Submitted:
06 June 2023
Posted:
25 June 2023
You are already at the latest version
Abstract
Jatropha curcas is a potential energy crop and has been identified as a significant resource for biodiesel. Retrotransposons occupy the plant genome in a large proportion, so the specific retrotransposon characterization will help improve understanding of the Jatropha genome evolution and organization. This research aimed to characterize and classify Ty1-copia retrotransposons in J. curcas using specific gene sequence and to detect the distribution of Ty1-copia in J. curcas and J. integerrima chromosomes using florescence in situ hybridization (FISH) method. Ty1-copia sequences from J. curcas were isolated using three degenerate primer sets specific to the conserved Ty1-copia reverse transcriptase gene for polymerase chain reaction (PCR). FISH technique using the Ty1-copia probes was used to map physically on J. curcas and J. integerrima chromosomes. Altogether, 164 Ty1-copia sequences were obtained to use for the phylogenetic analysis and were classified into the four families of TAR, Angela, Ale and Bianca. Physical map using FISH method of Angela and Ale families on J. curcas and J. integerrima chromosomes exhibited biases for specific regions around the centromeres or at the chromosome terminal regions. The study indicates the high variation of the Ty1-copia elements in the genome of Jatropha. These results will enhance understanding of retrotransposon chromosomal distribution and evolution of the J. curcas genome and also promotion of retrotransposon possible utilization in J. curcas improvement projects using retrotransposon-based markers.
Keywords:
florescence in situ hybridization
; Jatropha curcas
; phylogenetic tree analysis
; Ty1-copia retrotransposons
1. Introduction
Jatropha curcas L. or physic nut is an energy crop potential in the use of renewable energy [1], yield since the first year, the oil is not consumed as food and a high oil content in seeds making it possible to use J. curcas as a feedstock to produce biodiesel. It is highly resistant to drought [2]; therefore, planting does not need to compete with food crops that require water and fertile soil. Moreover, in the process of oil chests squeezing, the remaining residue can be used as raw material in the manufacture of fertilizers, plastics, synthetic fibers and animal feeds [3,4,5,6,7].
It is commonly known that the activities of retrotransposons can be transferred in the genome, resulting in differences in the plant genome. Retrotransposons are found widely distributed in the plant genome as one of two types of TE (transposable elements). They can replicate themselves via RNA intermediates and lead to the regulation of different gene expression. These cause retrotransposons to be a significant element in the plant genome evolution [8].
There are two types of retrotransposons, that are long terminal repeat (LTR) and non-long terminal repeat (non-LTR). Approximately 40-70% of retrotransposons found in the plant genome are the LTR type which has two important domains, including group-specific antigen (Gag) domain and polyprotein domain (Pol) [9]. The Pol domain consists of 4 gene regulating enzymes, including protease, integrase, reverse transcriptase and RNase H which serves to cut Pol polyprotein and causes retrotransposon movement. LTR retrotransposons contain two large groups: Ty1-copia and Ty3-gypsy [9,10].
Fluorescent in situ hybridization (FISH) technique as well as its variation have been widely utilized in plant karyotype characterizations [11]. The techniques generally consist of matching the probes, which can be DNA or RNA, that have a specific sequence into the target genome. The objective is to specify the exact location in the chromosome. FISH uses various probes, either tandem repeats/rDNAs, centromeric/telomeric satellites or another specific DNA sequences of interest. FISH studies in plant chromosomes can identify the distribution of retrotransposons on plant chromosomes. Ty1-copia tends to be found at the distances away from the centromere of the plant chromosome, while gypsy-type is often found at the centromere or maybe opposite in some plants [12]. The study of retrotransposons in the Jatropha chromosome will be useful for understanding Jatropha genome, leading to successful breeding of this plant.
The aim of the present study was to isolate and characterize Ty1-copia retrotransposons in J. curcas collected from Thailand and other countries and to use the FISH technique to detect the distribution of Ty1-copia retrotransposons in J. curcas and J. integerima chromosomes. The results from the study will give more information on the diversity of Ty1-copia retrotransposons in J. curcas that will be useful for DNA marker development associated with breeding as well as improvement of the understanding of Jatropha genome evolution and organization.
2. Results and Discussion
2.1. Characterization of Ty1-copia sequences in the J. curcas genome
Three degenerate primer pairs related to the conserved sequence of the Ty1-copia reverse transcriptase genes in higher plants [13,14,15] were used by PCR-based method to increase the amount of DNA fragments of about 251-280 bp from six samples of J. curcas collected from Thailand and three samples of J. curcas obtained from Madagascar, Indonesia and Taiwan (Table 1). All of the obtained PCR products were cloned and selected for at least 40 clones from the individual of the nine J. curcas samples. The inserted clones were selected by preliminary grouping based on restriction patterns after cutting the inserted PCR products by restriction enzymes AluI, EcoRI, HaeIII, BclI and NcoI before sequencing. All three degenerate primer pairs could amplify various Ty1-copia elements. Altogether, 164 resulting sequences were used for the phylogenetic tree analysis. This study indicates the high diversity of the Ty1-copia elements in the genome of Jatropha curcas.
