
Submitted:
27 June 2023
Posted:
29 June 2023
You are already at the latest version
Preprints on COVID-19 and SARS-CoV-2
Abstract
During the recent pandemics of COVID-19, sequencing technics became powerful tool for gaining information about the SARS-CoV-2 virus and using this knowledge in our advantage. Thanks to this advantage, scientists over the world were able to search for emerging variations, watching the virus evolving in real time. Assembly of the virus genomes is crucial part of getting this kind of useful information. In our study, we sequenced 79 samples from nasopharyngeal swabs of COVID-19 patients. Positivity on disease was evaluated by qRT-PCR. By studying assembly success rate, we noticed, that Ct-value of qRT-PCR can predict success of genome assembly and SARS-CoV-2 lineage assignment. In this work we described the relationship between Ct-value and further steps of genome construction and its assignment to specific viral strain.
Keywords:
SARS-CoV-2
; RNA genome assembly
; RT-qPCR
; SPAdes
1. Introduction
SARS-CoV-2 virus has appeared in the end of 2019, when it was found to cause a disease, today known as COVID-19 [1]. To date, a number of different lineages have emerged. Some of those variants that have emerged so far have become widely spread or even dominant for a time. The Alpha variant (B.1.1.7) from September 2020 carries 23 nucleotide substitutions; the Beta variant (B.1.351), which appeared in May 2020, was characterized by seven mutations in the spike protein. Later, the Gamma variant (P.1) appeared with 17 unique mutations, followed by the Delta variant (B.1.617.2) with 29 mutations in multiple proteins, and Omicron (B.1.1.529), which carried more than 50 new point mutations (30 associated with residue changes in the spike protein). Occasionally, coinfection can occur, leading to recombination and the appearance of recombinants among other variants that have actually been documented [2]. Scientists around the world have been collecting genomic or transcriptomic data on SARS-CoV-2 and infected patients, which has made it possible to study the infection and how the host responds to it at the molecular level. To date, 14,926,813 SARS-CoV-2 genome data have been submitted into the Gisaid system [3].
Assembling SARS-CoV-2 genome became standard procedure, since it is necessary for NGS data analysis. When it comes to the routinely used methods utilized in standard clinical sample processing during this pandemic mostly targeted enrichment strategies were used, based on either amplicon or hybridization-probe approaches. There are different designs or protocols that became routinely used, based on standardized academical research originated or commercially available solutions, e. g. ARTIC nCoV2019 protocol available also in standardized set of oligonucleotides from IDT as a result of ARTIC network initiative focused on SARS-CoV-2 [4] or Respiratory Virus Oligo Panel kit from Illumina. Such targeted solutions are perfect when it comes to the need of high throughput, short turnaround times for sample analysis as well as good economical effectiveness in cases when focused analysis - on detection of SARS-CoV-2 or panel of clinically relevant respiratory viruses, is the issue. But from scientific perspective this bring only limited amount of disease related information as the response of the patient to the viral infection is complex and should be studied in association with currently known relevant biological context. For studying the biological context at least local (or global) microbiome and local host transcriptome should be involved. Therefore, if we would like to go over this limitation (of focused viral detection analysis), we should use metatranscriptomic analyses as the method of choice [5].
In our study, virus genome was assembled by using only short read RNA-seq data directly from metatranscriptomic analyses originated from clinical samples represented by nasopharyngeal swabs.
2. Materials and Methods
2.1. Study Approval
Sample collection was performed as part of the clinical study approved by the Ethical Committee of Bratislava Self-Governing District under the identifier 03228/2021/HF from January 12, 2021. All patients have filled out the questionnaires with relevant information regarding their health status in relation to COVID-19 and signed informed consent.
2.2. Samples
Nasopharyngeal swabs from patients suspected of having COVID-19 were obtained in two primary regimens (79 patients). Patients hospitalized with severe symptoms of the disease at the collaborating hospitals were enrolled in the study. Patients with mild or any symptoms of the disease were recruited in mobile testing facilities for SARS-CoV-2 by a company providing routine laboratory diagnostics from the population during the COVID-19 pandemic.
