for human use. These limitations have therefore shifted efforts in this field toward the development of non-viral delivery systems such as EV(extracellular vesicles)- based systems, liposome and synthetic nanoparticles.
EV-based siRNA delivery
EVs are currently one of the most investigated shuttle system due to their potential to be easily loaded with small molecules cargos combined with their supposed ability to be selectively and specifically taken up by different cell types. Notably, the selective uptake of EVs by target cells could be further fine-tuned by chemically modifying the molecules expressed on the EVs membrane that are supposed to interact with their cognate counterparts present on the membranes of target cells.
OTHERS
Polymeric nanoparticles, metallic core nanoparticles, dendrimers and polymeric micelles have been extensively exploited in recent years and are currently under investigation as promising delivery systems. Indeed, these non-liposomal siRNA nanocarriers are quite stable, and their surface can be easily chemically modified to increase their transport and target efficiency.
A wide array of biocompatible and non-toxic materials, including chitosan, cyclodextrin, polyethyleneimine (PEI), polylactic-glycolic acid (PLGA) has been successfully tested and proven to be safe and efficient encapsulating nanoparticles for siRNA delivery [93].
Unfortunately, the downside of the nanocarriers relies on the difficulty to design a combination of suitable materials and, at the same time, make the necessary chemical modifications to obtain a reliable delivery system.
LIPID NANOPARTICLES (NP)
LNPs are widely used for siRNA delivery because the lipid envelope protects siRNA from nuclease cleavage thus allowing intact siRNA to reach targeted organs or cells.
On the basis of their charge properties, siRNA-binding lipids could also be classified into three main classes: cationic, neutral and ionizable LNPs [94].
Because ionizable and cationic LNPs interacts with circulating lipids, they are mainly accumulated in hepatocytes or in the endosomes and lysosomes compartments; however cationic LNPs are less potent and more toxic than ionizable LNPs due to their higher nonspecific binding with plasma proteins and their ability to induce immunogenic reactions.
These unwanted properties prompted efforts, either in private and public research institutions, to synthesize novel ionizable LNPs lacking of side effects and selectively designed to escape accumulation in hepatocytes. These considerations are especially instrumental when siRNA-LNPs are employed as shuttle for antitumoral agents where a stringent selectivity is required to target cancer cells and at the same time to spare healthy tissues.
Attempts to overcome these limitations involved the modification of LNPs surface with chemical moieties that could target molecules expressed on tumor cell outer membrane, in tumor extracellular matrix (ECM) or in other non-tumor cell types located in the tumor microenvironment (TME) such as endothelial cells or tumor associated macrophages (TAM).
The most employed and screened tumor targeting moieties, that facilitate siRNA-loaded LNP accumulation in cancer cells, include aptamers or other small molecules (hyaluronan or folate), short peptides (RGD-based peptides or octreotide) and large proteins or antibodies (transferrin).
Liposomal nanoparticles (NP) platforms have been investigated as siRNA carrier in a wide variety of cancer models to reduce cancer cell proliferation and their associated metastatic properties.
A combination of siRNAs targeting 3 genes (MDM2, c-myc and VEGF) expressed in a murine lung cancer model, encapsulated in PEGylated NP composed of carrier DNA and polycationic peptides, significantly reduced metastasis with little signs of systemic immunotoxicity or damage to other body compartments [95].
Other approaches have focused on the modification of liposomes combined with siRNAs to target multiple signaling transduction pathways underlying cancer cell metastatic processes. In metastatic breast cancer (MBC) the C-X-C chemokine receptor type 4 (CXCR4) and lipocalin-2(Lcn2) are two key molecules that mediate cell migration. Liposomes modified with anti-CXCR4 antibodies and engineered to release lipocalin-2 (Lcn2) siRNA elicited a synergistic effect in reducing cell migration in triple-negative human breast cancer cells [96].
A similar strategy exploits the chemical modifications of the surface of LNP with short peptides or chemical residues that are recognized by target cells, such as peptides incorporating the RGD sequence that binds several members of the integrins family overexpressed in a wide variety of cancer cells. An anti-angiogenic siRNA encapsulated in an RGD peptide-modified lipid nanoparticle (RGD-LNP) elicited a significant reduction of the vascular permeability in a lung-metastasized animal model [97]. Notably, the cumulative antiangiogenic effect induced by RGD-LNP in the metastasized lung was higher when compared to the analogous PEG-LNP, thus strongly indicating that this approach could be further refined to enhance the target specificity and cell permeability of surface modified LNP in the treatment of metastatic cancers.
The advantages of surface chemical modified LNP over non-modified ones are supported by another report [98]. Self-assembling LNPs composed by an amphiphilic low molecular weight heparin-ursolic acid conjugate were encapsulated in PEG-lipid containing anisamide, a targeting ligand for sigma receptor overexpressed in a lung metastasis model. The gene silencing activity of the targeted NP loaded with siRNA showed significantly higher silencing activity than the other formulations such as naked NP not PEGylated or PEGylated untargeted NP. In addition, confocal microscopy showed that, in contrast with the two other preparation devoid of ligand, the targeted NP are highly enriched in the lung metastasis without or with minimal interactions with other body compartments.
The conjugation with an antibody recognizing an antigen on the surface of target cell is another strategy for selective uptake into cancer cells. Antibody-conjugated mesoporous silica nanoparticle (MSNP) coated with cross-linked PEI/PEG polymers were loaded with polo-like kinase 1 siRNA (siPLK1) and tested in an animal model of highly metastatic triple-negative breast cancer (TNBC) [99]. The authors reported that MSNPs inhibit cancer cell invasion via its ROS- and NOX4-dependent mechanisms, reduce by 80% the level of PLK1 mRNA in metastatic breast cancer cells of mouse lungs and increase the overall survival of treated animals.
EXOSOMES
The discovery that cancer-derived exosomes have a preferential targeting to distant organs prompted researchers to pursue this strategy in animal model of cancers with high metastatic potential. Autologous serum-derived exosomes, obtained from mice injected with melanoma cells, loaded with siRNA directed against the heparan sulfate proteoglycans Glypica-3 (GPC3), significantly reduced the spread of tumor cells as assessed by the number of metastatic lung colonies. [100].
Another way to increase the selective targeting ability of autologous exosomes as siRNA drug delivery systems is the modification of biomimetic autologous exosomes. The application of this innovative approach leads to the development of CBSA/siS100A4-exosome, an exosome membrane coated lipid nanoparticles LNP, composed by cationic bovine serum albumin (CBSA) conjugated with siRNA directed against S100A4, an important metastasis-related protein that promotes tumor progression. These biomimetic exosomes showed higher degree of protection of siRNA from degradation and higher biocompatibility when compared to analogous conjugated LNP thus resulting in a pronounced gene-silencing effect of S100A4 that in turn reduces the proliferation of malignant triple-negative metastatic breast cancer (TNBC) cells [101].
Another study focused on the use of exosomes as siRNA carrier investigated the possibility to silence non canonical long non-coding RNA (lnRNA) involved in the progression of TNBC [102]. The overexpression of lnRNA DARS-AS1 modulates the migration and invasion of TNBC tumors by inhibiting miR-129-2-3p via upregulation of the CDK1 kinase. Treatment with DARS-AS1 siRNA-loaded exosomes substantially reduced TNBC cell growth and liver metastasis thus confirming that siRNA loaded exosomes represent a promising tool in contrasting highly metastatic tumors.
The encapsulation of siRNA into synthetic nanoparticles (SN), formed by PEG-polylactides, could represent an innovative tool to enhance efficacy of cancer therapy when combined with classical anticancer drugs. In a series of elegant in vitro and in vivo experiments carried out in a model of human pancreatic cancer, the authors of this study demonstrate that co-administration of the nanosystems encapsulating siRNA with arsenic-encapsulated vesicle led to a synergistic action of the mutant Kras siRNA silencing effect and of arsenic proapoptotic antiproliferative effects [103].
