
Submitted:
26 June 2023
Posted:
27 June 2023
You are already at the latest version
Abstract
Acute myeloid leukemia (AML) is the most common and incurable leukemia subtype. Despite extensive research into the disease's intricate molecular mechanisms, AML have not yet identified effective treatments or expanded diagnostic or prognostic markers. The morphological, immunophenotypic, cytogenetic, biomolecular, and clinical characteristics of AML patients are extensive and complex. Leukemia stem cells (LSC) consist of hematopoietic stem cells (HSC) and cancer cells transformed by the complex, finely-tuned interaction that causes the complexity of AML. Microenvironmental regulation of LSC dormancy, its diagnostic and therapeutic implications for identifying and targeting LSCs due to their significance in the pathogenesis of AML were discussed in this review. It is essential to perceive the relationship between the niche for LSCs and HSC, which together cause the progression of AML. Notably, methylation is a well-known epigenetic change that is significant in AML, and our data also reveals that microRNAs are a unique factor for LSC. Furthermore, multiple-targeted approaches to reduce the risk of epigenetic factors, such as the administration of natural compounds for the elimination of local LSC, may prevent potentially fatal relapses. Furthermore, the survival analysis of overlapping genes revealed that specific targets had significant effects on the survival and prognosis of patients. We predicted that the multiple-targeted effects of herbal products on epigenetic modification are governed by different mechanisms in AML and prevent potentially fatal relapses. Thus, these strategies can facilitate the incorporation of herbal medicine and natural compounds into the advanced drug discovery and development processes enabled achievable by Network Pharmacology research.
Keywords:
AML
; LSCs
; herbal medicine
; natural product
; miRNA
; epigenetic modifications
Introduction
AML has numerous morphological, immunophenotypic, cytogenetic, biomolecular, and clinical characteristics resulting from genomic and epigenetic alterations [1,2,3]. Thus far, the progress that has been made in treating AML, recurrence continues to be the most challenging problem. Even though 10–40% of AML patients younger than 60 years old are generally resistant to AML induction therapy, the rate for patients older than 60 years old is much greater, ranging from 40–60% [4]. The basic rationale for this approach is that AML has been recognized as the etiological agent that first provided development to the cancer stem cell. Thus, extensive research on the genetics of AML has led to the sustained use of stem cell therapy for the disease[5]. Furthermore, LSCs are derived from leukemia-initiating cells at various phases within primitive multipotent cells, which may cause the leukemic clone to relapse because leukemic cells are diverse [6,7,8]. Allogeneic stem cell transplantation (AlloSCT) is the only long-term cure for myeloid malignancies such as AML, chronic myeloid leukemia (CML), myelodysplastic syndromes (MDS), Chronic myelomonocytic leukemia (CMML), and juvenile myelomonocytic leukemia (JMML), especially in high-risk individuals [9]. Age and comorbidities, such as those calculated by the Hematopoietic Cell Transplantation-specific Comorbidity, affect transplant potential [10]. After a few cycles of induction chemotherapy (IC), 30% of patients with recently diagnosed AML do not experience morphological complete remission (CR) [11]. The fact that no one recognizes how to deal with AML that does not conform with IC makes this quite unsatisfactory [12]. There is no single approach that is commonly regarded as the gold standard for treating AML in the elderly. A few common ways to treat cancer are best supportive care (BSC) alone, standard IC, and low-dose Ara-C (LDAC). The National Comprehensive Cancer Network (NCCN) guidelines indicate that individuals with AML who are younger than 60 and have better prognostic factors should achieve IC. However, many elderly AML do not comply with these requirements [13,14]. The main problem with developing targeted inhibitors for therapeutic management of AML is that it differs so greatly between individuals. As a result, tailored therapy is the most effective way for efficiently controlling this disease [15]. Personalized treatment focuses on the main categories that each patient’s leukemia cells adhere to. Without functional research, genomic approaches are unable to explain how the disease pattern contributes to the disease; they can only identify the genes with significant defects [16]. AML’s classification, identification, prognosis, and treatment are determined by the gene mutation cause and cytogenetic profiling’s understanding of its association with LSC.
Characterization of Genetic mutation in AML
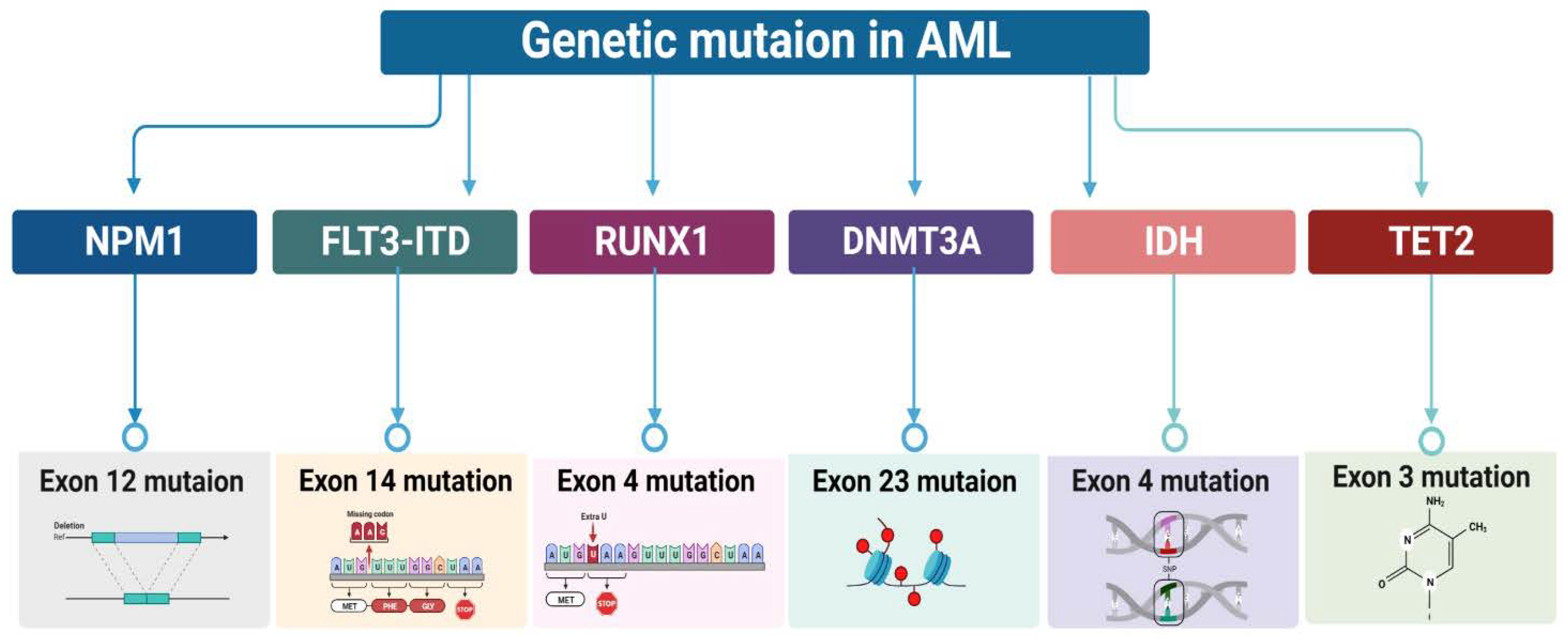
Through using next-generation sequencing, many recurrent somatic mutations were found in more than 90 % of AML patients [17,18]. Nucleophosmin (NPM1), FMS-like tyrosine kinase 3 (FLT3), Runt-related transcription factor (RUNX1), DNA methyltransferase 3A (DNMT3A), Isocitrate dehydrogenase (IDH), Ten-eleven translocation 2 (TET2), and CCAAT/enhancer-binding protein-alpha (CEBPA), are known to be altered [17,18,19]. In AML patients, exon 12 of the NPM1 gene on chromosome 5q35 is altered, causing frameshift and cytoplasmic protein elongation [20]. Deletion/insertion mutations in exon 12 of NPM1 (NPM1-DIM), typically seen in AML patients, modify the C-terminal amino acids and impair the protein’s nucleocytoplasmic shuttling function, causing disease pathogenesis[21]. Additionally, NPM1 mutations, which are present in about 30% of patients with de novo AML, are more frequent in the elderly than in the young [22]. Prognosis is determined by molecular subgroups that are therapeutically targetable. Sometimes exquisitely, single mutations and mutation combinations interact differentially in diploid karyotype patients. Approximately fifty percent of patients with a diploid karyotype and an NPM1 mutation acquired a FLT3 mutation, most notably FLT3 internal tandem duplication (FLT3-ITD)[23,24]. Consequently, the outcome was historically poor and was mostly driven by the FLT3 allelic ratio[25]. The FLT3 mutation is one of the most frequent somatic mutations in AML, affecting between 25 and 45 percent of patients [22]. At least in part, it has been suggested that FLT3 is a tyrosine kinase receptor that is constantly active in AML blasts due to the FLT3 gene mutation. This promotes leukemogenesis and controls how leukemic blasts function [26]. It’s interesting to note that a high incidence of in-frame mutations in FLT3-ITD of exon14, accounting for 70%, was found[27]. In AML patients, exon 12 of the NPM1 gene on chromosome 5q35 is altered, causing frameshift and cytoplasmic protein elongation [20]. Deletion/insertion mutations in exon 12 of NPM1 (NPM1-DIM), typically seen in AML patients, modify the C-terminal amino acids and impair the protein’s nucleocytoplasmic shuttling function, causing disease pathogenesis[21]. Acute erythroid leukemia, erythroid/myeloid type has been eliminated from the acute myeloid leukemia category, despite the fact that the subcategories of AML, not otherwise specified lack prognostic significance when cases are classified based on NPM1 mutation and CEBPA biallelic mutation status [28,29]. One of the most often unregulated genes in leukemia through chromosomal translocations and point mutations is the AML1/RUNX1 gene, also known as RUNX1, which has 10 exons (exons 1-6, 7A, 7B, 7C, and 8)[30,31]. An exon 4 (c.497_498insGA) frameshift RUNX1 mutation was unequivocally discovered in each and every one of the patient’s samples[32]. The fusion protein AML-ETO, or t(8;21)(q22;q22), is produced by its frequent translocation with the ETO/MTG 8/RUNX1T1 gene on 8q22. AML [33]. Most AML patients have recurrent DNMT3A mutations at many locations, including codon R882. Mutations have been identified on exons 8–23, with most, including R882, on exon 23 [34,35]. Important factor DNMT will be described in detail in subsequent contents, so that is all we will discuss in this section. Isocitrate dehydrogenase (IDH) is a three-isoform enzyme that transforms isocitrate to α-KG. IDH1 and IDH2 reside in the cytoplasm and mitochondria, respectively, whereas IDH3 resides in the mitochondrial matrix [36]. IDH1 and IDH2 mutations were found to reoccur in glioma [37,38] followed by AML, cholangiocarcinoma, chondrosarcoma, and chondromas [39]. Missense variants that cause a single amino acid substitution of arginine residues at codon 132 in exon 4 of the IDH1 gene and codons 140 or 172 in exon 4 of the IDH2 gene are the cause of recurrent IDH1/2 mutations[40]. It has been revealed that a germline-synonymous single-nucleotide polymorphism in exon 4 of the IDH1 gene, codon 105, has prognostic significance in AML[41,42]. Although IDH2 mutations are notably beneficial in all non-M3 or cytogenetically normal AML, IDH1 mutations are associated with lower overall survival and event-free survival, particularly for patients with normal cytogenetics [43,44]. Moreover, TET2 is also required for hematopoiesis and the correct differentiation of HSCs by oxidizing 5-methylcytosine to 5-hydroxymethylcytosine (5hmC), 5-formylcytosine, and 5-carboxylcytosine. TET2 exon 3 mutations cause AML by lowering 5hmC levels, promoting HSC self-renewal, and enlarging myeloid lineage cells[45]. Additionally, the CEBPA gene contains a GC-rich (more than 70%) coding sequence that is contained inside a single exon and aligns to chromosomal band 19q13.1[46]. A novel frameshift mutation, c.1590delC, was discovered in AML with biallelic CEBPA mutation [35]. Although the prognosis for young people with AML has substantially improved because of advancements in treatment, the majority of new cases involve elderly patients, and their prognosis is still extremely poor. The prognosis is depressing even with current therapies; the 5-year relative survival rate is about 30% to 50%[47]. No simple solution exists. For the same reasons, choosing the best approach is problematic. LSC characterization may assist in identifying patients who will gain advantage from a novel targeted therapy [48]. The focus of this review was to identify and characterize relapse and resistance due to dysregulation of pathway and environment with LSC in AML in order to better comprehend the therapeutic benefits of clinical trials for disease treatment. Thus, the purpose of this study is to determine how gene mutations influence biological subgroups and how diseases change between the point in time they are initially diagnosed and when they recur. Given this, it is evident that any potential treatment for AML is effective.
Figure 1.
An overview of the key molecules involved in the development of AML.
Significance of the phenomenon caused by HSC and AML’s initial interaction
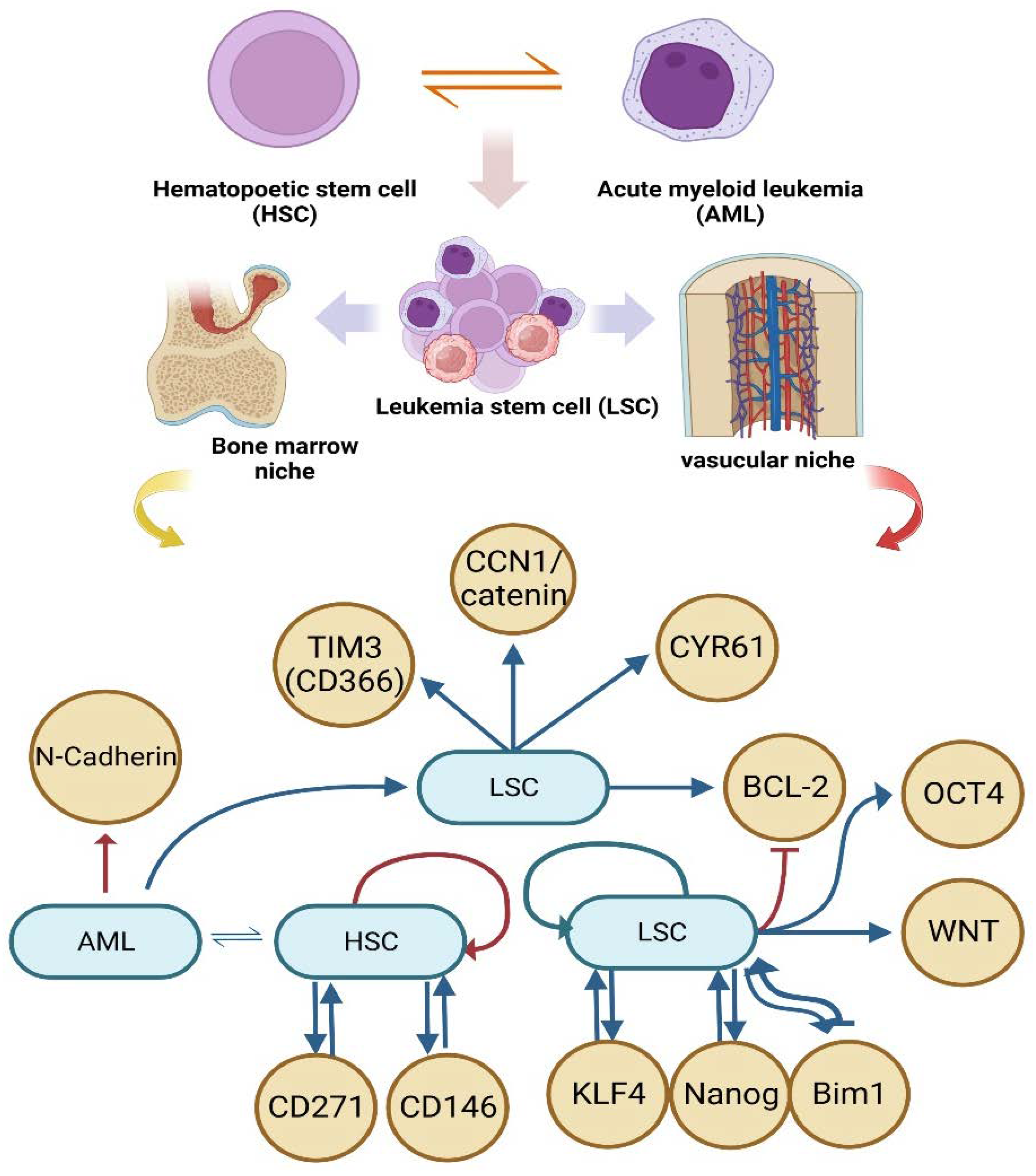
AML was one of the earliest cancers in which a stem cell was found. AML is characterized by the presence of numerous blast cells, including myeloblasts, monoblasts, and megakaryoblasts, in the bone marrow and/or peripheral circulation. Due to the rapid progression of AML, patients should undergo rapid cytogenetics and molecular analysis to determine optimal risks and treatments [49]. Furthermore, relapse is the greatest barrier to treating AML, as there are no effective treatments and 90% of people who experience it die. It is now well acknowledged that a persistent subset of AML cells called leukemia-initiating cells or LSCs are responsible for relapse [50]. AML in the initiation of the disease by stromal cells and immune cells through paracrine and autocrine signals for long-term hematopoietic regulation of [51] stem cells released factors and cytokines that defend AML cells against chemotherapeutic agents and promote drug resistance [52,53,54]. Although LSC are a very rare subgroup of AML cells, they have the unique ability to establish and sustain a cellular hierarchy, and their persistence throughout remission has been associated to a poor clinical prognosis[55,56,57]. It was interestingly revealed that circulating cancer cells target the "niche" in the bone marrow that is home to hematopoietic stem cells (HSCs) and, more importantly, that they compete with HSCs for occupancy of that niche[58]. HSCs are known to associate with at least two distinct niches in the bone marrow, the endosteal niche (which contains osteoblasts) and the vascular niche (which contains endothelial cells)[59]. Regulation of LSC and HSC contact-dependent (surface receptors and ligands) and contact-independent (cytokines, chemokines, growth factors, exosomes) interactions with the rest of the bone marrow microenvironment[60,61]. HSCs adapt blood production to the organism’s requirements by using cues from the bone marrow (BM) niche. Blood cancers alter the BM niche in a way that directly affects the LSCs that initiate the disease [62]. Cancer cells and leukocytes, including lymphocytes, tumor-associated macrophages (TAM), cancer-associated fibroblasts, endothelial cells, and pericytes, form a diverse population within the tumor microenvironment, contributing to a dynamic and complex ecology [63]. Atypical phenotypes caused by genetic and epigenetic changes to cells’ metabolism include adaptive modifications to signal transduction and transcriptional regulation, which supply the energy and biomass needed for cell proliferation [64]. When cancer cells first connect to the endothelium, N-cadherin plays a critical part in mediating cellular contact. This heterotypic contact is the initial event in the chain of events leading to the metastatic spread of cancer cells through transendothelial migration[65]. Here, it was observed that tumor stiffness modifies the CCN1/catenin/N-cadherin pathway, which, by making it easier for cancer cells to adhere to blood vessels, contributes to the metastatic cascade[66]. Cysteine-rich angiogenic inducer 61 (CYR61) is a matricellular protein expressed by endothelial cells in stiff surroundings that activates β-catenin to increase N-cadherin expression. This allows cancer cells to enter the bloodstream and metastasize by stable endothelium interaction [66,67]. Differentiating and targeting LSCs from HSCs has been produced possible by the identification of cell membrane surface markers, such as CD33[68], CD123[69], CLL-1[70], CD44[71], CD47[72], and CD96[73], which are expressed preferentially on LSCs compared to HSCs in AML[74]. The percentage of HSCs separated from a leukemia patient that carry at least the first mutation is what we refer to as the "preleukemic burden" in order to measure this heterogeneity. 39 individuals with AML were studied to determine the frequency of known leukemogenic driver mutations in HSCs, T cells, and blasts[75]. It has been predicted that TIM3 (CD366), a member of the T cell immunoglobulin mucin (TIM) family, is a novel, important, and differentiating membrane surface marker for LSCs [76,77,78,79,80] a negative regulator of T helper1 (Th1) cells immunity [81] and a prognostic factor in patients with AML[74]. High levels of TIM3 on T cells in the peripheral circulation mediate exhaustion/dysfunction of T cell in AML patients. This may be a crucial immune surveillance strategy for leukemia [82]. LSCs have many characteristics in common with their normally functioning counterparts, such as quiescence, which is characterized by low metabolic activity[83], high expression of anti-apoptotic proteins like Bcl-2[84], and resistance to cellular stress and cell death caused on by chemotherapeutics [50]. As previously discussed, cancer cells can enter a quiescent state and wait until their fitness and environmental conditions are conducive to growth in order to keep the disease undetected for a long time [67,85]. Stromal cells surround the sinusoidal blood vessels and are essential to HSC; it has been identified that CD271 and CD146 are markers for these cells [86,87,88]. By identifying these mutational patterns, it may be possible to cure or avoid these mutations using therapies such as HSCT, in addition to estimating patient outcomes based on AML subtype and prognostic variables [89]. Without cell division, bloodstream survival, or homecoming, LSCs increased. The self-renewal-related genes KLF4, Bmi-1, and Nanog are also expressed by LSCs, which have the ability to form sphere-like structures in vitro and weak tumors in vivo [90]. Additionally, tumor-promoting stem cell signaling pathways such Wnt, Nanog, and Oct4 were activated by extracellular matrix (ECM) components in LSCs, resulting in the spread of metastasis [91,92]. It is becoming obvious that the microenvironment interacts with leukemic blasts, develops malignant cells that support LSC immune escape mechanisms, and inhibits effector cell immunocompetence, such as T- and natural killer cells [93]. The perivascular niche is a tumor-promoting environment that is made up of a variety of micro vessels. It is responsible for regulating the dormancy of cancer cells that have spread from various primary tumors to the bone marrow, the lungs, and the brain [94,95,96,97]. The abundant availability of oxygen, nutrients, and paracrine substances found in perivascular niches makes them an ideal habitat for the growth of CSCs [98,99]. Dormant cells that have been adapted through genetic and epigenetic editing [85], may contribute to disease evolution in regards to phenotype and function by promoting TME remodeling, making it "fertile soil" for propagation [63]. Only cells grown in soft matrix express CSC markers [100], suggesting that a flexible microenvironment may enhance cancer cell stemness, an attractive hypothesis that needs in vivo validation [66]. Recent research has identified that CSCs can be relatively abundant (at least in some tumors), have the ability to switch between dormant and proliferating states, and are characterized by a high degree of heterogeneity and plasticity over space (that is, in different regions of the tumor) and time [101,102]. In general, this complex molecular cross-talk between the LSC and its gaps can be controlled by acquiring a comprehension of the interaction that maintains the LSC in a quiescent state. Epigenetic changes result in recurrence during the cytogenic compensated process. Althigh epigenetic modifications such as DNA methylation are crucial, it has been believed that identifying factors other than well-known mutations could bring the possibility of treatment closer.