2.3. Nucleic acid Extraction
Nasopharyngeal swabs specimens were collected from COVID-19 patients and controls and stored in viRNAtrap collection medium (GeneSpector, Czech Republic) at 4°C. Total nucleic acid was extracted using Sera-Xtracta Virus/Pathogen Kit (Cytiva, UK) according to manufacturer instructions. 400 μl of the nasopharyngeal swab medium was used for nucleic acid extraction with final elution to 50 μl of nuclease-free water. RNA was quantified with the Qubit™ RNA High Sensitivity Assay Kit (Invitrogen, USA). RNA isolates were stored at -80°C.
2.4. RT-qPCR
The presence of SARS-CoV-2 was determined by RT-qPCR using the COVID-19 Real Time Multiplex RT-PCR Kit (Labsystems Diagnostics, Finland) and RT-qPCR platform ABI QuantStudio 6 Real-Time PCR System (ThermoFisher, USA) utilizing the original manufacturers protocols. Amplification cycles threshold of Ct value <40 was needed to evaluate sample as positive.
2.5. RNA Library Preparation and Sequencing
The metatranscriptomic libraries were prepared using KAPA RNA HyperPrep Kit with RiboErase (HMR) (Kapa Biosystems, South Africa) according to the original protocol of manufacturer. For quantity and quality control of prepared libraries a Qubit 1X dsDNA High Sensitivity Assay Kit on Qubit 3.0 (Invitrogen) and Agilent High Sensitivity DNA Kit on Agilent 2100 Bioanalyzer (Agilent, USA) instruments were used. Sequencing of pooled libraries was performed on NextSeq 500 and NextSeq 2000 (Illumina, USA) platforms using 2x75 or 2x100 paired-end sequencing setup, respectively.
2.6. Data Quality Control and Preparation for Analysis
Quality control was done by FastQC v0.11.9 [6]. Reads were processed by Trimmomatic v0.39 (CROP:96 HEADCROP:10 LEADING:22 TRAILING:22 SLIDINGWINDOW:4:22 MINLEN:25 and our own set of adapter sequences were used in ILLUMINACLIP step) [7]. Parameters were chosen according to FastQC results. All mentioned tools were used as Linux command line instances with Conda.
2.7. Genome Assembly
Trimmed reads were mapped to the SARS-CoV-2 genome by BWA-MEM algorithm. Subsequently, those reads were extracted using samtools view from samtools v1.6 [8] and Picard SamToFastq from Picard v2.27.4 [9]. Reads were mapped as paired set, otherwise parameters of mapping were set to default. The assembly of SARS-CoV-2 was performed using coronaspades.py tool from Spades [10]. All mentioned tools were used as Linux command line instances with Conda.
2.8. SARS-CoV-2 Variants Identification
SARS-CoV-2 variant identification was done independently of genome assembly, by Galaxy pipeline „Mutation calling, viral genome reconstruction and lineage/clade assignment from SARS-CoV-2 sequencing data“ [11]. As an input we used reads mapping on SARS-CoV-2 genome. First step of the pipeline is variation analysis with key components: BWA-MEM for mapping, Lofreq for variant calling and SnpEff for variants’ annotation [12]. Then, next step is variant reporting. The third step is to generate consensus sequences and then identify SARS-CoV-2 clades/ lineages by Pangolin and Nextclade [13,14].
2.9. Statistics of Ct and Assembly
For the purpose of assembly quality estimation, LG50 value of assembly was computed by linux command line instance of Quast software [15]. To prove relationship between assembly and qRT-PCR, correlation coefficient was computed between qRT-PCR Ct values (E gene) and LG50 values. Correlation coefficient was computed by dividing the covariance by the product of the two variables' standard deviations:
Corr(X,Y) = Cov[X,Y] / ( StdDev(X) ∙ StdDev(Y) )
Same formula was used to get correlation coefficient between Ct-values and success of SARS-CoV-2 clade assignment (scored 0/1 for unassigned/assigned). Undetermined values of Ct-value were ignored, although for the purpose of visualization data on graph, value of 40 atop of maximal value was assigned.