Liver metastasis is the most critical health concern in patients affected by colorectal cancer (CRC) and therefore recent efforts to reduce the metastatic features of this type of cancer have been devoted to develop new formulations based on siRNA delivery [104].
The major issues in administered orally siRNA are avoiding gastrointestinal degradation and facilitating active transport thus obtaining the necessary bioavailability. The administration of AuNP−siRNA−glycolchitosan−taurocholic acid nanoparticle (AR-GT NPs) containing Akt2 siRNA resulted in the decrease of endogenous Akt levels, accompanied by a concomitant induction of apoptosis, in an animal model of colorectal-liver metastases (CLM) [105].
Other synthetic formulations for siRNA delivery, based on polysaccharides or derivatives, have been successfully validated in highly metastatic cancers such as osteosarcoma. In fact, siRNA directed against the astrocyte elevated gene-1 (AEG-1) gene, encapsulated in the polysaccharide Amy-g-PLLD, was found to decrease the proliferation and invasion of osteosarcoma tumors both in vitro and in vivo [106]. In addition, direct injection of Amy-g-PLLD/siAEG-1 complexes into the tumoral mass significantly inhibited lung metastasis in tumor-bearing mice with no apparent cytotoxicity.
Beyond classical delivery of chemically synthetized siRNA encapsulated in NP, an elegant alternative aimed at bypassing toxicity associated with NP transfection relies on the preparation of plasmids or viruses that contain precursor of short hairpin RNA (shRNA). Short hairpin RNA (shRNA) sequences are stem-loop stretches of RNA, usually encoded in a DNA vector that can be introduced into cells via plasmid transfection or viral transduction, that once formed indise the cell are then cleaved by endogenous cell RNAse to cut the loop thus releasing siRNAs. The Yes-associated protein (YAP) is overexpressed or constitutively activated in a wide variety of cancer types where it regulates several mechanisms involved in metastatic processes. Silencing of YAP by small-hairpin RNA (shRNA) induced tumor cell apoptosis and blocked tumor cell proliferation and angiogenesis in an animal model of gastric carcinoma [107]. Quite interestingly, YAP shRNA also decreases the expression of several other genes that support metastatic spreading thus highlighting the utility of the shRNA approach in enhancing the efficacy and safety of siRNA delivery systems.
The debate over the superiority of one siRNA delivery systems over others, in term of efficiency and toxicity, is still an open question seldom addressed in the siRNA literature. The comprehensive study of Kamerkar et al. [108] sought to fill this important gap by comparing two siRNA delivery platforms: exosomes versus liposomes. Exosomes obtained from the supernatant of normal fibroblast-like mesenchymal cells, carrying siRNA or shRNA directed against mutated oncogenic KRASG12D (iExosomes), exhibited superior evasion of phagocytic clearance by monocytes in the circulation together with enhanced efficacy compared to similar assembled liposomes. This iExosomes treatment, mediated by a CD47-dependent phagocytotic escape mechanism, significantly extended the overall survival of treated mice in a rodent model of pancreatic cancer.
The possibility for siRNAs to achieve clinical relevance in cancer therapy suggested by this study emphasizes the key role of proteins naturally present on the surface of heterologous non-cancer cells exosomes, not only in escaping clearing mechanisms, but especially in enhancing the target specificity of exosomes as delivery systems for siRNAs directed against undruggable targets.
Several clinical trials testing siRNA-based drugs against metastatic cancers are currently underway or completed; a search on the portal https://clinicaltrials.gov , updated on June 20,2023, querying with the entry “metastatic cancer and siRNA”, retrieved 7 clinical studies in US; among these 6 were completed and only one is still recruiting (phase 1). The only active recruiting phase I clinical trial will investigate in healthy subjects the best dose and side effects in of mesenchymal stromal cells-derived i-exosomes (iEXs) loaded with KrasG12D siRNA directed against the mutated form of KrasG12D to be then screened in patients with metastatic pancreatic cancer. A completed phase I study with metastatic melanoma was designed to test the safety of transfected modified siRNA, decorated with antigens derived from dendritic cells, targeting the inducible immunoproteasome subunits LMP2, LMP7, and MECL1. Another phase I trial studied the side effects and best dose of siRNA-transfected peripheral blood mononuclear cells APN401 (APN401), in treating patients with metastatic melanoma, kidney and pancreatic cancer. A first phase I trial assessed the safety, tolerability and toxicity of a single intravitreal injection of Sirna-027 (AGN211745), a siRNA targeting VEGFR-1, together with associated anatomical changes in the retina and in visual acuity in age-related macular degeneration. The purpose of another trial was to test the safety and tolerability of the new drug DCR-MYC, a novel synthetic double-stranded RNA formulated as lipid particle suspension, targeting the oncogene MYC whose activation regulates a wide array of cellular events deeply involved in many hematologic and solid metastatic tumors. In all these completed studies, no results were posted.
Despite these limited and poorly informative clinical trials, we do believe that the impact of siRNA-based drugs will become increasingly fundamental in the pharmacology against metastatic cancers in the near future. The recent improvements and refinements of new formulations [109], together with the development of a sequence-specific control of gene expression on a genomic wide scale, termed CRISPR-interference (CRISPRi) [110], will provide a very selective and complementary approach to siRNA in the targeted silencing of metastatic genes. Finally, increased knowledge of the key parameters involved in siRNA design, gained from the commercialization of several siRNA-based drugs [111] and from the technology used for the Sars-Covid19 mRNA vaccines encapsulated in LNP, will likely boost the development and approval of siRNA-based anticancer drugs.
CONCLUSION AND FUTURE PERSPECTIVES
Due to growing numbers of different classes of anticancer drugs, either in development or recently approved, the last decade have witnessed a spectacular improvement in survival rate of patients affected by several types of solid tumors and hematologic malignancies. However, this giant leap forward has not been mirrored by a concomitant advancement of effective treatments for metastatic cancer, that accounts for more 90% of mortality. This failure derives mainly by the still limited and vague knowledge of the many molecular mechanisms responsible of metastatic spread that makes it really difficult and tricky to identify suitable antimetastatic targets. Undoubtedly, the use of some of the SMIs (or their derivatives) reported in this account, either alone or in combination with other therapeutic innovative drugs, could represent a cornerstone in cancer therapy.
Nevertheless, the study of metastatic processes requires an enormous joint effort by the oncology community focused on developing novel approaches and refining existing advanced technologies in the cancer research. In our opinion, we do foresee that the greatest improvements will derive from the combination of two complementary and intertwined approaches; a) new types of functional ex vivo metastatic models and b) single-cell spatial multiomic analyses. A recent work of Ombrato et al. [112] proposed a very elegant and clever way to detect and identify early processes in the metastatic spread of cancer cells together with their ability to recruit and educate other non-cancer cells found in the premetastatic niche. The authors of this study engineered premetastatic cancer cells to express a releasable mCherry fluorescent protein that is then insert into a slice of non-tumor cells. The mCherry protein is released by premetastatic tumor cells and taken up by the contiguous surrounding cells within the local tumor microenvironment (TME).
Positive mCherry TME cells can be first isolated by fluorescence activated cell sorting (FACS) and then characterized by single cell transcriptomics and proteomics tools to identify changes in signaling pathways induced by the direct contact with metastatic cancer cells.
The exploitation of the full potential of this ex vivo model requires the implementation of reliable spatial single cell multiomic analyses thus allowing the characterization at genomic, transcriptomic and proteomic level of the different cluster of cells, together with their spatial localization, belonging to the metastatic niche and TME [113]. Recent improvements in analytical procedure have clearly shown that single-cell sequencing (SCS) is a potent and affordable high-throughput tool to investigate the molecular mechanisms underlying tumor metastasis at single cell level [114]. Indeed, SCS can be used to study tumor heterogeneity, drug resistance, changes in TME microenvironment, analysis of circulating tumor cells (CTCs) in liquid biopsy and, in combination with artificial intelligence (AI), to construct metastasis-related cell maps for predicting and monitoring the dynamics of metastasis.
