Submitted:
29 June 2023
Posted:
30 June 2023
You are already at the latest version
Abstract
Phytoestrogens (PEs) are estrogen-like nonsteroidal compounds derived from plants (e.g., nuts, seeds, fruits and vegetables) and fungi that are structurally similar to 17β-estradiol. PEs bind to all types of estrogen receptors, including ERα and ERβ receptors, the nuclear receptors, and a membrane-bound estrogen receptor known as the G protein-coupled estrogen receptor (GPER). As endocrine-disrupting chemicals (EDCs) with pro‐ or antiestrogenic properties, PEs can potentially disrupt hormonal regulation of homeostasis, resulting in developmental and reproductive abnormalities. However, the lack of PEs in the diet does not result in the development of deficiency symptoms. To properly assess the benefits and risks associated with the use of a PE-rich diet, it is necessary to distinguish between endocrine disruption (endocrine-mediated adverse effects) and nonspecific effects on the endocrine system. Endometriosis is an estrogen-dependent disease of unknown etiopathogenesis, in which tissue similar to the lining of the uterus (the endometrium) grows outside of the uterus with subsequent complications being manifested as a result of local inflammatory reactions. Endometriosis affects 10–15% of women of reproductive age and is associated with chronic pelvic pain, dysmenorrhoea, dyspareunia and infertility. In this review, the endocrine-disruptive actions of PEs are reviewed in the context of endometriosis to determine whether a PE-rich diet has a positive or negative effect on the risk and course of endometriosis.
Keywords:
endocrine disruption
; phytoestrogens
; endometriosis
; endocrine disrupting chemicals
; etiopathogenesis of endometriosis
; ectopic endometrium
; dietary phytoestrogen intake
; epigenetic factors
1. Endocrine disrupting chemicals (EDCs)
The endocrine system, in association with the nervous system and the immune system, regulates the body’s internal activities and interactions with the external environment to preserve the homeostasis of the internal environment [1,2]. Hormone-producing cells (both within endocrine glands or forming the disseminated endocrine system) secrete hormones (chemical messengers) that interact with specific targets (receptors), including those targets that are subjected to epigenetic modifications [2–4]. These interactions result in the regulation of a vast spectrum of functions, including development, growth, energy balance (metabolism), reproduction and regulation of body weight [3,4].
Organic compounds that (to varying degrees) resist photolytic, biological and chemical degradation are called persistent organic pollutants (POPs) [5]. POPs are often halogenated and characterized by low water solubility and high lipid solubility, thus leading to their bioaccumulation in fatty tissues [5,6]. Due to the semivolatility of POPs and the physico-chemical characteristics that permit these compounds to occur either in the vapor phase or being adsorbed on atmospheric particles, long-range transport of POPs through the atmosphere may be facilitated. Thus, POPs are ubiquitous throughout the world, even in regions where they have never been used [7]. The most commonly encountered POPs are organochlorine pesticides, such as DDT, industrial chemicals, polychlorinated biphenyls (PCBs) and unintentional byproducts of many industrial processes, especially polychlorinated dibenzo-p-dioxins (PCDDs) and dibenzofurans (PFDFs), which are commonly known as dioxins [8,9].
Many POPs are well known to interact with the endocrine system by mimicking, hindering, blocking and promoting the normal activity of hormones [8–11]. Thus, these endocrine-disrupting chemicals (EDCs) are compounds in the environment (air, soil or source of water), food, personal care products and manufactured products that possess the ability to interfere with the normal function of the endocrine system [12,13]. EDCs may interfere with the synthesis, secretion, transport, binding, action and metabolism of virtually all natural hormones in the body, including sex steroid hormones that correspondingly cause developmental and fertility problems, infertility and hormone-sensitive cancers in women and men [13–16]. Specifically, exposure to EDCs above the threshold dose causes carcinogenic, neurotoxic, hepatotoxic, nephrotoxic and immunotoxic effects, as well as teratogenic hazards with birth defects [17–23].
According to the Endocrine Society statement, endocrine disruptors can be defined as “an exogenous chemical, or mixture of chemicals, that can interfere with any aspect of hormone action” [24,25]. However, it is necessary to distinguish between endocrine disruption (endocrine-mediated adverse effects) and nonspecific effects on the endocrine system [26]. Endocrine disruption occurs as a consequence of the interaction of a chemical (classified as an EDC) with a specific molecular component of the endocrine system (for example, an estrogen receptor). In contrast, nonspecific effects on the endocrine system may be observed when systemic toxicity has a significant impact on homeostasis and indirectly perturbs endocrine signaling. When considering the integral nature of signaling pathways in the endocrine system, it is difficult to confidently distinguish endocrine disruption from transient fluctuations, adaptive/compensatory responses or adverse effects on the endocrine system caused by mechanisms outside of the endocrine system that use nonendocrine-mediated modes of action [26,27]. This situation is further complicated by the fact that some organs/tissues can be affected by both endocrine and nonendocrine erroneous/disrupting signals.
Given that EDCs originate from many different sources, people may be exposed in many ways, including the air that they breathe, the food that they eat and the water that they drink [25,28–30]. In addition, EDCs can enter the body via the intact skin and mucous membranes [31]. Dietary intake is the main entry route of POPs and other EDCs into the human body and accounts for more than 90% of the total chemical exposure [28,32]. Moreover, there is an increasing concern that permanent low-level exposure to EDCs may have adverse health impacts, particularly during fetal, neonatal and childhood development. Therefore, important human health hazards should be expected in relation to EDCs, especially in the event of increasing environmental pollution [33–36]. Furthermore, it has been demonstrated that in addition to EDC, estrogen is a persistent compound in the environment. Estrogen contamination was confirmed in both lake water used for drinking and sewage water used for irrigation at concentrations that could affect plant growth (e.g., alfalfa) and sexual differentiation in fish [37–39]. These findings of estrogen as an environmental pollutant have been repeated and confirmed throughout the world, thus indicating that sex hormones, including estrogen and testosterone, are present in several environmental compartments, including soil and groundwater [40–42].
Chemicals with hormonal activity that may induce endocrine disruption can be divided into three main groups: synthetic compounds used in industry, agriculture and consumer products, synthetic compounds used in the pharmaceutical industry (i.e., drugs), and natural compounds present in the food chain that contain phytoestrogens (PEs), i.e., compounds showing structural similarity to estradiol (E2) [14].
It should be clearly emphasized that, in this review, only the endocrine-disruptive actions of PEs will be reviewed in the context of endometriosis, which is an estrogen-dependent disease with still unknown etiology (see Chapter 2.2. Disruption in estrogen and P4 signaling). General considerations on the effects of PEs as endocrine disruptors and estrogen-mediated alterations in endometriosis are followed by the current data on the role of orally administered PEs in the etiopathogenesis and course of endometriosis.
1.1. Phytoestrogens (PEs)
PEs, also called "dietary estrogens", are estrogen-like nonsteroidal compounds derived from plants (e.g., nuts, seeds, fruits and vegetables) and fungi, which are structurally similar to 17β-estradiol [43,44]. The estrogenic activity of PEs was first demonstrated in 1926; however, for the next 20 years, until fertility problems in sheep on isoflavone-rich diets were reported in Western Australia, it was uncertain as to whether they could have any effect on human or animal metabolism [44–46].
Based on the chemical structure, six main classes of PEs can be distinguished: flavonoids, stilbens, enterolignans, coumestans, pterocarpans and mycotoxins [47] (Table 1). Over 5,000 naturally occurring flavonoids have been characterized from various plants. The main PEs derived from the diet are genistein, daidzein and glycitein, which belong to a subclass of flavonoids called isoflavones [48]. PEs do not participate in any essential biological processes, and the lack of PEs in the diet does not result in the development of deficiency symptoms. Therefore, PEs are not considered nutrients [49].
In humans, after consuming PEs, they are converted in the gastrointestinal tract by complex enzymatic processes to heterocyclic phenols that are structurally similar to E2 [44]. Subsequently, absorbed phytoestrogen metabolites enter into the enterohepatic circulation and may be excreted in the bile deconjugated by intestinal flora, reabsorbed, reconjugated by the liver and excreted in the urine [44,50,51]. Concentrations of the different phytoestrogen metabolites can vary widely between individuals, even when a controlled quantity of an isoflavone or lignan supplement is administered.
The structural similarity of PEs to endogenous estradiol E2 implies the presence of a phenolic ring that enables binding to estrogen receptors in humans. Other key structural elements that increase affinity for estrogen receptors and enable estrogen-like effects include low molecular weights similar to estrogens/E2 (MW = 272), optimal hydroxylation patterns and (in the case of isoflavones) similarities of the E2 distances between two hydroxyl groups at the nucleus [52–54]. Analogous to estradiol, PEs bind to all known types of estrogen receptors, including ERα (NR3A1) and ERβ (NR3A2) receptors (which are the members of the superfamily class of nuclear receptors located in either the cell cytoplasm or nucleus) and a membrane-bound estrogen receptor known as G protein-coupled estrogen receptor (GPER), which is also known as G protein-coupled receptor 30 (GPR30) [55–58].
1.1.1. Signaling via nuclear receptors
In humans, ERα is encoded by the gene ESR1, which is located on chromosome 6, locus 6q25.1, whereas ERβ is encoded by the ESR2 gene located on chromosome 14 (14q23–24) [59,60]. In addition to the full-length isoforms, several shorter isoforms of ERs have been identified as a result of the presence of alternate start codons or as products of alternative splicing. The six crucial structural and functional domains of both ERα and ERβ were distinguished within the N-terminus (NTD: A/B domains, AF-1), DNA binding domain (DBD or C domain), hinge (D domain) and C-terminal region containing the ligand binding domain (LBD: E/F domain, AF-2) (Figure 1) [61–63].
The main, well-documented signaling pathways of estrogens are shown in a simplified manner in Figure 2.
Classical ligand-dependent ER activation results in the regulation of gene transcription in the nucleus or the activation of kinases in the cytoplasm (Figure 2A). This form of signaling mediates long-term genomic effects in estrogen-responsive tissues, including the human endometrium [59].
Estrogen binding to ERα or ERβ leads to the removal of the polyprotein inhibitory complex from the LBD with the release of heat shock protein 90 (Hsp90) and the induction of a conformational change resulting in the homodimerization of the receptor. Crystallographic studies have shown that, in contrast to the classical binding characteristics of a substrate to its active site in an enzyme, the ligand binding domain of the ERs is larger than the E2 molecule, which explains why it can accommodate a range of different-sized molecules, including those corresponding to PEs. Afterwards, this signaling complex is translocated from the cytoplasm to the nucleus, where after the recruitment of other coregulators, ERs act as ligand-activated transcription factors [60,61]. This direct genomic activity is associated with binding of the DBD to the estrogen response element (ERE) on the target gene and subsequent cis-activation of the enhancer of the target gene regulatory region that promotes transcription. In the “tethered” signaling pathway, ligand-activated ERs interact with other transcription factor (TF) complexes and attach to these transcription factors, which enables the indirect binding of the DBD to the ERE as TF-ERE [61,62]. The transcriptional activities of ERs are mediated by the coordinated action of their two activation domains, including the constitutive activation domain AF-1 at the N-terminus and the hormone-dependent AF-2 at the LBD. ERs have more than 30 synergistic activation factors, many of which are shared by nuclear receptors. The indirect regulation of gene transcription via the activation of the extracellular signal-regulated kinase 1/2 (ERK1/2) cascade and the phosphatidylinositol 3' -kinase (PI3K) signaling pathways is also involved in ERα/ERβ signaling [61–63].
The estrogenic effects of PEs are primarily mediated via ERα and ERβ (with higher affinity for Erβ) and by acting as agonists, partial agonists and antagonists [69]. For example, isoflavone affinity to ERβ isoforms is approximately five times higher than the affinity to ERα isoforms, in contrast to E2, in which the affinities to both receptor types are generally the same [70–72].
Interesting results regarding phytoestrogen affinity to ERβ have originated by using molecular docking, which is a method that is frequently used in the process of computer-aided drug design (CADD) as a tool for the identification of novel and potent ligands, as well as for predicting the binding mode of already known ligands and for the comparative estimation/prediction of binding affinity [73]. In molecular docking, the most important aspect is the calculation of binding energy to fit a ligand in a binding site [74]. Comparisons between two or three complexes using the predicted binding energies as a criterion are commonly found in the literature [75,76]. Such studies have demonstrated that almost all popular herbal supplements contain phytochemical components that may bind to the human estrogen receptor and exhibit selective estrogen receptor modulation. For example, of the flavonoids, luteolin-8-propenoic acid has been shown to exhibit the strongest docking (most exothermic docking energies) to ERα, with a docking energy of −113.127 kJ/mol, which is more exothermic than those of E2, isoflavonoid genistein or mycotoxin zearalenone [78]. A common docking orientation for phenolic ligands in ERα is the hydrophobic acceptor pocket of Leu 387, Phe 404, Met 388 and Leu 391, along with edge-to-face π–π interactions with Phe 404 and hydrogen bonds between the phenolic –OH group and the guanidine group of Arg 394, as well as the carboxylate of Glu 356. The 7-OH group of this ligand can form an additional hydrogen bond with the carbonyl oxygen of Gly 521. No other flavonoid ligands showed notably strong docking with ERα [75]. However, without questioning the concept of phytoestrogen binding to ERs, some authors are concerned about the unreliability of binding energy comparisons between pairs of molecules using docking [79].
It has been proposed that the estrogenic or antiestrogenic activity of PEs may be determined by an individual's amount of circulating endogenous estrogens, as well as the amount of bioavailable PEs and the number and type of ERs [80–82]. The approximately 100-fold lower affinity of PEs to ERs compared to human estrogens may be compensated for by their potentially high concentrations. For phytoestrogen levels that are several times higher than the concentration of endogenous estrogens, this higher affinity for ERβ may be even stronger than that exhibited by steroidal estrogens, which additionally suggests that PEs may exert their actions through distinctly different pathways [83,84]. The broad spectrum of estrogenic/antiestrogenic activity of PEs is due to the obvious fact that ERs have different functions. For example, ERα acts in cell proliferation, including carcinogenesis, whereas ERβ is responsible for cell cycle arrest, the modulation of the expression of many ERα-regulated genes and the induction of multiple anticancer activities (e.g., apoptosis) [85–88]. Interestingly, some PEs have also demonstrated progesterone receptor activity [89].
ERα is predominantly expressed in the endometrium, breast cancer cells, ovarian stroma cells, efferent duct epithelium and hypothalamus, whereas ERβ is expressed in the kidney, brain, bone, heart, lungs, intestinal mucosa, prostate and endothelial cells [90–92]. Consequently, the preference of binding to ERα or ERβ by a given phytoestrogen may determine its tissue-selective biological effects, including endocrine disruption. Once bound, PEs exhibit selective ER modulator (SERMS) activity with a broad range of varying agonist/antagonist activities. The tissue-selective or tissue-specific effects depend significantly on the content and proportion of transcriptional coregulators (both coactivator and corepressor proteins) within the single cell. This indicates that in the case of predomination of coactivators in certain tissues, a given ligand may be an agonist of ERs, whereas a predominance of corepressors in another tissue releases the antagonistic effects of the same ligand [62]. Unlike the function of a cofactor to an enzyme, coregulators act as bridging or helper molecules that aid in forming large protein complexes to modulate appropriate activity on target gene chromatin. The detection of more than 200 coregulators for ER that are differentially expressed in many tissues can further confirm the tissue specificity of estrogen signaling [93]. Moreover, specific and unique conformational changes in the tertiary structure of the ER inherently resulting from phytoestrogen binding can modulate the recruitment of coregulator proteins. Both coactivators and corepressors are crucial for the subsequent transcriptional activity of ER after its dimerization and binding to specific response elements known as estrogen response elements (EREs), which are present in the promotor region of target genes [62,93]. For example, genistein acting on ERβ is more efficient in enhancing the transcriptional activity of ERs compared to the stimulation of ERα. The observed difference is derived from the more efficient recruitment of the p160 (SRC) steroid receptor coactivators TIF2 (SRC-2) and SRC-1a (NCoA-1) during ERβ activation. In general, the activation of ERβ has been shown to antagonize the cell growth-promoting effects of ERα. This scenario may be of importance in highly estrogen-sensitive tissues, especially in ERα-overexpressing cancers (e.g., breast tumors), wherein a potential protective action against estrogen-dependent cancer remains closely related to the ratio of active ERβ versus ERα [93–95]. PEs bound to ERs can also activate transcription at AP-1 binding sites that bind Jun/Fos transcription factors [96].
1.1.2. GPER signaling
The classic perception of ER receptors as ligand-activated transcription factors mediating long-term genomic effects in hormonally regulated tissues has changed, due to the fact that estrogens and PEs can also mediate rapid, nongenomic actions [97,98]. Such observations that the exposure of target tissue cells (including human endometrium) to estrogenic ligands can rapidly induce ion flows and the activation of various protein kinases across the plasma membrane independent of protein synthesis have led to the emergence of the concept of membrane ER [99]. Membrane-associated ER signaling pathways are typically associated with growth factor receptors and G protein–coupled receptors (GPCRs) [100,101]. A seven-transmembrane-domain receptor GPER (GPER1, first referred to as GPR30), which is a member of the G protein-coupled receptor (GPCR) superfamily, is one such first identified receptor that mediates estrogen-dependent kinase activation, as well as transcriptional responses [102,103]. Signaling through GPER occurs via the transactivation of the epidermal growth factor receptor (EGFR) and involves nonreceptor tyrosine kinases of the Src family [104]. The stimulation of GPER activates metalloproteinases and induces the release of heparin-binding epidermal growth factor-like growth factor (HB-EGF), which binds and activates EGFR, thus leading to the downstream activation of signaling molecules, such as the mitogen-activated protein kinases ERK1 and ERK2 [104–106]. In addition, 17β-estradiol-mediated activation of GPER stimulates cAMP production, intracellular calcium mobilization and PI3K activation [107,108]. The activation of signaling mechanisms involving cAMP, ERK and PI3K may be responsible for the indirect transcriptional activity of GPER, which represents another regulatory function in addition to the abovementioned rapid signaling events [97]. The indirect nongenomic signaling pathway via membrane-associated GPERs with the transactivation of EGFRs is shown in Figure 2B.
Several PEs, including flavones (e.g., quercetin), isoflavones (e.g., genistein), lignans, coumestans, saponins and stilbenes, can activate GPCRs [109]. For example, genistein and quercetin are able to stimulate c-fos expression in an ER-independent manner via GPER in ERβ-positive MCF7 and ERα-negative SKBR3 breast cancer cells [110]. However, PEs and mycoestrogens (e.g., zearalenone), even when displaying relatively high binding affinities for GPER and acting as agonists to increase cAMP synthesis, are more potent in activating ERα and ERβ [57,109]. In addition, some researchers have even suggested that the results obtained in vitro are not transferable to in vivo conditions; therefore, there is still a lack of evidence that GPER plays a significant role in mediating endogenous estrogen action in vivo [111]. The latter scenario may be due to the specificity of signaling via GPER and its intracellular localization. Namely, GPER is predominantly expressed on the membrane of the endoplasmic reticulum; thus, ligands must cross the plasma membrane to bind the receptor [112]. Thus, several studies have provided evidence demonstrating that a larger fraction of total cellular GPER is localized in intracellular compartments. These discrepancies regarding receptor localization may be partially caused by receptor trafficking between the endoplasmic reticulum and the plasma membrane during receptor biogenesis. The internalization of GPER in response to agonist stimulation should also be considered [113]. Moreover, GPER is made up of the same protein products of the genes that encode nuclear ERs. Specifically, membrane and nuclear ERs are derived from the same transcripts, but the former type is directed to the membrane via palmitoylation. The palmitoylation of the Cys447 residue of the ERα–ligand-binding domain (ERα-LBD) and Cys399 residue of ERβ-LBD through intermediary heat shock protein 27 enables the interaction of ERs with the caveolin-1 protein, which is required for the transport of GPER components to caveolae rafts within the cell membrane [63]. Palmitoylated ERs are translocated to the membrane as monomers, and the dimerization of GPER occurs within seconds of E2 exposure, which results in the activation of G protein α and βγ subunits (Gα and Gβγ, respectively) in a cell-type-dependent manner [63,114]. Subsequently, the depalmitoylation or weakening of the caveolin-1-receptor interaction causes the redistribution of ERs and their association with adaptors and/or signaling proteins, including proline-, glutamic acid-, and leucine-rich protein 1 (PELP1), which is also known as modulator of non-genomic activity of ER (MNAR), proto-oncogene tyrosine-protein kinase Src and tyrosine kinase receptors [63]. This correspondingly and ultimately contributes to the activation of the extracellular signal-regulated kinase/mitogen-activated protein kinase (ERK/MAPK) and phosphatidylinositol 3-kinase/serine-threonine kinase/mammalian target of rapamycin (PI3K/AKT/mTOR) signaling cascades, with respective effects on cellular proliferation, migration and other estrogen-dependent processes [115,116].
1.1.3. Signaling not mediated by ERs – a significant source of differences in bioactivity between E2 and PEs
Estrogens, including PEs, may also exert biological effects without interacting with ERs. The activation of ERs by ligand-independent mechanisms involves the recruitment of different sets of cofactors. In the ligand-independent signaling pathway, ERs are phosphorylated/activated by other active signaling cascades in a cell [66]. For example, growth factors or cyclic adenosine monophosphate (cAMP) activate receptor tyrosine kinases and intracellular kinase pathways, thus leading to MAPK activation with subsequent estrogen-independent phosphorylation of ERs (Figure 2C). This activation results in both direct ERE- and non-ERE-dependent genomic actions [67,68].
The activation of serotoninergic receptors and insulin-like growth factor receptor 1 (IGFR1), as well as the stimulation of free radical species binding and DNA methylation, are well-documented actions of PEs that do not involve ERs [46,55]. Moreover, in this mode of action of PEs, modified activities of tyrosine kinases, cycle adenosine monophosphate (cAMP), phosphatidylinositol-3 kinase/Akt and mitogen-activated protein (MAP) kinase transcription of nuclear factor-kappa β (NF-κB) should be expected. Together with the confirmed participation of PEs in the regulation of the cell cycle and apoptosis via ERs, these ER-independent activities cause PEs to possess antioxidant, antiproliferative, antimutagenic and antiangiogenic properties [117,118]. In clinical practice, this scenario translates into better or worse documented potential health benefits, including the alleviation of menopausal symptoms (e.g., hot flashes, night sweats, sleep problems and mood changes) and a reduced risk of osteoporosis, heart disease, neurodegenerative processes and breast cancer [46,119–122]. This last effect is still somewhat controversial because some clinical studies have reported of data that suggest that isoflavones may increase breast cancer incidence in sensitive individuals via their estrogenic and proliferative effects [123–125]. The use of PEs in the prevention and management of type 2 diabetes is also the subject of clinical research [126].
1.2. Phytoestrogens (PEs) as Endocrine Disrupting Chemicals (EDCs)
Adverse health effects should be expected following dietary intake of considerably high amounts of PEs because PEs may act as endocrine disruptors [127–130]. Consequently, the question of whether PEs are beneficial or harmful to human health remains unresolved. Given that the worldwide consumption of PEs is continually expanding, clarity on this subject is essential. The answer is likely complex and may depend on parameters such as age, health status and even the presence or absence of specific gut microflora [130,131].
Similar to other EDCs, PEs exhibit a wide spectrum of abilities for disrupting hormonal regulation of homeostasis. The most fundamental mechanisms of such potentially detrimental activity include: ❶ acting as a ligand at the binding sites of the hormone and mimicking the effects of the most specific endogenous ligand; ❷ antagonizing the effects of endogenous hormone by blocking its interaction at physiological binding sites; ❸ reacting directly and indirectly with a given hormone; ❹ altering the natural patterns of production and degradation of hormones; and ❺ disturbing cellular hormone receptor expression [132,133].
PEs behave as weak estrogen mimics or as antiestrogens. Despite the beneficial actions mentioned in the previous section, the supporting evidence that dietary intake of PEs is beneficial is indirect and inconsistent [47,48]. Moreover, it has been demonstrated that lifetime exposure to estrogen-like compounds, particularly during critical periods of development, has been associated with the formation of malignancies and several anomalies of the reproductive system [48]. PEs in maternal blood can pass through the placenta to the fetus in high amounts and can exert long-term effects, including adverse effects with consequences observed in postnatal life [134]. In addition, PEs are commonly found in pregnant women’s amniotic fluid. There is a sex difference in the concentrations, with higher levels observed in amniotic fluid containing female fetuses. This difference was not present in the maternal serum [135]. Moreover, soy ingestion increases amniotic fluid phytoestrogen concentrations in female and male fetuses [135]. The rapid transfer from the mother to the fetus was demonstrated for the phytoestrogen daidzein (which is an important representative of isoflavonoids in soya products) in pregnant rats. After the intravenous administration of daidzein to the mother, its concentration in the placental tissue and fetal liver amounted to 1/10 and 1/30 of the peak concentration of the maternal liver, respectively [134]. Exposure to a phytoestrogen-rich mesquite (Prosopis sp.) pod extract during the periconception and pregnancy periods in rats significantly affected the reproductive functions of male and female descendants. Furthermore, alterations in estrous cycles, decreased sexual behavior, estradiol and progesterone levels and increased uterine and vaginal epithelia were observed in females. In males, a decrease in sexual behavior, testosterone and sperm quality, as well as increased apoptosis in testicular cells, have been reported [134]. All of these effects were similar to those caused by daidzein. These results may indicate that prenatal exposure to mesquite pod extract or daidzein administered to females before and during pregnancy can disrupt normal organization, activation and behavioral programming with respect to reproductive physiology in female and male descendants [136,137].
The ingestion of genistein, which is a soybean-originated isoflavone, may modulate leptin hormone, C-reactive protein, tyrosine kinase activities and thyroid functions [138]. Similar to other EDCs, genistein produces a biphasic response in target cells. For example, depending on the concentration of genistein in the plasma of individuals consuming different amounts of soy dietary products (including soy supplements), cardioprotective (even if controversially reported) or cardiotoxic effects should be expected. The latter effects are related to much higher concentrations of genistein in the plasma (1–10 µM versus <1 µM) that produce potent inhibition of many membrane and cytosolic tyrosine kinases by competitively binding the ATP-binding sites of these kinases [138,139]. Soy PEs can also adversely affect thyroid function in susceptible individuals because in vitro studies have demonstrated that these compounds inhibit thyroid peroxidase (TPO), which is an enzyme involved in the synthesis of triiodothyronine (T3) and thyroxine (T4) [140,141]. In clinical settings, it has been established that patients with subclinical hypothyroidism receiving PEs in the diet are at higher risks of developing the overt form of the disease [142]. However, even with a higher utilized dose, a later study by the same team of researchers failed to confirm these findings [143]. In the most recently published study on rats, consumption of relevant doses of soy isoflavones during the peripubertal period in males induced subclinical hypothyroidism, with alterations in the regulation of the hypothalamic-pituitary-thyroid axis, modulation of thyroid hormone synthesis and peripheral alterations in thyroid hormone target organs being observed [144].
Genistein may also adversely affect fetoplacental development. It has been proposed that the fetoplacental growth disruption pathomechanism of genistein involves its interference with placental growth factor (PlGF) signaling [145]. In vitro data have shown that both genistein and daidzein may bind to uterine ERs and induce either anti-estrogenic or weak estrogenic effects (higher and lower concentrations, respectively), thus influencing uterine responsiveness to oxytocin (OT) and prostaglandin F2-alpha (PGF2-ɑ) and the corresponding contractility of the uterus [146]. The results of studies on human term trophoblast cells in vitro have shown that genistein and daidzein sufficiently reduce progesterone production in trophoblast cells via the disruption of estrogen receptor activity. Given that the blockade of progesterone is a possible mechanism involved in the initiation of labor, high doses of PEs at the feto-maternal unit could play a negative role in the maintenance of pregnancy. The compensatory mechanism observed in response to these PEs included higher estrogen production by trophoblast cells [147]. The clarification of whether a phytoestrogen-rich diet in pregnancy may pose an increased risk of preterm uterine contractions and subsequent preterm delivery requires further investigation.
Genistein exposure of infants may occur at physiologically relevant concentrations in the human diet that can be reached by using soy-based infant formulas. Infants consuming these products have serum genistein levels that are almost 20 times greater than those seen in vegetarian adults [148,149]. Importantly, the much weaker estrogenic activity of PEs can be compensated for by their high concentration in the body. For example, infants on soya formula can have plasma levels of isoflavones as high as 1000 ng/ml, which is 13,000–22,000 times higher than their own endogenous estrogen levels, as well as 50–100 times higher than estradiol levels in pregnant women and approximately 3,000 times higher than estradiol levels at ovulation [132,150,151]. Consistently, plasma isoflavone levels in infants fed cow’s milk formula or human breast milk were much lower (9.4 and 4.7 ng/ml, respectively) than those in soy-based infant formula consumers [132,149]. To date, there have been no extensive studies on the potential endocrine-disrupting adverse effects of soya products in infants; however, the problem should not be ignored. Most of the recent animal studies have shown that comparable exposures have adverse physiological effects [152]. A previous study on mammals has shown that individuals from a population subjected to a high consumption of isoflavones developed alterations in characteristics that may be of importance from an evolutionary perspective, such as epigenetic and morphometric characteristics or sexual maturation, which represents a life history characteristic [153]. It is likely that the most severe effects of hormonal disruption occur especially during a steroid hormone-sensitive period termed “minipuberty” when estrogenic chemical exposure (including isoflavone exposure) may alter normal reproductive tissue patterning and function [154]. Minipuberty is the transient sex-specific activation of the hypothalamic-pituitary-gonadal (HPG) axis during the first 6 months after birth in boys and during the first 2 years in girls. During the course of this important genital organ development period, increases in luteinizing hormone (LH), follicle-stimulating hormone (FSH), E2 and testosterone are observed [153,154]. There are more data supporting the hypothesis that disruption of development during this infant period in females may increase the risk of endometriosis in adulthood [153]. Moreover, developmental exposure to PEs may promote sensitivity to estrogen signaling diseases, including uterine fibroids and endometriosis. According to the results of population studies on soy phytoestrogen exposure, especially endometriosis, an estrogen-driven disease may have a developmental origin. In a study of 340 females diagnosed with endometriosis and 741 endometriosis-free, population-based controls, infant soy formula consumption was associated with over twice the risk of developing endometriosis relative to unexposed females [155,156]. The soy formula-exposed group was even at a higher risk of developing endometriosis compared to gestational parental exposure to diethylstilbestrol (DES), which is the compound with endometriosis induction efficacy that has been demonstrated in several epidemiological and animal studies [157–159]. There is still a need to understand the molecular mechanisms and to investigate how PEs can influence epigenetic patterns during development.