3. Results
This work is a part of a bigger project studying transcriptome and metatranscriptome of COVID-19 patients. We assembled genomes of SARS-CoV-2 from these samples and noticed the correlation between success rate of assemblies and measured Ct-value from RT-qPCR.
3.1. Assembly Performance
For assembly of the genome, we used SPAdes assembler (coronaspades mode) and it was done from SARS-CoV-2 mapped reads. Out of 79 samples, 36 genomes were assembled as one scaffold at least 28 000 nt long, while in another 6 samples, assembly was fragmented in 2 or more scaffolds, but its alignment covered almost the whole reference genome.
3.2. Presence of the Virus and Its Strain
Here, we report results on 79 samples from patients positive on COVID-19, whose positivity was proved by qRT-PCR. We assigned viral reads to specific WHO variants and clade. Results were correlating with assembly success. 24 samples were assigned as Alpha variant (clade 20I), 12 samples as Delta (clade 21J, in one sample 21I), 6 samples as Omicron (clade 21L and one sample with 22B), one sample was assigned to 20C clade and one was reported as recombinant.
3.3. RT-PCR and Assembly
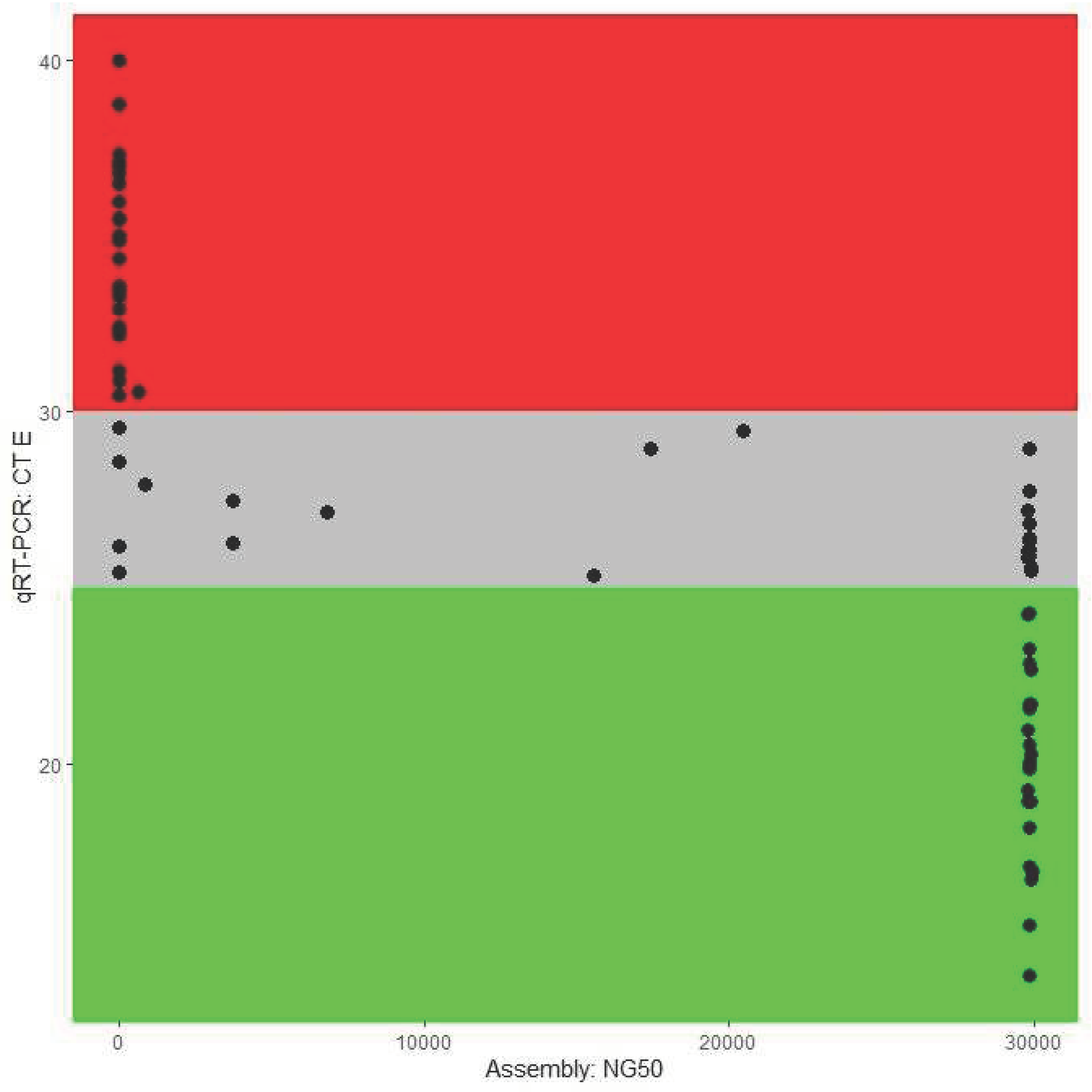
We examined Ct-values from RT-PCR and whether these values could predict assembly success. Samples with lower Ct-values (E gene) were mostly assembled. Median of Ct-value for samples with completely assembled genome 24.24, while in case of fragmented assembly it was 28.76. Median of samples with failed genome assembly was 32.95. Correlation coefficient between Ct-value and NG50 value of assembly was -0.81. In total, 8 samples had undetermined Ct-value and were left out from correlation statistics (7 with failed assembly and 1 with fragmented assembly). Relationship between qRT-PCR Ct-value and NG50 assembly quality metrics is shown on Figure 1, where three Ct-value zones of successful, unsuccessful and unsure assembly can be seen. Grey zone of unsure assembly is between Ct-values of 25-30, while everything what is lower was always successfully assembled and everything what is higher always failed.