Figure 2.
An overview of the molecular crosslinks produced by the interaction of the HSC and LSC.
Epigenetic modification of the LSC Microenvironment
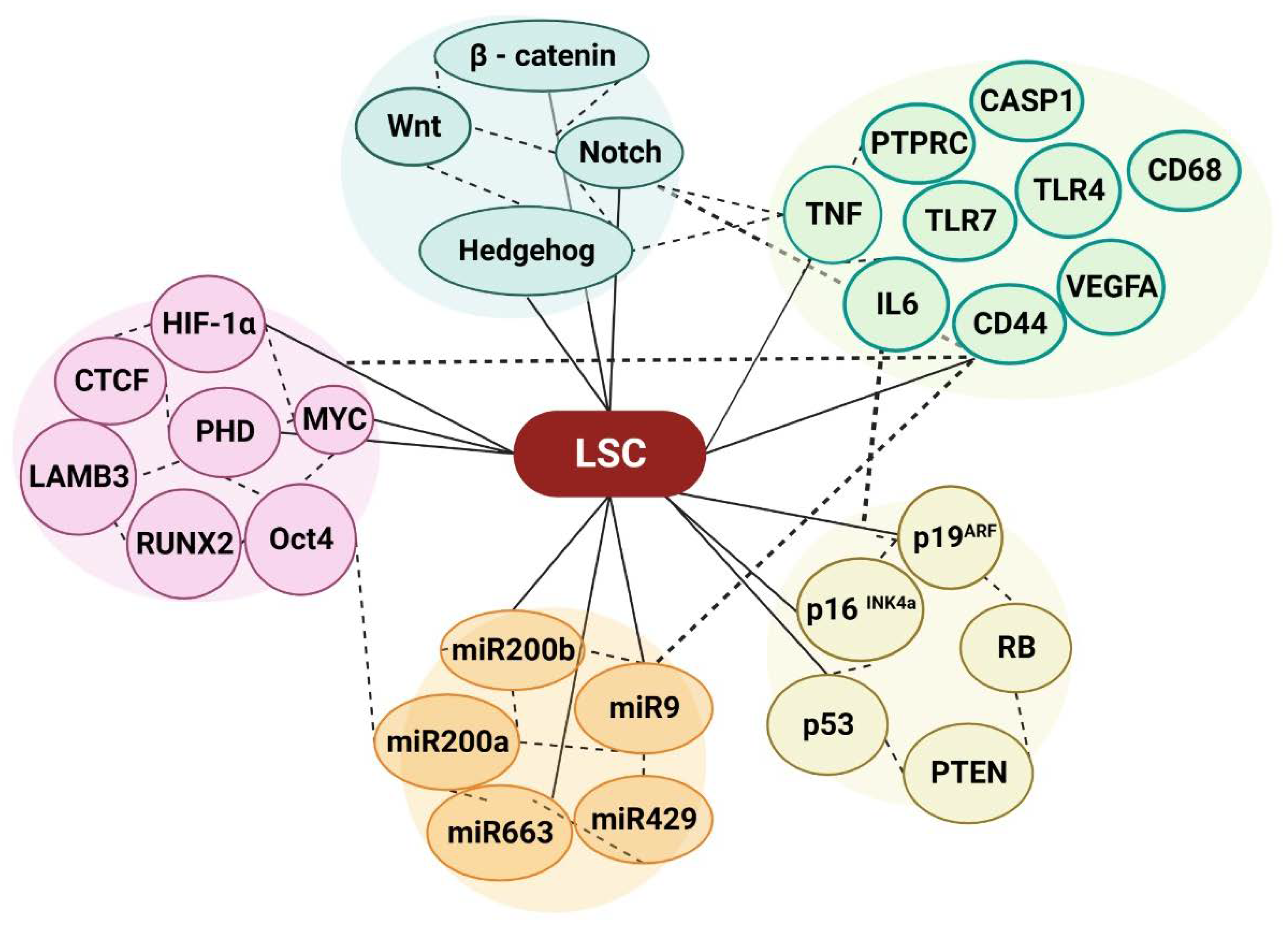
Cancer cells can acquire resistance to chemotherapy due to genetic alterations in tumor somatic cells. Multidrug resistance, apoptosis suppression, epigenetics metabolism, DNA repair, and gene amplification are some more mechanisms by which cancer cells might develop resistance to treatment [103]. Stem cell markers are present in both cancer stem cells and normal stem cells[104,105], therefore, genetic alterations that initiate cancer, such as the acquisition of oncogenes or the accumulation of DNA mutations, should be the primary determinant of differentiation [106,107]. We investigated into the extensive usage of epigenetic markers used by cancer cells to control gene expression, including DNA methylation and microRNA, despite decades of therapeutic success, the mystery surrounding progressive and recurring genes and non-coding expression patterns maintains. Several genes have been identified as either promoters or inhibitors of cancer cell proliferation; Wnt/ β - catenin, Hedgehog, and Notch signaling these genes also play important roles in the regulation of normal stem cells [108,109,110,111,112,113], Notably, mutations that open up regular stem cell pathways can promote the development of cancer stem cells. Self-renewal of hematopoietic and brain stem cells is regulated by Wnt and Bmi1-dependent pathways[114]. Conversely, p16 INK4a and p19ARF, retinoblastoma protein (RB), and p53 form a signaling network that regulates the entry and exit of the cell cycle. Cancers typically lack the activities of p16 INK4a and p19ARF, RB, and p53, indicating that metabolic processes mediated by these proteins must be impaired for normal cells to transition into cancer cells[115]. Furthermore, phosphatase and tensin homolog (PTEN) is frequently deleted or otherwise rendered inactive in a variety of cancers[116], such as hemopoietic malignancies[117,118,119]. Mutations in the aforementioned genes may alter gene expression, thereby transforming healthy stem cells into cancer stem cells that support tumor growth. Subsequently, gene mutations caused by RNA splicing, DNA methylation, chromatin remodeling, transcription factors, activated signaling, cohesion complexes, tumor suppressors, and nuclear phospholipids are associated with numerous instances of function-based subgroups [120,121,122]. Notably, it is well known that DNA methylation is essential for the hematopoiesis process [123,124]. During the onset and progression of hematological malignancies, the cellular epigenome endures multiple modifications, including hypo- or hypermethylation of CpG islands in promoter regions [125], well-established function of epigenetic regulation in carcinogenesis [126]. CpG islands imply differential methylation regulates miRNA genes. Several candidates had cancer-related hypermethylation and downregulation [127]. Bioinformatics predicted miRNA targets. GO and KEGG pathway enrichment analysis identified common target genes and differentially expressed genes. Immune response, xhemokine activity, immune system, and plasma membrane were the top enriched pathways, with 109 differentially expressed genes and 45 miRNAs. TNF, IL6, TLR4, VEGFA, PTPRC, TLR7, TLR1, CD44, CASP1, and CD68 were hub-genes[128]. Differentially methylated regions (DMRs) methylation at miRNA loci indirectly dysregulates several targets, including master cancer-related genes [129]. Epigenomic and transcriptome data sets have identified DMRs, CpG island methylator phenotypes (CIMPs), and cancer-associated miRNAs. Thus, DNA methylation is dynamic and reversible, which could affect cancer treatment[130]. Several transcription factors and DNA-binding proteins, including HIF-1α, CTCF motif, MYC, LAMB3, RUNX2, and Oct4, are known to be sensitive to CpG-methylated recognition site sequences, which may have direct or indirect effects on miRNAs through association with RNA pol II [131,132]. Recent research has shown that the expression of microRNAs is significantly influenced by DNA methylation and processing, cancer stem cell characteristics, and microRNA promoters. In addition, a recent study demonstrates that the miR-200 family, the miR-9 family, the miR-200b-200a-429 cluster, and their target mRNAs all maintain and regulate CSC DNA methylation[133,134,135,136,137]. It was found that the expression information for both mRNA and miRNA was included in all of the clinical and mutational data for all genomes. The investigation’s findings showed that the Fusion protein and DNA methylation patterns had a strong correlation with the AML sample’s expression signature and differentiation state [138,139]. The establishment of pluripotent states in early embryos and the erasure of parental-origin-specific marks in growing primordial germ cells (PGCs) depend on global DNA demethylation [140]. There is growing evidence that the rapid loss of 5mC during these two major waves of epigenetic reprogramming is unable to be fully explained by replication-dependent passive loss of fifth position of cytosine (5-methylcytosine, 5mC), indicating to the possibility of enzymatic processes which might actively remove or alter methyl groups on cytosines[141]. The DNA of the majority of plant, animal, and fungal models has been modified by 5mC, a widely conserved epigenetic modification [142]. It is well known as epigenetic modulator such as non-coding RNAs have been shown to be significant regulators of epithelial-to-mesenchymal transition (EMT), and CSCs, which overlaps cancer cell invasiveness [143]. Profiling microRNA expression and DNA methylation at microRNA promoters allowed us to examine EMT-induced stemness mediators. Supporting this observation, overexpression of miR-203 induces differentiation and reduces stem cell properties[133,144]. ATRA-induced HL-60 cell differentiation is similarly impacted by miR-663. According to our research, lentiviral-mediated miR-663 treatment can effectively treat hematological cancers [145]. Physiological and epigenetic modifications can lead to immunological escape or immunosurveillance, contributing to malignancies. This study discusses how cancer reprogramming of immune cells and tumor cells influences epigenetic changes such as DNA methylation and histone protein levels [146] as well as miRNA. As a result, in the current work, the epigenetic changes were explored in conjunction with miRNA and DNA methylation in AML signaling in order to explore the potential application of miRNA in AML therapy. The knowledge of how critical metabolites influence the epigenome via dynamically regulating the metabolic states of LSC is discussed in the following section.
Figure 3.
An overview of the LSC’s microenvironment in relation to epigenetic modification.