Due to its prevalence and well-known estrogen-like effects, another family of dietary EDCs produced by fungi called mycoestrogens should be mentioned. The compounds known as mycotoxins are found in poorly stored cereals. For example, natural products with estrogenic activities found in Fusarium crookwellnese (syn. Fusarium cerealis) include zearalenone, alpha-trans-zearalenol, beta-trans-zearalenol, fusarin, fusarenone X and nivalenol [160,161]. Zearalenone, which is a mycotoxin with a structure similar to that of naturally occurring estrogens, consists of a resorcinol moiety fused with a 14-member macrocyclic lactone and is the best-known representative of this group of EDCs [162]. Exposure to zearalenone and fusarin C has been linked to increased cancer rates. In in vitro studies, both fusarin C and zearalenone and its metabolites could stimulate the growth and proliferation of human breast tumor cells [163,164]. In addition, in vivo exposure of rats to environmental doses of zearalenone in the last two to three weeks of fetal development and in the first days after birth resulted in long-term changes in the development of the mammary gland, which was also associated with increased risks for the development of mammary tumors [47,165]. The ingestion of a sufficiently high dose of zearalenone in the diet may pose a risk to human health, not only because of its genotoxicity but also because of other adverse effects, including reprotoxicity and oxidative stress [166–168].
The results of studies on the involvement of zearalenone and other estrogenic mycotoxins, as well as Pes, in the etiopathogenesis of endometriosis are ambiguous [164,167,168,170]. In conjunction with the ability of PEs to induce anti-proliferative, anti-inflammatory and proapoptotic effects on cultured endometrial cells, beneficial effects have been reported in in vitro studies related to the inhibition of the spreading of endometriotic foci [46]. It has been proposed that this in vitro action of PEs involves the alteration of cell cycle proteins, the activation/inactivation of regulatory pathways and the modification of radical oxidative species levels [47,171]. However, in the case of zearalenone, a dual role and opposite effects on endometrial cells may be observed, which is dependent on the estrogen concentrations in the environment. Therefore, zearalenone acts as an antagonist and an inducer of apoptosis in endometriotic tissue when estrogen is sufficient; however, it transitions to estrogenic activity in the absence of estrogen during the development of endometriosis [170]. The results derived from animal models of endometriosis have generally supported a beneficial effect of the PEs in reducing lesion growth and development [169,171]. However, it is significant that the large amount of in vitro and in vivo animal findings did not correspond to a consistent literature regarding the women affected with endometriosis. Therefore, whether the experimental findings can be translated to women is currently unknown [47,159,169].
When regarding the etiopathogenesis of endometriosis, it may be important that endocrine disruption through GPER is linked to rapid epigenetic effects because the heritable, regulatory elements of a genome (exclusive of its primary DNA sequence) play an essential role in maintaining the correct, undisturbed development of the organism and influence its homeostasis [172,173]. Recently, evidence has emerged that epigenetics appears to be a common denominator for hormonal and immunological aberrations in endometriosis [174,175]. Moreover, the regulation of expression of all known estrogen-responsive and progesterone (P4)-responsive receptor types by epigenetics may be a critical factor for endometriosis [176].
1.2.1. Endocrine disruption and altered immune function
Interactions of PEs with estrogen receptors that correspond to endocrine disruption may influence any aspect of hormone action. It is becoming increasingly clear that EDCs (including Pes) not only affect endocrine function but also adversely affect immune system function [177]. Importantly, in endometriosis, which is an estrogen-dependent and progesterone-resistant chronic inflammatory disease, the immune system fails to recognize and target endometrial tissue growing in ectopic locations (outside of the uterine cavity) in the body. This failure may indicate that endometriosis is an immune disease [178,179].
In general, PEs can suppress the immune response both in vivo and in vitro. This effect is due to their ability to inhibit nuclear factor kappa-light-chain-enhancer of activated B cell (NF-κB) intracellular signaling pathways [180,181]. NF-κB is a crucial transcription factor that participates in a number of physiological and pathological conditions, including the immune response, apoptosis, carcinogenesis and inflammatory processes [182]. PEs (e.g., genistein) can suppress specific immune responses and lymphocyte proliferation [183]. Additionally, genistein can inhibit an allergic inflammatory response. In studies on mice, it has been shown that the administration of genistein in the diet produces reversible 46–67% decreases in the delayed-type hypersensitivity response, with reduced cell infiltrations in genetically treated animals compared with controls [184]. Genistein and daidzein, in particular, can suppress allergic inflammation by significantly reducing (by 25–30%) mast cell degranulation [185,186]. Consistently, the numbers of CD4+ and CD8+ T cells in normal lymph nodes were reduced on histopathological examinations. In contrast, it was demonstrated that genistein can increase cytokine production from T cells and enhance cytotoxic responses mediated by natural killers and cytotoxic T cells [187]. The treatment of activated dendritic cells (DCs) with genistein or daidzein led to increased NK-cell degranulation and cytotoxicity. This increased NK cell cytotoxicity was not influenced by other effects mediated by Pes, including reduced expression of IL-18 receptor alpha (IL-18Rα) and decreased production of interferon gamma (IFN-γ) in response to IL-12 and IL-18 [188].
Many studies have demonstrated that isoflavones and coumestrol can decrease the serum level of immunoglobulin G2a (IgG2a) antibodies. During experimental thyroiditis, low-dose coumestrol was able to decrease the titers of antigen-specific IgG1 and IgG3. Other isoflavones were effective in the suppression of IgE, thus possibly participating in the formation of the overall anti-allergic phenotype. Such a phenotype has been described in animal models, including airway and peanut sensitization models [185].
The vast majority of independent research has also demonstrated modulation concerning the inhibition of the innate immune system under the influence of PEs. Genistein, daidzein and glycitein are able to inhibit the production of IFN-γ, tumor necrosis factor alpha (TNF-α) and interleukins IL-9 and IL-13 by CD4+ T cells in response to interaction with DCs. Direct cytokine secretion from activated DCs was also inhibited by these PEs [189]. It was also shown in an intranasal allergic response model that PEs may temporarily block the cell surface expression of major histocompatibility complex class I (MHCI) (but not MHCII) molecules during the maturation of DCs. Thus, a significant delay in the immune response caused by altered antigen-presentation and effector-cell priming functions of DCs should be expected [185,188]. The anti-inflammatory action of PEs in DC lines is still under investigation, in conjunction with the dual response (either pro-inflammatory and anti-inflammatory) that is observed in NK cells.
Given that classically activated macrophages are products of a cell-mediated immune response, the proven anti-inflammatory phytoestrogen performance may be due to the fact that they make the full spectrum of macrophage activation more difficult [189,190]. Genistein and daidzein can decrease the synthesis of nitric oxide and the expression of inducible nitric oxide synthase (iNOS) with the accompanying increase in superoxide dismutase and catalase activities. Moreover, it has been demonstrated that genistein administration may alter macrophage polarization toward the noninflammatory M2 phenotype with a subsequent decrease in inflammatory cytokine concentrations [191]. M2 macrophages are necessary for the regulation of the resolution phase of inflammation and the repair of damaged tissues. In addition, genistein produces a strong expression of interleukin 10 (IL-10) in macrophages, which can limit the host immune response to pathogens, thereby preventing damage to the host and maintaining normal tissue homeostasis [192].
The complex action of PEs in relation to the innate immune system may explain the well-documented systemic anti-inflammatory effects of these xenoestrogens, including decreased allergic responses and decreased autoreactive immune responses [183,184,193]. The consumption of soy is growing at a significant rate, and its immune effect is extended. As the immune system influences basic physiological processes, including metabolic health, it seems likely that evolutionary alterations will be observed. It is important to monitor this situation and, if necessary, to prevent possible long-term detrimental consequences because quantitatively or qualitatively enormous amounts of PEs may cause pathological and epigenetically inherited alterations/dysfunction to the immune system.
2. Endometriosis
2.1. General characteristics of the disease
The name "endometriosis" refers to the condition in which endometrial tissue grows outside of the uterine cavity [194]. Depending on the location of the endometriotic foci, an endopelvic or extrapelvic form of endometriosis is distinguished [195]. Abnormally implanted endometrial tissue is primarily found in the pelvis, including the ovaries, ovarian fossa, fallopian tubes, uterine wall (endometriosis genitalis interna or adenomyosis), broad ligaments, round ligaments, uterosacral ligaments, appendix, large bowel, ureters, bladder or rectovaginal septum [194,196,197]. Extrapelvic localization of endometriosis is uncommon, and the disease is still underdiagnosed. Nevertheless, several cases of endometriosis of the upper abdomen, abdominal wall, abdominal scar tissue, diaphragm, pleura, pericardium, liver, pancreas, lower and upper respiratory tract tissues (or even brain) have been reported [198–200].
Endometriosis affects 10–15% of women between the ages of 15-44 years and is associated with chronic pelvic pain, dysmenorrhoea, dyspareunia and infertility. Endometriotic foci contain tissue that is virtually the same in terms of biological properties as basal intrauterine endometrial tissue [201]. This tissue contains stromal cells, glands and smooth muscles and is innervated and vascularized, with the presence of blood and lymphatic microvessels [201,202]. The cells within endometriotic lesions express all of the receptors for estrogens (ERα, ERβ and GPER) and progesterone (PR-A and PR-B). Therefore, they react to hormonal changes during the menstrual cycle and are subjected to cyclical changes analogous to the endometrium, ranging from re-epithelization and proliferation to breakdown and desquamation. In the uterine cycle, this corresponds to the phases of proliferation, secretion and menstruation [203,204]. The lack of blood outflow from the extrauterine “trapped” endometrial cells may predispose patients to internal bleeding that remains on site. Such bleeding may be the starting point of the local inflammatory response, accompanied by pain and the development of more serious fibrosis-based complications [205]. Due to pain, the quality of life of women suffering from endometriosis may be significantly compromised. Additionally, fibrosis and scarring with the formation of adhesions will be elicited as a result of repair processes within inflamed endometriotic tissue and its vicinity [194,199,205]. The question that needs to be resolved is whether the inflammatory process favors the development of endometriosis foci or whether endometriosis foci induce the inflammatory process [206,207]. In addition to pain-related dysmenorrhea and dyspareunia, the disease makes it difficult to get pregnant and to have a successful pregnancy outcome [208,209]. Moreover, a higher incidence of cancer and autoimmune diseases has been linked to endometriosis [210].
Despite several decades of intensive investigation into the underlying etiology and pathogenesis of endometriosis, the current understanding of the disease remains unclear. Several theories for the pathogenesis of endometriosis have been elaborated or updated in recent years, including implantation (retrograde menstruation) and metaplasia of Müllerian-type epithelium (coelomic metaplasia) theories, as well as the induction theory (a combination of the previous two theories) that emphasizes the impact of unidentified substances released from shed endometrium that induce the formation of endometriotic tissue from undifferentiated mesenchyme [211,212]. The implantation theory has been supplemented with new data indicating that the endometrium contains a particular population of cells with clonogenic activity that resembles the properties of mesenchymal stem cells, in which the dysfunction of these cells may lead to the formation of initial endometrial lesions [213]. It has also been proposed that stem cells derived from bone marrow may be a primary source of endometriotic cells [214,215]. The most recent hypothesis suggests that endometriosis risk is driven by relatively low levels of prenatal and postnatal testosterone. Testosterone affects the developing hypothalamic-pituitary-ovarian (HPO) axis; moreover, at low levels, it can result in an altered trajectory of reproductive and physiological phenotypes that, in extreme cases, can mediate the symptoms of endometriosis [216]. In summary, endometriosis is a multifactorial disease with the involvement of genetic, immunological, hormonal, anatomical and environmental factors in different proportions [206,207] (Figure 3).
2.2. Disruption in estrogen and P4 signaling
Hormone release dynamics and the interplay between the main female sex steroid hormones, including estradiol (E2) and progesterone (P4), govern the periodic growth and regression of the endometrium. Thus, such a balance between E2- and P4-responsive signaling pathways creates an extraordinary environment for controlled tissue remodeling during the menstrual cycle. In normal endometrium, where estrogen and P4 signaling coordination is tightly regulated, this remodeling plays a key role in decidualization to allow for implantation during the window of receptivity, as well as, in the absence of fertilization, for the disintegration of the endometrium, thus leading to menstruation [217].
According to the implantation theory of endometriosis, which assumes the spreading out of endometrial stromal cells (EnSCs) with the menstrual blood to establish ectopic growth (endometriotic foci), there is a significant disruption in estrogen and P4 signaling, which commonly results in P4 resistance and E2 dominance [218]. Thus, a hormonal imbalance caused by the actual or relative excess of E2 throughout the menstrual cycle and the expression of their cognate nuclear receptors, the progesterone receptors (PR-A and PR-B) and estrogen receptors (ERɑ and ERβ) deserves attention [219]. Moreover, the mutual affinity of nuclear receptors for the main female sex steroid hormones is necessary, given that the interaction of two domains of the P4 receptor with ER is required for P4 activation of the proto-oncogene tyrosine-protein kinase Src/extracellular signal-regulated kinase (c-Src/ERK) pathway in mammalian cells [220]. Additionally, sex steroid membrane receptors that are responsible for rapid nongenomic signaling/responses have garnered attention, also in the context of endometriosis. It has been demonstrated that P4 affects cell proliferation and survival via nongenomic effects. In this process, membrane progesterone receptors (mPRα, mPRβ, mPRγ, mPRδ and mPRε) were identified as being putative G protein-coupled receptors (GPCRs) for progesterone [221]. Similarly, the G protein-coupled estrogen receptor (GPER) is a seven-transmembrane-domain receptor that mediates nongenomic estrogen-related signaling. After ligand activation, GPER triggers multiple downstream pathways that exert diverse biological effects on the regulation of cell growth, migration and programmed cell death in a variety of tissues, including the human endometrium [109,204].
It is worth noting that chronic stress and inflammation also lead to a further imbalance between P4 and estrogen, thus exacerbating the course of preexisting endometriosis [222].
2.2.1. Estrogen dominance
The symptoms of estrogen excess and estrogen dependence in endometriosis are striking. This observation is limited to endometrial tissue and ectopic endometrial foci because the intratissue estrogen concentrations do not reflect the corresponding serum levels [219,223].
Absolute or relative hyperestrogenism, which is well documented in endometriosis, can also confirm the fact that estrogen-dependent endometriosis is rarely diagnosed after menopause when the symptoms and endometriotic lesions are typically relieved [224]. Similarly, during pregnancy, when estrogen action is oversuppressed by the influence of P4 or while taking hormonal contraceptives (e.g., via the use of ethinylestradiol-containing pills) that cause pharmacological suppression of endogenous estrogen synthesis, the severity of the disease usually decreases [225,226].
2.2.1.1. Aromatase activity
Aromatase (EC 1.14.14.1), which is also known as estrogen synthetase or estrogen synthase, is a unique rate-limiting enzyme that transforms androgen precursors into estrogens via aromatization. This member of the cytochrome P450 family (CYP) and the product of the CYP19A1 gene is responsible for the conversion of androstenedione, testosterone and 16-hydroxytestosterone into estrone (E1), estradiol (E2) and estriol (E3), respectively [227]. The most potent endogenous estrogen E2 exhibits extremely strong mitogenic properties in endometriotic tissue. Hence, any alterations in aromatase activity will produce a shift in the balance between estrogenic and androgenic effects within responsive tissues. Not coincidentally, the growth of ectopic endometrial tissue requires high aromatase activity induction, which is normally not detectable in eutopic (located in the proper place as the inner lining of the uterus) endometrium [228]. In contrast to normal endometrium, where estrogens are not locally produced, endometrial stromal cells (EnSCs) isolated from women with pelvic endometriosis exhibit significantly high P450 aromatase mRNA expression levels [229].
Analogous to breast cancer, abnormally expressed aromatase in EnSCs within endometriotic foci may be stimulated by prostaglandin E2 (PGE2) via the promoter II region of the aromatase gene. When considering the fact that PGE2 is one of the best-known mediators of inflammation and pain, the local production of estrogens will be accompanied by the typical pain of the disease. Moreover, a positive feedback loop (aromatase-PGE2-aromatase) is established because estrogen itself upregulates cyclooxygenase 2 (COX-2) and subsequently stimulates PGE2 formation [227–229].
It has been documented that the hyperestrogenic nature of the microenvironment within endometriotic lesions is the derivative of an epigenetic regulatory mechanism action involving the aromatase gene (CYP19A1), which is located on chromosome 15q21. Thus, endocrine disruption by dietary PEs may be important as an epigenetic modulator of estrogen signaling at the level of endometrial foci. Additionally, multiple exons of CYP19A1 may be alternatively used in endometriotic cells corresponding to EnSCs that exploit identical aromatase promoters (promoters II, I.3 and I.6) as aromatase-negative eutopic endometrial cells [175,230,231]. Given that endometriotic stromal cells are equipped with the same set of promoters as normal eutopic EnSCs, the differences in aromatase gene expression may be caused by an epigenetic regulatory mechanism that inhibits aromatase gene expression in healthy endometrium, whereas this effect is not present in endometriosis. The confirmation of the abovementioned effect may be the fact that CpG islands (the regions of the genome that are rich in promoters) are hypomethylated in endometriotic cells and hypermethylated in endometrial cells [232]. DNA methylation is strictly linked to histone modifications and the recruitment of histone deacetylases (HDACs), followed by chromatin condensation. It is generally accepted that hypomethylated genes possess an increased potential for expression compared to hypermethylated genes [233]. Thus, the differential expression of the aromatase gene between normal intrauterine endometrium and endometriotic foci may be due to the absence or presence, respectively, of the transcription factor known as steroidogenic factor 1 (SF-1). It has been found that methylation of CpG islands in the SF-1 gene, which spans from exon II to intron III, positively regulates its expression in EnSCs in endometriosis, whereas hypomethylation of SF-1 gene CpG islands in eutopic endometrium drastically decreases SF-1 levels [234,235].
Deficient 17β-hydroxysteroid dehydrogenase type 2 (17β-HSD2) expression is another abnormality that has been reported in endometriosis, and it predisposes afflicted individuals to hyperestrogenism. Normally, the accumulation of increasing quantities of E2 in target tissues is counteracted by the conversion of adequate levels of 17β-estradiol to much less potent estrone (E1) [236]. This pathway of E2 inactivation is disrupted in ectopic EnSCs via hypermethylation of the 17β-HSD2 gene, thus resulting in insufficient 17β-HSD2 activity within endometrial lesions [237]. Although of unknown importance in endometriosis, it should be mentioned that the same epigenetic mechanism (e.g., DNA methylation) is likely to influence the activity of 17β-hydroxysteroid dehydrogenases type 1 and 4 (17β-HSD1 and 17β-HSD4, respectively), which are enzymes present in the human endometrium and EnSCs [238,239].
All of these interrelationships between epigenetic modulators of aromatase activity and hyperestrogenism are summarized in Figure 4.
2.2.2. The importance of epigenetic factors
The epigenome is defined as the complete description of all of the chemical modifications to DNA and histone proteins that regulate the expression (activity) of genes within the genome without interfering with the DNA nucleotide sequences, and it encompasses both small and long noncoding RNAs (miRNAs and lncRNAs, respectively) [240,241]. Epigenetic changes occur regularly and naturally in response to aging, the environment/lifestyle and disease states. Furthermore, this phenomenon aims to maintain genomic integrity [242,243].
The properties of cellular targets for epigenetic factors in endometriosis are very particular because EnSCs with clonogenic potential constitute the most abundant population of cells within the endometrium and endometriotic tissue that resemble the properties of mesenchymal stem cells (MSCs) [244]. The unique nature of stem cells involves the ability to divide and renew themselves for long periods of time, as well as unspecialization and the capability of differentiating into specialized cell types [245]. Therefore, stem cell plasticity causes the precise control of both metabolism and gene expression to be rapidly adjusted to varying conditions (e.g., hormonal status and the phase of the menstrual cycle), including environmental factors related to dietary intake of PEs and other compounds with endocrine-disrupting potential [169,175,246,247].
The failure of epigenetic homeostasis in the endometrial tissue may demonstrate local intrauterine abnormalities or a generalized systemic disorder during repeated menstrual cycles or pregnancies [215,248]. Research results from recent years have determined that the regulation of ERs and P4 receptor expression by epigenetics may be a critical factor for endometriosis [176,249,250]. Specifically, disrupted estrogen and P4 signaling that correspond to increased estrogen activity and P4 resistance, respectively, are the main substrates of the disease, wherein environmental factors contribute to the inflammatory response and debilitating symptoms, including pain and infertility.
2.2.2.1. Epigenetic modulation of ERs in endometriosis
It has been demonstrated that ERs in EnSCs are subjected to the same epigenetic regulation as in other estrogen-reactive tissues [235,251,252]. In human endometriotic stromal cells corresponding to EnSCs, markedly higher levels of ERβ and lower levels of ERα have been reported compared to EnSCs obtained from eutopic endometrium [253,254]. Such overexpression of ERβ in endometriosis has been linked to significantly pathologically reduced methylation of a CpG island in the promoter region of the ERβ gene (ESR2). Conversely, bisulfite sequencing of this region has identified significantly higher methylation in primary endometrial cells versus endometriotic cells [255]. Consequently, the experimental use of a demethylating agent can significantly increase ERβ mRNA levels in endometrial cells. Moreover, the overexpression of ERβ in endometriosis correspondingly suppresses ERα expression and response to E2 in EnSCs by binding to nonclassical DNA motifs in alternatively used ERα promoters [203]. Thus, the normal response pertaining to ERα expression in endometriotic lesions is suppressed by both abnormally high quantities of E2 resulting from local aromatase overactivity and the epigenetic upregulation of ERβ in stromal cells [256]. When considering that the P4 receptor (PR) gene is induced in reproductive tissues by estrogen acting via ERα, the decreased expression of ERα observed in endometriosis may contribute to P4 resistance, which is a typical feature in women suffering from this disorder [203,257].
The proliferation of endometriotic lesions can also be linked to severely increased ERβ mRNA levels in EnSC- and/or MSC-derived endometriotic cells following DNA demethylation because ERβ signaling stimulates cell cycle progression [258].
Extraordinarily higher ERβ and significantly lower ERα and PR expression in endometriotic stromal cells compared with endometrial stromal cells may be caused by another epigenetic mechanism related to small (19–25 nucleotides long), single-stranded noncoding RNAs (miRNAs) that regulate gene expression. This dominant pool of RNA does not code for proteins but is processed to produce functional RNAs, and miRNAs are crucial regulators of gene expression in E2-treated human endothelial cells [259,260].
Based on animal models and human studies, ER expression during the different phases of the menstrual (endometrial) cycle is modulated by miRNAs [259,261]. These data relate especially to the numerous miRNAs that directly target ERα, whereas less information is available for miRNAs modulating ERβ and GPER [262–265].
Nevertheless, results indicating that GPER-mediated downregulation of miR-148a expression through the GPER/miR-148a/HLA-G signaling pathway may mediate the development of ovarian endometriosis have recently been published [266]. In addition, the epigenetic regulation of ER expression by miRNAs coexists with opposing mechanisms that act in parallel, such as the ER-mediated regulation of miRNA expression. For example, E2-treated human umbilical vein endothelial cells (HUVECs) have differentially regulated specific miRNAs via pathways related to both classical ERs (ERα and ERβ) and membrane-bound ERs (GPER) [260]. Among the most modified miRNAs, miR-30b-5p, miR-487a-5p, miR-4710 and miR-501-3p were overexpressed after E2 treatment, whereas miR-378 h and miR-1244 were downregulated [260].
In addition to miRNAs, researchers studying the epigenetic regulation of estrogen signaling have recently focused on the role of some transcripts longer than 200 nucleotides that lack protein coding potential and transcribed by RNA polymerase II (RNA Pol II), which are known as long noncoding RNAs (lncRNAs) [267]. Together with the research progress on lncRNAs, there is increasing evidence that by regulating the epigenetic status of protein-coding genes, lncRNAs are involved in the pathogenesis of endometriosis [268]. For example, the upregulation of lncRNA HOTAIR is caused by E2 binding to ERα and ERβ. Moreover, coregulators, including histone methyltransferases (MLL1 and MLL3) and histone acetylases in the p300–CBP family, are recruited together with ERs to bind estrogen response elements in the HOTAIR promoter in response to E2; additionally, they are necessary for the upregulation of HOTAIR [269].
As was previously mentioned (see Chapter 1.1.1. Signaling via nuclear receptors), estrogen signaling involves the recruitment of many coregulator proteins (coactivators and corepressors) that interact with many members of nuclear receptor-related multifunctional protein complexes, thus resulting in both transcriptional and epigenetic changes. The latter changes include (but are likely not limited to) chromatin density changes, histone modifications by acetylation/deacetylation and DNA methylation/demethylation, as well as noncoding RNAs. Therefore, the expression of ERs in health and disease may depend on the recruitment of comodulators that are crucial for the activities of the respective acetyltransferases (e.g., p300-CBP and its paralog p300; GNAT or GCN5-related N-acetyltransferase, nuclear receptor coactivator-NCOA-related histone acetyltransferase) and methyltransferases (e.g., histone-lysine N-methyl-transferases and histone-arginine N-methyltransferases) [270–272].
Interestingly, being classified as a lncRNA, steroid receptor RNA activator (SRA), which acts as the nuclear receptor coactivator, can influence the activities of both ERα and ERβ [273]. High expression levels of SRA lncRNA and ERβ (but relatively low expression levels of SRA and Erα) have been demonstrated in ovarian endometriotic tissues compared to normal endometrium. In conjunction with the abovementioned findings, SRA1-small interfering RNA treatment significantly increased ERα levels but reduced ERβ levels in EnSCs. Such a treatment with interfering RNA reduced proliferation within ovarian endometriotic foci and promoted the early onset of apoptosis in endometriotic cells [274].
ER activity may be regulated by sirtuins (SIRTs), which possess histone deacetylase (HDAC) activities and act as comodulators of both estrogen-regulated gene silencers and inhibitors of ligand-dependent activation of ERα [275]. The overexpression of SIRT1 may contribute to both the pathomechanism of endometriosis and P4 resistance [276]. Interestingly, eutopic end ectopic endometrial tissues obtained from the same patient differ in the content of SIRT1. Significantly decreased levels of SIRT1 mRNA were demonstrated in eutopic EnSCs compared to EnSCs from endometriotic lesions [277].
When considering that complex and nonuniform mechanisms of estrogen/ER signaling within endometrial cells are subjected to significant modulation by epigenetic factors, endocrine disruptors may induce pathological regulatory mechanisms that are responsible for ectopic EnSC persistence and the development of endometriotic foci [278–281].
2.3. Estrogen-dependent immune system interactions in endometriosis
First, the immune system is responsible for eliminating cells that are located in ectopic sites (endometriotic foci). The failure of this elimination in endometriosis may be due to both resistance of ectopic cells to be eliminated by immune cells and a deficit in the immune response [209,211]. Numerous studies have demonstrated that endometriosis is associated with aberrant growth and loss of sensitivity to apoptosis of endometrial tissue cells [179]. This effect may be confirmed by an increase in the expression of anti-apoptotic proteins, such as Bcl-2, c-IAP1 and c-IAP2, in ectopic endometrial cells compared to eutopic endometrial cells [282]. Thus, apoptosis-inducing processes that are mainly related to interactions with immune cells (e.g., cytotoxic T lymphocytes [CTLs], also known as killer T cells) may be suppressed, thus promoting the survival and development of endometriotic lesions [283,284]. Estrogen excess observed in endometriosis can activate both epithelial and stromal cells that constitute the population of endometriotic cells, thus causing the anti-apoptotic status of the respective ectopic tissue [179,285]. This scenario is facilitated by the impact of estrogen excess on CD4 T-helper development and function, especially with regard to the profile of the produced cytokines [286]. The immunosuppressive functions of Tregs are widely acknowledged and have been extensively studied [287,288]. Altered CD4 T lymphocytes may lead to disturbances in the coordination of the immune response by inappropriately stimulating other immune cells, such as macrophages, B lymphocytes (B cells) and CD8 T lymphocytes (CD8 cells), to fight ectopic endometrial foci development [178,287,289].
It should be noted that at the current stage of research, it is not possible to distinguish to what extent observed alterations are intrinsic to the endometriotic cells or are induced by their ectopic location [284,290,291]. Moreover, it has been demonstrated in previous studies on cancer cells that estrogen acting through different ER isoforms can induce opposing mechanisms (i.e., antiapoptotic types that promote tumor growth and proapoptotic types that promote programmed cell death). Accordingly, it has been shown that the E2/ERα complex activates multiple pathways involved in both cell cycle progression and apoptotic cascade prevention, whereas the E2/ERβ complex in many cases directs the cells to apoptosis [292].
Excess estrogen has a strong effect on the immune response because the immune system is a natural target for these classes of sex steroid hormones, and immune cells express all types of currently known receptors [293]. Although the cause of sex differences in the immune system has not been definitively identified, possible causes should be investigated, including different sex hormone profiles (estrogens, androgens and differential sex hormone receptor-mediated pathways), X-chromosomes, microbiome and epigenetic factors. Females tend to have a more responsive and powerful immune system than members of the opposite sex. The consequence of the abovementioned scenario is a more aggressive response to self-antigens and a more frequent prevalence of autoimmune diseases among women [294,295]. For example, extremely higher estrogen concentrations in females compared to males drive increased T-cell IFNγ production and, in this manner, predispose females to IFNγ–mediated autoimmune conditions [296]. To date, clinicians do not consider endometriosis an autoimmune disease; however, it resembles an autoimmune condition in many aspects [179,297].
It has been well established that E2 signaling participates in the precise control of proinflammatory signal/pathway-related phenomena of the immune system [298–301]. Estrogen regulates key genes that are responsible for the innate and adaptive immune systems, and the list of immune cells that are subject to this regulation is almost complete, including granulocytes (neutrophils), monocytes (macrophages and monocyte-derived dendritic cells) and lymphocytes (T cells and B cells) [293]. For example, within the innate immune response, estrogen signaling modulates neutrophil numbers, migration, infiltration and activation via genes coding cytokine-induced neutrophil chemoattractant proteins 1-3 (CINC-1, CINC-2 and CINC-3) TNFα, IL-1ß and IL-6 [302–304]. In contrast, in macrophages, estrogen signaling may modify chemotaxis, phagocytic activity and induction of cytokines, iNOS and nitric oxide by affecting genes IL-6, TNFα, iNOS and NO, respectively [305–308]. In terms of the adaptive response, estrogen signaling modulates all subtypes of T cells, including CD4+ (Th1, Th2, Th17 and Tregs) and cytotoxic CD8+ cells (CTLs) [293,309,310]. For example, this modulation pertains to genes encoding interferon gamma (IFNγ) in Th1 cells, IL-4 in Th2 cells and FoxP3, PD-1 and CTLA-4 in Tregs [310–315]. Thus, there is no doubt that estrogen plays a major role in shaping T-cell responses. This action is observed independently of the direct disruptive effect on gene transcriptional programs of T cells and involves T-cell maturation, activation and differentiation [315,316]. Moreover, B-cell (B lymphocyte) differentiation, activity, function and survival are also highly dependent on estrogen, which can modify the expression of genes such as CD22, SHP-1, Bcl-2, and VCAM-1 [317,318]. In certain states, estrogen acting through either ERα or ERβ may contribute significantly to autoimmune disorders because a study on autoimmune mice subjected to estrogen demonstrated increased plasma cell and autoantibody-producing cell numbers [319]. However, signaling via ERα is crucial in altered cell maturation coexisting with autoimmunity [320].
Estrogens can indirectly inhibit NF-κB DNA binding, as they have been shown to inhibit IKK activation, increase IkappaB protein expression and decrease its phosphorylation [321–324]. ERα and GPER1 signaling is commonly associated with anti-inflammatory phenotypes, whereas data on ERβ signaling are not consistent, thus indicating both anti-inflammatory roles similar to ERα and GPER1 and proinflammatory effects in the case of an increased ratio of ERβ [293,325]. It may be important in the context of endometriosis that 17β-estradiol signaling via overexpressed ERα may inhibit inflammatory activation mediated by NF-κB and JNK via PI3K/AKT [326]. However, it is likely that reported differences in the effects of estrogen on the immune system are related to the timing at which such effects are observed following estrogen exposure, as well as variations in the respective type of ER expression in various cells and during different physiological or pathological conditions [284,293,324].
