
Submitted:
27 June 2023
Posted:
29 June 2023
You are already at the latest version
Abstract
The multitarget therapeutic strategy, as opposed to the more traditional ‘one disease-one tar-get-one drug’, may hold promise in treating multifactorial neurodegenerative syndromes, such as Alzheimer’s disease (AD) and related dementias. Recently, combining a photopharmacology approach with the multitarget-directed ligand (MTDL) design strategy, we disclosed a novel donepezil-like compound, namely 2-(4-((diethylamino)methyl)benzylidene)-5-methoxy- 2,3-dihydro-1H-inden-1-one (1a), which in the E diastereomeric form (and about tenfold less in the UV-B photo-induced isomer Z) showed the best activity as dual inhibitor of the AD-related targets acetylcholinesterase (AChE) and monoamine oxidase B (MAO-B). Herein, we investigated further photoisomerizable 2-benzylideneindan-1-one analogs 1b-h with the unconjugated tertiary amino moiety bearing alkyls of different bulkiness and lipophilicity. For each compound the thermal stable E diastereoisomer, along with the E/Z mixture as produced by UV-B light irradia-tion in the photostationary state (PSS, 75% Z), was investigated for the inhibition of human ChEs and MAOs. The pure E-diastereoisomer of the N-benzyl(ethyl)amino analog 1h achieved low nanomolar AChE and high nanomolar MAO-B inhibition potencies (IC50s 39 and 355 nM, re-spectively), whereas photoisomerization to the Z isomer (75% Z in the PSS mixture) resulted in a decrease (about 30%) of AChE inhibitory potency, and not in the MAO-B one. Molecular docking studies were performed to rationalize the different E/Z diastereoselectivity of 1h toward the two target enzymes.
Keywords:
Neurodegenerative diseases
; cholinesterase inhibitors
; monoaminoxidase inhibitors
; photo-pharmacology
1. Introduction
The incidence of neurodegenerative diseases in the ageing world population, particularly Alzheimer's disease (AD) and Parkinson's disease (PD), is influenced by the increase in average life expectancy and unregulated lifestyle, as highlighted in the recent World Alzheimer Reports.1 In the absence of disease-modifying drugs, AD and PD are considered incurable, and this serious health threat is expected to increase in the near future.2 By limiting oxidative stress and the associated inflammatory state, as well as by fine-tuning the concentration of neurotransmitters, it is possible to apply some palliative pharmacological therapies to slow down the progression of neurodegenerative diseases and control their symptoms. Among the various pharmacological targets identified, monoamine oxidases (MAOs) and cholinesterases (ChEs) are of particular interest.
MAOs are mitochondrial enzymes responsible for the oxidative catabolism of endogenous monoamines and xenobiotics. Their levels increase with age, resulting in increased production of reactive oxygen species (ROS) as by-products of the oxidation process, which ultimately leads to neuroinflammation and subsequent brain tissue damage. In humans, two enzyme isoforms, MAOs A and B, have been identified,3 both playing roles in the inactivation of endogenous monoaminergic neurotransmitters, albeit with different specificities. MAO-A is mainly responsible for the catabolism of norepinephrine, serotonin, and epinephrine, whereas MAO-B is more selective for tyramine and dopamine.4-5 Selective MAO-B inhibitors (MAO-B Is) showed neuroprotective effects, due to a reduction in ROS formation, and improved cognitive abilities in patients with neurodegenerative diseases by enhancing the level of monoamine neurotransmitters in the CNS.6 The MAO-B Is selegiline, safinamide and rasagiline, whose structure is incorporated into that of ladostigil (Figure 1), are used for the treatment of PD.7
Cholinesterase exists in two isoforms, acetylcholinesterase (AChE) and butyrylcholinesterase (BChE), which catalyze the hydrolysis of acetylcholine (ACh), that activates cholinergic synapses and receptors.8 The impairment of cholinergic neurotransmission is a downstream process in the cognitive deficit associated to AD. To date, cholinesterases inhibitors (ChE Is) are among the few approved drugs for the symptomatic treatment of AD (Figure 1), that are able to restore cortical cholinergic neurotransmission with consequent beneficial effects on cognition in both AD and PD-related dementia.9 Among them, rivastigmine, donepezil and galantamine are currently indicated in patients with mild-to-moderate AD.10,11
Due to the multifactorial nature of AD, as well as the frequent comorbidity of various neurodegenerative diseases, the design and optimization of multitarget-directed ligands (MTDLs) represents a noteworthy pharmaceutical development strategy.12-15 In particular, compounds targeting AChE and MAO-B could be useful tools for the treatment of neurodegenerative diseases. The first concrete result was achieved with ladostigil (Figure 1), a dual inhibitor of AChE (reversible) and MAO-B (irreversible), currently in clinical trials (NCT01354691) as anti-AD drug.16
Because of the extreme individual variability of the neurodegenerative processes during the different stages of the disease, the search for innovative pharmacological tools that can be used in precision medicine is a real challenge. Controlling the activity of a drug by appropriate external stimuli could represent an effective strategy in the development of “individualizable molecules”. Among the external stimuli, there is growing interest in light, which can affect a tissue with high spatial and temporal precision in a noninvasive and easily controlled manner.17
In the last decade, the concept of photopharmacology has been widely developed using bioactive molecules that can change their biological effect as a result of light irradiation.17 In an initial phase, the development of photoactive molecules was based on photolabile functional groups that irreversibly detached from the photodrug and released the bioactive molecule into the irradiated area. Later, with the advent of light-controlled molecular switches (notably azobenzene) and aimed at developing more adaptable molecules, photodrug design shifted to the conjugation of bioactive molecules with molecular photoswitches that could change their geometry in response to an appropriate light stimulus, and thus reversibly switch the pharmacological activity on or off.18-19,20
Several case studies have been proposed for molecules with photomodulable activity against targets such as ion channels,21-23 enzymes,24,25 and G-protein-coupled receptors (GPCRs).18,20,26 However, despite the crucial role of AChE and MAO-B in the neuronal network, which proceeds with extremely fast kinetics, there are few examples of photomodulable inhibitors for these enzymes.27,28
In this context, we have combined the expertise in the synthesis and characterization of biomimetic light-controlled molecular switches and motors,29-35 and in design of ChE-Is,36-38 to recently report a small series of photoisomerizable donepezil-like compounds with on-off inhibitory activity towards AChE and MAO-B.39 These molecules consist of a diversely decorated indanone moiety linked to an N-benzyl-N-ethylethanamine group via an isomerizable carbon-carbon double bond. Their irradiation leads to spatial repositioning of the key pharmacophore groups responsible for the interactions in the binding sites of the target enzymes.
Among these molecules, 1a (Figure 1), bearing the 5-methoxyindan-1-one moiety, showed the most interesting combination of photophysical and biological profile. The diastereoisomer E-1a achieved noteworthy inhibitory activity in the high nanomolar range against both enzymes (AChE IC50 = 0.113 μM, MAO-B IC50 = 0.260 μM), showing selectivity over the BChE and MAO-A isoforms of two orders of magnitude. Irradiation of E-1a with UV-B light produced an E/Z mixture composed by 75% of the Z-diastereoisomer in the photostationary state (PSS), and the inhibitory effect of Z-1a on both AChE and MAO-B decreased by one order of magnitude (AChE IC50 = 1.15 μM, MAO-B IC50 = 1.94 μM), demonstrating off-activity behavior.
Based on these findings, we extend herein the series of 5-methoxyindanone derivatives, by synthesizing new compounds in which the tertiary amino residue of 1a bears alkyls with different bulkiness and lipophilicity (Figure 1). Aim of this study is the investigation of the effect of the amino/ammonium head on the diastereoisomerism-dependent activity, by evaluating the difference in inhibitory capacity of the mixtures at the PSS, which mainly consist of the photoinduced diastereoisomer Z (75%), on AChE and MAO-B.
2. Design and Synthesis
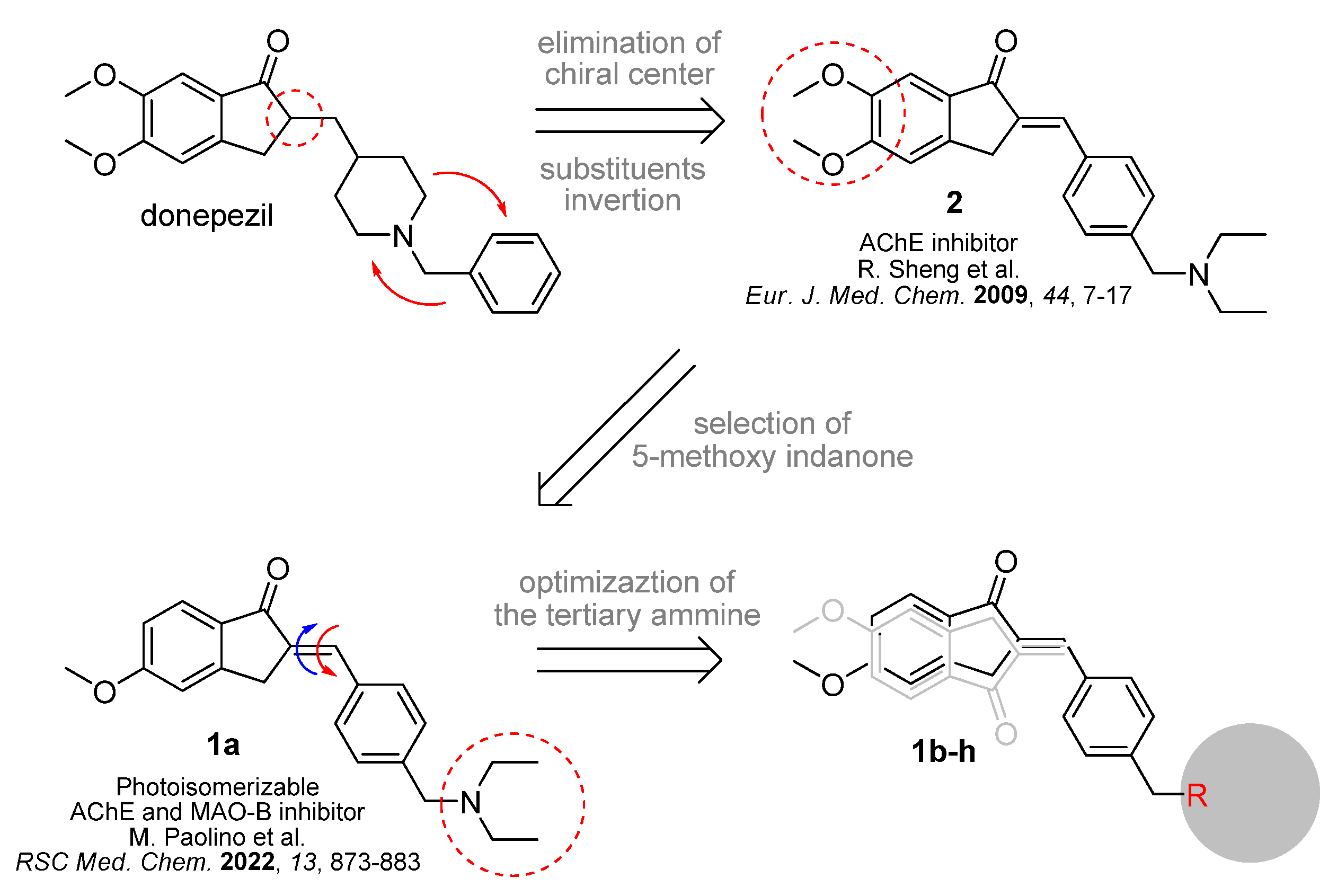
From a pharmacological point of view, the structure of parent compound 1a can be traced back to the well-known anti-Alzheimer drug donepezil. This molecule has a 5,6-methoxyindanone unit linked to an N-benzylpiperidine via a chiral center. By manipulating this structure, Sheng and coworkers discovered non-chiral compounds with AChE-inhibitory activity comparable to donepezil.40 Specifically, they replaced the chiral center with an exocyclic double bond and reversed the position of the benzyl ring with the basic center (e.g., compound 2, Figure 2). In our previous study, focusing on dual AChE/MAO-B inhibitors, the effect of methoxy groups on the interaction with the two enzymes was investigated. We found that removing the 6-OMe group of the indanone moiety resulted in a molecule with submicromolar inhibitory activity for both target enzymes.39 Thanks to the presence of the hemiindigoid chromophore,41,42 this molecule is able to reversibly photoisomerize by the application of UV light and spatially shift the functional groups responsible for the interaction in the binding sites of the two enzymes. Among the key pharmacophore features, the tertiary amino residue plays a first-rate role by interacting at the catalytic anionic site (CAS) site of AChE, anchoring the inhibitor in the enzyme gorge. Photoisomerization of the exocyclic double bond leads to the displacement of the indanone carbonyl and the 5-OMe group alters their interactions in the middle of the binding gorge and at the peripheral anionic site (PAS).
In this study, the ethyl residues on the tertiary amine were replaced by various alkyl chains (linear or branched) of different lipophilicity and bulkiness. These substitutions could have favorable or unfavorable effects on the interaction of the tertiary amine in the CAS region of the AChE gorge and, conversely, on other interactions in the middle gorge and in PAS. On the other hand, the increase of lipophilicity around the basic head could improve the inhibition potency against the MAO-B enzyme.
Compounds 1b-h were prepared by aldol condensation of 5-methoxyindan-1-one (3) with the corresponding aldehyde derivative 4b-h using KOH in methanol, as shown in Scheme 1A. In this reaction, only the E diastereoisomer was obtained for each compound, according to the assignment by 1H NMR analysis. In the 1H NMR spectra of compounds 1b-h, the vinyl proton “b” (see Scheme 1A for numbering) shows a chemical shift of 7.5 ppm, consistent with the deshielding effect of the carbonyl group, as already observed in the spectrum of 1a.
The aldehyde derivatives 4b-h were synthesized according to the reactions shown in Scheme 1B. Bromide derivative 5 was reacted with the corresponding secondary amine in DCM at refluxing temperature. The ester function of the resulting tertiary amines 6b-h was reduced with LiAlH4 in anhydrous THF to afford the primary alcohol derivatives 7b-h, which were oxidized to the corresponding aldehydes 4b-h with MnO2 in refluxing 1,4-dioxane.
3. Photophysical and Photochemical Characterization
The substitutions at the amino group in compounds 1b-h do not change the structure of the chromophore compared to their precursor 1a. The absorption spectra registered in methanol at concentrations of about 1×10-5 M (Figure 3) are indeed very similar to the UV-vis spectrum of 1a. For each compound, the absorption spectrum is dominated by an intense peak at about 334-340 nm (with an absorption tail up to about 390 nm) that largely overlaps with the emission spectrum from UV-B lamps. Therefore, this light emission was used for the photoisomerization of the new compounds in 1H NMR and UV-Vis absorption experiments.
Using 1H NMR spectroscopy, the formation of the Z diastereoisomer can be determined with confidence. Specifically, as previously done for 1a, methanol-d4 solutions of compounds 1b-e at a concentration of about 1 mM were irradiated with UV-B light in a Pyrex NMR tube and observed until the photostationary state (PSS) was reached.