Comparison of all 164 obtained sequences to the GenBank-NCBI database using the BLAST network service at http://www.ncbi.nlm.nih.gov/BLAST/ was performed and the analysis using SIB ExPASy Bioinformatic Resources portal at https://web.expasy.org/translate/ was done. Amino acid sequences of approximately 83-93 amino acids in length derived from all isolated Ty1-copia fragments, together with retrotransposons from other species, Phaeodactylum tricornutum (GenBank: EU432487.1) as an outgroup, were aligned and then used to construct a phylogenetic tree based on maximum likelihood (ML) method (Figure 1). Consistent with the high heterogeneity level reported among the reverse transcriptase (RT) sequences of Ty1-copia in plants [16], most of our isolated RT sequences were found different from each other and were found distributed into 12 clusters, which corresponds to Ty1-copia families of Jatropha curcas. The identified Ty1-copia sequences were classified in to four distinct groups (TAR, Angela, Ale and Bianca) with high bootstrap values, which indicated that they belong to at least four distinct Ty1-copia families in the Jatropha genome. Family TAR were found in a large number of isolated Ty1-copia clones (46.01%) that further classified into five subfamilies, followed by family Angela (34.97%) and family Ale (19.02%) which both could be further divided into 3 subfamilies, while Bianca possessed the smallest number of clone (0.61%) as shown in Figure 1. The isolated retrotransposon clone numbers in the individual family were counted and the patterns of retrotransposon family distribution were identified among the nine Jatropha curcas samples. We found that distribution patterns of Ty1-copia families were different among the nine Jatropha samples (Table 1).
The consensus reverse transcriptase gene sequence of individual family was analysed and used to estimate relative copy number of the individual family in the Jatropha genome by the hit number of BLAST search in the database of Jatropha Genome at http://www.kazusa.or.jp/jatropha/ [17]. Average of Hit numbers of the TAR (75 clones), Angela (57 clones), Ale (31 clones) and Bianca (only 1 clone) were 61, 69, 64 and 11 respectively.
Molecular markers, for example SSR, AFLP and RAPD, have been widely used to evaluate genetic relationship among plants. Using these markers in J. curcas, almost very low genetic divergence was detected among the J. curcas varieties currently cultivated in Asia and America [18,19]. Therefore, other types of DNA markers, such as retrotransposon-based markers are of importance in order to use to identify and classify genetic characteristics of plants with narrow genetic base. This will increase the breeding efficiency in the Jatropha population.
Retrotransposons have been known to be one of the important components of the eukaryotic genome. This should be related to the variation and also evolution of the species. The differences within the inactive elements can contribute to plant genome evolution, gene duplication events and new properties in the retrotransposition mechanisms [20]. Detection of the retrotransposon presence or absence in an appropriate method will enhance characterization a population with low genetic diversity.
Sizes of plant genome are reported to be positively related to both differences and copy numbers of retrotransposon families [21]. The Ty1-copia in the plant kingdom have been characterized into six major families: lvana, Maximus, Ale, TAR, Angela and Bianca [22]. Ty1-copia families are reported to be more evolutionarily scattered and smaller in size than the Ty3-gypsy [23]. Different plant species carry different number of retrotransposon elements. Arabidopsis, having a small genome size of about 157 Mb, contains LTR retrotransposons in a very limited number of 5.60% [24], whereas rice with genome size of about 389 Mb is composed of 22% LTR retrotransposon sequences [25]. Maize, having large-sized genome of about 2045 Mb, has 74.6% of the LTR retrotransposons elements [26], while J. curcas with genome size of about 370 Mb contains 36% of transposable elements, comprising of Ty1-copia and Ty3-gypsy by 8.0% and 19.6%, respectively [27]. In this study on Ty1-copia characterization in J. curcas, we found four families of Ty1-copia, including TAR, Angela, Ale and Bianca, and distribution patterns of the four families were different among the nine Jatropha samples studied. TAR was found in a large number (46.01%), followed by Angela and Ale, whereas Bianca was found in a small number (0.61%), with only 1 sequence found from J. curcas from Indonesia. The result is consistent with previous reports on soybeans in which Ivana has the largest number of Ty1-copia families and Bianca was not found [28]. Tuntipaiboontana et al. (2018) [29] reported Ty1-copia in waterlilies and found Ale to be abundant and diverse, whereas Angela and TAR were found to be conserved.
Among LTR retrotransposons in plant species, the Ty1-copia superfamily has been reported as a multitude component of both angiosperm and gymnosperm genomes [30]. In our study, a degenerated primer method was used to evaluate the existence of Ty1-copia retrotransposons in J. curcas. The results indicated the existence of copia-related sequences in all genomes of nine J. curcas samples. This suggested that all of the nine J. curcas samples share this repetitive elements in their genomes. In addition, the deduced amino acid sequences support the presence of J. curcas Ty1-copia sequences, where the consensus domain SLYGLKQA (Figure 2), which is the Ty1-copia characteristic of plant genome, was found to be highly conserved.