During the menstrual cycle of healthy women, increased concentrations of cytotoxic (CD8+) T lymphocytes (CTLs) and HLA-DR- activated T cells were observed in peripheral blood during the luteal phase compared to the follicular phase. These fluctuations in the concentrations of cytotoxic and activated peripheral blood lymphocytes are not present during the menstrual cycle of women with endometriosis [327]. Moreover, there has only been a marked increase in Treg concentration in the peripheral blood of women with endometriosis, which was positively correlated with the serum levels of cortisol [327]. In addition, a significant reduction in the cytotoxic/proapoptotic potential of CTLs was demonstrated in endometriosis, wherein the number of perforin+ CTLs among CD8+ T cells in the menstrual effluent was decreased compared to healthy controls. Perforin is a glycoprotein mediator of cytolysis that is responsible for pore formation in cell membranes of target cells, thereby causing the initiation of programmed cell death [328,329]. Perforin mRNA levels correlate with the methylation status and accessibility of the promoter at the 5′ flanking region of its gene. Thus, the defective apoptotic process may be caused by DNA hypermethylation and changed chromatin structure that negatively affects perforin gene expression in T cells [179,330].
2.3.1. Estrogen and mast cells (MCs) in endometriotic lesions
MCs express estrogen (ERα, ERβ and GPER) and P4 receptors (PR-A and PR-B) and further respond to these hormones, which causes changes in the MC cell number, distribution and functional state in various tissues [331,332]. It should be noted that E2 is implicated in the immune response as an enhancer, including MC activation and the subsequent release of mediators stored in the secretory granules (degranulation) [333]. Among the ERs, GPER is responsible for the various running fast nongenomic effects of estrogens, including the degranulation of MCs [334]. The activation and degranulation of MCs significantly modulates many aspects of physiological and pathological conditions in various settings. MC secretory granules are lysosome-like organelles that contain a large panel of preformed bioactive constituents, including lysosomal hydrolases (e.g., carboxypeptidase A, chymase and tryptase), amines (histamine), cytokines (interleukin [IL]-1, IL-2, IL-3, IL-4, IL-5, IL-6, granulocyte-macrophage colony stimulating factor, interferon-γ [IFN-γ] and tumor necrosis factor-α [TNF-α]) and proteoglycans (e.g., heparin) [335,336]. These mediators are responsible for many of the acute signs and symptoms of MC-mediated allergic inflammatory reactions, including edema, bronchoconstriction and increased vascular permeability [336,337]. In addition, MCs are involved in angiogenesis, fibrosis and pain, and a significant increase in MC numbers within endometriotic lesions has been demonstrated compared to matched eutopic endometrium from the same patients [338,339]. Furthermore, endometriotic tissue specimens demonstrate significantly higher expression of stem cell factor (SCF), which is a potent growth factor critical for MC expansion, differentiation and survival for MCs localized in connective tissue [340]. Following pretreatment with estrogen in mice, the endometriotic foci demonstrated a higher density of Alcian blue-stained MCs. In patients with endometroid endometrial cancer, MC density was positively correlated with angiogenesis, as assessed by local microvascular density [341].
The abovementioned results indicate that the conditions that are characteristic of the disease (particularly, the abnormal hyperestrogenic/P4 resistant endocrine microenvironment within endometriotic lesions) promote recruitment and differentiation of MCs. As a result, MCs may release a diverse spectrum of mediators that contribute to inflammation, chronic pelvic pain and local angiogenesis, thus resulting in disease progression [339,340,342]. Due to the fact that MCs are very prevalent in endometriotic tissue, it has been proposed that this population of MCs represents a therapeutic target in endometriosis to assure better control of disease inhibition and symptom relief [343].
The multifunctional nature of MCs includes their involvement in the regulation of innate and adaptive immune responses. Increasing evidence has suggested that MCs play a regulatory role in inflammatory diseases (such as endometriosis) by regulating T-cell activities. In addition to serving as effector cells, MCs are able to induce T-cell activation, recruitment, proliferation and cytokine secretion in an antigen (autoantigen)-dependent manner and to impact regulatory T cells [344,345].
Estrogen-dependent immune responses related to MCs in endometriotic foci are shown in Figure 5.
3. Dietary PEs and endometriosis
3.1. PE intake and the risk of endometriosis – interactions at the level of gut microbiota
The gut microbiota or gut microbiome consists of microorganisms, including bacteria, archaea, fungi and viruses, living in a state of dynamic equilibrium in the digestive tract. The aggregate of all of the genomes of the gut microbiota is known as the gastrointestinal metagenome, and it is very large [347]. For example, the microbiota “organ” is the central bioreactor of the gastrointestinal tract, and it is populated by a total of 1014 bacteria and characterized by a genomic content (microbiome), which represents more than 100 times the human genome. Bacteria account for up to 60% of the dry weight of the feces [348,349]. Due to this scenario, symbiosis and dysbiosis of this dynamic ecosystem play an important role in health and disease, respectively [349]. Colonization of bacteria that make up the microbiome has a broad impact on resistance to pathogens, whereby it maintains the intestinal epithelium, metabolizes dietary and pharmaceutical compounds, controls immune function and even (to some extent) controls behavior through the gut-brain axis [350–352]. Moreover, when considering the plasticity of the gut microbiota, diet has emerged as a main contributor to the microbiota composition and functional capacity. The number of studies showing that food/nutrient-microbiota interactions are important modulators of host physiology and pathophysiology is constantly increasing [353]. Accordingly, a phytoestrogen-rich diet affects the composition of the gut microbial community (gut homeostasis) as an environmental epigenetic factor and provides metabolites that influence host physiology, including endocrine balance and the potential risks of endocrine disruption [137]. The microbiota undoubtedly functions as a full-fledged endocrine organ influencing the reproductive endocrine system throughout a woman’s lifetime by interacting with estrogen, androgens, insulin and other hormones [354]. For example, the mammalian PEs enterolactone and enterodiol are formed in the colon by the action of bacteria on plant lignans by matairesinol and secoisolariciresinol, which exist in various whole-grain cereals (barley, rye, and wheat), seeds, nuts, legumes and vegetables. Both enterolactone and enterodiol have been shown to possess weakly estrogenic and antiestrogenic activities, and it has been suggested that the increased production of these antiestrogenic mammalian lignans in the gut may serve to protect against breast cancer in women and prostate cancer in men [355]. Furthermore, the protective effects of these mammalian lignans may be due to their ability to compete with E2 for ERs, as well as to induce sex hormone binding globulin (SHBG), to inhibit aromatase and to act as antioxidants [356].
Due to the fact that the gut flora affects immune health by controlling inflammatory responses and plays an important role in estrogen metabolism and in the regulation of estrogen cycling, the onset and progression of endometriosis may be a consequence of gut dysbiosis. Such dysbiosis may modulate the estrobolome (the collection of genes encoding estrogen-metabolizing enzymes in the gut microbiome) with a subsequent increase in the levels of circulating estrogen, which may markedly stimulate the growth and cyclic bleeding within endometriotic foci [357]. The results of recent preclinical and clinical studies suggest that altered interactions between gut permeability and intestinal (as well as extraintestinal) bacteria collectively contribute to systemic inflammation and metabolism. According to the "leaky gut" concept, disturbances in the composition of the intestinal microflora may change gut permeability and predispose to alterations in different types of immune cells and inflammatory factors (e.g., increased levels of inflammatory cytokines in the peritoneal fluid and serum) in endometriosis [358,359]. When referring to the pathomechanism of endometriosis, once the balance between estrogen levels in the circulation and the gut microbiome is disrupted, increased estrogen exposure due to a phytoestrogen-rich diet can stimulate the development and progression of endometriotic lesions [360]. A strong interrelationship between immunological processes and endometriosis is confirmed by the observation that the risk of inflammatory bowel disease (IBD) in women with endometriosis increases by 50% [361]. In a murine model of endometriosis, dysbiosis of the gut microbiota was manifested by an elevated Firmicutes/Bacteroidetes ratio and an increased ratio of Bifidobacterium [362]. Another previous study in mice showed the effectiveness of broad-spectrum antibiotic therapy in limiting the inflammatory response and in inhibiting the growth of endometriotic tissue. Oral gavage of feces from mice with endometriosis restored endometriotic lesion growth and inflammation, thus indicating that gut bacteria may promote endometriosis progression in mice [363]. Additionally, a higher prevalence of intestinal inflammation coexisting with dysbiosis of gut microflora (lower lactobacilli concentrations and higher gram-negative bacterial load) was also documented in experimental endometriosis in rhesus macaques (Macaca mulatta) [364]. In a previous human study, it was found that women with stage 3/4 endometriosis excrete more Escherichia/Shigella in their stool compared to the control group, whereas the vaginal, cervical and gut microbiota compositions were similar [360].
In summary, the unmistakable connections between changes in the intestinal microbiome and gut homeostasis, intestinal permeability and inflammation deserve to be pursued in future research in the context of estrogen excess in endometriosis [354,365].
3.2. PE oral intake and the course of endometriosis – the results in animal models
The results of research obtained on animal models of endometriosis (with the exception of higher primates) should be treated with great reserve because the estrous cycle is not equal to the menstrual cycle; in addition, a typical, plant-based diet in rodents in the natural environment has a much higher phytoestrogen content than a typical nonvegan, nonvegetarian human diet [366,367]. Translational animal models for endometriosis developed in mice (female BALB/C) and rats (female Sprague Dawley or Wistar-Albino) conducted endometriotic cell transplantation into sites on the periteum or intestinal mesentery via intraperitoneal injections or homologous uterine horn transplantations [366]. Based on the review of the obtained results for different PEs, it can be concluded that these compounds that are orally administered can cause the regression of endometriotic implants.
Resveratrol is one of the most studied compounds. This nonflavonoid polyphenol that naturally occurs as a phytoalexin inhibited the development of experimental endometriosis in mice and reduced endometrial stromal cell invasiveness in vitro. Mice treated orally with resveratrol (6 mg/mouse; n = 20) for 18-20 days exhibited both statistically significant decreases in the number of endometrial implants and in the total volume of lesions (by 60% and 80% per mouse, respectively). It is worth noting that human endometrial stromal cells were used to induce endometriosis [368]. In another study on BALB/C mice with surgically induced endometriosis, resveratrol (40 mg/kg/day; n = 10) that was orally administered for 4 weeks inhibited angiogenesis in peritoneal and mesenteric endometriotic lesions, as indicated by a significantly reduced microvessel density when compared with controls. Decreased proliferating activity of CD31(+)-positive cells in the newly developing microvasculature of the lesions was also confirmed. This scenario coexisted with lower numbers of proliferating cell nuclear antigen- and Ki67-positive stromal and glandular cells. The authors noted limitations in translating the results into human conditions, which was caused by the mouse model that was used in the study [369]. Similar results have also been demonstrated in a study on the effects of resveratrol and another polyphenol known as epigallocatechin-3-gallate (EGCG) on the development of endometriosis in a BALB/C mouse model. Both treatments significantly reduced the mean number and volume of established lesions with corresponding diminished cell proliferation, reduced vascular density and increased apoptosis within the lesions [370]. The ability of dietary resveratrol to inhibit angiogenesis and inflammation in endometriosis was demonstrated in a study on 24 female Wistar-Albino rats medicated for 21 days. After the treatment, significant reductions in the mean areas of the endometriotic implants and mean VEGF-staining scores of the endometriotic implants were confirmed. Moreover, the plasma fluid and serum levels of VEGF and MCP-1 were also significantly lower in the resveratrol-fed group [371]. The effect of polydatin (PLD, which is a natural potent stilbenoid polyphenol that is a natural precursor of resveratrol), which was orally administered in comicronized form with palmitoylethanolamide (PEA, which is an endogenous fatty acid amide possessing anti-inflammatory activity, but unlike resveratrol, having no free radical scavenging activity), was examined in an autologous rat model of surgically induced endometriosis. After 28 days of micronized (PEA/PLD) treatment at 10 mg/kg/day, the rats (n = 10) displayed a smaller cyst diameter, with an improved fibrosis score and decreased mast cell number. The combined use of PEA and PLD resulted in decreased angiogenesis (vascular endothelial growth factor), nerve growth factor, intercellular adhesion molecule, matrix metalloproteinase 9 expression and lymphocyte accumulation. Furthermore, an anti-inflammatory effect was documented, as markers of inflammation were reduced, such as peroxynitrite formation, (poly-ADP)ribose polymerase activation, IκBα phosphorylation and nuclear factor-κB translocation in the nucleus [372].
Injections of the highest doses of genistein (50 μg/g and 16.6 μg/g of the body weight) sustained intestinal mesentery implants of uterine (endomteriotic) tissue in rats, whereas dietary genistein (250 or 1,000 mg/kg) and a lower dose (5.0 μg/g of the body weight) of this phytoestrogen did not support the implants. These results, which were obtained after 3 weeks of daily injections or exposure to dietary genistein, may indicate that ER modulation and genistein bioavailability play a critical role in the maintenance of endometriotic implants [373]. In another previous study on female Wistar-Albino rats, in which endometriotic implants were induced by transplanting autologous uterine tissue to ectopic sites on the peritoneum, the results in the study group (n = 10) subjected to the oral administration of genistein at 500 mg/kg per day exhibited statistically significant regression of the endometriosis. After 3 weeks, the decrease in the surface area of the endometriotic implants was confirmed during histopathologic examinations with morphometry [374].
In the mouse model of endometriosis established by transplanting donor-mouse uterine fragments into recipient mice, the administration of a diet containing a mixture of principal isoflavonoids of soy (daidzein + genistein + glycitein) significantly decreased the number, weight and Ki-67 proliferative activity of endometriosis-like lesions. According to the results of a parallel study in vitro on the effect of the combined administration of daizein, genistein and glycitein on stromal cells isolated from ovarian endometrioma, the indicated anti-endometriotic effects may be related to the reduced expression of IL-6, IL-8, COX-2 and aromatase, as well as reduced aromatase activity, serum glucocorticoid-regulated kinase levels and PGE2 levels [375].
Puerarin, which is a hydroxyisoflavone glycoside originally isolated from Pueraria lobata (Willd.), is an isoflavone substituted by hydroxy groups at positions 7 and 4', as well as a beta-D-glucopyranosyl residue at position 8 via a C-glycosidic linkage. The ability of this phytoestrogen to treat endometriosis was examined in female Sprague‒Dawley rats with endometriotic implants during 4 weeks of administration via oral gavage at doses of 600, 200 or 60 mg/kg per day. The endometriotic tissue weight and serum estrogen levels were significantly lower in the high-, medium- and low-dose puerarin treatment groups than in the control group. Moreover, even low-dose puerarin inhibited aromatase cytochrome P450 (P450AROM) expression and reduced estrogen levels in endometriotic tissue. Furthermore, three doses of puerarin had no adverse effects on the liver, kidney and ovary, whereas high-dose puerarin administration caused thinner bone trabeculae with distortion and breakage [376].
Xanthohumol is a prenylated flavonoid isolated from hops, and its effectiveness in the treatment of endometriosis was tested in BALB/C mice with surgically induced peritoneal and mesenteric endometriosis by uterine tissue transplantation into the abdominal cavity. After 28 days of daily treatment with 100 µM xanthohumol (n = 8) via the drinking water, a marked reduction in the diameter of the endometriotic lesions was observed (regardless of their location within the peritoneal cavity) compared with the control. This effect was accompanied by a reduced level of phosphoinositide 3-kinase (PI3K) protein. A significantly lower microvessel density documented within the xanthohumol-treated lesions indicates an inhibitory effect of this flavonoid on angiogenesis. Moreover, additional analyses demonstrated that treatment with xanthohumol did not affect the histomorphology, proliferation or vascularization of the uterine horns and ovaries. The lack of serious side effects in the reproductive organs may be an advantage when considering the treatment of endometriosis [377].
The beneficial effects of silymarin, which is a compound with potent phytoestrogenic, proapoptotic and antioxidative properties, have been confirmed in a prospective study on rats with experimentally induced endometriosis (n = 12). After 28 days of oral silymarin administration (50 mg/kg per day), a significant decrease in the establishment and size of endometriotic lesions was noted with decreased mRNA levels of glial cell-derived neurotrophic factor (GDNF) and its essential receptor component GFRα1, as well as the proto-oncogenes B-cell lymphoma 6 (Bcl-6b) and Bcl-2. The number of GDNF-, GFRα1-, Bcl-6b- and Bcl-2-positive cell distribution/mm2 was remarkably diminished within endometriotic foci in the silymarin-treated group vs. the control. Moreover, silymarin promoted the apoptosis pathway by enhancing extracellular regulator kinase (ERK1/2) expression and by suppressing Bcl-2 expression. The authors of the study concluded that silymarin downregulates the angiogenesis ratio, accelerates apoptosis and consequently induces severe fibrosis in endometriotic-like lesions [378].
A significant regression of surgically induced endometriotic foci in Wistar albino rats was observed after oral administration of the terpene nerolidol (trans-nerolidol) and the flavone glycoside hesperidin. Both PEs are potent antioxidants. In addition to a reduction in the average volume of the lesions in rats treated with hesperidin and nerolidol, malondialdehyde levels (the marker of oxidative stress) were significantly reduced in the nerolidol-treated group, and glutathione levels and superoxide dismutase activity (the first line defense antioxidants) were significantly elevated in the endometriotic foci of both the hesperidin- and nerolidol-treated groups compared with the control endometriosis group [379].
The action of isoliquiritigenin, which is a natural flavonoid isolated from the root of licorice (Glycyrrhiza uralensis) and shallot (Allium cepa) with documented antioxidant, anti-inflammatory, antiproliferation and antitumor activities, was examined in female BALB/C mice that were surgically induced to have endometriosis by transplanting uterine tissue into the abdominal cavity. Four weeks of oral administration of isoliquiritigenin reduced the volume and weight of endometriotic lesions, decreased serum and lesion inflammatory cytokines, induced apoptosis of the lesions and inhibited the epithelial-mesenchymal transition (EMT) [380]. The latter effect should be emphasized because EMT, which is a process in which epithelial cells lose polarized organization of the cytoskeleton and cell-to-cell contacts, thus acquiring the high motility of mesenchymal cells, seems to be a prerequisite for the original establishment of endometriotic lesions [381].
Naringenin is a plant-derived flavonoid with anti-proliferative, anti-inflammatory and anti-angiogenic properties in chronic and metabolic diseases. The therapeutic potential of orally administered naringenin in endometriosis was evaluated in a rat model of the disease. The endometrial lesion volumes, weight, serum TNF-α level and histopathologic scores were significantly reduced in the naringenin-treated group compared to the endometriotic control group. Accordingly, naringenin ameliorated the expression of various proteins involved in the development and progression of endometriotic cells, such as p21-activated kinase 1 (PAK1), transforming growth factor β-activated kinase 1 (TAK1), VAGF and proliferating cell nuclear antigen (PCNA). Moreover, in an in vitro study, naringenin caused a dose-dependent loss of mitochondrial membrane potential, induced apoptosis and inhibited the proliferation/invasiveness of endometriotic cells with a corresponding downregulation of matrix metalloproteinase-2 (MMP-2) and 9 (MMP-9). The induction of reactive oxygen species (ROS)-mediated apoptosis was demonstrated by analyzing the effect of naringenin on the factor erythroid 2-related factor 2 (Nrf2)/heme oxygenase-1 (HO-1)/NADPH-quinone oxidoreductase-1 (NQO1)/Kelch-like ECH associated protein 1 (KEAP1) axis. This axis is a fundamental signaling cascade that controls multiple cytoprotective responses via the induction of a complex transcriptional program that finally provides endometriotic cells with increased resistance to oxidative, metabolic and therapeutic stress. Naringenin significantly inhibited this transmission by modulating the expression of Nrf2 and its downstream effector molecules [382].
Similar results regarding the effect on the course of endometriosis were obtained by using oral flavonoid phytoestrogen-rich plant extracts in animal models, including surgically induced endometriosis in mice (female BALB/C) and female Sprague Dawley or Wistar-Albino rats. Significant regressions of the endometriotic foci were demonstrated after 3-4 weeks of administration of the extracts prepared from U. dioica L. (leaves and roots), the aerial parts of Achillea millefolium L., Achillea biebersteinii Afan and Achillea cretics L, Rosmarinus officinalis (leaves), Scutellaria baicalensis (root), Anthemis austriaca Jacq. (flowers) and Melilotus officinalis (L.) Pall. (aerial parts) [383–388]. The antiproliferative effects on the cells of the ectopic endometrium were accompanied by anti-inflammatory effects, which were manifested by decreased proinflammatory cytokine levels in the peritoneal fluid, including TNF-α, VEGF and IL-6 levels [383,384,387,388].
3.3. PE oral intake and the course of endometriosis – the results obtained in human studies
The studies on the effect of orally administered PEs in women on the risk of onset and the course of endometriosis are summarized in Table 2. For this purpose, the electronic databases PubMed, EMBASE and MEDLINE were searched until April 2023. The number of studies including randomized controlled trials that were identified in this manner was surprisingly few, especially considering the numerous studies that have been conducted in animals. In contrast, the number of comprehensive reviews oriented toward the role of diet in human health (particularly in endometriosis) is striking [169,171,389–393].
The results of most of the ten studies cited in Table 2 demonstrated some beneficial effects of a PE-rich diet or a diet supplemented with various doses of particular PE/PEs (i.e., resveratrol, daidzein + genistein or quercetin + curcumin + parthenium) in endometriosis. This scenario applies to both the risk of disease onset and its course [394–403]. For example, the results of 15 years of two case‒control studies (n = 504) have shown that a PE-rich diet decreases the risk of endometriosis compared to a low-PE diet [398]. The same effect was shown regarding a decreased risk of endometriosis for isoflavones, lignans and coumestrol, although this was observed in a much smaller 12-month case‒control study on dietary data (n = 78) [402]. Moreover, increased urinary levels of soy isoflavones (daidzein and denistein) were inversely associated with both the risk of advanced endometriosis (stage III-IV) and the severity of endometriosis. Importantly, the authors of this case‒control study (n = 79) with 24 months of the recruiting period noted that the ERβ gene ESR2 RsaI polymorphism significantly modified these effects of genistein for advanced endometriosis [401]. In addition, resveratrol appeared in three small studies [394–396] (Table 2). In the retrospective study (n = 26), 82% of patients reported complete resolution of dysmenorrhea and pelvic pain after 2 months of treatment with 30 mg oral administration of resveratrol per day. This effect was accompanied by reduced expression of both COX-2 and aromatase in the eutopic endometrium [395]. In a placebo-controlled, randomized, double-blind clinical trial (n = 17), the dose of resveratrol was considerably higher (400 mg daily for 12-14 weeks). Resveratrol has been shown to have anti-inflammatory effects by affecting the metalloproteinases MMP-2 and MMP-9. Furthermore, the decrease in the mRNA and protein levels of both MMP-2 and MMP-9 was significant. As a consequence, decreased concentrations of MMP-2 and MMP-9 in the serum and endometrial fluid were confirmed [394].
In contrast, the ineffectiveness of resveratrol in endometriosis pain relief was demonstrated in a short randomized clinical trial. After 42 days of treatment with a dose of 40 mg, resveratrol was not superior to placebo (no difference in median pain scores observed between the groups) [396].
Research by another team has provided new information on the role of dietary factors in the development of endometriosis [400]. The results of this 60-month, population-based case control study involving 300 patients in the study group demonstrated that an increased risk of endometriosis is positively correlated with β-carotene consumption and servings/d of fruit. Unlike fruits served twice a day, vegetable intake was not associated with endometriosis risk. These findings were not consistent with the reduced risk of endometriosis associated with the consumption of both green vegetables and fresh fruit reported by Parazzini et al. [398]. The authors realized that their results require confirmation or even validation because the risk of misinterpretation of the data is high due to the number of included variables. The study by Parazzini demonstrated partial confirmation in a paper published by Harris et al. [403], in which a reduced risk of endometriosis was reported when consuming significant amounts of fruit (especially citrus) in the diet (but not for vegetables). Moreover, due to the much larger size of the cohort, these results may be more convincing. To date, no similar prospective studies with PEs have been conducted in humans. A more detailed understanding of the impact of dietary PEs, including mycotoxins and dietary patterns, on the risk of endometriosis is urgently needed [48,49,148,166,167]. Furthermore, it may help to develop population-based strategies to prevent this chronic disease with a high impact of environmental (possibly dietary) triggers. A key question for those individuals already suffering from endometriosis may be whether a diet rich in phytoestrogens is truly beneficial [169].
The inconclusive or (less commonly) opposing results are understandable with so few human studies and their varied methodologies (e.g., differences in the amount and content of PEs in the diet, different duration of the study and/or size of the sample, imprecise inclusion/exclusion criteria and interpretation of results of measured outcomes in nonconsistent manners) [404].
4. Concluding remarks
There is no doubt that PEs may have antiestrogenic activities typical of substances from the group of endocrine disruptors [46,137]. If absolute or relative excess estrogen plays a key role in endometriosis, the antiestrogenic effect of PEs may provide therapeutic benefits. In vitro studies in cell cultures and experiments on animals using various types of endometriosis models are widely represented, as well as in the latest literature. In addition to zearalenone and other mycotoxins, the most commonly studied phytoestrogens in this manner have been resveratrol, curcumin, soy isoflavones (daidzein and genistein), lignans, coumestrol, quercetin, epigallocatechin-3-gallate, parthenolide (parthenium), puerarin, ginsenosides (steroid-like saponins), xanthohumol and cannabinoids (apigenin) [405,406]. The characteristics of the detected pleiotropic effects of these xenoestrogens are related to a variety of known signaling effectors, including ERs, GPER, COX-2, IL-1, IL-6, TNFα, VEGF, ROS, MMPs, NF-κB and apoptosis-related proteins (e.g., Bax, Bcl-2, Caspase-3, Caspase-9, p53 and β-actin) [406,407]. In most such experimental models, solid evidence about the anti-inflammatory, proapoptotic, antioxidant and immunomodulatory functions of phenolic compounds (e.g., flavonoids and phenolic acids) have translated into an effect of inhibiting the development of endometriosis via modulation of estrogen activity [393].
However, one should be fully aware that these promising results are based on in vitro and animal models of endometriosis. As the summary in Table 2 shows, there are virtually no randomized controlled trials in this area. Thus, to achieve conclusive results regarding the potential benefits of oral (dietary) phytoestrogens, properly designed clinical trials are essential. When developing schemes for such studies, several (often underestimated) issues should also be considered (Figure 6).
Research has demonstrated a connection between diet (which is the most important environmental epigenetic factor) and the incidence of estrogen-dependent diseases (e.g., breast and endometrial cancer) [408,409]. With regard to endometriosis, it has been established that fish oil capsules in combination with vitamin B12 can be helpful in relieving endometriosis symptoms (especially dysmenorrhea), whereas alcohol and increased consumption of red meat and trans fats are associated with exacerbation of the disease [184]. However, the existing information about the effect of dietary phytoestrogens on endometriosis in humans is still incomplete, inconclusive or ambiguous. This is an important topic to explore because endometriosis affects up to 10% of women, and diet is a modifiable risk factor for this chronic disease, both in terms of onset and management. The analysis of the results cannot ignore the possible conflict of interest caused by the source of funding for such research [169,410].
An evaluation of the effects of PEs should consider differences between populations and different nutritional patterns. For example, the prevalence of endometriosis appears to be higher in Asian women (e.g., Filipino, Indian, Japanese and Korean women) than in Caucasian women and African Americans. Although the utilization of health care may account for some of the observed differences, the incidence of endometriosis is estimated at 5-10% in Western populations, compared with 15-18% in Asian female populations [411–413]. This result is puzzling because a PE-rich diet is the basis in most Asian countries. For example, in the context of the beneficial effects of PEs on endometriosis, it is surprising that India (the country with the highest percentage of vegans/vegetarians) has a high incidence of endometriosis [412,414,415]. It is suggested that a genetic polymorphism predisposing to endometriosis and/or environmental pollution with other EDCs (e.g., pesticides used in plant cultivation) may be of importance in this scenario [412].
It should be noted that the family of PEs includes compounds of different strength and specificity of action on receptors and signaling pathways. These actions are sometimes contradictory. For example, resveratrol inhibits aromatase, thus lowering the concentration of estrogens, whereas genistein has the opposite effect on aromatase activity in human endometrial stromal cells [247,416].
In accordance with the principle known from toxicology that "the dose defines the poison", to distinguish genotoxic from beneficial effects, we need to know the bioavailability of PEs in a given person with a specific health condition [417]. Correspondingly, the bioavailability, bioactivity and health effects of dietary PEs are strongly determined by the intestinal bacteria of each individual [418]. The gut microbiota regulates estrogenic activity in the body through the secretion of β-glucuronidase, which is an enzyme that deconjugates estrogens into their active forms. Notably, the "carrier" of PEs is a diet rich in fiber. Dietary fiber has been shown to affect the absorption, reabsorption and excretion of estrogens and PEs by influencing the ß-glucosidase and ß-glucuronidase activities of the intestinal microflora [419]. The swelling of the fiber leads to the dilution of the intestinal bacterial flora and hydrophobic bonding, particularly of nonconjugated compounds, which contributes to a reduced absorption of PEs and reabsorption of endogenous estrogens. Therefore, the antiestrogenic effects of PEs may be variable and secondary to dietary fiber content [420]. High dietary fiber intake may lead to the partial disruption of enterohepatic circulation of estrogens within the estrogen-gut microbiome axis [421,422]. Accordingly, vegetarians generally have higher fecal weights than omnivores and lower fecal bacterial ß-glucuronidase activity [423]. There is evidence that a dysbiotic gut microbiota and dysfunctional estrobolome (which represents the aggregate of all of the enteric bacteria capable of metabolizing estrogen) are associated with multiple gynecologic conditions, with mounting data supporting an association between the intestinal bacteria and endometriosis and infertility [357]. In such cases (including endometriosis), bacterial-derived/induced metabolites may interact with cells of the immune system and nervous system, thus modulating the actions of these systems [177,184].
In summary, due to the lack of relevant studies in humans, it is impossible to unequivocally determine the benefits or adverse effects of using a diet rich in PEs in relation to the risk of endometriosis or its course [169,424–426].