The same procedure was followed for compounds 1f-h. However, due to the poor solubility in methanol at the concentrations required for 1H NMR analysis, 1f-h were irradiated and monitored in acetone-d6 solution. The diastereomeric E/Z ratio of the obtained PSS-1b-h was determined by calculating the area of the easily distinguishable signals assigned to the E and Z isomers (Figure 4).
In all experiments, the PSS resulting from UV-B irradiation consisted mainly of the Z diastereoisomer (75%) together with the E diastereoisomer (25%), confirming that the replacement of the nonconjugated amino group by the hemiindigoid chromophore did not affect the photochemical properties of the new compounds. All PSS-1b-h solutions were stored at room temperature in the dark (or even in visible ambient light) for 2 days and reexamined by 1H NMR. As with Z-1a, for which a half-life in MeOH at room temperature of 47 days was calculated, no significant change in E/Z composition was observed.
The effect of UV-B irradiation was also monitored by UV-Vis absorption spectroscopy (Figure 5). Methanolic solutions (1×10-5 M) of 1b-h at the PSS showed a drastic decrease in the absorption bands attributed to the E diastereomers and the appearance of new blue-shifted absorption bands attributed to the presence of the Z diastereoisomers, consistent with a decrease in π-π electron conjugation.43
4. Biological Evaluation
The pure E diastereoisomers 1b-h, along with E-1a retested in this study, and the related E/Z mixtures at the PSS (75% Z isomer) were evaluated for their inhibitory activity against AChE and BChE, and MAO-A and B. The inhibition data summarized in Table 1 show that all compounds, without any noteworthy structure-dependent difference, are selective toward AChE and MAO-B over BChE and MAO-A, respectively.
ChEs inhibitory activities were determined by applying the Ellman’s assay, with slight modifications.44,45 All the pure distreoisomers E-1a-h selectively inhibit AChE, with IC50 values falling in the submicromolar concentration range. Weaker inhibition of BChE was observed for all the compounds, with only E-1f-h reaching finite IC50s ≤ 10 μM. E-1h (IC50 = 39 nM) turned out almost equipotent with donepezil (IC50 = 21 nM), taken as positive control together with the dual AChE/BChE inhibitor tacrine. All the E/Z mixtures (75% Z) were found to be less active toward AChE than the corresponding pure E isomers, suggesting that the Z isomers have a lower binding affinity compared to the E isomers, as previously observed for the pure Z-1a.39
Pairwise comparison of the AChE inhibition data with the physicochemical features of the tertiary amino head would suggest that the hydrophobicity/bulkiness (not sharply distinguishable within this series) of the -NR2 may affect the AChE inhibitory potency of the examined compounds. However, the pure E isomers cover an IC50 range of just 1.4 log units, and with the exception of the most active (1h, R = -N(Et)Bn) and the least active (1d, R = -NnBu2) inhibitors, the activity data for the other six compounds span a range of only 0.5 log units. Nevertheless, within the limits of the biological and lipophilicity space explored, a nonlinear (likely parabolic) correlation trend (not shown) between -logIC50 and the calculated log P for the -NR2 fragment appears to hold for six of eight compounds. Two apparent outliers with respect to the parabolic relationship trend were the less-active-than-predicted 1f (likely, higher steric hindrance of iPr compared to nPr groups in 1c) and the more-active-than-predicted 1h (additional aromatic interactions of the Bn group compared to Et in 1a).
The mechanism of AChE inhibition by the most potent inhibitor E-1h was investigated. Lineweaver-Burk curves were generated with a fixed amount of AChE and substrate concentrations ranging between 25 and 300 mM, in the absence or presence of the inhibitor at different concentrations (ranging from 5 to 500 nM). The binding of E-1h to AChE changed both Vmax and Km values, a trend generally attributed to the mixed-type inhibition (Figure 6). The replot of the slopes versus the corresponding inhibitor concentrations provided a Ki value of 100 nM.
Taking into account our previous findings39 and literature data on structurally similar compounds,46,47 MAO-A/B inhibition was evaluated for pure E isomers and E/Z mixtures. All the compounds in the pure E form proved to be MAO-B-selective inhibitors with all submicromolar IC50 values. Only in two cases, namely E/Z 1c (R = -NnPr2) and 1f (R = -NiPr2), finite IC50 values around 3 μM were achieved against MAO-A, with the Z isomers being slightly more potent than the E isomers.
The IC50 values of MAO-B are in a narrow concentration range (just 0.6 log units), with no apparent lipophilicity-dependent effect. Even in the case of MAO-B inhibition, the Z isomers were found to be less potent than the E isomers, with the notable exception of 1h (R = -N(Et)Bn) whose E/Z mixture in the PSS (75% Z) achieving the same IC50 as the pure E-1h.
Combining the inhibition data on the two enzymes, it is interesting to note that the replacement of the -NEt2 group of compound 1a with a more hindered dissymmetrical substituent as in the case of compound 1e (R = -N(Me)Bu) led to an improvement of the MAO-B IC50 of the diastereoisomer E (IC50 E-1a = 0.260 μM vs E-1e = 0.178 μM) at the expense of the diastereoisomeric difference (IC50 PSS E/Z-1a = 1.59 μM vs PSS E/Z-1e 0.651 μM). In contrast, the inhibitory capacity of E-1e towards AChE remained almost unchanged with respect to 1a, while significantly increased the deactivating effect of light (IC50 PSS E/Z-1a = 0.78 μM vs PSS E/Z-1e 1.36 μM).
Regarding the potent AChE/MAO-B dual inhibitor 1h, the E isomer is slightly more potent than the Z isomer as AChE inhibitor, whereas no diastereoselectivity was observed for MAO-B inhibition.
5. Molecular Docking Analysis
To explain the observed inhibition data of the diastereomers E-1h and Z-1h against human AChE and MAO-B at the molecular level, docking calculations were performed on crystal structures of hAChE complexed with donepezil (PDB code: 4EY7) and hMAO-B in complex with safinamide (PDB code: 2V5Z). Considering the top-scored docking pose, both E-1h and Z-1h diastereoisomers accommodate in the gorge of the active site, in the middle of the PAS and the CAS.48
In particular, the indanone group of E-1h, in its top-scored pose (Figure 7), occupies the PAS by establishing a π–π interaction with W286 and the gorge through a backbone H-bond with F295 mediated by the carbonyl group; additionally, it interact inside the CAS forming a cation-π interaction which involves W86 and the protonated tertiary amino group. Otherwise, the 1h in Z geometry locates the indanone core away from the PAS and contacts the CAS through π–π interactions with W86; moreover, the Z-diastereoisomer engages the aromatic residues of the gorge, and, more specifically, Y341 and Y337 by π–π stacking and cation-π interactions, respectively. More importantly, the docking-based comparison of E and Z diasteroisomers show that the former best complies with the X-ray donepezil bioactive conformation (Figure 8). The indanone cores of donepezil and E-1h are well superimposed and both make interactions with F295 and W286. Furthermore, although lacking the piperidine nitrogen as donepezil which forms cation-π interaction with Y337, E-1h has a protonable benzylamino group which may engage double cation-π interactions with W86, as the cation-π interaction and π–π stacking found out in the AChE inhibitor drug. Noteworthy, the higher inhibitor potency of the E-1h diastereoisomer is in agreement with its higher docking score and ligand efficiency (LE) values.
As far as docking calculation on hMAO-B is concerned, the top-scored poses of both E-1h and Z-1h diastereoisomers shows the indanone moiety facing the FAD moiety (Figure 9). As experienced by the cognate ligand, a H-bond is established between the carbonyl backbone of I199 and the protonated amino as well as π–π stacking interactions.
Unlike safinamide, the indanone scaffold achieves π–π interaction with Y398 for both the E and Z diastereoisomers while the benzyl group the E diasteroisomer engages Y326, a MAO-B selective residue.49 In the case of the top-scored docking poses of 1h complexed with MAO-B, the Glide docking score and the calculated ligand efficiency slightly favors the Z isomer over the E isomer, likely agreeing with the experimental IC50 values, considering that even the PSS E/Z mixture containing 75% Z isomer showed a similar inhibition potency of the pure E isomer.
6. Materials and Methods
Synthesis. All chemicals used were of reagent grade. Yields refer to purified products and are not optimized. Merck silica gel 60 (230−400 mesh) was used for column chromatography. Merck TLC plates and silica gel 60 F254 were used for TLC. NMR spectra were obtained with a Bruker 400 AVANCE spectrometer in the indicated solvents. Melting points were determined in open capillaries in a Gallenkamp apparatus and are uncorrected. The chemical shifts are referenced to the residual not deuterated solvent signal (CHD2OD: δ (1H) = 3.31 ppm, δ (13C) = 49.86 ppm). The values of the chemical shifts are expressed in ppm and the coupling constants (J) in Hz. An Agilent 1100 LC/MSD operating with an electrospray source was used in mass spectrometry experiments. Purity of compounds 1b-h was assessed by RP-HPLC (Agilent 1100 series) and was found to be higher than 95%. A Zorbax Eclipse XDB-C8 column (4.6×150 mm, 5μm) was used in the HPLC analysis with methanol-H2O (0.1% formic acid) (80:20) as the mobile phase at a flow rate of 0.5 mL/min. UV detection was achieved at 280 nm. The absorption spectra were recorded with a PerkinElmer Lambda 900 in the indicated solvent. UV-B irradiations were conducted using a Multyrays chamber equipped with 2 GT15T8 Hg UV-B tube (2 x 15 Watt) in continuous rotation.
6.1. Chemistry
6.1.1. General Procedure for the Synthesis of Compounds 1b-h.
Compounds 1b-h were prepared by optimizing a previously reported procedure.40 To a solution 5-methoxy indanone (1 eq.) in methanol (5 mL/mmol), KOH (1 eq.) and the appropriate aldehyde 4b-h (1 eq.) were added. The resulting mixture was stirred at room temperature under nitrogen atmosphere for 2-6 h. Subsequently, the solvent was removed under reduced pressure to obtain a solid residue, which was dissolved in ethyl acetate and washed with brine. The organic phase was dried over anhydrous sodium sulfate, filtered, and concentrated under reduced pressure. The residue was purified by flash chromatography with the indicate solvent as the eluent to afford an off-white solid corresponding to the desired compound 1b-h as E diastereoisomer. NMR data of the Z diastereoisomers of each compound were derived after UV-B light irradiation of the corresponding E diastereoisomer until the PSS.
(E)-2-(4-((Dimethylamino)methyl)benzylidene)-5-methoxy-2,3-dihydro-1H-inden-1-one (E-1b).
Compound E-1b (0.058 g, yield 53%, m.p. 122-123 °C) was obtained as a pale yellow solid from aldehyde 4b (0.06 g, 0.36 mmol) after purification with a mixture of ethyl acetate/methanol (9:1) as the eluent. 1H NMR (400 MHz, CD3OD): 2.29 (s, 6H), 3.56 (s, 2H), 3.96 (s, 3H), 4.09 (s, 2H), 7.04 (dd, J = 8.6, 2.3, 1H), 7.20 (d, J = 2.0, 1H), 7.48 (d, J = 8.2, 2H), 7.58 (t, J = 2.0, 1H), 7.74 (d, J = 8.2, 2H), 7.80 (d, J = 8.6, 1H). 13C NMR (400 MHz, CD3OD): 32.0, 43.9, 55.0, 63.2, 109.4, 115.5, 125.5, 129.9, 130.4, 130.6, 132.3, 134.5, 135.4, 139.4, 153.6, 166.1, 193.6. MS(ESI): m/z 308.1 [M+H]+.
(Z)-2-(4-((Dimethylamino)methyl)benzylidene)-5-methoxy-2,3-dihydro-1H-inden-1-one (Z-1b).
1H NMR (400 MHz, CD3OD): 2.31 (s, 6H), 3.56 (s, 2H), 3.90 (s, 2H), 3.93 (s, 3H), 7.01 (dd, J = 8.6, 2.3, 1H), 7.08 (m, 2H), 7.37 (d, J = 8.2, 2H), 7.72 (d, J = 8.6, 1H), 8.07 (d, J = 8.2, 2H).
(E)-2-(4-((Dipropylamino)methyl)benzylidene)-5-methoxy-2,3-dihydro-1H-inden-1-one (E-1c).
Compound E-1c (0.12 g, yield 60%, m.p. 73.5-75.5 °C) was obtained as a pale yellow solid from aldehyde 4c (0.12 g, 0.55 mmol) after purification with a mixture of petroleum ether/ethyl acetate (1:1) as the eluent. 1H NMR (400 MHz, CD3OD): 0.90 (t, J = 7.4, 6H), 1.55 (m, 4H), 2.44 (t, J = 7.6, 4H), 3.64 (s, 2H), 3.94 (s, 3H), 4.05 (s, 2H), 7.02 (dd, J = 8.5, 2.2, 1H), 7.17 (d, J = 2.0, 1H), 7.48 (d, J = 8.2, 2H), 7.55 (t, J = 1.8, 1H), 7.69 (d, J = 8.2, 2H), 7.78 (d, J = 8.5, 1H). 13C NMR (100 MHz, CD3OD): 10.8, 19.5, 32.0, 55.0, 55.7, 58.0, 109.4, 115.5, 125.4, 129.4, 130.3, 130.6, 132.6, 134.0, 135.0, 141.4, 153.6, 166.0, 193.7. MS(ESI): m/z 364.2 [M+H]+.
(Z)-2-(4-((Dipropylamino)methyl)benzylidene)-5-methoxy-2,3-dihydro-1H-inden-1-one (Z-1c).
1H NMR (400 MHz, CD3OD): 0.91 (t, J = 7.4, 6H), 1.55 (m, 4H), 2.46 (t, J = 7.6, 4H), 3.65 (s, 2H), 3.90 (s, 2H), 3.93 (s, 3H), 7.01 (dd, J = 8.6, 2.2, 1H), 7.08 (m, 2H), 7.38 (d, J = 8.2, 2H), 7.72 (d, J = 8.7, 1H), 8.06 (d, J = 8.2, 2H).
(E)-2-(4-((Dibutylamino)methyl)benzylidene)-5-methoxy-2,3-dihydro-1H-inden-1-one (E-1d).
Compound E-1d (0.04 g, yield 64%, m.p. 74.5-75.5 °C) was obtained as a pale yellow solid from aldehyde 4d (0.04 g, 0.16 mmol) after purification with a mixture of petroleum ether/ethyl acetate (2:1) as the eluent. 1H NMR (400 MHz, CD3OD): 0.93 (t, J = 7.3, 6H), 1.34 (m, 4H), 1.52 (m, 4H), 2.48 (t, J = 7.5, 4H), 3.64 (s, 2H), 3.95 (s, 3H), 4.08 (s, 2H), 7.03 (dd, J = 8.5, 2.2, 1H), 7.19 (d, J = 1.9, 1H), 7.48 (d, J = 8.1, 2H), 7.56 (s, 1H), 7.70 (d, J = 8.2, 2H), 7.79 (d, J = 8.6, 1H). 13C NMR (400 MHz, CD3OD): 13.0, 20.3, 28.6, 32.0, 53.3, 55.0, 58.0, 109.4, 115.5, 125.4, 129.5, 130.3, 130.67, 132.6, 134.0, 135.0, 143.6, 153.6, 166.0, 193.7. MS(ESI): m/z 392.2 [M+H]+.