2.2. Molecular cytogenetic evaluation of the Ty1-copia in Jatropha based on FISH
DAPI-stained chromosomes in primary root of J. curcas at metaphase did not show clear constriction or distinguishing features among chromosomes in this study (Figure 3A). However, Kikuchi et al. (2011) [31] performed cytogenetic approach and reported that chromosomes of J. curcas are mostly metacentric or submetacentric and of similar size. In addition, in our study the relaxed prometaphase chromosomes of J. curcas showed obvious of centromere (primary constriction), secondary constriction and satellite (Figure 3B). The condensation patterns observed in chromosomes at the prometaphase stage of J. curcas were similar to those in the chromosomes of coffee, quinoa and lemon, in which the centromeric and telomeric regions mostly consist of the highly repetitive elements [32,33,34].
The chromosome distribution of our two identified families of Ty1-copia, Ale and Angela, was evaluated using FISH technique on mitotic chromosomes of both J. curcas and J. integerrima, Results suggested that Ale and Angela Ty1-copia families are dominantly clustered around the centromeres or at the chromosome terminal regions of Jatropha chromosomes (Figure 4A,B). The FISH result confirms the existence of Ty1-copia, Ale and Angela, in genomes of both J. curcas and J. integerrima in this study. Patterns of Ty1-copia distribution in the chromosomes of Jatropha were the same as those in slash pine, lemon and oat, in that telomeric regions contained mostly repetitive retrotransposons [34,35]. In addition, the results were supported by the analysis of flanking regions in the EMBL database under AJ269530 and AJ269531 accession numbers [36].
During normal development, transposable elements have been known to be silenced, but under abiotic and biotic stress, they will be motivated. The stimulators of stress, for example wounding, pathogen attack, allopolyploidization tissue culture conditions and unfavourable environmental conditions, including suboptimal temperature or water and nutrient availability, can cause transposon activation [37,38]. Under stress, transcripts of plant retrotransposons have been reported to be found in many plants, for example pine, Orobanche and Phelipanche [27,39]. This supports the genomic reorganization role predicted by McClintock for transposable elements in stress response [40]. Stress-activated transposable elements can provide plants more variability, making them possible to survive in stressful environments. This may be due to various genes can be amplified and combined by the activity of transposable elements. New genetic variability induced by the action of transposable elements can facilitate quick adaptation [41]. Therefore, transposable elements are the potential source of genetic variation that can contribute to differences in both genome structure and gene expression. They are an important factor for genome plasticity and genome evolution in plant [42].
3. Materials and Methods
3.1. Isolation and characterization of Ty1-copia retrotransposons in J. curcas
Genomic DNA was extracted individually from young leaves of nine J. curcas accessions, including four high yield accessions from KU Bio-diesel project (KUBP) and two accessions from salinity soil area, representing salt-tolerant cultivars, from Thailand and 3 accessions of J. curcas obtained from Madagascar, Indonesia and Taiwan. DNA extraction was performed following the cetyl trimethyl ammonium bromide (CTAB) extraction method with some modifications [43]. The quality and quantity of the DNA were evaluated based on 0.8% agarose gel electrophoresis.
Three degenerate primer sets corresponding to the highly conserved peptide sequence of Ty1-copia reverse transcriptase (RT) gene were used in this study. The primers include F: 5′–ACNGCNTTYYTNCAYGG–3′ and 5′– ARCATRTCRTCNACRTA–3′ (13), V: 5′–CARATGGAYGTNAARAC–3′ and 5′–CATRTCRTCNACRTA–3′ (14), H: 5′– GAYGTNAARACVGNTTYY–3′ and 5′–AYRTRTCNACRTANARNA–3′ (15). PCR reaction was performed in a volume of 25 µL reaction mixture, containing final concentrations of 100 ng genomic DNA, 2mM MgCl2, 200 µM dNTPs, 50 pmol forward and reverse primer and 2 U Taq DNA polymerase (Invitrogen Life Technology, Brazil). PCR amplification was done in a thermal cycler (PCR-100TM; MJ Research, Inc.; USA) with a PCR program of initial denaturation at 94 °C for 4 min, 35 cycles of denaturation at 94 °C for 1 min, primer annealing at 45 °C for 30 s, extension at 72 °C for 2 min and the final extension at 72 °C for 7 min.
The amplified products were electrophoresed on 1% agarose gel using 1X TBE buffer at 120 V for 35 min. Then, the gel was stained with ethidium bromide (0.5 µg/mL) and was recorded using the Gel Doc XR+ imaging system. PCR products were purified from the agarose gel, cloned into the pGEM®-T Vector (Promega, USA) using T4 DNA ligase (400,000 U/µl, Fermentas, USA), and then the recombination plasmids were transformed into E. coli strain (XL1-Blue) followed by screening white colonies in selective LB/IPTG/X-gal/Ampicillin/agar plates. The transformed clones were screened by PCR analysis (colony PCR) using corresponding M13 primers and were grouped based on restriction patterns produced by five restriction enzymes (NcoI, BclI, HaeIII, EcoRI and AluI). The colonies containing various groups of DNA fragments were selected for extraction of their plasmids using a High-Speed Plasmid Mini Kit (Geneaid, Taiwan). Then the plasmids were sent for sequencing at 1st BASE, Malaysia.
The obtained nucleotide sequences were trimmed and amino acid sequences were deduced using SIB ExPASy Bioinformatic Resources portal [44] with a consideration of spontaneous frameshift mutations. Multiple sequence alignments were performed using Clustal Omega online software [45]. The phylogenetic trees were constructed using maximum likelihood method.