3.4. RT-PCR and Lineage/Clade Assignment
For samples with lower Ct-values (E gene), SARS-CoV-2 clade was mostly assigned (correlation coefficient with successful clade assignment = -0.78, while 8 Ct-values were undetermined, left out of correlation statistics, all unassigned). For samples, where the assembly was not successful enough to assign a clade to the virus, the median Ct-value was 33.54. The minimum value was 25.46. The Ct-value was not determined for 8 samples. The median of the determined samples was 24.24 with a maximum value of 30.57. Samples with a Ct value < 25 appear to be viable for both virus clade assignment and assembly.
4. Discussion
Alhough we originally performed our study with focus on identification of potential novel biomarkers resulting from human transcriptome and metatranscriptome analysis of samples from COVID-19 patients with different severity of the disease, here, we reported dependency of SARS-CoV-2 genome assembly and lineage assignment on Ct value from diagnostic qRT-PCR. This work is a collateral output of the complex research but is an independent and specifically focused part of it and a brief report of this finding.
We performed multiple steps of data analysis to achieve the observation. First, assembly of the genome was necessary. There are various assemblers fit for a job. Benchmarking study of Gupta & Kumar (2022) recommends SPAdes, IDBA, and ABySS out of 8 assemblers they tested [16]. We tested performance of SPAdes ourselves, trying various strategies and also Trinity, which was not compared by Gupta & Kumar (2022). These results are about to be published as “Possibilities of assembling whole genome of clinically relevant pathogens directly from clinical samples sequenced as metatranscriptomic data” (Hadzega et al., 2023). For presenting results here, we chose the best performing assembly strategy.
It was observed, that samples with Ct value under 25 were always assembled, samples between Ct value of 25-30 are in kind of grey zone, where assembly might be successful, but not always and samples over Ct value of 30 were never successfully assembled.