References
- Suhail, Y.; Cain, M.P.; Vanaja, K.; Kurywchak, P.A.; Levchenko, A.; Kalluri, R.; Kshitiz. Systems Biology of Cancer Metastasis. Cell Syst. 2019, 9, 109–127. [Google Scholar] [CrossRef]
- Fares, J.; Fares, M.Y.; Khachfe, H.H.; Salhab, H.A.; Fares, Y. Molecular Principles of Metastasis: A Hallmark of Cancer Revisited. Signal Transduct. Target. Ther. 2020, 5, 28. [Google Scholar] [CrossRef] [PubMed]
- Yin, W.; Wang, J.; Jiang, L.; James Kang, Y. Cancer and Stem Cells. Exp. Biol. Med. 2021, 246, 1791–1801. [Google Scholar]
- Yang, L.; Shi, P.; Zhao, G.; Xu, J.; Peng, W.; Zhang, J.; Zhang, G.; Wang, X.; Dong, Z.; Chen, F.; Cui, H. Targeting Cancer Stem Cell Pathways for Cancer Therapy. Signal Transduct. Target. Ther. 2020, 5, 8. [Google Scholar] [CrossRef]
- Becker, A.; Thakur, B.K.; Weiss, J.M.; Kim, H.S.; Peinado, H.; Lyden, D. Extracellular Vesicles in Cancer: Cell-to-Cell Mediators of Metastasis. Cancer Cell 2016, 30, 836–848. [Google Scholar] [CrossRef]
- Sleeman, J.; Steeg, P.S. Cancer Metastasis as a Therapeutic Target. Eur. J. Cancer 2010, 46, 1177–1180. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Wang, M.; Xu, C.; Li, B.; Chen, J.; Chen, J.; Wang, Z. Immune Checkpoint Inhibitor Therapy for Bone Metastases: Specific Microenvironment and Current Situation. J. Immunol. Res. 2021, 2021, 1–18. [Google Scholar]
- Lange, A.; Prenzler, A.; Frank, M.; Kirstein, M.; Vogel, A.; Von Der Schulenburg, J.M. A Systematic Review of Cost-Effectiveness of Monoclonal Antibodies for Metastatic Colorectal Cancer. Eur. J. Cancer 2014, 50, 40–49. [Google Scholar] [CrossRef]
- Bedard, P.L.; Hyman, D.M.; Davids, M.S.; Siu, L.L. Small Molecules, Big Impact: 20 Years of Targeted Therapy in Oncology. The Lancet 2020, 395, 1078–1088. [Google Scholar]
- Kobelt, D.; Dahlmann, M.; Dumbani, M.; Güllü, N.; Kortüm, B.; Vílchez, M.E.A.; Stein, U.; Walther, W. Small Ones to Fight a Big Problem—Intervention of Cancer Metastasis by Small Molecules. Cancers 2020, 12, 1454. [Google Scholar] [CrossRef]
- Zhong, L.; Li, Y.; Xiong, L.; Wang, W.; Wu, M.; Yuan, T.; Yang, W.; Tian, C.; Miao, Z.; Wang, T.; Yang, S. Small Molecules in Targeted Cancer Therapy: Advances, Challenges, and Future Perspectives. Signal Transduct. Target. Ther. 2021, 6, 201. [Google Scholar]
- Zhong, S.; Jeong, J.-H.; Chen, Z.; Chen, Z.; Luo, J.-L. Targeting Tumor Microenvironment by Small-Molecule Inhibitors. Transl. Oncol. 2020, 13, 57–69. [Google Scholar] [CrossRef]
- Gandalovičová, A.; Rosel, D.; Fernandes, M.; Veselý, P.; Heneberg, P.; Čermák, V.; Petruželka, L.; Kumar, S.; Sanz-Moreno, V.; Brábek, J. Migrastatics—Anti-Metastatic and Anti-Invasion Drugs: Promises and Challenges. Trends Cancer 2017, 3, 391–406. [Google Scholar] [CrossRef]
- Tan, H.-Y.; Wang, N.; Lam, W.; Guo, W.; Feng, Y.; Cheng, Y.-C. Targeting Tumour Microenvironment by Tyrosine Kinase Inhibitor. Mol. Cancer 2018, 17, 43. [Google Scholar] [CrossRef]
- Huang, L.; Jiang, S.; Shi, Y. Tyrosine Kinase Inhibitors for Solid Tumors in the Past 20 Years (2001–2020). J. Hematol. Oncol.J Hematol Oncol 2020, 13, 143. [Google Scholar] [CrossRef]
- Angeli, E.; Bousquet, G. Brain Metastasis Treatment: The Place of Tyrosine Kinase Inhibitors and How to Facilitate Their Diffusion across the Blood–Brain Barrier. Pharmaceutics 2021, 13, 1446. [Google Scholar] [PubMed]
- Esteban-Villarrubia, J.; Soto-Castillo, J.J.; Pozas, J.; San Román-Gil, M.; Orejana-Martín, I.; Torres-Jiménez, J.; Carrato, A.; Alonso-Gordoa, T.; Molina-Cerrillo, J. Tyrosine Kinase Receptors in Oncology. Int. J. Mol. Sci. 2020, 21, 8529. [Google Scholar] [CrossRef] [PubMed]
- Petrelli, F.; Ghidini, M.; Lonati, V.; Tomasello, G.; Borgonovo, K.; Ghilardi, M.; Cabiddu, M.; Barni, S. The Efficacy of Lapatinib and Capecitabine in HER-2 Positive Breast Cancer with Brain Metastases: A Systematic Review and Pooled Analysis. Eur. J. Cancer 2017, 84, 141–148. [Google Scholar] [CrossRef] [PubMed]
- Tevaarwerk, A.J.; Kolesar, J.M. Lapatinib: A Small-Molecule Inhibitor of Epidermal Growth Factor Receptor and Human Epidermal Growth Factor Receptor—2 Tyrosine Kinases Used in the Treatment of Breast Cancer. Clin. Ther. 2009, 31, 2332–2348. [Google Scholar] [CrossRef] [PubMed]
- Hackshaw, M.D.; Danysh, H.E.; Henderson, M.; Wang, E.; Tu, N.; Islam, Z.; Ladner, A.; Ritchey, M.E.; Salas, M. Prognostic Factors of Brain Metastasis and Survival among HER2-Positive Metastatic Breast Cancer Patients: A Systematic Literature Review. BMC Cancer 2021, 21, 967. [Google Scholar] [CrossRef]
- Sartore-Bianchi, A.; Trusolino, L.; Martino, C.; Bencardino, K.; Lonardi, S.; Bergamo, F.; Zagonel, V.; Leone, F.; Depetris, I.; Martinelli, E.; et al. Dual-Targeted Therapy with Trastuzumab and Lapatinib in Treatment-Refractory, KRAS Codon 12/13 Wild-Type, HER2-Positive Metastatic Colorectal Cancer (HERACLES): A Proof-of-Concept, Multicentre, Open-Label, Phase 2 Trial. Lancet Oncol. 2016, 17, 738–746. [Google Scholar] [PubMed]
- Kaur, N.; Sharma, P. ; Mimansa; Jaganathan, M.; Munawara, R.; Aggarwal, A.; Shanavas, A. Glycol Chitosan Stabilized Nanomedicine of Lapatinib and Doxorubicin for the Management of Metastatic Breast Tumor. Drug Deliv. Transl. Res. 2023. [Google Scholar] [CrossRef] [PubMed]
- Lavi, O. Redundancy: A Critical Obstacle to Improving Cancer Therapy. Cancer Res. 2015, 75, 808–812. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Bernards, R. Feedback and Redundancy in Receptor Tyrosine Kinase Signaling: Relevance to Cancer Therapies. Trends Biochem. Sci. 2014, 39, 465–474. [Google Scholar]
- Takigawa, H.; Kitadai, Y.; Shinagawa, K.; Yuge, R.; Higashi, Y.; Tanaka, S.; Yasui, W.; Chayama, K. Multikinase Inhibitor Regorafenib Inhibits the Growth and Metastasis of Colon Cancer with Abundant Stroma. Cancer Sci. 2016, 107, 601–608. [Google Scholar] [CrossRef]
- Chen, D.; Wei, L.; Yu, J.; Zhang, L. Regorafenib Inhibits Colorectal Tumor Growth through PUMA-Mediated Apoptosis. Clin. Cancer Res. 2014, 20, 3472–3484. [Google Scholar] [CrossRef] [PubMed]