3.2. FISH technique for study of chromosomal distribution of Ty1-copia elements
3.2.1. Chromosome preparation
Seeds of J. curcas and J. integerrima were germinated at 25 °C in a growth chamber. Root-tips of about 0.3-0.5 mm long were collected and pretreated with 2mM 8-hydroxyquinoline at 20 °C for 3 h and then fixed in 3:1 ethanol: acetic acid for 16 h. Root-tips were washed with distilled water and incubated in enzymatic mixture (1% cellulase, 2.5% pectolyase) at 37 °C for 1 h 30 min to 2 h. They were washed for 1 min in distilled water. The meristem was excised and squashed in ethanol: acetic acid (3:1), then exposed to steam and dried in air.
3.2.2. Probe labeling
Total genomic DNA was extracted from young fresh leaves of J. curcas (2n=2x=22) and J. integerrima (2n=2x=22). Based on sequence alignment, the primers specific to the two lineages of Ty1-copia retrotransposons; Ale: 5′-AYGGAGTGTTAAAGGAAGATG-3′ and 5′-CRTACAGAAGCAATACTGCC-3′ and Angela: 5′-AYGGTGAYTTAGAGGOAGGAAG-3′ and 5′-ACTAACATRATGAGCTTGCTC-3′, were developed. PCR products were labeled with Cy3-dUTP (Cytiva) for Ale and digoxigenin-dUTP (Roche) for Angela by nick translation, according to the manufacturer’s protocol. FISH was conducted according to the procedures of Ohmido and Fukui (1997) [46] and Ohmido et al. (1998) [47] with minor modifications. After the cell spreads were dried on slides, they were post-fixed in 1% formaldehyde/1X PBS buffer for 5 min at room temperature, followed by washing the slides with distilled water and then the slides were dried before hybridization.
3.2.3. Hybridization
The hybridization mixture contains labeled probes in hybridization buffer, consisting of 50% formamide, 2X SSC and 10% dextran sulfate. The hybridization mixture was denatured at 80 °C for 10 min and immediately cooled down on ice. 100 µL of 50% formamide in 2X SSC was applied to the chromosomal DNA, covered with a glass cover slip (24 x 45 mm) and then denatured at 70 °C for 4 min. Slides were dehydrated in the ethanol series (70% and 100%) at -20 °C for 5 min each and then the slides were dried. 10 µL of hybridization mixture was applied to each slide, covered with a cover slip (20x20 mm) and sealed with sealing film. Then, slides were incubated in a moist chamber at 37 °C for 18-48 h. After hybridization, the cover slips were removed carefully and washed three times in 20% formamide/2X SSC and 0.1X SSC respectively for 5 min each, at 42 °C. After washing, 100 µL of anti-digoxigenin-fluorescein isothiocyanate (FITC) (Roche) in 4X SSC was applied for 1-2 h at 37 °C in a humid dark box. The slides were washed in 2X SSC at room temperature three times for 5 min each, then air-dried. 70 µL of DAPI (4′6-Diamidino-2-Phenylindole, Dihydrochloride; 0.5 µg/mL) was added and the slides were mounted with a parafilm for 10 min. The slides were washed in water for 5 min and then dried. After washing, 10µL of Vectashield (mounting medium, Vector) was added to slides and the slides were mounted with a glass cover slip (18 x 18 mm).
4. Conclusions
In the present study, Ty1-copia sequences from J. curcas were isolated and characterized using three degenerate primer sets corresponding to the conserved Ty1-copia reverse transcriptase gene. In total, 164 Ty1-copia amino acid sequences were used for the phylogenetic tree construction and were classified into four families of TAR (46.01%), Angela (34.97%), Ale (19.02%) and Bianca (0.61%). FISH technique was used to map physically Angela and Ale families on to J. curcas and J. integerrima chromosomes. They tend to exhibit biases for the specific regions around the centromeres or at the chromosome ends. This study indicates the high diversity of the Ty1-copia elements in the Jatropha genome.
Funding
This research was funded by the Research and Researcher for Industry (RRi) PhD level, grant number PHD57I0043 and by Ladda Co., Ltd.
Data Availability Statement
Data available within the article.
