
Submitted:
02 July 2023
Posted:
03 July 2023
You are already at the latest version
Abstract
The possibility of treating more patients for a longer time, has exploded the concept of anticipation in the systemic treatment of metastatic prostate cancer (mPC). The genetic analysis of Damage DNA Repair (DDR) mutations in pathogenetic variants (PV) and the development of poly (adenosine diphosphate-ribose) polymerase (PARP) inhibitors led to the first valid precision medicine option tailored for mPC. PARP inhibitors are repeating the same evolutionary path experienced for androgen receptor signaling inhibitors (ARSI), starting from the late stage of second line mCRPC in a limited population and time frame. Inevitably also PARP inhibitors will be absorbed by the concept of anticipation and intensification of care. The anticipation of PARP inhibitors in the first line mCRPC therapy is already underway and further one in mHSPC will soon be verified. According to the crosstalk between androgen receptors (AR) and DNA repair, the new message that is emerging is that the combination of PARP inhibitors with ARSI, disconnects PARP from the genetic analysis. Most of the recent trials analyzing PARP inhibitors plus abiraterone or enzalutamide, enrolled first line mCRPC irrespectively to gene PV. The conclusion of the PROPEL trial was that the advantage was independent to PV status, however the highest advantage was reported in BRCA1/2 mutated subgroup. The conclusion of the MAGNITUDE trial was different, with a significant advantage only in the DDR mutated subgroup and the DDR non-mutated was closed for further enrollment. The combination of PARP inhibitors with ARSI represents a significant strategy in the concept of anticipation and intensification of care in mPC. However, it should not nullify the advantages of precision medicine linked to the genetic analysis of DDR.
Keywords:
prostate cancer
; PARP inhibitors
; DDR gene
; CRPC
1. Introduction: the concept of anticipation in the systemic therapy of prostate cancer
For a long time, the natural history of metastatic prostate cancer (mPC) has been simply divided in two separated phases: 1. Hormone sensitive (HSPC) and 2. Hormone refractory (HRPC), subsequently better defined as Castration resistant (CRPC). These two phases were considered as two distinct pathologies with different therapeutic approaches, the first subjected to different lines of manipulation of the classic androgen deprivation (ADT), the second to chemotherapy (CHT) with taxanes. The systemic treatment of mPC has undergone an epochal positive evolution in recent years, in particular with the development of new generation hormone manipulations and androgen receptor signaling inhibitors (ARSI). Most of these new therapies were initially indicated in the late stages of the disease (post CHT-mCRPC) [1,2] where options and patient survival were limited. However, several clinical trials have shown that ARSI and CHT are equally effective in earlier stages, such as first-line mHSPC [3,4,5,6]. The intention of treating more patients for a longer time has led to the concept of anticipation in the systemic treatment of mPC. With anticipation came several changes in therapeutic strategies, in particular: 1. The use of both docetaxel and ARSI in mHSPC; 2. The stratification of mPC in classes such as high and low volume, high and low risk, or de novo and progressive tumors; 3. The competition in the use of these drugs between the mHSPC and mCRPC phases; 4. The shift from a clear distinction between the HSPC and CRPC phases and their management to a merger of the two. At present, the management of mCRPC represents a second line of therapy, sharing several of the recommended drugs with mHSPC. The concept of anticipation of therapy has merged with that of intensification of care, leading to first-line therapeutic schemes with doublets or triplets of drugs in mHSPC. As a matter of fact, ADT alone no longer represents a therapeutic option in mHSPC and the current recommendation is for an association with ARSI, docetaxel or both [3,4,5,6]. In patients with high volume/high risk de novo mHSPC, the recent trials PEACE1 [7] and ARASENS [8] introduced the concept of triplets, with abiraterone or daralutamide in combination with docetaxel and ADT.
