
Submitted:
23 June 2023
Posted:
26 June 2023
You are already at the latest version
Abstract
This work aimed to develop an easy-to-use smartphone-based electrochemical biosensor to quickly assess coffee blend's total polyphenol (Phs) content at industrial and individual level. The device is based on a commercial carbon-based screen-printed electrode (SPE) modified with multi-walled carbon nanotubes (CNTs) and gold nanoparticles (GNPs). At the same time, the biotransducer, Laccase from Trametes versicolor, TvLac, was immobilized on the sensor surface by using glutaraldehyde (GA) as cross-linking agent. The platform was electrochemically characterized to ascertain the influence of the SPE surface modification on its performance. The working electrode (WE) surface morphology characterization was obtained by scanning electron microscopy (SEM) and Fourier-transform infrared (FT-IR) imaging. All the measurements were carried out with a micro-potentiostat Sensit Smart by PalmSens connected to a smartphone. The developed biosensor provided a sensitivity of 0.12 μA/μM, a linear response ranging from 5 to 70 μM, and a lower detection limit (LOD) of 2.99 μM. Afterwards, the biosensor was tested for quantifying the total Phs content in coffee blends, evaluating the influence of both the variety and the roasting degree. The smartphone-based electrochemical biosensor performance has been validated though the Folin-Ciocâlteu standard method.
Keywords:
polyphenols
; smartphone-based electrochemical biosensors
; TAC
; amperometric biosensors
; free radicals
; coffee
1. Introduction
In recent years, the growing demand for devices detecting specific substances has led to looking for new technologies with quick response and detection characteristics. In this field, smartphone-based electrochemical biosensors are emerging as powerful tools for the fast detection of markers in healthcare, environmental monitoring, and food safety [2]. These biosensors can provide rapid, sensitive, and selective analysis of food matrices with minimal sample preparation and low cost. Moreover, smartphone-based electrochemical biosensors can be integrated with wireless communication and cloud computing to enable real-time data processing, storage, and sharing [3].
Smartphone-based electrochemical biosensors have been mainly applied to point-of-care (POC) diagnostic tools for health monitoring [4]. At the same time, still, few works in the literature are focused on food quality evaluation [5]. Analyzing the characteristics of food products results of crucial importance for the determination of their quality (e.g., pH and temperature standards [6]), nutritional value (e.g., nutrients level [7]), authenticity (e.g., adulterants [8] detection) and safety (e.g. pathogens, allergens, contaminants detection [9]).
Currently, particular attention is given to promoting body wellness by following optimal dietary protocols specific to the individual, providing the proper micronutrient intake and preventing chronic diseases such as diabetes, obesity and heart diseases [10,11]. A key role is played by polyphenols (Phs), non-enzymatic exogenous antioxidants present in natural phytochemical compounds (e.g. coffee, tea, fruits) able to direct inactivate free radicals, which in the human body are responsible for cellular oxidative stress, involving the damage of membranes, proteins, DNA and lipids [12]. Phs showed the ability to promote immunity towards foreign pathogens through numerous biological activities [13,14], reducing risks of arteriosclerosis, cancer, neurodegenerative diseases, and osteoporosis [15], as well as regulating intestinal mucosal immune responses and allergic diseases [16,17,18].
Coffee beverage is widely consumed worldwide, representing one of the principal sources of Phs in the human diet [19]. Several scientific studies directly correlate coffee intake with a lower risk of type 2 diabetes mellitus, kidney stones, Parkinson's disease, liver cancer or fibrosis etc. [20], making the Phs content an important index of the nutritional quality of coffee at industrial and commercial scale.
Phs concentration in coffee primarily depends on its variety. Hundred coffee species are available and two of them are the most cultivated in the world, "Arabica" (Coffea Arabica L) and "Robusta" (Coffea Canephora) [21]. When the coffee is not roasted, it is called "green", specifically "Robusta" quality, in this case, shows the maximum polyphenol content (7-14.4 % of the dry weight), which is higher than that for Arabica variety (6-7% of the dry weight) [22,23]. The roasting degree of the beans is also an important parameter since both time and T directly affect the Phs content and the airflow manipulation in commercial coffee-roasting setups [24]. Green coffee has the highest range of Phs [25]. In contrast, high-temperature treatments induce Phs degradation, sugar decomposition, lipid oxidations and pyrolysis, determining the color, aroma and flavor, and its bioactive compounds content.
Typically measuring the polyphenol content in coffee samples is done in laboratories using expensive and complex instruments. Smartphone-based electrochemical biosensors offer a low-cost, portable and user-friendly alternative for polyphenol detection in various settings [26]. Herein, to the best of our knowledge, we demonstrate for the first time a low-cost smartphone-based amperometric biosensor for the on-site quantification of total Phs content in coffee blends. The device is based on a previous electrochemical platform developed by our laboratory [26], where a carbon screen-printed electrode (SPE) was modified via drop-casting with multi-walled carbon nanotubes (CNTs) and gold nanoparticles (GNPs). After that, Laccase enzyme from Trametes versicolor (TvLac) was immobilized through PVA-SbQ photopolymerization. In the present work, a commercial SPE already modified at the surface by CNTs-GNPs nanostructures was employed, quickly immobilizing the TvLac via glutaraldehyde (GA) cross-linking. As well known, this enzyme is able to catalyze the oxidation of ortho- and para-diphenols, aminophenols, polyphenols, polyamines, lignins and aryl diamines [27]. The developed biosensor is optimized for TvLac and GA concentrations by monitoring the enzyme kinetic efficiency through electrochemical tests. The commercial electrodes modifications were investigated by scanning electron microscopy (SEM) and Fourier-transform infrared (FT-IR) measurements, highlighting improvements of electroactive area (Ael), surface roughness index (ρ) and also homogeneity as TvLac is immobilized through the formation of GA cross-linked aggregates. The smartphone-based device proposed provides a sensitivity of 0.12 μA/μM, a linear response ranging between 5 and 70 μM and a limit of detection (LOD) of 2.99 μM towards Phs content in coffee samples. The influence of the coffee variety and the roasting degree, both time of roasting and temperature, are further evaluated. The results obtained are validated through the Folin-Ciocâlteu standard method. These biosensors have shown promising results in terms of accuracy, sensitivity, specificity and reproducibility, and have potential applications in food quality control.
2. Results and Discussions
Laccase from Trametes versicolor (TvLac) is a blue multicopper oxidase enzyme (BMCO) of the oxidoreductase family, with an active site consisting of three copper centers, namely T1Cu, T2Cu, T3Cu. The T1Cu site is responsible for the oxidation of the substrate, while T2Cu-T3Cu center binds the oxygen, which is converted into water through a four-electron reduction process [28]. Thus, we based our biosensor on the activity of TvLac that can mediate the reduction of O2 to H2O, which is possible through the catalytic oxidation of specific substrates such as Phs. It is possible to measure the concentration of Phs from the cathodic current produced by the Phs(ox) reduction during the enzyme regeneration. The TvLac has been immobilized through GA intermolecular cross-linking and physical adsorption, evaluating the optimal concentration in order not to obstacle the substrate movement through the membrane and improve the sensor stability over time.
Improvement of biosensor sensitivity and enzyme loading are obtained by testing commercial SPEs already modified with different nanomaterials combinations: i) unmodified carbon SPE (DRP-110), ii) multi-walled carbon nanotubes (CNTs) modified carbon SPE (DRP-110CNT), iii) gold nanoparticles (GNPs) modified carbon SPE (DRP-110GNP), iv) CNTs and GNPs modified carbon SPE (DRP-110CNT-GNP).
The optimal amount of TvLac to immobilize was assessed onto the DRP-110CNT-GNP SPE by testing three different amounts of the enzyme: 1, 1.25 and 1.5 enzyme units (U). The catalytic current, Figure 1a, increases with increasing the immobilized enzyme units onto the working electrode (WE) surface. However, despite 1.50 U provide for the highest catalytic current, 1.25 U was chosen as the optimal amout, giving a current magnitude almost comparable to that obtained with 1.50 U, but giving a greater reproducibility.
GA concentration was also evaluated, namely 0.8%, 1.3% and 1.6% v/v. Chronoamperometry tests (CA) were performed applying a constant potential of -0.1 V vs. silver pseudo-reference electrode (AgPRE). In Figure 1b is shown that 0.8% v/v GA is responsible for a less stability of the current over time, losing the CA response at 6 μM of catechol. This is probably due to a weaker cross-linking, exposing more the enzyme towards the solution and inducing eventually the bioreceptor loss.
The biosensors modified with GA at 1.3% and 1.6% v/v showed comparable responses towards the substrate however, in the case of GA at 1.6% v/v the stronger cross-linking produced a denser GA matrix, hindering the substrate diffusion and causing the saturation of the signal at a catechol concentration of 20 μM. The 1.3% v/v GA solution was selected as optimal, ensuring both a wider concentration range of analysis (0-2000 μM) and good sensitivity.