Acknowledgments
We would like to thank Mr. Suksan Suttipolpaiboon and Mr. Pichai Maneechote for supplying Jatropha samples. The Petroleum Authority of Thailand (PTT) funded research on breeding Jatropha using interspecific hybridization under the KU-Biodiesel Project and the Kasetsart University Research and Development Institute (KURDI), Bangkok, Thailand supported research on J. curcas. This work was also supported by JSPS Bilateral Program Number JPJSBP 120203507 and JPJSBP 120223504 to N.O.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Ranade, S.A.; Srivastava, A.P.; Rana, T. S.; Srivastava, J.; Tuli, R. Easy assessment of diversity in Jatropha curcas L. plants using two single-primer amplification reaction (SPAR) methods. Biomass Bioenergy 2008, 32, 533–540. [Google Scholar] [CrossRef]
- Maes, W.H.; Trabucco, A.; Achten, W.M.J.; Muys, B. Climatic growing condition of Jatropha curcas. Biomass Bioenergy 2009, 33, 1481–1485. [Google Scholar] [CrossRef]
- Openshaw, H. A. review of Jatropha curcas: an oil plant of unfulfilled promise. Biomass Bioenergy 2000, 19, 1–15. [Google Scholar] [CrossRef]
- Augustus, G.D.P.S.; Jayabalan, M; Seiler, G. J. Evaluation and bioinduction of energy components of Jatropha curcas. Biomass Bioenergy 2002, 23, 161–164. [Google Scholar] [CrossRef]
- Kaushik, N.; Kumar, K.; Kumar, S.; Kaushik, N.; Roy, S. Genetic variability and divergence studies in seed traits and oil content of Jatropha (Jatropha curcas L.) accessions. Biomass Bioenergy 2007, 31, 497–502. [Google Scholar] [CrossRef]
- Sirisomboon, P.; Kitchaiya, P.; Pholpho, T.; Mahuttanyavanitch, W. Physical and mechanical properties of Jatropha curcas L. fruits, nuts and kernels. Biosyst. Eng. 2007, 97, 201–207. [Google Scholar] [CrossRef]
- Sujatha, M. Biotechnological intervention for improving Jatropha and castor for biofuels. In Petrotech, New Delhi, India, 11-15 January 2009. [Google Scholar]
- Finnegan, D.J. Transposable elements and DNA transposition in eukaryotes. Curr. Opin. Cell Biol. 1990, 2, 471–477. [Google Scholar] [CrossRef]
- Kumar, A.; Bennetzen, J.L. Plant retrotransposons. Annu. Rev. Genet. 1999, 33, 479–532. [Google Scholar] [CrossRef]
- Habibollahi, H.; Noormohammadi, Z.; Sheidai, M.; Farahani, F.; Talebi, S.M.; Torabizadeh, E. Assessments of genetic diversity in Iranian flax populations using retrotransposon microsatellite amplification polymorphisms (REMAP) markers. The Nucleus 2017, 61, 55–60. [Google Scholar] [CrossRef]
- Ohmido, N.; Fukui, K.; Kinoshita, T. Recent advances in rice genome and Chromosome structure research by fluorescence in situ hybridization (FISH). Proc. Jpn. Acad. B. 2010, 86, 103–116. [Google Scholar] [CrossRef]
- Heslop-Harrison, J.S.; Brandes, A.; Taketa, S.; Schmidt, T.; Vershinin, A.V.; Alkhimova, E.G.; Kamm, A.; Doudrick, R.L.; Schwarzacher, T.; Katsiotis, A.; Kubis, S.; Kumar, A.; Pearce, S.R.; Flavell, A.J.; Harrison, G.E. The chromosomal distribution of Ty1-copia group retrotransposable elements in higher plants and their implication for genome evolution. Genetica. 1997, 100, 197–204. [Google Scholar] [CrossRef] [PubMed]
- Flavell, A.J.; Dunbar. E.; Anderson, R.; Pearce, S.R.; Hartley, R.; Kumar, A. Ty1-copia group retrotransposons are ubiquitous and heterogeneous in higher plants. Nucleic Acids Res. 1992, 20, 3639–3644. [Google Scholar] [CrossRef]
- Voytas, D.F.; Cummings, M.P.; Konieczy, A.; Ausubel, F.M. Copia-like retrotransposons are ubiquitous among plants. Proc. Natl. Acad. Sci. USA. 1992, 89, 7124–7128. [Google Scholar] [CrossRef] [PubMed]