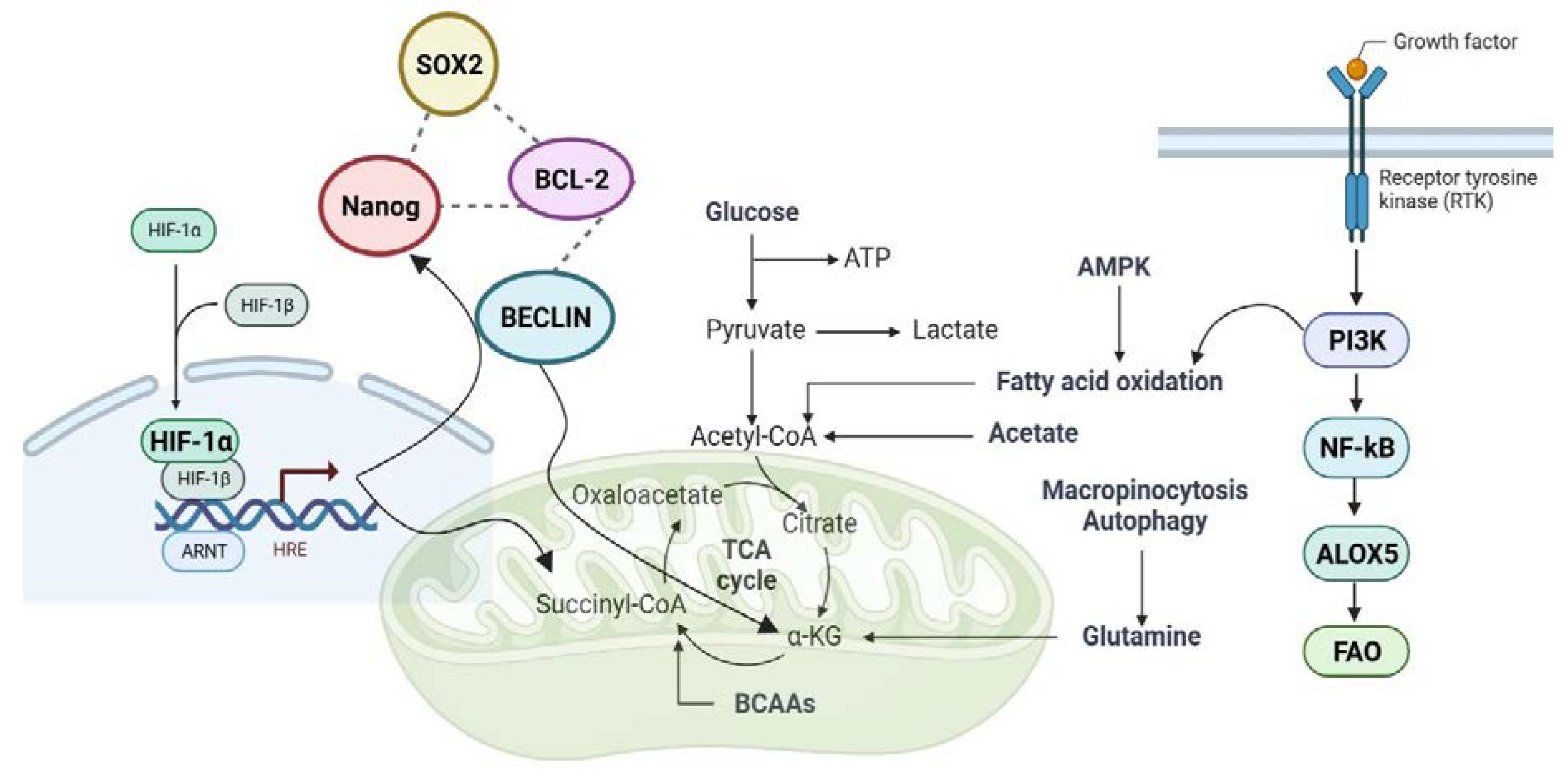
Intelligent application: let the fat out and use it as fuel
Surprisingly, the majority of LSCs that can be identified functionally are "ROS-low." BCL-2 inhibition decreased oxidative phosphorylation and eliminated quiescent LSCs in ROS-low LSCs [147]. Despite the fact that it has been presumed, at least in part, that the biology of LSCs is similar to that of normal tissues, the majority of LSCs in patients with AML are not quiescent (G0/G1: 80-90% with 0.5-10% in G0)[148,149]. When CSCs transition from a quiescent stage to a highly proliferating stage, glycolysis appears to be crucial[150]. Thus, it was found that amino acid or fatty acid oxidation (FAO) creates the high-energy hydrogens (NADH+H+ and FADH2) that drive oxidative phosphorylation (OXPHOS) in AML, which also requires efficient mitochondria[151,152]. LSCs sustain lipolysis in adipocytes, which generates free fatty acids that enhance β-oxidation in AML cells, thereby enhancing their survival, proliferation, and transformation[153]. Thus, AML bone marrow continuously assimilated glucose, whereas pyruvate, lactate, and glycerol-3-phosphate were inversely related to survival [154]. Here, leukemia increases lipoxygenases arachidonate 15-lipoxygenase (Alox15/15-LO (ALOX15) and ALOX5, which metabolize polyunsaturated fatty acids [107]. Increased mitochondrial mass1 and altered metabolic characteristics, such as a greater reliance on OXPHOS [155,156], and FAO [157], are characteristics of this altered phenotype. When compared to normal tissues, cancer cells have a highly stimulated de novo production of these elongated FA chains, which are structural elements of membranes[158,159]. Moreover, many critical signaling pathways, such as p53[160], Nuclear factor-κB (NF-κB) and Phosphoinositide 3-kinases (PI3Ks) [161], have been connected to Alox5 function[162]. Furthermore, the identification of IDH1 and IDH2 mutations in cancer further emphasizes the direct relationship between metabolic instability and carcinogenesis. These alterations join loss-of-function variants that target fumarate hydratase (FH) and various succinate dehydrogenase (SDH) subunits[163,164]. The metabolic enzymes FH, SDH, and IDH have been altered by cancer. Scientists believed that abnormal cancer cell proliferation was brought on by metabolic problems. According to this hypothesis, metabolic reprogramming in CSCs can cause cancer without DNA changes in cancer genes[165]. Given that mutations targeting FH and SDH result in similar increases in prolyl hydroxylases (PHDs), Hypoxia-inducible factor-1 alpha (HIF-1 α) is highly intriguing [166,167]. Multiple studies have shown that Nanog and Sox2, two of the most important transcription factors in cancer stem cells, induce cancer cell metastasis in response to hypoxia. Nanog binds directly to the enhancer region of the BNIP3L (BCL2 Interacting Protein 3 Like) promoter during hypoxia, thereby releasing BECLIN from Bcl-2 (B-cell lymphoma 2), increasing its expression, and promoting one of the hypoxia-induced responses of cancer cells[143,168]. AML patients have altered succinate levels and dysregulated TCA cycle activity. Succinate modifies epigenetic activity by inhibiting 2-oxoglutarate (2-OG)-dependent histone and DNA methylation enzymes [169]. Between succinyl-CoA and isocitrate, alpha-ketoglutarate (KG) is a critical intermediary in the tricarboxylic acid (TCA) cycle. As a crucial component of the anaplerotic process, KG controls ATP synthesis and decreases equivalent (NAD+/NADH) production in the TCA cycle, which affects the level of ROS and immune system homeostasis[170]. Fe (II)/ α-KG-dependent dioxygenases, which are widely distributed in all living organisms[171,172], are thought to use α-KG as a co-substrate. Only a few of the biological processes in which these enzymes play a key role include the post-translational modification of collagen, fatty acid metabolism, oxygen sensing, DNA and RNA repair, and demethylations associated to epigenetic regulation[173]. According to research, alpha-KG maintains the pluripotency of embryonic stem cells (ESCs) [174]. Moreover, α-KG is a primary source of glutamine and glutamate, which are required for the synthesis of both amino acid and collagen [175]. Glutamine is important for cancer stem cell maintenance because it repairs DNA damage through nucleotide biosynthesis and a redox-mediated mechanism [176]. Based on the above mentioned, it has been determined that AML influences the corresponding process, FAs metabolism [177,178] and mitochondrial oxidative phosphorylation [179,180,181,182] during the disease by requesting preferential lipid metabolization over glucose or glutamine [177,178]. Understanding LSC- and HSC-quiescence is crucial for selecting future AML treatment targets, as quiescent AML cells are responsible for the majority of relapses [152]. Reprogramming cells into cancer stem cells exacerbated relapse, drug resistance, and metastasis in a variety of patient cancers. This analysis provides a greater understanding of HSCs and LSCs, enabling researchers to target the LSC population based on the characteristics of AML.
Figure 4.
An overview of the LSC mechanism employs fat as fuel.
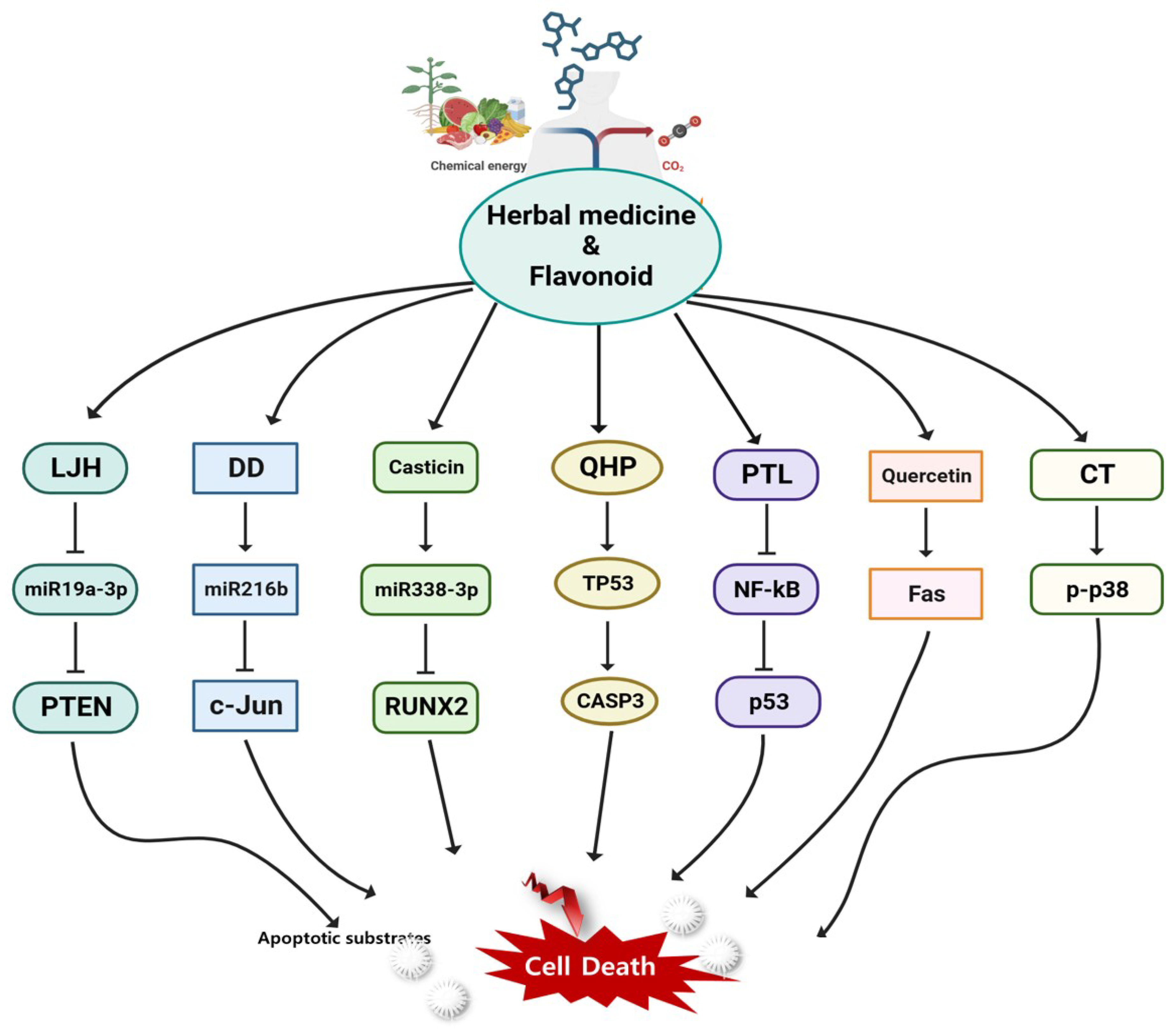
Herbal Treatment Strategies for AML Employing System Biology
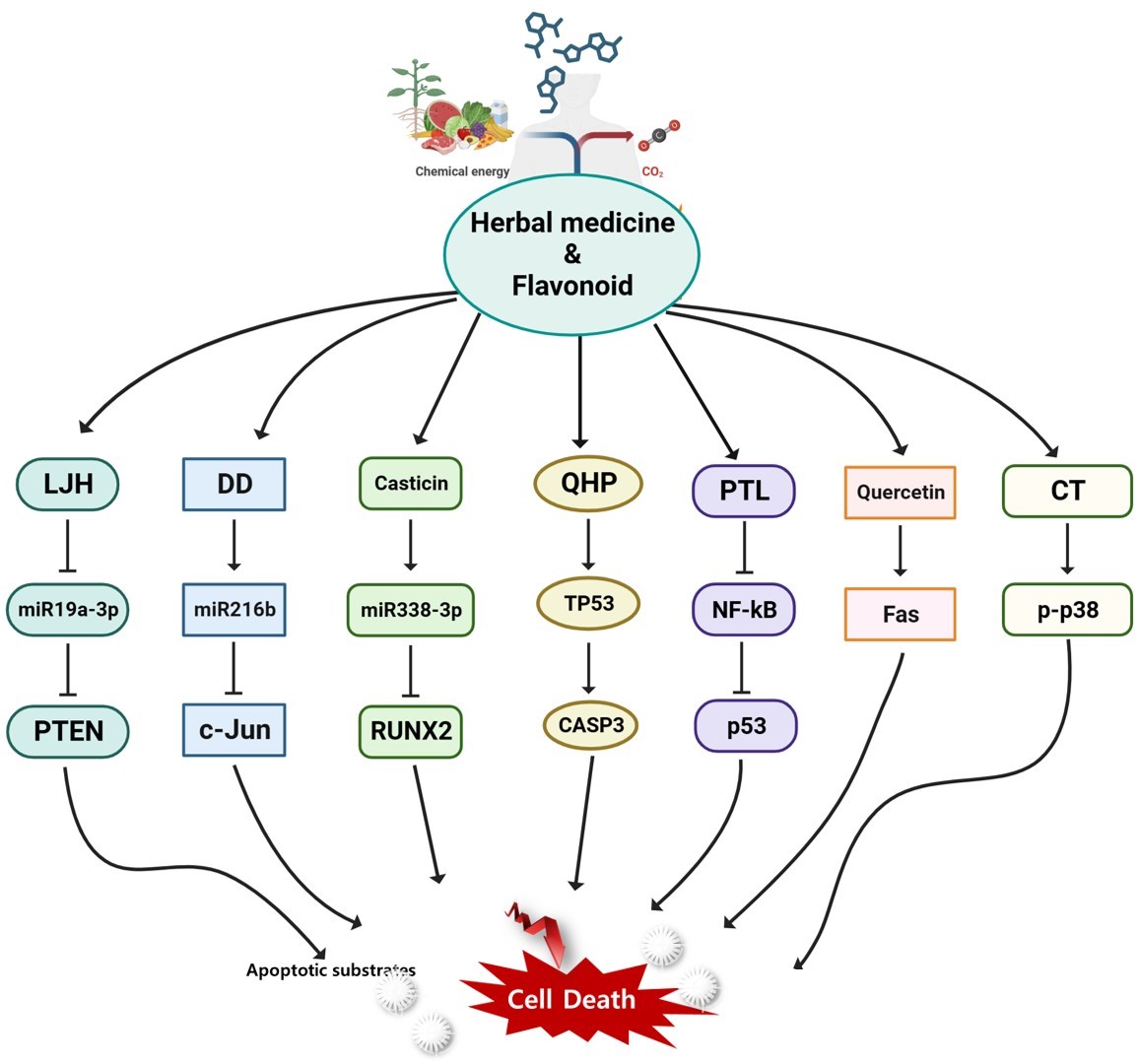
Rydapt, an inhibitor of Fms-related receptor tyrosine kinase 3 (FLT3), and venetoclax, an inhibitor of Bcl-2, are two examples of inhibitory biological therapies used to treat AML[183,184]. Nowadays, genetic and epigenetic pharmaceuticals can treat the genetic etiology of disease as opposed to the gene mutation[185]. Given the complexity of AML, we suggest a candidate group that uses herbal medicines, including the aforementioned components, to target many factors at once. Due to their various efficient constituents, herbal remedies have "multitarget and multipath effects". Numerous studies have demonstrated the synergistic effects that formulations frequently have, which are likely to have an impact on a variety of biological functions and intracellular pathway networks[186,187]. Drug screening is sped up, made more affordable, and improved through computer modeling of biological systems [188]. It facilitates in drugs discovery through herbal and pharmacological understanding [189,190]. Recent studies have shown that miRNAs have a crucial role in controlling the production of oncogenes or tumor suppressors, which in turn controls biological processes such apoptosis, proliferation, and differentiation in hematopoietic cells [191,192]. If we can determine which herbal medicine mechanism modulates miRNA and AML-related target factor, we provide a candidate group for a long-term AML therapeutic alternative. Over 300 prescriptions contain Leonurus japonicus Houttuyn (LJH) in traditional Korean medicine. LJH increased reactive oxygen species, and miR-19a-3p was suppressed in direct relation to PTEN, which increased apoptosis[193]. Dragon's blood, also called Daemonorops draco Blume (DD), is a traditional Korean medicine used to relieve pain from wound infections. DD increased ROS generation and miR-216b expression, while downregulating c-Jun [194]. Casticin has anti-inflammatory and anti-carcinogenic properties that are used in traditional herbal medicine. MicroRNAs, which control oncogenes and cancer suppressors, are dysregulated in diseases. Through upregulating miR-338-3p, which targets RUNX2 and inhibits the PI3K-Akt signaling pathway, casticin increases AML cell death but reduces cell proliferation in vitro and tumor growth in vivo [195]. Qinghuang (QHP) powder and various mixtures are utilized to treat blood cancers such as AML. QHPs were associated with important targets such as AKT1, MAPK1, MAPK3, PIK3CG, CASP3, CASP9, TNF, TGFB1, MAPK8, and TP53 using systematic docking and molecular docking visualization [196]. A-PACs got rid of primary AML blast and progenitor/stem cells without hurting normal cells. Parthenolide (PTL) from fever few extracts kill both primary human LSCs and bulk leukemic cells [197,198]. PTL inhibits the expression of the NF-kB and IL-6 genes in U937 cells, which inhibits apoptosis in a variety of blood malignancies [199,200,201,202]. Quercetin is effective against malignancy in numerous tumor cell lines, including AML[203,204,205]. Quercetin increased the apoptotic process in AML. Both internal mitochondrial routes and external mechanisms under the control of Fas were used for this [203,206,207]. Quercetin is effective against malignancy in numerous tumor cell lines, including AML[203,204,205]. Parts of the fruit of the medicinal tree Melia azedarach have been identified as having anti-cancer properties. 1-Cinnamoyltrichilinin (CT) significantly decreased the viability of AML cells. CT to induce apoptosis in AML cells and activate the p38 pathway [208]. Parts of the fruit of the medicinal tree Melia azedarach have been identified as having anti-cancer properties. 1-Cinnamoyltrichilinin (CT) significantly decreased the viability of AML cells. CT to induce apoptosis in AML cells and activate the p38 pathway [208]. Using the peculiar and complex molecular characteristics of leukemia cells, we provide adequate support for herbal medicine, a potential group that may be involved in the selective targeting of LSCs via a number of strategies. However, our limited understanding of the mechanisms of action of many of these natural compounds is one of the primary obstacles preventing the widespread adoption of herbal medicine in contemporary healthcare[209]. Vitality and homeostasis are returned to the body through herbal medicine[210]. Until now, drug research and development targets one target—a receptor, ion channel, enzyme, or regulatory protein—to treat a disease or symptom. Conventional drug is increasingly embracing polypharmacology as a supplement to using a single active ingredient or component to treat a specific target because the human body is a complex, interrelated system of cells, tissues, organs, and the whole body[211,212]. Systems biology techniques, however still in their earliest stages, may offer a mathematical framework for usefully combining the growing quantity of data and information obtained during cancer diagnosis and help design logical defenses to herbal treatment[213]. The research and application of herbal medicine, which has numerous chemical constituents and molecular targets, is ideal for network pharmacology because it emphasizes multi-channel modulation of signaling pathways[214]. Thus, herbal medicines and natural substances demonstrated potential as AML inhibitors in pharmaceutical research. Additionally, our investigation revealed the effects of chemoprevention, which have been extensively studied over the past decade. In the following section, clinical trials are conducted to determine how adding additional medications to HMA treatments can improve them.
Figure 5.
An overview of the anti-cancer effects of herbal medicine signaling pathways in AML.