The surface modification of SPE electrodes was investigated through FT-IR/imaging analysis (Figure 2) and scanning electron microscopy (SEM) (Figure S1). Specifically, FT-IR measurements have been performed by collecting 25 different spots in a 5x5 grid array area on the electrode surface (25 pixels). Figure 2a reports the FT-IR responses of DRP-110, DRP-110CNT and DRP-110CNT-GNP electrodes, at grid central spot. The DRP-110 electrode shows two characteristic broad bands between 879-1103 cm-1 and 1261-1458 cm-1, corresponding to the alkoxy C–O stretching and C–C stretching respectively. After introducing CNTs at the electrode surface (DRP-110CNT) there is a slight increase of the aforementioned bands, while the presence of GNPs (DRP-110CNT-GNP) enhances the absorbance response inducing a surface IR enhancement effect (SEIRS) [29]. GNPs presence further promote intensity of response and also appearance of two broad bands at 2728-2983 cm-1 and 3056-3446 cm-1, associated to the C–H asymmetric and symmetric stretching and alcohol/carboxylic O–H stretching respectively.
The FT-IR profile when TvLac is immobilized onto the WE surface by physical adsorption shows five characteristic bands, Figure 2b, namely the C – O – C asymmetric and symmetric stretching at 1014 cm-1 and 1002 cm-1, the C – N stretching at 1373 cm-1, the CO – NH stretching of peptide linkage at 1648 cm-1, the alkylic C – H stretching at 2932 cm-1 and N – H/O – H stretching at 3310 cm-1 [30,31,32]. FT-IR tests are also performed after depositing TvLac onto the DRP-110CNT-GNP electrode. The presence of hydrophobic forces normally induces a difficulty in the identification of the C–O–C and CO–NH FT-IR spectrum [31]. However, in our case the presence of a TvLac multilayer onto the electrode, where the outer protein layer has no direct interaction with the carbon-based electrode, it is possible to distinguish vibration of the C–O–C and CO–NH functionalities (1002-1140 cm-1 and 1648 cm-1 respectively). By integrating the area of the CO–NH stretching band over the entire electrode surface (i.e., considering the 25 different collected spectra), it is possible to map the surface distribution of TvLac, Figure 2c,d. The CO–NH stretching band was selected as reference signal for the FT-IR imaging, being the peptide linkage present in the whole TvLac structure. In fact, the choose of other functionalities present only in specific regions of the protein could only give information about the TvLac enzyme orientation respect to the electrode surface. Since the CO–NH stretching vibration is directly influenced by the hydrophobic interaction strength between the protein and the carbon surface, the less the amount of TvLac onto a specific region, the less the possibility to observe the CO–NH stretching band when the FT-IR spectrum is collected in that spot. Therefore, by observing the map an inhomogeneous distribution of the CO–NH stretching vibration intensity highlights not homogeneous deposition of enzyme on the electrode surface, thus the protein doesn’t cover the electrode homogeneously when immobilized in absence of GA. Differently, when TvLac is immobilized through the formation of GA cross-linked aggregates, intensity of FT-IR absorbance is higher. In this case, the O–H stretching vibration band is influenced by different contributes: i) the vibration of the OH present in the protein structure; ii) the vibration of the OH related to the incorporated water molecules; iii) the vibration of the OH present in the GA structure [33]. Also, the alkylic C – H stretching at 2932 cm-1 is due to both GA and TvLac. The remaining vibrational modes are only related to the protein; therefore, the increase of the spectrum intensity is due to a higher concentration of the biomolecule in the central spot, which suggests a different distribution over the electrode surface respect to the previous described. This is confirmed by the distribution map in Figure 2d, where the vibration intensity of the CO–NH stretching mode is strong in the central region of the electrode surface, index of a homogeneous immobilization of TvLac via GA cross-linking.
CV tests, Figure S2, were performed to calculate the electroactive area (Ael) of the four SPEs. Specifically, CV at different scan rates have been performed in a solution of 1 mM [Fe (CN)6]3−/4− with KCl 0.1 M. According to the Randles-Ševčík equation and the obtained plot of Ip,a vs v1/2 [34] the Ael values have been calculated and summarized in Table S1 with the relative roughness factors (ρ) defined by the electroactive/geometric area ratio (Ael/Ageo). Both Ael and ρ increase in the order DRP-110 < DRP-110GNP < DRP-110CNT < DRP-110CNT-GNP. Further CV measurements have been conducted to evaluate the catalytic response of TvLac depending on the specific surface modification, Figure 3, in sodium acetate buffer (AcONa) 0.01 mM, pH 5 with catechol 0.02 mM, in presence and absence of enzyme.
When the enzyme is employed the voltammograms showed diffusion controlled anodic and cathodic peaks for the oxidation and reduction of the catechol at the electrode surface. In the presence of TvLac, if the catalysis regularly takes place, the voltammograms shapes become characteristic sigmoidal curves. Because of the oxidation of the catechol is catalyzed by the enzyme, the anodic peak disappears, while the cathodic peak becomes broader, indicating higher amount of catechol reduced at the electrode surface [35]. Following the steady state catalytic current and the sigmoid morphology, the performance of each electrode was evaluated. The platform DRP-110 showed a cathodic current of 0.8 μA, while the additional presence of CNTs in DRP-110CNT (Figure 3a,b) induces improvement of current to 1.7 μA, broadening the cathodic peak. In fact, the higher electronic conductivity and electroactive area provided by the presence of CNTs result in a faster electron transfer (ET) at the electrode surface.
The DRP-110GNP electrode showed an increased catalytic current of 3 μA, Figure 3c, despite the lower electroactive area in comparison with the DRP-110CNT electrode. This behavior is due to a stronger direct ET communication between the TvLac and the WE surface promoted by GNPs, known to work as highly efficient electron-conducting tunnels between WE and enzymes [36]. In addition, GNPs have remarkable affinity with proteins, allowing a suitable surface coverage and preserving the catalytic activity at the same time [37,38,39,40]. By combining the GNPs and CNTs in the DRP-110CNT-GNP platform, the highest catalytic current of 4.05 μA is reached, Figure 3d. The catalytic behavior is further investigated through CA tests, carried out measuring the cathodic catalytic current produced through successive additions of equal volumes of a catechol solution at 255 mM over time, Figure 4a,b.
The DRP-110/TvLac+GA sensor showed the lowest signal-to-noise ratio (S/N) in the CA profile and the less stability of signal over time, Figure 4a in grey, as the catalytic current was constant after 20 μM of catechol, impeding the production of an acceptable Michaelis-Menten hyperbole and the kinetic parameters calculation. This behavior can be explained as consequence of three factors: i) immobilization of lower amount of enzyme as the smallest electroactive area and thus the smallest roughness and porosity of the unmodified carbon electrode surface (Ael=2.04 mm2, ρ=0.16); ii) consequent fast saturation of enzyme active sites; iii) possible enzyme loss in solution as weaker physical adsorption on electrodes material and the poor adhesion of the glutaraldehyde matrix on the same. The DRP-110GNP/TvLac+GA platform revealed better sensitivity to the substrate as the higher slope of CA profile in respect to that of DRP-110, Figure 4a in black, as highlighted before. However, there is a low stability over time of the CA signal and the S/N ratio, probably meaning that the roughness (ρ=0.68) and porosity of the WE surface are not suitable enough to guarantee a long permanence of the GA cross-linked aggregate. Also in this case the kinetic parameters could not be determined. Differently, the presence of CNTs in DRP-110CNT/TvLac+GA significantly favors the stability of the catalytic activity over time till 400 μM of catechol, Figure 4b in grey. In fact, the well-known high porosity of CNTs plays a dramatic role in the physical adsorption mechanism as well as in the immobilization stability [41,42,43,44,45]. However, catalytic current increases slower in comparison to DRP-110GNP/TvLac+GA electrode, index of lower sensitivity. The co-presence of GNPs and CNTs in DRP-110CNT-GNP/TvLac+GA, Figure 4b in black, guarantees an optimal direct ET at the surface, high enzyme loading and good immobilization stability at the same time, providing for a sensitive and stable CA response over time. For the last two platforms the Michaelis-Menten hyperboles were obtained (Figure 4c), calculating the kinetic parameters and range of linear response (Figure 4d) for both.
The DRP-110CNT-GNP/TvLac+GA was chosen for the realization of the smartphone-based biosensor for Phs detection in coffee blends, with a linear range of 5-70 μM, analytical sensitivity (a) equal to 0.124 μA/μM and limit of detection (LOD) of 2.99 μM. LOD calculated from the analytical sensitivity through the following equation: LOD = kσB/a, where σB is the standard deviation of the blank measurements and k is chosen relatively to the confidence level required [46], in this work k = 3. Also, the stability over time of the biosensor was evaluated giving a 94% retention of the response after 21 days (Figure S4). Total Phs content was then evaluated in seven different coffee blends (Table S2), differing on roasting degrees and origin. More specifically, the roasting degree influence was studied by testing a green coffee sample treated in oven under different temperatures and times. All coffee blends were analysed using the biosensor device and recording a CA with constant applied potential of -0.1 V vs. AgPRE.