Abbreviations
| 17β-hydroxysteroid dehydrogenases type 1 and 4 (, respectively), 17β-HSD1, 17β-HSD2, 17β-HSD4 – 17β-hydroxysteroid dehydrogenase type 1, 2, and 4, respectively AKT – protein kinase B AP-1 – activator protein 1 ATP – adenosine triphosphate Bcl-2 – anti-apoptotic B-cell lymphoma-2 protein c-IAP1, c-IAP2 – cellular inhibitors of apoptosis 1 and 2, respectively CADD – computer aided drug design cAMP – cyclic adenosine monophosphate CINC-1, CINC-2, CINC-3 – cytokine-induced neutrophil chemoattractant proteins 1-3 COX-2 – cyclooxygenase 2 c-Src/ERK pathway – Src/extracellular signal-regulated kinase pathway CTLs – cytotoxic T lymphocytes, also known as killer T cells DBD – DNA binding domain (or C domain) DCs – dendritic cells DDT – dichlorodiphenyltrichloroethane E1, E2, E3 – estrone, estradiol and estriol, respectively estradiol and estriol, respectively E2 – estradiol EDCs – endocrine disrupting chemicals EGCG – polyphenol epigallocatechin-3-gallate EGFR – epidermal growth factor receptor EMT – epithelial-mesenchymal-transition EnSCs – endometrial stromal cells ERα, ERβ – estrogen receptors α and β, also known as NR3A1 and NR3A2, respectively ERE – estrogen response element ERK1, ERK2 – mitogen-activated protein-serine/threonine kinases ERs – estrogen receptors ESR1, ESR2 – genes encoding estrogen receptors ERα and ERβ, respectively FSH – follicle-stimulating hormone GDNF – glial cell line derived neurotrophic factor GFRα1 – glial cell line derived neurotrophic factor (GDNF) family receptor alpha 1 GM-CSF – granulocyte-macrophage colony stimulating factor GPCRs – G protein-coupled receptors GPER – G protein-coupled estrogen receptor, also known as G protein-coupled receptor 30 (GPR30) HB-EGF – heparin-binding epidermal growth factor (EGF)-like growth factor HDACs – histone deacetylases HLA-G – human leukocyte antigen G HLA-DR – major histocompatibility complex (MHC) II cell surface receptor HO-1 – heme oxygenase-1 HPG axis – hypothalamic-pituitary-gonadal axis HPO axis – hypothalamic-pituitary-ovarian axis HSP90 – heat shock protein 90 HUVECs – human umbilical vein endothelial cells IBD – inflammatory bowel disease IGFR1 – insulin-like growth factor receptor 1 IL-1, IL-1ß, IL-2, IL-3, IL-4, IL-5, IL-6, IL-9, IL-10, IL-12, IL-13, IL-18 – interleukins: 1, 1ß, 2, 3, 4, 5, 6, 9, 10, 12, 13, and 18 IL-18Rα – interleukin 18 receptor alpha IFN-γ – interferon gamma IKK – IκB kinase iNOS – inducible nitric oxide synthase JNK – cJun NH(2)-terminal kinase KEAP1 – Kelch-like ECH associated protein 1 LBD – ligand binding domain LH – luteinizing hormone lncRNAs – long non-coding RNAs MAP – mitogen-activated protein MAPK – mitogen-activated protein kinase MCP-1 – monocyte chemoattractant protein-1 MCs – mast cells MHCI – major histocompatibility complex class I MHCII – major histocompatibility complex class II MMP-2, MMP-9 – matrix metalloproteinases 2 and 9 MMPs – matrix metalloproteinases MNAR – modulator of non-genomic activity of estrogen receptor, also known as proline-, glutamate- and leucine-rich protein 1 (PELP1) mPRα, mPRβ, mPRγ, mPRδ, mPRε – membrane progesterone receptors MSCs – mesenchymal stem cells MW – molecular weight mTOR – mammalian target of rapamycin (a serine-threonine protein kinase) NADPH – nicotinamide adenine dinucleotide phosphate NF-κB – nuclear factor kappa-light-chain-enhancer of activated B cells NK-cell – natural killer cell NO – nitric oxide NQO1 – nicotinamide adenine dinucleotide phosphate (NADPH)-quinone oxidoreductase-1 Nrf2 – factor erythroid 2-related factor 2 NTD – N-terminal domain OT - oxytocin P4 – progesterone P450AROM – aromatase cytochrome P450 PAK1 – p21-activated kinase 1 PCB – polychlorinated biphenyls PCDD – polychlorinated dibenzo-p-dioxins PCDF – polychlorinated dibenzofurans PCNA – proliferating cell nuclear antigen PEA – palmitoylethanolamide PELP1 – proline-, glutamate- and leucine-rich protein 1, also known as modulator of non-genomic activity of estrogen receptor (MNAR) PEs – phytoestrogens PGE2 – prostaglandin E2 PGF2-ɑ – prostaglandin F2-alpha PI3K – phosphatidylinositol-3-kinase PLD – polydatin (natural precursor of resveratrol) PlGF – placental growth factor POPs – persistent organic pollutants PR-A, PR-B – progesterone receptors type A and B, respectively RNA Pol II – RNA polymerase II ROS – reactive oxygen species RTKs – receptor tyrosine kinases SCF – stem cell factor SERMS – selective estrogen receptor (ER) modulators SF-1 – steroidogenic factor 1 SIRTs – sirtuins SRA – steroid receptor RNA activator Src – non-receptor tyrosine kinase (proto-oncogene tyrosine-protein kinase Src) SRC – steroid receptor coactivator SRC-2 – steroid receptor coactivator-2, also known as transcriptional mediators/intermediary factor 2 (TIF2) T3, T4 – triiodothyronine, thyroxine (tetraiodothyronine) TAK1 – transforming growth factor β-activated kinase 1 TIF2 – transcriptional mediators/intermediary factor 2, also known as (SRC-2) TF – transcription factor Th1, Th2, Th17 cells – T helper cell subtypes Tregs – regulatory T cells TNF-α – tumor necrosis factor alpha TPO – thyroid peroxidase VCAM-1 – vascular cell adhesion molecule 1, also known as vascular cell adhesion protein 1 VEGF – vascular endothelial growth factor |
References
- Milling S. Beyond cytokines: Influences of the endocrine system on human immune homeostasis. Immunology. 2021;163(2):113-114. [CrossRef]
- Verburg-van Kemenade BML, Cohen N, Chadzinska M. Neuroendocrine-immune interaction: Evolutionarily conserved mechanisms that maintain allostasis in an ever-changing environment. Dev Comp Immunol. 2017;66:2-23. [CrossRef]
- Tanida T. Molecular dynamics of estrogen-related receptors and their regulatory proteins: roles in transcriptional control for endocrine and metabolic signaling. Anat Sci Int. 2022; 97(1):15-29. [CrossRef]
- Baumbach JL, Zovkic IB. Hormone-epigenome interactions in behavioural regulation. Horm Behav. 2020;118:104680. [CrossRef]
- Ruzzin J. Public health concern behind the exposure to persistent organic pollutants and the risk of metabolic diseases. BMC Public Health. 2012;12:298. [CrossRef]
- Li QQ, Loganath A, Chong YS, Tan J, Obbard JP. Persistent organic pollutants and adverse health effects in humans. J Toxicol Environ Health A. 2006;69(21):1987-2005. [CrossRef]
- Xie Z, Zhang P, Wu Z, Zhang S, Wei L, Mi L, Kuester A, Gandrass J, Ebinghaus R, Yang R, Wang Z, Mi W. Legacy and emerging organic contaminants in the polar regions. Sci Total Environ. 2022;835:155376. [CrossRef]
- Jayaraj R, Megha P, Sreedev P. Organochlorine pesticides, their toxic effects on living organisms and their fate in the environment. Interdiscip Toxicol. 2016;9(3-4):90-100. [CrossRef]
- Guo W, Pan B, Sakkiah S, Yavas G, Ge W, Zou W, Tong W, Hong H. Persistent Organic Pollutants in Food: Contamination Sources, Health Effects and Detection Methods. Int J Environ Res Public Health. 2019;16(22):4361. [CrossRef]
- Organochlorine Pollutants in Plasma, Blood Pressure, and Hypertension in a Longitudinal Study. Hypertension. 2018;71(6):1258-1268. [CrossRef]
- Wilson J, Berntsen HF, Zimmer KE, Verhaegen S, Frizzell C, Ropstad E, Connolly L. Do persistent organic pollutants interact with the stress response? Individual compounds, and their mixtures, interaction with the glucocorticoid receptor. Toxicol Lett. 2016;241:121-32. [CrossRef]
- La Merrill MA, Vandenberg LN, Smith MT, Goodson W, Browne P, Patisaul HB, Guyton KZ, Kortenkamp A, Cogliano VJ, Woodruff TJ, Rieswijk L, Sone H, Korach KS, Gore AC, Zeise L, Zoeller RT. Consensus on the key characteristics of endocrine-disrupting chemicals as a basis for hazard identification. Nat Rev Endocrinol. 2020;16(1):45-57. [CrossRef]
- Yilmaz B, Terekeci H, Sandal S, Kelestimur F. Endocrine disrupting chemicals: exposure, effects on human health, mechanism of action, models for testing and strategies for prevention. Rev Endocr Metab Disord. 2020;21(1):127-147. [CrossRef]
- Gregoraszczuk EL, Ptak A. Endocrine-Disrupting Chemicals: Some Actions of POPs on Female Reproduction. Int J Endocrinol. 2013;2013:828532. [CrossRef]
- Kowalczyk A, Wrzecińska M, Czerniawska-Piątkowska E, Araújo JP, Cwynar P. Molecular consequences of the exposure to toxic substances for the endocrine system of females. Biomed Pharmacother. 2022;155:113730. [CrossRef]
- Thambirajah AA, Wade MG, Verreault J, Buisine N, Alves VA, Langlois VS, Helbing CC. Disruption by stealth - Interference of endocrine disrupting chemicals on hormonal crosstalk with thyroid axis function in humans and other animals. Environ Res. 2022 ;203: 111906. [CrossRef]
- Buoso E, Masi M, Racchi M, Corsini E. Endocrine-Disrupting Chemicals' (EDCs) Effects on Tumour Microenvironment and Cancer Progression: Emerging Contribution of RACK1. Int J Mol Sci. 2020;21(23):9229. [CrossRef]
- Masuo Y, Ishido M. Neurotoxicity of endocrine disruptors: possible involvement in brain development and neurodegeneration. J Toxicol Environ Health B Crit Rev. 2011;14(5-7):346-69. [CrossRef]
- Rahman MS, Pang WK, Amjad S, Ryu DY, Adegoke EO, Park YJ, Pang MG. Hepatic consequences of a mixture of endocrine-disrupting chemicals in male mice. J Hazard Mater. 2022;436:129236. [CrossRef]
- Singh RD, Koshta K, Tiwari R, Khan H, Sharma V, Srivastava V. Developmental Exposure to Endocrine Disrupting Chemicals and Its Impact on Cardio-Metabolic-Renal Health. Front Toxicol. 2021;3:663372. [CrossRef]
- Sabuz Vidal O, Deepika D, Schuhmacher M, Kumar V. EDC-induced mechanisms of immunotoxicity: a systematic review. Crit Rev Toxicol. 2021;51(7):634-652. [CrossRef]
- Meeker JD. Exposure to environmental endocrine disruptors and child development. Arch Pediatr Adolesc Med. 2012;166(6):E1-7. [CrossRef]
- Lakshmanan MD, Shaheer K. Endocrine disrupting chemicals may deregulate DNA repair through estrogen receptor mediated seizing of CBP/p300 acetylase. J Endocrinol Invest. 2020; 43(9):1189-1196. [CrossRef]
- Zoeller RT, Brown TR, Doan LL, Gore AC, Skakkebaek NE, Soto AM, Woodruff TJ, Vom Saal FS. Endocrine-disrupting chemicals and public health protection: a statement of principles from The Endocrine Society. Endocrinology. 2012;153(9):4097-110. [CrossRef]
- Lisco G, Giagulli VA, Iovino M, Guastamacchia E, Pergola G, Triggiani V. Endocrine-Disrupting Chemicals: Introduction to the Theme. Endocr Metab Immune Disord Drug Targets. 2022;22(7):677-685. [CrossRef]
- Marty MS, Borgert C, Coady K, Green R, Levine SL, Mihaich E, Ortego L, Wheeler JR, Yi KD, Zorrilla LM. Distinguishing between endocrine disruption and non-specific effects on endocrine systems. Regul Toxicol Pharmacol. 2018;99:142-158. [CrossRef]
- Keller DA, Juberg DR, Catlin N, Farland WH, Hess FG, Wolf DC, Doerrer NG. Identification and characterization of adverse effects in 21st century toxicology. Toxicol Sci. 2012;126(2):291-7. [CrossRef]
- McKone TE, Daniels JI. Estimating human exposure through multiple pathways from air, water, and soil. Regul Toxicol Pharmacol. 1991;13(1):36-61. [CrossRef]
- Anwer F, Chaurasia S, Khan AA. Hormonally active agents in the environment: a state-of-the-art review. Rev Environ Health. 2016;31(4):415-433. [CrossRef]
- Yue B, Ning S, Miao H, Fang C, Li J, Zhang L, Bao Y, Fan S, Zhao Y, Wu Y. Human exposure to a mixture of endocrine disruptors and serum levels of thyroid hormones: A cross-sectional study. J Environ Sci (China). 2023;125:641-649. [CrossRef]
- Darbre PD, Harvey PW. Paraben esters: review of recent studies of endocrine toxicity, absorption, esterase and human exposure, and discussion of potential human health risks. J Appl Toxicol. 2008;28(5):561-78. [CrossRef]
- He D, Ye X, Xiao Y, Zhao N, Long J, Zhang P, Fan Y, Ding S, Jin X, Tian C, Xu S, Ying C. Dietary exposure to endocrine disrupting chemicals in metropolitan population from China: a risk assessment based on probabilistic approach. Chemosphere. 2015;139:2-8. [CrossRef]
- Chen Y, Yang J, Yao B, Zhi D, Luo L, Zhou Y. Endocrine disrupting chemicals in the environment: Environmental sources, biological effects, remediation techniques, and perspective. Environ Pollut. 2022;310:119918. [CrossRef]
- Sakali AK, Bargiota A, Fatouros IG, Jamurtas A, Macut D, Mastorakos G, Papagianni M. Effects on Puberty of Nutrition-Mediated Endocrine Disruptors Employed in Agriculture. Nutrients. 2021;13(11):4184. [CrossRef]
- Beszterda M, Frański R. Endocrine disruptor compounds in environment: As a danger for children health. Pediatr Endocrinol Diabetes Metab. 2018;24(2):88-95. [CrossRef]
- Li N, Li J, Zhang Q, Gao S, Quan X, Liu P, Xu C. Effects of endocrine disrupting chemicals in host health: Three-way interactions between environmental exposure, host phenotypic responses, and gut microbiota. Environ Pollut. 2021;271:116387. [CrossRef]
- Shore LS, Shemesh M. Estrogen as an Environmental Pollutant. Bull Environ Contam Toxicol. 2016;97(4):447-8. [CrossRef]
- Mheidli N, Malli A, Mansour F, Al-Hindi M. Occurrence and risk assessment of pharmaceuticals in surface waters of the Middle East and North Africa: A review. Sci Total Environ. 2022;851(Pt 2):158302. [CrossRef]
- Atkinson S, Atkinson MJ, Tarrant AM. Estrogens from sewage in coastal marine environments. Environ Health Perspect. 2003;111(4):531-5. [CrossRef]
- Adeel M, Song X, Wang Y, Francis D, Yang Y. Environmental impact of estrogens on human, animal and plant life: A critical review. Environ Int. 2017;99:107-119. [CrossRef]
- Shore LS, Bar-El CK. The environmental compartments of environmental hormones. Rev Environ Health. 2010;25(4):345-50. [CrossRef]
- Varticovski L, Stavreva DA, McGowan A, Raziuddin R, Hager GL. Endocrine disruptors of sex hormone activities. Mol Cell Endocrinol. 2022;539:111415. [CrossRef]
- Thompson LU, Robb P, Serraino M, Cheung F. Mammalian lignan production from various foods. Nutr Cancer. 1991;16(1):43-52. [CrossRef]
- Murkies AL, Wilcox G, Davis SR. Clinical review 92: Phytoestrogens. J Clin Endocrinol Metab. 1998;83(2):297-303. [CrossRef]
- Bennetts HW, Underwood EJ, Shier FL. A specific breeding problem of sheep on subterranean clover pastures in Western Australia. Aust Vet J. 1946;22(1):2-12. [CrossRef]
- Rietjens IMCM, Louisse J, Beekmann K. The potential health effects of dietary phytoestrogens. Br J Pharmacol. 2017;174(11):1263-1280. [CrossRef]
- Lecomte S, Demay F, Ferrière F, Pakdel F. Phytochemicals Targeting Estrogen Receptors: Beneficial Rather Than Adverse Effects? Int J Mol Sci. 2017;18(7):1381. [CrossRef]
- Bar-El DS, Reifen R. Soy as an endocrine disruptor: cause for caution? J Pediatr Endocrinol Metab. 2010;23(9):855-61. [CrossRef]
- Beekmann K. The potential health effects of dietary phytoestrogens. Br J Pharmacol. 2017; 174(11):1263-1280. [CrossRef]
- van Duursen MBM. Modulation of estrogen synthesis and metabolism by phytoestrogens in vitro and the implications for women's health. Toxicol Res (Camb). 2017;6(6):772-794. [CrossRef]
- Gaya P, Medina M, Sánchez-Jiménez A, Landete JM. Phytoestrogen Metabolism by Adult Human Gut Microbiota. Molecules. 2016;21(8):1034. [CrossRef]
- Kuiper GG, Lemmen JG, Carlsson B, Corton JC, Safe SH, van der Saag PT, van der Burg B, Gustafsson JA. Interaction of estrogenic chemicals and phytoestrogens with estrogen receptor beta. Endocrinology. 1998;139(10):4252-63. [CrossRef]
- Kiyama R. Estrogenic flavonoids and their molecular mechanisms of action. J Nutr Biochem. 2022;114:109250. [CrossRef]
- Han DH, Denison MS, Tachibana H, Yamada K. Relationship between estrogen receptor-binding and estrogenic activities of environmental estrogens and suppression by flavonoids. Biosci Biotechnol Biochem. 2002;66(7):1479-87. [CrossRef]
- Rietjens IM, Sotoca AM, Vervoort J, Louisse J. Mechanisms underlying the dualistic mode of action of major soy isoflavones in relation to cell proliferation and cancer risks. Mol Nutr Food Res. 2013;57(1):100-13. [CrossRef]
- Paterni I, Granchi C, Katzenellenbogen JA, Minutolo F. Estrogen receptors alpha (ERα) and beta (ERβ): subtype-selective ligands and clinical potential. Steroids. 2014;90:13-29. [CrossRef]
- Thomas P, Dong J. Binding and activation of the seven-transmembrane estrogen receptor GPR30 by environmental estrogens: a potential novel mechanism of endocrine disruption. J Steroid Biochem Mol Biol. 2006;102(1-5):175-9. [CrossRef]
- Liu EYL, Xu ML, Jin Y, Wu Q, Dong TTX, Tsim KWK. Genistein, a Phytoestrogen in Soybean, Induces the Expression of Acetylcholinesterase via G Protein-Coupled Receptor 30 in PC12 Cells. Front Mol Neurosci. 2018;11:59. [CrossRef]
- Sundermann EE, Maki PM, Bishop JR. A review of estrogen receptor alpha gene (ESR1) polymorphisms, mood, and cognition. Menopause. 2010;17(4):874-86. [CrossRef]
- Li J, Liu Q, Jiang C. Signal Crosstalk and the Role of Estrogen Receptor beta (ERβ) in Prostate Cancer. Med Sci Monit. 2022;28:e935599. [CrossRef]
- Arao Y, Korach KS. The physiological role of estrogen receptor functional domains. Essays Biochem. 2021;65(6):867-875. [CrossRef]
- Fuentes N, Silveyra P. Estrogen receptor signaling mechanisms. Adv Protein Chem Struct Biol. 2019;116:135-170. [CrossRef]
- Yaşar P, Ayaz G, User SD, Güpür G, Muyan M. Molecular mechanism of estrogen-estrogen receptor signaling. Reprod Med Biol. 2016;16(1):4-20. [CrossRef]
- George AJ, Hannan RD, Thomas WG. Unravelling the molecular complexity of GPCR-mediated EGFR transactivation using functional genomics approaches. FEBS J. 2013; 280(21):5258-68. [CrossRef]
- Yu X, Stallone JN, Heaps CL, Han G. The activation of G protein-coupled estrogen receptor induces relaxation via cAMP as well as potentiates contraction via EGFR transactivation in porcine coronary arteries. PLoS One. 2018;13(1):e0191418. [CrossRef]
- Kato S, Endoh H, Masuhiro Y, Kitamoto T, Uchiyama S, Sasaki H, Masushige S, Gotoh Y, Nishida E, Kawashima H, Metzger D, Chambon P. Activation of the estrogen receptor through phosphorylation by mitogen-activated protein kinase. Science. 1995;270(5241):1491-4. [CrossRef]
- Coleman KM, Smith CL. Intracellular signaling pathways: nongenomic actions of estrogens and ligand-independent activation of estrogen receptors. Front Biosci. 2001;6: D1379-91. [CrossRef]
- Atanaskova N, Keshamouni VG, Krueger JS, Schwartz JA, Miller F, Reddy KB. MAP kinase/estrogen receptor cross-talk enhances estrogen-mediated signaling and tumor growth but does not confer tamoxifen resistance. Oncogene. 2002;21(25):4000-8. [CrossRef]
- Visser K, Mortimer M, Louw A. Cyclopia extracts act as ERα antagonists and ERβ agonists, in vitro and in vivo. PLoS One. 2013;8(11):e79223. Erratum in: PLoS One. 2013;8(12). [CrossRef]
- Vitale DC, Piazza C, Melilli B, Drago F, Salomone S. Isoflavones: estrogenic activity, biological effect and bioavailability. Eur J Drug Metab Pharmacokinet. 2013;38(1):15-25. [CrossRef]
- Messina M, Kucuk O, Lampe JW. An overview of the health effects of isoflavones with an emphasis on prostate cancer risk and prostate-specific antigen levels. J AOAC Int. 2006; 89(4):1121-34.
- Křížová L, Dadáková K, Kašparovská J, Kašparovský T. Isoflavones. Molecules. 2019; 24(6):1076. [CrossRef]
- Bartuzi D, Kaczor AA, Targowska-Duda KM, Matosiuk D. Recent Advances and Applications of Molecular Docking to G Protein-Coupled Receptors. Molecules. 2017;22(2): 340. [CrossRef]
- Meng XY, Zhang HX, Mezei M, Cui M. Molecular docking: a powerful approach for structure-based drug discovery. Curr Comput Aided Drug Des. 2011;7(2):146-57. [CrossRef]
- Fukunishi Y, Yamashita Y, Mashimo T, Nakamura H. Prediction of Protein-compound Binding Energies from Known Activity Data: Docking-score-based Method and its Applications. Mol Inform. 2018;37(6-7):e1700120. [CrossRef]
- Sulimov VB, Kutov DC, Sulimov AV. Advances in Docking. Curr Med Chem. 2019;26(42):7555-7580. [CrossRef]
- Sahayarayan JJ, Rajan KS, Vidhyavathi R, Nachiappan M, Prabhu D, Alfarraj S, Arokiyaraj S, Daniel AN. In-silico protein-ligand docking studies against the estrogen protein of breast cancer using pharmacophore based virtual screening approaches. Saudi J Biol Sci. 2021;28(1):400-407. [CrossRef]
- Powers CN, Setzer WN. A molecular docking study of phytochemical estrogen mimics from dietary herbal supplements. In Silico Pharmacol. 2015;3:4. [CrossRef]
- Ramírez D, Caballero J. Is It Reliable to Use Common Molecular Docking Methods for Comparing the Binding Affinities of Enantiomer Pairs for Their Protein Target? Int J Mol Sci. 2016;17(4):525. [CrossRef]
- Cassidy A, Bingham S, Setchell KD. Biological effects of a diet of soy protein rich in isoflavones on the menstrual cycle of premenopausal women. Am J Clin Nutr. 1994;60333- 340.
- Jang WY, Kim MY, Cho JY. Antioxidant, Anti-Inflammatory, Anti-Menopausal, and Anti-Cancer Effects of Lignans and Their Metabolites. Int J Mol Sci. 2022;23(24):15482. [CrossRef]
- Liu Y, Hassan S, Kidd BN, Garg G, Mathesius U, Singh KB, Anderson JP. Ethylene Signaling Is Important for Isoflavonoid-Mediated Resistance to Rhizoctonia solani in Roots of Medicago truncatula. Mol Plant Microbe Interact. 2017;30(9):691-700. [CrossRef]
- Setchell KD. Phytoestrogens: the biochemistry, physiology, and implications for human health of soy isoflavones. Am J Clin Nutr. 1998;68(6 Suppl):1333S-1346S. [CrossRef]
- Zhu Y, Kawaguchi K, Kiyama R. Differential and directional estrogenic signaling pathways induced by enterolignans and their precursors. PLoS One. 2017;12(2):e0171390. [CrossRef]
- JavanMoghadam S, Weihua Z, Hunt KK, Keyomarsi K. Estrogen receptor alpha is cell cycle-regulated and regulates the cell cycle in a ligand-dependent fashion. Cell Cycle. 2016;15(12):1579-90. [CrossRef]
- Livezey M, Kim JE, Shapiro DJ. A New Role for Estrogen Receptor α in Cell Proliferation and Cancer: Activating the Anticipatory Unfolded Protein Response. Front Endocrinol (Lausanne). 2018;9:325. [CrossRef]
- Thomas C, Gustafsson JÅ. The different roles of ER subtypes in cancer biology and therapy. Nat Rev Cancer. 2011;11(8):597-608. [CrossRef]
- Yang ZM, Yang MF, Yu W, Tao HM. Molecular mechanisms of estrogen receptor β-induced apoptosis and autophagy in tumors: implication for treating osteosarcoma. J Int Med Res. 2019;47(10):4644-4655. [CrossRef]
- Zava DT, Dollbaum CM, Blen M. Estrogen and progestin bioactivity of foods, herbs, and spices. Proc Soc Exp Biol Med. 1998;217(3):369-78. [CrossRef]
- Gruber CJ, Tschugguel W, Schneeberger C, Huber JC. Production and actions of estrogens. N Engl J Med. 2002;346(5):340-52. [CrossRef]
- Cassidy A. Potential tissue selectivity of dietary phytoestrogens and estrogens. Curr Opin Lipidol. 1999;10(1):47-52. [CrossRef]
- Patel S, Homaei A, Raju AB, Meher BR. Estrogen: The necessary evil for human health, and ways to tame it. Biomed Pharmacother. 2018;102:403-411. [CrossRef]
- Manavathi B, Samanthapudi VS, Gajulapalli VN. Estrogen receptor coregulators and pioneer factors: the orchestrators of mammary gland cell fate and development. Front Cell Dev Biol. 2014;2:34. [CrossRef]
- Zhou Y, Liu X. The role of estrogen receptor beta in breast cancer. Biomark Res. 2020; 8: 39. [CrossRef]
- Bhat SS, Prasad SK, Shivamallu C, Prasad KS, Syed A, Reddy P, Cull CA, Amachawadi RG. Genistein: A Potent Anti-Breast Cancer Agent. Curr Issues Mol Biol. 2021;43(3):1502-1517. [CrossRef]
- Kushner PJ, Agard DA, Greene GL, Scanlan TS, Shiau AK, Uht RM, Webb P. Estrogen receptor pathways to AP-1. J Steroid Biochem Mol Biol. 2000;74(5):311-7. [CrossRef]
- Prossnitz ER, Barton M. The G-protein-coupled estrogen receptor GPER in health and disease. Nat Rev Endocrinol. 2011;7(12):715-26. [CrossRef]
- Puglisi R, Mattia G, Carè A, Marano G, Malorni W, Matarrese P. Non-genomic Effects of Estrogen on Cell Homeostasis and Remodeling With Special Focus on Cardiac Ischemia/Reperfusion Injury. Front Endocrinol (Lausanne). 2019;10:733. [CrossRef]
- Yu K, Huang ZY, Xu XL, Li J, Fu XW, Deng SL. Estrogen Receptor Function: Impact on the Human Endometrium. Front Endocrinol (Lausanne). 2022;13:827724. [CrossRef]
- Filardo EJ, Quinn JA, Frackelton AR Jr, Bland KI. Estrogen action via the G protein-coupled receptor, GPR30: stimulation of adenylyl cyclase and cAMP-mediated attenuation of the epidermal growth factor receptor-to-MAPK signaling axis. Mol Endocrinol. 2002;16(1):70-84. [CrossRef]
- Albanito L, Sisci D, Aquila S, Brunelli E, Vivacqua A, Madeo A, Lappano R, Pandey DP, Picard D, Mauro L, Andò S, Maggiolini M. Epidermal growth factor induces G protein-coupled receptor 30 expression in estrogen receptor-negative breast cancer cells. Endocrinology. 2008;149(8):3799-808. [CrossRef]
- Pupo M, Maggiolini M, Musti AM. GPER Mediates Non-Genomic Effects of Estrogen. Methods Mol Biol. 2016;1366:471-488. [CrossRef]
- Barton M, Filardo EJ, Lolait SJ, Thomas P, Maggiolini M, Prossnitz ER. Twenty years of the G protein-coupled estrogen receptor GPER: Historical and personal perspectives. J Steroid Biochem Mol Biol. 2018;176:4-15. [CrossRef]
- Filardo EJ, Quinn JA, Bland KI, Frackelton AR Jr. Estrogen-induced activation of Erk-1 and Erk-2 requires the G protein-coupled receptor homolog, GPR30, and occurs via trans-activation of the epidermal growth factor receptor through release of HB-EGF. Mol Endocrinol. 2000;14(10):1649-60. [CrossRef]
- Prenzel N, Zwick E, Daub H, Leserer M, Abraham R, Wallasch C, Ullrich A. EGF receptor transactivation by G-protein-coupled receptors requires metalloproteinase cleavage of proHB-EGF. Nature. 1999;402(6764):884-8. [CrossRef]
- Edwin F, Wiepz GJ, Singh R, Peet CR, Chaturvedi D, Bertics PJ, Patel TB. A historical perspective of the EGF receptor and related systems. Methods Mol Biol. 2006;327:1-24. [CrossRef]
- Fan DX, Yang XH, Li YN, Guo L. 17β-Estradiol on the Expression of G-Protein Coupled Estrogen Receptor (GPER/GPR30) Mitophagy, and the PI3K/Akt Signaling Pathway in ATDC5 Chondrocytes In Vitro. Med Sci Monit. 2018;24:1936-1947. [CrossRef]
- Prossnitz ER, Maggiolini M. Mechanisms of estrogen signaling and gene expression via GPR30. Mol Cell Endocrinol. 2009;308(1-2):32-8. [CrossRef]
- Xu S, Yu S, Dong D, Lee LTO. G Protein-Coupled Estrogen Receptor: A Potential Therapeutic Target in Cancer. Front Endocrinol (Lausanne). 2019;10:725. [CrossRef]
- Maggiolini M, Vivacqua A, Fasanella G, Recchia AG, Sisci D, Pezzi V, Montanaro D, Musti AM, Picard D, Andò S. The G protein-coupled receptor GPR30 mediates c-fos up-regulation by 17beta-estradiol and phytoestrogens in breast cancer cells. J Biol Chem. 2004;279(26): 27008-16. [CrossRef]
- Luo J, Liu D. Does GPER Really Function as a G Protein-Coupled Estrogen Receptor in vivo? Front Endocrinol (Lausanne). 2020;11:148. [CrossRef]
- Roque C, Baltazar G. G protein-coupled estrogen receptor 1 (GPER) activation triggers different signaling pathways on neurons and astrocytes. Neural Regen Res. 2019;14(12): 2069-2070. [CrossRef]
- Conn PM, Ulloa-Aguirre A. Trafficking of G-protein-coupled receptors to the plasma membrane: insights for pharmacoperone drugs. Trends Endocrinol Metab. 2010;21(3):190-7. [CrossRef]
- Acconcia F, Bocedi A, Ascenzi P, Marino M. Does palmitoylation target estrogen receptors to plasma membrane caveolae? IUBMB Life. 2003;55(1):33-5. [CrossRef]
- Mendoza MC, Er EE, Blenis J. The Ras-ERK and PI3K-mTOR pathways: cross-talk and compensation. Trends Biochem Sci. 2011;36(6):320-8. [CrossRef]
- Moriarty K, Kim KH, Bender JR. Minireview: estrogen receptor-mediated rapid signaling. Endocrinology. 2006;147(12):5557-63. [CrossRef]
- Sirotkin AV, Harrath AH. Phytoestrogens and their effects. Eur J Pharmacol. 2014; 741: 230-6. [CrossRef]
- Cederroth CR, Zimmermann C, Nef S. Soy, phytoestrogens and their impact on reproductive health. Mol Cell Endocrinol. 2012;355(2):192-200. [CrossRef]
- Moreira AC, Silva AM, Santos MS, Sardão VA. Phytoestrogens as alternative hormone replacement therapy in menopause: What is real, what is unknown. J Steroid Biochem Mol Biol. 2014;143:61-71. [CrossRef]
- Domańska A, Orzechowski A, Litwiniuk A, Kalisz M, Bik W, Baranowska-Bik A. The Beneficial Role of Natural Endocrine Disruptors: Phytoestrogens in Alzheimer's Disease. Oxid Med Cell Longev. 2021;2021:3961445. [CrossRef]
- Nguyen M, Osipo C. Targeting Breast Cancer Stem Cells Using Naturally Occurring Phytoestrogens. Int J Mol Sci. 2022;23(12):6813. [CrossRef]
- Scherbakov AM, Andreeva OE. Apigenin Inhibits Growth of Breast Cancer Cells: The Role of ERα and HER2/neu. Acta Naturae. 2015;7(3):133-9.