Previously published studies have shown that use of different approaches for SARS-CoV-2 genome sequencing is capable, although targeted resequencing (either amplicon or hybridization -based) with subsequent full genome assembly were working horses when it comes to massively applied diagnostic and epidemiologic surveillance studies. Additionally, short- as well as long-read sequencing solutions were used with acceptable ratio when it comes to turn-around-times, throughput and cost-effectiveness of such testing [5,17]. But when it comes to the research potential with focus not only on diagnostics but also complex study of viral infection and local and potentially global host reaction the most relevant strategy is the metatranscriptomic analysis starting directly from clinical samples. With this approach host transcriptome, microbiome as well as viral genome could be analyzed in parallel if there is enough of viral genome also full viral genome could be assembled. In a comprehensive study of Xiao et al. complete genomes, inter-individual and intra-individual variations of SARS-CoV-2 from serials dilutions of a cultured isolate as well as clinical samples covering a range of sample types and viral loads were analyzed and in the case of metatranscriptomic analyses similar findings as in our study were reported. Their analysis performed on only 8 clinical samples with focus on correlation between Ct values and successful SARS-CoV-2 genome assembly showed the limiting Ct value for the completeness of assembly at the Ct value ≤ 24.5 (corresponding to conc. ≥ 1E+05 viral genome copies per milliliter) [5].
In our study, we used Quast’s NG50 value as quality metrics of assembly. In comparison with more standard N50 value, it compares assembled contigs to reference genome, not to assembly itself. With this value, it was possible to evaluate assembly success relation with Ct value. In computing correlation coefficient, we ignored undetermined Ct-values from qRT-PCR. By this simplification, we meant to eliminate risk of incorrect methodology if undetermined values would be replaced by number, although it probably causes slight underestimation of result number for correlation coefficient. Our study strength was in number of samples, which we believe is enough for presented statistics.
5. Conclusions
This section is not mandatory but can be added to the manuscript if the discussion is unusually long or complex.
Author Contributions
Conceptualization, DH, KB, MH, NJ; methodology, DH, MH, GM; software, DH; formal analysis, PJ, GM; investigation, DH, NJ, KB, MH; resources, PS, PJ, GM; data curation, KB, MH, PS; writing—original draft preparation, DH; writing—review and editing, GM, MH, PS, PJ; visualization, DH; supervision, GM; project administration, PJ; funding acquisition, PJ, GM.
Funding
This research was funded by: OP Integrated Infrastructure for the project: Serious diseases of civilization and Covid19, ITMS: 313011AVH7, co-financed by the European Regional Development Fund.".
Institutional Review Board Statement
The study was conducted in accordance with the Declaration of Helsinki, and approved by the Ethics Committee of Bratislava self-governing region (12.1.2021).
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Acknowledgments
We thank Dr. Lassan Stefan (Hospital Ružinov, Bratislava), Dr.Jackuliak Peter (Hospital Ružinov, Bratislava) for providing clinical samples, medical documentation and informed consent.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.-M.; Wang, W.; Song, Z.-G.; Hu, Y.; Tao, Z.-W.; Tian, J.-H.; Pei, Y.-Y.; Yuan, M.-L.; Zhang, Y.-L.; Dai, F.-H.; Liu, Y.; Wang, Q.-M.; Zheng, J.-J.; Xu, L.; Holmes, E. C.; Zhang, Y.-Z. A New Coronavirus Associated with Human Respiratory Disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef] [PubMed]
- Tosta, S.; Moreno, K.; Schuab, G.; Fonseca, V.; Segovia, F. M. C.; Kashima, S.; Elias, M. C.; Sampaio, S. C.; Ciccozzi, M.; Alcantara, L. C. J.; Slavov, S. N.; Lourenço, J.; Cella, E.; Giovanetti, M. Global SARS-CoV-2 Genomic Surveillance: What We Have Learned (so Far). Infection, Genetics and Evolution 2023, 108, 105405. [Google Scholar] [CrossRef] [PubMed]
- https://gisaid.org/.
- https://artic.network/.
- Xiao, M.; Liu, X.; Ji, J.; Li, M.; Li, J.; Yang, L.; Sun, W.; Ren, P.; Yang, G.; Zhao, J.; Liang, T.; Ren, H.; Chen, T.; Zhong, H.; Song, W.; Wang, Y.; Deng, Z.; Zhao, Y.; Ou, Z.; Wang, D.; Cai, J.; Cheng, X.; Feng, T.; Wu, H.; Gong, Y.; Yang, H.; Wang, J.; Xu, X.; Zhu, S.; Chen, F.; Zhang, Y.; Chen, W.; Li, Y.; Li, J. Multiple Approaches for Massively Parallel Sequencing of SARS-CoV-2 Genomes Directly from Clinical Samples. Genome Med 2020, 12, 57. [Google Scholar] [CrossRef] [PubMed]
- Andrews, S. http://www.bioinformatics.babraham.ac.uk/projects/fastqc (Accessed 23.05. 2023). 17. FastQC: a quality control tool for high throughput sequence data.