The method used for the analysis was the Standard Addition Method: the current intensity value of the blank (I0) is recorded and, sequentially, the unknown sample followed by the standard catechol solutions at 0, 25, 40, 50, 65 μM were added. As a reference, the CA profile for the analysis of the coffee blend II and the corresponding graph of standard additions are reported in Figure 5a,b. The Icat related to each standard sample was plotted vs the corresponding catechol standard concentration and the one related to the unknown sample was plotted in the graph at [catechol]=0 μM. The Phs concentration was determined by extrapolation of the line passing through the points, taking the dilution factor into account. The reference method employed for the validation of the biosensor is the Folin-Ciocâlteu spectrophotometric assay (calibration curve in Figure 5c) by diluting each sample 200:1. All the values of Phs content obtained with the two methods are reported in Table S3. The experimental data were finally converted into mg/g defined as the total concentration of Phs expressed as gallic acid on the total grams (7g) of fresh coffee used to prepare the sample. The analysis of the actual samples thus led to satisfactory results in agreement with work in the literature [19,47], in which the concentration of polyphenols was determined by spectrophotometric methods.
The values determined using the optimized biosensor resulted in great agreement with those obtained by Folin-Ciocâlteu, with a mean recovery of 95%. Looking at the results, the dependence of Phs content on both the coffee variety (C. robusta or C. arabica) and roasting degree (light, medium, dark) was confirmed. In fact, the roasting temperature causes the chlorogenic acid degradation, reducing the amounts of malic and citric acid thus quinic acid concentration [48,49]. Specifically, by considering the blends I, II and III, characterized by the same variety (100% C. arabica), a higher roasting degree led to greater degradation of Phs resulting in a lower content. The samples IV, V and VI belong to the same manufacturer are characterized by different percentages of C. arabica and C. robusta. The highest Phs concentrations were found in coffee blends with higher amount of C. robusta, as reported in the literature [50,51]. The sample VI and II, despite belonging to different manufacturers, showed similar Phs concentrations, as characterized by a medium roasting degree and a composition of 100% C. arabica. Interesting the case of coffee blend VII, which is composed by 98% of C. arabica variety and 2% of C. robusta green, presenting a higher Phs content than blends II and I, but similar to blend III; in fact, the presence of green coffee in a very low percentage did not significantly influence the Phs content. The last blend analyzed is the VIII, a green coffee 100% C. arabica, roasted under different temperature and time conditions. The results reported in Table S3 and Figure 5d besides being in good agreement with those obtained through Folin-Ciocâlteu reference method, are also consistent with the behavior largely treated in literature [47,52]. The light roasted samples generally show the highest Phs content [53], even compared with the relative unroasted samples. In fact, thermal treatments are responsible for both the antioxidants degradation and the production of new antioxidants species such as heterocyclic compounds obtained through the Maillard reaction. However, as breakdown of cellular components during thermal processes is responsible for the release of bound phenolic acids, the higher the roasting time the lower the total Phs content, under all roasting temperatures.
3. Materials and Methods
3.1. Reagentes and Samples
Catechol, potassium chloride (KCl), sodium acetate (AcONa), glutaraldehyde 25% v/v, laccase from Trametes versicolor (TvLac) were purchased by Merck Life Science (Milan, Italy). All solutions were prepared using Milli-Q water (R = 18.2 MΩ cm at 25 °C; TOC < 10 μg L−1, Millipore, Molsheim, France). TvLac was solubilized in acetate buffer (AcONa buffer) 0,01 M pH 5, stored at -20 °C and catechol was solubilized in AcONa buffer 0,01 M pH 5, freshly prepared for each experiment.
3.2. Electrochemical and Surface Characterization Apparatus
All the CV and CA measurements were performed by using a portable potentiostat, the Sensit Smart potentiostat by PalmSens, controlled by PSTouch on the smartphone. The experiments were carried out through the three-electrode miniaturized cell of a screen-printed electrode (SPE), with a pseudo-reference electrode (PRE) made of silver and an auxiliary electrode (AE) made of carbon ink in a 7 mL electrochemical cell at RT (Figure 6a,b).
Four commercial SPEs with different working electrodes (WE) have been tested: carbon (DRP-110), carbon modified with GNPs (DRP-110GNP), carbon modified with CNTs (DRP-110CNT) and carbon modified with CNTs and GNPs (DRP-110CNT-GNP). All the SPEs already modified were purchased by Metrohm Italiana (Formello, Italy), having a WE with a diameter of 4 mm.
Scanning electron microscopy (SEM) was performed for the characterization of the electrode surface morphology using a Dual Beam Auriga Zeiss instrument located at the Sapienza Nanoscience & Nanotechnology Labs (SNN-Lab). The chemical imaging for spatially resolve the chemical properties of the different platforms used in this work has been performed through FT-IR measurements using a Bruker Lumos II microscope.
3.3. Real Sample Preparation and Treatment
Eight coffee blends with a medium-fine grind for bar machines, belonging to different brands, were analyzed (Table S2). The effect of roasting on polyphenolic content was assessed by analyzing three coffee blends (blend I, II and III) purchased in Rome at an historical famous artisanal roasting and blending facility, obtained from the selection of the best arabica coffee qualities from four different countries (Brazil, Vietnam, Colombia and Indonesia).The first coffee blend (blend I) is dark roasted (240-250 °C), the coffee blend II is medium roasted (210-220 °C) and the coffee blend III is clear roasted (180-205 °C). The different polyphenolic content in Coffea arabica and Coffea canephora (also called C. robusta) varieties was assessed by testing three coffee blends (blend IV, V, VI), obtained by blending arabica and robusta varieties in different percentages but maintaining the same medium roasting degree (210-220 °C). In general, the values described in the literature for total polyphenols may vary from 4 to 8,4 % for C. arabica, and from 7 to 14,4 % for C. robusta [54]. The C. arabica variety contains less content in polyphenols, prefers high-altitude cultivation between 1,000 and 2,000 metres and comes from global producers such as Brazil, Vietnam, Colombia and Indonesia; C. robusta, on the other hand, has a higher polyphenols content, grows at altitudes below 700 metres and comes from Mexico, Guatemala, Honduras, Nicaragua, El Salvador, Ethiopia, India and Ecuador [55]. Coffee blend IV is medium roasted and the coffee is obtained from a skillful blend of both varieties, 80% C. robusta which gives body and intensity and 20% C. arabica with a fine and characteristic aroma. The coffee blend V is obtained from a blend of 40% C. arabica and 60% C. robusta. Finally, the coffee blend VI is 100% arabica, obtained from the careful blending of the best selections of coffee from Central and South America, Africa and East Asia Moreovera blend of green coffee obtained from 'raw' coffee beans which have not undergone the roasting process but only the drying process was also analyzed. The coffee obtained contains a high quality of the polyphenol chlorogenic acid normally reduced during the roasting process [56]. Coffee blend VII (98% arabica, 2% robusta green) comes from a small community of native Indians in Central America. Green coffee (blend VIII, 100 % arabica) was then roasted in the laboratory oven at three different temperatures (180, 200, 250 °C) to examine the impact of roasting on polyphenol concentration. Every sample was taken off the oven at regular temperature intervals (10, 15, 20 min) to obtain coffee beans with various levels of roasting.
Coffee making refers to the coffee-making standard for sensory analysis on ISO 6668:2008, as much as 7 grams of brewed coffee used in 100 ml of boiling water of 92°C-96°C with unfiltered pour-over method. After extraction (3 min), the samples were filtered through a Whatman5 paper filter 15 cm in diameter (Vetroscientifica srl, Italy). Finally, cooled samples were diluted 1:3000 with 10 mM PBS pH 7.4.
3.4. Folin-Ciocâlteu Assay
As a reference method to measure the total polyphenol content in coffee samples, the Folin-Ciocâlteu method was employed. This colorimetric assay consists of using the Folin-Ciocâlteu reagent, composed of tungstophosphate and molybdophosphate, which can react with phenolic compounds in an alkaline medium causing a change in color, which is proportional to the concentration of phenolic compounds in the sample [57]. The determination of the total polyphenol content is expressed through gallic acid equivalents. For this purpose, a calibration curve was made with standard solutions of gallic acid prepared at known concentrations (0, 25, 100, 300 µg/mL) using the BioQuoChem® kit for the spectrophotometric determination. Specifically, 500 µL of the Folin-Ciocâlteu reagent and 400 µL of a carbonate and hydrogen carbonate buffer solution were added to 100 µL of each standard solution for a total volume of 1 mL [58]. The obtained solutions were mixed and incubated for 15 min at room temperature. The absorbance of standard solutions in quartz cuvettes was measured by a T60U (UV-Visible) Spectrophotometer from PG Instruments Ltd at a wavelength of 700 nm. The same procedure was followed with the coffee samples of unknown polyphenol concentration, previously diluted 1:3000 with PBS 10 mM pH 7.4 so that the absorbance at 700 nm falls within the limits of the standard curve.