(Z)-2-(4-((Dibutylamino)methyl)benzylidene)-5-methoxy-2,3-dihydro-1H-inden-1-one (Z-1d).
1H NMR (400 MHz, CD3OD): 2.31 (s, 6H), 3.56 (s, 2H), 3.90 (s, 2H), 3.93 (s, 3H), 7.01 (dd, J = 8.6, 2.3, 1H), 7.08 (m, 2H), 7.37 (d, J = 8.2, 2H), 7.72 (d, J = 8.6, 1H), 8.07 (d, J = 8.2, 2H).
(E)-2-(4-((Butyl(methyl)amino)methyl)benzylidene)-5-methoxy-2,3-dihydro-1H-inden-1-one (E-1e).
Compound E-1e (0.071 g, yield 79%, m.p. 62.4-63.0 °C) was obtained as a pale yellow solid from aldehyde 4e (0.05 g, 0.26 mmol) after purification with a mixture of petroleum ether/ethyl acetate (1:1) as the eluent. 1H NMR (400 MHz, CD3OD): 0.95 (t, J = 7.4, 3H), 1.35 (m, 2H), 1.56 (m, 2H), 2.24 (s, 3H), 2.43 (t, J = 7.8, 2H), 3.58 (s, 2H), 3.95 (s, 3H), 4.06 (s, 2H), 7.03 (dd, J = 8.6, 2.2, 1H), 7.18 (d, J = 2.0, 1H), 7.47 (d, J = 8.2, 2H), 7.55 (t, J = 1.9, 1H), 7.71 (d, J = 8.2, 2H), 7.78 (d, J = 8.6, 1H). 13C NMR (100 MHz, CD3OD): 12.9, 20.3, 28.8, 32.0, 41.1, 55.0, 56.9, 61.4, 109.4, 115.5, 125.5, 129.8, 130.4, 130.6, 132.4, 134.3, 135.3, 140.0, 153.6, 166.1, 193.6. MS(ESI): m/z 350.2 [M+H]+.
(Z)-2-(4-((Butyl(methyl)amino)methyl)benzylidene)-5-methoxy-2,3-dihydro-1H-inden-1-one (Z-1e).
1H NMR (400 MHz, CD3OD): 0.95 (t, J = 7.4, 3H), 1.36 (m, 2H), 1.57 (m, 2H), 2.25 (s, 3H), 2.44 (t, J = 7.8, 2H), 3.59 (s, 2H), 3.90 (s, 2H), 3.93 (s, 3H), 7.01 (dd, J = 8.6, 2.3, 1H), 7.08 (m, 2H), 7.37 (d, J = 8.2, 2H), 7.72 (d, J = 8.6, 1H), 8.07 (d, J = 8.2, 2H).
(E)-2-(4-((Diisopropylamino)methyl)benzylidene)-5-methoxy-2,3-dihydro-1H-inden-1-one (E-1f).
Compound E-1f (0.022 g, yield 45%, m.p. 74.5-75.5 °C) was obtained as a off-white solid from aldehyde 4f (0.03 g, 0.14 mmol) after purification with a mixture of petroleum ether/ethyl acetate (8:2) as the eluent. 1H NMR (400 MHz, (CD3)2CO): 1.07 (d, J = 6.6, 12H), 3.07 (m, 2H), 3.75 (s, 2H), 3.96 (s, 3H), 4.09 (s, 2H), 7.04 (dd, J = 8.5, 2.2, 1H), 7.20 (d, J = 1.9, 1H), 7.50 (t, J = 2.0, 1H), 7.54 (d, J = 8.2, 2H), 7.71 (d, J = 8.2, 2H), 7.75 (d, J = 8.5, 1H). 1H NMR (400 MHz, (CD3)2SO): 1.01 (d, J = 6.6 Hz, 12H), 3.04 – 2.87 (m, 2H), 3.67 (s, 2H), 3.90 (s, 3H), 4.07 (s, 2H), 7.04 (dd, J = 8.5, 2.2 Hz, 1H), 7.19 (d, J = 1.9 Hz, 1H), 7.44 (s, 1H), 7.48 (d, J = 8.1 Hz, 1H), 7.69 (d, J = 8.2 Hz, 1H), 7.72 (t, J = 6.0 Hz, 1H). 13C NMR (100 MHz, (CD3)2SO): 21.1, 32.5, 48.1, 48.7, 56.3, 110.7, 115.9, 125.9, 128.6, 130.9, 131.1, 132.1, 133.6, 135.2, 145.7, 153.4, 165.4, 192.1. MS(ESI): m/z 364.2 [M+H]+.
(Z)-2-(4-((Diisopropylamino)methyl)benzylidene)-5-methoxy-2,3-dihydro-1H-inden-1-one (Z-1f).
1H NMR (400 MHz, (CD3)2CO): 1.07 (d, J = 6.6, 12H), 3.07 (m, 2H), 3.73 (s, 2H), 3.91 (s, 2H), 3.95 (s, 3H), 6.99-7.06 (m, 2H), 7.10 (d, J = 1.9, 1H), 7.44 (d, J = 8.2, 2H), 7.71 (d, J = 8.6, 1H), 8.19 (d, J = 8.2, 2H).
(E)-2-(4-((Benzyl(methyl)amino)methyl)benzylidene)-5-methoxy-2,3-dihydro-1H-inden-1-one (E-1g).
Compound E-1g (0.055 g, yield 62%, m.p. 126.5-127.5 °C) was obtained as a off-white solid from aldehyde 4g (0.06 g, 0.23 mmol) after purification with a mixture of petroleum ether/ethyl acetate (1:1) as the eluent. 1H NMR (400 MHz, (CD3)2CO): 2.19 (s, 3H), 3.58 (s, 2H), 3.60 (s, 2H), 3.96 (s, 3H), 4.10 (s, 2H), 7.04 (dd, J = 8.5, 2.2, 1H), 7.20 (d, J = 1.9 1H), 7.27 (t, J = 7.3, 1H), 7.35 (t, J = 7.5, 2H), 7.43 (d, J = 7.4, 2H), 7.50 (t, J = 2.0, 1H), 7.55 (d, J = 8.1, 2H), 7.75 (d, J = 8.2, 3H). 1H NMR (400 MHz, (CD3)2SO): 2.11 (s, 3H), 3.53 (s, 2H), 3.56 (s, 2H), 3.90 (s, 3H), 4.08 (s, 2H), 7.04 (dd, J = 8.5, 1.9 Hz, 1H), 7.19 (s, 1H), 7.26 (t, J = 6.5 Hz, 1H), 7.42 – 7.30 (m, 4H), 7.45 (s, 1H), 7.49 (d, J = 8.0 Hz, 2H), 7.74 (m, 3H). 13C NMR (100MHz, (CD3)2SO): 32.5, 42.2, 56.3, 61.1, 61.5, 110.7, 115.9, 125.9, 127.4, 128.7, 129.1, 129.6, 131.1, 131.8, 133.4, 134.2, 135.7, 139.5, 141.5, 153.4, 165.4, 192.1. MS(ESI): m/z 384.2 [M+H]+.
(Z)-2-(4-((Benzyl(methyl)amino)methyl)benzylidene)-5-methoxy-2,3-dihydro-1H-inden-1-one (Z-1g).
1H NMR (400 MHz, (CD3)2CO): 2.18 (s, 3H), 3.57 (s, 2H), 3.59 (s, 2H), 3.91 (s, 2H), 3.94 (s, 3H), 7.02 (dd, J = 8.5, 2.2, 1H), 7.04 (s, 1H), 7.10 (s, 1H), 7.26 (t, J = 7.3, 1H), 7.35 (t, J = 7.5, 2H), 7.44 (m, 4H), 7.70 (d, J = 8.5, 1H), 8.22 (d, J = 8.2, 2H).
(E)-2-(4-((Benzyl(ethyl)amino)methyl)benzylidene)-5-methoxy-2,3-dihydro-1H-inden-1-one (E-1h).
Compound E-1h (0.06 g, yield 50%, m.p. 118.5-119.5 °C) was obtained as a off-white solid from aldehyde 4h (0.07 g, 0.28 mmol) after purification with a mixture of petroleum ether/ethyl acetate (8:2) as the eluent. 1H NMR (400 MHz, ((CD3)2CO): 1.10 (t, J = 7.1, 3H), 2.53 (q, J = 7.1, 2H), 3.62 (s, 2H), 3.64 (s, 2H), 3.96 (s, 3H), 4.09 (s, 2H), 7.04 (dd, J = 8.5, 2.1, 1H), 7.19 (d, J = 2.0, 1H), 7.25 (t, J = 7.3, 1H), 7.34 (t, J = 7.5, 2H), 7.44 (d, J = 7.4, 2H), 7.49 (s, 1H), 7.55 (d, J = 8.1, 2H), 7.73 (d, J = 8.1, 2H), 7.75 (d, J = 8.2, 1H). 1H NMR (400 MHz, ((CD3)2SO): 1.03 (t, J = 7.1 Hz, 3H), 2.45 (q, J = 7.1 Hz, 2H), 3.57 (s, 2H), 3.60 (s, 2H), 3.90 (s, 3H), 4.08 (s, 2H), 7.04 (dd, J = 8.5, 2.3 Hz, 1H), 7.19 (d, J = 2.0 Hz, 1H), 7.25 (ddd, J = 8.5, 3.0, 1.5 Hz, 1H), 7.31-7.41 (m, 4H), 7.45 (s, 1H), 7.49 (d, J = 8.1 Hz, 2H), 7.73 (d, J = 8.4 Hz, 3H). 13C NMR (400 MHz, (CD3)2SO): 12.1, 32.5, 47.0, 56.3, 57.2, 57.5, 110.7, 115.9, 125.9, 127.3, 128.7, 128.9, 129.4, 131.0, 131.1, 131.9, 134.1, 135.6, 140.0, 142.2, 153.4, 165.4, 192.1. MS(ESI): m/z 398.2 [M+H]+.
(Z)-2-(4-((Benzyl(ethyl)amino)methyl)benzylidene)-5-methoxy-2,3-dihydro-1H-inden-1-one (Z-1h).
1H NMR (400 MHz, (CD3)2CO): 1.10 (t, J = 7.1, 3H), 2.52 (q, J = 7.1, 2H), 3.61 (s, 2H), 3.63 (s, 2H), 3.91 (s, 2H), 3.94 (s, 3H), 6.99-7.04 (m, 1H), 7.10 (d, J = 2.0, 1H), 7.24 (t, J = 7.3, 1H), 7.34 (t, J = 7.5, 2H), 7.45 (m, 3H), 7.70 (d, J = 8.5, 1H), 8.21 (d, J = 8.2, 2H).
6.1.2. General Procedure for the Synthesis of Compounds 6b-h.
To a solution of ester 5 (1 equivalent) in dichloromethane (10 mL/mmol), the appropriate secondary ammine (3 equivalents) was added. The mixture was stirred at refluxed temperature for 4-5 h. Then, the solvent was removed under reduced pressure and the resulting oily residue was diluted with dichloromethane and washed with water. The organic phase was dried over anhydrous sodium sulfate, filtered, and concentrated under reduced pressure. The resulting oily residue was purified by flash chromatography using the indicated solvent as the eluent to obtain the desired tertiary ammine derivatives 6b-h.
Methyl 4-((dimethylamino)methyl)benzoate (6b)40
Compound 6b was obtained as a yellow oil (0.42 g, yield 98%) by using a 1 M solution of dimethyl ammine in THF (6.6 mL, 6.60 mmol) after purification with a mixture of petroleum ether/ethyl acetate (1:1) as the eluent. 1H NMR (400 MHz, CD3OD): 2.28 (s, 6H), 3.57 (s, 2H), 3.92 (s, 3H), 7.46 (d, J = 8.4, 2H), 8.01 (d, J = 8.4, 2H). MS(ESI): m/z 194.1 [M+H]+.
Methyl 4-((dipropylamino)methyl)benzoate (6c)50
Compound 6c was obtained as a yellow oil (0.54 g, yield 99%) by using dipropyl ammine (0.9 mL, 6.51 mmol) after purification with a mixture of petroleum ether/ethyl acetate (8:2) as the eluent. 1H NMR (400 MHz, CD3OD): 0.89 (t, J = 7.4, 6H), 1.53 (m, 4H), 2.43 (t, J = 7.5, 4H), 3.67 (s, 2H), 3.92 (s, 3H), 7.48 (d, J = 8.3, 2H), 7.98 (d, J = 8.3, 2H). MS(ESI): m/z 250.1 [M+H]+.
Methyl 4-((dibutylamino)methyl)benzoate (6d)51
Compound 6d was obtained as a yellow oil (0.59 g, yield 94%) by using dibutyl ammine (1.1 mL, 6.81 mmol) after purification with a mixture of petroleum ether/ethyl acetate (9:1) as the eluent. 1H NMR (400 MHz, CD3OD): 0.91 (t, J = 7.3, 6H), 1.33 (m, 4H), 1.49 (m, 4H), 2.45 (t, J = 7.5, 4H), 3.64 (s, 2H), 3.91 (s, 3H), 7.47 (d, J = 8.3, 2H), 7.98 (d, J = 8.3, 2H). MS(ESI): m/z 278.2 [M+H]+.
Methyl 4-((butyl(methyl)amino)methyl)benzoate (6e).
Compound 6e was obtained as a yellow oil (0.51 g, yield 99%) by using N-methylbutan-1-amine (0.8 mL, 6.50 mmol) after purification with a mixture of petroleum ether/ethyl acetate (1:1) as the eluent. 1H NMR (400 MHz, CD3OD): 0.94 (t, J = 7.4, 3H), 1.35 (m, 2H), 1.55 (m, 2H), 2.22 (s, 3H), 2.41 (t, J = 7.7, 2H), 3.59 (s, 2H), 3.92 (s, 3H), 7.46 (d, J = 8.1, 2H), 8.00 (d, J = 8.2, 2H). MS(ESI): m/z 236.1 [M+H]+.
Methyl 4-((diisopropylamino)methyl)benzoate (6f)52
Compound 6f was obtained as a yellow oil (0.33 g, yield 60%) by using diisopropyl ammine (0.9 mL, 6.53 mmol) after purification with petroleum ether as the eluent. 1H NMR (400 MHz, CD3OD): 1.08 (d, J = 6.6, 12H), 3.07 (m, 2H), 3.75 (s, 2H), 3.91 (s, 3H), 7.51 (d, J = 8.6, 2H), 7.94 (d, J = 8.4, 2H). MS(ESI): m/z 250.1 [M+H]+.
Methyl 4-((benzyl(methyl)amino)methyl)benzoate (6g)53
Compound 6g was obtained as a yellow oil (0.58 g, yield 99%) by using N-methyl-1-phenylmethanamine (0.8 mL, 6.54 mmol) after purification with a mixture of petroleum ether/ethyl acetate (9:1) as the eluent. 1H NMR (400 MHz, CD3OD): 2.19 (s, 3H), 3.56 (s, 2H), 3.60 (s, 2H), 3.91 (s, 3H), 7.27 (m, 1H), 7.35 (m, 4H), 7.50 (d, J = 8.4, 2H), 8.00 (d, J = 8.3, 2H). MS(ESI): m/z 270.1 [M+H]+.
Methyl 4-((benzyl(ethyl)amino)methyl)benzoate (6h).