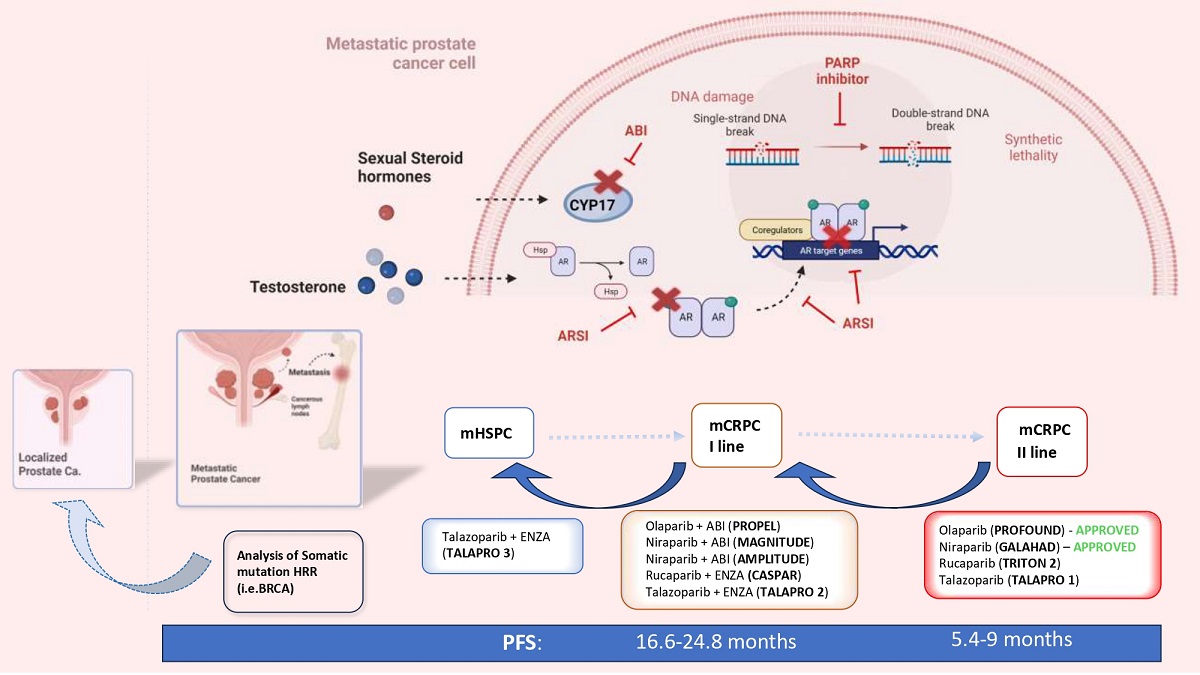
The main criticalities associated with the concepts of anticipation and intensification of care are: 1. An uncontrolled progression of anticipation in the already ongoing non-metastatic PC; 2. The development of early drug resistance with limited choice of treatment for late mCRPC and the need to repeat the same combination of drugs used as first-line therapies in HSPC. The key point of failure in this process, accentuated by the numerous therapeutic options available, is the identification of valid prognostic indicators of treatment response and the development of tailored precision therapies for each patient. The stratification of mPC on the basis of volume, risk and development of the disease in not sufficient for precision medicine. The genetic analysis of pathogenetic variants (PV) of genes involved in the DNA Damage Repair (DDR) mechanisms and the development of Poly ADP ribose polymesare (PARP) inhibitors (PARPi) led to the first valid precision medicine option tailored for mPC. PARPi are following the same evolutionary path traced by ARSI, expanding their indication starting from the late stage of second line mCRPC treatment in a limited population and time frame. Inevitably, also PARP inhibitors will be absorbed by the concept of anticipation and intensification of care. Mounting evidence in favour of the anticipation of PARPi in the first-line mCRPC therapy is already emerging and further one in mHSPC is underway. (Figure 1).
The purpose of this review is to analyze how the genetic basis of DDR PV and the clinical results with PARPi currently available can guarantee an anticipation process, always in compliance with precision medicine and tailored indications.