- Hirochika, H; Hirochika, R. Ty1-copia group retrotransposons as ubiquitous components of plant genomes. Jpn. J. Genet. 1993, 68, 35–46. [Google Scholar] [CrossRef] [PubMed]
- Ambrozova, K.; Mandakova, T.; Bures, P.; Neumann, P.; Leitch, I.J.; Kobizkova, I.; Macas, J.; Lysak, M.A. Diverse retrotransposon families and AT-rich satellite DNA revealed in giant genomes of Fritillaria lilies. Ann. Bot. 2011, 107, 255–268. [Google Scholar] [CrossRef]
- Sato, S.; Hirakawa, H.; Isobe, S.; Fukai, E.; Watanabe, A.; Kato, M.; Kawashima, K.; Minami, C.; Muraki, A.; Nakazaki, N.; Takahashi, C. Sequence analysis of the genome of an oil-bearing tree, Jatropha curcas L. DNA Res. 2011, 18, 65–76. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.B.; Li, L.F.; Li, Y.; Wu, G.J.; Ge, X.J. SSR and AFLP Markers Reveal Low Genetic Diversity in the Biofuel Plant Jatropha curcas in China. Crop Sci. 2008, 48, 1865–1871. [Google Scholar] [CrossRef]
- Soonthornyatara, S.; Sripichitt, P.; Kaveeta, R.; Hongtrakul, V. Assessment of genetic diversity of Jatropha curcas L. using AFLP and ISSR markers. Chiang Mai J. Sci. 2015, 42, 614–625. [Google Scholar]
- Woodrow, P.; Pontecorvo, G.; Fantaccione, S.; Fuggi, A.; Kafantaris, I.; Parisi, D.; Carillo, P. Polymorphism of a new Ty1-copia retrotransposon in durum wheat under salt and light stresses. Theor. Appl. Genet. 2010, 121, 311–322. [Google Scholar] [CrossRef]
- Vitte, C.; Bennetzen, J.L. Analysis of retrotransposon structural diversity uncovers properties and propensities in angiosperm genome evolution. Proc. Natl. Acad. Sci. USA. 2006, 103, 17638–17643. [Google Scholar] [CrossRef]
- Wicker, T.; Keller, B. Genome-wide comparative analysis of copia retrotransposons in Triticeae, rice, and Arabidopsis reveals conserved ancient evolutionary lineages and distinct dynamics of individual copia families. Genome Res. 2007, 17, 1072–1081. [Google Scholar] [CrossRef] [PubMed]
- Vitte, C.; Panaud, O.; Quesneville, H. LTR retrotransposons in rice (Oryza sativa, L.): recent burst amplifications followed by rapid DNA loss. BMC Genom. 2007, 8, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Pereira, V. Insertion bias and purifying selection of retrotransposons in the Arabidopsis thaliana genome. Genome Biol. 2004, 5, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Devos, K.M.; Bennetzen, J.L. Analyses of LTR-retrotransposon structures reveal recent and rapid genomic DNA loss in rice. Genome Res. 2004, 14, 860–869. [Google Scholar] [CrossRef] [PubMed]
- Baucom, R.S.; Estill, J.C.; Chaparro, C.; Upshaw, N.; Jogi, A.; Deragon, J.M.; Westerman, R.P.; SanMiguel, P.J.; Bennetzen, J.L. Exceptional diversity, non-random distribution, and rapid evolution of retroelements in the B73 maize genome. PLoS Genet. 2009, 5, e1000732. [Google Scholar] [CrossRef] [PubMed]
- Hirakawa, H.; Tsuchimoto, S.; Sakai, H.; Nakayama, S.; Fujishiro, T.; Kishida, Y.; Kohara, M.; Watanabe, A.; Yamada, M.; Aizu, T.; Toyoda, A.; Fujiyama, A.; Tabata, S.; Fukui, K.; Sato, T. Upgraded genomic information of Jatropha curcas L. Plant Biotechnol. 2012, 29, 123–130. [Google Scholar] [CrossRef]
- Du, J.; Tian, Z.; Hans, C.S.; Laten, H.M.; Cannon, S.B.; Jackson, S.A.; Shoemaker, R.C.; Ma, J. Evolutionary conservation, diversity and specificity of LTR-retrotransposons in flowering plants: insights from genome-wide analysis and multi-specific comparison. Plant J. 2010, 63, 584–598. [Google Scholar] [CrossRef]
- Tuntipaiboontana, R.; Kuleung, C.; Hongtrakul, V. Diverse Ty1-copia retrotransposons found in waterlilies of the genus Nymphaea. Hort. J. 2018, 87, 524–531. [Google Scholar] [CrossRef]