Compound 6h was obtained as a yellow oil (0.61 g, yield 99%) by using N-ethyl-1-phenylmethanamine (1.0 mL, 6.60 mmol) after purification with a mixture of petroleum ether/ethyl acetate (9:1) as the eluent. 1H NMR (400 MHz, CD3OD): 1.09 (t, J = 7.1, 3H), 2.50 (q, J = 7.1, 2H), 3.57 (s, 2H), 3.62 (s, 2H), 3.90 (s, 3H), 7.23 (t, J = 7.2, 1H), 7.31 (t, J = 7.3, 2H), 7.37 (d, J = 7.9, 2H), 7.48 (d, J = 8.1, 2H), 7.96 (d, J = 8.3, 2H). MS(ESI): m/z 284.1 [M+H]+.
6.1.3. General Procedure for the Synthesis of the Alcohol Derivatives 7b-h.
To a solution of the appropriate ester 6b-h (1 equivalent) in dry THF (5 mL/mmol), a 1 M solution of LiAlH4 in THF (1 equivalent) was added. The reaction mixture was stirred at room temperature under nitrogen atmosphere for 12 h and, subsequently was concentrated under reduced pressure. The oily residue was dissolved in dichloromethane and then washed with water. The organic phase was dried over anhydrous sodium sulfate, filtered, and concentrated under reduced pressure. The resulting oily residue was purified by flash chromatography using the indicated eluent to obtain the desired alcohol derivatives 7b-h.
(4-((Dimethylamino)methyl)phenyl)methanol (7b)54
Compound 7b was obtained as a yellow oil (0.37 g, yield 82%) starting from ester 6b (0.52 g, 2.71 mmol) after purification with ethyl acetate as the eluent. 1H NMR (400 MHz, CD3OD): 2.26 (s, 6H), 3.49 (s, 2H), 4.63 (s, 2H), 7.34 (m, 4H). MS(ESI): m/z 194.1 [M+H]+.
(4-((Dipropylamino)methyl)phenyl)methanol (7c).
Compound 7c was obtained as a yellow oil (0.29 g, yield 67%) starting from ester 6c (0.49 g, 1.98 mmol) after purification with a mixture of petroleum ether/ethyl acetate (8:2) as the eluent. 1H NMR (400 MHz, CD3OD): 0.88 (t, J = 7.4, 6H), 1.53 (m, 4H), 2.41 (t, J = 7.6, 4H), 3.59 (s, 2H), 4.60 (s, 2H), 7.32 (m, 4H). MS(ESI): m/z 222.2 [M+H]+.
(4-((Dibutylamino)methyl)phenyl)methanol (7d).
Compound 7d was obtained as a yellow oil (0.16 g, yield 33%) starting from ester 6d (0.55 g, 1.98 mmol) after purification with a mixture of petroleum ether/ethyl acetate (8:2) as the eluent. 1H NMR (400 MHz, CD3OD): 0.92 (t, J = 7.3, 6H), 1.31 (m, 4H), 1.50 (m, 4H), 2.44 (t, J = 7.6, 4H), 3.59 (s, 2H), 4.61 (s, 2H), 7.32 (m, 4H). MS(ESI): m/z 250.2 [M+H]+.
(4-((Butyl(methyl)amino)methyl)phenyl)methanol (7e).
Compound 7e was obtained as a yellow oil (0.40 g, yield 83%) starting from ester 6e (0.55 g, 2.35 mmol) after purification with a mixture of petroleum ether/ethyl acetate (8:2) as the eluent. 1H NMR (400 MHz, CD3OD): 0.94 (t, J = 7.4, 3H), 1.34 (m, 2H), 1.54 (m, 2H), 2.21 (s, 3H), 2.40 (t, J = 7.8, 2H), 3.54 (s, 2H), 4.62 (s, 2H), 7.34 (m, 4H). MS(ESI): m/z 208.1 [M+H]+.
(4-((Diisopropylamino)methyl)phenyl)methanol (7f).
Compound 7f was obtained as a yellow oil (0.09 g, yield 31%) starting from ester 6f (0.33 g, 1.32 mmol) after purification with a mixture of petroleum ether/ethyl acetate (9:1) as the eluent. 1H NMR (400 MHz, CD3OD): 1.06 (d, J = 6.6, 12H), 3.05 (m, 2H), 3.67 (s, 2H), 4.58 (s, 2H), 7.28 (d, J = 8.1, 2H), 7.36 (d, J = 8.2, 2H). MS(ESI): m/z 222.1 [M+H]+.
(4-((Benzyl(methyl)amino)methyl)phenyl)methanol (7g).
Compound 7g was obtained as a yellow oil (0.38 g, yield 80%) starting from ester 6g (0.53 g, 1.98 mmol) after purification with a mixture of petroleum ether/ethyl acetate (8:2) as the eluent. 1H NMR (400 MHz, CD3OD): 2.18 (s, 3H), 3.53 (s, 2H), 3.54 (s, 2H), 4.61 (s, 2H), 7.28-7.38 (m, 9H). MS(ESI): m/z 242.1 [M+H]+.
(4-((Benzyl(ethyl)amino)methyl)phenyl)methanol (7h).
Compound 7h was obtained as a yellow oil (0.49 g, yield 98%) starting from ester 6h (0.56 g, 1.98 mmol) after purification with a mixture of petroleum ether/ethyl acetate (8:2) as the eluent. 1H NMR (400 MHz, CD3OD): 1.09 (t, J = 7.1, 3H), 2.50 (q, J = 7.1, 2H), 3.56 (s, 2H), 3.57 (s, 2H), 4.60 (s, 2H), 7.23 (t, J = 7.1, 1H), 7.33 (m, 8H). MS(ESI): m/z 256.2 [M+H]+.
6.1.4. General Procedure for the Synthesis of Aldehyde Derivatives 4b-h.
To a solution of the appropriate alcohol derivative 7b-h (1 equivalent) in 1,4-dioxane (3 mL/mmol), MnO2 (10 equivalents) was added. The reaction mixture was stirred at reflux temperature for 2-4 h and, subsequently, was filtered on celite. The filtrate was concentrated under reduced pressure and the resulting residue was purified by flash chromatography with the indicated solvent to give the desired aldehyde derivatives 4b-h.
4-((Dimethylamino)methyl)benzaldehyde (4b)54
Compound 4b was obtained as a pale yellow oil (0.09 g, yield 24%) starting from alcohol derivative 7b (0.37 g, 2.22 mmol) and was purified using ethyl acetate as the eluent. 1H NMR (400 MHz, CD3OD): 2.26 (s, 6H), 3.58 (s, 2H), 7.56 (d, J = 8.1, 2H), 7.91 (d, J = 8.2, 2H), 10.0 (s, 1H). MS(ESI): m/z 164.1 [M+H]+.
4-((Dipropylamino)methyl)benzaldehyde (4c)
Compound 4c was obtained as a pale yellow oil (0.15 g, yield 62%) starting from alcohol derivative 7c (0.25 g, 1.12 mmol) and was purified using a mixture of petroleum ether/ethyl acetate (1:1) as the eluent. 1H NMR (400 MHz, CD3OD): 0.90 (t, J = 7.4, 6H), 1.54 (m, 4H), 2.43 (t, J = 7.5, 4H), 3.69 (s, 2H), 7.58 (d, J = 8.1, 2H), 7.88 (d, J = 8.2, 2H), 9.98 (s, 1H). MS(ESI): m/z 220.1 [M+H]+.
4-((Dibutylamino)methyl)benzaldehyde (4d)
Compound 4d was obtained as a pale yellow oil (0.04 g, yield 36%) starting from alcohol derivative 7d (0.12 g, 0.48 mmol) and was purified using a mixture of petroleum ether/ethyl acetate (2:1) as the eluent. 1H NMR (400 MHz, CD3OD): 0.91 (t, J = 7.3, 6H), 1.32 (m, 4H), 1.50 (m, 4H), 2.46 (t, J = 7.4, 4H), 3.67 (s, 2H), 7.58 (d, J = 8.1, 2H), 7.88 (d, J = 8.2, 2H), 9.98 (s, 1H). MS(ESI): m/z 248.2 [M+H]+.
4-((Butyl(methyl)amino)methyl)benzaldehyde (4e)
Compound 4e was obtained as a pale yellow oil (0.78 g, yield 76%) starting from alcohol derivative 7e (1.02 g, 5.02 mmol) and was purified using a mixture of petroleum ether/ethyl acetate (8:2) as the eluent. 1H NMR (400 MHz, CD3OD): 0.94 (t, J = 7.4, 3H), 1.36 (m, 2H), 1.55 (m, 2H), 2.23 (s, 3H), 2.42 (t, J = 7.6, 2H), 3.61 (s, 2H), 7.57 (d, J = 8.0, 2H), 7.90 (d, J = 8.2, 2H), 10.00 (s, 1H). MS(ESI): m/z 206.1 [M+H]+.
4-((Diisopropylamino)methyl)benzaldehyde (4f)55
Compound 4f was obtained as a pale yellow oil (0.03 g, yield 34%) starting from alcohol derivative 7f (0.09 g, 0.41 mmol) and was purified using a mixture of petroleum ether/ethyl acetate (8:2) as the eluent. 1H NMR (400 MHz, CD3OD): 1.07 (d, J = 6.6, 12H), 3.06 (m, 2H), 3.78 (s, 2H), 7.62 (d, J = 8.1, 2H), 7.85 (d, J = 8.2, 2H), 9.96 (s, 1H). MS(ESI): m/z 220.1 [M+H]+.
4-((Benzyl(methyl)amino)methyl)benzaldehyde (4g)56
Compound 4g was obtained as a pale yellow oil (0.13 g, yield 38%) starting from alcohol derivative 7g (0.34 g, 1.41 mmol) and was purified using a mixture of petroleum ether/ethyl acetate (7:3) as the eluent. 1H NMR (400 MHz, CD3OD): 2.21 (s, 3H), 3.57 (s, 2H), 3.63 (s, 2H), 7.27 (t, J = 7.0, 1H), 7.31-7.42 (m, 4H), 7.60 (d, J = 8.0, 2H), 7.90 (d, J = 8.2, 2H), 9.99 (s, 1H). MS(ESI): m/z 240.1 [M+H]+.
4-((Benzyl(ethyl)amino)methyl)benzaldehyde (4h)56
Compound 4h was obtained as a pale yellow oil (0.07 g, yield 55%) starting from alcohol derivative 7h (0.17 g, 0.87 mmol) and was purified using a mixture of petroleum ether/ethyl acetate (8:2) as the eluent. 1H NMR (400 MHz, CD3OD): 1.11 (t, J = 7.1, 3H), 2.53 (q, J = 7.1, 2H), 3.60 (s, 2H), 3.67 (s, 2H), 7.24 (t, J = 7.2, 1H), 7.35 (t, J = 7.5, 2H), 7.38 (m, 2H), 7.61 (d, J = 8.1, 2H), 7.88 (d, J = 8.2, 2H), 9.97 (s, 1H). MS(ESI): m/z 254.1 [M+H]+.
6.2. Inhibition of Cholinesterases and Monoamine Oxidases
Human isoforms of ChEs (human recombinant AChE and BChE from human serum) and MAOs (from baculovirus-infected insect cells), purchased from Sigma Aldrich (Milan, Italy), were used for inhibition assays. Experiments were performed in 96-well plates (Greiner Bio-One, Kremsmünster, Austria) on the Infinite M1000 Pro plate reader (Tecan, Cernusco s.N., Italy), using already published protocols.57,58,59,-60 Inhibition data and constants (IC50s and Kis) were calculated with Prism (version 5.01 for Windows; GraphPad Software, San Diego, CA, USA).
The inhibition of human recombinant AChE or BChE from human serum was determined by applying Ellman’s spectrophotometric method as described in previously reported protocols.58,59, The AChE activity was determined in an assay solution containing AChE (0.09 U/mL), 5,50-dithiobis(2-nitrobenzoic acid) (i.e., the Ellman’s reagent, 0.33 mM), the test compound (10 μM concentration, or seven scalar concentrations for compounds achieving > 60% enzyme inhibition at 10 μM), in 0.1 M PBS pH 8.0. After 20 min incubation at 25 °C, the substrate acetylthiocholine iodide (5 μM) was added, and its hydrolysis rates were monitored for 5.0 min at 412 nm. The BChE inhibitory activity was similarly determined by using BChE (0.09 U/mL) and butyrylthiocholine iodide (5 μM) as the substrate. IC50 value, determined by the nonlinear regression method ‘log[inhibitor] vs. response’, or the % inhibition at 10 μM, is expressed as the mean ± SD of three independent measurements, each one performed in duplicate. The IC50 values, Michaelis–Menten curve fitting and inhibition constant (Ki) were calculated by nonlinear regression, using Prism software.
In MAOs’ inhibition assays, each test compound, at 10 μM concentration, was preincubated for 20 min at 37 °C with 50 μM kynuramine as the substrate in 0.1 M phosphate buffer solution (PBS) pH 8.0 made 0.39 osmolar with KCl. After the addition of human recombinant MAO A (250 U/mg) or MAO B (59 U/mg) and a further 30 min of incubation, NaOH was added, and the fluorescence read at 310/400 nm excitation/emission wavelength. For compounds achieving at least 60% inhibition of MAO at 10 μM concentration, seven scalar concentrations of each inhibitor were tested and the concentration producing 50% inhibition of the MAO activity (IC50) was calculated by nonlinear regression. IC50 is expressed as mean ± SD of three independent measurements, each one performed in duplicate. For the kinetic study on the inhibition mechanism of MAO B, three diverse scalar concentrations of the inhibitor and seven concentrations of kynuramine were used.
6.3. Molecular Docking Calculations
The three-dimensional (3D) structures of hAChE complexed with donepezil (PDB ID: 4EY7) and hMAO-B in complex with safinamide (PDB ID: 2V5Z) were taken from the Protein Data Bank. The protein preparation wizard available in the Schrödinger Suite (Schrödinger Release 2022-4) was employed to optimize X-ray crystal structures: missing side chains have been reconstructed and protonation states at pH = 7.4 ± 0.0 have been predicted. Finally, energy minimization on the crystal structures were applied using OPLS_2005 force field (Banks et al., 2005). Starting from SMILES annotations (Weininger, 1988), 3D structures of E-1h and Z-1h diastereoisomers have been generated and their protonation states at pH = 7.4 ± 0.0 were computed by LigPrep tool of Schrödinger Suite using OPLS_2005 force field. The same steps have been reiterated for hAchE and hMAO-B cognate ligands (i.e., donepezil and safinamide, respectively). Glide v9.1 (Schrödinger Release 2022-4: Glide, Schrödinger, LLC, New York, NY, 2022) was adopted to perform docking simulations upon both targets applying OPLS_2005 force field. Docking calibration on cognate ligands was carried out in order to find out the most suitable docking protocol for each target: the Root Mean Square Deviation (RMSD) on heavy atoms between the docking pose and the X-ray coordinates of the cognate ligand was computed. In this respect, a cubic grid box of 15 Å for AchE (RMSD = 0.372) and a cubic grid box of 12 Å for MAO-B (RMSD = 0.467), both placed in the center of mass of the X-ray cognate ligands, were used to perform docking simulations. Standard Precision (SP) docking precision and OPLS_2005 force field were applied. The top-ranked docking poses of E-1h and Z-1h diastereoisomers were chosen to examinate their target interactions and to compare them with the cognate ligands. All pictures have been made with Pymol (Schrodinger, LLC. 2010. The PyMOL Molecular Graphics System, Version 2.4.0).