- Wong, A.L.A.; Lim, J.S.J.; Sinha, A.; Gopinathan, A.; Lim, R.; Tan, C.-S.; Soh, T.; Venkatesh, S.; Titin, C.; Sapari, N.S.; et al. Tumour Pharmacodynamics and Circulating Cell Free DNA in Patients with Refractory Colorectal Carcinoma Treated with Regorafenib. J. Transl. Med. 2015, 13, 57. [Google Scholar] [CrossRef]
- Crona, D.J.; Keisler, M.D.; Walko, C.M. Regorafenib: A Novel Multitargeted Tyrosine Kinase Inhibitor for Colorectal Cancer and Gastrointestinal Stromal Tumors. Ann. Pharmacother. 2013, 47, 1685–1696. [Google Scholar] [CrossRef]
- Granito, A.; Marinelli, S.; Forgione, A.; Renzulli, M.; Benevento, F.; Piscaglia, F.; Tovoli, F. Regorafenib Combined with Other Systemic Therapies: Exploring Promising Therapeutic Combinations in HCC. J. Hepatocell. Carcinoma 2021, 8, 477–492. [Google Scholar] [CrossRef]
- Tahara, M.; Kiyota, N.; Hoff, A.O.; Badiu, C.; Owonikoko, T.K.; Dutcus, C.E.; Suzuki, T.; Ren, M.; Wirth, L.J. Impact of Lung Metastases on Overall Survival in the Phase 3 SELECT Study of Lenvatinib in Patients with Radioiodine-Refractory Differentiated Thyroid Cancer. Eur. J. Cancer 2021, 147, 51–57. [Google Scholar] [CrossRef]
- Navarro-Gonzalez, E. Use of Multikinase Inhibitors/Lenvatinib Concomitant with Antiresorptive Therapy for Bone Metastases from Radioiodine-resistant Differentiated Thyroid Cancer. Cancer Med. 2022, 11 (S1), 54–58. [Google Scholar]
- Grünwald, V.; Powles, T.; Choueiri, T.K.; Hutson, T.E.; Porta, C.; Eto, M.; Sternberg, C.N.; Rha, S.Y.; He, C.S.; Dutcus, C.E.; Smith, A.; Dutta, L.; Mody, K.; Motzer, R.J. Lenvatinib plus Everolimus or Pembrolizumab versus Sunitinib in Advanced Renal Cell Carcinoma: Study Design and Rationale. Future Oncol. 2019, 15, 929–941. [Google Scholar] [PubMed]
- Rehman, O.; Jaferi, U.; Padda, I.; Khehra, N.; Atwal, H.; Mossabeh, D.; Bhangu, R. Overview of Lenvatinib as a Targeted Therapy for Advanced Hepatocellular Carcinoma. Clin. Exp. Hepatol. 2021, 7, 249–257. [Google Scholar] [CrossRef] [PubMed]
- Zheng, C.; Zhang, W.; Wang, J.; Zhai, Y.; Xiong, F.; Cai, Y.; Gong, X.; Zhu, B.; Zhu, H.H.; Wang, H.; Li, Y.; Zhang, P. Lenvatinib- and Vadimezan-Loaded Synthetic High-Density Lipoprotein for Combinational Immunochemotherapy of Metastatic Triple-Negative Breast Cancer. Acta Pharm. Sin. B 2022, 12, 3726–3738. [Google Scholar] [CrossRef]
- Rosenzweig, S.A. Acquired Resistance to Drugs Targeting Tyrosine Kinases. In Advances in Cancer Research; Elsevier, 2018; Vol. 138, pp 71–98.
- Lovly, C.M.; Shaw, A.T. Molecular Pathways: Resistance to Kinase Inhibitors and Implications for Therapeutic Strategies. Clin. Cancer Res. 2014, 20, 2249–2256. [Google Scholar]
- Jiao, Q.; Bi, L.; Ren, Y.; Song, S.; Wang, Q.; Wang, Y. Advances in Studies of Tyrosine Kinase Inhibitors and Their Acquired Resistance. Mol. Cancer 2018, 17, 36. [Google Scholar] [CrossRef]
- Baek, M.; DiMaio, F.; Anishchenko, I.; Dauparas, J.; Ovchinnikov, S.; Lee, G.R.; Wang, J.; Cong, Q.; Kinch, L.N.; Schaeffer, R.D.; et al. Accurate Prediction of Protein Structures and Interactions Using a Three-Track Neural Network. Science 2021, 373, 871–876. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly Accurate Protein Structure Prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Yang, Y.; Li, S.; Wang, Y.; Zhao, Y.; Li, Q. Protein Tyrosine Kinase Inhibitor Resistance in Malignant Tumors: Molecular Mechanisms and Future Perspective. Signal Transduct. Target. Ther. 2022, 7, 329. [Google Scholar]
- Desgrosellier, J.S.; Cheresh, D.A. Integrins in Cancer: Biological Implications and Therapeutic Opportunities. Nat. Rev. Cancer 2010, 10, 9–22. [Google Scholar] [CrossRef]
- Hatley, R.J.D.; Macdonald, S.J.F.; Slack, R.J.; Le, J.; Ludbrook, S.B.; Lukey, P.T. An Av-RGD Integrin Inhibitor Toolbox: Drug Discovery Insight, Challenges and Opportunities. Angew. Chem. Int. Ed. 2018, 57, 3298–3321. [Google Scholar] [CrossRef]
- Slack, R.J.; Macdonald, S.J.F.; Roper, J.A.; Jenkins, R.G.; Hatley, R.J.D. Emerging Therapeutic Opportunities for Integrin Inhibitors. Nat. Rev. Drug Discov. 2022, 21, 60–78. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.; Dong, B.; He, X.; Qiu, Z.; Zhang, J.; Zhang, M.; Liu, H.; Pang, X.; Cui, Y. The Challenges and Opportunities of Avβ3-Based Therapeutics in Cancer: From Bench to Clinical Trials. Pharmacol. Res. 2023, 189, 106694. [Google Scholar] [CrossRef]
- Mas-Moruno, C.; Rechenmacher, F.; Kessler, H. Cilengitide: The First Anti-Angiogenic Small Molecule Drug Candidate. Design, Synthesis and Clinical Evaluation. Anticancer Agents Med. Chem. 2010, 10, 753–768. [Google Scholar]
- Stupp, R.; Hegi, M.E.; Gorlia, T.; Erridge, S.C.; Perry, J.; Hong, Y.-K.; Aldape, K.D.; Lhermitte, B.; Pietsch, T.; Grujicic, D.; et al. Cilengitide Combined with Standard Treatment for Patients with Newly Diagnosed Glioblastoma with Methylated MGMT Promoter (CENTRIC EORTC 26071-22072 Study): A Multicentre, Randomised, Open-Label, Phase 3 Trial. Lancet Oncol. 2014, 15, 1100–1108. [Google Scholar]
- Reynolds, A.R.; Hart, I.R.; Watson, A.R.; Welti, J.C.; Silva, R.G.; Robinson, S.D.; Da Violante, G.; Gourlaouen, M.; Salih, M.; Jones, M.C.; Jones, D.T.; Saunders, G.; Kostourou, V.; Perron-Sierra, F.; Norman, J.C.; Tucker, G.C.; Hodivala-Dilke, K.M. Stimulation of Tumor Growth and Angiogenesis by Low Concentrations of RGD-Mimetic Integrin Inhibitors. Nat. Med. 2009, 15, 392–400. [Google Scholar] [CrossRef]
- Li, J.; Fukase, Y.; Shang, Y.; Zou, W.; Muñoz-Félix, J.M.; Buitrago, L.; Van Agthoven, J.; Zhang, Y.; Hara, R.; et al. Novel Pure AVβ3 Integrin Antagonists That Do Not Induce Receptor Extension, Prime the Receptor, or Enhance Angiogenesis at Low Concentrations. ACS Pharmacol. Transl. Sci. 2019, 2, 387–401. [Google Scholar]
- Paolillo, M.; Serra, M.; Schinelli, S. Integrins in Glioblastoma: Still an Attractive Target? Pharmacol. Res. 2016, 113, 55–61. [Google Scholar]
- Arosio, D.; Casagrande, C. Advancement in Integrin Facilitated Drug Delivery. Adv. Drug Deliv. Rev. 2016, 97, 111–143. [Google Scholar] [CrossRef]