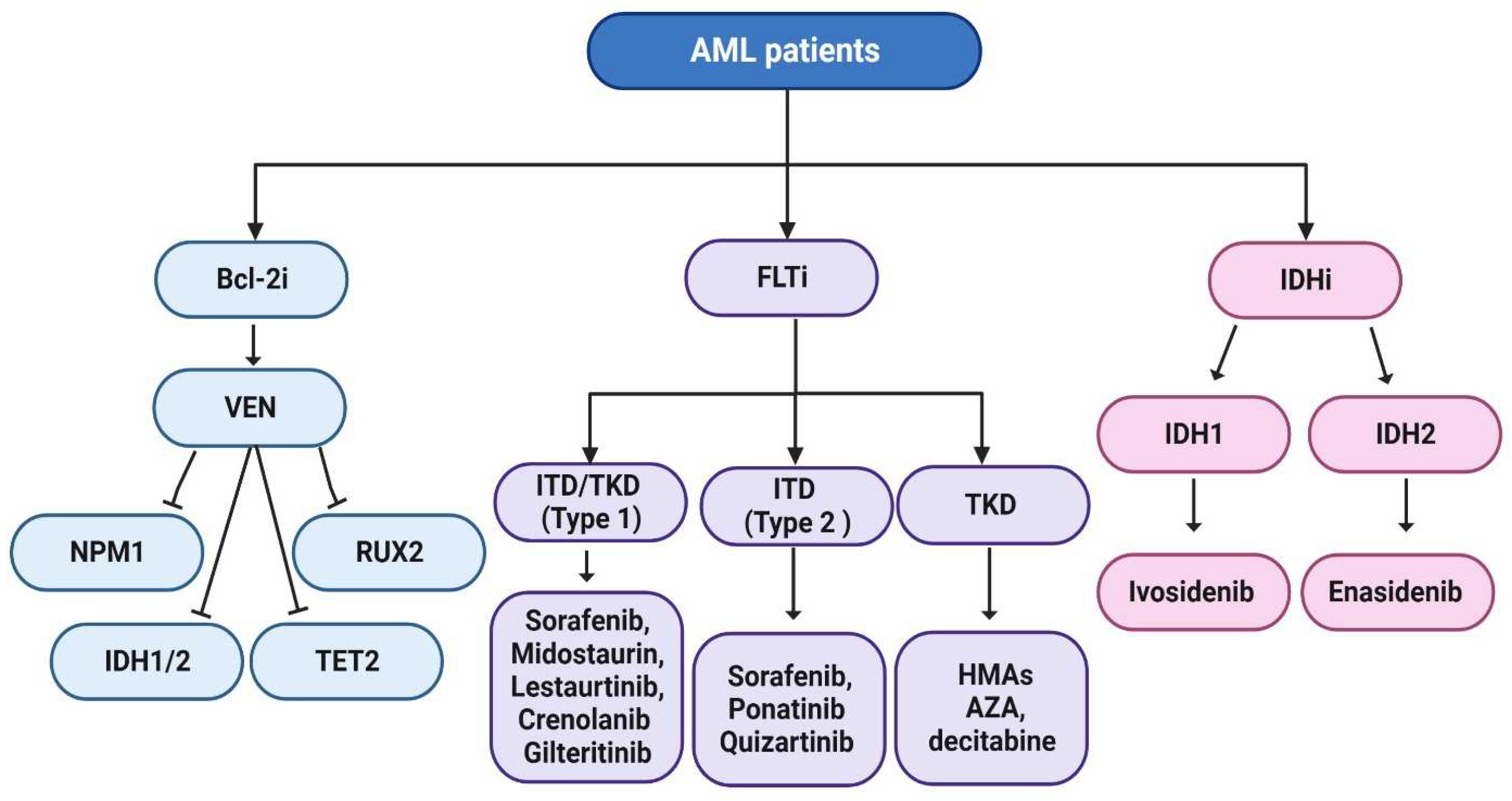
Clinical research and AML treatment
For decades, therapy has been divided into two categories: intensive and non-intensive. In intensive chemotherapy (IC), anthracycline + cytosine arabinoside (araC) backbones are used, followed by consolidation with further chemotherapy or allogeneic stem cell transplantation (SCT)[215,216]. AZA and decitabine are recommended by the NCCN for older adults with newly diagnosed AML. Recent European approval of decitabine for over-65s with newly diagnosed AML (20% BM blasts) who are not candidates for standard IC[217]. Thus, AZA and a BCL-2 inhibitor (venetoclax;VEN) can disrupt the TCA cycle, decrease OXPHOS, and impair energy production[218]. Notably, the efficacy of VEN-based therapy for AML patients was compared to standard chemotherapy, and factors and mechanisms involved in VEN sensitivity and resistance in AML (stem) cells were examined to identify response biomarkers and combination therapies that could increase AML cell sensitivity to VEN. VEN works better for NPM1-, IDH1/2-, TET2-, and RUNX1-m Mutated NPM1 is called NPM1c and localizes in the cytoplasm instead of nucleoli[219,220]. NPM1c AML has a good prognosis without FLT3-ITD mutations[221]. FLT3 is still an appropriate target for the treatment of AML, although the therapeutic efficacy of single medicines has been constrained by transient responses and resistance[222]. Due to their widespread availability and poor prognosis, FLT3 inhibitor therapy for people with FLT3-ITD mutations has gained favor over the past ten years [223]. Based on their capacity to inhibit FLT3 with either an ITD or TKD mutation (type 1 inhibitors) or just an ITD mutation (type 2 inhibitors), FLT3 inhibitors (FLT3i) are classified as either type 1 or type 2 inhibitors. The type 1 inhibitor Sorafenib, Midostaurin, Lestaurtinib, Crenolanib and Gilteritinib and the type 2 inhibitor Sorafenib, Ponatinib and Quizartinib are both components of first-generation FLT3i[223,224]. According to preliminary studies, people who use these drugs may eventually develop a FLT3 TKD mutation as a kind of resistance or an escape mechanism [225]. Treatment options for FLT3-mutated AML include hypomethylating agents (HMAs; AZA, decitabine)/low-intensity treatment or aggressive chemotherapy [226]. Ivosidenib (AG-120)[227] specific inhibitor for IDH1-mutation and enasidenib (AG-221) specific inhibitor for IDH2-mutation [228,229] were recently approved by the FDA for the treatment of AML patients with their respective mutations[230]. However, as most AML patients are over 65, they may be greater at risk for early mortality and induction failure. The majority of younger patients with newly diagnosed AML get intensive therapy with high-dose araC (HiDAC) and anthracyclines. Greater unfavorable characteristics, comorbidities, and early organ damage were present in older AML patients[14]. Ongoing research and recently approved AML agents are intriguing, especially in combination with VEN, HMAs, and epigenetic therapy in elderly patients [226]. In older persons with newly diagnosed AML, this phase III trial compared decitabine to SC and low-dose cytarabine. Due to the risks associated with standard medications, older AML patients have few treatment options[231]. It is likely that TP53-mutated patients have a diminished capacity to undergo apoptosis in response to DNA damage, which diminishes their sensitivity to chemotherapy[232,233]. Previously, HMA monotherapy was used to treat these patients. HMA and VEN together have increased complete (CR) rates, but overall survival (OS) remains poor[234]. Unfortunately, there aren't many choices for treating older patients with TP53-mutated AML with currently approved medications[235]. Resolving the ongoing clinical challenges experienced by elderly AML patients is challenging. Due to the increased prevalence of medical comorbidities and disease symptoms among the elderly [224]. However, they are more common in patients with complicated cytogenetics or AML associated to therapy. Given the frequency of mutations in AML, the knowledge gained via the use of these inhibitors in AML may alter the therapeutic options for older AML patients with these mutations.
Figure 6.
Approach to treating an AML patient.
Conclusions and Perspectives
Due to its high toxicity, patients and doctors frequently choose palliative care over standard chemotherapy reinduction [236]. Multiple genes and signaling pathways are involved in early and late AML due to its complexity [237]. Utilizing cell lines for decades has enabled extensive, reproducible, and cost-effective AML biology research. Abnormal cell lines facilitate research on diseases, AML subtypes, and gene mutations[238]. Given the complexity and diversity of AML genes, system biology is particularly beneficial in network pharmacology in this instance. Because it can analyze biological networks and select signal nodes (Nodes) for the development of drugs with multiple targets. Pharmacology networks regulate multichannel signal pathways. This improves drug efficacy and decreases adverse effects, which increases clinical trial success and decreases drug development costs [239,240]. It improves pharmaceutical understanding and drug discovery [189,190]. A unique approach to understanding herbal medicine efficacy Regulation, targets, and diseases is developing alongside network pharmacology in AML [241]. Network pharmacology research may uncover drug-disease interactions, such as a herbal heal with several active ingredients targeting a wide network. [242]. Thus, herbal medicine generated from plants capable of treat a variety of diseases produces efficacious natural components for commercial use [209]. Traditional research is limited in its scope. Since the majority of tertiary academic cancer patients were from the west, conventional treatment was less beneficial for eastern cancer patients. Expression profiling is one of the best molecular methods for prognosis and prediction, particularly when combined with other data, despite its limitations [243]. It is unknown if patients who obtain remission have deeper remissions and can tolerate additional consolidation therapies, despite the fact that the rate of remission induction for FDA-approved drugs is extraordinarily high [244,245]. Different metabolic reactions are present in AML because of its complexity. The characteristics of the patient and the disease should direct the treatment. Herbal medicines possess significant bioactivity and minimal cytotoxicity because of the variety of compounds they contain. Innovative treatments based on natural products are required for AML and its effects. This review demonstrates that network pharmacology can provide a reliable evaluation of drug candidate function[246]. The utilization of herbal medicines and natural chemicals can then be established by investigating their absorption and bioavailability in animals. A number of compounds with anti-cancer characteristics were found in the extract after chemical analysis, opening up interesting new study directions. As plants are an important source for the development of novel chemotherapeutic drugs, this study paves the way for the identification of numerous natural chemicals that have the ability to prevent and treat cancer[247]. Using network pharmacology to investigate the pharmacological effects of herbal medicines and natural compounds on AML, the researcher anticipates to achieve a major breakthrough toward improved older AML patients’ therapies.
Funding
This research was supported by Graduate School Innovation office, Kyung Hee University, a grant from Kyung Hee University in 2023 (KHU-20230914), Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (NRF-2020R1I1A2066868), the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (No. 2020R1A5A2019413), a grant of the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health & Welfare, Republic of Korea (grant number : HF20C0038), and the innovation network support Program through the INNOPOLIS funded by Ministry of Science and ICT (2022-IT-RD-0205-01-101).
References
- Li, S.; Mason, C.E.; Melnick, A. Genetic and epigenetic heterogeneity in acute myeloid leukemia. Curr Opin Genet Dev 2016, 36, 100–106. [Google Scholar] [CrossRef]
- Marcucci, G.; Haferlach, T.; Döhner, H. Molecular genetics of adult acute myeloid leukemia: Prognostic and therapeutic implications. J Clin Oncol 2011, 29, 475–486. [Google Scholar] [CrossRef]
- Yang, X.; Wang, J. Precision therapy for acute myeloid leukemia. J Hematol Oncol 2018, 11, 3. [Google Scholar] [CrossRef]
- Thol, F.; Ganser, A. Treatment of Relapsed Acute Myeloid Leukemia. Curr Treat Options Oncol 2020, 21, 66. [Google Scholar] [CrossRef] [PubMed]
- Andresen, V.; Gjertsen, B.T. Clinical Trials of Repurposing Medicines in Acute Myeloid Leukemia: Limitations and Possibilities in the Age of Precision Therapy. Cancer J 2019, 25, 153–163. [Google Scholar] [CrossRef] [PubMed]
- Zeijlemaker, W.; Gratama, J.W.; Schuurhuis, G.J. Tumor heterogeneity makes AML a "moving target" for detection of residual disease. Cytometry B Clin Cytom 2014, 86, 3–14. [Google Scholar] [CrossRef] [PubMed]
- McCulloch, E.A. Stem cells in normal and leukemic hemopoiesis (Henry Stratton Lecture, 1982). Blood 1983, 62, 1–13. [Google Scholar] [CrossRef]
- Bonnet, D. Normal and leukaemic stem cells. Br J Haematol 2005, 130, 469–479. [Google Scholar] [CrossRef]
- Arfons, L.M.; Tomblyn, M.; Rocha, V.; Lazarus, H.M. Second hematopoietic stem cell transplantation in myeloid malignancies. Curr Opin Hematol 2009, 16, 112–123. [Google Scholar] [CrossRef] [PubMed]
- Sorror, M.L.; Maris, M.B.; Storb, R.; Baron, F.; Sandmaier, B.M.; Maloney, D.G.; Storer, B. Hematopoietic cell transplantation (HCT)-specific comorbidity index: A new tool for risk assessment before allogeneic HCT. Blood 2005, 106, 2912–2919. [Google Scholar] [CrossRef]
- Burnett, A.K.; Russell, N.H.; Hills, R.K.; Kell, J.; Freeman, S.; Kjeldsen, L.; Hunter, A.E.; Yin, J.; Craddock, C.F.; Dufva, I.H.; et al. Addition of gemtuzumab ozogamicin to induction chemotherapy improves survival in older patients with acute myeloid leukemia. J Clin Oncol 2012, 30, 3924–3931. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, P.; Hills, R.K.; Grech, A.; Betteridge, S.; Kjeldsen, L.; Dennis, M.; Vyas, P.; Goldstone, A.H.; Milligan, D.; Clark, R.E.; et al. An operational definition of primary refractory acute myeloid leukemia allowing early identification of patients who may benefit from allogeneic stem cell transplantation. Haematologica 2016, 101, 1351–1358. [Google Scholar] [CrossRef] [PubMed]
- Dombret, H.; Seymour, J.F.; Butrym, A.; Wierzbowska, A.; Selleslag, D.; Jang, J.H.; Kumar, R.; Cavenagh, J.; Schuh, A.C.; Candoni, A.; et al. International phase 3 study of azacitidine vs conventional care regimens in older patients with newly diagnosed AML with >30% blasts. Blood 2015, 126, 291–299. [Google Scholar] [CrossRef]
- Kantarjian, H.; O'Brien, S.; Cortes, J.; Giles, F.; Faderl, S.; Jabbour, E.; Garcia-Manero, G.; Wierda, W.; Pierce, S.; Shan, J.; et al. Results of intensive chemotherapy in 998 patients age 65 years or older with acute myeloid leukemia or high-risk myelodysplastic syndrome: Predictive prognostic models for outcome. Cancer 2006, 106, 1090–1098. [Google Scholar] [CrossRef]
- Burd, A.; Levine, R.L.; Ruppert, A.S.; Mims, A.S.; Borate, U.; Stein, E.M.; Patel, P.; Baer, M.R.; Stock, W.; Deininger, M.; et al. Precision medicine treatment in acute myeloid leukemia using prospective genomic profiling: Feasibility and preliminary efficacy of the Beat AML Master Trial. Nat Med 2020, 26, 1852–1858. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, S.; Eiring, A.M.; Khorashad, J.S. Genomic Abnormalities as Biomarkers and Therapeutic Targets in Acute Myeloid Leukemia. Cancers 2021, 13, 5055. [Google Scholar] [CrossRef] [PubMed]
- Papaemmanuil, E.; Gerstung, M.; Bullinger, L.; Gaidzik, V.I.; Paschka, P.; Roberts, N.D.; Potter, N.E.; Heuser, M.; Thol, F.; Bolli, N.; et al. Genomic Classification and Prognosis in Acute Myeloid Leukemia. N Engl J Med 2016, 374, 2209–2221. [Google Scholar] [CrossRef] [PubMed]
- Angenendt, L.; Röllig, C.; Montesinos, P.; Martínez-Cuadrón, D.; Barragan, E.; García, R.; Botella, C.; Martínez, P.; Ravandi, F.; Kadia, T.; et al. Chromosomal Abnormalities and Prognosis in NPM1-Mutated Acute Myeloid Leukemia: A Pooled Analysis of Individual Patient Data From Nine International Cohorts. J Clin Oncol 2019, 37, 2632–2642. [Google Scholar] [CrossRef]
- Richard-Carpentier, G.; DiNardo, C.D. Single-agent and combination biologics in acute myeloid leukemia. Hematology Am Soc Hematol Educ Program 2019, 2019, 548–556. [Google Scholar] [CrossRef]
- Falini, B.; Mecucci, C.; Tiacci, E.; Alcalay, M.; Rosati, R.; Pasqualucci, L.; La Starza, R.; Diverio, D.; Colombo, E.; Santucci, A. Cytoplasmic nucleophosmin in acute myelogenous leukemia with a normal karyotype. New England Journal of Medicine 2005, 352, 254–266. [Google Scholar] [CrossRef]
- Du Pisani, L.A.; Shires, K. Development of a flow cytometric method to detect the presence of mutated nucleophosmin 1 in acute myeloid leukemia. Hematol Oncol Stem Cell Ther 2015, 8, 106–114. [Google Scholar] [CrossRef]
- Renneville, A.; Roumier, C.; Biggio, V.; Nibourel, O.; Boissel, N.; Fenaux, P.; Preudhomme, C. Cooperating gene mutations in acute myeloid leukemia: A review of the literature. leukemia 2008, 22, 915–931. [Google Scholar] [CrossRef]
- Pratcorona, M.; Brunet, S.; Nomdedéu, J.; Ribera, J.M.; Tormo, M.; Duarte, R.; Escoda, L.; Guàrdia, R.; Queipo de Llano, M.P.; Salamero, O.; et al. Favorable outcome of patients with acute myeloid leukemia harboring a low-allelic burden FLT3-ITD mutation and concomitant NPM1 mutation: Relevance to post-remission therapy. Blood 2013, 121, 2734–2738. [Google Scholar] [CrossRef] [PubMed]
- Ho, A.D.; Schetelig, J.; Bochtler, T.; Schaich, M.; Schäfer-Eckart, K.; Hänel, M.; Rösler, W.; Einsele, H.; Kaufmann, M.; Serve, H.; et al. Allogeneic Stem Cell Transplantation Improves Survival in Patients with Acute Myeloid Leukemia Characterized by a High Allelic Ratio of Mutant FLT3-ITD. Biol Blood Marrow Transplant 2016, 22, 462–469. [Google Scholar] [CrossRef] [PubMed]
- Döhner, H.; Estey, E.; Grimwade, D.; Amadori, S.; Appelbaum, F.R.; Büchner, T.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Larson, R.A.; et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood 2017, 129, 424–447. [Google Scholar] [CrossRef] [PubMed]