- Petrakis NL, Barnes S, King EB, Lowenstein J, Wiencke J, Lee MM, Miike R, Kirk M, Coward L. Stimulatory influence of soy protein isolate on breast secretion in pre- and postmenopausal women. Cancer Epidemiol Biomarkers Prev. 1996;5(10):785-94.
- Hargreaves DF, Potten CS, Harding C, Shaw LE, Morton MS, Roberts SA, Howell A, Bundred NJ. Two-week dietary soy supplementation has an estrogenic effect on normal premenopausal breast. J Clin Endocrinol Metab. 1999;84(11):4017-24. [CrossRef]
- Lee AW, Poynor V, McEligot AJ. Urinary Phytoestrogen Levels Are Associated with Female Hormonal Cancers: An Analysis of NHANES Data From 1999 to 2010. Nutr Cancer. 2022;74(8):2748-2756. [CrossRef]
- Talaei M, Pan A. Role of phytoestrogens in prevention and management of type 2 diabetes. World J Diabetes. 2015;6(2):271-83. [CrossRef]
- Jefferson WN, Patisaul HB, Williams CJ. Reproductive consequences of developmental phytoestrogen exposure. Reproduction. 2012;143(3):247-60. [CrossRef]
- Swathi Krishna S, Kuriakose BB, Lakshmi PK. Effects of phytoestrogens on reproductive organ health. Arch Pharm Res. 2022;45(12):849-864. [CrossRef]
- Solano F, Hernández E, Juárez-Rojas L, Rojas-Maya S, López G, Romero C, Casillas F, Betancourt M, López A, Heidari R, Ommati MM, Retana-Márquez S. Reproductive disruption in adult female and male rats prenatally exposed to mesquite pod extract or daidzein. Reprod Biol. 2022;22(3):100683. [CrossRef]
- Patisaul HB, Jefferson W. The pros and cons of phytoestrogens. Front Neuroendocrinol. 2010;31(4):400-19. [CrossRef]
- Frankenfeld CL. Cardiometabolic risk and gut microbial phytoestrogen metabolite phenotypes. Mol Nutr Food Res. 2017;61(1). [CrossRef]
- Gore AC, Chappell VA, Fenton SE, Flaws JA, Nadal A, Prins GS, Toppari J, Zoeller RT. EDC-2: The Endocrine Society's Second Scientific Statement on Endocrine-Disrupting Chemicals. Endocr Rev. 2015;36(6):E1-E150. [CrossRef]
- Frye CA, Bo E, Calamandrei G, Calzà L, Dessì-Fulgheri F, Fernández M, Fusani L, Kah O, Kajta M, Le Page Y, Patisaul HB, Venerosi A, Wojtowicz AK, Panzica GC. Endocrine disrupters: a review of some sources, effects, and mechanisms of actions on behaviour and neuroendocrine systems. J Neuroendocrinol. 2012;24(1):144-59. [CrossRef]
- Degen GH, Janning P, Diel P, Michna H, Bolt HM. Transplacental transfer of the phytoestrogen daidzein in DA/Han rats. Arch Toxicol. 2002;76(1):23-9. [CrossRef]
- Jarrell J, Foster WG, Kinniburgh DW. Phytoestrogens in human pregnancy. Obstet Gynecol Int. 2012;2012:850313. [CrossRef]
- Patisaul HB, Adewale HB. Long-term effects of environmental endocrine disruptors on reproductive physiology and behavior. Front Behav Neurosci. 2009;3:10. [CrossRef]
- Patisaul HB. Endocrine disruption by dietary phyto-oestrogens: impact on dimorphic sexual systems and behaviours. Proc Nutr Soc. 2017;76(2):130-144. [CrossRef]
- Akiyama T, Ishida J, Nakagawa S, Ogawara H, Watanabe S, Itoh N, Shibuya M, Fukami Y. Genistein, a specific inhibitor of tyrosine-specific protein kinases. J Biol Chem. 1987;262(12):5592-5.
- Harvey PA, Leinwand LA. Dietary phytoestrogens present in soy dramatically increase cardiotoxicity in male mice receiving a chemotherapeutic tyrosine kinase inhibitor. Mol Cell Endocrinol. 2015;399:330-5. [CrossRef]
- Divi RL, Doerge DR. Inhibition of thyroid peroxidase by dietary flavonoids. Chem Res Toxicol. 1996;9(1):16-23. [CrossRef]
- Divi RL, Chang HC, Doerge DR. Anti-thyroid isoflavones from soybean: isolation, characterization, and mechanisms of action. Biochem Pharmacol. 1997;54(10):1087-96. [CrossRef]
- Sathyapalan T, Manuchehri AM, Thatcher NJ, Rigby AS, Chapman T, Kilpatrick ES, Atkin SL. The effect of soy phytoestrogen supplementation on thyroid status and cardiovascular risk markers in patients with subclinical hypothyroidism: a randomized, double-blind, crossover study. J Clin Endocrinol Metab. 2011;96(5):1442-9. [CrossRef]
- Sathyapalan T, Dawson AJ, Rigby AS, Thatcher NJ, Kilpatrick ES, Atkin SL. The Effect of Phytoestrogen on Thyroid in Subclinical Hypothyroidism: Randomized, Double Blind, Crossover Study. Front Endocrinol (Lausanne). 2018;9:531. [CrossRef]
- Dal Forno GO, Oliveira IM, Cavallin MD, Santos TIA, Sleiman HK, Falbo MK, Romano MA, Romano RM. Peripubertal soy isoflavone consumption leads to subclinical hypothyroidism in male Wistar rats. J Dev Orig Health Dis. 2022:1-14. [CrossRef]
- Awobajo FO, Medobi EF, Abdul MW, Aminu BB, Ojimma CT, Dada OG. The effect of genistein on IGF-1, PlGF, sFLT-1 and fetoplacental development. Gen Comp Endocrinol. 2022;329:114122. [CrossRef]
- Picherit C, Dalle M, Néliat G, Lebecque P, Davicco MJ, Barlet JP, Coxam V. Genistein and daidzein modulate in vitro rat uterine contractile activity. J Steroid Biochem Mol Biol. 2000;75(2-3):201-8. [CrossRef]
- Richter DU, Mylonas I, Toth B, Scholz C, Briese V, Friese K, Jeschke U. Effects of phytoestrogens genistein and daidzein on progesterone and estrogen (estradiol) production of human term trophoblast cells in vitro. Gynecol Endocrinol. 2009;25(1):32-8. [CrossRef]
- Badger TM, Ronis MJ, Hakkak R, Rowlands JC, Korourian S. The health consequences of early soy consumption. J Nutr. 2002;132(3):559S-565S. [CrossRef]
- Franke AA, Custer LJ, Tanaka Y. Isoflavones in human breast milk and other biological fluids. Am J Clin Nutr. 1998;68(6 Suppl):1466S-1473S. [CrossRef]
- Setchell KD, Zimmer-Nechemias L, Cai J, Heubi JE. Exposure of infants to phyto-oestrogens from soy-based infant formula. Lancet. 1997;350(9070):23-7. [CrossRef]
- Setchell KD, Zimmer-Nechemias L, Cai J, Heubi JE. Isoflavone content of infant formulas and the metabolic fate of these phytoestrogens in early life. Am J Clin Nutr. 1998; 68(6 Suppl):1453S-1461S. [CrossRef]
- Suk An E, Park D, Ban YH, Jieun C, Seo DW, Bok Lee Y, Shon MY, Choi EK, Kim YB. Effects of a soybean milk product on feto-neonatal development in rats. J Biomed Res. 2017;32(1):51–7. [CrossRef]
- Guerrero-Bosagna CM, Sabat P, Valdovinos FS, Valladares LE, Clark SJ. Epigenetic and phenotypic changes result from a continuous pre and postnatal dietary exposure to phytoestrogens in an experimental population of mice. BMC Physiol. 2008;8:17. [CrossRef]
- Becker M, Hesse V. Minipuberty: Why Does it Happen? Horm Res Paediatr. 2020;93(2):76-84. [CrossRef]
- Suen AA, Kenan AC, Williams CJ. Developmental exposure to phytoestrogens found in soy: New findings and clinical implications. Biochem Pharmacol. 2022;195: 114848. [CrossRef]
- Upson K, Sathyanarayana S, Scholes D, Holt VL. Early-life factors and endometriosis risk. Fertil Steril. 2015;104(4):964-971.e5. [CrossRef]
- Ottolina J, Schimberni M, Makieva S, Bartiromo L, Fazia T, Bernardinelli L, Viganò P, Candiani M, Gentilini D. Early-life factors, in-utero exposures and endometriosis risk: a meta-analysis. Reprod Biomed Online. 2020;41(2):279-289. [CrossRef]
- Gao M, Allebeck P, Mishra GD, Koupil I. Developmental origins of endometriosis: a Swedish cohort study. J Epidemiol Community Health. 2019;73(4):353-359. [CrossRef]
- Krishnamoorthy SP, Kalimuthu V, Chandran Manimegalai S, Arulanandu AM, Thiyagarajan R, Balamuthu K. Evaluation of the potential role of diethylstilbestrol on the induction of endometriosis in a rat model - An alternative approach. Biochem Biophys Res Commun. 2022;617(Pt 2):18-24. [CrossRef]
- Golinski P, Vesonder RF, Latus-Zietkiewicz D, Perkowski J. Formation of fusarenone X, nivalenol, zearalenone, alpha-trans-zearalenol, beta-trans-zearalenol, and fusarin C by Fusarium crookwellense. Appl Environ Microbiol. 1988;54(8):2147-8. [CrossRef]
- Bryła M, Pierzgalski A, Zapaśnik A, Uwineza PA, Ksieniewicz-Woźniak E, Modrzewska M, Waśkiewicz A. Recent Research on Fusarium Mycotoxins in Maize-A Review. Foods. 2022;11(21):3465. [CrossRef]
- Ropejko K, Twarużek M. Zearalenone and Its Metabolites-General Overview, Occurrence, and Toxicity. Toxins (Basel). 2021;13(1):35. [CrossRef]
- Sondergaard TE, Hansen FT, Purup S, Nielsen AK, Bonefeld-Jørgensen EC, Giese H, Sørensen JL. Fusarin C acts like an estrogenic agonist and stimulates breast cancer cells in vitro. Toxicol Lett. 2011;205(2):116-21. [CrossRef]
- Zheng W, Wang B, Li X, Wang T, Zou H, Gu J, Yuan Y, Liu X, Bai J, Bian J, Liu Z. Zearalenone Promotes Cell Proliferation or Causes Cell Death? Toxins (Basel). 2018;10(5):184. [CrossRef]
- Belli P, Bellaton C, Durand J, Balleydier S, Milhau N, Mure M, Mornex JF, Benahmed M, Le Jan C. Fetal and neonatal exposure to the mycotoxin zearalenone induces phenotypic alterations in adult rat mammary gland. Food Chem Toxicol. 2010;48(10):2818-26. [CrossRef]
- Jing S, Liu C, Zheng J, Dong Z, Guo N. Toxicity of zearalenone and its nutritional intervention by natural products. Food Funct. 2022;13(20):10374-10400. [CrossRef]
- Kinkade CW, Rivera-Núñez Z, Gorcyzca L, Aleksunes LM, Barrett ES. Impact of Fusarium-Derived Mycoestrogens on Female Reproduction: A Systematic Review. Toxins (Basel). 2021;13(6):373. [CrossRef]
- Gao X, Sun L, Zhang N, Li C, Zhang J, Xiao Z, Qi D. Gestational Zearalenone Exposure Causes Reproductive and Developmental Toxicity in Pregnant Rats and Female Offspring. Toxins (Basel). 2017;9(1):21. [CrossRef]
- Bartiromo L, Schimberni M, Villanacci R, Ottolina J, Dolci C, Salmeri N, Viganò P, Candiani M. Endometriosis and Phytoestrogens: Friends or Foes? A Systematic Review. Nutrients. 2021;13(8):2532. [CrossRef]
- Yan WK, Liu YN, Song SS, Kang JW, Zhang Y, Lu L, Wei SW, Xu QX, Zhang WQ, Liu XZ, Wu Y, Su RW. Zearalenone affects the growth of endometriosis via estrogen signaling and inflammatory pathways. Ecotoxicol Environ Saf. 2022;241:113826. [CrossRef]
- Cai X, Liu M, Zhang B, Zhao SJ, Jiang SW. Phytoestrogens for the Management of Endometriosis: Findings and Issues. Pharmaceuticals (Basel). 2021;14(6):569. [CrossRef]
- Rosenfeld CS, Cooke PS. Endocrine disruption through membrane estrogen receptors and novel pathways leading to rapid toxicological and epigenetic effects. J Steroid Biochem Mol Biol. 2019;187:106-117. [CrossRef]
- Feng JX, Riddle NC. Epigenetics and genome stability. Mamm Genome. 2020;31(5-6): 181-195. [CrossRef]
- Zubrzycka A, Zubrzycki M, Perdas E, Zubrzycka M. Genetic, Epigenetic, and Steroidogenic Modulation Mechanisms in Endometriosis. J Clin Med. 2020;9(5):1309. [CrossRef]
- Szukiewicz D, Stangret A, Ruiz-Ruiz C, Olivares EG, Soriţău O, Suşman S, Szewczyk G. Estrogen- and Progesterone (P4)-Mediated Epigenetic Modifications of Endometrial Stromal Cells (EnSCs) and/or Mesenchymal Stem/Stromal Cells (MSCs) in the Etiopathogenesis of Endometriosis. Stem Cell Rev Rep. 2021;17(4):1174-1193. [CrossRef]
- Chen H, Malentacchi F, Fambrini M, Harrath AH, Huang H, Petraglia F. Epigenetics of Estrogen and Progesterone Receptors in Endometriosis. Reprod Sci. 2020;27(11):1967-1974. [CrossRef]
- Bansal A, Henao-Mejia J, Simmons RA. Immune System: An Emerging Player in Mediating Effects of Endocrine Disruptors on Metabolic Health. Endocrinology. 2018; 159(1):32-45. [CrossRef]
- Abramiuk M, Grywalska E, Małkowska P, Sierawska O, Hrynkiewicz R, Niedźwiedzka-Rystwej P. The Role of the Immune System in the Development of Endometriosis. Cells. 2022;11(13):2028. [CrossRef]
- Szukiewicz D. Epigenetic regulation and T-cell responses in endometriosis - something other than autoimmunity. Front Immunol. 2022;13:943839. [CrossRef]
- Li Y, Zhang JJ, Chen RJ, Chen L, Chen S, Yang XF, Min JW. Genistein mitigates oxidative stress and inflammation by regulating Nrf2/HO-1 and NF-κB signaling pathways in hypoxic-ischemic brain damage in neonatal mice. Ann Transl Med. 2022;10(2):32. [CrossRef]
- Chen Y, Peng F, Xing Z, Chen J, Peng C, Li D. Beneficial effects of natural flavonoids on neuroinflammation. Front Immunol. 2022;13:1006434. [CrossRef]
- Dąbek J, Kułach A, Gąsior Z. Nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB): a new potential therapeutic target in atherosclerosis? Pharmacol Rep. 2010; 62(5):778-83. [CrossRef]
- Desmawati D, Sulastri D. Phytoestrogens and Their Health Effect. Open Access Maced J Med Sci. 2019;7(3):495-499. [CrossRef]
- Cady N, Peterson SR, Freedman SN, Mangalam AK. Beyond Metabolism: The Complex Interplay Between Dietary Phytoestrogens, Gut Bacteria, and Cells of Nervous and Immune Systems. Front Neurol. 2020;11:150. [CrossRef]
- Masilamani M, Wei J, Bhatt S, Paul M, Yakir S, Sampson HA. Soybean isoflavones regulate dendritic cell function and suppress allergic sensitization to peanut. J Allergy Clin Immunol. 2011;128(6):1242-1250.e1. [CrossRef]
- Chiang SS, Pan TM. Beneficial effects of phytoestrogens and their metabolites produced by intestinal microflora on bone health. Appl Microbiol Biotechnol. 2013;97(4):1489-500. [CrossRef]
- Mace TA, Ware MB, King SA, Loftus S, Farren MR, McMichael E, Scoville S, Geraghty C, Young G, Carson WE 3rd, Clinton SK, Lesinski GB. Soy isoflavones and their metabolites modulate cytokine-induced natural killer cell function. Sci Rep. 2019;9(1):5068. [CrossRef]
- Wei J, Bhatt S, Chang LM, Sampson HA, Masilamani M. Isoflavones, genistein and daidzein, regulate mucosal immune response by suppressing dendritic cell function. PLoS One. 2012;7(10):e47979. [CrossRef]
- Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol. 2008;8(12):958-69. [CrossRef]
- Dia VP, Berhow MA, Gonzalez De Mejia E. Bowman-Birk inhibitor and genistein among soy compounds that synergistically inhibit nitric oxide and prostaglandin E2 pathways in lipopolysaccharide-induced macrophages. J Agric Food Chem. 2008;56(24):11707-17. [CrossRef]
- Abron JD, Singh NP, Price RL, Nagarkatti M, Nagarkatti PS, Singh UP. Genistein induces macrophage polarization and systemic cytokine to ameliorate experimental colitis. PLoS One. 2018;13(7):e0199631. [CrossRef]
- Iyer SS, Cheng G. Role of interleukin 10 transcriptional regulation in inflammation and autoimmune disease. Crit Rev Immunol. 2012;32(1):23-63. [CrossRef]
- Csaba G. Effect of endocrine disruptor phytoestrogens on the immune system: Present and future. Acta Microbiol Immunol Hung. 2018;65(1):1-14. [CrossRef]
- Saunders PTK, Horne AW. Endometriosis: Etiology, pathobiology, and therapeutic prospects. Cell. 2021;184(11):2807-2824. [CrossRef]
- International working group of AAGL, ESGE, ESHRE and WES; Tomassetti C, Johnson NP, Petrozza J, Abrao MS, Einarsson JI, Horne AW, Lee TTM, Missmer S, Vermeulen N, Zondervan KT, Grimbizis G, De Wilde RL. An International Terminology for Endometriosis, 2021. J Minim Invasive Gynecol. 2021;28(11):1849-1859. [CrossRef]
- Sampson JA. Metastatic or Embolic Endometriosis, due to the Menstrual Dissemination of Endometrial Tissue into the Venous Circulation. Am J Pathol. 1927;3(2):93-110.43.
- Mikhaleva LM, Radzinsky VE, Orazov MR, Khovanskaya TN, Sorokina AV, Mikhalev SA, Volkova SV, Shustova VB, Sinelnikov MY. Current Knowledge on Endometriosis Etiology: A Systematic Review of Literature. Int J Womens Health. 2021;13:525-537. [CrossRef]
- Signorile PG, Viceconte R, Baldi A. New Insights in Pathogenesis of Endometriosis. Front Med (Lausanne). 2022;9:879015. [CrossRef]
- Chamié LP, Ribeiro DMFR, Tiferes DA, Macedo Neto AC, Serafini PC. Atypical Sites of Deeply Infiltrative Endometriosis: Clinical Characteristics and Imaging Findings. Radiographics. 2018;38(1):309-328. [CrossRef]
- Machairiotis N, Stylianaki A, Dryllis G, Zarogoulidis P, Kouroutou P, Tsiamis N, Katsikogiannis N, Sarika E, Courcoutsakis N, Tsiouda T, Gschwendtner A, Zarogoulidis K, Sakkas L, Baliaka A, Machairiotis C. Extrapelvic endometriosis: a rare entity or an under diagnosed condition? Diagn Pathol. 2013;8:194. [CrossRef]
- Kamergorodsky G, Ribeiro PA, Galvão MA, Abrão MS, Donadio N, Lemos NL, Aoki T. Histologic classification of specimens from women affected by superficial endometriosis, deeply infiltrating endometriosis, and ovarian endometriomas. Fertil Steril. 2009;92(6):2074-7. [CrossRef]
- Al-Jefout M, Dezarnaulds G, Cooper M, Tokushige N, Luscombe GM, Markham R, Fraser IS. Diagnosis of endometriosis by detection of nerve fibres in an endometrial biopsy: a double blind study. Hum Reprod. 2009;24(12):3019-24. [CrossRef]
- Bulun SE, Cheng YH, Pavone ME, Xue Q, Attar E, Trukhacheva E, Tokunaga H, Utsunomiya H, Yin P, Luo X, Lin Z, Imir G, Thung S, Su EJ, Kim JJ. Estrogen receptor-beta, estrogen receptor-alpha, and progesterone resistance in endometriosis. Semin Reprod Med. 2010;28(1):36-43. [CrossRef]
- Plante BJ, Lessey BA, Taylor RN, Wang W, Bagchi MK, Yuan L, Scotchie J, Fritz MA, Young SL. G protein-coupled estrogen receptor (GPER) expression in normal and abnormal endometrium. Reprod Sci. 2012;19(7):684-93. [CrossRef]
- Kim JH, Han E. Endometriosis and Female Pelvic Pain. Semin Reprod Med. 2018;36(2): 143-151. [CrossRef]
- Burney RO, Giudice LC. Pathogenesis and pathophysiology of endometriosis. Fertil Steril. 2012;98(3):511-9. [CrossRef]
- Patel BG, Lenk EE, Lebovic DI, Shu Y, Yu J, Taylor RN. Pathogenesis of endometriosis: Interaction between Endocrine and inflammatory pathways. Best Pract Res Clin Obstet Gynaecol. 2018;50:50-60. [CrossRef]
- Tomassetti C, D'Hooghe T. Endometriosis and infertility: Insights into the causal link and management strategies. Best Pract Res Clin Obstet Gynaecol. 2018;51:25-33. [CrossRef]
- Zondervan KT, Becker CM, Koga K, Missmer SA, Taylor RN, Viganò P. Endometriosis. Nat Rev Dis Primers. 2018;4(1):9. [CrossRef]
- Kajiyama H, Suzuki S, Yoshihara M, Tamauchi S, Yoshikawa N, Niimi K, Shibata K, Kikkawa F. Endometriosis and cancer. Free Radic Biol Med. 2019;133:186-192. [CrossRef]
- Klemmt PAB, Starzinski-Powitz A. Molecular and Cellular Pathogenesis of Endometriosis. Curr Womens Health Rev. 2018;14(2):106-116. [CrossRef]
- Rolla E. Endometriosis: advances and controversies in classification, pathogenesis, diagnosis, and treatment. F1000Res. 2019;8:F1000 Faculty Rev-529. [CrossRef]
- Logan PC, Yango P, Tran ND. Endometrial Stromal and Epithelial Cells Exhibit Unique Aberrant Molecular Defects in Patients With Endometriosis. Reprod Sci. 2018;25(1):140-159. [CrossRef]
- Maruyama T, Yoshimura Y. Stem cell theory for the pathogenesis of endometriosis. Front Biosci (Elite Ed). 2012;4(8):2754-63. [CrossRef]
- Gargett CE, Schwab KE, Brosens JJ, Puttemans P, Benagiano G, Brosens I. Potential role of endometrial stem/progenitor cells in the pathogenesis of early-onset endometriosis. Mol Hum Reprod. 2014;20(7):591-8. [CrossRef]
- Dinsdale N, Nepomnaschy P, Crespi B. The evolutionary biology of endometriosis. Evol Med Public Health. 2021;9(1):174-191. [CrossRef]
- Ng SW, Norwitz GA, Pavlicev M, Tilburgs T, Simón C, Norwitz ER. Endometrial Decidualization: The Primary Driver of Pregnancy Health. Int J Mol Sci. 2020;21(11):4092. [CrossRef]
- Xu Y, Zhu H, Zhao D, Tan J. Endometrial stem cells: clinical application and pathological roles. Int J Clin Exp Med. 2015;8(12):22039-44.