- Bolger, A. M.; Lohse, M.; Usadel, B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Danecek, P.; Bonfield, J. K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M. O.; Whitwham, A.; Keane, T.; McCarthy, S. A.; Davies, R. M.; Li, H. Twelve Years of SAMtools and BCFtools. Gigascience 2021, 10. [Google Scholar] [CrossRef] [PubMed]
- Broad Institute. https://broadinstitute.github.io/picard/. “Picard Toolkit.” .
- Prjibelski, A.; Antipov, D.; Meleshko, D.; Lapidus, A.; Korobeynikov, A. Using SPAdes De Novo Assembler. Curr Protoc Bioinformatics 2020, 70. [Google Scholar] [CrossRef] [PubMed]
- Maier, W.; Batut, B. https://training.galaxyproject.org/training-material/topics/variant-analysis/tutorials/sars-cov-2-variant-discovery/tutorial.html. Mutation calling, viral genome reconstruction and lineage/clade assignment from SARS-CoV-2 sequencing data (Galaxy Training Materials).
- Cingolani, P.; Platts, A.; Wang, L. L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S. J.; Lu, X.; Ruden, D. M. A Program for Annotating and Predicting the Effects of Single Nucleotide Polymorphisms, SnpEff. Fly (Austin) 2012, 6, 80–92. [Google Scholar] [CrossRef] [PubMed]
- O’Toole, Á.; Scher, E.; Underwood, A.; Jackson, B.; Hill, V.; McCrone, J. T.; Colquhoun, R.; Ruis, C.; Abu-Dahab, K.; Taylor, B.; Yeats, C.; du Plessis, L.; Maloney, D.; Medd, N.; Attwood, S. W.; Aanensen, D. M.; Holmes, E. C.; Pybus, O. G.; Rambaut, A. Assignment of Epidemiological Lineages in an Emerging Pandemic Using the Pangolin Tool. Virus Evol 2021, 7. [Google Scholar] [CrossRef] [PubMed]
- Aksamentov, I.; Roemer, C.; Hodcroft, E.; Neher, R. Nextclade: Clade Assignment, Mutation Calling and Quality Control for Viral Genomes. J Open Source Softw 2021, 6, 3773. [Google Scholar] [CrossRef]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality Assessment Tool for Genome Assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A. K.; Kumar, M. Benchmarking and Assessment of Eight De Novo Genome Assemblers on Viral Next-Generation Sequencing Data, Including the SARS-CoV-2. OMICS 2022, 26, 372–381. [Google Scholar] [CrossRef] [PubMed]
- González-Recio, O.; Gutiérrez-Rivas, M.; Peiró-Pastor, R.; Aguilera-Sepúlveda, P.; Cano-Gómez, C.; Jiménez-Clavero, M. Á.; Fernández-Pinero, J. Sequencing of SARS-CoV-2 Genome Using Different Nanopore Chemistries. Appl Microbiol Biotechnol 2021, 105, 3225–3234. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Dot-plot graph showing relationship between SARS-CoV-2 assembly success (NG50 value) and qRT-PCR Ct-value. Here we can see 3 highlighted zones – green zone under Ct-value < 25, where all samples were assembled into complete genome, grey zone between Ct-values of 25 and 30 where genomes were assembled completely, frag-mented or assembly failed. None of the samples over Ct-value 30 were assembled into SARS-CoV-2 genome.
Figure 1.
Dot-plot graph showing relationship between SARS-CoV-2 assembly success (NG50 value) and qRT-PCR Ct-value. Here we can see 3 highlighted zones – green zone under Ct-value < 25, where all samples were assembled into complete genome, grey zone between Ct-values of 25 and 30 where genomes were assembled completely, frag-mented or assembly failed. None of the samples over Ct-value 30 were assembled into SARS-CoV-2 genome.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