- Bruserud, O.; Hovland, R.; Wergeland, L.; Huang, T.-s.; Gjertsen, B.T. Flt3-mediated signaling in human acute myelogenous leukemia (AML) blasts: A functional characterization of Flt3-ligand effects in AML cell populations with and without genetic Flt3 abnormalities. haematologica 2003, 88, 416–428. [Google Scholar] [PubMed]
- Gilliland, D.G.; Griffin, J.D. The roles of FLT3 in hematopoiesis and leukemia. Blood 2002, 100, 1532–1542. [Google Scholar] [CrossRef]
- Walter, R.B.; Othus, M.; Burnett, A.K.; Löwenberg, B.; Kantarjian, H.M.; Ossenkoppele, G.J.; Hills, R.K.; Van Montfort, K.G.; Ravandi, F.; Evans, A. Significance of FAB subclassification of “acute myeloid leukemia, NOS” in the 2008 WHO classification: Analysis of 5848 newly diagnosed patients. Blood, The Journal of the American Society of Hematology 2013, 121, 2424–2431. [Google Scholar] [CrossRef]
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef]
- Osato, M. Point mutations in the RUNX1/AML1 gene: Another actor in RUNX leukemia. Oncogene 2004, 23, 4284–4296. [Google Scholar] [CrossRef]
- Yamagata, T.; Maki, K.; Mitani, K. Runx1/AML1 in normal and abnormal hematopoiesis. International journal of hematology 2005, 82, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Kunadt, D.; Herold, S.; Poitz, D.; Wagenführ, L.; Kretschmann, T.; Sockel, K.; Ruhnke, L.; Brückner, S.; Sommer, U.; Meier, F.; et al. Spatial heterogeneity and differential treatment response of acute myeloid leukemia and relapsed/refractory extramedullary disease after allogeneic hematopoietic cell transplantation. Ther Adv Hematol 2022, 13, 20406207221115005. [Google Scholar] [CrossRef] [PubMed]
- Saultz, J.N.; Garzon, R. Acute myeloid leukemia: A concise review. Journal of clinical medicine 2016, 5, 33. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, A.F.T.; Pratcorona, M.; Erpelinck-Verschueren, C.; Rockova, V.; Sanders, M.; Abbas, S.; Figueroa, M.E.; Zeilemaker, A.; Melnick, A.; Löwenberg, B. Mutant DNMT3A: A marker of poor prognosis in acute myeloid leukemia. Blood, The Journal of the American Society of Hematology 2012, 119, 5824–5831. [Google Scholar] [CrossRef] [PubMed]
- Park, D.J.; Kwon, A.; Cho, B.S.; Kim, H.J.; Hwang, K.A.; Kim, M.; Kim, Y. Characteristics of DNMT3A mutations in acute myeloid leukemia. Blood Res 2020, 55, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Reitman, Z.J.; Yan, H. Isocitrate dehydrogenase 1 and 2 mutations in cancer: Alterations at a crossroads of cellular metabolism. J Natl Cancer Inst 2010, 102, 932–941. [Google Scholar] [CrossRef]
- Parsons, D.W.; Jones, S.; Zhang, X.; Lin, J.C.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Siu, I.M.; Gallia, G.L.; et al. An integrated genomic analysis of human glioblastoma multiforme. Science 2008, 321, 1807–1812. [Google Scholar] [CrossRef]
- Yan, H.; Parsons, D.W.; Jin, G.; McLendon, R.; Rasheed, B.A.; Yuan, W.; Kos, I.; Batinic-Haberle, I.; Jones, S.; Riggins, G.J.; et al. IDH1 and IDH2 mutations in gliomas. N Engl J Med 2009, 360, 765–773. [Google Scholar] [CrossRef]
- Liu, X.; Gong, Y. Isocitrate dehydrogenase inhibitors in acute myeloid leukemia. Biomarker Research 2019, 7, 22. [Google Scholar] [CrossRef]
- Marcucci, G.; Maharry, K.; Wu, Y.Z.; Radmacher, M.D.; Mrózek, K.; Margeson, D.; Holland, K.B.; Whitman, S.P.; Becker, H.; Schwind, S.; et al. IDH1 and IDH2 gene mutations identify novel molecular subsets within de novo cytogenetically normal acute myeloid leukemia: A Cancer and Leukemia Group B study. J Clin Oncol 2010, 28, 2348–2355. [Google Scholar] [CrossRef]
- Willander, K.; Falk, I.J.; Chaireti, R.; Paul, E.; Hermansson, M.; Gréen, H.; Lotfi, K.; Söderkvist, P. Mutations in the isocitrate dehydrogenase 2 gene and IDH1 SNP 105C > T have a prognostic value in acute myeloid leukemia. Biomark Res 2014, 2, 18. [Google Scholar] [CrossRef] [PubMed]
- Wagner, K.; Damm, F.; Göhring, G.; Görlich, K.; Heuser, M.; Schäfer, I.; Ottmann, O.; Lübbert, M.; Heit, W.; Kanz, L.; et al. Impact of IDH1 R132 mutations and an IDH1 single nucleotide polymorphism in cytogenetically normal acute myeloid leukemia: SNP rs11554137 is an adverse prognostic factor. J Clin Oncol 2010, 28, 2356–2364. [Google Scholar] [CrossRef] [PubMed]
- Montalban-Bravo, G.; DiNardo, C.D. The role of IDH mutations in acute myeloid leukemia. Future Oncology 2018, 14, 979–993. [Google Scholar] [CrossRef]
- Chou, W.; Lei, W.; Ko, B.; Hou, H.; Chen, C.; Tang, J.; Yao, M.; Tsay, W.; Wu, S.; Huang, S. The prognostic impact and stability of Isocitrate dehydrogenase 2 mutation in adult patients with acute myeloid leukemia. Leukemia 2011, 25, 246–253. [Google Scholar] [CrossRef] [PubMed]
- Das, A.B.; Smith-Díaz, C.C.; Vissers, M.C.M. Emerging epigenetic therapeutics for myeloid leukemia: Modulating demethylase activity with ascorbate. Haematologica 2021, 106, 14–25. [Google Scholar] [CrossRef]
- Antonson, P.; Xanthopoulos, K.G. Molecular cloning, sequence, and expression patterns of the human gene encoding CCAAT/enhancer binding protein α (C/EBPα). Biochemical and biophysical research communications 1995, 215, 106–113. [Google Scholar] [CrossRef]
- Ravandi, F.; Burnett, A.K.; Agura, E.D.; Kantarjian, H.M. Progress in the treatment of acute myeloid leukemia. Cancer 2007, 110, 1900–1910. [Google Scholar] [CrossRef]
- Ishikawa, F.; Yoshida, S.; Saito, Y.; Hijikata, A.; Kitamura, H.; Tanaka, S.; Nakamura, R.; Tanaka, T.; Tomiyama, H.; Saito, N.; et al. Chemotherapy-resistant human AML stem cells home to and engraft within the bone-marrow endosteal region. Nat Biotechnol 2007, 25, 1315–1321. [Google Scholar] [CrossRef]
- Döhner, H.; Estey, E.H.; Amadori, S.; Appelbaum, F.R.; Büchner, T.; Burnett, A.K.; Dombret, H.; Fenaux, P.; Grimwade, D.; Larson, R.A.; et al. Diagnosis and management of acute myeloid leukemia in adults: Recommendations from an international expert panel, on behalf of the European LeukemiaNet. Blood 2010, 115, 453–474. [Google Scholar] [CrossRef]
- O'Reilly, E.; Zeinabad, H.A.; Nolan, C.; Sefy, J.; Williams, T.; Tarunina, M.; Hernandez, D.; Choo, Y.; Szegezdi, E. Recreating the Bone Marrow Microenvironment to Model Leukemic Stem Cell Quiescence. Front Cell Dev Biol 2021, 9, 662868. [Google Scholar] [CrossRef]
- Su, Y.C.; Li, S.C.; Wu, Y.C.; Wang, L.M.; Chao, K.S.; Liao, H.F. Resveratrol downregulates interleukin-6-stimulated sonic hedgehog signaling in human acute myeloid leukemia. Evid Based Complement Alternat Med 2013, 2013, 547430. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.T.; Hernandez, D.; Alonso, S.; Gao, M.; Su, M.; Ghiaur, G.; Levis, M.J.; Jones, R.J. Role of CYP3A4 in bone marrow microenvironment-mediated protection of FLT3/ITD AML from tyrosine kinase inhibitors. Blood Adv 2019, 3, 908–916. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Zhang, G.; Kong, L.; Xu, S.; Wang, Y.; Dong, M. Leukemia-derived exosomes induced IL-8 production in bone marrow stromal cells to protect the leukemia cells against chemotherapy. Life Sci 2019, 221, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Thomas, C.M.; Campbell, P. FLT3 inhibitors in acute myeloid leukemia: Current and future. J Oncol Pharm Pract 2019, 25, 163–171. [Google Scholar] [CrossRef]
- Terwijn, M.; Zeijlemaker, W.; Kelder, A.; Rutten, A.P.; Snel, A.N.; Scholten, W.J.; Pabst, T.; Verhoef, G.; Löwenberg, B.; Zweegman, S.; et al. Leukemic stem cell frequency: A strong biomarker for clinical outcome in acute myeloid leukemia. PLoS ONE 2014, 9, e107587. [Google Scholar] [CrossRef]
- Pollyea, D.A.; Jordan, C.T. Therapeutic targeting of acute myeloid leukemia stem cells. Blood 2017, 129, 1627–1635. [Google Scholar] [CrossRef]
- Zeijlemaker, W.; Grob, T.; Meijer, R.; Hanekamp, D.; Kelder, A.; Carbaat-Ham, J.C.; Oussoren-Brockhoff, Y.J.M.; Snel, A.N.; Veldhuizen, D.; Scholten, W.J.; et al. CD34(+)CD38(-) leukemic stem cell frequency to predict outcome in acute myeloid leukemia. Leukemia 2019, 33, 1102–1112. [Google Scholar] [CrossRef]
- Shiozawa, Y.; Pedersen, E.A.; Havens, A.M.; Jung, Y.; Mishra, A.; Joseph, J.; Kim, J.K.; Patel, L.R.; Ying, C.; Ziegler, A.M.; et al. Human prostate cancer metastases target the hematopoietic stem cell niche to establish footholds in mouse bone marrow. J Clin Invest 2011, 121, 1298–1312. [Google Scholar] [CrossRef]
- Jung, Y.; Wang, J.; Song, J.; Shiozawa, Y.; Wang, J.; Havens, A.; Wang, Z.; Sun, Y.X.; Emerson, S.G.; Krebsbach, P.H.; et al. Annexin II expressed by osteoblasts and endothelial cells regulates stem cell adhesion, homing, and engraftment following transplantation. Blood 2007, 110, 82–90. [Google Scholar] [CrossRef]
- Nwajei, F.; Konopleva, M. The bone marrow microenvironment as niche retreats for hematopoietic and leukemic stem cells. Adv Hematol 2013, 2013, 953982. [Google Scholar] [CrossRef]
- Tabe, Y.; Konopleva, M. Role of Microenvironment in Resistance to Therapy in AML. Curr Hematol Malig Rep 2015, 10, 96–103. [Google Scholar] [CrossRef] [PubMed]
- Schepers, K.; Campbell, T.B.; Passegué, E. Normal and leukemic stem cell niches: Insights and therapeutic opportunities. Cell Stem Cell 2015, 16, 254–267. [Google Scholar] [CrossRef] [PubMed]
- Linde, N.; Fluegen, G.; Aguirre-Ghiso, J.A. The Relationship Between Dormant Cancer Cells and Their Microenvironment. Adv Cancer Res 2016, 132, 45–71. [Google Scholar] [CrossRef] [PubMed]
- Mesbahi, Y.; Trahair, T.N.; Lock, R.B.; Connerty, P. Exploring the metabolic landscape of AML: From haematopoietic stem cells to myeloblasts and leukaemic stem cells. Frontiers in Oncology 2022, 12, 281. [Google Scholar] [CrossRef]
- Qi, J.; Chen, N.; Wang, J.; Siu, C.H. Transendothelial migration of melanoma cells involves N-cadherin-mediated adhesion and activation of the beta-catenin signaling pathway. Mol Biol Cell 2005, 16, 4386–4397. [Google Scholar] [CrossRef]
- Reid, S.E.; Kay, E.J.; Neilson, L.J.; Henze, A.T.; Serneels, J.; McGhee, E.J.; Dhayade, S.; Nixon, C.; Mackey, J.B.; Santi, A.; et al. Tumor matrix stiffness promotes metastatic cancer cell interaction with the endothelium. Embo j 2017, 36, 2373–2389. [Google Scholar] [CrossRef]
- Sistigu, A.; Musella, M.; Galassi, C.; Vitale, I.; De Maria, R. Tuning Cancer Fate: Tumor Microenvironment's Role in Cancer Stem Cell Quiescence and Reawakening. Front Immunol 2020, 11, 2166. [Google Scholar] [CrossRef]
- Stasi, R.; Evangelista, M.L.; Buccisano, F.; Venditti, A.; Amadori, S. Gemtuzumab ozogamicin in the treatment of acute myeloid leukemia. Cancer treatment reviews 2008, 34, 49–60. [Google Scholar] [CrossRef]
- Jin, L.; Lee, E.M.; Ramshaw, H.S.; Busfield, S.J.; Peoppl, A.G.; Wilkinson, L.; Guthridge, M.A.; Thomas, D.; Barry, E.F.; Boyd, A. Monoclonal antibody-mediated targeting of CD123, IL-3 receptor α chain, eliminates human acute myeloid leukemic stem cells. Cell stem cell 2009, 5, 31–42. [Google Scholar] [CrossRef]
- Van Rhenen, A.; Van Dongen, G.A.; Kelder, A.; Rombouts, E.J.; Feller, N.; Moshaver, B.; Walsum, M.S.-v.; Zweegman, S.; Ossenkoppele, G.J.; Jan Schuurhuis, G. The novel AML stem cell–associated antigen CLL-1 aids in discrimination between normal and leukemic stem cells. Blood, The Journal of the American Society of Hematology 2007, 110, 2659–2666. [Google Scholar] [CrossRef]
- Jin, L.; Hope, K.J.; Zhai, Q.; Smadja-Joffe, F.; Dick, J.E. Targeting of CD44 eradicates human acute myeloid leukemic stem cells. Nature medicine 2006, 12, 1167–1174. [Google Scholar] [CrossRef]
- Jaiswal, S.; Jamieson, C.H.; Pang, W.W.; Park, C.Y.; Chao, M.P.; Majeti, R.; Traver, D.; van Rooijen, N.; Weissman, I.L. CD47 is upregulated on circulating hematopoietic stem cells and leukemia cells to avoid phagocytosis. Cell 2009, 138, 271–285. [Google Scholar] [CrossRef] [PubMed]
- Hosen, N.; Park, C.Y.; Tatsumi, N.; Oji, Y.; Sugiyama, H.; Gramatzki, M.; Krensky, A.M.; Weissman, I.L. CD96 is a leukemic stem cell-specific marker in human acute myeloid leukemia. Proceedings of the National Academy of Sciences 2007, 104, 11008–11013. [Google Scholar] [CrossRef]
- Mohamed, M.M.I.; Aref, S.; Agdar, M.A.; Mabed, M.; El-Sokkary, A.M.A. Leukemic Stem Cell (CD34+/CD38–/TIM3+) Frequency in Patients with Acute Myeloid Leukemia: Clinical Implications. Clinical Lymphoma Myeloma and Leukemia 2021, 21, 508–513. [Google Scholar] [CrossRef] [PubMed]
- Corces, M.R.; Buenrostro, J.D.; Wu, B.; Greenside, P.G.; Chan, S.M.; Koenig, J.L.; Snyder, M.P.; Pritchard, J.K.; Kundaje, A.; Greenleaf, W.J.; et al. Lineage-specific and single-cell chromatin accessibility charts human hematopoiesis and leukemia evolution. Nature Genetics 2016, 48, 1193–1203. [Google Scholar] [CrossRef] [PubMed]
- Freeman, G.J.; Casasnovas, J.M.; Umetsu, D.T.; DeKruyff, R.H. TIM genes: A family of cell surface phosphatidylserine receptors that regulate innate and adaptive immunity. Immunological reviews 2010, 235, 172–189. [Google Scholar] [CrossRef]
- Kikushige, Y.; Shima, T.; Takayanagi, S.-i.; Urata, S.; Miyamoto, T.; Iwasaki, H.; Takenaka, K.; Teshima, T.; Tanaka, T.; Inagaki, Y. TIM-3 is a promising target to selectively kill acute myeloid leukemia stem cells. Cell stem cell 2010, 7, 708–717. [Google Scholar] [CrossRef]
- Jan, M.; Chao, M.P.; Cha, A.C.; Alizadeh, A.A.; Gentles, A.J.; Weissman, I.L.; Majeti, R. Prospective separation of normal and leukemic stem cells based on differential expression of TIM3, a human acute myeloid leukemia stem cell marker. Proceedings of the National Academy of Sciences 2011, 108, 5009–5014. [Google Scholar] [CrossRef]
- Kikushige, Y.; Miyamoto, T. TIM-3 as a novel therapeutic target for eradicating acute myelogenous leukemia stem cells. International journal of hematology 2013, 98, 627–633. [Google Scholar] [CrossRef]
- Zhu, C.; Anderson, A.C.; Schubart, A.; Xiong, H.; Imitola, J.; Khoury, S.J.; Zheng, X.X.; Strom, T.B.; Kuchroo, V.K. The Tim-3 ligand galectin-9 negatively regulates T helper type 1 immunity. Nature immunology 2005, 6, 1245–1252. [Google Scholar] [CrossRef]
- Monney, L.; Sabatos, C.A.; Gaglia, J.L.; Ryu, A.; Waldner, H.; Chernova, T.; Manning, S.; Greenfield, E.A.; Coyle, A.J.; Sobel, R.A. Th1-specific cell surface protein Tim-3 regulates macrophage activation and severity of an autoimmune disease. Nature 2002, 415, 536–541. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Yu, X.; Gu, C.; Ma, C.; Liu, H.; Tong, W.; Zou, Q.; Ye, L.; Chen, X.; Depei, W. The expression and mechanism of Tim3 and PD-1 in patients with acute myeloid leukemia. American Society of Hematology Washington, DC: 2015.