- Alipour, M.; Baneshi, M.; Hosseinkhani, S.; Mahmoudi, R.; Jabari Arabzadeh, A.; Akrami, M.; Mehrzad, J.; Bardania, H. Recent Progress in Biomedical Applications of RGD-based Ligand: From Precise Cancer Theranostics to Biomaterial Engineering: A Systematic Review. J. Biomed. Mater. Res. A 2020, 108, 839–850. [Google Scholar] [PubMed]
- Battistini, L.; Bugatti, K.; Sartori, A.; Curti, C.; Zanardi, F. RGD Peptide-Drug Conjugates as Effective Dual Targeting Platforms: Recent Advances. Eur. J. Org. Chem. 2021, 2021, 2506–2528. [Google Scholar]
- Paolillo, M.; Russo, M.; Serra, M.; Colombo, L.; Schinelli, S. Small Molecule Integrin Antagonists in Cancer Therapy. Mini-Rev. Med. Chem. 2009, 9, 1439–1446. [Google Scholar] [CrossRef]
- Khashper, A.; Lubell, W.D. Design, Synthesis, Conformational Analysis and Application of Indolizidin-2-One Dipeptide Mimics. Org Biomol Chem 2014, 12, 5052–5070. [Google Scholar] [CrossRef] [PubMed]
- Arosio, D.; Belvisi, L.; Colombo, L.; Colombo, M.; Invernizzi, D.; Manzoni, L.; Potenza, D.; Serra, M.; Castorina, M.; Pisano, C.; Scolastico, C. A Potent Integrin Antagonist from a Small Library of Cyclic RGD Pentapeptide Mimics Including Benzyl-Substituted Azabicycloalkane Amino Acids. ChemMedChem 2008, 3, 1589–1603. [Google Scholar] [CrossRef]
- Serra, M.; Peviani, E.G.; Bernardi, E.; Colombo, L. Synthesis of Variously Functionalized Azabicycloalkane Scaffolds by Domino Metathesis Reactions. J. Org. Chem. 2017, 82, 11091–11101. [Google Scholar] [CrossRef] [PubMed]
- Serra, M.; Bernardi, E.; Lorenzi, E.D.; Colombo, L. Synthesis of Functionalized 6,5- and 7,5-Azabicycloalkane Amino Acids by Metathesis Reactions. J. Org. Chem. 2019, 84, 15726–15734. [Google Scholar] [CrossRef]
- Serra, M.; Tambini, S.M.; Di Giacomo, M.; Peviani, E.G.; Belvisi, L.; Colombo, L. Synthesis of Easy-to-Functionalize Aza bicycloalkane Scaffolds as Dipeptide Turn Mimics En Route to CRGD-Based Bioconjugates: Synthesis of Easily Functionalizable Azabicycloalkane Scaffolds. Eur. J. Org. Chem. 2015, 2015, 7557–7570. [Google Scholar] [CrossRef]
- Serra, M.; Terreni, M.; Bernardi, E.; Colombo, L. Synthesis of Functionalized Proline-Derived Azabicycloalkane Amino Acids and Their Applications in Drug Discovery: Recent Advances. Eur. J. Org. Chem. 2023, 26. [Google Scholar]
- Pilkington-Miksa, M.; Arosio, D.; Battistini, L.; Belvisi, L.; De Matteo, M.; Vasile, F.; Burreddu, P.; Carta, P.; Rassu, G.; Perego, P.; et al. Design, Synthesis, and Biological Evaluation of Novel CRGD–Paclitaxel Conjugates for Integrin-Assisted Drug Delivery. Bioconjug. Chem. 2012, 23, 1610–1622. [Google Scholar] [CrossRef]
- Lanzardo, S.; Conti, L.; Brioschi, C.; Bartolomeo, M.P.; Arosio, D.; Belvisi, L.; Manzoni, L.; Maiocchi, A.; Maisano, F.; Forni, G. A New Optical Imaging Probe Targeting AVβ3 Integrin in Glioblastoma Xenografts: OPTICAL IMAGING OF AVβ3 INTEGRIN. Contrast Media Mol. Imaging 2011, 6, 449–458. [Google Scholar]
- Manzoni, L.; Belvisi, L.; Arosio, D.; Bartolomeo, M.P.; Bianchi, A.; Brioschi, C.; Buonsanti, F.; Cabella, C.; Casagrande, C.; Civera, M.; De Matteo, M.; Fugazza, L.; Lattuada, L.; Maisano, F.; Miragoli, L.; Neira, C.; Pilkington-Miksa, M.; Scolastico, C. Synthesis of Gd and 68 Ga Complexes in Conjugation with a Conformationally Optimized RGD Sequence as Potential MRI and PET Tumor-Imaging Probes. ChemMedChem 2012, 7, 1084–1093. [Google Scholar] [PubMed]
- Battistini, L.; Burreddu, P.; Sartori, A.; Arosio, D.; Manzoni, L.; Paduano, L.; D’Errico, G.; Sala, R.; Reia, L.; Bonomini, S.; Rassu, G.; Zanardi, F. Enhancement of the Uptake and Cytotoxic Activity of Doxorubicin in Cancer Cells by Novel CRGD-Semipeptide-Anchoring Liposomes. Mol. Pharm. 2014, 11, 2280–2293. [Google Scholar] [CrossRef] [PubMed]
- Bari, E.; Serra, M.; Paolillo, M.; Bernardi, E.; Tengattini, S.; Piccinini, F.; Lanni, C.; Sorlini, M.; Bisbano, G.; Calleri, E.; Torre, M.L.; Perteghella, S. Silk Fibroin Nanoparticle Functionalization with Arg-Gly-Asp Cyclopentapeptide Promotes Active Targeting for Tumor Site-Specific Delivery. Cancers 2021, 13, 1185. [Google Scholar] [CrossRef] [PubMed]
- Spinello, A.; Barone, G.; Grunenberg, J. Molecular Recognition of Naphthalene Diimide Ligands by Telomeric Quadruplex-DNA: The Importance of the Protonation State and Mediated Hydrogen Bonds. Phys. Chem. Chem. Phys. 2016, 18, 2871–2877. [Google Scholar]
- Doria, F.; Salvati, E.; Pompili, L.; Pirota, V.; D’Angelo, C.; Manoli, F.; Nadai, M.; Richter, S.N.; Biroccio, A.; Manet, I.; Freccero, M. Dyads of G-Quadruplex Ligands Triggering DNA Damage Response and Tumour Cell Growth Inhibition at Subnanomolar Concentration. Chem. – Eur. J. 2019, 25, 11085–11097. [Google Scholar] [CrossRef]
- Micco, M.; Collie, G.W.; Dale, A.G.; Ohnmacht, S.A.; Pazitna, I.; Gunaratnam, M.; Reszka, A.P.; Neidle, S. Structure-Based Design and Evaluation of Naphthalene Diimide G-Quadruplex Ligands As Telomere Targeting Agents in Pancreatic Cancer Cells. J. Med. Chem. 2013, 56, 2959–2974. [Google Scholar] [CrossRef]
- Ahmed, A.A.; Angell, R.; Oxenford, S.; Worthington, J.; Williams, N.; Barton, N.; Fowler, T.G.; O’Flynn, D.E.; Sunose, M.; McConville, M.; Vo, T.; Wilson, W.D.; Karim, S.A.; Morton, J.P.; Neidle, S. Asymmetrically Substituted Quadruplex-Binding Naphthalene Diimide Showing Potent Activity in Pancreatic Cancer Models. ACS Med. Chem. Lett. 2020, 11, 1634–1644. [Google Scholar]
- Pirota, V.; Nadai, M.; Doria, F.; Richter, S. Naphthalene Diimides as Multimodal G-Quadruplex-Selective Ligands. Molecules 2019, 24, 426. [Google Scholar] [CrossRef]
- Pirota, V.; Bisbano, G.; Serra, M.; Torre, M.L.; Doria, F.; Bari, E.; Paolillo, M. CRGD-Functionalized Silk Fibroin Nanoparticles: A Strategy for Cancer Treatment with a Potent Unselective Naphthalene Diimide Derivative. Cancers 2023, 15, 1725. [Google Scholar]
- Information about past and ongoing clinical trials are available on the U.S. National Library of Medicine’s clinical trials website https://www.clinicaltrials.gov/ct2/home.