- Brandes, A.; Heslop-Harrison, J.S.; Kamm, A.; Kubis, S.; Doudrick, R.L.; Schmidt, T. Comparative analysis of the chromosomal and genomic organization of Ty1-copia-like retrotransposons in pteridophytes, gymnosperms and angiosperms. Plant Mol. Biol. 1997, 33, 11–21. [Google Scholar] [CrossRef]
- Kikuchi, S.; Tsujimoto, H.; Sassa, H.; Koba, T. JcSat1, a novel subtelomeric repeat of Jatropha curcas L. and its use in karyotyping. Chromosome Sci. 2011, 13, 11–16. [Google Scholar]
- Herrera, J.C.; Camayo, G.; Torre, G.D.L.T.; Galeano, N.; Salcedo, E.; Rivera, L.F.; Duran, A. Identification and chromosomal distribution of copia-like retrotransposon sequences in the coffee (Coffea L.) genome. Agron. Colomb. 2013, 31, 269–278. [Google Scholar]
- Kolano, B.; Bednara, E.; Weiss-Schneeweiss, H. Isolation and characterization of reverse transcriptase fragments of LTR retrotransposons from the genome of Chenopodium quinoa (Amaranthaceae). Plant Cell Rep. 2013, 32, 1575–1588. [Google Scholar] [PubMed]
- Felice, B.D.; Wilson, R.R.; Argenziano, C.; Kafantaris, I; Conicella, C. A transcriptionally active copia-like retroelement in citrus limon. Cell. Mol. Biol. Lett. 2009, 14, 289–304. [Google Scholar]
- Kamm, A.; Doudrick, R.L.; Heslop-Harrison, J.S.; Schmidt, T. The genomic and physical organization of Ty1-copia-like sequence as a component of large genomes in Pinus elliottii var. elliottii and other gymnosperms. Proc. Natl. Acad. Sci. USA. 1996, 93, 2708–2713. [Google Scholar]
- Linares, C.; Loarce, Y.; Serna, A.; Fominaya, A. Isolation and characterization of two novel retrotransposons of the Ty1-copia group in oat genomes. Chromosoma. 2001, 110, 25. [Google Scholar]
- Ito, H. Environmental stress and transposons in plants. Genes Genet. Syst. 2022, 97, 169–175. [Google Scholar] [CrossRef]
- Wessler, S.R. Plant retrotransposons: turned on by stress. Curr. Biol. 1996, 6, 959–961. [Google Scholar] [CrossRef] [PubMed]
- Elsik, C.G.; Williams, C.G. Retroelements contribute to the excess low-copy-number DNA in pine. Mol. Gen. Genet. 2000, 264, 47–55. [Google Scholar] [CrossRef]
- McClintock, B. The significance of responses of the genome to challenge. Science. 1984, 226, 792–801. [Google Scholar] [CrossRef]
- Kidwell, M.G.; Lisch, D.R. Perspective: transposable elements, parasitic DNA and genome evolution. Evol. 2001, 2001. 55, 1–24. [Google Scholar]
- Kidwell, M.G.; Lisch, D. Transposable elements as sources of variation in animals and plants. Proc. Natl. Acad. Sci. USA. 1997, 94, 7704–7711. [Google Scholar] [CrossRef] [PubMed]
- Doyle, J.J.; Doyle, J.L. Isolation of plant DNA from fresh tissue. Focus. 1990, 12, 13–15. [Google Scholar]
- Artimo, P.; Jonnalagedda, M.; Arnold, K.; Baratin, D.; Csardi, G.; de Castro, E.; Duvaud, S.; Flegel, V.; Fortier, A.; Gasteiger, E.; Grosdidier, A.; Hernandez, C.; Ioannidis, V.; Kuznetsov, D.; Liechti, R.; Moretti, S.; Mostaguir, K.; Redaschi, N.; Rossier, G.; Xenarios, I.; Stockinger, H. ExPASy: SIB Bioinformatics Resource Potal. Nucleic Acids Res. 2012, 40, 597–603. [Google Scholar] [CrossRef]
- McWilliam, H.; Li, W.; Uludag, M.; Squizzato, S.; Park, Y.M.; Buso, N.; Cowley, A.P.; Lopez, R. Analysis tool web services from EMBL-EBI. Nucleic Acids Res. 2013, 14, W597–W600. [Google Scholar] [CrossRef]
- Ohmido, N.; Fukui, K. Visual verification of close disposition between a rice A genome-specific DNA sequence (TrsA) and the telomere sequence. Plant Mol. Biol. 1997, 35, 963–968. [Google Scholar] [CrossRef] [PubMed]
- Ohmido, N.; Akiyama, Y.; Fukui, K. Physical mapping of unique nucleotide sequences on identified rice chromosomes. Plant Mol. Biol. 1998, 38, 1041–1052. [Google Scholar] [CrossRef]