7. Conclusions
Investigating the AChE/MAO-B dual inhibitory activity of some newly synthesized donepezil-like 2-benzylideneindan-1-one derivatives, which differ for bulkiness/lipophilicity of the unconjugated tertiary amino head, we gathered new clues about the photoswitchable E/Z isomerization controlling inhibitors’ binding to the two AD-related target enzymes. Confirming our previous study,39 all the new compounds were obtained exclusively as E diastereoisomers. Thanks to the photoswitching properties of the 2-benzylideneindan-1-one chromophore, Z diastereoisomers were generated in photostationary mixtures (75% of Z in all the examined cases) by exposure to UV-B radiation. The pure E-diastereoisomers showed submicromolar IC50 values on human AChE, in most cases with a good selectivity over the BChE isoform. Interestingly, among the donepezil-like molecules synthesized sofar, 1h bearing the N-benzyl(ethyl)amino group at the para position of the benzylidene moiety proved to be a potent nanomolar AChE inhibitor, achieving an IC50 of 39 nM (i.e., threefold more potent of the parent E-1a). The irradiation, and consequent enrichment in the Z diastereoisomer (75% Z in the PSS), led to a decrease of activity (IC50 of PSS-1h equals 53 nM), less sharp than that observed with the parent compound 1a. This suggests that the replacement of one ethyl group in E-1a with a more lipophilic benzyl moiety in E-1h, due to additional hydrophobic and aromatic interactions with residues into the enzyme binding pockets (e.g., Phe295 as shown by docking calculation), may improve the inhibitory potency of the E-1h compared to E-1a, at the same time flattening the differences in IC50s between the diastereomer E and the PSS E/Z mixture. No other compound showed an increase in inhibitory activity as appreciable as 1h, whereas the N-Me(nBu) analog 1e showed the highest difference in AChE inhibition potency between the thermal steady state E-1e (IC50 = 0.102 μM) and the photoinduced one PSS-1e (IC50 = 1.36 μM).
The modifications carried out on the tertiary amino head of the parent compound 1a seems well tolerated by MAO-B; indeed, all compounds in the E geometry achieved submicromolar IC50 values and discrete selectivity over MAO-A. The effect of the E-to-Z photoinduced transition in MAO-B inhibition was more modest (no diastereoselectivity with 1h) than that observed in AChE. Molecular docking studies performed with the potent dual inhibitor 1h helped in highlighting the binding modes of the two diastereoisomers in AChE and MAO-B binding sites.
The data discussed herein represent the basis for the optimization of a donepezil-like molecule capable of synergistically inhibiting two target enzymes, namely AChE and MAO-B, involved in neurodegenerative diseases, with the added value of the photomodulation of the pharmacological effect, a tool of interest for the personalized medicine of the future.
Supplementary Materials
The following materials are available online. 1H and 13C NMR spectra of the newly synthesized compounds 1b-h.
Author Contributions
Conceptualization, M.P., M.d.C., F.C., and C.D.A.; methodology, M.P., M.d.C., R.P., M.C., M.S., A.C., A.B., and C.M.; software, A.R.T. and O.N.; validation, M.d.C., R.P., and M.C.; writing—original draft preparation, M.P., M.d.C., A.R.T., O.N., F.C., and C.D.A.; writing—review and editing, all the co-authors; funding acquisition, M.P. and C.D.A. All authors have read and agreed to the published version of the manuscript.
Funding
M.P. and M.S. acknowledge the MUR for the financial support under the project ECS00000017 Tuscany- Health Ecosystem – CUP B63C2200068007 spoke 6 for Mission 4 Component 2 (M4C2) – investment 1.5 of the National Recovery and Resilience Plan (PNRR) funded by the European Union “Next Generation EU”. M.d.C., R.P., M.C. and C.D.A. acknowledge the financial support of the Italian Ministry of Education, Universities and Research (PRIN, Grant 201744BN5T_004).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
All data presented in this study are available in the article and in Supplementary Information.
Conflicts of Interest
The authors declare no conflict of interest.
Sample Availability
Samples of the compounds presented are available from the authors.
References
- Available online: https://www.alzint.org/resource/world-alzheimer-report-2022/.
- Gerrow, K.; Triller, A. Synaptic stability and plasticity in a floating world. Curr. Opin. Neurobiol. 2010, 20, 631–639. [Google Scholar] [CrossRef] [PubMed]
- Finberg, J.P.M.; Rabey, J.M. Inhibitors of MAO-A and MAO-B in Psychiatry and Neurology. Front. Pharmacol. 2016, 7, 340. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z. Monoamine oxidase inhibitors: Promising therapeutic agents for Alzheimer’s disease (Review). Mol. Med. Rep. 2014, 9, 1533–1541. [Google Scholar] [CrossRef]
- Carradori, S.; Silvestri, R. New Frontiers in Selective Human MAO-B Inhibitors. J. Med. Chem. 2015, 58, 6717–6732. [Google Scholar] [CrossRef]
- Kumar, B.; Sheetal; Mantha, A. K.; Kumar, V. Recent developments on the structure–activity relationship studies of MAO inhibitors and their role in different neurological disorders. RSC Adv. 2016, 6, 42660–42683. [Google Scholar] [CrossRef]
- Cereda, E.; Cilia, R.; Canesi, M.; Tesei, S.; Mariani, C.B.; Zecchinelli, A.L.; Pezzoli, G. Efficacy of rasagiline and selegiline in Parkinson’s disease: a head-to-head 3-year retrospective case–control study. J. Neurol. 2017, 264, 1254–1263. [Google Scholar] [CrossRef]
- Picciotto, M.R.; Higley, M.J.; Mineur, Y.S. Acetylcholine as a Neuromodulator: Cholinergic Signaling Shapes Nervous System Function and Behavior. Neuron 2012, 76, 116–129. [Google Scholar] [CrossRef] [PubMed]
- Marucci, G.; Buccioni, M.; Dal Ben, D.; Lambertucci, C.; Volpini, R.; Amenta, F. Efficacy of acetylcholinesterase inhibitors in Alzheimer’s disease. Neuropharmacology 2021, 190, 108352. [Google Scholar] [CrossRef]
- Spencer, C.M.; Noble, S. Rivastigmine. Drugs Aging 1998, 13, 391–411. [Google Scholar] [CrossRef]
- Dooley, M.; Lamb, H.M. Donepezil. Drugs Aging 2000, 16, 199–226. [Google Scholar] [CrossRef]
- Maramai, S.; Benchekroun, M.; Gabr, M.T.; Yahiaoui, S. Multitarget Therapeutic Strategies for Alzheimer’s Disease: Review on Emerging Target Combinations. Biomed Res. Int. 2020, 2020, 5120230. [Google Scholar] [CrossRef] [PubMed]
- Rullo, M.; Cipolloni, M.; Catto, M.; Colliva, C.; Miniero, D.V.; Latronico, T.; de Candia, M.; Benicchi, T.; Linusson, A.; Giacchè, N.; Altomare, C.D.; Pisani, L. Probing Fluorinated Motifs onto Dual AChE-MAO-B Inhibitors: Rational Design, Synthesis, Biological Evaluation, and Early-ADME Studies. J. Med. Chem. 2022, 65, 3962–3977. [Google Scholar] [CrossRef] [PubMed]
- Mathew, B.; Oh, J.M.; Baty, R.S.; Batiha, G.E.-S.; Parambi, D.G.T.; Gambacorta, N.; Nicolotti, O.; Kim, H. Piperazine-substituted chalcones: a new class of MAO-B, AChE, and BACE-1 inhibitors for the treatment of neurological disorders. Environ. Sci. Pollut. Res. 2021, 28, 38855–38866. [Google Scholar] [CrossRef] [PubMed]
- Guieu, B.; Lecoutey, C.; Legay, R.; Davis, A.; Sopkova de Oliveira Santos, J.; Altomare, C.D.; Catto, M.; Rochais, C.; Dallemagne, P. First Synthesis of Racemic Trans Propargylamino-Donepezil, a Pleiotrope Agent Able to Both Inhibit AChE and MAO-B, with Potential Interest against Alzheimer's Disease. Molecules 2020, 26, 80. [Google Scholar] [CrossRef] [PubMed]
- Ramsay, R.R.; Popovic-Nikolic, M.R.; Nikolic, K.; Uliassi, E.; Bolognesi, M.L. A perspective on multi-target drug discovery and design for complex diseases. Clin. Transl. Med. 2018, 7, 3. [Google Scholar] [CrossRef]
- Velema, W.A.; Szymanski, W.; Feringa, B.L. Photopharmacology: Beyond Proof of Principle. J. Am. Chem. Soc. 2014, 136, 2178–2191. [Google Scholar] [CrossRef]
- Duran-Corbera, A.; Catena, J.; Otero-Viñas, M.; Llebaria, A.; Rovira, X. Photoswitchable Antagonists for a Precise Spatiotemporal Control of β2-Adrenoceptors. J. Med. Chem. 2020, 63, 8458–8470. [Google Scholar] [CrossRef]
- Fuchter, M.J. On the Promise of Photopharmacology Using Photoswitches: A Medicinal Chemist's Perspective. J. Med. Chem. 2020, 63, 11436–11447. [Google Scholar] [CrossRef]
- Rodríguez-Soacha, D.A.; Fender, J.; Ramírez, Y.A.; Collado, J.A.; Muñoz, E.; Maitra, R.; Sotriffer, C.; Lorenz, K.; Decker, M. "Photo-Rimonabant": Synthesis and Biological Evaluation of Novel Photoswitchable Molecules Derived from Rimonabant Lead to a Highly Selective and Nanomolar "Cis-On" CB1R Antagonist. ACS Chem. Neurosci. 2021, 12, 1632–1647. [Google Scholar] [CrossRef]
- Qiao, Z.; Fu, W.; Zhang, Y.; Chen, R.; Xu, Z.; Li, Z.; Shao, X. Azobenzene-Semicarbazone Enables Optical Control of Insect Sodium Channels and Behavior. J. Agric. Food Chem. 2021, 69, 15554–15561. [Google Scholar] [CrossRef]
- Yue, L.; Pawlowski, M.; Dellal, S.S.; Xie, A.; Feng, F.; Otis, T.S.; Bruzik, K.S.; Qian, H.; Pepperberg, D.R. Robust photoregulation of GABAA receptors by allosteric modulation with a propofol analogue. Nat. Commun. 2012, 3, 1095. [Google Scholar] [CrossRef] [PubMed]
- Mourot, A.; Kienzler, M.A.; Banghart, M.R.; Fehrentz, T.; Huber, F.M.E.; Stein, M.; Kramer, R.H.; Trauner, D. Tuning Photochromic Ion Channel Blockers. ACS Chem. Neurosci. 2011, 2, 536–543. [Google Scholar] [CrossRef]
- Szymanski, W.; Ourailidou, M.E.; Velema, W.A.; Dekker, F.J.; Feringa, B.L. Light-Controlled Histone Deacetylase (HDAC) Inhibitors: Towards Photopharmacological Chemotherapy. Chem. – A Eur. J. 2015, 21, 16517–16524. [Google Scholar] [CrossRef] [PubMed]
- Reisinger, B.; Kuzmanovic, N.; Löffler, P.; Merkl, R.; König, B.; Sterner, R. Exploiting Protein Symmetry To Design Light-Controllable Enzyme Inhibitors. Angew. Chem. Int. Ed. 2014, 53, 595–598. [Google Scholar] [CrossRef] [PubMed]
- Rovira, X.; Trapero, A.; Pittolo, S.; Zussy, C.; Faucherre, A.; Jopling, C.; Giraldo, J.; Pin, J.-P.; Gorostiza, P.; Goudet, C.; et al. OptoGluNAM4.1, a Photoswitchable Allosteric Antagonist for Real-Time Control of mGlu4 Receptor Activity. Cell Chem. Biol. 2016, 23, 929–934. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Wehle, S.; Kuzmanovic, N.; Merget, B.; Holzgrabe, U.; König, B.; Sotriffer, C.A.; Decker, M. Acetylcholinesterase Inhibitors with Photoswitchable Inhibition of β-Amyloid Aggregation. ACS Chem. Neurosci. 2014, 5, 377–389. [Google Scholar] [CrossRef]
- Broichhagen, J.; Jurastow, I.; Iwan, K.; Kummer, W.; Trauner, D. Optical Control of Acetylcholinesterase with a Tacrine Switch. Angew. Chem. Int. Ed. 2014, 53, 7657–7660. [Google Scholar] [CrossRef]
- Paolino, M.; Gueye, M.; Pieri, E.; Manathunga, M.; Fusi, S.; Cappelli, A.; Latterini, L.; Pannacci, D.; Filatov, M.; Léonard, J.; et al. Design, Synthesis, and Dynamics of a Green Fluorescent Protein Fluorophore Mimic with an Ultrafast Switching Function. J. Am. Chem. Soc. 2016, 138, 9807–9825. [Google Scholar] [CrossRef]
- Paolino, M.; Giovannini, T.; Manathunga, M.; Latterini, L.; Zampini, G.; Pierron, R.; Léonard, J.; Fusi, S.; Giorgi, G.; Giuliani, G.; et al. On the Transition from a Biomimetic Molecular Switch to a Rotary Molecular Motor. J. Phys. Chem. Lett. 2021, 12, 3875–3884. [Google Scholar] [CrossRef]
- Tassone, G.; Paolino, M.; Pozzi, C.; Reale, A.; Salvini, L.; Giorgi, G.; Orlandini, M.; Galvagni, F.; Mangani, S.; Yang, X.; et al. Xanthopsin-Like Systems via Site-Specific Click-Functionalization of a Retinoic Acid Binding Protein. ChemBioChem 2022, 23, e202100449. [Google Scholar] [CrossRef]
- Gueye, M.; Manathunga, M.; Agathangelou, D.; Orozco, Y.; Paolino, M.; Fusi, S.; Haacke, S.; Olivucci, M.; Léonard, J. Engineering the vibrational coherence of vision into a synthetic molecular device. Nature Communications 2018, 9, 313. [Google Scholar] [CrossRef] [PubMed]
- Pagano, K.; Paolino, M.; Fusi, S.; Zanirato, V.; Trapella, C.; Giuliani, G.; Cappelli, A.; Zanzoni, S.; Molinari, H.; Ragona, L.; Olivucci, M. Bile acid binding protein functionalization leads to a fully synthetic rhodopsin mimic. J. Phys. Chem. Lett. 2019, 10, 2235–2243. [Google Scholar] [CrossRef] [PubMed]
- Filatov, M.; Paolino, M.; Pierron, R.; Cappelli, A.; Giorgi, G.; Léonard, J.; Huix-Rotllant, M.; Ferré, N.; Yang, X.; Kaliakin, D.; Blanco-González, A.; Olivucci, M. Towards the engineering of a photon-only two-stroke rotary molecular motor. Nature Communications 2022, 13, 6433. [Google Scholar] [CrossRef] [PubMed]
- Paolino, M.; Saletti, M.; Reale, A.; Licciardi, M.; Varvarà, P.; Marquette, A.; Léonard, J.; Bonechi, C.; Donati, A.; Giorgi, G.; Giuliani, G.; Carlotti, B.; Ortica, F.; Latterini, L.; Gentile, M.; Paccagnini, E.; Olivucci, M.; Cappelli, A. Chem. Eur. J. 2022, 28, e2022014.