- Cirkel, G.A.; Kerklaan, B.M.; Vanhoutte, F.; Der Aa, A.V.; Lorenzon, G.; Namour, F.; Pujuguet, P.; Darquenne, S.; De Vos, F.Y.F.; Snijders, T.J.; Voest, E.E.; et al. A Dose Escalating Phase I Study of GLPG0187, a Broad Spectrum Integrin Receptor Antagonist, in Adult Patients with Progressive High-Grade Glioma and Other Advanced Solid Malignancies. Invest. New Drugs 2016, 34, 184–192. [Google Scholar] [CrossRef]
- Rosenthal, M.A.; Davidson, P.; Rolland, F.; Campone, M.; Xue, L.; Han, T.H.; Mehta, A.; Berd, Y.; He, W.; Lombardi, A. Evaluation of the Safety, Pharmacokinetics and Treatment Effects of an α ν β 3 Integrin Inhibitor on Bone Turnover and Disease Activity in Men with Hormone-Refractory Prostate Cancer and Bone Metastases. Asia Pac. J. Clin. Oncol. 2010, 6, 42–48. [Google Scholar]
- Zhou, X.; Zhang, J.; Haimbach, R.; Zhu, W.; Mayer-Ezell, R.; Garcia-Calvo, M.; Smith, E.; Price, O.; Kan, Y.; Zycband, E.; Zhu, Y.; Hoek, M.; Cox, J.M.; Ma, L.; Kelley, D.E.; Pinto, S. An Integrin Antagonist (MK-0429) Decreases Proteinuria and Renal Fibrosis in the ZSF1 Rat Diabetic Nephropathy Model. Pharmacol. Res. Perspect. 2017, 5, e00354. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.M.; et al. Composition and methods for treating chronic kidney disease. US 20190307735 2019. [Google Scholar]
- Stoeltzing, O.; Liu, W.; Reinmuth, N.; Fan, F.; Parry, G.C.; Parikh, A.A.; McCarty, M.F.; Bucana, C.D.; Mazar, A.P.; Ellis, L.M. Inhibition of Integrin ?5?1 Function with a Small Peptide (ATN-161) plus Continuous 5-FU Infusion Reduces Colorectal Liver Metastases and Improves Survival in Mice. Int. J. Cancer 2003, 104, 496–503. [Google Scholar] [CrossRef]
- Khalili, P.; Arakelian, A.; Chen, G.; Plunkett, M.L.; Beck, I.; Parry, G.C.; Doñate, F.; Shaw, D.E.; Mazar, A.P.; Rabbani, S.A. A Non–RGD-Based Integrin Binding Peptide (ATN-161) Blocks Breast Cancer Growth and Metastasis in Vivo. Mol. Cancer Ther. 2006, 5, 2271–2280. [Google Scholar] [CrossRef]
- Doñate, F.; Parry, G.C.; Shaked, Y.; Hensley, H.; Guan, X.; Beck, I.; Tel-Tsur, Z.; Plunkett, M.L.; Manuia, M.; Shaw, D.E.; Kerbel, R.S.; Mazar, A.P. Pharmacology of the Novel Antiangiogenic Peptide ATN-161 (Ac-PHSCN-NH2): Observation of a U-Shaped Dose-Response Curve in Several Preclinical Models of Angiogenesis and Tumor Growth. Clin. Cancer Res. 2008, 14, 2137–2144. [Google Scholar] [CrossRef]
- Beddingfield, B.J.; Iwanaga, N.; Chapagain, P.P.; Zheng, W.; Roy, C.J.; Hu, T.Y.; Kolls, J.K.; Bix, G.J. The Integrin Binding Peptide, ATN-161, as a Novel Therapy for SARS-CoV-2 Infection. JACC Basic Transl. Sci. 2021, 6, 1–8. [Google Scholar]
- Makowski, L.; Olson-Sidford, W.; W. Weisel, J. Biological and Clinical Consequences of Integrin Binding via a Rogue RGD Motif in the SARS CoV-2 Spike Protein. Viruses 2021, 13, 146. [Google Scholar] [CrossRef]
- Sigrist, C.J.; Bridge, A.; Le Mercier, P. A Potential Role for Integrins in Host Cell Entry by SARS-CoV-2. Antiviral Res. 2020, 177, 104759. [Google Scholar] [CrossRef]
- Hailemichael, Y.; Vanderslice, P.; Market, R.V.; Biediger, R.J.; Woodside, D.G.; Marathi, U.K.; Overwijk, W.W. Abstract 5010: Potentiating Immune Checkpoint Blockade Therapeutic Efficacy Using a Small Molecule Activator of Integrin Cell Adhesion Receptors. Cancer Res. 2019, 79 (13_Supplement), 5010. [Google Scholar]
- Hills Pharma’s Clinical-stage Novel Immunostimulant 7HP349 Granted FDA Fast Track Designation for Anti-PD-1-resistant Metastatic Melanoma. News Release. Biospace. March 8, 2022. 8 March.