3.5. TvLac-Based Biosensor Fabrication
Each SPE was easily modified by drop casting of a solution composed of 2 μL of GA 8% v/v and 10 μL of TvLac 125 U/mL, to have final concentrations of GA and TvLac of 1.33% v/v and 1.25 U/mL respectively. Afterwards, the electrode was left in the fridge at 4° C for 60 min to promote a stable intermolecular crosslinking among the enzyme molecules [59,60] and then left drying for 30 min at RT, allowing the physical adsorption of the crosslinked laccase molecules onto the WE surface.
3.6. Electrochemical Measurements
CV measurements were carried out in 7 mL solution of catechol 0.02 mM, prepared in AcONa buffer 10 mM, pH = 5, in presence and absence of enzyme, from 0.6 V to -0.2 V vs AgPRE, scan rate 5 mV/s. The CA measurements were carried out under constant stirring, by applying a fixed potential equal to -0.1 V to the WE according to literature [35], in 7 mL of AcONa buffer solution 10 mM, pH = 5, and by adding equal volumes of 75 mM concentrated catechol solution, prepared in AcONa buffer 10 mM, pH = 5. All the experiments were carried out at RT.
4. Conclusions
In this work, for the first time, a smartphone-based electrochemical biosensor for the rapid, sensitive and user-friendly determination of total Phs content in coffee blends was realized. TvLac and GA concentrations have been optimized, to reach the best result in terms of sensitivity and stability. FT-IR imaging characterization highlighted the improved homogeneity of TvLac-GA layer on the WE central surface. The biosensor was calibrated by chronoamperometry employing catechol as standard solution. The linearity response is ranging between 5 and 70 μM with a lower detection limit of 2.99 and sensitivity as 0.124 μA/μM. The lifetime stability showed a signal retention of the 94% after 21 days. Finally, the proposed biosensor was tested for the determination of the total Phs content in real coffee samples with different roasting degrees and blends. The results were in good agreement with those obtained through the Foline-Ciocâlteu reference method, with a mean recovery of 95%. The good results obtained with the developed smartphone-based biosensor suggest its possible use for the quick and cheap determination of Phs in coffee blends.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
Conceptualization, F.M. and G.F.; formal analysis, C.D., C.C., R.Z., F.P., L.S. and F.S.; validation, C.D., C.C., R.Z.; investigation, R.Z. and C.D; data curation, R.Z. and C.R.; writing—original draft preparation, R.Z. and C.D.; writing—review and editing, R.Z., F.M., F.P., G.F., M.A. and S.B.; All authors have read and agreed to the published version of the manuscript.
Funding
This research was partially funded by Angelini Holding S.p.A.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Shen, M.; Rusling, J.F.; Dixit, C.K. Site-selective orientated immobilization of antibodies and conjugates for immunodiagnostics development. Methods 2017, 116, 95–111. https://doi.org/10.1016/j.ymeth.2016.11.010. [CrossRef]
- Younis, M.R.; Wang, C.; Younis, M.A.; Xia, X. Smartphone-Based Biosensors. Nanobiosensors From Des. to Appl. 2020, 357–387.
- Xu, K.; Chen, Q.; Zhao, Y.; Ge, C.; Lin, S.; Liao, J. Cost-effective, wireless, and portable smartphone-based electrochemical system for on-site monitoring and spatial mapping of the nitrite contamination in water. Sensors Actuators B Chem. 2020, 319, 128221. https://doi.org/10.1016/j.snb.2020.128221. [CrossRef]
- Sun, A.C.; Hall, D.A. Point-of-care smartphone-based electrochemical biosensing. Electroanalysis 2019, 31, 2–16. https://doi.org/10.1002/elan.201800474. [CrossRef]
- Lu, Y.; Shi, Z.; Liu, Q. Smartphone-based biosensors for portable food evaluation. Curr. Opin. Food Sci. 2019, 28, 74–81. https://doi.org/10.1016/j.cofs.2019.09.003. [CrossRef]
- Yousefi, H.; Su, H.-M.; Imani, S.M.; Alkhaldi, K.; M. Filipe, C.D.; Didar, T.F. Intelligent food packaging: A review of smart sensing technologies for monitoring food quality. ACS sensors 2019, 4, 808–821. https://doi.org/10.1021/acssensors.9b00440. [CrossRef]
- Wang, M.; Yang, Y.; Min, J.; Song, Y.; Tu, J.; Mukasa, D.; Ye, C.; Xu, C.; Heflin, N.; McCune, J.S. A wearable electrochemical biosensor for the monitoring of metabolites and nutrients. Nat. Biomed. Eng. 2022, 1–11. https://doi.org/10.1038/s41551-022-00916-z. [CrossRef]
- Irannejad, N.; Rezaei, B. Electrochemical sensors for food adulterants. In Biosensing and Micro-Nano Devices: Design Aspects and Implementation in Food Industries; Springer, 2022; pp. 69–90.
- Wang, K.; Lin, X.; Zhang, M.; Li, Y.; Luo, C.; Wu, J. Review of Electrochemical Biosensors for Food Safety Detection. Biosensors 2022, 12, 959. https://doi.org/10.3390/bios12110959. [CrossRef]
- Viswanathan, V.; Krishnan, D.; Kalra, S.; Chawla, R.; Tiwaskar, M.; Saboo, B.; Baruah, M.; Chowdhury, S.; Makkar, B.M.; Jaggi, S. Insights on medical nutrition therapy for type 2 diabetes mellitus: an Indian perspective. Adv. Ther. 2019, 36, 520–547. https://doi.org/10.1007/s12325-019-0872-8. [CrossRef]
- Nestel, P.J.; Beilin, L.J.; Clifton, P.M.; Watts, G.F.; Mori, T.A. Practical guidance for food consumption to prevent cardiovascular disease. Hear. Lung Circ. 2021, 30, 163–179. https://doi.org/10.1016/j.hlc.2020.08.022. [CrossRef]
- Valko, M.; Leibfritz, D.; Moncol, J.; Cronin, M.T.D.; Mazur, M.; Telser, J. Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell Biol. 2007, 39, 44–84. https://doi.org/10.1016/j.biocel.2006.07.001. [CrossRef]
- Chiva-Blanch, G.; Visioli, F. Polyphenols and health: Moving beyond antioxidants. J. Berry Res. 2012, 2, 63–71. https://doi.org/10.3233/JBR-2012-028. [CrossRef]
- Ding, S.; Jiang, H.; Fang, J. Review Article Regulation of Immune Function by Polyphenols. J. Immunol. Res. 2018, 2018.
- Scalbert, A.; Johnson, I.T.; Saltmarsh, M. Polyphenols: antioxidants and beyond. Am. J. Clin. Nutr. 2005, 81, 215–217. https://doi.org/10.1093/ajcn/81.1.215s. [CrossRef]
- Okazaki, Y.; Han, Y.; Kayahara, M.; Watanabe, T.; Arishige, H.; Kato, N. Consumption of curcumin elevates fecal immunoglobulin A, an index of intestinal immune function, in rats fed a high-fat diet. J. Nutr. Sci. Vitaminol. (Tokyo). 2010, 56, 68–71. https://doi.org/10.3177/jnsv.56.68. [CrossRef]
- Pérez-Berezo, T.; Franch, A.; Castellote, C.; Castell, M.; Pérez-Cano, F.J. Mechanisms involved in down-regulation of intestinal IgA in rats by high cocoa intake. J. Nutr. Biochem. 2012, 23, 838–844. https://doi.org/10.1016/j.jnutbio.2011.04.008. [CrossRef]
- Di Meo, F.; Lemaur, V.; Cornil, J.; Lazzaroni, R.; Duroux, J.-L.; Olivier, Y.; Trouillas, P. Free radical scavenging by natural polyphenols: atom versus electron transfer. J. Phys. Chem. A 2013, 117, 2082–2092. https://doi.org/10.1021/jp3116319. [CrossRef]
- Król, K.; Gantner, M.; Tatarak, A.; Hallmann, E. The content of polyphenols in coffee beans as roasting, origin and storage effect. Eur. Food Res. Technol. 2020, 246, 33–39. https://doi.org/10.1007/s00217-019-03388-9. [CrossRef]
- Kolb, H.; Kempf, K.; Martin, S. Health effects of coffee: mechanism unraveled? Nutrients 2020, 12, 1842. https://doi.org/10.3390/nu12061842. [CrossRef]
- Tarigan, E.B.; Wardiana, E.; Hilmi, Y.S.; Komarudin, N.A. The changes in chemical properties of coffee during roasting: A review. IOP Conf. Ser. Earth Environ. Sci. 2022, 974. https://doi.org/10.1088/1755-1315/974/1/012115. [CrossRef]
- Farah, A.; Monteiro, M.C.; Calado, V.; Franca, A.S.; Trugo, L.C. Correlation between cup quality and chemical attributes of Brazilian coffee. Food Chem. 2006, 98, 373–380. https://doi.org/10.1016/j.foodchem.2005.07.032. [CrossRef]
- Perdani, C.G.; Pranowo, D. Total phenols content of green coffee (Coffea arabica and Coffea canephora) in East Java. In Proceedings of the IOP Conference Series: Earth and Environmental Science; IOP Publishing, 2019; Vol. 230, p. 12093.