- de Candia, M.; Zaetta, G.; Denora, N.; Tricarico, D.; Majellaro, M.; Cellamare, S.; Altomare, C.D. New azepino[4,3-b]indole derivatives as nanomolar selective inhibitors of human butyrylcholinesterase showing protective effects against NMDA-induced neurotoxicity. Eur. J. Med. Chem. 2017, 125, 288–298. [Google Scholar] [CrossRef] [PubMed]
- Purgatorio, R.; de Candia, M.; Catto, M.; Carrieri, A.; Pisani, L.; De Palma, A.; Toma, M.; Ivanova, O.A.; Voskressensky, L.G.; Altomare, C.D. Investigating 1,2,3,4,5,6-hexahydroazepino[4,3-b]indole as scaffold of butyrylcholinesterase-selective inhibitors with additional neuroprotective activities for Alzheimer’s disease. Eur. J. Med. Chem. 2019, 177, 414–424. [Google Scholar] [CrossRef]
- Purgatorio, R.; Gambacorta, N.; de Candia, M.; Catto, M.; Rullo, M.; Pisani, L.; Nicolotti, O.; Altomare, C.D. First-in-Class Isonipecotamide-Based Thrombin and Cholinesterase Dual Inhibitors with Potential for Alzheimer Disease. Molecules 2021, 26, 5208. [Google Scholar] [CrossRef]
- Paolino, M.; Rullo, M.; Maramai, S.; de Candia, M.; Pisani, L.; Catto, M:; Mugnaini, C. ; Brizzi, A.; Cappelli, A.; Olivucci, M.; Corelli, F.; Altomare, C.D. Design, synthesis and biological evaluation of light-driven on-off multitarget AChE and MAO-B inhibitors. RSC Med. Chem. 2022, 13, 873–883. [Google Scholar] [CrossRef]
- Sheng, R.; Xu, Y.; Hu, C.; Zhang, J.; Lin, X.; Li, J.; Yang, B.; He, Q.; Hu, Y. Design, synthesis and AChE inhibitory activity of indanone and aurone derivatives. Eur. J. Med. Chem. 2009, 44, 7–17. [Google Scholar] [CrossRef]
- Petermayer, C.; Thumser, S.; Kink, F.; Mayer, P.; Dube, H. Hemiindigo: highly bistable photoswitching at the biooptical window. J. Am. Chem. Soc. 2017, 139, 15060–15067. [Google Scholar] [CrossRef]
- Lazinski, L.M.; Royal, G.; Robin, M.; Maresca, M.; Haudecoeur, R. Bioactive aurones, indanones, and other hemiindigoid scaffolds: medicinal chemistry and photopharmacology perspectives. J. Med. Chem. 2022, 65, 12594–12625. [Google Scholar] [CrossRef] [PubMed]
- Salum, M.L.; Arroyo Mañez, P.; Luque, F.J.; Erra-Balsells, R. Combined experimental and computational investigation of the absorption spectra of E- and Z-cinnamic acids in solution: The peculiarity of Z-cinnamics. J. Photochem. Photobiol. B Biol. 2015, 148, 128–135. [Google Scholar] [CrossRef] [PubMed]
- Purgatorio, R.; de Candia, M.; Catto, M.; Rullo, M.; Pisani, L.; Denora, N.; Carrieri, A.; Nevskaya, A.A.; Voskressensky, L.G.; Altomare, C.D. Evaluation of Water-Soluble Mannich Base Prodrugs of 2,3,4,5-Tetrahydroazepino[4,3-b]indol-1(6H)-one as Multitarget-Directed Agents for Alzheimer's Disease. ChemMedChem 2021, 16, 589–598. [Google Scholar] [CrossRef]
- Purgatorio, R.; Gambacorta, N.; Samarelli, F.; Lopopolo, G.; de Candia, M.; Catto, M.; Nicolotti, O.; Altomare, C.D. Assessing the Role of a Malonamide Linker in the Design of Potent Dual Inhibitors of Factor Xa and Cholinesterases. Molecules 2022, 27, 4269. [Google Scholar] [CrossRef]
- Nel, M.S.; Petzer, A.; Petzer, J.P.; Legoabe, L.J. 2-Benzylidene-1-indanone derivatives as inhibitors of monoamine oxidase. Bioorg. Med. Chem. Lett. 2016, 26, 4599–4605. [Google Scholar] [CrossRef]
- Affini, A.; Hagenow, S.; Zivkovic, A.; Marco-Contelles, J.; Stark, H. Novel indanone derivatives as MAO-B/H3R dual-targeting ligands for treatment of Parkinson’s disease. Eur. J. Med. Chem. 2018, 148, 487–497. [Google Scholar] [CrossRef] [PubMed]
- Pourshojaei, Y.; Abiri, A.; Eskandari, K.; Haghighijoo, Z.; Edraki, N.; Asadipour, A. Phenoxyethyl Piperidine/Morpholine Derivatives as PAS and CAS Inhibitors of Cholinesterases: Insights for Future Drug Design. Sci. Rep. 2019, 9, 19855. [Google Scholar] [CrossRef] [PubMed]
- Alagöz, M.A.; Oh, J M. ; Zenni, Y.N.; Özdemir, Z.; Abdelgawad, M.A.; Naguib, I.A.; Ghoneim, M.M.; Gambacorta, N.; Nicolotti, O.; Kim, H.; Mathew, B. Development of a Novel Class of Pyridazinone Derivatives as Selective MAO-B Inhibitors. Molecules 2022, 27, 3801. [Google Scholar] [CrossRef] [PubMed]
- Jaafar, H.; Li, H.; Misal Castro, L.C.; Zheng, J.; Roisnel, T.; Dorcet, V.; Sortais, J.-B.; Darcel, C. Phosphane-Pyridine Iron Complexes: Synthesis, Characterization and Application in Reductive Amination through the Hydrosilylation Reaction. Eur. J. Inorg. Chem. 2012, 3546–3550. [Google Scholar] [CrossRef]
- Kuwano, R.; Kondo, Y.; Matsuyama, Y. Palladium-Catalyzed Nucleophilic Benzylic Substitutions of Benzylic Esters. J. Am. Chem. Soc. 2003, 125, 12104–12105. [Google Scholar] [CrossRef]
- Das, S.; Karmakar, H.; Bhattacharjee, J.; Panda, T.K. Aluminium complex as an efficient catalyst for the chemo-selective reduction of amides to amines. Dalton Trans. 2019, 48, 11978–11984. [Google Scholar] [CrossRef]
- Lator, A.; Gaignard Gaillard, Q.; Mérel, D.S.; Lohier, J.-F.; Gaillard, S.; Poater, A.; Renaud, J.-L. Room-Temperature Chemoselective Reductive Alkylation of Amines Catalyzed by a Well-Defined Iron(II) Complex Using Hydrogen. J. Org. Chem. 2019, 84, 6813–6829. [Google Scholar] [CrossRef] [PubMed]
- Horiuchi, T.; Nagata, M.; Kitagawa, M.; Akahane, K.; Uoto, K. Discovery of novel thieno[2,3-d]pyrimidin-4-yl hydrazone-based inhibitors of cyclin D1-CDK4: synthesis, biological evaluation and structure-activity relationships. Part 2. Bioorg. Med. Chem. 2009, 17, 7850–7860. [Google Scholar] [CrossRef] [PubMed]
- Claffey, J.; Müller-Bunz, H.; Tacke, M. Benzyl-substituted titanocene dichloride anticancer drugs: From lead to hit. J. Organometal. Chem. 2010, 695, 2105–2117. [Google Scholar] [CrossRef]
- Rizzo, S.; Bartolini, M.; Ceccarini, L.; Piazzi, L.; Gobbi, S.; Cavalli, A.; Recanatini, M.; Andrisano, V.; Rampa, A. Targeting Alzheimer's disease: Novel indanone hybrids bearing a pharmacophoric fragment of AP2238. Bioorg. Med. Chem. 2010, 18, 1749–1760. [Google Scholar] [CrossRef] [PubMed]
- Purgatorio, R.; de Candia, M.; De Palma, A.; De Santis, F.; Pisani, L.; Campagna, F.; Cellamare, S.; Altomare, C.D.; Catto, M. Insights into Structure-Activity Relationships of 3-Arylhydrazonoindolin-2-One Derivatives for Their Multitarget Activity on β-Amyloid Aggregation and Neurotoxicity. Molecules 2018, 23, 1544. [Google Scholar] [CrossRef]
- Purgatorio, R.; de Candia, M.; Catto, M.; Rullo, M.; Pisani, L.; Denora, N.; Carrieri, A.; Nevskaya, A.A.; Voskressensky, L.G.; Altomare, C.D. Evaluation of Water-Soluble Mannich Base Prodrugs of 2,3,4,5-Tetrahydroazepino[4,3-b]indol-1(6H)-one as Multitarget-Directed Agents for Alzheimer's Disease. ChemMedChem 2021, 16, 589–598. [Google Scholar] [CrossRef]
- Purgatorio, R.; Kulikova, L.N.; Pisani, L.; Catto, M.; Candia, M.; Carrieri, A.; Cellamare, S.; De Palma, A.; Beloglazkin, A.A.; Reza Raesi, G.; et al. Scouting around 1,2,3,4-tetrahydrochromeno[3,2-c]pyridin-10-ones for single- and multitarget ligands directed towards relevant Alzheimer’s targets. ChemMedChem 2020, 15, 1947–1955. [Google Scholar] [CrossRef]
- Titov, A.A.; Purgatorio, R.; Obydennik, A.Y.; Listratova, A.V.; Borisova, T.N.; de Candia, M.; Catto, M.; Altomare, C.D.; Varlamov, A.V.; Voskressensky, L.G. Synthesis of isomeric 3-Benzazecines decorated with endocyclic allene moiety and exocyclic conjugated double bond and evaluation of their anticholinesterase activity. Molecules 2022, 27, 6276. [Google Scholar] [CrossRef]
Figure 1.
Photoisomerizable dual AChE/MAO-B inhibitors synthesized in this study, aiming at
optimizing the tertiary amino head of the hit compound 1a; the E/Z diastereoisomers of 1a are
surrounded by known AChE-Is, MAO-B-Is, and synthetic light-gated molecular switches and rotors.
Figure 1.
Photoisomerizable dual AChE/MAO-B inhibitors synthesized in this study, aiming at
optimizing the tertiary amino head of the hit compound 1a; the E/Z diastereoisomers of 1a are
surrounded by known AChE-Is, MAO-B-Is, and synthetic light-gated molecular switches and rotors.
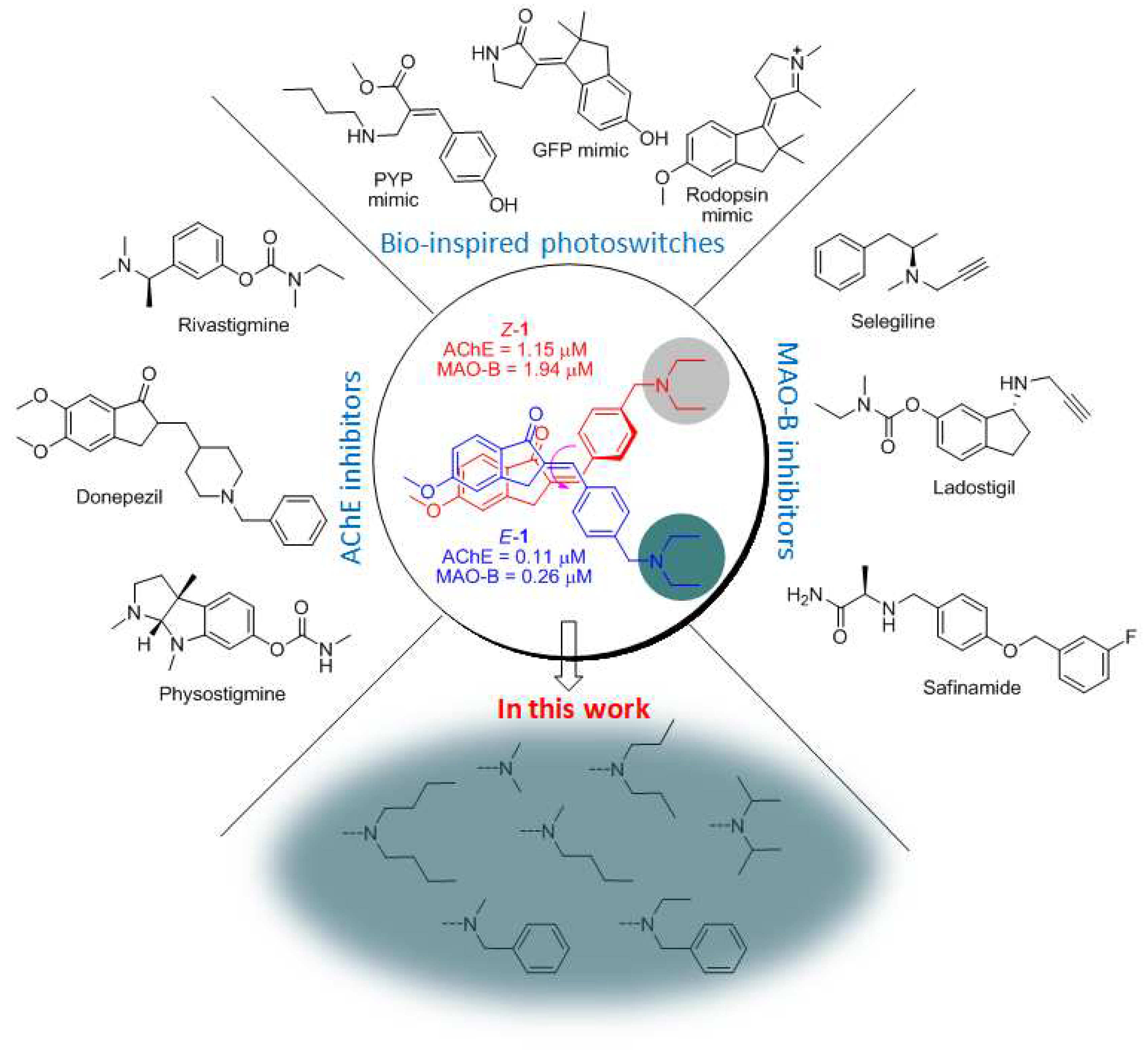
Figure 2.
Design of the new photoisomerizable dual AChE/MAO-B inhibitors.
Scheme 1.
Synthesis of compounds E-1b-h (A) and aldehyde derivatives 4b-h (B). Reagents and conditions: i) CH3OH, KOH, r.t., 2-6 h; ii) appropriate secondary ammine, CH2Cl2, reflux, 4-5 h; iii) LiAlH4, dry THF, 12 h; iv) MnO2, 1,4 dioxane, reflux, 2-4 h.
Scheme 1.
Synthesis of compounds E-1b-h (A) and aldehyde derivatives 4b-h (B). Reagents and conditions: i) CH3OH, KOH, r.t., 2-6 h; ii) appropriate secondary ammine, CH2Cl2, reflux, 4-5 h; iii) LiAlH4, dry THF, 12 h; iv) MnO2, 1,4 dioxane, reflux, 2-4 h.