- Ranasinghe, P.; Addison, M.L.; Dear, J.W.; Webb, D.J. Small Interfering RNA: Discovery, Pharmacology and Clinical Development—An Introductory Review. Br. J. Pharmacol. 2022; bph.15972. [Google Scholar]
- Friedrich, M.; Aigner, A. Therapeutic SiRNA: State-of-the-Art and Future Perspectives. BioDrugs 2022, 36, 549–571. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Zhong, L.; Weng, Y.; Peng, L.; Huang, Y.; Zhao, Y.; Liang, X.-J. Therapeutic SiRNA: State of the Art. Signal Transduct. Target. Ther. 2020, 5, 101. [Google Scholar]
- Selvam, C.; Mutisya, D.; Prakash, S.; Ranganna, K.; Thilagavathi, R. Therapeutic Potential of Chemically Modified SiRNA: Recent Trends. Chem. Biol. Drug Des. 2017, 90, 665–678. [Google Scholar] [CrossRef]
- Deleavey, G.F.; Watts, J.K.; Damha, M.J. Chemical Modification of SiRNA. Curr. Protoc. Nucleic Acid Chem. 2009, 39. [Google Scholar] [CrossRef] [PubMed]
- Tatiparti, K.; Sau, S.; Kashaw, S.; Iyer, A. SiRNA Delivery Strategies: A Comprehensive Review of Recent Developments. Nanomaterials 2017, 7, 77. [Google Scholar] [CrossRef]
- Haraszti, R.A.; Roux, L.; Coles, A.H.; Turanov, A.A.; Alterman, J.F.; Echeverria, D.; Godinho, B.M.D.C.; Aronin, N.; Khvorova, A. 5΄-Vinylphosphonate Improves Tissue Accumulation and Efficacy of Conjugated SiRNAs in Vivo. Nucleic Acids Res. 2017, 45, 7581–7592. [Google Scholar] [CrossRef]
- Chernikov, I.V.; Vlassov, V.V.; Chernolovskaya, E.L. Current Development of SiRNA Bioconjugates: From Research to the Clinic. Front. Pharmacol. 2019, 10, 444. [Google Scholar] [CrossRef]
- Nguyen, G.N.; Everett, J.K.; Kafle, S.; Roche, A.M.; Raymond, H.E.; Leiby, J.; Wood, C.; Assenmacher, C.-A.; Merricks, E.P.; Long, C.T.; Kazazian, H.H.; Nichols, T.C.; Bushman, F.D.; Sabatino, D.E. A Long-Term Study of AAV Gene Therapy in Dogs with Hemophilia A Identifies Clonal Expansions of Transduced Liver Cells. Nat. Biotechnol. 2021, 39, 47–55. [Google Scholar] [CrossRef]
- Dong, Y.; Siegwart, D.J.; Anderson, D.G. Strategies, Design, and Chemistry in SiRNA Delivery Systems. Adv. Drug Deliv. Rev. 2019, 144, 133–147. [Google Scholar] [CrossRef]
- Kulkarni, J.A.; Witzigmann, D.; Chen, S.; Cullis, P.R.; Van Der Meel, R. Lipid Nanoparticle Technology for Clinical Translation of SiRNA Therapeutics. Acc. Chem. Res. 2019, 52, 2435–2444. [Google Scholar] [CrossRef] [PubMed]
- Li, S.-D.; Chono, S.; Huang, L. Efficient Oncogene Silencing and Metastasis Inhibition via Systemic Delivery of SiRNA. Mol. Ther. 2008, 16, 942–946. [Google Scholar] [CrossRef]
- Guo, P.; You, J.-O.; Yang, J.; Jia, D.; Moses, M.A.; Auguste, D.T. Inhibiting Metastatic Breast Cancer Cell Migration via the Synergy of Targeted, PH-Triggered SiRNA Delivery and Chemokine Axis Blockade. Mol. Pharm. 2014, 11, 755–765. [Google Scholar] [CrossRef]
- Sakurai, Y.; Hada, T.; Kato, A.; Hagino, Y.; Mizumura, W.; Harashima, H. Effective Therapy Using a Liposomal SiRNA That Targets the Tumor Vasculature in a Model Murine Breast Cancer with Lung Metastasis. Mol. Ther. - Oncolytics 2018, 11, 102–108. [Google Scholar] [CrossRef] [PubMed]
- Li, S.-D.; Chono, S.; Huang, L. Efficient Gene Silencing in Metastatic Tumor by SiRNA Formulated in Surface-Modified Nanoparticles. J. Controlled Release 2008, 126, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Morry, J.; Ngamcherdtrakul, W.; Gu, S.; Reda, M.; Castro, D.J.; Sangvanich, T.; Gray, J.W.; Yantasee, W. Targeted Treatment of Metastatic Breast Cancer by PLK1 SiRNA Delivered by an Antioxidant Nanoparticle Platform. Mol. Cancer Ther. 2017, 16, 763–772. [Google Scholar] [CrossRef] [PubMed]
- Hazekawa, M.; Nishinakagawa, T.; Hosokawa, M.; Ishibashi, D. Development of an Organ-Directed Exosome-Based SiRNA-Carrier Derived from Autologous Serum for Lung Metastases and Testing in the B16/BL6 Spontaneous Lung Metastasis Model. Pharmaceutics 2022, 14, 815. [Google Scholar] [CrossRef]
- Zhao, L.; Gu, C.; Gan, Y.; Shao, L.; Chen, H.; Zhu, H. Exosome-Mediated SiRNA Delivery to Suppress Postoperative Breast Cancer Metastasis. J. Controlled Release 2020, 318, 1–15. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, G.; Yu, T.; He, J.; Liu, J.; Chai, X.; Zhao, G.; Yin, D.; Zhang, C. Exosomes Deliver LncRNA DARS-AS1 SiRNA to Inhibit Chronic Unpredictable Mild Stress-Induced TNBC Metastasis. Cancer Lett. 2022, 543, 215781. [Google Scholar] [CrossRef]
- Zeng, L.; Li, J.; Wang, Y.; Qian, C.; Chen, Y.; Zhang, Q.; Wu, W.; Lin, Z.; Liang, J.; Shuai, X.; Huang, K. Combination of SiRNA-Directed Kras Oncogene Silencing and Arsenic-Induced Apoptosis Using a Nanomedicine Strategy for the Effective Treatment of Pancreatic Cancer. Nanomedicine Nanotechnol. Biol. Med. 2014, 10, 463–472. [Google Scholar] [CrossRef]
- Xie, J.; Wang, S. Small Interfering RNA in Colorectal Cancer Liver Metastasis Therapy. Technol. Cancer Res. Treat. 2022, 21, 153303382211033. [Google Scholar] [CrossRef]
- Kang, S.H.; Revuri, V.; Lee, S.-J.; Cho, S.; Park, I.-K.; Cho, K.J.; Bae, W.K.; Lee, Y. Oral SiRNA Delivery to Treat Colorectal Liver Metastases. ACS Nano 2017, 11, 10417–10429. [Google Scholar] [CrossRef]
- Wang, F.; Pang, J.; Huang, L.; Wang, R.; Jiang, Q.; Zhang, L.; Sun, K. Inhibition of Osteosarcoma Growth and Metastasis Using a Polysaccharide Derivative of Amy-g-PLLD for the Delivery of AEG-1 SiRNA. Nano Res. 2018, 11, 3886–3898. [Google Scholar] [CrossRef]
- Zhou, Z.; Zhu, J.-S.; Gao, C.-P.; Li, L.-P.; Zhou, C.; Wang, H.; Liu, X.-G. SiRNA Targeting YAP Gene Inhibits Gastric Carcinoma Growth and Tumor Metastasis in SCID Mice. Oncol. Lett. 2016, 11, 2806–2814. [Google Scholar] [CrossRef] [PubMed]
- Kamerkar, S.; LeBleu, V.S.; Sugimoto, H.; Yang, S.; Ruivo, C.F.; Melo, S.A.; Lee, J.J.; Kalluri, R. Exosomes Facilitate Therapeutic Targeting of Oncogenic KRAS in Pancreatic Cancer. Nature 2017, 546, 498–503. [Google Scholar] [CrossRef]
- Rossi, J.J.; Rossi, D.J. SiRNA Drugs: Here to Stay. Mol. Ther. 2021, 29, 431–432. [Google Scholar] [CrossRef]
- Larson, M.H.; Gilbert, L.A.; Wang, X.; Lim, W.A.; Weissman, J.S.; Qi, L.S. CRISPR Interference (CRISPRi) for Sequence-Specific Control of Gene Expression. Nat. Protoc. 2013, 8, 2180–2196. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.M.; Bahal, R.; Rasmussen, T.P.; Manautou, J.E.; Zhong, X. The Growth of SiRNA-Based Therapeutics: Updated Clinical Studies. Biochem. Pharmacol. 2021, 189, 114432. [Google Scholar] [CrossRef] [PubMed]
- Ombrato, L.; Nolan, E.; Kurelac, I.; Mavousian, A.; Bridgeman, V.L.; Heinze, I.; Chakravarty, P.; Horswell, S.; Gonzalez-Gualda, E.; Matacchione, G.; Weston, A.; Kirkpatrick, J.; Husain, E.; Speirs, V.; Collinson, L.; Ori, A.; Lee, J.-H.; Malanchi, I. Metastatic-Niche Labelling Reveals Parenchymal Cells with Stem Features. Nature 2019, 572, 603–608. [Google Scholar] [CrossRef]
- Passaro, D.; Garcia-Albornoz, M.; Diana, G.; Chakravarty, P.; Ariza-McNaughton, L.; Batsivari, A.; Borràs-Eroles, C.; Abarrategi, A.; Waclawiczek, A.; Ombrato, L.; Malanchi, I.; Gribben, J.; Bonnet, D. Integrated OMICs Unveil the Bone-Marrow Microenvironment in Human Leukemia. Cell Rep. 2021, 35, 109119. [Google Scholar] [CrossRef]
- Han, Y.; Wang, D.; Peng, L.; Huang, T.; He, X.; Wang, J.; Ou, C. Single-Cell Sequencing: A Promising Approach for Uncovering the Mechanisms of Tumor Metastasis. J. Hematol. Oncol.J Hematol Oncol 2022, 15, 59. [Google Scholar] [CrossRef]
Scheme 1.