- Kwak, H.S.; Ji, S.; Jeong, Y. The effect of air flow in coffee roasting for antioxidant activity and total polyphenol content. Food Control 2017, 71, 210–216. https://doi.org/10.1016/j.foodcont.2016.06.047. [CrossRef]
- Ludwig, I.A.; Clifford, M.N.; Lean, M.E.J.; Ashihara, H.; Crozier, A. Coffee: biochemistry and potential impact on health. Food Funct. 2014, 5, 1695–1717. https://doi.org/10.1039/c4fo00042k. [CrossRef]
- Bunney, J.; Williamson, S.; Atkin, D.; Jeanneret, M.; Cozzolino, D.; Chapman, J.; Power, A.; Chandra, S. Current Research in Nutrition and Food Science The Use of Electrochemical Biosensors in Food Analysis. 2017, 5.
- Unal, Y.D.; Pazarlioglu, N.K. Production and Gelatin Entrapment of Laccase from Trametes versicolor and its Application to Quantitative Determination of Phenolic Contents of Commercial Fruit Juices. Food Biotechnol. 2011, 25, 351–368. https://doi.org/10.1080/08905436.2011.617258. [CrossRef]
- Castrovilli, M.C.; Bolognesi, P.; Chiarinelli, J.; Avaldi, L.; Calandra, P.; Antonacci, A.; Scognamiglio, V. The convergence of forefront technologies in the design of laccase-based biosensors–An update. TrAC Trends Anal. Chem. 2019, 119, 115615. https://doi.org/10.1016/j.trac.2019.07.026. [CrossRef]
- Aroca, R.F.; Ross, D.J.; Domingo, C. Surface-Enhanced Infrared Spectroscopy. Appl. Spectrosc. 2004, 58, 324A-338A.
- Romero-Arcos, M.; Garnica-Romo, M.G.; Martínez-Flores, H.E. Electrochemical study and characterization of an amperometric biosensor based on the immobilization of laccase in a nanostructure of TiO2 synthesized by the sol-gel method. Materials (Basel). 2016, 9, 8–13. https://doi.org/10.3390/ma9070543. [CrossRef]
- Tavares, A.P.M.; Silva, C.G.; Dražić, G.; Silva, A.M.T.; Loureiro, J.M.; Faria, J.L. Laccase immobilization over multi-walled carbon nanotubes: Kinetic, thermodynamic and stability studies. J. Colloid Interface Sci. 2015, 454, 52–60. https://doi.org/10.1016/j.jcis.2015.04.054. [CrossRef]
- Latif, A.; Maqbool, A.; Sun, K.; Si, Y. Immobilization of Trametes versicolor laccase on Cu-alginate beads for biocatalytic degradation of bisphenol A in water: Optimized immobilization, degradation and toxicity assessment. J. Environ. Chem. Eng. 2022, 10, 107089. https://doi.org/10.1016/j.jece.2021.107089. [CrossRef]
- Patro, T.U.; Wagner, H.D. Influence of graphene oxide incorporation and chemical cross-linking on structure and mechanical properties of layer-by-layer assembled poly(Vinyl alcohol)-Laponite free-standing films. J. Polym. Sci. Part B Polym. Phys. 2016, 54, 2377–2387. https://doi.org/10.1002/polb.24226. [CrossRef]
- García-Miranda Ferrari, A.; Foster, C.W.; Kelly, P.J.; Brownson, D.A.C.; Banks, C.E. Determination of the electrochemical area of screen-printed electrochemical sensing platforms. Biosensors 2018, 8, 53. https://doi.org/10.3390/bios8020053. [CrossRef]
- Tortolini, C.; Di Fusco, M.; Frasconi, M.; Favero, G.; Mazzei, F. Laccase-polyazetidine prepolymer-MWCNT integrated system: Biochemical properties and application to analytical determinations in real samples. Microchem. J. 2010, 96, 301–307. https://doi.org/10.1016/j.microc.2010.05.004. [CrossRef]
- Chen, X.; Li, D.; Li, G.; Luo, L.; Ullah, N.; Wei, Q.; Huang, F. Facile fabrication of gold nanoparticle on zein ultrafine fibers and their application for catechol biosensor. Appl. Surf. Sci. 2015, 328, 444–452. https://doi.org/10.1016/j.apsusc.2014.12.070. [CrossRef]
- Kizling, M.; Dzwonek, M.; Wieckowska, A.; Bilewicz, R. Gold nanoparticles in bioelectrocatalysis – The role of nanoparticle size. Curr. Opin. Electrochem. 2018, 12, 113–120. https://doi.org/10.1016/j.coelec.2018.05.021. [CrossRef]
- Kizling, M.; Dzwonek, M.; Więckowska, A.; Bilewicz, R. Size Does Matter—Mediation of Electron Transfer by Gold Clusters in Bioelectrocatalysis. ChemCatChem 2018, 10, 1988–1992. https://doi.org/10.1002/cctc.201800032. [CrossRef]
- Pankratov, D.; Sundberg, R.; Suyatin, D.B.; Sotres, J.; Barrantes, A.; Ruzgas, T.; Maximov, I.; Montelius, L.; Shleev, S. The influence of nanoparticles on enzymatic bioelectrocatalysis. RSC Adv. 2014, 4, 38164–38168. https://doi.org/10.1039/c4ra08107b. [CrossRef]
- Silveira, C.M.; Zumpano, R.; Moreira, M.; de Almeida, M.P.; Oliveira, M.J.; Bento, M.; Montez, C.; Paixão, I.; Franco, R.; Pereira, E.; et al. Star-Shaped Gold Nanoparticles as Friendly Interfaces for Protein Electrochemistry: the Case Study of Cytochrome c. ChemElectroChem 2019, 6, 4696–4703. https://doi.org/10.1002/celc.201901393. [CrossRef]
- Mohamad, N.R.; Marzuki, N.H.C.; Buang, N.A.; Huyop, F.; Wahab, R.A. An overview of technologies for immobilization of enzymes and surface analysis techniques for immobilized enzymes. Biotechnol. Biotechnol. Equip. 2015, 29, 205–220. https://doi.org/10.1080/13102818.2015.1008192. [CrossRef]
- Zhou, W.; Zhang, W.; Cai, Y. Laccase immobilization for water purification: A comprehensive review. Chem. Eng. J. 2021, 403, 126272. https://doi.org/10.1016/j.cej.2020.126272. [CrossRef]
- Hanefeld, U.; Gardossi, L.; Magner, E. Understanding enzyme immobilisation. Chem. Soc. Rev. 2009, 38, 453–468. https://doi.org/10.1039/b711564b. [CrossRef]
- Ashkan, Z.; Hemmati, R.; Homaei, A.; Dinari, A.; Jamlidoost, M.; Tashakor, A. Immobilization of enzymes on nanoinorganic support materials: An update. Int. J. Biol. Macromol. 2021, 168, 708–721. https://doi.org/10.1016/j.ijbiomac.2020.11.127. [CrossRef]
- Ciogli, L.; Zumpano, R.; Poloznikov, A.A.; Hushpulian, D.M.; Tishkov, V.I.; Andreu, R.; Gorton, L.; Mazzei, F.; Favero, G.; Bollella, P. Highly Sensitive Hydrogen Peroxide Biosensor Based on Tobacco Peroxidase Immobilized on p-Phenylenediamine Diazonium Cation Grafted Carbon Nanotubes: Preventing Fenton-like Inactivation at Negative Potential. ChemElectroChem 2021, 8, 2495–2504. https://doi.org/10.1002/celc.202100341. [CrossRef]
- Lavín, Á.; de Vicente, J.; Holgado, M.; Laguna, M.F.; Casquel, R.; Santamaría, B.; Maigler, M.V.; Hernández, A.L.; Ramírez, Y. On the determination of uncertainty and limit of detection in label-free biosensors. Sensors (Switzerland) 2018, 18. https://doi.org/10.3390/s18072038. [CrossRef]
- Dybkowska, E.; Sadowska, A.; Rakowska, R.; Dębowska, M.; Świderski, F.; Świąder, K. Assessing polyphenols content and antioxidant activity in coffee beans according to origin and the degree of roasting. Rocz. Panstw. Zakl. Hig. 2017, 68, 347–353.
- Várady, M.; Hrušková, T.; Popelka, P. Effect of preparation method and roasting temperature on total polyphenol content in coffee beverages. Czech J. Food Sci. 2020, 38, 417–421. https://doi.org/10.17221/122/2020-CJFS. [CrossRef]
- Król, K.; Gantner, M.; Tatarak, A.; Hallmann, E. The effect of roasting, storage, origin on the bioactive compounds in organic and conventional coffee (Caffea arabica). Eur. Food Res. Technol 2019, 246, 33–39.