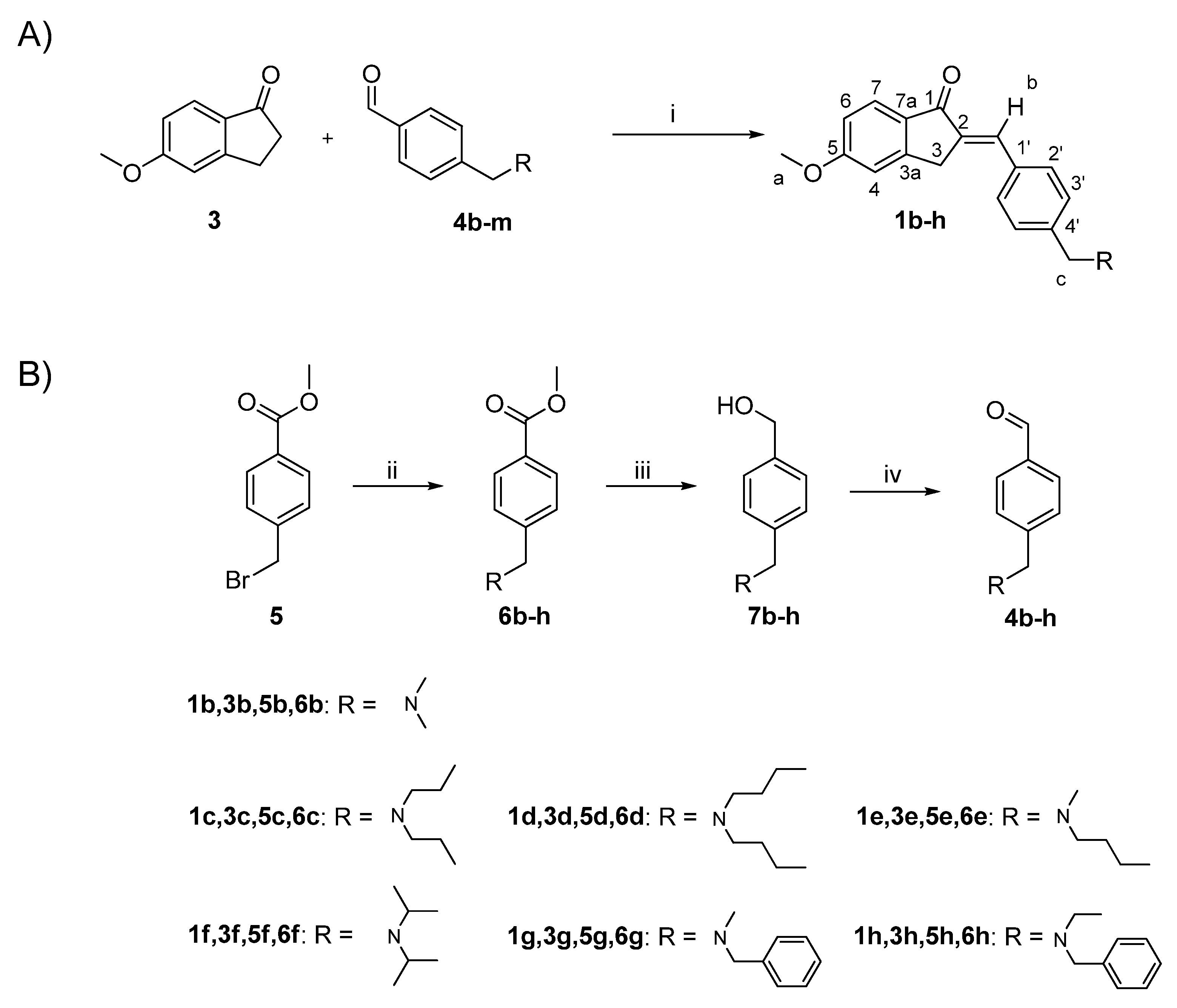
Figure 3.
Normalized UV-Vis absorption spectra of methanolic solutions of compounds E-1a-h.
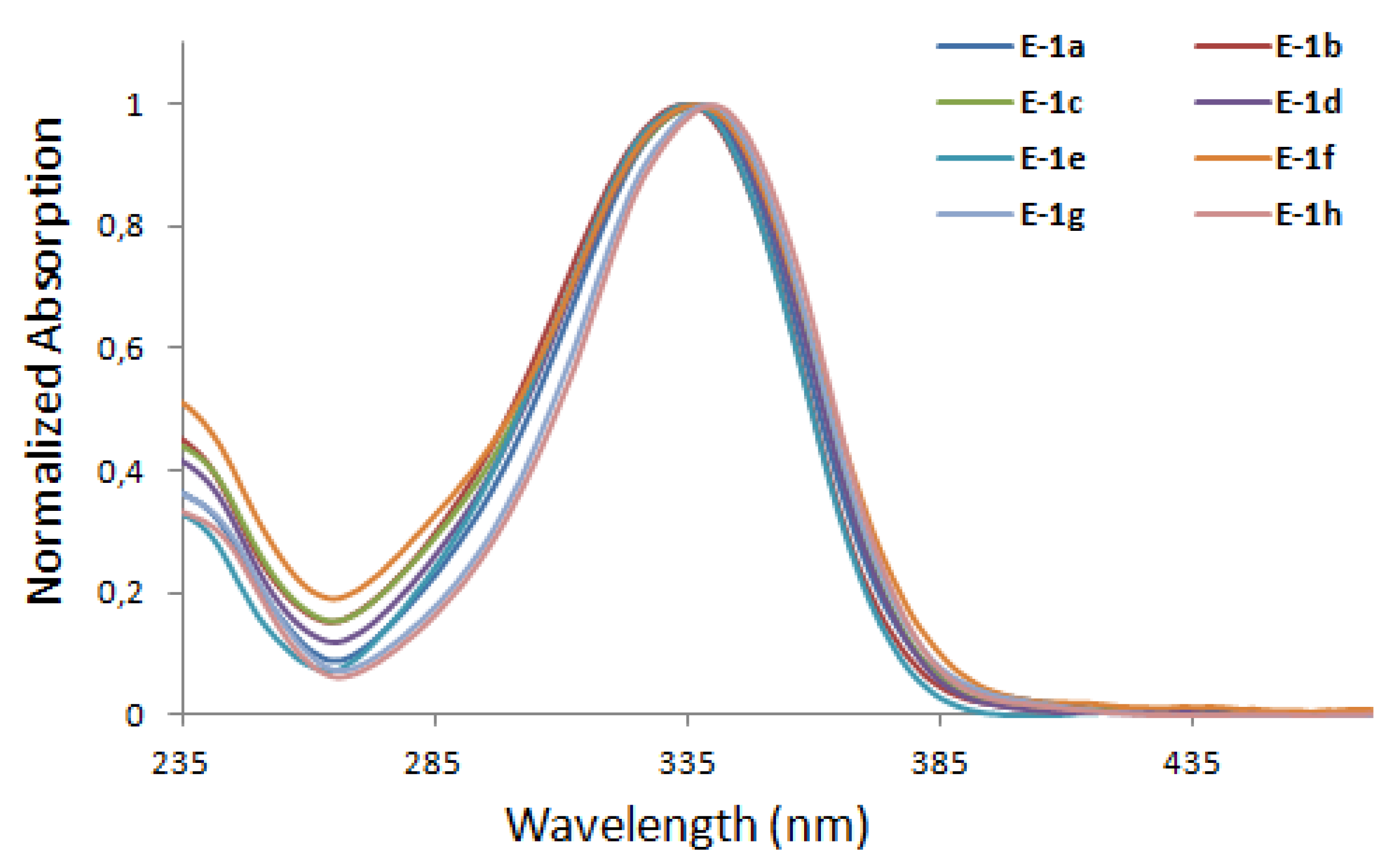
Figure 4.
1H NMR spectra (400 MHz) of the E/Z mixtures obtained at the PSSs after irradiation with UV-B light of compounds 1a-e (spectra A-E) in methanol-d4 and of compounds 1f-h (spectra F-H) in acetone-d6. Blue numbering was used for assignment of the E diastereomers, while red numbering was used for the Z diastereomers.
Figure 4.
1H NMR spectra (400 MHz) of the E/Z mixtures obtained at the PSSs after irradiation with UV-B light of compounds 1a-e (spectra A-E) in methanol-d4 and of compounds 1f-h (spectra F-H) in acetone-d6. Blue numbering was used for assignment of the E diastereomers, while red numbering was used for the Z diastereomers.

Figure 5.
Normalized UV-Vis absorption spectra of methanolic solution of compound 1a-h at PSS.
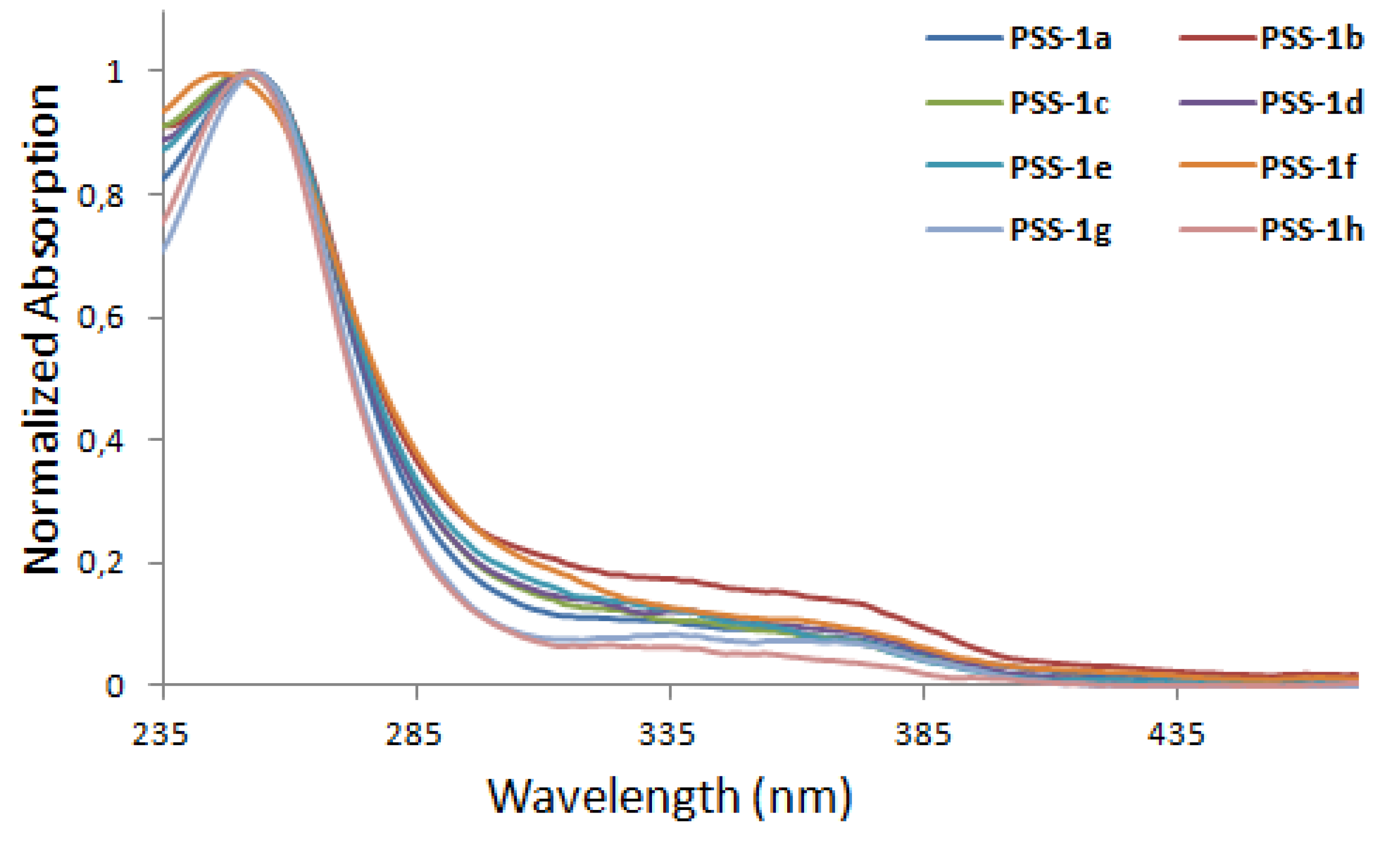
Figure 6.
Inhibition kinetics (left) and Lineweaver–Burk plot (right), r2 = 0.983–0.999 for hAChE (0.2 U/mL) and E-1h (0–500 nM), using different substrate (acetylthiocholine iodide) concentrations (50–300 μM). The replot (r2 = 0.997) of the slopes versus [I] determined the Ki (100 nM) as the x-axis intercept (see Supplementary Information). (○) no inhibitor, (●) 10 nM, (■) 25 nM, (◆) 50 nM, (◇) 100 nM, (▲) 200 nM, (△) 500 nM.
Figure 6.
Inhibition kinetics (left) and Lineweaver–Burk plot (right), r2 = 0.983–0.999 for hAChE (0.2 U/mL) and E-1h (0–500 nM), using different substrate (acetylthiocholine iodide) concentrations (50–300 μM). The replot (r2 = 0.997) of the slopes versus [I] determined the Ki (100 nM) as the x-axis intercept (see Supplementary Information). (○) no inhibitor, (●) 10 nM, (■) 25 nM, (◆) 50 nM, (◇) 100 nM, (▲) 200 nM, (△) 500 nM.
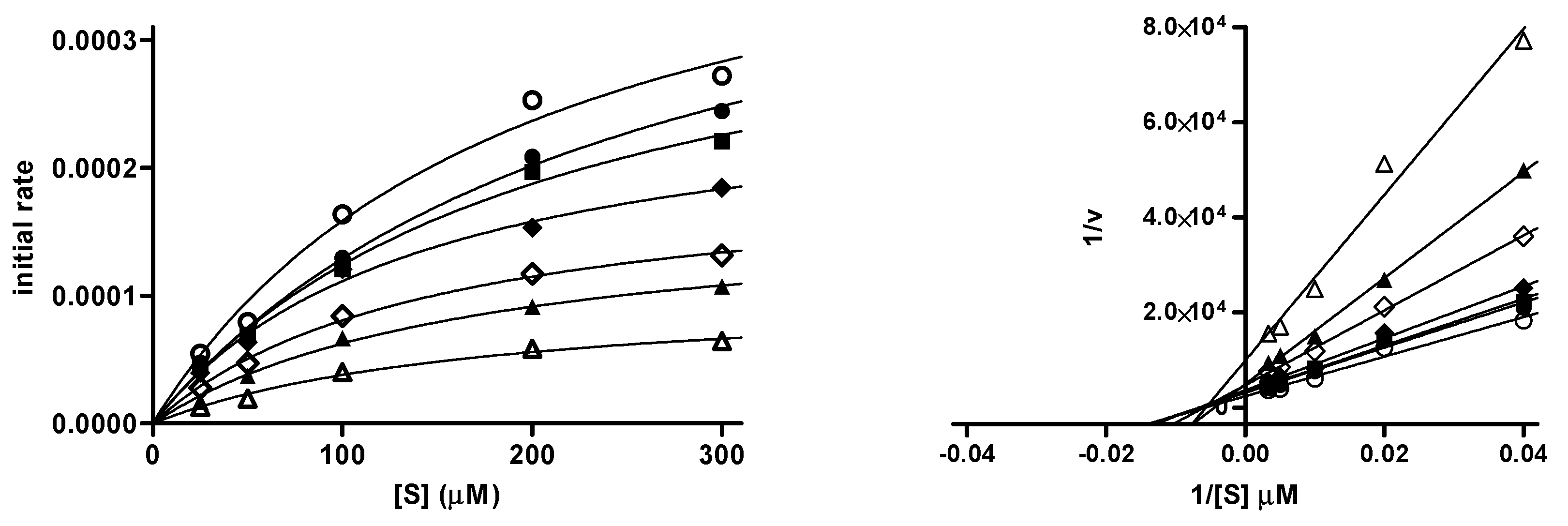
Figure 7.