- de Almeida, M.J.; Luchsinger, L.L.; Corrigan, D.J.; Williams, L.J.; Snoeck, H.W. Dye-Independent Methods Reveal Elevated Mitochondrial Mass in Hematopoietic Stem Cells. Cell Stem Cell 2017, 21, 725–729. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Pollyea, D.A. Shutting Down Acute Myeloid Leukemia and Myelodysplastic Syndrome with BCL-2 Family Protein Inhibition. Curr Hematol Malig Rep 2018, 13, 256–264. [Google Scholar] [CrossRef]
- Vitale, I.; Sistigu, A.; Manic, G.; Rudqvist, N.P.; Trajanoski, Z.; Galluzzi, L. Mutational and Antigenic Landscape in Tumor Progression and Cancer Immunotherapy. Trends Cell Biol 2019, 29, 396–416. [Google Scholar] [CrossRef]
- Tormin, A.; Li, O.; Brune, J.; Walsh, S.; Ehinger, M.; Ditzel, N.; Kassem, M.; Scheding, S. Human Primary CD271+/CD45−/CD146−/Low and CD271+/CD45−/CD146+ Bone Marrow Cells Are Developmentally Closely-Related Stroma Stem Cells with Similar Functional Properties but Different In-Situ Localization. Blood 2010, 116, 2594. [Google Scholar] [CrossRef]
- Matsuoka, Y.; Nakatsuka, R.; Sumide, K.; Kawamura, H.; Takahashi, M.; Fujioka, T.; Uemura, Y.; Asano, H.; Sasaki, Y.; Inoue, M.; et al. Prospectively Isolated Human Bone Marrow Cell-Derived MSCs Support Primitive Human CD34-Negative Hematopoietic Stem Cells. Stem Cells 2015, 33, 1554–1565. [Google Scholar] [CrossRef]
- Harkness, L.; Zaher, W.; Ditzel, N.; Isa, A.; Kassem, M. CD146/MCAM defines functionality of human bone marrow stromal stem cell populations. Stem Cell Res Ther 2016, 7, 4. [Google Scholar] [CrossRef]
- Sumbly, V.; Landry, I.; Sneed, C.; Iqbal, Q.; Verma, A.; Dhokhar, T.; Masood, A.; Amaraneni, A. Leukemic stem cells and advances in hematopoietic stem cell transplantation for acute myeloid leukemia: A narrative review of clinical trials. Stem Cell Investig 2022, 9, 10. [Google Scholar] [CrossRef]
- Shiozawa, Y.; Berry, J.E.; Eber, M.R.; Jung, Y.; Yumoto, K.; Cackowski, F.C.; Yoon, H.J.; Parsana, P.; Mehra, R.; Wang, J.; et al. The marrow niche controls the cancer stem cell phenotype of disseminated prostate cancer. Oncotarget 2016, 7, 41217–41232. [Google Scholar] [CrossRef]
- Malanchi, I.; Santamaria-Martínez, A.; Susanto, E.; Peng, H.; Lehr, H.-A.; Delaloye, J.-F.; Huelsken, J. Interactions between cancer stem cells and their niche govern metastatic colonization. Nature 2012, 481, 85–89. [Google Scholar] [CrossRef] [PubMed]
- Oskarsson, T.; Batlle, E.; Massagué, J. Metastatic stem cells: Sources, niches, and vital pathways. Cell stem cell 2014, 14, 306–321. [Google Scholar] [CrossRef] [PubMed]
- Sendker, S.; Reinhardt, D.; Niktoreh, N. Redirecting the immune microenvironment in acute myeloid leukemia. Cancers 2021, 13, 1423. [Google Scholar] [CrossRef] [PubMed]
- Ghajar, C.M.; Peinado, H.; Mori, H.; Matei, I.R.; Evason, K.J.; Brazier, H.; Almeida, D.; Koller, A.; Hajjar, K.A.; Stainier, D.Y.; et al. The perivascular niche regulates breast tumour dormancy. Nat Cell Biol 2013, 15, 807–817. [Google Scholar] [CrossRef] [PubMed]
- Kienast, Y.; von Baumgarten, L.; Fuhrmann, M.; Klinkert, W.E.; Goldbrunner, R.; Herms, J.; Winkler, F. Real-time imaging reveals the single steps of brain metastasis formation. Nat Med 2010, 16, 116–122. [Google Scholar] [CrossRef] [PubMed]
- Cao, Z.; Ding, B.S.; Guo, P.; Lee, S.B.; Butler, J.M.; Casey, S.C.; Simons, M.; Tam, W.; Felsher, D.W.; Shido, K.; et al. Angiocrine factors deployed by tumor vascular niche induce B cell lymphoma invasiveness and chemoresistance. Cancer Cell 2014, 25, 350–365. [Google Scholar] [CrossRef]
- Price, T.T.; Burness, M.L.; Sivan, A.; Warner, M.J.; Cheng, R.; Lee, C.H.; Olivere, L.; Comatas, K.; Magnani, J.; Kim Lyerly, H.; et al. Dormant breast cancer micrometastases reside in specific bone marrow niches that regulate their transit to and from bone. Sci Transl Med 2016, 8, 340ra373. [Google Scholar] [CrossRef]
- Butler, J.M.; Kobayashi, H.; Rafii, S. Instructive role of the vascular niche in promoting tumour growth and tissue repair by angiocrine factors. Nat Rev Cancer 2010, 10, 138–146. [Google Scholar] [CrossRef]
- Fessler, E.; Dijkgraaf, F.E.; De Sousa, E.M.F.; Medema, J.P. Cancer stem cell dynamics in tumor progression and metastasis: Is the microenvironment to blame? Cancer Lett 2013, 341, 97–104. [Google Scholar] [CrossRef]
- Schrader, J.; Gordon-Walker, T.T.; Aucott, R.L.; van Deemter, M.; Quaas, A.; Walsh, S.; Benten, D.; Forbes, S.J.; Wells, R.G.; Iredale, J.P. Matrix stiffness modulates proliferation, chemotherapeutic response, and dormancy in hepatocellular carcinoma cells. Hepatology 2011, 53, 1192–1205. [Google Scholar] [CrossRef]
- Talukdar, S.; Bhoopathi, P.; Emdad, L.; Das, S.; Sarkar, D.; Fisher, P.B. Dormancy and cancer stem cells: An enigma for cancer therapeutic targeting. Adv Cancer Res 2019, 141, 43–84. [Google Scholar] [CrossRef]
- Crea, F.; Nur Saidy, N.R.; Collins, C.C.; Wang, Y. The epigenetic/noncoding origin of tumor dormancy. Trends Mol Med 2015, 21, 206–211. [Google Scholar] [CrossRef] [PubMed]
- Mansoori, B.; Mohammadi, A.; Davudian, S.; Shirjang, S.; Baradaran, B. The Different Mechanisms of Cancer Drug Resistance: A Brief Review. Adv Pharm Bull 2017, 7, 339–348. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, D.; Dick, J.E. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med 1997, 3, 730–737. [Google Scholar] [CrossRef] [PubMed]
- Lapidot, T.; Sirard, C.; Vormoor, J.; Murdoch, B.; Hoang, T.; Caceres-Cortes, J.; Minden, M.; Paterson, B.; Caligiuri, M.A.; Dick, J.E. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature 1994, 367, 645–648. [Google Scholar] [CrossRef]
- Bowie, M.B.; Kent, D.G.; Dykstra, B.; McKnight, K.D.; McCaffrey, L.; Hoodless, P.A.; Eaves, C.J. Identification of a new intrinsically timed developmental checkpoint that reprograms key hematopoietic stem cell properties. Proc Natl Acad Sci U S A 2007, 104, 5878–5882. [Google Scholar] [CrossRef]
- Chen, Y.; Hu, Y.; Zhang, H.; Peng, C.; Li, S. Loss of the Alox5 gene impairs leukemia stem cells and prevents chronic myeloid leukemia. Nat Genet 2009, 41, 783–792. [Google Scholar] [CrossRef]
- Pardal, R.; Clarke, M.F.; Morrison, S.J. Applying the principles of stem-cell biology to cancer. Nat Rev Cancer 2003, 3, 895–902. [Google Scholar] [CrossRef]
- Reya, T.; Morrison, S.J.; Clarke, M.F.; Weissman, I.L. Stem cells, cancer, and cancer stem cells. Nature 2001, 414, 105–111. [Google Scholar] [CrossRef]
- Molofsky, A.V.; Pardal, R.; Morrison, S.J. Diverse mechanisms regulate stem cell self-renewal. Curr Opin Cell Biol 2004, 16, 700–707. [Google Scholar] [CrossRef]
- Zhao, C.; Blum, J.; Chen, A.; Kwon, H.Y.; Jung, S.H.; Cook, J.M.; Lagoo, A.; Reya, T. Loss of beta-catenin impairs the renewal of normal and CML stem cells in vivo. Cancer Cell 2007, 12, 528–541. [Google Scholar] [CrossRef]
- Pardal, R.; Clarke, M.F.; Morrison, S.J. Applying the principles of stem-cell biology to cancer. Nature Reviews Cancer 2003, 3, 895–902. [Google Scholar] [CrossRef] [PubMed]
- Molofsky, A.V.; Pardal, R.; Morrison, S.J. Diverse mechanisms regulate stem cell self-renewal. Current Opinion in Cell Biology 2004, 16, 700–707. [Google Scholar] [CrossRef] [PubMed]
- Molofsky, A.V.; He, S.; Bydon, M.; Morrison, S.J.; Pardal, R. Bmi-1 promotes neural stem cell self-renewal and neural development but not mouse growth and survival by repressing the p16Ink4a and p19Arf senescence pathways. Genes Dev 2005, 19, 1432–1437. [Google Scholar] [CrossRef] [PubMed]
- Lowe, S.W.; Sherr, C.J. Tumor suppression by Ink4a–Arf: Progress and puzzles. Current Opinion in Genetics & Development 2003, 13, 77–83. [Google Scholar] [CrossRef]
- Di Cristofano, A.; Pandolfi, P.P. The multiple roles of PTEN in tumor suppression. Cell 2000, 100, 387–390. [Google Scholar] [CrossRef]
- Aggerholm, A.; Grønbaek, K.; Guldberg, P.; Hokland, P. Mutational analysis of the tumour suppressor gene MMAC1/PTEN in malignant myeloid disorders. Eur J Haematol 2000, 65, 109–113. [Google Scholar] [CrossRef]
- Dahia, P.L.; Aguiar, R.C.; Alberta, J.; Kum, J.B.; Caron, S.; Sill, H.; Marsh, D.J.; Ritz, J.; Freedman, A.; Stiles, C.; et al. PTEN is inversely correlated with the cell survival factor Akt/PKB and is inactivated via multiple mechanismsin haematological malignancies. Hum Mol Genet 1999, 8, 185–193. [Google Scholar] [CrossRef]
- Cheong, J.W.; Eom, J.I.; Maeng, H.Y.; Lee, S.T.; Hahn, J.S.; Ko, Y.W.; Min, Y.H. Phosphatase and tensin homologue phosphorylation in the C-terminal regulatory domain is frequently observed in acute myeloid leukaemia and associated with poor clinical outcome. Br J Haematol 2003, 122, 454–456. [Google Scholar] [CrossRef]
- Eisfeld, A.K.; Kohlschmidt, J.; Mims, A.; Nicolet, D.; Walker, C.J.; Blachly, J.S.; Carroll, A.J.; Papaioannou, D.; Kolitz, J.E.; Powell, B.E.; et al. Additional gene mutations may refine the 2017 European LeukemiaNet classification in adult patients with de novo acute myeloid leukemia aged <60 years. Leukemia 2020, 34, 3215–3227. [Google Scholar] [CrossRef]
- Bullinger, L.; Döhner, K.; Döhner, H. Genomics of Acute Myeloid Leukemia Diagnosis and Pathways. J Clin Oncol 2017, 35, 934–946. [Google Scholar] [CrossRef]
- Ley, T.J.; Miller, C.; Ding, L.; Raphael, B.J.; Mungall, A.J.; Robertson, A.; Hoadley, K.; Triche, T.J., Jr.; Laird, P.W.; Baty, J.D.; et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med 2013, 368, 2059–2074. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Wong, M.P.M.; Ng, R.K. Aberrant DNA Methylation in Acute Myeloid Leukemia and Its Clinical Implications. Int J Mol Sci 2019, 20. [Google Scholar] [CrossRef] [PubMed]
- Celik, H.; Kramer, A.; Challen, G.A. DNA methylation in normal and malignant hematopoiesis. Int J Hematol 2016, 103, 617–626. [Google Scholar] [CrossRef]
- Dexheimer, G.M.; Alves, J.; Reckziegel, L.; Lazzaretti, G.; Abujamra, A.L. DNA Methylation Events as Markers for Diagnosis and Management of Acute Myeloid Leukemia and Myelodysplastic Syndrome. Dis Markers 2017, 2017, 5472893. [Google Scholar] [CrossRef]
- Varley, K.E.; Gertz, J.; Bowling, K.M.; Parker, S.L.; Reddy, T.E.; Pauli-Behn, F.; Cross, M.K.; Williams, B.A.; Stamatoyannopoulos, J.A.; Crawford, G.E.; et al. Dynamic DNA methylation across diverse human cell lines and tissues. Genome Res 2013, 23, 555–567. [Google Scholar] [CrossRef]
- Lehmann, U.; Hasemeier, B.; Christgen, M.; Müller, M.; Römermann, D.; Länger, F.; Kreipe, H. Epigenetic inactivation of microRNA gene hsa-mir-9-1 in human breast cancer. J Pathol 2008, 214, 17–24. [Google Scholar] [CrossRef]
- Fard, A.A.; Rahimi, H.; Shams, Z.; Ghoraeian, P. Screening and in Silico Functional Analysis of MiRNAs Associated with Acute Myeloid Leukemia Relapse. Microrna 2022, 11, 227–244. [Google Scholar] [CrossRef] [PubMed]
- Baer, C.; Claus, R.; Plass, C. Genome-wide epigenetic regulation of miRNAs in cancer. Cancer Res 2013, 73, 473–477. [Google Scholar] [CrossRef]
- Sasheva, P.; Grossniklaus, U. Differentially Methylated Region-Representational Difference Analysis (DMR-RDA): A Powerful Method to Identify DMRs in Uncharacterized Genomes. Methods Mol Biol 2017, 1456, 113–125. [Google Scholar] [CrossRef]
- Sheppard, T.L. lncRNA express delivery. Nature Chemical Biology 2014, 10, 794. [Google Scholar] [CrossRef]
- Zhou, J.; Yang, L.; Zhong, T.; Mueller, M.; Men, Y.; Zhang, N.; Xie, J.; Giang, K.; Chung, H.; Sun, X.; et al. H19 lncRNA alters DNA methylation genome wide by regulating S-adenosylhomocysteine hydrolase. Nat Commun 2015, 6, 10221. [Google Scholar] [CrossRef] [PubMed]
- Taube, J.H.; Malouf, G.G.; Lu, E.; Sphyris, N.; Vijay, V.; Ramachandran, P.P.; Ueno, K.R.; Gaur, S.; Nicoloso, M.S.; Rossi, S.; et al. Epigenetic silencing of microRNA-203 is required for EMT and cancer stem cell properties. Sci Rep 2013, 3, 2687. [Google Scholar] [CrossRef]
- Gwak, J.M.; Kim, H.J.; Kim, E.J.; Chung, Y.R.; Yun, S.; Seo, A.N.; Lee, H.J.; Park, S.Y. MicroRNA-9 is associated with epithelial-mesenchymal transition, breast cancer stem cell phenotype, and tumor progression in breast cancer. Breast Cancer Res Treat 2014, 147, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Sauer, M.A.; Hussein, S.G.; Yang, J.; Tenen, D.G.; Chai, L. SALL4 and microRNA: The Role of Let-7. Genes (Basel) 2021, 12. [Google Scholar] [CrossRef] [PubMed]
- Neves, R.; Scheel, C.; Weinhold, S.; Honisch, E.; Iwaniuk, K.M.; Trompeter, H.I.; Niederacher, D.; Wernet, P.; Santourlidis, S.; Uhrberg, M. Role of DNA methylation in miR-200c/141 cluster silencing in invasive breast cancer cells. BMC Res Notes 2010, 3, 219. [Google Scholar] [CrossRef]
- Vrba, L.; Muñoz-Rodríguez, J.L.; Stampfer, M.R.; Futscher, B.W. miRNA gene promoters are frequent targets of aberrant DNA methylation in human breast cancer. PLoS ONE 2013, 8, e54398. [Google Scholar] [CrossRef]
- Payton, J.E.; Grieselhuber, N.R.; Chang, L.W.; Murakami, M.; Geiss, G.K.; Link, D.C.; Nagarajan, R.; Watson, M.A.; Ley, T.J. High throughput digital quantification of mRNA abundance in primary human acute myeloid leukemia samples. J Clin Invest 2009, 119, 1714–1726. [Google Scholar] [CrossRef] [PubMed]
- Di Croce, L.; Raker, V.A.; Corsaro, M.; Fazi, F.; Fanelli, M.; Faretta, M.; Fuks, F.; Lo Coco, F.; Kouzarides, T.; Nervi, C.; et al. Methyltransferase recruitment and DNA hypermethylation of target promoters by an oncogenic transcription factor. Science 2002, 295, 1079–1082. [Google Scholar] [CrossRef]
- Feng, S.; Jacobsen, S.E.; Reik, W. Epigenetic reprogramming in plant and animal development. Science 2010, 330, 622–627. [Google Scholar] [CrossRef]
- Wu, S.C.; Zhang, Y. Active DNA demethylation: Many roads lead to Rome. Nat Rev Mol Cell Biol 2010, 11, 607–620. [Google Scholar] [CrossRef]
- Law, J.A.; Jacobsen, S.E. Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat Rev Genet 2010, 11, 204–220. [Google Scholar] [CrossRef] [PubMed]