cRGD derivatives with application in anticancer therapy.
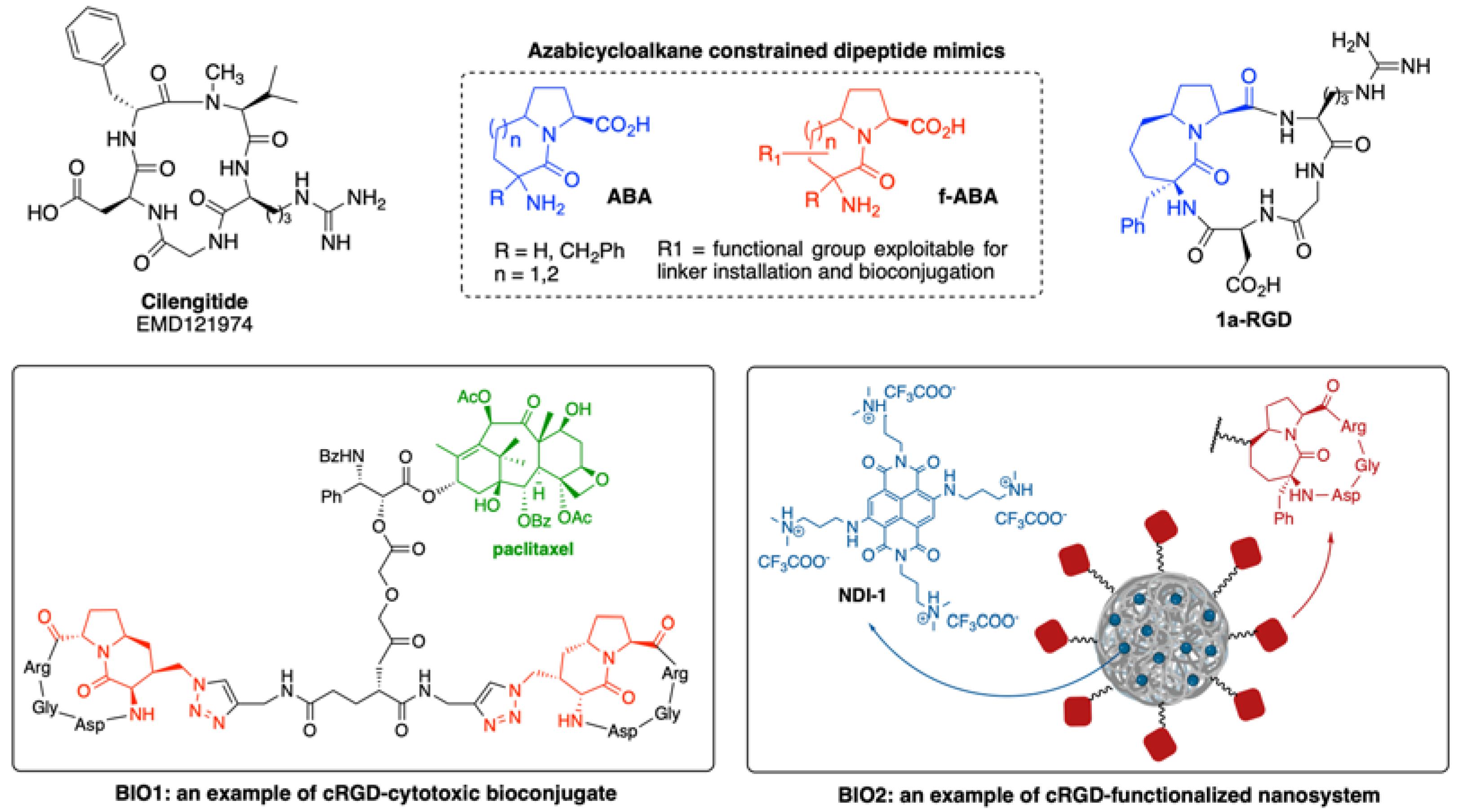
Figure 1.
siRNA modifications used for cancer treatment.
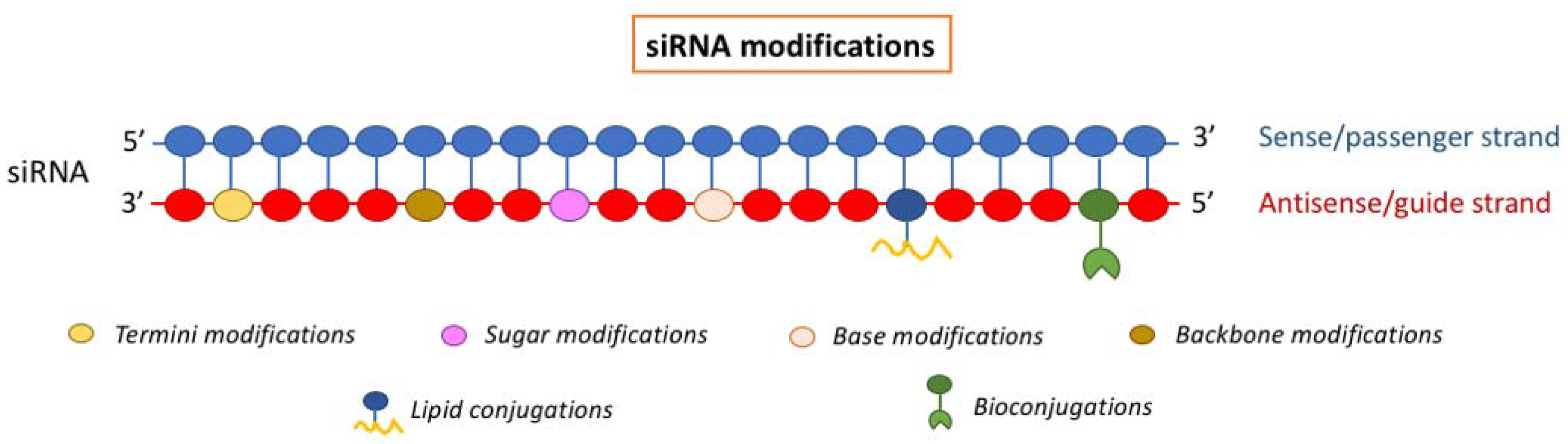
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