- Sunarharum, W.B.; Yuwono, S.S.; Aziza, O.F. Study on the effect of roasting temperature on antioxidant activity of early-roasted Java coffee powder (Arabica and Robusta). IOP Conf. Ser. Earth Environ. Sci. 2019, 230. https://doi.org/10.1088/1755-1315/230/1/012045. [CrossRef]
- Martín, M.J.; Pablos, F.; González, A.G. Discrimination between arabica and robusta green coffee varieties according to their chemical composition. Talanta 1998, 46, 1259–1264. https://doi.org/10.1016/S0039-9140(97)00409-8. [CrossRef]
- Cho, A.R.; Park, K.W.; Kim, K.M.; Kim, S.Y.; Han, J. Influence of Roasting Conditions on the Antioxidant Characteristics of C olombian Coffee (C offea arabica L.) Beans. J. Food Biochem. 2014, 38, 271–280. https://doi.org/10.1111/jfbc.12045. [CrossRef]
- Somporn, C.; Kamtuo, A.; Theerakulpisut, P.; Siriamornpun, S. Effects of roasting degree on radical scavenging activity, phenolics and volatile compounds of Arabica coffee beans (Coffea arabica L. cv. Catimor). Int. J. Food Sci. Technol. 2011, 46, 2287–2296. https://doi.org/10.1111/j.1365-2621.2011.02748.x. [CrossRef]
- Farah, A.; Donangelo, C.M. Phenolic compounds in coffee. Brazilian J. Plant Physiol. 2006, 18, 23–36. https://doi.org/10.1590/S1677-04202006000100003. [CrossRef]
- Bicho, N.C.; Lidon, F.C.; Ramalho, J.C.; Leitão, A.E. Quality assessment of Arabica and Robusta green and roasted coffees - A review. Emirates J. Food Agric. 2013, 25, 945–950. https://doi.org/10.9755/ejfa.v25i12.17290. [CrossRef]
- Bobková, A.; Hudáček, M.; Jakabová, S.; Belej, Ľ.; Capcarová, M.; Čurlej, J.; Bobko, M.; Árvay, J.; Jakab, I.; Čapla, J.; et al. The effect of roasting on the total polyphenols and antioxidant activity of coffee. J. Environ. Sci. Heal. - Part B Pestic. Food Contam. Agric. Wastes 2020, 55, 495–500. https://doi.org/10.1080/03601234.2020.1724660. [CrossRef]
- Folin, O.; Denis, W. on Phosphotungstic-Phosphomolybdic Compounds As Color Reagents. J. Biol. Chem. 1912, 12, 239–243. https://doi.org/10.1016/s0021-9258(18)88697-5. [CrossRef]
- A Agbor, G.; Vinson, J.A.; Donnelly, P.E. Folin-Ciocalteau Reagent for Polyphenolic Assay. Int. J. Food Sci. Nutr. Diet. 2014, 147–156. https://doi.org/10.19070/2326-3350-1400028. [CrossRef]
- Ovemair Barbosa, Claudia Ortiz, Angel Berenguer-Murcia, Rodrigo Torres, Rafael C. Rodrigues, R.F.-L. GLUTARALDEHYDE IN BIO-CATALYSTS DESIGN: A useful crosslinker and a versatile tool in enzyme immobilization. RSC Adv. 2014, 4, 1583–1600. https://doi.org/10.1039/c3ra45991h. [CrossRef]
- Migneault, I.; Dartiguenave, C.; Bertrand, M.J.; Waldron, K.C. Glutaraldehyde: behavior in aqueous solution, reaction with proteins, and application to enzyme crosslinking. BioTecniques 2004, 37, 790–802. https://doi.org/10.1039/C6TA07211A.
Figure 1.
(a) Catalytic currents obtained through CV measurements by modifying the DRP-110GNP-CNT platform with 1, 1.25 and 1.5 U/mL of TvLac. (b) CA responses of the DRP-110GNP-CNT platform modified with 1.25 U of TvLac, cross-linked with GA at 0.8%, 1.3% and 1.6% v/v.
Figure 1.
(a) Catalytic currents obtained through CV measurements by modifying the DRP-110GNP-CNT platform with 1, 1.25 and 1.5 U/mL of TvLac. (b) CA responses of the DRP-110GNP-CNT platform modified with 1.25 U of TvLac, cross-linked with GA at 0.8%, 1.3% and 1.6% v/v.
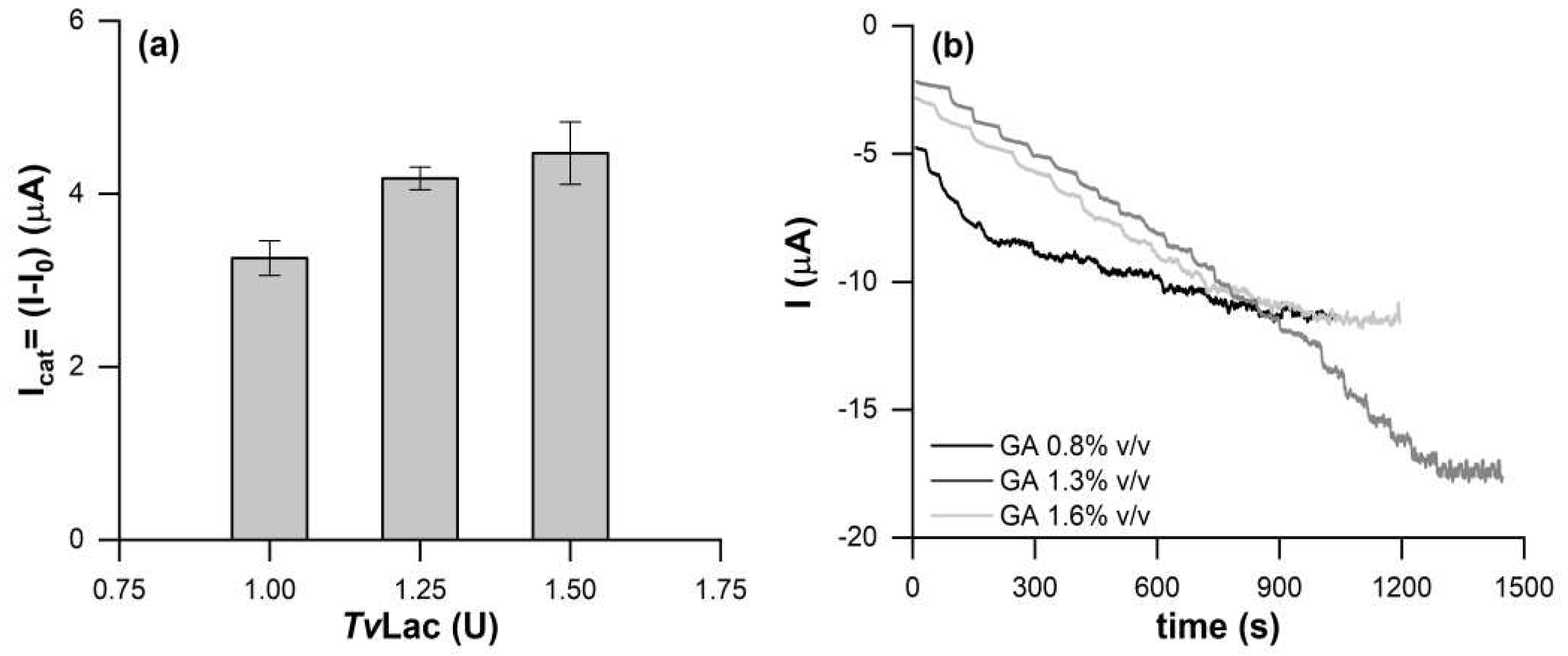
Figure 2.
Surface analysis of electrodes: FT-IR spectra performed for the different platforms: (a) DRP-110, DRP-110CNT, DRP-110CNT-GNP; (b) DRP-110CNT-GNP/TvLac, DRP-110CNT-GNP/TvLac+GA. The comparison among the spectra is related to the grid central spot. Distribution maps of the CO – NH amide I bond; (c) Integration of the band 1601.20-1731.20 cm-1 over the DRP-110CNT-GNP/TvLac surface; (d) Integration of the band 1601.20-1731.20 cm-1 over the DRP-110CNT-GNP/TvLac+GA surface.
Figure 2.
Surface analysis of electrodes: FT-IR spectra performed for the different platforms: (a) DRP-110, DRP-110CNT, DRP-110CNT-GNP; (b) DRP-110CNT-GNP/TvLac, DRP-110CNT-GNP/TvLac+GA. The comparison among the spectra is related to the grid central spot. Distribution maps of the CO – NH amide I bond; (c) Integration of the band 1601.20-1731.20 cm-1 over the DRP-110CNT-GNP/TvLac surface; (d) Integration of the band 1601.20-1731.20 cm-1 over the DRP-110CNT-GNP/TvLac+GA surface.