- Babaei, G.; Aziz, S.G.; Jaghi, N.Z.Z. EMT, cancer stem cells and autophagy; The three main axes of metastasis. Biomed Pharmacother 2021, 133, 110909. [Google Scholar] [CrossRef] [PubMed]
- Elenbaas, B.; Spirio, L.; Koerner, F.; Fleming, M.D.; Zimonjic, D.B.; Donaher, J.L.; Popescu, N.C.; Hahn, W.C.; Weinberg, R.A. Human breast cancer cells generated by oncogenic transformation of primary mammary epithelial cells. Genes Dev 2001, 15, 50–65. [Google Scholar] [CrossRef] [PubMed]
- Jian, P.; Li, Z.W.; Fang, T.Y.; Jian, W.; Zhuan, Z.; Mei, L.X.; Yan, W.S.; Jian, N. Retinoic acid induces HL-60 cell differentiation via the upregulation of miR-663. J Hematol Oncol 2011, 4, 20. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Zhang, H.; Gao, P. Metabolic reprogramming and epigenetic modifications on the path to cancer. Protein Cell 2022, 13, 877–919. [Google Scholar] [CrossRef]
- Lagadinou, E.D.; Sach, A.; Callahan, K.; Rossi, R.M.; Neering, S.J.; Minhajuddin, M.; Ashton, J.M.; Pei, S.; Grose, V.; O'Dwyer, K.M.; et al. BCL-2 inhibition targets oxidative phosphorylation and selectively eradicates quiescent human leukemia stem cells. Cell Stem Cell 2013, 12, 329–341. [Google Scholar] [CrossRef]
- Sarry, J.E.; Murphy, K.; Perry, R.; Sanchez, P.V.; Secreto, A.; Keefer, C.; Swider, C.R.; Strzelecki, A.C.; Cavelier, C.; Récher, C.; et al. Human acute myelogenous leukemia stem cells are rare and heterogeneous when assayed in NOD/SCID/IL2Rγc-deficient mice. J Clin Invest 2011, 121, 384–395. [Google Scholar] [CrossRef]
- Guan, Y.; Gerhard, B.; Hogge, D.E. Detection, isolation, and stimulation of quiescent primitive leukemic progenitor cells from patients with acute myeloid leukemia (AML). Blood 2003, 101, 3142–3149. [Google Scholar] [CrossRef]
- Shibuya, K.; Okada, M.; Suzuki, S.; Seino, M.; Seino, S.; Takeda, H.; Kitanaka, C. Targeting the facilitative glucose transporter GLUT1 inhibits the self-renewal and tumor-initiating capacity of cancer stem cells. Oncotarget 2015, 6, 651. [Google Scholar] [CrossRef]
- Jones, C.L.; Stevens, B.M.; D'Alessandro, A.; Reisz, J.A.; Culp-Hill, R.; Nemkov, T.; Pei, S.; Khan, N.; Adane, B.; Ye, H. Inhibition of amino acid metabolism selectively targets human leukemia stem cells. Cancer cell 2018, 34, 724–740. [Google Scholar] [CrossRef]
- O'Reilly, E.; Zeinabad, H.A.; Szegezdi, E. Hematopoietic versus leukemic stem cell quiescence: Challenges and therapeutic opportunities. Blood Reviews 2021, 50, 100850. [Google Scholar] [CrossRef]
- Schuringa, J.J. Smart niche usage: Release its fat and burn it! Blood 2017, 129, 1239–1240. [Google Scholar] [CrossRef] [PubMed]
- Anselmi, L.; Bertuccio, S.N.; Lonetti, A.; Prete, A.; Masetti, R.; Pession, A. Insights on the interplay between cells metabolism and signaling: A therapeutic perspective in pediatric acute leukemias. International Journal of Molecular Sciences 2020, 21, 6251. [Google Scholar] [CrossRef] [PubMed]
- Skrtić, M.; Sriskanthadevan, S.; Jhas, B.; Gebbia, M.; Wang, X.; Wang, Z.; Hurren, R.; Jitkova, Y.; Gronda, M.; Maclean, N.; et al. Inhibition of mitochondrial translation as a therapeutic strategy for human acute myeloid leukemia. Cancer Cell 2011, 20, 674–688. [Google Scholar] [CrossRef]
- Farge, T.; Saland, E.; de Toni, F.; Aroua, N.; Hosseini, M.; Perry, R.; Bosc, C.; Sugita, M.; Stuani, L.; Fraisse, M.; et al. Chemotherapy-Resistant Human Acute Myeloid Leukemia Cells Are Not Enriched for Leukemic Stem Cells but Require Oxidative Metabolism. Cancer Discov 2017, 7, 716–735. [Google Scholar] [CrossRef] [PubMed]
- Samudio, I.; Harmancey, R.; Fiegl, M.; Kantarjian, H.; Konopleva, M.; Korchin, B.; Kaluarachchi, K.; Bornmann, W.; Duvvuri, S.; Taegtmeyer, H.; et al. Pharmacologic inhibition of fatty acid oxidation sensitizes human leukemia cells to apoptosis induction. J Clin Invest 2010, 120, 142–156. [Google Scholar] [CrossRef]
- Kuhajda, F.P. Fatty-acid synthase and human cancer: New perspectives on its role in tumor biology. Nutrition 2000, 16, 202–208. [Google Scholar] [CrossRef]
- Mashima, T.; Seimiya, H.; Tsuruo, T. De novo fatty-acid synthesis and related pathways as molecular targets for cancer therapy. Br J Cancer 2009, 100, 1369–1372. [Google Scholar] [CrossRef]
- Catalano, A.; Rodilossi, S.; Caprari, P.; Coppola, V.; Procopio, A. 5-Lipoxygenase regulates senescence-like growth arrest by promoting ROS-dependent p53 activation. Embo j 2005, 24, 170–179. [Google Scholar] [CrossRef]
- Wymann, M.P.; Schneiter, R. Lipid signalling in disease. Nat Rev Mol Cell Biol 2008, 9, 162–176. [Google Scholar] [CrossRef]
- Yilmaz, O.H.; Valdez, R.; Theisen, B.K.; Guo, W.; Ferguson, D.O.; Wu, H.; Morrison, S.J. Pten dependence distinguishes haematopoietic stem cells from leukaemia-initiating cells. Nature 2006, 441, 475–482. [Google Scholar] [CrossRef] [PubMed]
- Tomlinson, I.P.; Alam, N.A.; Rowan, A.J.; Barclay, E.; Jaeger, E.E.; Kelsell, D.; Leigh, I.; Gorman, P.; Lamlum, H.; Rahman, S.; et al. Germline mutations in FH predispose to dominantly inherited uterine fibroids, skin leiomyomata and papillary renal cell cancer. Nat Genet 2002, 30, 406–410. [Google Scholar] [CrossRef]
- Baysal, B.E.; Ferrell, R.E.; Willett-Brozick, J.E.; Lawrence, E.C.; Myssiorek, D.; Bosch, A.; van der Mey, A.; Taschner, P.E.; Rubinstein, W.S.; Myers, E.N.; et al. Mutations in SDHD, a mitochondrial complex II gene, in hereditary paraganglioma. Science 2000, 287, 848–851. [Google Scholar] [CrossRef]
- Lee, E.; Yang, J.; Ku, M.; Kim, N.; Park, Y.; Park, C.; Suh, J.; Park, E.; Yook, J.; Mills, G. Metabolic stress induces a Wnt-dependent cancer stem cell-like state transition. Cell death & disease 2015, 6, e1805. [Google Scholar]
- Isaacs, J.S.; Jung, Y.J.; Mole, D.R.; Lee, S.; Torres-Cabala, C.; Chung, Y.L.; Merino, M.; Trepel, J.; Zbar, B.; Toro, J.; et al. HIF overexpression correlates with biallelic loss of fumarate hydratase in renal cancer: Novel role of fumarate in regulation of HIF stability. Cancer Cell 2005, 8, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Pollard, P.J.; Brière, J.J.; Alam, N.A.; Barwell, J.; Barclay, E.; Wortham, N.C.; Hunt, T.; Mitchell, M.; Olpin, S.; Moat, S.J.; et al. Accumulation of Krebs cycle intermediates and over-expression of HIF1alpha in tumours which result from germline FH and SDH mutations. Hum Mol Genet 2005, 14, 2231–2239. [Google Scholar] [CrossRef] [PubMed]
- Hasmim, M.; Janji, B.; Khaled, M.; Noman, M.Z.; Louache, F.; Bordereaux, D.; Abderamane, A.; Baud, V.; Mami-Chouaib, F.; Chouaib, S. Cutting Edge: NANOG Activates Autophagy under Hypoxic Stress by Binding to BNIP3L Promoter. The Journal of Immunology 2017, 198, 1423–1428. [Google Scholar] [CrossRef]
- Stockard, B.; Garrett, T.; Guingab-Cagmat, J.; Meshinchi, S.; Lamba, J. Distinct metabolic features differentiating FLT3-ITD AML from FLT3-WT childhood acute myeloid leukemia. Scientific reports 2018, 8, 5534. [Google Scholar] [CrossRef]
- Zdzisińska, B.; Żurek, A.; Kandefer-Szerszeń, M. Alpha-Ketoglutarate as a Molecule with Pleiotropic Activity: Well-Known and Novel Possibilities of Therapeutic Use. Arch Immunol Ther Exp (Warsz) 2017, 65, 21–36. [Google Scholar] [CrossRef]
- Xiao, M.; Yang, H.; Xu, W.; Ma, S.; Lin, H.; Zhu, H.; Liu, L.; Liu, Y.; Yang, C.; Xu, Y.; et al. Inhibition of α-KG-dependent histone and DNA demethylases by fumarate and succinate that are accumulated in mutations of FH and SDH tumor suppressors. Genes Dev 2012, 26, 1326–1338. [Google Scholar] [CrossRef]
- Meier, J.L. Metabolic mechanisms of epigenetic regulation. ACS Chem Biol 2013, 8, 2607–2621. [Google Scholar] [CrossRef]
- Martinez, S.; Hausinger, R.P. Catalytic Mechanisms of Fe(II)- and 2-Oxoglutarate-dependent Oxygenases. J Biol Chem 2015, 290, 20702–20711. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Deng, P.; Liu, Y.; Wu, Y.; Chen, Y.; Guo, Y.; Zhang, S.; Zheng, X.; Zhou, L.; Liu, W.; et al. Alpha-ketoglutarate ameliorates age-related osteoporosis via regulating histone methylations. Nat Commun 2020, 11, 5596. [Google Scholar] [CrossRef]
- Xiao, D.; Zeng, L.; Yao, K.; Kong, X.; Wu, G.; Yin, Y. The glutamine-alpha-ketoglutarate (AKG) metabolism and its nutritional implications. Amino Acids 2016, 48, 2067–2080. [Google Scholar] [CrossRef]
- Shen, Y.A.; Hong, J.; Asaka, R.; Asaka, S.; Hsu, F.C.; Suryo Rahmanto, Y.; Jung, J.G.; Chen, Y.W.; Yen, T.T.; Tomaszewski, A.; et al. Inhibition of the MYC-Regulated Glutaminase Metabolic Axis Is an Effective Synthetic Lethal Approach for Treating Chemoresistant Ovarian Cancers. Cancer Res 2020, 80, 4514–4526. [Google Scholar] [CrossRef]
- German, N.J.; Yoon, H.; Yusuf, R.Z.; Murphy, J.P.; Finley, L.W.; Laurent, G.; Haas, W.; Satterstrom, F.K.; Guarnerio, J.; Zaganjor, E. PHD3 loss in cancer enables metabolic reliance on fatty acid oxidation via deactivation of ACC2. Molecular cell 2016, 63, 1006–1020. [Google Scholar] [CrossRef] [PubMed]
- Tabe, Y.; Yamamoto, S.; Saitoh, K.; Sekihara, K.; Monma, N.; Ikeo, K.; Mogushi, K.; Shikami, M.; Ruvolo, V.; Ishizawa, J. Bone marrow adipocytes facilitate fatty acid oxidation activating AMPK and a transcriptional network supporting survival of acute monocytic leukemia cells. Cancer research 2017, 77, 1453–1464. [Google Scholar] [CrossRef]
- Cunningham, I.; Kohno, B. 18FDG-PET/CT: 21st century approach to leukemic tumors in 124 cases. American journal of hematology 2016, 91, 379–384. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.-L.; Wang, J.-H.; Zhao, A.-H.; Xu, X.; Wang, Y.-H.; Chen, T.-L.; Li, J.-M.; Mi, J.-Q.; Zhu, Y.-M.; Liu, Y.-F. A distinct glucose metabolism signature of acute myeloid leukemia with prognostic value. Blood, The Journal of the American Society of Hematology 2014, 124, 1645–1654. [Google Scholar] [CrossRef]
- Ye, H.; Adane, B.; Khan, N.; Alexeev, E.; Nusbacher, N.; Minhajuddin, M.; Stevens, B.M.; Winters, A.C.; Lin, X.; Ashton, J.M. Subversion of systemic glucose metabolism as a mechanism to support the growth of leukemia cells. Cancer cell 2018, 34, 659–673. [Google Scholar] [CrossRef]
- Kreitz, J.; Schönfeld, C.; Seibert, M.; Stolp, V.; Alshamleh, I.; Oellerich, T.; Steffen, B.; Schwalbe, H.; Schnütgen, F.; Kurrle, N. Metabolic plasticity of acute myeloid leukemia. Cells 2019, 8, 805. [Google Scholar] [CrossRef] [PubMed]
- Gallogly, M.M.; Lazarus, H.M.; Cooper, B.W. Midostaurin: A novel therapeutic agent for patients with FLT3-mutated acute myeloid leukemia and systemic mastocytosis. Ther Adv Hematol 2017, 8, 245–261. [Google Scholar] [CrossRef] [PubMed]
- Jonas, B.A.; Pollyea, D.A. How we use venetoclax with hypomethylating agents for the treatment of newly diagnosed patients with acute myeloid leukemia. Leukemia 2019, 33, 2795–2804. [Google Scholar] [CrossRef] [PubMed]
- Bulaklak, K.; Gersbach, C.A. The once and future gene therapy. Nat Commun 2020, 11, 5820. [Google Scholar] [CrossRef]
- Lu, Q.; He, Y.; Wang, Y.; Gao, L.; Zheng, Y.; Zhang, Z.; Cao, B.; Wang, Q.; Mao, X.; Hu, S. Saponins From Paris forrestii (Takht.) H. Li Display Potent Activity Against Acute Myeloid Leukemia by Suppressing the RNF6/AKT/mTOR Signaling Pathway. Front Pharmacol 2018, 9, 673. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Hui, H.; Yang, H.; Zhao, K.; Qin, Y.; Gu, C.; Wang, X.; Lu, N.; Guo, Q. Wogonoside induces cell cycle arrest and differentiation by affecting expression and subcellular localization of PLSCR1 in AML cells. Blood 2013, 121, 3682–3691. [Google Scholar] [CrossRef]
- Rubio-Perez, C.; Tamborero, D.; Schroeder, M.P.; Antolín, A.A.; Deu-Pons, J.; Perez-Llamas, C.; Mestres, J.; Gonzalez-Perez, A.; Lopez-Bigas, N. In silico prescription of anticancer drugs to cohorts of 28 tumor types reveals targeting opportunities. Cancer Cell 2015, 27, 382–396. [Google Scholar] [CrossRef]
- Zhang, R.Z.; Yu, S.J.; Bai, H.; Ning, K. TCM-Mesh: The database and analytical system for network pharmacology analysis for TCM preparations. Sci Rep 2017, 7, 2821. [Google Scholar] [CrossRef]
- Chen, L.; Cao, Y.; Zhang, H.; Lv, D.; Zhao, Y.; Liu, Y.; Ye, G.; Chai, Y. Network pharmacology-based strategy for predicting active ingredients and potential targets of Yangxinshi tablet for treating heart failure. J Ethnopharmacol 2018, 219, 359–368. [Google Scholar] [CrossRef]
- Li, H.; Xing, C.; Zhou, B.; Ye, H.; Feng, J.; Wu, J.; Gao, S. A regulatory circuitry between miR-193a/miR-600 and WT1 enhances leukemogenesis in acute myeloid leukemia. Exp Hematol 2018, 61, 59–68. [Google Scholar] [CrossRef]
- Bi, L.; Zhou, B.; Li, H.; He, L.; Wang, C.; Wang, Z.; Zhu, L.; Chen, M.; Gao, S. A novel miR-375-HOXB3-CDCA3/DNMT3B regulatory circuitry contributes to leukemogenesis in acute myeloid leukemia. BMC Cancer 2018, 18, 182. [Google Scholar] [CrossRef] [PubMed]
- Park, M.N.; Um, E.S.; Rahman, M.A.; Kim, J.W.; Park, S.S.; Cho, Y.; Song, H.; Son, S.R.; Jang, D.S.; Kim, W.; et al. Leonurus japonicus Houttuyn induces reactive oxygen species-mediated apoptosis via regulation of miR-19a-3p/PTEN/PI3K/AKT in U937 and THP-1 cells. J Ethnopharmacol 2022, 291, 115129. [Google Scholar] [CrossRef]
- Park, M.N.; Jeon, H.W.; Rahman, M.A.; Park, S.S.; Jeong, S.Y.; Kim, K.H.; Kim, S.H.; Kim, W.; Kim, B. Daemonorops draco Blume Induces Apoptosis Against Acute Myeloid Leukemia Cells via Regulation of the miR-216b/c-Jun. Front Oncol 2022, 12, 808174. [Google Scholar] [CrossRef] [PubMed]
- Yu, K.; Wang, J.; Hou, J.; Zhang, L.; Liang, H. miR-338-3p Plays a Significant Role in Casticin-Induced Suppression of Acute Myeloid Leukemia via Targeting PI3K/Akt Pathway. Biomed Res Int 2022, 2022, 9214130. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Y.J.; Wu, M.; Zhang, H.; Wu, X.P.; Zhou, L.; Wan, N.; Wu, Z.H. Effects of Qinghuang Powder on Acute Myeloid Leukemia Based on Network Pharmacology, Molecular Docking, and In Vitro Experiments. Evid Based Complement Alternat Med 2021, 2021, 6195174. [Google Scholar] [CrossRef]