Figure 1.
Phylogenetic tree based on reverse transcriptase gene sequences for Ty1-copia from nine Jatropha samples. TAR, Angela, Ale and Bianca sequences are marked with yellow, blue, pink and purple circles (I to V represent subfamilies of TAR; VI to VIII represent subfamilies of Angela; IX to XI represent subfamilies of Ale and XII represents family Bianca, containing one sequence belongs to J. curcas from Indonesia and the other sequence belongs to an outgroup: Phaeodactylum tricornutum, CoDi5.2).
Figure 1.
Phylogenetic tree based on reverse transcriptase gene sequences for Ty1-copia from nine Jatropha samples. TAR, Angela, Ale and Bianca sequences are marked with yellow, blue, pink and purple circles (I to V represent subfamilies of TAR; VI to VIII represent subfamilies of Angela; IX to XI represent subfamilies of Ale and XII represents family Bianca, containing one sequence belongs to J. curcas from Indonesia and the other sequence belongs to an outgroup: Phaeodactylum tricornutum, CoDi5.2).
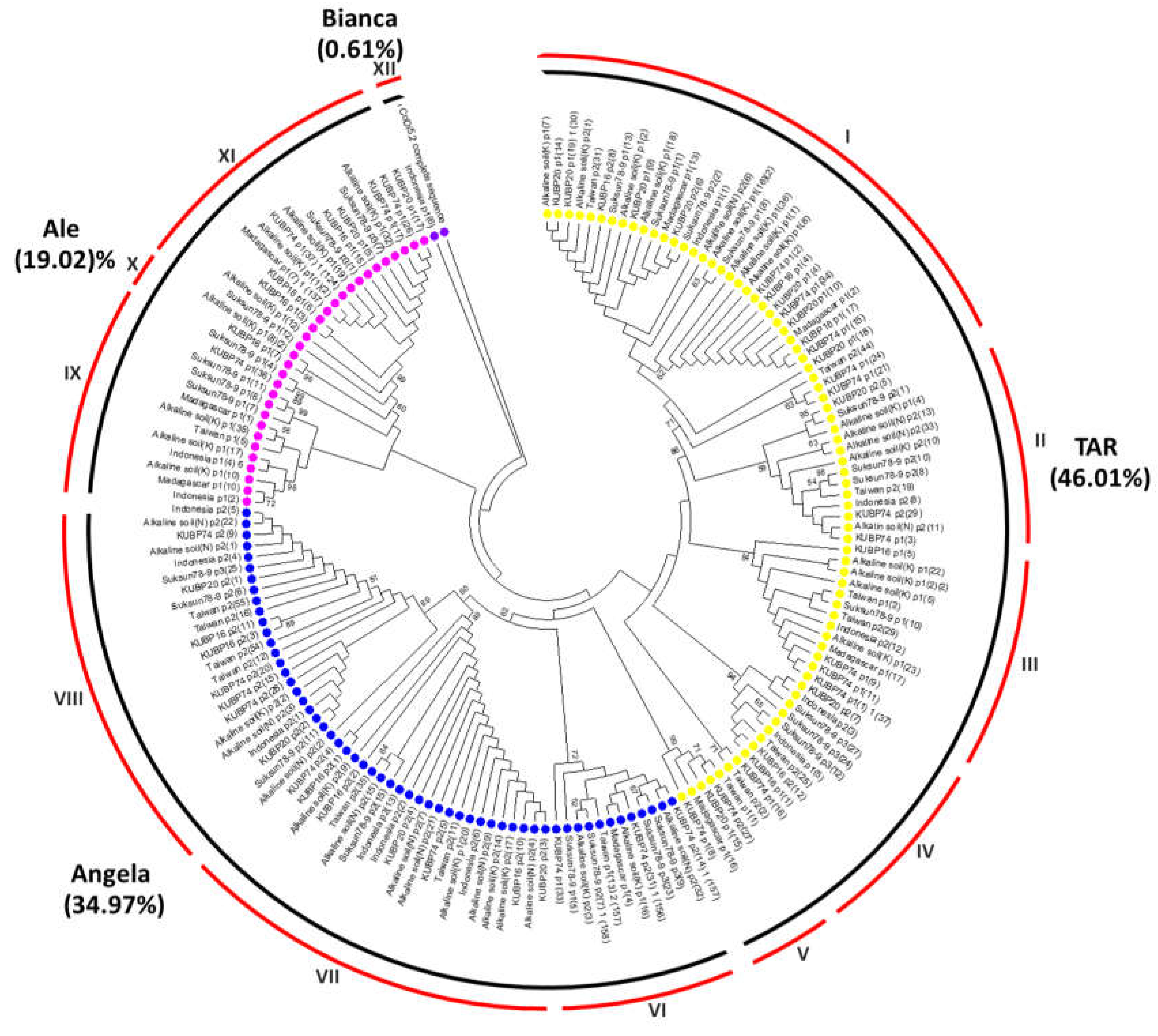
Figure 2.
Alignment of some amino acid sequences corresponding to the fragments of Ty1-copia reverse transcriptase gene isolated from J. curcas. Gaps are indicated as (-) while the consensus domain SLYGLKQA in amino acid sequences are indicated within the two black lines.
Figure 2.
Alignment of some amino acid sequences corresponding to the fragments of Ty1-copia reverse transcriptase gene isolated from J. curcas. Gaps are indicated as (-) while the consensus domain SLYGLKQA in amino acid sequences are indicated within the two black lines.

Figure 3.
DAPI-stained somatic J. curcas chromosomes in primary root (2n = 22). (A) Metaphase chromosomes are mostly observed without clear constrictions. (B) Relaxed prometaphase chromosomes exhibit obviously of centromere, secondary constriction and satellite.
Figure 3.
DAPI-stained somatic J. curcas chromosomes in primary root (2n = 22). (A) Metaphase chromosomes are mostly observed without clear constrictions. (B) Relaxed prometaphase chromosomes exhibit obviously of centromere, secondary constriction and satellite.

Figure 4.
Chromosomes of J. curcas were counterstained with DAPI in light blue color. Red and green color signals represent Ale and Angela Ty1-copia with Cy3-dUTP and Dig-dUTP, respectively. (A) J. curcas, Ale and Angela are found on different chromosomes. (B) J. integerrima, Ale and Angela are found on the same chromosome.
Figure 4.
Chromosomes of J. curcas were counterstained with DAPI in light blue color. Red and green color signals represent Ale and Angela Ty1-copia with Cy3-dUTP and Dig-dUTP, respectively. (A) J. curcas, Ale and Angela are found on different chromosomes. (B) J. integerrima, Ale and Angela are found on the same chromosome.


Table 1.
Distribution of isolated Ty1-copia sequences.
| J. curcas plant samples | Origin | TAR | Angela | Ale | Bianca | Total |
|---|---|---|---|---|---|---|
| KUBP78-9 | Thailand | 11 | 8 | 7 | - | 26 |
| KUBP74 | Thailand | 13 | 9 | 4 | - | 26 |
| KUBP20 | Thailand | 11 | 4 | 2 | - | 17 |
| KUBP16 | Thailand | 6 | 5 | 4 | - | 15 |
| Salt-tolerant #1 from None-Sung district of Nakhon Ratchasima province | Thailand | 4 | 10 | - | - | 14 |
| Salt-tolerant #2 from Kham-Talay-Sor district of Nakhon Ratchasima province | Thailand | 14 | 7 | 8 | - | 29 |
| Madagascar | Madagascar | 4 | 1 | 3 | - | 8 |
| Taiwan | Taiwan | 7 | 7 | 1 | - | 15 |
| Indonesia | Indonesia | 5 | 6 | 2 | 1 | 14 |
| Total | 75 (46.01%) |
57 (34.97%) |
31 (19.02%) |
1 (0.61%) |
164 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