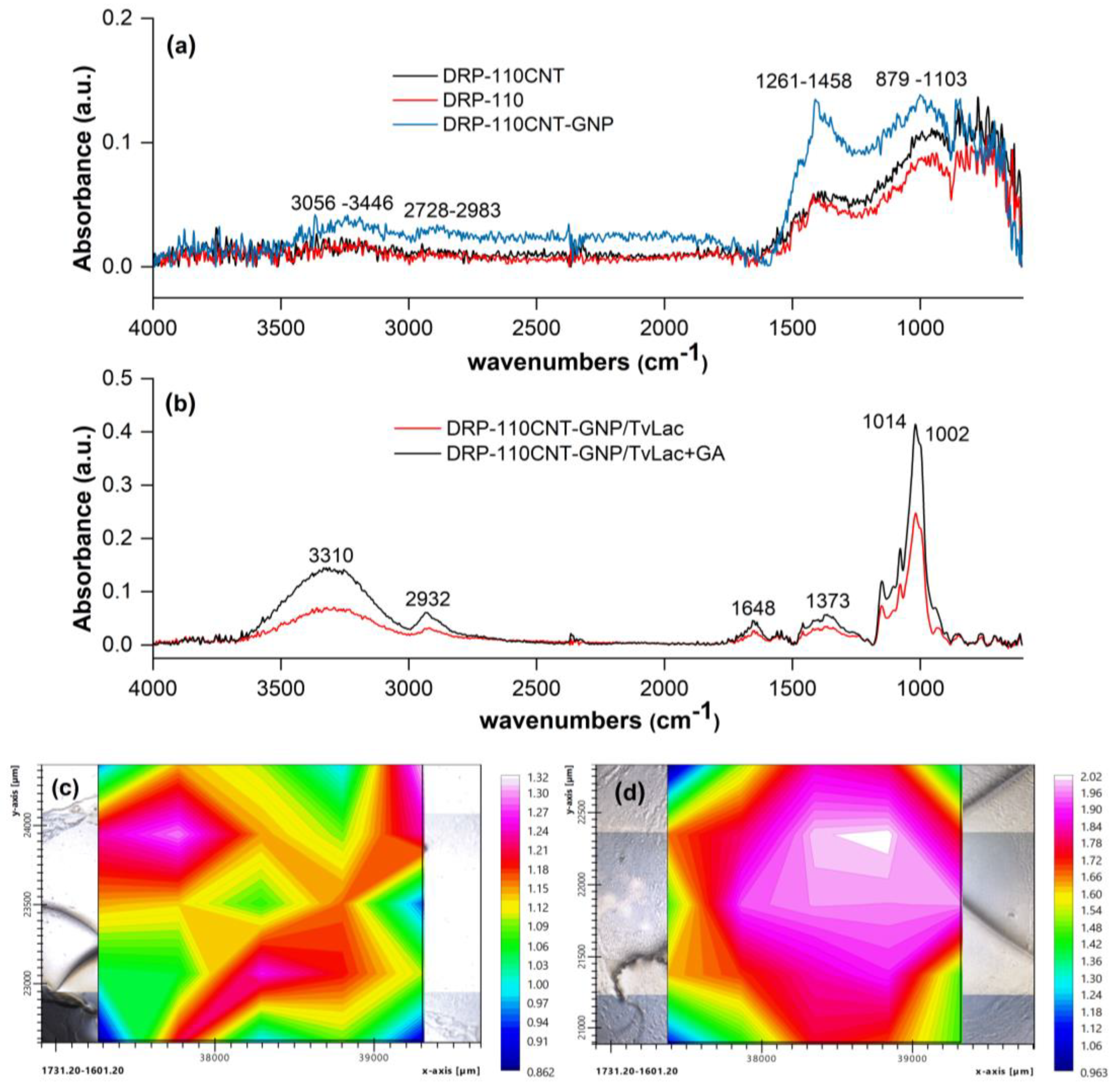
Figure 3.
CVs measurements carried out in a solution of catechol 0.02 mM, AcONa buffer 0.01 mM, pH 5, scan rate 5 mV/s for (a) DRP-110/ TvLac+GA; (b) DRP-110CNT/TvLac+GA; (c) DRP-110GNP/TvLac+GA; (d) DRP-110GNP-CNT/ TvLac+GA in presence (red curves) and absence (black curves) of TvLac.
Figure 3.
CVs measurements carried out in a solution of catechol 0.02 mM, AcONa buffer 0.01 mM, pH 5, scan rate 5 mV/s for (a) DRP-110/ TvLac+GA; (b) DRP-110CNT/TvLac+GA; (c) DRP-110GNP/TvLac+GA; (d) DRP-110GNP-CNT/ TvLac+GA in presence (red curves) and absence (black curves) of TvLac.
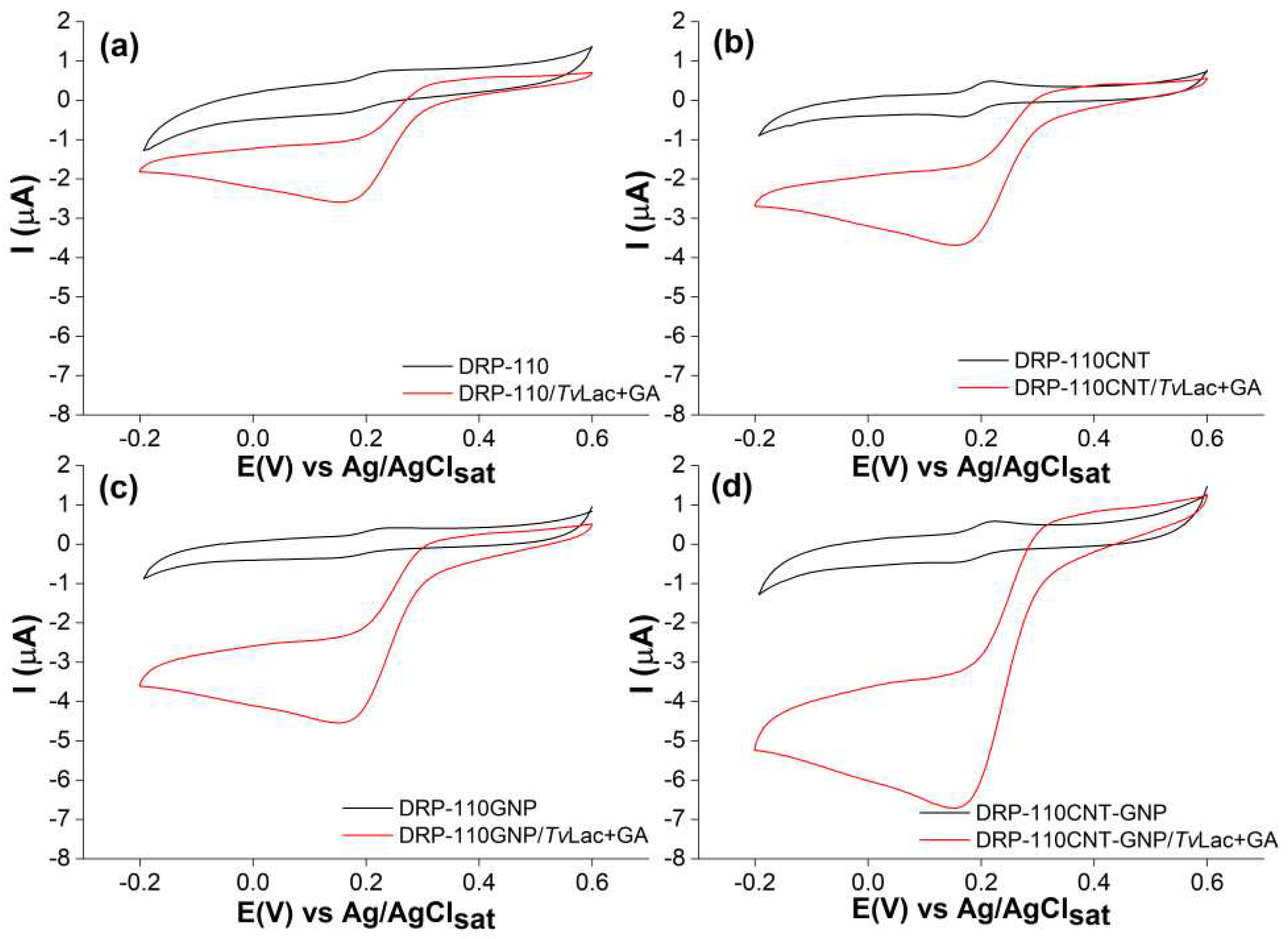
Figure 4.
CAs measurements carried out in AcONa buffer 0.01 M, pH 5, Edc = -0.1 V, tinterval = 0.1 s, by adding equal volumes of a catechol solution at 255 mM, under constant stirring. (a) DRP-110/TvLac+GA (grey); DRP-110GNP/TvLac+GA (black); (b) DRP-110CNT/TvLac+GA (grey); DRP-110CNT-GNP/TvLac+GA (black); (c) Michaelis-Menten curves obtained by fitting the Icat data vs catechol concentration (where Icat = I0 – I) related to the CA profiles reported in Figure 4b; (d) Linear ranges of response for DRP-110CNT/TvLac+GA and DRP-110CNT/TvLac+GA.
Figure 4.