- Guzman, M.L.; Rossi, R.M.; Karnischky, L.; Li, X.; Peterson, D.R.; Howard, D.S.; Jordan, C.T. The sesquiterpene lactone parthenolide induces apoptosis of human acute myelogenous leukemia stem and progenitor cells. Blood 2005, 105, 4163–4169. [Google Scholar] [CrossRef]
- Neelakantan, S.; Nasim, S.; Guzman, M.L.; Jordan, C.T.; Crooks, P.A. Aminoparthenolides as novel anti-leukemic agents: Discovery of the NF-kappaB inhibitor, DMAPT (LC-1). Bioorg Med Chem Lett 2009, 19, 4346–4349. [Google Scholar] [CrossRef]
- Zhao, W.; Wang, L.; Zhang, M.; Wang, P.; Zhang, L.; Yuan, C.; Qi, J.; Qiao, Y.; Kuo, P.C.; Gao, C. NF-κB- and AP-1-mediated DNA looping regulates osteopontin transcription in endotoxin-stimulated murine macrophages. J Immunol 2011, 186, 3173–3179. [Google Scholar] [CrossRef]
- Samant, R.S.; Clark, D.W.; Fillmore, R.A.; Cicek, M.; Metge, B.J.; Chandramouli, K.H.; Chambers, A.F.; Casey, G.; Welch, D.R.; Shevde, L.A. Breast cancer metastasis suppressor 1 (BRMS1) inhibits osteopontin transcription by abrogating NF-kappaB activation. Mol Cancer 2007, 6, 6. [Google Scholar] [CrossRef]
- Inoue, K.; Sugiyama, H.; Ogawa, H.; Yamagami, T.; Azuma, T.; Oka, Y.; Miwa, H.; Kita, K.; Hiraoka, A.; Masaoka, T.; et al. Expression of the interleukin-6 (IL-6), IL-6 receptor, and gp130 genes in acute leukemia. Blood 1994, 84, 2672–2680. [Google Scholar] [CrossRef]
- Libermann, T.A.; Baltimore, D. Activation of interleukin-6 gene expression through the NF-kappa B transcription factor. Mol Cell Biol 1990, 10, 2327–2334. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.J.; Hsiao, M.; Chang, J.L.; Yang, S.F.; Tseng, T.H.; Cheng, C.W.; Chow, J.M.; Lin, K.H.; Lin, Y.W.; Liu, C.C.; et al. Quercetin induces mitochondrial-derived apoptosis via reactive oxygen species-mediated ERK activation in HL-60 leukemia cells and xenograft. Arch Toxicol 2015, 89, 1103–1117. [Google Scholar] [CrossRef]
- Srivastava, S.; Somasagara, R.R.; Hegde, M.; Nishana, M.; Tadi, S.K.; Srivastava, M.; Choudhary, B.; Raghavan, S.C. Quercetin, a Natural Flavonoid Interacts with DNA, Arrests Cell Cycle and Causes Tumor Regression by Activating Mitochondrial Pathway of Apoptosis. Sci Rep 2016, 6, 24049. [Google Scholar] [CrossRef]
- Ren, M.X.; Deng, X.H.; Ai, F.; Yuan, G.Y.; Song, H.Y. Effect of quercetin on the proliferation of the human ovarian cancer cell line SKOV-3 in vitro. Exp Ther Med 2015, 10, 579–583. [Google Scholar] [CrossRef]
- Lee, W.J.; Chen, Y.R.; Tseng, T.H. Quercetin induces FasL-related apoptosis, in part, through promotion of histone H3 acetylation in human leukemia HL-60 cells. Oncol Rep 2011, 25, 583–591. [Google Scholar] [CrossRef] [PubMed]
- Avci, C.B.; Yilmaz, S.; Dogan, Z.O.; Saydam, G.; Dodurga, Y.; Ekiz, H.A.; Kartal, M.; Sahin, F.; Baran, Y.; Gunduz, C. Quercetin-induced apoptosis involves increased hTERT enzyme activity of leukemic cells. Hematology 2011, 16, 303–307. [Google Scholar] [CrossRef]
- Jeong, H.; Park, S.; Kim, S.Y.; Cho, S.H.; Jeong, M.S.; Kim, S.R.; Seo, J.B.; Kim, S.H.; Kim, K.N. 1-Cinnamoyltrichilinin from Melia azedarach Causes Apoptosis through the p38 MAPK Pathway in HL-60 Human Leukemia Cells. Int J Mol Sci 2020, 21. [Google Scholar] [CrossRef]
- Kim, H.U.; Ryu, J.Y.; Lee, J.O.; Lee, S.Y. A systems approach to traditional oriental medicine. Nat Biotechnol 2015, 33, 264–268. [Google Scholar] [CrossRef] [PubMed]
- Cheung, F. TCM: Made in China. Nature 2011, 480, S82–S83. [Google Scholar] [CrossRef]
- Dar, A.C.; Das, T.K.; Shokat, K.M.; Cagan, R.L. Chemical genetic discovery of targets and anti-targets for cancer polypharmacology. Nature 2012, 486, 80–84. [Google Scholar] [CrossRef]
- Gujral, T.S.; Peshkin, L.; Kirschner, M.W. Exploiting polypharmacology for drug target deconvolution. Proc Natl Acad Sci U S A 2014, 111, 5048–5053. [Google Scholar] [CrossRef] [PubMed]
- Stylianopoulos, T.; Munn, L.L.; Jain, R.K. Reengineering the Physical Microenvironment of Tumors to Improve Drug Delivery and Efficacy: From Mathematical Modeling to Bench to Bedside. Trends Cancer 2018, 4, 292–319. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Huang, Q.; Zhao, R.; Ma, Z. A network pharmacology study on the Tripteryguim wilfordii Hook for treatment of Crohn's disease. BMC Complement Med Ther 2020, 20, 95. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, F.; Barosi, G.; Venditti, A.; Angelucci, E.; Gobbi, M.; Pane, F.; Tosi, P.; Zinzani, P.; Tura, S. Consensus-based definition of unfitness to intensive and non-intensive chemotherapy in acute myeloid leukemia: A project of SIE, SIES and GITMO group on a new tool for therapy decision making. Leukemia 2013, 27, 997–999. [Google Scholar] [CrossRef] [PubMed]
- Burnett, A.K.; Russell, N.H.; Hills, R.K.; Hunter, A.E.; Kjeldsen, L.; Yin, J.; Gibson, B.E.; Wheatley, K.; Milligan, D. Optimization of chemotherapy for younger patients with acute myeloid leukemia: Results of the medical research council AML15 trial. J Clin Oncol 2013, 31, 3360–3368. [Google Scholar] [CrossRef]
- Fenaux, P.; Mufti, G.J.; Hellström-Lindberg, E.; Santini, V.; Gattermann, N.; Germing, U.; Sanz, G.; List, A.F.; Gore, S.; Seymour, J.F.; et al. Azacitidine prolongs overall survival compared with conventional care regimens in elderly patients with low bone marrow blast count acute myeloid leukemia. J Clin Oncol 2010, 28, 562–569. [Google Scholar] [CrossRef]
- Pollyea, D.A.; Stevens, B.M.; Jones, C.L.; Winters, A.; Pei, S.; Minhajuddin, M.; D'Alessandro, A.; Culp-Hill, R.; Riemondy, K.A.; Gillen, A.E.; et al. Venetoclax with azacitidine disrupts energy metabolism and targets leukemia stem cells in patients with acute myeloid leukemia. Nat Med 2018, 24, 1859–1866. [Google Scholar] [CrossRef]
- Falini, B.; Mecucci, C.; Tiacci, E.; Alcalay, M.; Rosati, R.; Pasqualucci, L.; La Starza, R.; Diverio, D.; Colombo, E.; Santucci, A.; et al. Cytoplasmic nucleophosmin in acute myelogenous leukemia with a normal karyotype. N Engl J Med 2005, 352, 254–266. [Google Scholar] [CrossRef]
- Heath, E.M.; Chan, S.M.; Minden, M.D.; Murphy, T.; Shlush, L.I.; Schimmer, A.D. Biological and clinical consequences of NPM1 mutations in AML. Leukemia 2017, 31, 798–807. [Google Scholar] [CrossRef]
- Ostronoff, F.; Othus, M.; Lazenby, M.; Estey, E.; Appelbaum, F.R.; Evans, A.; Godwin, J.; Gilkes, A.; Kopecky, K.J.; Burnett, A.; et al. Prognostic significance of NPM1 mutations in the absence of FLT3-internal tandem duplication in older patients with acute myeloid leukemia: A SWOG and UK National Cancer Research Institute/Medical Research Council report. J Clin Oncol 2015, 33, 1157–1164. [Google Scholar] [CrossRef]
- Knapper, S. The clinical development of FLT3 inhibitors in acute myeloid leukemia. Expert Opin Investig Drugs 2011, 20, 1377–1395. [Google Scholar] [CrossRef] [PubMed]
- Daver, N.; Schlenk, R.F.; Russell, N.H.; Levis, M.J. Targeting FLT3 mutations in AML: Review of current knowledge and evidence. Leukemia 2019, 33, 299–312. [Google Scholar] [CrossRef] [PubMed]
- Saxena, K.; Konopleva, M. New Treatment Options for Older Patients with Acute Myeloid Leukemia. Curr Treat Options Oncol 2021, 22, 39. [Google Scholar] [CrossRef] [PubMed]
- Moore, A.S.; Faisal, A.; Gonzalez de Castro, D.; Bavetsias, V.; Sun, C.; Atrash, B.; Valenti, M.; de Haven Brandon, A.; Avery, S.; Mair, D.; et al. Selective FLT3 inhibition of FLT3-ITD+ acute myeloid leukaemia resulting in secondary D835Y mutation: A model for emerging clinical resistance patterns. Leukemia 2012, 26, 1462–1470. [Google Scholar] [CrossRef]
- Kadia, T.M.; Ravandi, F.; Cortes, J.; Kantarjian, H. Toward Individualized Therapy in Acute Myeloid Leukemia: A Contemporary Review. JAMA Oncol 2015, 1, 820–828. [Google Scholar] [CrossRef]
- Popovici-Muller, J.; Lemieux, R.M.; Artin, E.; Saunders, J.O.; Salituro, F.G.; Travins, J.; Cianchetta, G.; Cai, Z.; Zhou, D.; Cui, D. Discovery of AG-120 (Ivosidenib): A first-in-class mutant IDH1 inhibitor for the treatment of IDH1 mutant cancers. ACS medicinal chemistry letters 2018, 9, 300–305. [Google Scholar] [CrossRef]
- Yen, K.; Travins, J.; Wang, F.; David, M.D.; Artin, E.; Straley, K.; Padyana, A.; Gross, S.; DeLaBarre, B.; Tobin, E. AG-221, a First-in-Class Therapy Targeting Acute Myeloid Leukemia Harboring Oncogenic IDH2 MutationsAG-221 Therapy for IDH2-Mutant AML. Cancer discovery 2017, 7, 478–493. [Google Scholar] [CrossRef]
- Stein, E.M.; DiNardo, C.D.; Pollyea, D.A.; Fathi, A.T.; Roboz, G.J.; Altman, J.K.; Stone, R.M.; DeAngelo, D.J.; Levine, R.L.; Flinn, I.W. Enasidenib in mutant IDH2 relapsed or refractory acute myeloid leukemia. Blood, The Journal of the American Society of Hematology 2017, 130, 722–731. [Google Scholar] [CrossRef]
- DiNardo, C.D.; Stein, E.M.; de Botton, S.; Roboz, G.J.; Altman, J.K.; Mims, A.S.; Swords, R.; Collins, R.H.; Mannis, G.N.; Pollyea, D.A.; et al. Durable Remissions with Ivosidenib in IDH1-Mutated Relapsed or Refractory AML. N Engl J Med 2018, 378, 2386–2398. [Google Scholar] [CrossRef]
- Kantarjian, H.M.; Thomas, X.G.; Dmoszynska, A.; Wierzbowska, A.; Mazur, G.; Mayer, J.; Gau, J.P.; Chou, W.C.; Buckstein, R.; Cermak, J.; et al. Multicenter, randomized, open-label, phase III trial of decitabine versus patient choice, with physician advice, of either supportive care or low-dose cytarabine for the treatment of older patients with newly diagnosed acute myeloid leukemia. J Clin Oncol 2012, 30, 2670–2677. [Google Scholar] [CrossRef]
- Bykov, V.J.; Issaeva, N.; Shilov, A.; Hultcrantz, M.; Pugacheva, E.; Chumakov, P.; Bergman, J.; Wiman, K.G.; Selivanova, G. Restoration of the tumor suppressor function to mutant p53 by a low-molecular-weight compound. Nat Med 2002, 8, 282–288. [Google Scholar] [CrossRef]
- O'Connor, P.M.; Jackman, J.; Bae, I.; Myers, T.G.; Fan, S.; Mutoh, M.; Scudiero, D.A.; Monks, A.; Sausville, E.A.; Weinstein, J.N.; et al. Characterization of the p53 tumor suppressor pathway in cell lines of the National Cancer Institute anticancer drug screen and correlations with the growth-inhibitory potency of 123 anticancer agents. Cancer Res 1997, 57, 4285–4300. [Google Scholar] [PubMed]
- DiNardo, C.D.; Jonas, B.A.; Pullarkat, V.; Thirman, M.J.; Garcia, J.S.; Wei, A.H.; Konopleva, M.; Döhner, H.; Letai, A.; Fenaux, P.; et al. Azacitidine and Venetoclax in Previously Untreated Acute Myeloid Leukemia. N Engl J Med 2020, 383, 617–629. [Google Scholar] [CrossRef] [PubMed]
- Wei, A.H.; Döhner, H.; Pocock, C.; Montesinos, P.; Afanasyev, B.; Dombret, H.; Ravandi, F.; Sayar, H.; Jang, J.H.; Porkka, K.; et al. Oral Azacitidine Maintenance Therapy for Acute Myeloid Leukemia in First Remission. N Engl J Med 2020, 383, 2526–2537. [Google Scholar] [CrossRef] [PubMed]
- Sievers, E.L. Antibody-targeted chemotherapy of acute myeloid leukemia using gemtuzumab ozogamicin (Mylotarg). Blood Cells, Molecules, and Diseases 2003, 31, 7–10. [Google Scholar] [CrossRef]
- Carter, J.L.; Hege, K.; Yang, J.; Kalpage, H.A.; Su, Y.; Edwards, H.; Hüttemann, M.; Taub, J.W.; Ge, Y. Targeting multiple signaling pathways: The new approach to acute myeloid leukemia therapy. Signal Transduct Target Ther 2020, 5, 288. [Google Scholar] [CrossRef]
- Skopek, R.; Palusińska, M.; Kaczor-Keller, K.; Pingwara, R.; Papierniak-Wyglądała, A.; Schenk, T.; Lewicki, S.; Zelent, A.; Szymański, Ł. Choosing the Right Cell Line for Acute Myeloid Leukemia (AML) Research. Int J Mol Sci 2023, 24. [Google Scholar] [CrossRef]
- Liu, Z.H.; Sun, X.B. [Network pharmacology: New opportunity for the modernization of traditional Chinese medicine]. Yao Xue Xue Bao 2012, 47, 696–703. [Google Scholar]
- Fang, T.; Liu, L.; Liu, W. Network pharmacology-based strategy for predicting therapy targets of Tripterygium wilfordii on acute myeloid leukemia. Medicine (Baltimore) 2020, 99, e23546. [Google Scholar] [CrossRef]
- Huang, S.; Zhang, Z.; Li, W.; Kong, F.; Yi, P.; Huang, J.; Mao, D.; Peng, W.; Zhang, S. Network Pharmacology-Based Prediction and Verification of the Active Ingredients and Potential Targets of Zuojinwan for Treating Colorectal Cancer. Drug Des Devel Ther 2020, 14, 2725–2740. [Google Scholar] [CrossRef]
- Huang, Z.; Yang, Y.; Fan, X.; Ma, W. Network pharmacology-based investigation and experimental validation of the mechanism of scutellarin in the treatment of acute myeloid leukemia. Front Pharmacol 2022, 13, 952677. [Google Scholar] [CrossRef] [PubMed]
- Barretina, J.; Caponigro, G.; Stransky, N.; Venkatesan, K.; Margolin, A.A.; Kim, S.; Wilson, C.J.; Lehár, J.; Kryukov, G.V.; Sonkin, D.; et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012, 483, 603–607. [Google Scholar] [CrossRef] [PubMed]
- De Angelo, D.; Schiffer, C.; Stone, R.; Amrein, P.; Fernandez, H.; Bradstock, K.; Tallman, M.; Foran, J.; Juliusson, G.; Liu, D. Interim analysis of a phase II study of the safety and efficacy of gemtuzumab ozogamicin (Mylotarg (R)) given in combination with cytarabine and daunorubicin to patients< 60 years old with untreated acute myeloid leukemia. Blood 2002, 100, 745. [Google Scholar]
- Bassan, R.; Intermesoli, T.; Masciulli, A.; Pavoni, C.; Boschini, C.; Gianfaldoni, G.; Marmont, F.; Cavattoni, I.; Mattei, D.; Terruzzi, E.; et al. Randomized trial comparing standard vs sequential high-dose chemotherapy for inducing early CR in adult AML. Blood Adv 2019, 3, 1103–1117. [Google Scholar] [CrossRef] [PubMed]
- Luo, T.T.; Lu, Y.; Yan, S.K.; Xiao, X.; Rong, X.L.; Guo, J. Network Pharmacology in Research of Chinese Medicine Formula: Methodology, Application and Prospective. Chin J Integr Med 2020, 26, 72–80. [Google Scholar] [CrossRef]
- Haykal, T.; Younes, M.; El Khoury, M.; Ammoury, C.; Tannous, S.; Hodroj, M.H.; Sarkis, R.; Gasilova, N.; Menin, L.; Rizk, S. The pro-apoptotic properties of a phytonutrient rich infusion of A. cherimola leaf extract on AML cells. Biomedicine & Pharmacotherapy 2021, 140, 111592. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