- da Costa e Silva Rde C, Moura KK, Ribeiro Júnior CL, Guillo LA. Estrogen signaling in the proliferative endometrium: implications in endometriosis. Rev Assoc Med Bras. (1992). 2016;62(1):72-7. [CrossRef]
- Ballaré C, Uhrig M, Bechtold T, Sancho E, Di Domenico M, Migliaccio A, Auricchio F, Beato M. Two domains of the progesterone receptor interact with the estrogen receptor and are required for progesterone activation of the c-Src/Erk pathway in mammalian cells. Mol Cell Biol. 2003;23(6):1994-2008. [CrossRef]
- Kasubuchi M, Watanabe K, Hirano K, Inoue D, Li X, Terasawa K, Konishi M, Itoh N, Kimura I. Membrane progesterone receptor beta (mPRβ/Paqr8) promotes progesterone-dependent neurite outgrowth in PC12 neuronal cells via non-G protein-coupled receptor (GPCR) signaling. Sci Rep. 2017;7(1):5168. [CrossRef]
- Appleyard CB, Flores I, Torres-Reverón A. The Link Between Stress and Endometriosis: from Animal Models to the Clinical Scenario. Reprod Sci. 2020;27(9):1675-1686. [CrossRef]
- Huhtinen K, Desai R, Ståhle M, Salminen A, Handelsman DJ, Perheentupa A, Poutanen M. Endometrial and endometriotic concentrations of estrone and estradiol are determined by local metabolism rather than circulating levels. J Clin Endocrinol Metab. 2012;97(11):4228-35. [CrossRef]
- Streuli I, Gaitzsch H, Wenger JM, Petignat P. Endometriosis after menopause: physiopathology and management of an uncommon condition. Climacteric. 2017;20(2):138-143. [CrossRef]
- Leone Roberti Maggiore U, Ferrero S, Mangili G, Bergamini A, Inversetti A, Giorgione V, Viganò P, Candiani M. A systematic review on endometriosis during pregnancy: diagnosis, misdiagnosis, complications and outcomes. Hum Reprod Update. 2016;22(1):70-103. [CrossRef]
- Jeng CJ, Chuang L, Shen J. A comparison of progestogens or oral contraceptives and gonadotropin-releasing hormone agonists for the treatment of endometriosis: a systematic review. Expert Opin Pharmacother. 2014;15(6):767-73. [CrossRef]
- Stocco C. Tissue physiology and pathology of aromatase. Steroids. 2012;77(1-2):27-35. [CrossRef]
- Bulun SE, Fang Z, Imir G, Gurates B, Tamura M, Yilmaz B, Langoi D, Amin S, Yang S, Deb S. Aromatase and endometriosis. Semin Reprod Med. 2004;22(1):45-50. [CrossRef]
- Noble LS, Takayama K, Zeitoun KM, Putman JM, Johns DA, Hinshelwood MM, Agarwal VR, Zhao Y, Carr BR, Bulun SE. Prostaglandin E2 stimulates aromatase expression in endometriosis-derived stromal cells. J Clin Endocrinol Metab. 1997;82(2):600-6. [CrossRef]
- Bulun SE, Takayama K, Suzuki T, Sasano H, Yilmaz B, Sebastian S. Organization of the human aromatase p450 (CYP19) gene. Semin Reprod Med. 2004;22(1):5-9. [CrossRef]
- Izawa M, Harada T, Taniguchi F, Ohama Y, Takenaka Y, Terakawa N. An epigenetic disorder may cause aberrant expression of aromatase gene in endometriotic stromal cells. Fertil Steril. 2008;89(5 Suppl):1390-6. [CrossRef]
- Izawa M, Taniguchi F, Uegaki T, Takai E, Iwabe T, Terakawa N, Harada T. Demethylation of a nonpromoter cytosine-phosphate-guanine island in the aromatase gene may cause the aberrant up-regulation in endometriotic tissues. Fertil Steril. 2011;95(1):33-9. [CrossRef]
- Moore LD, Le T, Fan G. DNA methylation and its basic function. Neuropsychopharmacology. 2013;38(1):23-38. [CrossRef]
- Xue Q, Zhou YF, Zhu SN, Bulun SE. Hypermethylation of the CpG island spanning from exon II to intron III is associated with steroidogenic factor 1 expression in stromal cells of endometriosis. Reprod Sci. 2011;18(11):1080-4. [CrossRef]
- Koukoura O, Sifakis S, Spandidos DA. DNA methylation in endometriosis (Review). Mol Med Rep. 2016;13(4):2939-48. [CrossRef]
- Zeitoun K, Takayama K, Sasano H, Suzuki T, Moghrabi N, Andersson S, Johns A, Meng L, Putman M, Carr B, Bulun SE. Deficient 17beta-hydroxysteroid dehydrogenase type 2 expression in endometriosis: failure to metabolize 17beta-estradiol. J Clin Endocrinol Metab. 1998;83(12):4474-80. [CrossRef]
- Yamagata Y, Nishino K, Takaki E, Sato S, Maekawa R, Nakai A, Sugino N. Genome-wide DNA methylation profiling in cultured eutopic and ectopic endometrial stromal cells. PLoS One. 2014;9(1):e83612. [CrossRef]
- Husen B, Psonka N, Jacob-Meisel M, Keil C, Rune GM. Differential expression of 17beta-hydroxysteroid dehydrogenases types 2 and 4 in human endometrial epithelial cell lines. J Mol Endocrinol. 2000;24(1):135-44. [CrossRef]
- He W, Gauri M, Li T, Wang R, Lin SX. Current knowledge of the multifunctional 17β-hydroxysteroid dehydrogenase type 1 (HSD17B1). Gene. 2016;588(1):54-61. [CrossRef]
- Allis CD, Jenuwein T. The molecular hallmarks of epigenetic control. Nat Rev Genet. 2016;17(8):487-500. [CrossRef]
- Loganathan T, Doss C GP. Non-coding RNAs in human health and disease: potential function as biomarkers and therapeutic targets. Funct Integr Genomics. 2023;23(1):33. [CrossRef]
- Trerotola M, Relli V, Simeone P, Alberti S. Epigenetic inheritance and the missing heritability. Hum Genomics. 2015;9(1):17. [CrossRef]
- Wątroba M, Dudek I, Skoda M, Stangret A, Rzodkiewicz P, Szukiewicz D. Sirtuins, epigenetics and longevity. Ageing Res Rev. 2017;40:11-19. [CrossRef]
- Cousins FL, Filby CE, Gargett CE. Endometrial Stem/Progenitor Cells-Their Role in Endometrial Repair and Regeneration. Front Reprod Health. 2022;3:811537. [CrossRef]
- Gurusamy N, Alsayari A, Rajasingh S, Rajasingh J. Adult Stem Cells for Regenerative Therapy. Prog Mol Biol Transl Sci. 2018;160:1-22. [CrossRef]
- Papatsenko D, Waghray A, Lemischka IR. Feedback control of pluripotency in embryonic stem cells: Signaling, transcription and epigenetics. Stem Cell Res. 2018;29:180-188. [CrossRef]
- Edmunds KM, Holloway AC, Crankshaw DJ, Agarwal SK, Foster WG. The effects of dietary phytoestrogens on aromatase activity in human endometrial stromal cells. Reprod Nutr Dev. 2005;45(6):709-20. [CrossRef]
- Koninckx PR, Ussia A, Adamyan L, Wattiez A, Gomel V, Martin DC. Pathogenesis of endometriosis: the genetic/epigenetic theory. Fertil Steril. 2019;111(2):327-340. [CrossRef]
- Retis-Resendiz AM, González-García IN, León-Juárez M, Camacho-Arroyo I, Cerbón M, Vázquez-Martínez ER. The role of epigenetic mechanisms in the regulation of gene expression in the cyclical endometrium. Clin Epigenetics. 2021;13(1):116. [CrossRef]
- Szukiewicz D. Aberrant epigenetic regulation of estrogen and progesterone signaling at the level of endometrial/endometriotic tissue in the pathomechanism of endometriosis. Vitam Horm. 2023;122:193-235. [CrossRef]
- Vrtačnik P, Ostanek B, Mencej-Bedrač S, Marc J. The many faces of estrogen signaling. Biochem Med (Zagreb). 2014;24(3):329-42. [CrossRef]
- Zhou Q, Shaw PG, Davidson NE. Epigenetics meets estrogen receptor: regulation of estrogen receptor by direct lysine methylation. Endocr Relat Cancer. 2009;16(2):319-23. [CrossRef]
- Brandenberger AW, Lebovic DI, Tee MK, Ryan IP, Tseng JF, Jaffe RB, Taylor RN. Oestrogen receptor (ER)-alpha and ER-beta isoforms in normal endometrial and endometriosis-derived stromal cells. Mol Hum Reprod. 1999;5(7):651-5. [CrossRef]
- Simmen RC, Kelley AS. Reversal of fortune: estrogen receptor-β in endometriosis. J Mol Endocrinol. 2016;57(2):F23-7. [CrossRef]
- Xue Q, Lin Z, Cheng YH, Huang CC, Marsh E, Yin P, Milad MP, Confino E, Reierstad S, Innes J, Bulun SE. Promoter methylation regulates estrogen receptor 2 in human endometrium and endometriosis. Biol Reprod. 2007;77(4):681-7. [CrossRef]
- Bulun SE, Zeitoun KM, Takayama K, Simpson E, Sasano H. Aromatase as a therapeutic target in endometriosis. Trends Endocrinol Metab. 2000;11(1):22-7. [CrossRef]
- Patel BG, Rudnicki M, Yu J, Shu Y, Taylor RN. Progesterone resistance in endometriosis: origins, consequences and interventions. Acta Obstet Gynecol Scand. 2017;96(6):623-632. [CrossRef]
- Trukhacheva E, Lin Z, Reierstad S, Cheng YH, Milad M, Bulun SE. Estrogen receptor (ER) beta regulates ERalpha expression in stromal cells derived from ovarian endometriosis. J Clin Endocrinol Metab. 2009;94(2):615-22. [CrossRef]
- Ferlita A, Battaglia R, Andronico F, Caruso S, Cianci A, Purrello M, Pietro CD. Non-Coding RNAs in Endometrial Physiopathology. Int J Mol Sci. 2018;19(7):2120. [CrossRef]
- Vidal-Gómez X, Pérez-Cremades D, Mompeón A, Dantas AP, Novella S, Hermenegildo C. MicroRNA as Crucial Regulators of Gene Expression in Estradiol-Treated Human Endothelial Cells. Cell Physiol Biochem. 2018;45(5):1878-1892. [CrossRef]
- Cai H, Zhu XX, Li ZF, Zhu YP, Lang JH. MicroRNA Dysregulation and Steroid Hormone Receptor Expression in Uterine Tissues of Rats with Endometriosis during the Implantation Window. Chin Med J (Engl). 2018;131(18):2193-2204. [CrossRef]
- Klinge CM. miRNAs and estrogen action. Trends Endocrinol Metab. 2012;23(5):223-33. [CrossRef]
- Pandey DP, Picard D. miR-22 inhibits estrogen signaling by directly targeting the estrogen receptor alpha mRNA. Mol Cell Biol. 2009;29(13):3783-90. [CrossRef]
- Lin Y, Xiao L, Zhang Y, Li P, Wu Y, Lin Y. MiR-26b-3p regulates osteoblast differentiation via targeting estrogen receptor α. Genomics. 2019;111(5):1089-1096. [CrossRef]
- Al-Nakhle H, Burns PA, Cummings M, Hanby AM, Hughes TA, Satheesha S, Shaaban AM, Smith L, Speirs V. Estrogen receptor {beta}1 expression is regulated by miR-92 in breast cancer. Cancer Res. 2010;70(11):4778-84. [CrossRef]
- He SZ, Li J, Bao HC, Wang MM, Wang XR, Huang X, Li FH, Zhang W, Xu AL, Fang HC, Sheng YX. G protein-coupled estrogen receptor/miR-148a/human leukocyte antigen-G signaling pathway mediates cell apoptosis of ovarian endometriosis. Mol Med Rep. 2018; 18(1):1141-1148. [CrossRef]
- Knoll M, Lodish HF, Sun L. Long non-coding RNAs as regulators of the endocrine system. Nat Rev Endocrinol. 2015;11(3):151-60. [CrossRef]
- Yan W, Hu H, Tang B. Progress in understanding the relationship between long noncoding RNA and endometriosis. Eur J Obstet Gynecol Reprod Biol X. 2019;5:100067. [CrossRef]
- Bhan A, Hussain I, Ansari KI, Kasiri S, Bashyal A, Mandal SS. Antisense transcript long noncoding RNA (lncRNA) HOTAIR is transcriptionally induced by estradiol. J Mol Biol. 2013;425(19):3707-22. [CrossRef]
- Trisciuoglio D, Di Martile M, Del Bufalo D. Emerging Role of Histone Acetyltransferase in Stem Cells and Cancer. Stem Cells Int. 2018;2018:8908751. [CrossRef]
- Zhang J, Jing L, Li M, He L, Guo Z. Regulation of histone arginine methylation/demethylation by methylase and demethylase (Review). Mol Med Rep. 2019; 19(5):3963-3971. [CrossRef]
- Yokoyama A, Fujiki R, Ohtake F, Kato S. Regulated histone methyltransferase and demethylase complexes in the control of genes by nuclear receptors. Cold Spring Harb Symp Quant Biol. 2011;76:165-73. [CrossRef]
- Liu C, Wu HT, Zhu N, Shi YN, Liu Z, Ao BX, Liao DF, Zheng XL, Qin L. Steroid receptor RNA activator: Biologic function and role in disease. Clin Chim Acta. 2016;459:137-146. [CrossRef]
- Lin K, Zhan H, Ma J, Xu K, Wu R, Zhou C, Lin J. Silencing of SRA1 Regulates ER Expression and Attenuates the Growth of Stromal Cells in Ovarian Endometriosis. Reprod Sci. 2017;24(6):836-843. [CrossRef]
- Moore RL, Dai Y, Faller DV. Sirtuin 1 (SIRT1) and steroid hormone receptor activity in cancer. J Endocrinol. 2012;213(1):37-48. [CrossRef]
- Yoo JY, Kim TH, Fazleabas AT, Palomino WA, Ahn SH, Tayade C, Schammel DP, Young SL, Jeong JW, Lessey BA. KRAS Activation and over-expression of SIRT1/BCL6 Contributes to the Pathogenesis of Endometriosis and Progesterone Resistance. Sci Rep. 2017; 7(1):6765. [CrossRef]
- Xiaomeng X, Ming Z, Jiezhi M, Xiaoling F. Aberrant histone acetylation and methylation levels in woman with endometriosis. Arch Gynecol Obstet. 2013;287(3):487-94. [CrossRef]
- Han SJ, O'Malley BW. The dynamics of nuclear receptors and nuclear receptor coregulators in the pathogenesis of endometriosis. Hum Reprod Update. 2014;20(4):467-84. [CrossRef]
- Mahajan V, Farquhar C, Ponnampalam AP. Could DNA hydroxymethylation be crucial in influencing steroid hormone signaling in endometrial biology and endometriosis? Mol Reprod Dev. 2020;87(1):7-16. [CrossRef]
- Klinge CM. Estrogen action: Receptors, transcripts, cell signaling, and non-coding RNAs in normal physiology and disease. Mol Cell Endocrinol. 2015;418 Pt 3:191-2. [CrossRef]
- Grimstad FW, Decherney A. A Review of the Epigenetic Contributions to Endometriosis. Clin Obstet Gynecol. 2017;60(3):467-476. [CrossRef]
- Nasu K, Nishida M, Kawano Y, Tsuno A, Abe W, Yuge A, Takai N, Narahara H. Aberrant expression of apoptosis-related molecules in endometriosis: a possible mechanism underlying the pathogenesis of endometriosis. Reprod Sci. 2011;18(3):206-18. [CrossRef]
- Chopyak VV, Koval HD, Havrylyuk AM, Lishchuk-Yakymovych KA, Potomkina HA, Kurpisz MK. Immunopathogenesis of endometriosis - a novel look at an old problem. Cent Eur J Immunol. 2022;47(1):109-116. [CrossRef]
- Vallvé-Juanico J, Houshdaran S, Giudice LC. The endometrial immune environment of women with endometriosis. Hum Reprod Update. 2019; 25(5):564-591. [CrossRef]
- Kobayashi H, Imanaka S. Understanding the molecular mechanisms of macrophage polarization and metabolic reprogramming in endometriosis: A narrative review. Reprod Med Biol. 2022;21(1):e12488. [CrossRef]
- Pernis AB. Estrogen and CD4+ T cells. Curr Opin Rheumatol. 2007;19(5):414-20. [CrossRef]
- Wan YY. Regulatory T cells: immune suppression and beyond. Cell Mol Immunol. 2010;7(3):204-10. [CrossRef]
- Bayati F, Mohammadi M, Valadi M, Jamshidi S, Foma AM, Sharif-Paghaleh E. The Therapeutic Potential of Regulatory T Cells: Challenges and Opportunities. Front Immunol. 2021;11:585819. [CrossRef]
- de Barros IBL, Malvezzi H, Gueuvoghlanian-Silva BY, Piccinato CA, Rizzo LV, Podgaec S. "What do we know about regulatory T cells and endometriosis? A systematic review". J Reprod Immunol. 2017;120:48-55. Erratum in: J Reprod Immunol. 2017;121:34. [CrossRef]
- Camboni A, Marbaix E. Ectopic Endometrium: The Pathologist's Perspective. Int J Mol Sci. 2021;22(20):10974. [CrossRef]
- McKinnon B, Mueller M, Montgomery G. Progesterone Resistance in Endometriosis: an Acquired Property? Trends Endocrinol Metab. 2018;29(8):535-548. [CrossRef]
- Chimento A, De Luca A, Avena P, De Amicis F, Casaburi I, Sirianni R, Pezzi V. Estrogen Receptors-Mediated Apoptosis in Hormone-Dependent Cancers. Int J Mol Sci. 2022;23(3):1242. [CrossRef]
- Khan D, Ansar Ahmed S. The Immune System Is a Natural Target for Estrogen Action: Opposing Effects of Estrogen in Two Prototypical Autoimmune Diseases. Front Immunol. 2016;6:635. [CrossRef]
- Hewagama A, Patel D, Yarlagadda S, Strickland FM, Richardson BC. Stronger inflammatory/cytotoxic T-cell response in women identified by microarray analysis. Genes Immun. 2009;10(5):509-16. [CrossRef]
- Priyanka HP, Krishnan HC, Singh RV, Hima L, Thyagarajan S. Estrogen modulates in vitro T cell responses in a concentration- and receptor-dependent manner: effects on intracellular molecular targets and antioxidant enzymes. Mol Immunol. 2013;56(4):328-39. [CrossRef]
- Greenbaum H, Galper BL, Decter DH, Eisenberg VH. Endometriosis and autoimmunity: Can autoantibodies be used as a non-invasive early diagnostic tool? Autoimmun Rev. 2021;20(5):102795. [CrossRef]
- Giannoni E, Guignard L, Knaup Reymond M, Perreau M, Roth-Kleiner M, Calandra T, Roger T. Estradiol and progesterone strongly inhibit the innate immune response of mononuclear cells in newborns. Infect Immun. 2011;79(7):2690-8. [CrossRef]
- Harding AT, Heaton NS. The Impact of Estrogens and Their Receptors on Immunity and Inflammation during Infection. Cancers (Basel). 2022;14(4):909. [CrossRef]
- Harding AT, Goff MA, Froggatt HM, Lim JK, Heaton NS. GPER1 is required to protect fetal health from maternal inflammation. Science. 2021;371(6526):271-276. [CrossRef]
- Fan Z, Che H, Yang S, Chen C. Estrogen and estrogen receptor signaling promotes allergic immune responses: Effects on immune cells, cytokines, and inflammatory factors involved in allergy. Allergol Immunopathol (Madr). 2019;47(5):506-512. [CrossRef]
- Hsu JT, Kan WH, Hsieh CH, Choudhry MA, Schwacha MG, Bland KI, Chaudry IH. Mechanism of estrogen-mediated attenuation of hepatic injury following trauma-hemorrhage: Akt-dependent HO-1 up-regulation. J Leukoc Biol. 2007;82(4):1019-26. [CrossRef]
- Xing D, Feng W, Miller AP, Weathington NM, Chen YF, Novak L, Blalock JE, Oparil S. Estrogen modulates TNF-alpha-induced inflammatory responses in rat aortic smooth muscle cells through estrogen receptor-beta activation. Am J Physiol Heart Circ Physiol. 2007; 292(6):H2607-12. [CrossRef]
- Dai R, Cowan C, Heid B, Khan D, Liang Z, Pham CT, Ahmed SA. Neutrophils and neutrophil serine proteases are increased in the spleens of estrogen-treated C57BL/6 mice and several strains of spontaneous lupus-prone mice. PLoS One. 2017;12(2):e0172105. [CrossRef]
- Biswas P, Delfanti F, Bernasconi S, Mengozzi M, Cota M, Polentarutti N, Mantovani A, Lazzarin A, Sozzani S, Poli G. Interleukin-6 induces monocyte chemotactic protein-1 in peripheral blood mononuclear cells and in the U937 cell line. Blood. 1998;91(1): 258-65.
- Michlewska S, Dransfield I, Megson IL, Rossi AG. Macrophage phagocytosis of apoptotic neutrophils is critically regulated by the opposing actions of pro-inflammatory and anti-inflammatory agents: key role for TNF-alpha. FASEB J. 2009 ;23(3): 844-54. [CrossRef]
- Azenabor AA, Yang S, Job G, Adedokun OO. Expression of iNOS gene in macrophages stimulated with 17beta-estradiol is regulated by free intracellular Ca2+. Biochem Cell Biol. 2004;82(3):381-90. [CrossRef]
- Karpuzoglu E, Ahmed SA. Estrogen regulation of nitric oxide and inducible nitric oxide synthase (iNOS) in immune cells: implications for immunity, autoimmune diseases, and apoptosis. Nitric Oxide. 2006;15(3):177-86. [CrossRef]
- Lélu K, Laffont S, Delpy L, Paulet PE, Périnat T, Tschanz SA, Pelletier L, Engelhardt B, Guéry JC. Estrogen receptor α signaling in T lymphocytes is required for estradiol-mediated inhibition of Th1 and Th17 cell differentiation and protection against experimental autoimmune encephalomyelitis. J Immunol. 2011;187(5):2386-93. [CrossRef]
- Robinson DP, Hall OJ, Nilles TL, Bream JH, Klein SL. 17β-estradiol protects females against influenza by recruiting neutrophils and increasing virus-specific CD8 T cell responses in the lungs. J Virol. 2014;88(9):4711-20. [CrossRef]
- Karpuzoglu-Sahin E, Hissong BD, Ansar Ahmed S. Interferon-gamma levels are upregulated by 17-beta-estradiol and diethylstilbestrol. J Reprod Immunol. 2001;52(1-2):113-27. [CrossRef]
- Dragin N, Nancy P, Villegas J, Roussin R, Le Panse R, Berrih-Aknin S. Balance between Estrogens and Proinflammatory Cytokines Regulates Chemokine Production Involved in Thymic Germinal Center Formation. Sci Rep. 2017;7(1):7970. Erratum in: Sci Rep. 2018;8(1):8118. [CrossRef]
- Tang S, Han H, Bajic VB. ERGDB: Estrogen Responsive Genes Database. Nucleic Acids Res. 2004;32(Database issue):D533-6. [CrossRef]
- Polanczyk MJ, Carson BD, Subramanian S, Afentoulis M, Vandenbark AA, Ziegler SF, Offner H. Cutting edge: estrogen drives expansion of the CD4+CD25+ regulatory T cell compartment. J Immunol. 2004;173(4):2227-30. [CrossRef]
- Tai P, Wang J, Jin H, Song X, Yan J, Kang Y, Zhao L, An X, Du X, Chen X, Wang S, Xia G, Wang B. Induction of regulatory T cells by physiological level estrogen. J Cell Physiol. 2008; 214(2):456-64. [CrossRef]
- Brown MA, Su MA. An Inconvenient Variable: Sex Hormones and Their Impact on T Cell Responses. J Immunol. 2019;202(7):1927-1933. [CrossRef]
- Hosokawa H, Rothenberg EV. How transcription factors drive choice of the T cell fate. Nat Rev Immunol. 2021;21(3):162-176. [CrossRef]
- Grimaldi CM, Cleary J, Dagtas AS, Moussai D, Diamond B. Estrogen alters thresholds for B cell apoptosis and activation. J Clin Invest. 2002;109(12):1625-33. [CrossRef]
- Grimaldi CM, Jeganathan V, Diamond B. Hormonal regulation of B cell development: 17 beta-estradiol impairs negative selection of high-affinity DNA-reactive B cells at more than one developmental checkpoint. J Immunol. 2006;176(5):2703-10. [CrossRef]
- Verthelyi DI, Ahmed SA. Estrogen increases the number of plasma cells and enhances their autoantibody production in nonautoimmune C57BL/6 mice. Cell Immunol. 1998;189(2):125-34. [CrossRef]
- Hill L, Jeganathan V, Chinnasamy P, Grimaldi C, Diamond B. Differential roles of estrogen receptors α and β in control of B-cell maturation and selection. Mol Med. 2011;17(3-4):211-20. [CrossRef]
- Stice JP, Mbai FN, Chen L, Knowlton AA. Rapid activation of nuclear factor κB by 17β-estradiol and selective estrogen receptor modulators: pathways mediating cellular protection. Shock. 2012;38(2):128-36. [CrossRef]
- Xing D, Oparil S, Yu H, Gong K, Feng W, Black J, Chen YF, Nozell S. Estrogen modulates NFκB signaling by enhancing IκBα levels and blocking p65 binding at the promoters of inflammatory genes via estrogen receptor-β. PLoS One. 2012;7(6):e36890. [CrossRef]
- Stice JP, Knowlton AA. Estrogen, NFkappaB, and the heat shock response. Mol Med. 2008;14(7-8):517-27. [CrossRef]
- Monteiro R, Teixeira D, Calhau C. Estrogen signaling in metabolic inflammation. Mediators Inflamm. 2014;2014:615917. [CrossRef]
- Pelekanou V, Kampa M, Kiagiadaki F, Deli A, Theodoropoulos P, Agrogiannis G, Patsouris E, Tsapis A, Castanas E, Notas G. Estrogen anti-inflammatory activity on human monocytes is mediated through cross-talk between estrogen receptor ERα36 and GPR30/GPERJ Leukoc Biol. 2016;99(2):333-47. [CrossRef]
- Liu CJ, Lo JF, Kuo CH, Chu CH, Chen LM, Tsai FJ, Tsai CH, Tzang BS, Kuo WW, Huang CY. Akt mediates 17beta-estradiol and/or estrogen receptor-alpha inhibition of LPS-induced tumor necresis factor-alpha expression and myocardial cell apoptosis by suppressing the JNK1/2-NFkappaB pathway. J Cell Mol Med. 2009;13(9B):3655-67. [CrossRef]
- Slabe N, Meden-Vrtovec H, Verdenik I, Kosir-Pogacnik R, Ihan A. Cytotoxic T-Cells in Peripheral Blood in Women with Endometriosis. Geburtshilfe Frauenheilkd. 2013;73(10): 1042-1048. [CrossRef]
- Bolitho P, Voskoboinik I, Trapani JA, Smyth MJ. Apoptosis induced by the lymphocyte effector molecule perforin. Curr Opin Immunol. 2007;19(3):339-47. [CrossRef]
- Osińska I, Popko K, Demkow U. Perforin: an important player in immune response. Cent Eur J Immunol. 2014;39(1):109-15. [CrossRef]
- Lu Q, Wu A, Ray D, Deng C, Attwood J, Hanash S, Pipkin M, Lichtenheld M, Richardson B. DNA methylation and chromatin structure regulate T cell perforin gene expression. J Immunol. 2003;170(10):5124-32. [CrossRef]
- Zierau O, Zenclussen AC, Jensen F. Role of female sex hormones, estradiol and progesterone, in mast cell behavior. Front Immunol. 2012;3:169. [CrossRef]
- Szukiewicz D, Wojdasiewicz P, Watroba M, Szewczyk G. Mast Cell Activation Syndrome in COVID-19 and Female Reproductive Function: Theoretical Background vs. Accumulating Clinical Evidence. J Immunol Res. 2022;2022:9534163. [CrossRef]
- Cutolo M, Capellino S, Sulli A, Serioli B, Secchi ME, Villaggio B, Straub RH. Estrogens and autoimmune diseases. Ann N Y Acad Sci. 2006;1089:538-47. [CrossRef]
- Jacenik D, Krajewska WM. Significance of G Protein-Coupled Estrogen Receptor in the Pathophysiology of Irritable Bowel Syndrome, Inflammatory Bowel Diseases and Colorectal Cancer. Front Endocrinol (Lausanne). 2020;11:390. [CrossRef]
- Moon TC, Befus AD, Kulka M. Mast cell mediators: their differential release and the secretory pathways involved. Front Immunol. 2014;5:569. [CrossRef]
- Theoharides TC, Alysandratos KD, Angelidou A, Delivanis DA, Sismanopoulos N, Zhang B, Asadi S, Vasiadi M, Weng Z, Miniati A, Kalogeromitros D. Mast cells and inflammation. Biochim Biophys Acta. 2012;1822(1):21-33. [CrossRef]
- Valent P, Hartmann K, Bonadonna P, Niedoszytko M, Triggiani M, Arock M, Brockow K. Mast Cell Activation Syndromes: Collegium Internationale Allergologicum Update Int Arch Allergy Immunol. 2022;183(7):693-705. [CrossRef]
- Szewczyk G, Pyzlak M, Klimkiewicz J, Smiertka W, Miedzińska-Maciejewska M, Szukiewicz D. Mast cells and histamine: do they influence placental vascular network and development in preeclampsia? Mediators Inflamm. 2012;2012:307189. [CrossRef]
- Kempuraj D, Papadopoulou N, Stanford EJ, Christodoulou S, Madhappan B, Sant GR, Solage K, Adams T, Theoharides TC. Increased numbers of activated mast cells in endometriosis lesions positive for corticotropin-releasing hormone and urocortin. Am J Reprod Immunol. 2004;52(4):267-75. [CrossRef]
- McCallion A, Nasirzadeh Y, Lingegowda H, Miller JE, Khalaj K, Ahn S, Monsanto SP, Bidarimath M, Sisnett DJ, Craig AW, Young SL, Lessey BA, Koti M, Tayade C. Estrogen mediates inflammatory role of mast cells in endometriosis pathophysiology. Front Immunol. 2022;13:961599. [CrossRef]
- Pansrikaew P, Cheewakriangkrai C, Taweevisit M, Khunamornpong S, Siriaunkgul S. Correlation of mast cell density, tumor angiogenesis, and clinical outcomes in patients with endometrioid endometrial cancer. Asian Pac J Cancer Prev. 2010;11(3):623-6.