CAs measurements carried out in AcONa buffer 0.01 M, pH 5, Edc = -0.1 V, tinterval = 0.1 s, by adding equal volumes of a catechol solution at 255 mM, under constant stirring. (a) DRP-110/TvLac+GA (grey); DRP-110GNP/TvLac+GA (black); (b) DRP-110CNT/TvLac+GA (grey); DRP-110CNT-GNP/TvLac+GA (black); (c) Michaelis-Menten curves obtained by fitting the Icat data vs catechol concentration (where Icat = I0 – I) related to the CA profiles reported in Figure 4b; (d) Linear ranges of response for DRP-110CNT/TvLac+GA and DRP-110CNT/TvLac+GA.
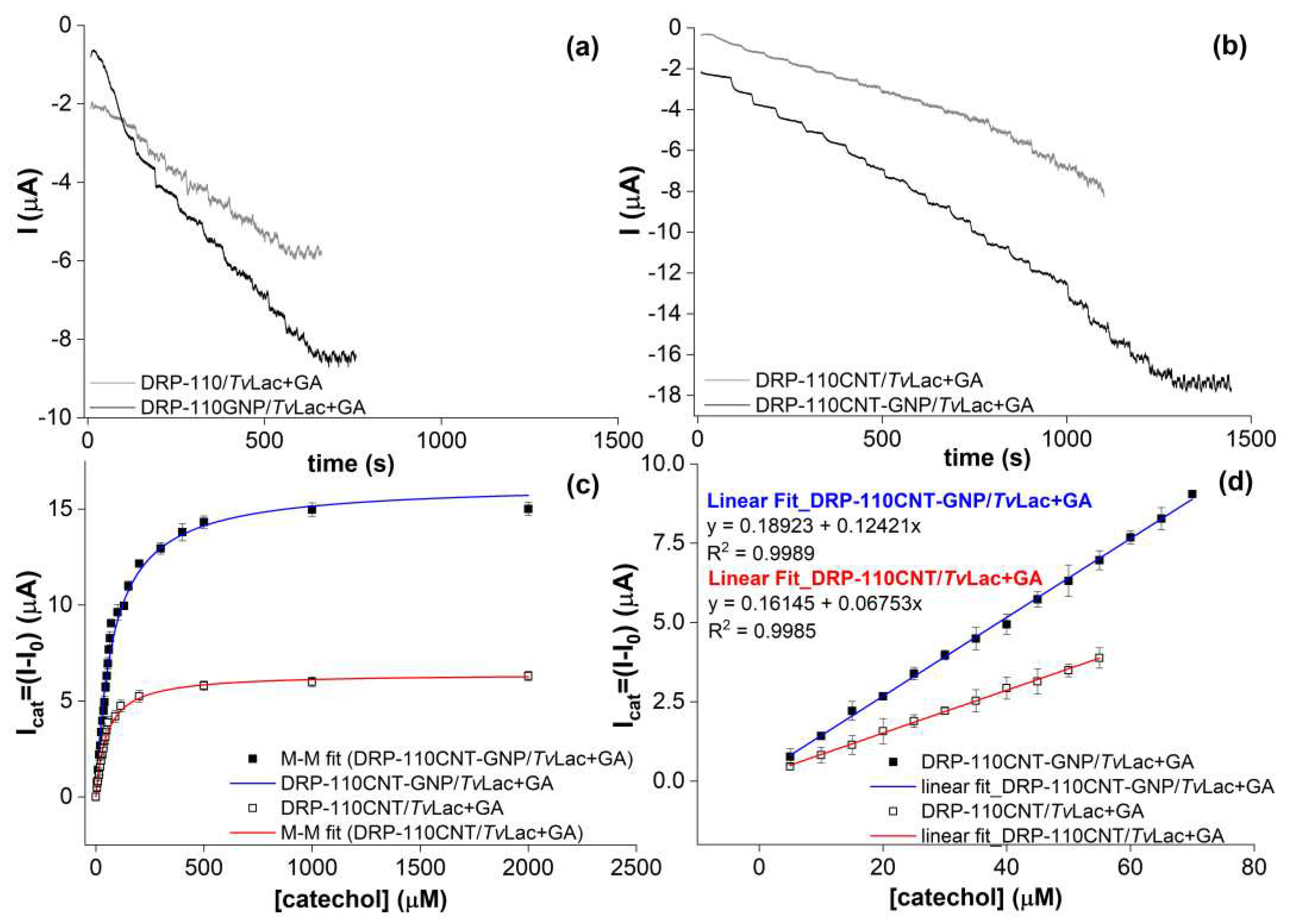
Figure 5.
(a) Chronoamperogram for the analysis of the coffee blend II and (b) the corresponding graph of standard additions equation y=0.1306x+1.7263 r2=0.9978; (c) Standard calibration curve for Folin-Ciocâlteu method; (d) Phs content variation vs roasting degree and time; (e) Correlation between Phs concentration for the different coffee blends tested with the smartphone-based electrochemical biosensor (EC) and Foline-Ciocâlteu standard method (FC).
Figure 5.
(a) Chronoamperogram for the analysis of the coffee blend II and (b) the corresponding graph of standard additions equation y=0.1306x+1.7263 r2=0.9978; (c) Standard calibration curve for Folin-Ciocâlteu method; (d) Phs content variation vs roasting degree and time; (e) Correlation between Phs concentration for the different coffee blends tested with the smartphone-based electrochemical biosensor (EC) and Foline-Ciocâlteu standard method (FC).
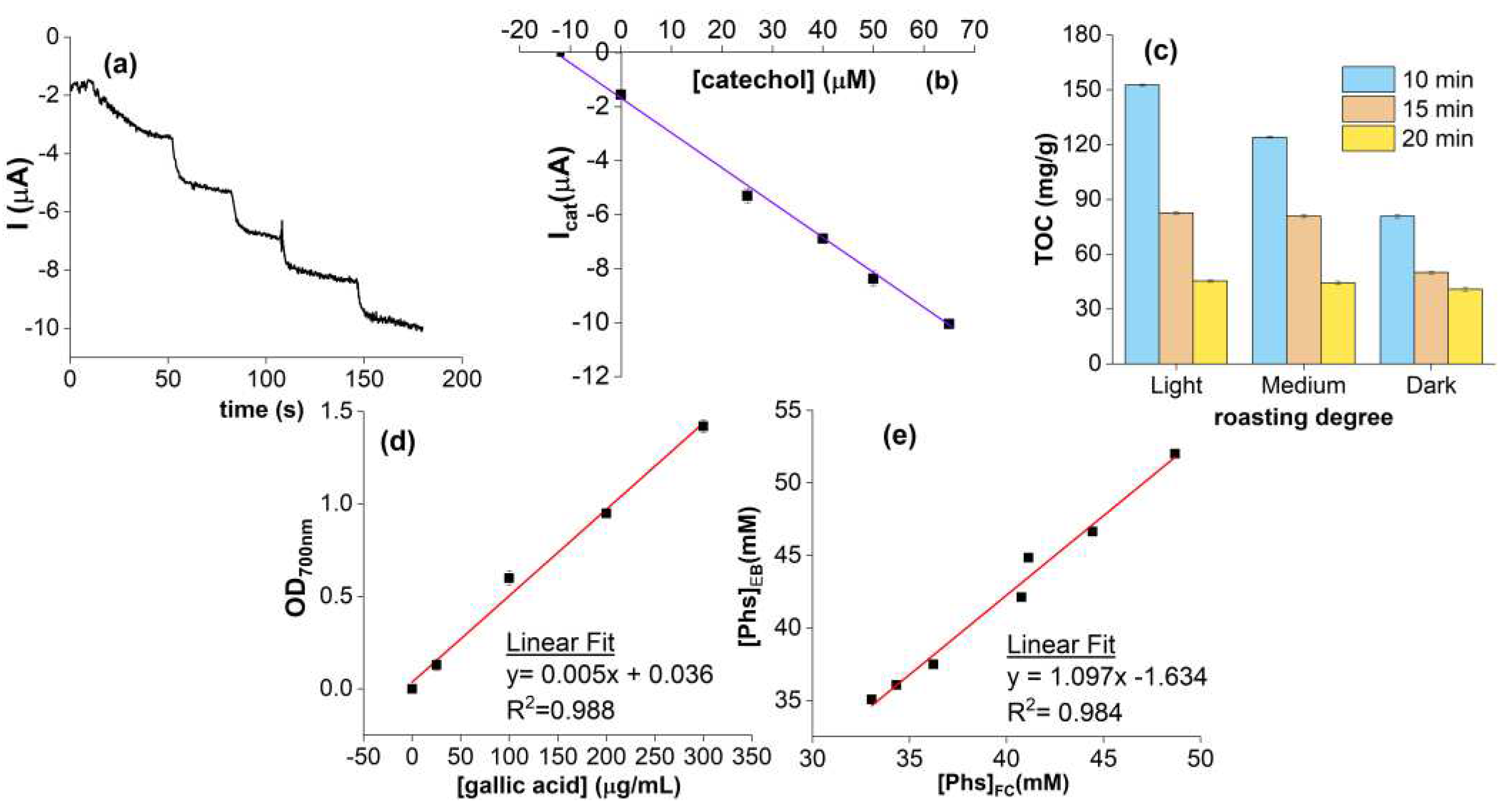
Figure 6.
(a) Measurement setup; (b) catechol solution addition.

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