Glide top-ranked docking poses of (a) E-1h (docking score = -13.172 kcal/mol, ligand efficiency = -0.439) and (b) Z-1h (docking score = -11.494 kcal/mol, ligand efficiency = -0.383) in complex with hAChE (PDB 4EY7; donepezil: docking score = -13.117 kcal/mol, ligand efficiency = -0.468). The diastereoisomer ligands are depicted as sticks; the interacting residues are rendered as lines, while protein is represented as cartoon. Dashed black lines represent H-bond, meanwhile π-π and π-cation interactions are indicated by dashed cyan lines.
Figure 7.
Glide top-ranked docking poses of (a) E-1h (docking score = -13.172 kcal/mol, ligand efficiency = -0.439) and (b) Z-1h (docking score = -11.494 kcal/mol, ligand efficiency = -0.383) in complex with hAChE (PDB 4EY7; donepezil: docking score = -13.117 kcal/mol, ligand efficiency = -0.468). The diastereoisomer ligands are depicted as sticks; the interacting residues are rendered as lines, while protein is represented as cartoon. Dashed black lines represent H-bond, meanwhile π-π and π-cation interactions are indicated by dashed cyan lines.
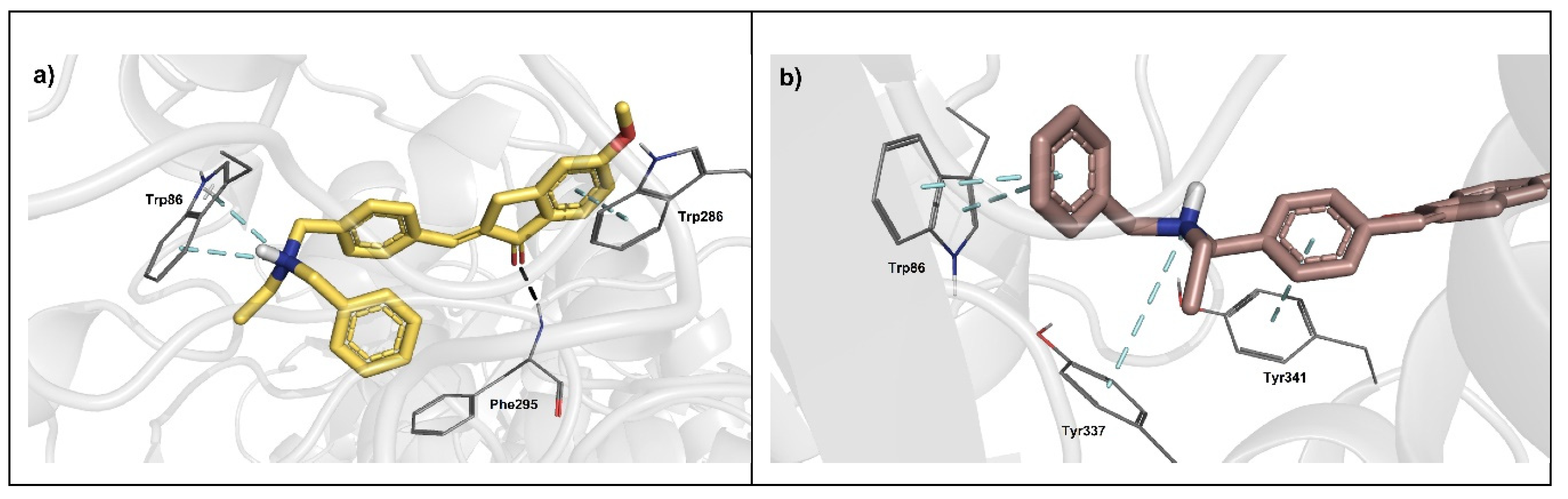
Figure 8.
Superimposition of E-1h (yellow sticks), Z-1h (violet sticks) and donepezil (green sticks) as docked in the top-scored poses (E/Z-1h) or established by X-ray crystal structure (donepezil) within the AChE binding site.
Figure 8.
Superimposition of E-1h (yellow sticks), Z-1h (violet sticks) and donepezil (green sticks) as docked in the top-scored poses (E/Z-1h) or established by X-ray crystal structure (donepezil) within the AChE binding site.
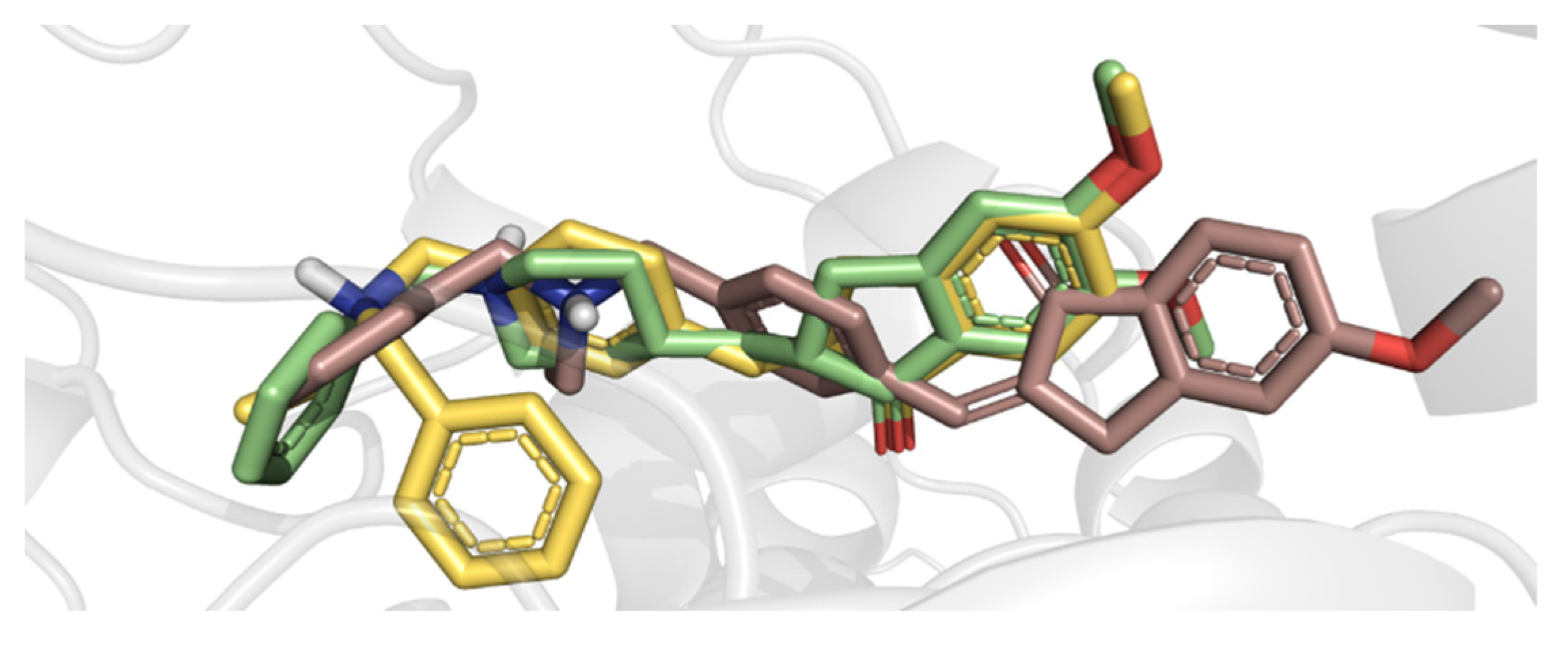
Figure 9.
Glide top-ranked docking poses of (a) E-1h (grey stick, docking score = -10.129 kcal/mol, ligand efficiency = -0.338) and (b) Z-1h (blue stick, docking score = -12.256 kcal/mol, ligand efficiency = -0.409) in complex with hMAO-B (PDB 2V5Z; safinamide: docking score = -10.428 kcal/mol, ligand efficiency = -0.474). The diastereoisomer ligands are depicted as sticks; the interacting residues are rendered as lines, while protein is represented as cartoon. Dashed black lines represent H-bonds, meanwhile π-π and π-cation interactions are represented as dashed cyan lines. FAD coenzyme is shown in orange sticks.
Figure 9.
Glide top-ranked docking poses of (a) E-1h (grey stick, docking score = -10.129 kcal/mol, ligand efficiency = -0.338) and (b) Z-1h (blue stick, docking score = -12.256 kcal/mol, ligand efficiency = -0.409) in complex with hMAO-B (PDB 2V5Z; safinamide: docking score = -10.428 kcal/mol, ligand efficiency = -0.474). The diastereoisomer ligands are depicted as sticks; the interacting residues are rendered as lines, while protein is represented as cartoon. Dashed black lines represent H-bonds, meanwhile π-π and π-cation interactions are represented as dashed cyan lines. FAD coenzyme is shown in orange sticks.
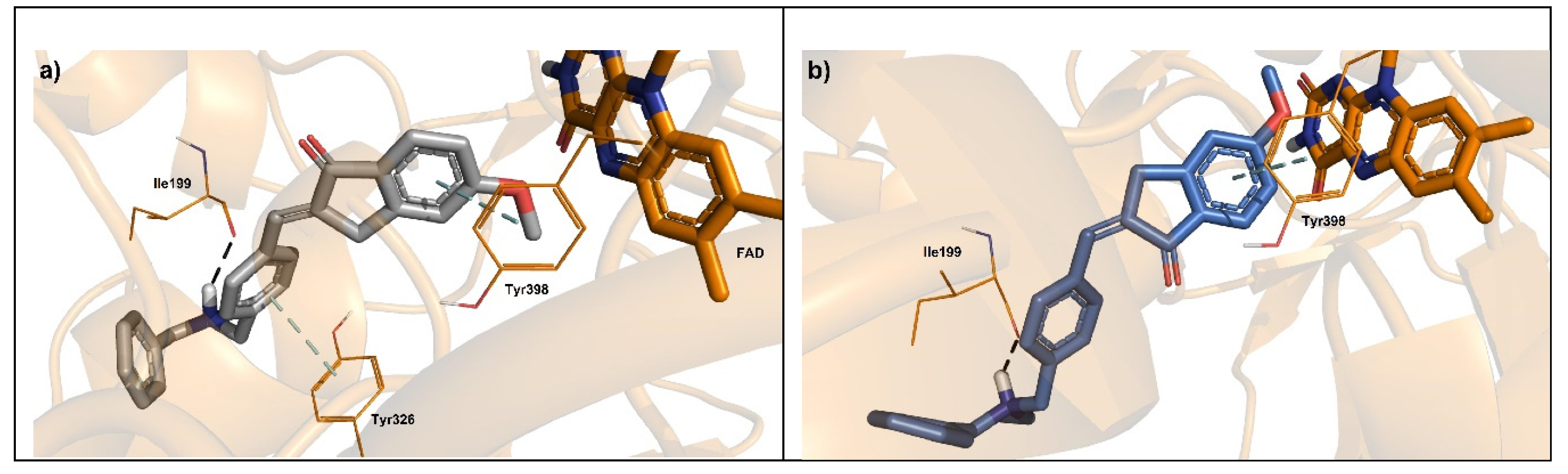

Table 1.
Inhibition data of recombinant human cholinesterases (AChE and BChE) and monoamine oxidases (MAOs A and B) by E isomers and E/Z mixtures of compounds 1a-h.
Table 1.
Inhibition data of recombinant human cholinesterases (AChE and BChE) and monoamine oxidases (MAOs A and B) by E isomers and E/Z mixtures of compounds 1a-h.
| Cmpdsa | IC50 (μM) or % inhibition at 10 μMb | |||
|---|---|---|---|---|
| AChEc | BChEc | MAO-Ac | MAO-Bc | |
| E-1a | 0.105±0.005f | 10.0±0.15f | (33±3)f | 0.260±0.012f |
| E/Z-1a | 0.780±0.013f | (28±6%)f | (45±5)f | 1.59±0.04f |
| E-1b | 0.155±0.010 | n.a. | (33±1) | 0.686±0.059 |
| E/Z-1b | 0.915±0.138 | (25±10%) | (32±2) | 0.839±0.123 |
| E-1c | 0.174±0.017 | (28±7%) | (15±1) | 0.479±0.081 |
| E/Z-1c | 0.295±0.045 | (20±7%) | 2.98±0.08 | 0.743±0.048 |
| E-1d | 0.990±.050 | (28±9%) | (12±4) | 0.338±0.036 |
| E/Z-1d | 2.28±0.21 | n.a. | (37±2) | 0.382±0.006 |
| E-1e | 0.102±0.019 | 5.00±0.45 | (20±5) | 0.178±0.086 |
| E/Z-1e | 1.36±0.06 | n.a. | (25±5) | 0.651±0.061 |
| E-1f | 0.261±0.014 | 10.0±0.85 | (22±2) | 0.178±0.086 |
| E/Z-1f | 1.41±0.09 | (20±3%) | 3.19±0.10 | 0.241±0.026 |
| E-1g | 0.115±0.004 | 9.90±0.11 | (39±7) | 0.292±0.060 |
| E/Z-1g | 0.135±0.062 | (40±2%) | (49±1) | 0.354±0.070 |
| E-1h | 0.039±0.001 | 7.00±0.01 | (13±6) | 0.355±0.134 |
| E/Z-1h | 0.053±0.006 | 10.0±0.32 | (39±5) | 0.358±0.008 |
| Donepezild | 0.021±0.005 | 4.80±1.00 | ||
| Tacrined | 0.090±0.005 | 0.025±0.003 | ||
| Clorgylinee | 0.0022±0.0002 | 2.48±0.43 | ||
| Safinamidee | (18±3%) | 0.028±0.001 | ||
aThe diastereomeric E/Z mixtures’ ratios (25:75) at the photostationary state after UV-B light irradiation of the E methanolic solutions was established for all compounds by 1H NMR spectrometry. bIC50 values determined by interpolation of the sigmoidal dose–response curves as obtained by regression with GraphPad Prism software (ver.5.01) of at least seven data points, or percent inhibition in parentheses for samples achieving less than 50% inhibition at 10 μM concentration; n.a. = not active; data are means ± SD of three independent measurements. cHuman cholinesterases and monoamine oxidase isoforms. dDonepezil and tacrine were used as positive controls in AChE and BChE inhibition assays, respectively. eClorgyline and safinamide were used as positive control in MAO-A and MAO-B, inhibition assays, respectively. fData re-determined in this work, agreeing with those previously reported.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