- Mercorio A, Giampaolino P, Romano A, Dällenbach P, Pluchino N. Is intracrinology of endometriosis relevant in clinical practice? A systematic review on estrogen metabolism. Front Endocrinol (Lausanne). 2022;13:950866. [CrossRef]
- Binda MM, Donnez J, Dolmans MM. Targeting mast cells: a new way to treat endometriosis. Expert Opin Ther Targets. 2017;21(1):67-75. [CrossRef]
- Bulfone-Paus S, Bahri R. Mast Cells as Regulators of T Cell Responses. Front Immunol. 2015; 6:394. [CrossRef]
- Krystel-Whittemore M, Dileepan KN, Wood JG. Mast Cell: A Multi-Functional Master Cell. Front Immunol. 2016;6:620. [CrossRef]
- Khan D, Ansar Ahmed S. The Immune System Is a Natural Target for Estrogen Action: Opposing Effects of Estrogen in Two Prototypical Autoimmune Diseases. Front Immunol. 2016;6:635. [CrossRef]
- Walker ME, Simpson JB, Redinbo MR. A structural metagenomics pipeline for examining the gut microbiome. Curr Opin Struct Biol. 2022;75:102416. [CrossRef]
- Putignani L, Del Chierico F, Petrucca A, Vernocchi P, Dallapiccola B. The human gut microbiota: a dynamic interplay with the host from birth to senescence settled during childhood. Pediatr Res. 2014;76(1):2-10. [CrossRef]
- Stephen AM, Cummings JH. The microbial contribution to human faecal mass. J Med Microbiol. 1980;13(1):45-56. [CrossRef]
- Kho ZY, Lal SK. The Human Gut Microbiome - A Potential Controller of Wellness and Disease. Front Microbiol. 2018;9:1835. [CrossRef]
- Valdes AM, Walter J, Segal E, Spector TD. Role of the gut microbiota in nutrition and health. BMJ. 2018;361:k2179. [CrossRef]
- Kasarello K, Cudnoch-Jedrzejewska A, Czarzasta K. Communication of gut microbiota and brain via immune and neuroendocrine signaling. Front Microbiol. 2023;14:1118529. [CrossRef]
- Nova E, Gómez-Martinez S, González-Soltero R. The Influence of Dietary Factors on the Gut Microbiota. Microorganisms. 2022;10(7):1368. [CrossRef]
- Qi X, Yun C, Pang Y, Qiao J. The impact of the gut microbiota on the reproductive and metabolic endocrine system. Gut Microbes. 2021;13(1):1-21. [CrossRef]
- Wang LQ. Mammalian phytoestrogens: enterodiol and enterolactone. J Chromatogr B Analyt Technol Biomed Life Sci. 2002;777(1-2):289-309. [CrossRef]
- Mukhija M, Joshi BC, Bairy PS, Bhargava A, Sah AN. Lignans: a versatile source of anticancer drugs. Beni Suef Univ J Basic Appl Sci. 2022;11(1):76. [CrossRef]
- Salliss ME, Farland LV, Mahnert ND, Herbst-Kralovetz MM. The role of gut and genital microbiota and the estrobolome in endometriosis, infertility and chronic pelvic pain. Hum Reprod Update. 2021;28(1):92-131. [CrossRef]
- Anderson G. Endometriosis Pathoetiology and Pathophysiology: Roles of Vitamin A, Estrogen, Immunity, Adipocytes, Gut Microbiome and Melatonergic Pathway on Mitochondria Regulation. Biomol Concepts. 2019;10(1):133-149. [CrossRef]
- Chakaroun RM, Massier L, Kovacs P. Gut Microbiome, Intestinal Permeability, and Tissue Bacteria in Metabolic Disease: Perpetrators or Bystanders? Nutrients. 2020;12(4): 1082. [CrossRef]
- Ata B, Yildiz S, Turkgeldi E, Brocal VP, Dinleyici EC, Moya A, Urman B. The Endobiota Study: Comparison of Vaginal, Cervical and Gut Microbiota Between Women with Stage 3/4 Endometriosis and Healthy Controls. Sci Rep. 2019;9(1):2204. [CrossRef]
- Jess T, Frisch M, Jørgensen KT, Pedersen BV, Nielsen NM. Increased risk of inflammatory bowel disease in women with endometriosis: a nationwide Danish cohort study. Gut. 2012; 61(9):1279-83. [CrossRef]
- Yuan M, Li D, Zhang Z, Sun H, An M, Wang G. Endometriosis induces gut microbiota alterations in mice. Hum Reprod. 2018;33(4):607-616. [CrossRef]
- Chadchan SB, Cheng M, Parnell LA, Yin Y, Schriefer A, Mysorekar IU, Kommagani R. Antibiotic therapy with metronidazole reduces endometriosis disease progression in mice: a potential role for gut microbiota. Hum Reprod. 2019;34(6):1106-1116. [CrossRef]
- Bailey MT, Coe CL. Endometriosis is associated with an altered profile of intestinal microflora in female rhesus monkeys. Hum Reprod. 2002;17(7):1704-8. [CrossRef]
- Qin R, Tian G, Liu J, Cao L. The gut microbiota and endometriosis: From pathogenesis to diagnosis and treatment. Front Cell Infect Microbiol. 2022;12:1069557. [CrossRef]
- Laganà AS, Garzon S, Franchi M, Casarin J, Gullo G, Ghezzi F. Translational animal models for endometriosis research: a long and windy road. Ann Transl Med. 2018;6(22):431. [CrossRef]
- Grümmer R. Animal models in endometriosis research. Hum Reprod Update. 2006;12(5):641-9. [CrossRef]
- Bruner-Tran KL, Osteen KG, Taylor HS, Sokalska A, Haines K, Duleba AJ. Resveratrol inhibits development of experimental endometriosis in vivo and reduces endometrial stromal cell invasiveness in vitro. Biol Reprod. 2011;84(1):106-12. [CrossRef]
- Rudzitis-Auth J, Menger MD, Laschke MW. Resveratrol is a potent inhibitor of vascularization and cell proliferation in experimental endometriosis. Hum Reprod. 2013;28 (5):1339-47. [CrossRef]
- Ricci AG, Olivares CN, Bilotas MA, Bastón JI, Singla JJ, Meresman GF, Barañao RI. Natural therapies assessment for the treatment of endometriosis. Hum Reprod. 2013;28(1):178-88. [CrossRef]
- Ozcan Cenksoy P, Oktem M, Erdem O, Karakaya C, Cenksoy C, Erdem A, Guner H, Karabacak O. A potential novel treatment strategy: inhibition of angiogenesis and inflammation by resveratrol for regression of endometriosis in an experimental rat model. Gynecol Endocrinol. 2015;31(3):219-24. [CrossRef]
- Di Paola R, Fusco R, Gugliandolo E, Crupi R, Evangelista M, Granese R, Cuzzocrea S. Co-micronized Palmitoylethanolamide/Polydatin Treatment Causes Endometriotic Lesion Regression in a Rodent Model of Surgically Induced Endometriosis. Front Pharmacol. 2016;7:382. [CrossRef]
- Cotroneo MS, Lamartiniere CA. Pharmacologic, but not dietary, genistein supports endometriosis in a rat model. Toxicol Sci. 2001;61(1):68-75. [CrossRef]
- Yavuz E, Oktem M, Esinler I, Toru SA, Zeyneloglu HB. Genistein causes regression of endometriotic implants in the rat model. Fertil Steril. 2007;88(4 Suppl):1129-34. [CrossRef]
- Takaoka O, Mori T, Ito F, Okimura H, Kataoka H, Tanaka Y, Koshiba A, Kusuki I, Shigehiro S, Amami T, Kitawaki J. Daidzein-rich isoflavone aglycones inhibit cell growth and inflammation in endometriosis. J Steroid Biochem Mol Biol. 2018;181:125-132. [CrossRef]
- Chen Y, Chen C, Shi S, Han J, Wang J, Hu J, Liu Y, Cai Z, Yu C. Endometriotic implants regress in rat models treated with puerarin by decreasing estradiol level. Reprod Sci. 2011;18 (9):886-91. [CrossRef]
- Rudzitis-Auth J, Körbel C, Scheuer C, Menger MD, Laschke MW. Xanthohumol inhibits growth and vascularization of developing endometriotic lesions. Hum Reprod. 2012 ;27(6): 1735-44. [CrossRef]
- Nahari E, Razi M. Silymarin amplifies apoptosis in ectopic endometrial tissue in rats with endometriosis; implication on growth factor GDNF, ERK1/2 and Bcl-6b expression. Acta Histochem. 2018;120(8):757-767. [CrossRef]
- Melekoglu R, Ciftci O, Eraslan S, Cetin A, Basak N. The beneficial effects of nerolidol and hesperidin on surgically induced endometriosis in a rat model. Gynecol Endocrinol. 2018; 34(11):975-980. [CrossRef]
- Hsu YW, Chen HY, Chiang YF, Chang LC, Lin PH, Hsia SM. The effects of isoliquiritigenin on endometriosis in vivo and in vitro study. Phytomedicine. 2020;77:153214. [CrossRef]
- Yang YM, Yang WX. Epithelial-to-mesenchymal transition in the development of endometriosis. Oncotarget. 2017;8(25):41679-41689. [CrossRef]
- Kapoor R, Sirohi VK, Gupta K, Dwivedi A. Naringenin ameliorates progression of endometriosis by modulating Nrf2/Keap1/HO1 axis and inducing apoptosis in rats. J Nutr Biochem. 2019;70:215-226. [CrossRef]
- Ilhan M, Ali Z, Khan IA, Taştan H, Küpeli Akkol E. Bioactivity-guided isolation of flavonoids from Urtica dioica L. and their effect on endometriosis rat model. J Ethnopharmacol. 2019;243:112100. [CrossRef]
- Demirel MA, Suntar I, Ilhan M, Keles H, Kupeli Akkol E. Experimental endometriosis remission in rats treated with Achillea biebersteinii Afan.: histopathological evaluation and determination of cytokine levels. Eur J Obstet Gynecol Reprod Biol. 2014;175:172-7. [CrossRef]
- Ferella L, Bastón JI, Bilotas MA, Singla JJ, González AM, Olivares CN, Meresman GF. Active compounds present inRosmarinus officinalis leaves andScutellaria baicalensis root evaluated as new therapeutic agents for endometriosis. Reprod Biomed Online. 2018;37(6): 769-782. [CrossRef]
- Ilhan M, Ali Z, Khan IA, Taştan H, Küpeli Akkol E. Promising activity of Anthemis austriaca Jacq. on the endometriosis rat model and isolation of its active constituents. Saudi Pharm J. 2019;27(6):889-899. [CrossRef]
- Bina F, Daglia M, Santarcangelo C, Baeeri M, Abdollahi M, Nabavi SM, Tabarrai M, Rahimi R. Phytochemical profiling and ameliorative effects of Achillea cretica L. on rat model of endometriosis. J Ethnopharmacol. 2020;254:112747. [CrossRef]
- Ilhan M, Ali Z, Khan IA, Taştan H, Küpeli Akkol E. The regression of endometriosis with glycosylated flavonoids isolated from Melilotus officinalis (L.) Pall. in an endometriosis rat model. Taiwan J Obstet Gynecol. 2020;59(2):211-219. [CrossRef]
- Machairiotis N, Vasilakaki S, Kouroutou P. Natural products: Potential lead compounds for the treatment of endometriosis. Eur J Obstet Gynecol Reprod Biol. 2020;245:7-12. [CrossRef]
- Ilhan M, Gürağaç Dereli FT, Akkol EK. Novel Drug Targets with Traditional Herbal Medicines for Overcoming Endometriosis. Curr Drug Deliv. 2019;16(5):386-399. [CrossRef]
- Bina F, Soleymani S, Toliat T, Hajimahmoodi M, Tabarrai M, Abdollahi M, Rahimi R. Plant-derived medicines for treatment of endometriosis: A comprehensive review of molecular mechanisms. Pharmacol Res. 2019;139:76-90. [CrossRef]
- Parazzini F, Viganò P, Candiani M, Fedele L. Diet and endometriosis risk: a literature review. Reprod Biomed Online. 2013;26(4):323-36. [CrossRef]
- Della Corte L, Noventa M, Ciebiera M, Magliarditi M, Sleiman Z, Karaman E, Catena U, Salvaggio C, Falzone G, Garzon S. Phytotherapy in endometriosis: an up-to-date review. J Complement Integr Med. 2020;17(3). [CrossRef]
- Kodarahmian M, Amidi F, Moini A, Kashani L, Shabani Nashtaei M, Pazhohan A, Bahramrezai M, Berenjian S, Sobhani A. The modulating effects of Resveratrol on the expression of MMP-2 and MMP-9 in endometriosis women: a randomized exploratory trial. Gynecol Endocrinol. 2019;35(8):719-726. [CrossRef]
- Maia H Jr, Haddad C, Pinheiro N, Casoy J. Advantages of the association of resveratrol with oral contraceptives for management of endometriosis-related pain. Int J Womens Health. 2012;4:543-9. [CrossRef]
- Mendes da Silva D, Gross LA, Neto EPG, Lessey BA, Savaris RF. The Use of Resveratrol as an Adjuvant Treatment of Pain in Endometriosis: A Randomized Clinical Trial. J Endocr Soc. 2017;1(4):359-369. [CrossRef]
- Nagata C, Takatsuka N, Kawakami N, Shimizu H. Soy product intake and premenopausal hysterectomy in a follow-up study of Japanese women. Eur J Clin Nutr. 2001;55(9):773-7. [CrossRef]
- Parazzini F, Chiaffarino F, Surace M, Chatenoud L, Cipriani S, Chiantera V, Benzi G, Fedele L. Selected food intake and risk of endometriosis. Hum Reprod. 2004;19(8):1755-9. [CrossRef]
- Signorile PG, Viceconte R, Baldi A. Novel dietary supplement association reduces symptoms in endometriosis patients. J Cell Physiol. 2018;233(8):5920-5925. [CrossRef]
- Trabert B, Peters U, De Roos AJ, Scholes D, Holt VL. Diet and risk of endometriosis in a population-based case-control study. Br J Nutr. 2011;105(3):459-67. [CrossRef]
- Tsuchiya M, Miura T, Hanaoka T, Iwasaki M, Sasaki H, Tanaka T, Nakao H, Katoh T, Ikenoue T, Kabuto M, Tsugane S. Effect of soy isoflavones on endometriosis: interaction with estrogen receptor 2 gene polymorphism. Epidemiology. 2007;18(3):402-8. [CrossRef]
- Youseflu S, Jahanian Sadatmahalleh SH, Mottaghi A, Kazemnejad A. Dietary Phytoestrogen Intake and The Risk of Endometriosis in Iranian Women: A Case-Control Study. Int J Fertil Steril. 2020;13(4):296-300. [CrossRef]
- Harris HR, Eke AC, Chavarro JE, Missmer SA. Fruit and vegetable consumption and risk of endometriosis. Hum Reprod. 2018;33(4):715-727. [CrossRef]
- Mirmiran P, Bahadoran Z, Gaeini Z. Common Limitations and Challenges of Dietary Clinical Trials for Translation into Clinical Practices. Int J Endocrinol Metab. 2021;19(3): e108170. [CrossRef]
- Dull AM, Moga MA, Dimienescu OG, Sechel G, Burtea V, Anastasiu CV. Therapeutic Approaches of Resveratrol on Endometriosis via Anti-Inflammatory and Anti-Angiogenic Pathways. Molecules. 2019;24(4):667. [CrossRef]
- Meresman GF, Götte M, Laschke MW. Plants as source of new therapies for endometriosis: a review of preclinical and clinical studies. Hum Reprod Update. 2021; 27(2):367-392. [CrossRef]
- Afrin S, AlAshqar A, El Sabeh M, Miyashita-Ishiwata M, Reschke L, Brennan JT, Fader A, Borahay MA. Diet and Nutrition in Gynecological Disorders: A Focus on Clinical Studies. Nutrients. 2021;13(6):1747. [CrossRef]
- Chapadgaonkar SS, Bajpai SS, Godbole MS. Gut microbiome influences incidence and outcomes of breast cancer by regulating levels and activity of steroid hormones in women. Cancer Rep (Hoboken). 2023:e1847. [CrossRef]
- Lathigara D, Kaushal D, Wilson RB. Molecular Mechanisms of Western Diet-Induced Obesity and Obesity-Related Carcinogenesis-A Narrative Review. Metabolites. 2023; 13(5):675. [CrossRef]
- Saguyod SJU, Kelley AS, Velarde MC, Simmen RC. Diet and endometriosis-revisiting the linkages to inflammation. Journal of Endometriosis and Pelvic Pain Disorders. 2018;10(2):51-58. [CrossRef]
- Arumugam K, Templeton AA. Endometriosis and race. Aust N Z J Obstet Gynaecol. 1992;32:164–165. [CrossRef]
- Yen CF, Kim MR, Lee CL. Epidemiologic Factors Associated with Endometriosis in East Asia. Gynecol Minim Invasive Ther. 2019;8(1):4-11. [CrossRef]
- Yamamoto A, Johnstone EB, Bloom MS, Huddleston HG, Fujimoto VY. A higher prevalence of endometriosis among Asian women does not contribute to poorer IVF outcomes. J Assist Reprod Genet. 2017;34(6):765-774. [CrossRef]
- Gajbhiye RK. Endometriosis and inflammatory immune responses: Indian experience. Am J Reprod Immunol. 2023;89(2):e13590. [CrossRef]
- Moradi Y, Shams-Beyranvand M, Khateri S, Gharahjeh S, Tehrani S, Varse F, Tiyuri A, Najmi Z. A systematic review on the prevalence of endometriosis in women. Indian J Med Res. 2021;154(3):446-454. [CrossRef]
- Chottanapund S, Van Duursen MB, Navasumrit P, Hunsonti P, Timtavorn S, Ruchirawat M, Van den Berg M. Anti-aromatase effect of resveratrol and melatonin on hormonal positive breast cancer cells co-cultured with breast adipose fibroblasts. Toxicol In Vitro. 2014; 28(7): 1215-21. [CrossRef]
- Klein CB, King AA. Genistein genotoxicity: critical considerations of in vitro exposure dose. Toxicol Appl Pharmacol. 2007;224(1):1-11. [CrossRef]
- Landete JM, Arqués J, Medina M, Gaya P, de Las Rivas B, Muñoz R. Bioactivation of Phytoestrogens: Intestinal Bacteria and Health. Crit Rev Food Sci Nutr. 2016;56(11):1826-43. [CrossRef]
- Ervin SM, Li H, Lim L, Roberts LR, Liang X, Mani S, Redinbo MR. Gut microbial β-glucuronidases reactivate estrogens as components of the estrobolome that reactivate estrogens. J Biol Chem. 2019;294(49):18586-18599. [CrossRef]
- Tew BY, Xu X, Wang HJ, Murphy PA, Hendrich S. A diet high in wheat fiber decreases the bioavailability of soybean isoflavones in a single meal fed to women. J Nutr. 1996; 126(4):871-7. [CrossRef]
- Baldi S, Tristán Asensi M, Pallecchi M, Sofi F, Bartolucci G, Amedei A. Interplay between Lignans and Gut Microbiota: Nutritional, Functional and Methodological Aspects. Molecules. 2023;28(1):343. [CrossRef]
- Baker JM, Al-Nakkash L, Herbst-Kralovetz MM. Estrogen-gut microbiome axis: Physiological and clinical implications. Maturitas. 2017;103:45-53. [CrossRef]
- Adlercreutz H, Höckerstedt K, Bannwart C, Bloigu S, Hämäläinen E, Fotsis T, Ollus A. Effect of dietary components, including lignans and phytoestrogens, on enterohepatic circulation and liver metabolism of estrogens and on sex hormone binding globulin (SHBG). J Steroid Biochem. 1987;27(4-6):1135-44. [CrossRef]
- Nap A, de Roos N. Endometriosis and the effects of dietary interventions: what are we looking for? Reprod Fertil. 2022;3(2):C14-C22. [CrossRef]
- Gołąbek A, Kowalska K, Olejnik A. Polyphenols as a Diet Therapy Concept for Endometriosis-Current Opinion and Future Perspectives. Nutrients. 2021;13(4):1347. [CrossRef]
- Amro B, Ramirez Aristondo ME, Alsuwaidi S, Almaamari B, Hakim Z, Tahlak M, Wattiez A, Koninckx PR. New Understanding of Diagnosis, Treatment and Prevention of Endometriosis. Int J Environ Res Public Health. 2022;19(11):6725. [CrossRef]
Figure 1.
Structural and functional domains of estrogen nuclear receptors: ERα vs. ERβ. Both receptors have six crucial different domains marked with the letters A to F: N-terminal domain (NTD, A/B domain containing AF-1 domain), DNA binding domain (DBD, C domain), D domain (hinge), and ligand binding domain (LBD, E/F domain containing AF-2 domain) at the C-terminal region. Homology between ERα and ERβ, understood as a percentage (%) of amino acid identity within the respective domains, is also shown [61–63].
Figure 1.
Structural and functional domains of estrogen nuclear receptors: ERα vs. ERβ. Both receptors have six crucial different domains marked with the letters A to F: N-terminal domain (NTD, A/B domain containing AF-1 domain), DNA binding domain (DBD, C domain), D domain (hinge), and ligand binding domain (LBD, E/F domain containing AF-2 domain) at the C-terminal region. Homology between ERα and ERβ, understood as a percentage (%) of amino acid identity within the respective domains, is also shown [61–63].
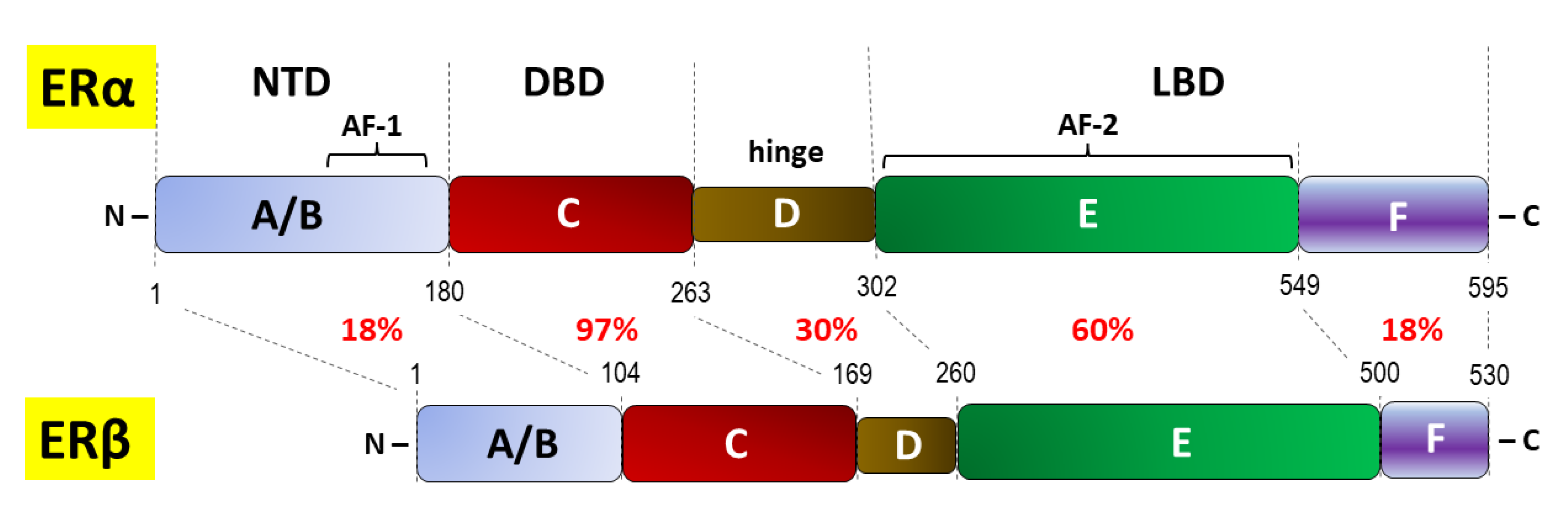
Figure 2.
Mechanisms of estrogen signaling. A. “Classic” ligand-dependent genomic signaling via nuclear receptors ERα/ ERβ: estrogen binding to the ligand binding domain (LBD) unblocks the receptor with the release of “inhibitory” heat shock protein 90 (Hsp90) and subsequent ER dimerization; ER is then translocated from the cytoplasm to the nucleus and activated by phosphorylation (P); in the nucleus ER acts as ligand-activated transcription factor and exerts both direct and indirect genomic activity through binding of DNA binding domain (DBD) to the estrogen response element (ERE) on the target gene, and via interaction with transcription factor (TF) that enables binding of DBD to the ERE as TF-ERE, respectively [61–63]. B. Indirect, rapid, non-genomic signaling via membrane-associated G protein–coupled estrogen receptor (GPER) and transactivation of the epidermal growth factor receptor (EGFR): GPER stimulation activates non-receptor tyrosine kinase (proto-oncogene tyrosine-protein kinase Src, Src), which increases the concentration of matrix metalloproteinases (MMPs) resulting in EGFR ligand cleavage; indirect transcriptional activity may occur, because released heparin-binding EGF-like growth factor (HB-EGF) produces downstream activation of mitogen-activated protein-serine/threonine kinases (ERK1 and ERK2) and phosphatidylinositol-3-kinase (PI3K) pathways [64,65]. C. The ligand independent pathway on the example of growth factors signaling through receptor tyrosine kinases (RTKs); growth factor receptor-specific ligands bind to the extracellular regions of RTKs and interacting with cAMP to activate RTKs (activate the receptor tyrosine kinases) and a mitogen-activated protein kinase (MAPK); MAPK can then phosphorylate and activate ERα/ ERβ either independent of E2 or in synergy with E2 [66–68].
Figure 2.
Mechanisms of estrogen signaling. A. “Classic” ligand-dependent genomic signaling via nuclear receptors ERα/ ERβ: estrogen binding to the ligand binding domain (LBD) unblocks the receptor with the release of “inhibitory” heat shock protein 90 (Hsp90) and subsequent ER dimerization; ER is then translocated from the cytoplasm to the nucleus and activated by phosphorylation (P); in the nucleus ER acts as ligand-activated transcription factor and exerts both direct and indirect genomic activity through binding of DNA binding domain (DBD) to the estrogen response element (ERE) on the target gene, and via interaction with transcription factor (TF) that enables binding of DBD to the ERE as TF-ERE, respectively [61–63]. B. Indirect, rapid, non-genomic signaling via membrane-associated G protein–coupled estrogen receptor (GPER) and transactivation of the epidermal growth factor receptor (EGFR): GPER stimulation activates non-receptor tyrosine kinase (proto-oncogene tyrosine-protein kinase Src, Src), which increases the concentration of matrix metalloproteinases (MMPs) resulting in EGFR ligand cleavage; indirect transcriptional activity may occur, because released heparin-binding EGF-like growth factor (HB-EGF) produces downstream activation of mitogen-activated protein-serine/threonine kinases (ERK1 and ERK2) and phosphatidylinositol-3-kinase (PI3K) pathways [64,65]. C. The ligand independent pathway on the example of growth factors signaling through receptor tyrosine kinases (RTKs); growth factor receptor-specific ligands bind to the extracellular regions of RTKs and interacting with cAMP to activate RTKs (activate the receptor tyrosine kinases) and a mitogen-activated protein kinase (MAPK); MAPK can then phosphorylate and activate ERα/ ERβ either independent of E2 or in synergy with E2 [66–68].
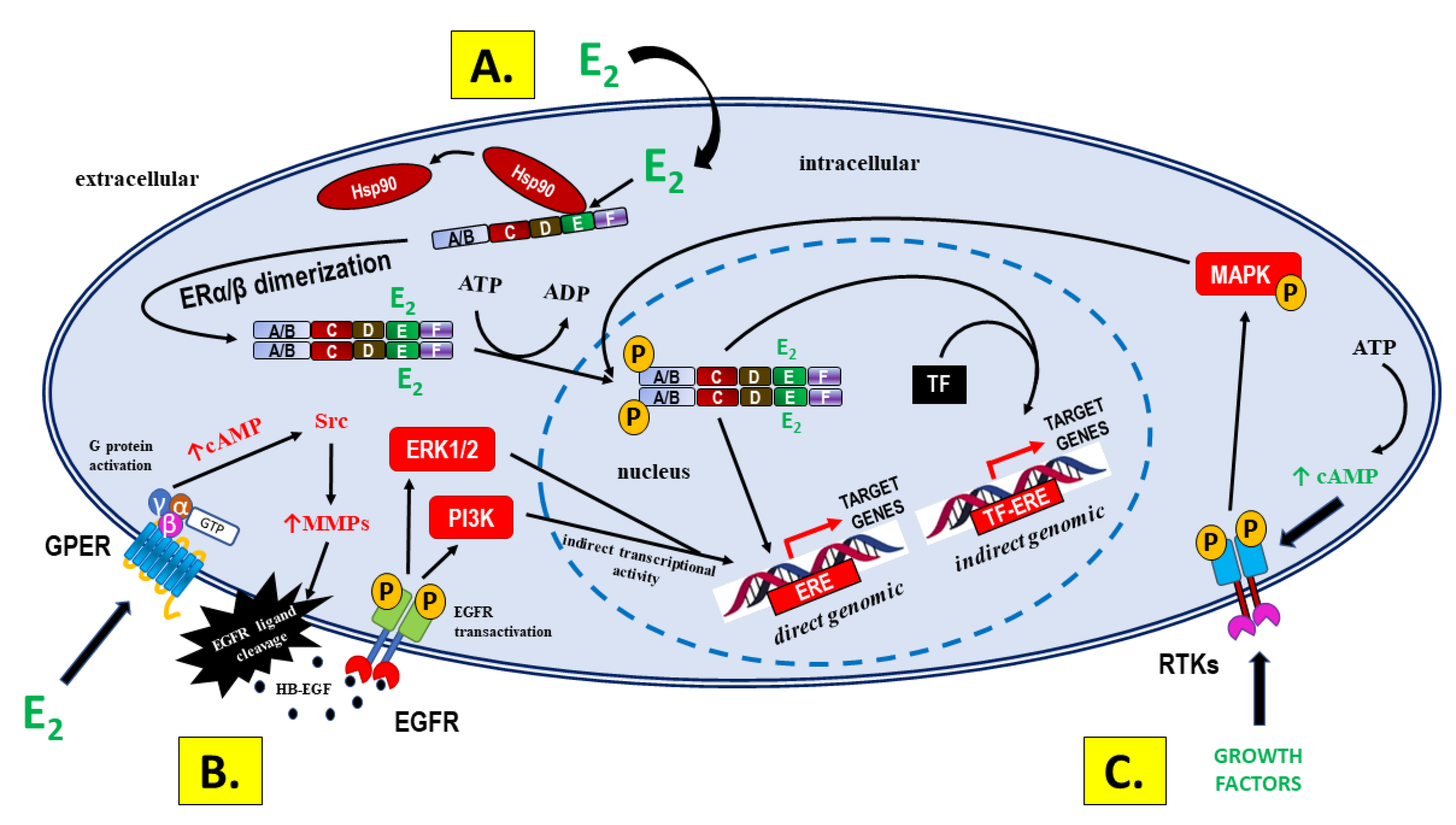
Figure 3.
Theories on etiopathogenesis of endometriosis. It is assumed that the development of endometriotic foci is a consequence of dissemination and transplantation of the cells with clonogenic activity or gradual transformation of embryonic duct remnants. The specific system of interactions between genetic, endogenous and environmental factors determines the occurrence of the disease [206,207,211–215].
Figure 3.
Theories on etiopathogenesis of endometriosis. It is assumed that the development of endometriotic foci is a consequence of dissemination and transplantation of the cells with clonogenic activity or gradual transformation of embryonic duct remnants. The specific system of interactions between genetic, endogenous and environmental factors determines the occurrence of the disease [206,207,211–215].
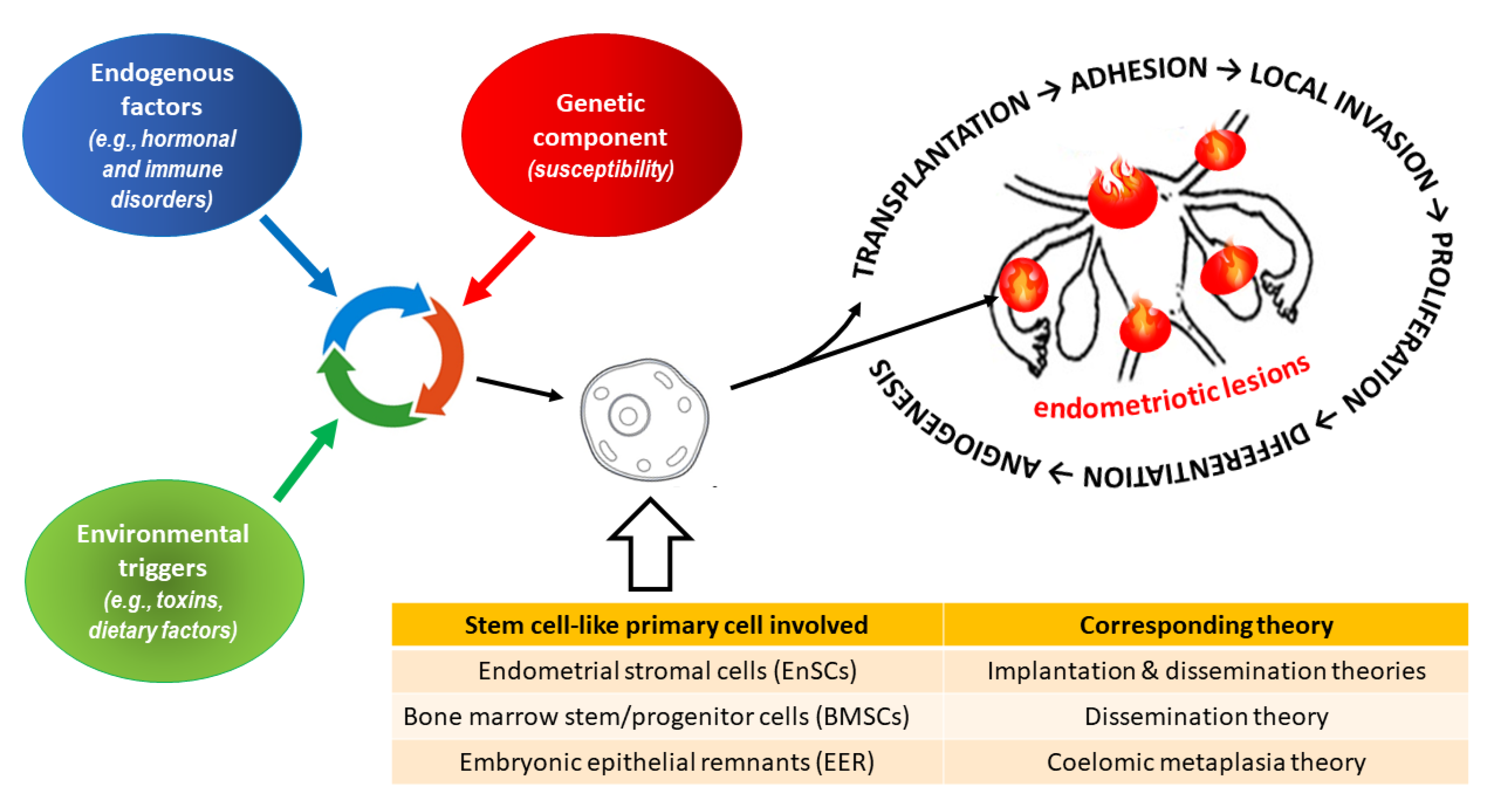
Figure 4.
Modulation of aromatase by epigenetic factors and estrogenic activity: normal endometrium (pathways in green) vs. endometriotic lesion (pathways in red). Estrogenic hyperactivity in endometriosis is caused by: ❶- hypermethylation of CpG island in the transcriptor factor steroidogenic factor 1 (SF-1) gene; ❷ - deficient 17β-hydroxysteroid dehydrogenases expression due to hypermethylation of the respective genes (HSD17B2, HSD17B1, HSD17B4); ❸ - aromatase [EC 1.14.14.1] gene (CYP19A1) activation due to CpG islands hypomethylation; ❹ - positive feedback: estrogens → cyclooxygenase 2 (COX-2) → prostaglandin E2 (PGE2) → aromatase activity in menstrual blood stem cells (MBSC).
Figure 4.
Modulation of aromatase by epigenetic factors and estrogenic activity: normal endometrium (pathways in green) vs. endometriotic lesion (pathways in red). Estrogenic hyperactivity in endometriosis is caused by: ❶- hypermethylation of CpG island in the transcriptor factor steroidogenic factor 1 (SF-1) gene; ❷ - deficient 17β-hydroxysteroid dehydrogenases expression due to hypermethylation of the respective genes (HSD17B2, HSD17B1, HSD17B4); ❸ - aromatase [EC 1.14.14.1] gene (CYP19A1) activation due to CpG islands hypomethylation; ❹ - positive feedback: estrogens → cyclooxygenase 2 (COX-2) → prostaglandin E2 (PGE2) → aromatase activity in menstrual blood stem cells (MBSC).
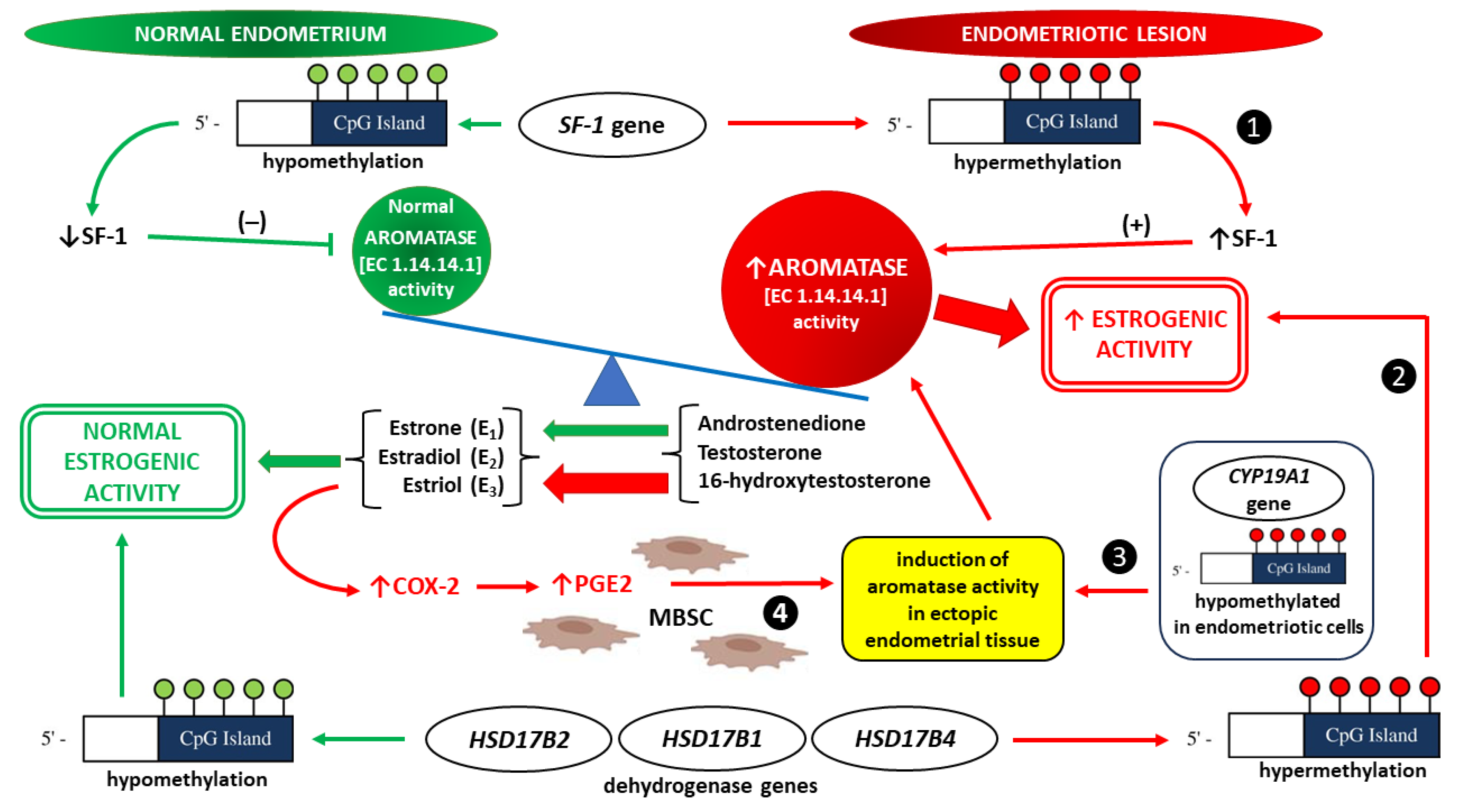
Figure 5.
Estrogen-dependent innate and adaptive immune responses related to mast cells (MCs) in endometriotic foci. ❶ - MCs develop from mast cell progenitors (MCp), which migrate to the peripheral tissues, including endometriotic foci, via the blood circulation. ❷ - When stimulated by estrogens, e.g. estradiol (E2) or phytoestrogens (PEs), endometrial-like tissue within the endometriotic lession produces increased amounts of stem cel factor (SCF), a potent growth factor critical for MC expansion, differentiation, and survival for tissue resident MCs. ❸ - Among the estrogen receptors, G protein-coupled receptor (GPER) is responsible for the various running fast nongenomic effects of estrogens, including activation and degranulation of MCs. ❹ - MCs may release a diverse spectrum of mediators that contribute to inflammation, chronic pelvic pain, and local angiogenesis resulting in the disease progression. ❺ - The release of granular and secreted mediators from MCs modulate innate immune response (IIR). An altered adaptive immune response (AIR) is observed, mainly due to MC-dependent enhancement of T cell activation. Moreover, estrogens have been shown to modulate all subsets of T cells that include CD4+ (Th1, Th2, Th17, and Tregs) and CD8+ cells (cytotoxic T lymphocytes, or CTLs) [346].
Figure 5.
Estrogen-dependent innate and adaptive immune responses related to mast cells (MCs) in endometriotic foci. ❶ - MCs develop from mast cell progenitors (MCp), which migrate to the peripheral tissues, including endometriotic foci, via the blood circulation. ❷ - When stimulated by estrogens, e.g. estradiol (E2) or phytoestrogens (PEs), endometrial-like tissue within the endometriotic lession produces increased amounts of stem cel factor (SCF), a potent growth factor critical for MC expansion, differentiation, and survival for tissue resident MCs. ❸ - Among the estrogen receptors, G protein-coupled receptor (GPER) is responsible for the various running fast nongenomic effects of estrogens, including activation and degranulation of MCs. ❹ - MCs may release a diverse spectrum of mediators that contribute to inflammation, chronic pelvic pain, and local angiogenesis resulting in the disease progression. ❺ - The release of granular and secreted mediators from MCs modulate innate immune response (IIR). An altered adaptive immune response (AIR) is observed, mainly due to MC-dependent enhancement of T cell activation. Moreover, estrogens have been shown to modulate all subsets of T cells that include CD4+ (Th1, Th2, Th17, and Tregs) and CD8+ cells (cytotoxic T lymphocytes, or CTLs) [346].
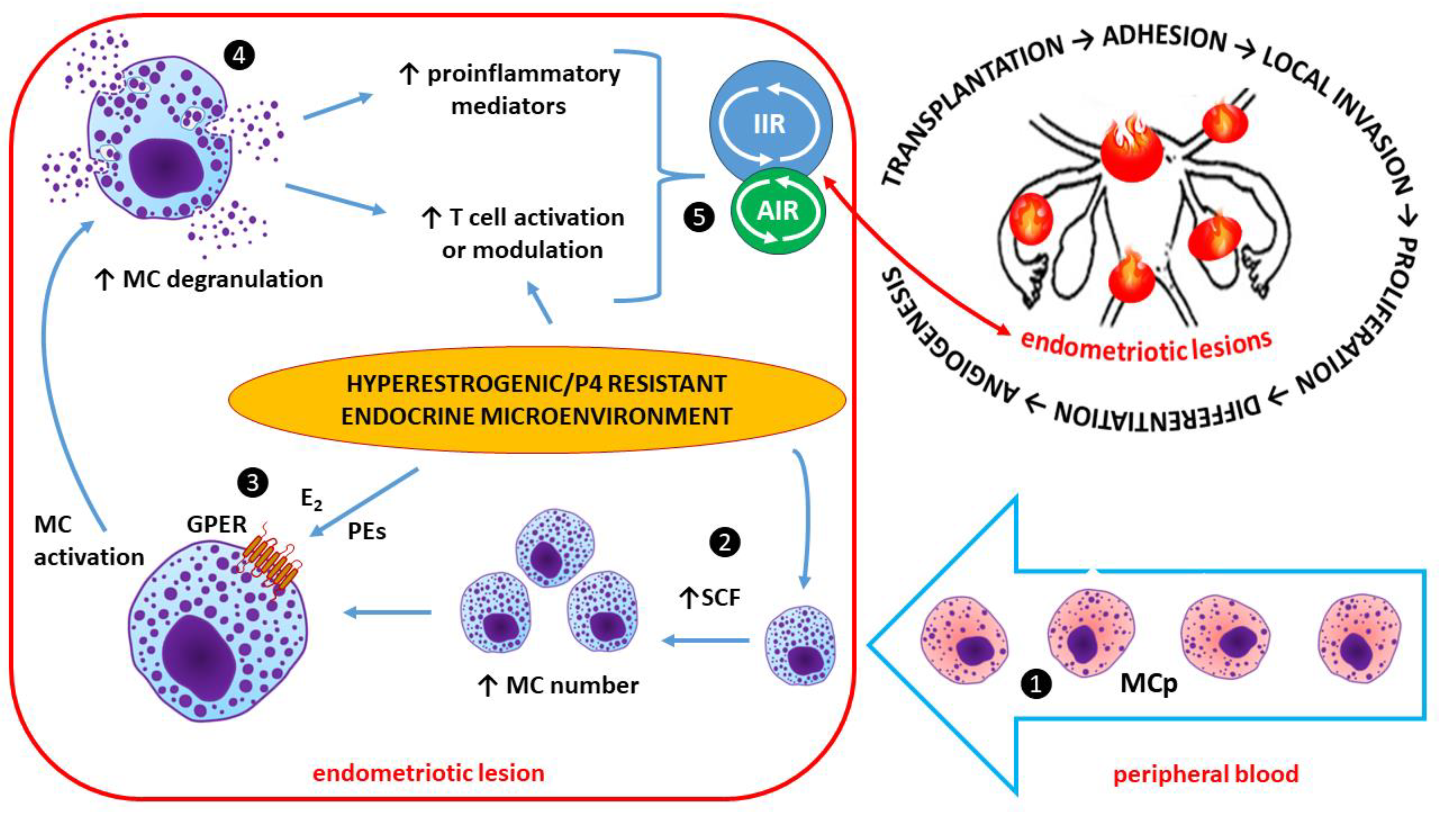
Figure 6.
Key issues and factors potentially affecting the beneficial, neutral or adverse effects of phytoestrogens (PEs) in endometriosis. Due to the lack of adequate randomized controlled clinical trials on a representative sample of women, a difficult unequivocal answer regarding the recommendation of PEs in endometriosis remains unclear. Due to expected clinically significant heterogeneity in response to PEs, some patients will experience more or less benefit from the treatment of endometriosis than the averages, while other patients may experience an exacerbation of the disease.
Figure 6.
Key issues and factors potentially affecting the beneficial, neutral or adverse effects of phytoestrogens (PEs) in endometriosis. Due to the lack of adequate randomized controlled clinical trials on a representative sample of women, a difficult unequivocal answer regarding the recommendation of PEs in endometriosis remains unclear. Due to expected clinically significant heterogeneity in response to PEs, some patients will experience more or less benefit from the treatment of endometriosis than the averages, while other patients may experience an exacerbation of the disease.
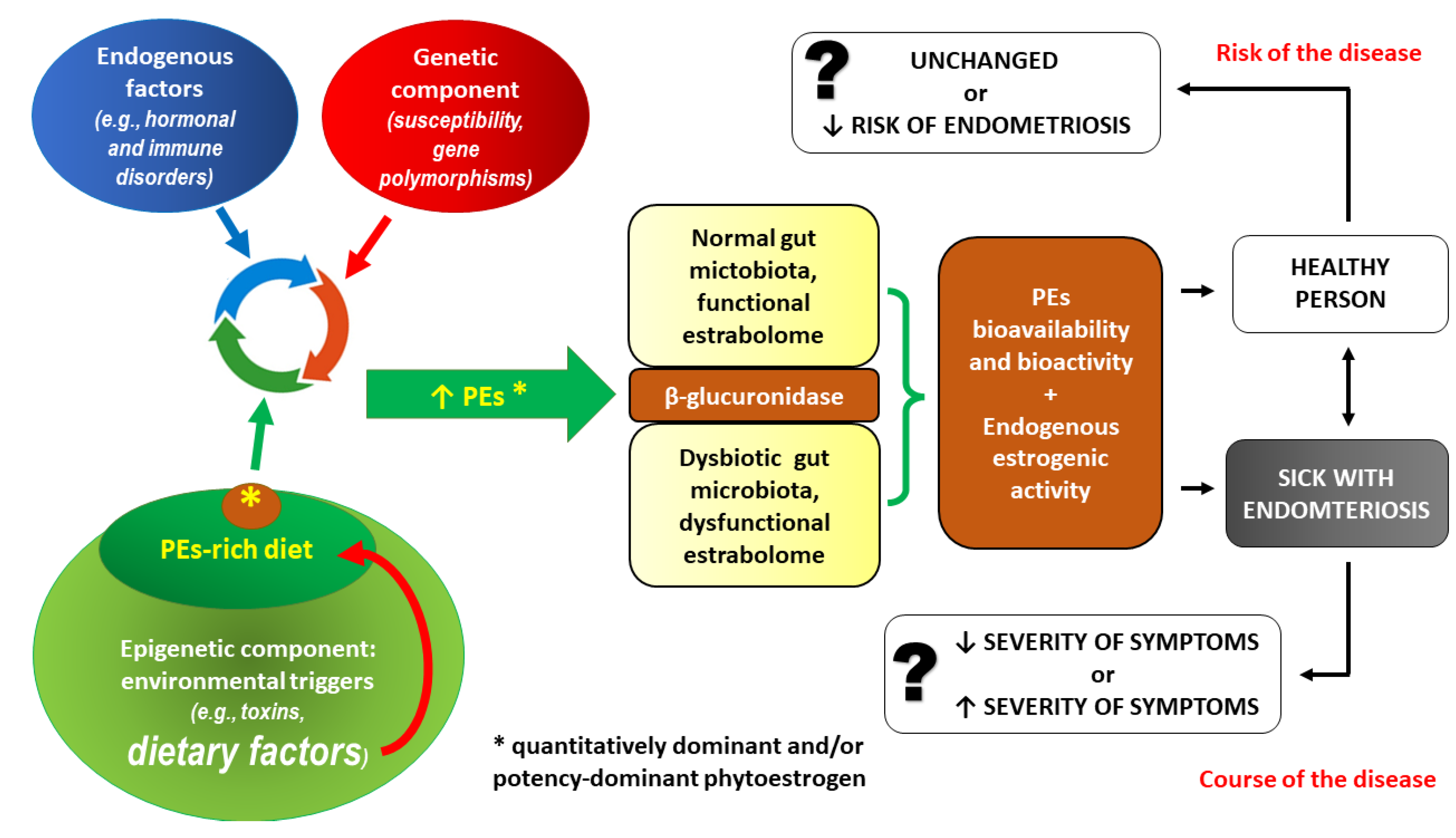

Table 1.
Phytoestrogens (PEs) – an overview of the family of naturally occurring polycyclic phenols.
Table 1.
Phytoestrogens (PEs) – an overview of the family of naturally occurring polycyclic phenols.
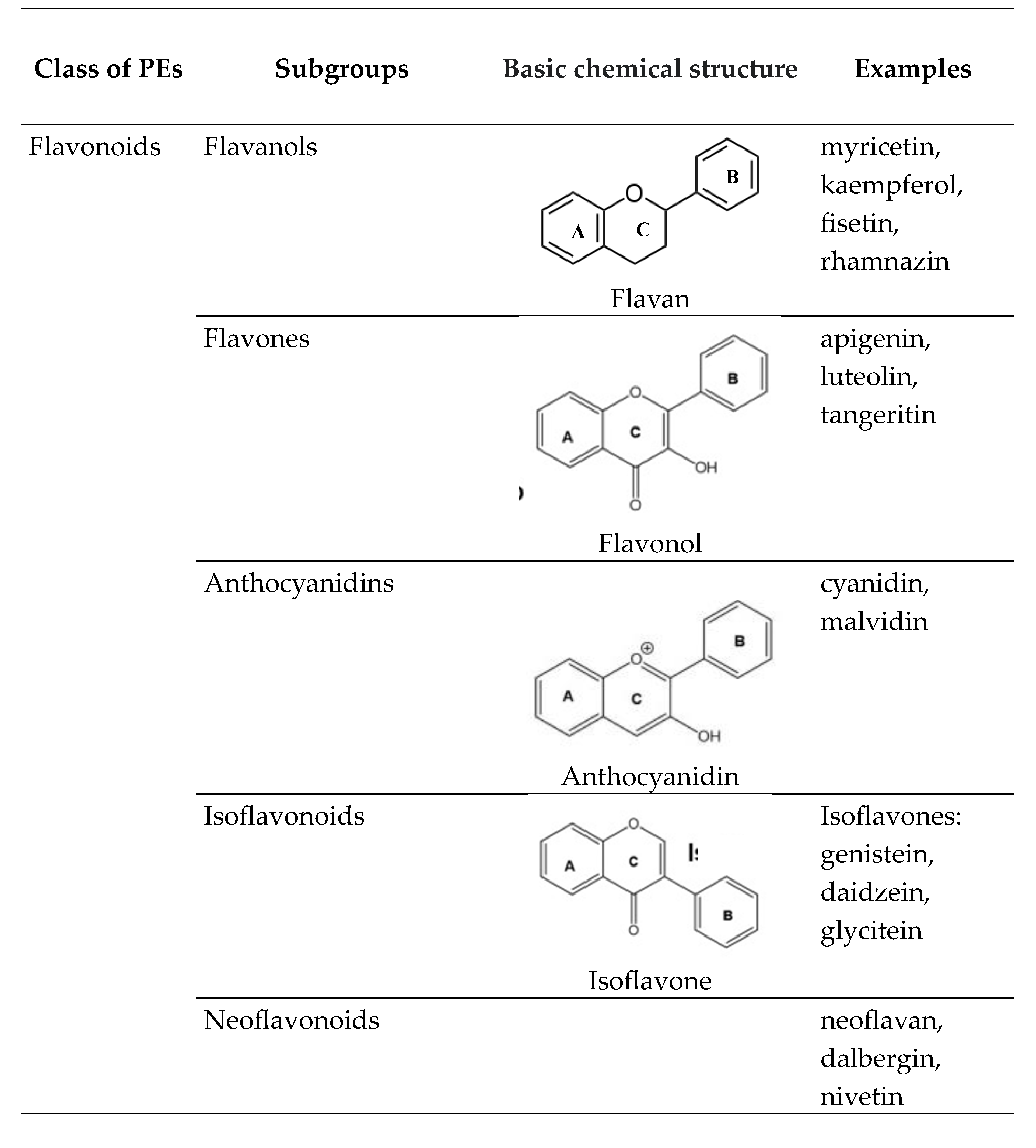
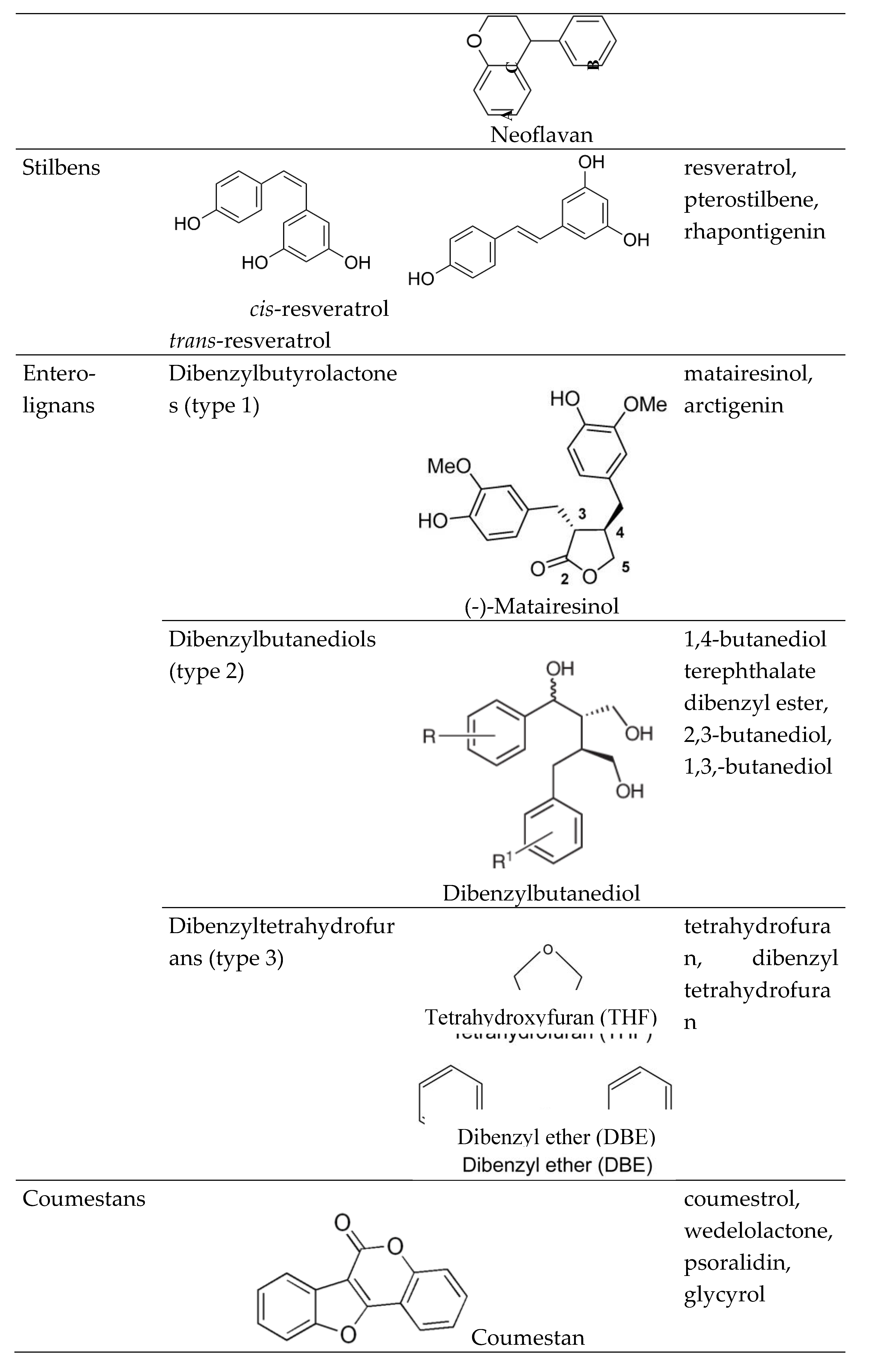
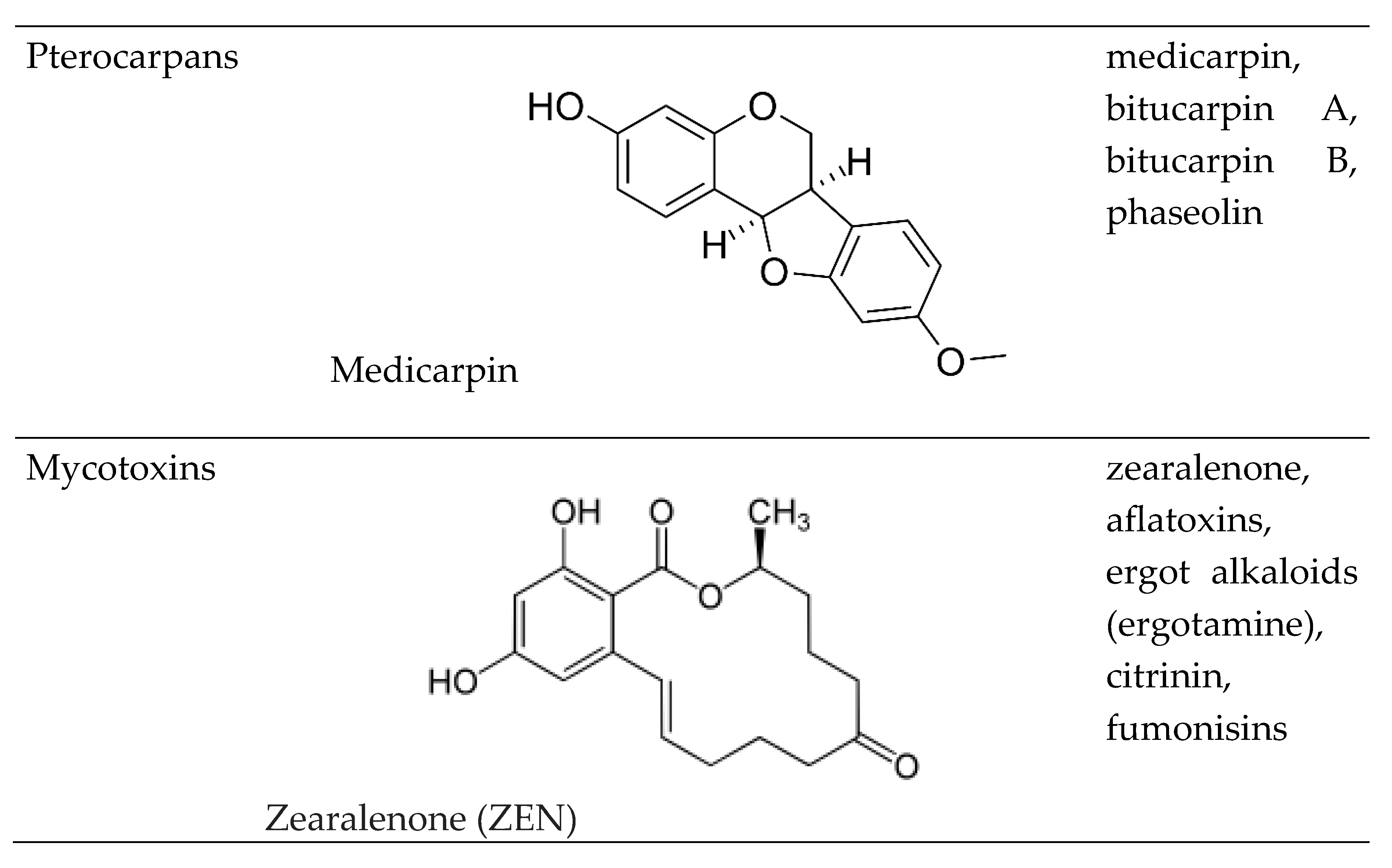
Table 2.
Human studies on the effect of phytoestrogens (PEs) administered orally in endometriosis [169]. Two studies were included where PEs were analyzed together with other nutrients in the diet [398,400]. In such studies, only endometriosis-related outcomes are summarized. * Level of evidence (LoE) for clinical studies, according to the Levels of Evidence for Primary Research Question adopted by the North American Spine Society January 2005; CI – confidence interval; COX-2 – cyclooxygenase-2; ER2 – estrogen receptror-2; MMP-2, MMP-9 – matrix metalloproteinases 2 and 9, respectively; MOC – monophasic oral contraceptive; N/A – not applicable; NP – not provided; OC – oral contraceptive; OR – odds ratio; RR – rate ratio;
Table 2.
Human studies on the effect of phytoestrogens (PEs) administered orally in endometriosis [169]. Two studies were included where PEs were analyzed together with other nutrients in the diet [398,400]. In such studies, only endometriosis-related outcomes are summarized. * Level of evidence (LoE) for clinical studies, according to the Levels of Evidence for Primary Research Question adopted by the North American Spine Society January 2005; CI – confidence interval; COX-2 – cyclooxygenase-2; ER2 – estrogen receptror-2; MMP-2, MMP-9 – matrix metalloproteinases 2 and 9, respectively; MOC – monophasic oral contraceptive; N/A – not applicable; NP – not provided; OC – oral contraceptive; OR – odds ratio; RR – rate ratio;
| Authors | Year | Type of the study | Compound(s), duration | Sample size (n) | Age range (years, mean) | Control (n) | Main results (p < 0.05) | LoE * |
| Kodarahmian et al. [394] | 2019 | Placebo-controlled, randomized, double-blind clinical trial | resveratrol 400 mg; 12 – 14 weeks | 17 | 18-37 (30.19 ±2.4) | 17 (placebo) | - ↓ level of mRNA and protein of both MMP-2 and MMP-9; - ↓ concentration of MMP-2 and MMP-9 in the serum and the endome-trial fluid | II |
| Maia Jr et al. [395] | 2012 | retrospective study | resveratrol 30 mg; 2 – 6 months | 26 using OC | 24-40 (31 ±4.0) | 16 using OC | - ↓ pain (82% of patients reporting complete resolution of dysmenorrhea and pelvic pain after 2 months); - ↓ expressions of both COX-2 and aromatase in eutopic endometrium | II |
| Mendes da Silva et al. [396] | 2017 | randomized clinical trial | resveratrol 40 mg; 42 days | 22 using MOC | 20-50 (35.4 ±7.1) | 22 using MOC (placebo) | - NO DIFFERENCE in median pain scores between the groups; resveratrol is not superior to placebo for treatment of pain in endometriosis | III |
| Nagata et al. [397] | 2001 | prospective cohort study | soy isoflavones: daidzein and genistein; 6 years | 1172 | 35-54 (42.9 ±4.4) | N/A | - ↓ risk of premenopausal hysterectomy: RR (95% CI) 0.35 (0.13-0.97) | II |
| Parazzini et al. [398] | 2004 | two case-control studies | PE-rich vs. low-PE diet; 15-year data | 504 | Cases: 20-65 (33 ±3.3) Controls: 20-61 (34 ± 2.9) | 504 | - ↓ risk of endometriosis for PE-rich diet (OR = 0.3 for the highest tertile of intake for green vegetables, and OR = 0.6 for fresh fruit) | III |
| Signorile et al. [399] | 2018 | Prospective, placebo-controlled, cohort study | dietary supplement containing quer-cetin (200 mg), curcumin (turmeric curcumin 20 mg), parthenium (19.5 mg); 3 months | 34 | NP | 30 (placebo) | - ↓ symptoms in endometriosis: dysmenorrhea and chronic pelvic pain (both from 62% to 18 %), dyspareunia (from 30% to 15%); - ↓ serum levels of PGE2 and CA-125 | III |
| Trabert B et al. [400] | 2011 | population-based case control study | overall intake of fruits and vegetables, dairy, vegetable, fruit (excluding fruit juice), whole grains, legumes, red meat, poultry, fatty fish, nonfatty fish and seafood; 60 months | 284 | Cases: 18-49 (NP) Controls: 18-49 (NP) |
660 (randomly selected, without a history of endometriosis) | - ↑ risk of endometriosis positively correlated with β-carotene consumption and servings/d of fruit, whereas - vegetable intake was NOT ASSOCIATED with endometriosis risk |
I |
| Tsuchiya et al. [401] | 2007 | case-control study | urinary levels of soy isoflavones: daidzein and genistein; 24 months of recruiting period | 79 (stage I-II: 31) (stage III-IV: 48) |
20-45 (stage I-II: 32.3 ±3.2) (stage III-IV: 32.6 ±3.7) | 59 | - ↑ urinary level of isoflavones was inversely associated with both the risk of advanced endometriosis (stage III-IV) and severity of endometriosis; - for advanced endometriosis, ER2 gene RsaI polymorphism significantly modifies the effects of genistein | III |
| Youseflu et al. [402] | 2020 | case-control study on dietary data | isoflavones, lignans, and coumestrol; 12 months | 78 | 15-45 (31.01 ±6.56) | 78 | - ↓ risk of endometriosis for isoflavones, lignans, and coumestrol | III |
| Harris et al. [403] | 2018 | prospective cohort study | intake of fruits and vegetables; 22-year follow-up period | 70835 | 25-42 (NP) | N/A | - ↓ risk of endometriosis for higher fruit consumption, especially for citrus fruits - ↓ risk of endometriosis was positively correlated with β-Cryptoxanthin intake - No association between total vegetable intake and endometriosis risk. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