2. The rational and the genetic profile that sustain PARP inhibitors in PC
To address the underlying issue of genomic erosion, a complex network of DDR mechanisms has evolved [9,10]. Genetic abnormalities in DDR response systems are associated with life-threatening diseases such as immune deficiency, premature aging and cancer susceptibility. There are six major DNA repair pathways in humans: base excision repair (BER), nucleotide excision repair (NER), single strand break repair (SSR), homologous recombination repair (HRR), non-homologous end joining (NHEJ), mismatch repair (MMR). When the lesion is situated in only one strand of DNA, the undamaged complementary strand acts as a template for the NER and BER repair mechanisms. When a damaging agent induces DNA double-strand breaks (DSBs) MMR, HRR and NHEJ come into the picture [11,12]
PARP is a multifunctional protein that plays a critical role in SSR and BER mechanisms, primarily by recruiting DNA repair proteins to the sites of damage. So far, eighteen members of the PARP family have been found, among which PARP-1 is the most important [13]. PARP's main enzymatic function is to add ADP-ribose to substrate proteins by cleaving NAD+ and releasing nicotinamide, thus activating a complex cascade which ends in the recruitment of different proteins, such as DNA polymerase theta (POLQ), DNA ligase I and XRCC1, to the site of damage. Despite being well-known for its role in SSR and BER, recent evidence suggests that PARP can also affect DSB repair, mainly via controlling the expression of key HR genes BRCA1 and RAD51 [14,15,16].
Defects in the DDR pathways can result in genomic instability, gene mutations, and, eventually, lead to the development of cancer. On the other hand, germline or acquired mutations in DDR also provide cancer-specific vulnerabilities that can be targeted by synthetic lethality-based therapies [17]. PARPi are the first successful example of a targeted therapy that uses synthetic lethality to kill cancerous cells with DNA-repairing deficiencies (e.g., BRCA1/2 mutation). These drugs act by competing with NAD+ for the catalytically active site of PARP molecules, interfering with SSB repair by BER. The unrepaired SSB can be converted to DSB, and the primary mechanism to repair such lesions during cell cycle is HRR. While HRR-proficient cells can repair the DSB originated from the SSB, thus ensuring genetic stability and cell survival, HRR-deficient cells are unable to do so, resulting in apoptosis and ultimately cell death [16,17,18]. Olaparib was the first drug to be developed in this group. Initially approved in 2014 for patients with ovarian cancer with germline BRCA PV and subsequently in breast, pancreatic and prostate cancer [19].
Germline HRR PV increase the risk of developing PC by 8 times at the age of 65 years and are linked to more aggressive PC, higher risk of lymph-node invasion and distant metastasis at the time of diagnosis [20]. Moreover, germline BRCA 1/2 PV are associated with higher probability of failure in patients with localized disease undergoing active surveillance and higher risk of recurrence in patients who underwent curative treatment [21,22]. Several studies have analyzed the prevalence of germline and somatic mutations in HRR genes, both in metastatic and localized PC. A 2019 systematic review by Lang et al. [23] found a median prevalence of germline HRR PV and specifically of BRCA2 PV in patients with familiar PC of 29.3% (range, 7.3-91.67%) and 3.7% (range, 1.3-7.9%) respectively. In the same study, for unselected patients, the frequency of somatic and germline mutations was, respectively, 3.9% and 1.5% for ATM, 1.1% and 0.6% for BRCA1, 4.9% and 1.1% for BRCA2, 1.3% and 0.5% for PALB2, 1.5% and 0.5% for RAD51C. The overall prevalence of somatic DDR genes PV ranged between 4.9 and 22%, while the rate of germline DDR PV was 17.2-19%.
Regarding metastatic PC, the incidence of germline PV in HRR genes was found between 11% and 33% in a pan-cancer analysis of whole genomes [23]. Similarly, 8% of germline and 23% of somatic HRR PV were found in 150 metastatic PC by the International Stand Up to Cancer/Prostate Cancer Foundation team (SU2C-PCF) [24]. In this cohort, BRCA2 was the most prevalent mutation (13%) followed by ATM (7.3%), MSH2 (2%) and BRCA1. More specifically, in mCRPC, the incidence of somatic HRR PV was 24% (BRCA 13%, ATM 7.3%, MSH2 2%, BRCA1 0.3%) in a study by Eeles et al. [25] and 28% in the Profound study, which analyzed the results of 2792 biopsies of mCRPC patients.
Considering localized PC, the rates of PV in HRR seem to be lower than those seen in mPC. Despite that, a 2019 study by Kim et al. [26] found an overall incidence of HRR pathways alteration in localized PC of 29.9%, higher than suggested by a previous study by Marshall et al. [27] (11% in Gleason Grade Group 5 and 15.8% in cT3 patients).
In the Profound study, a total of 4,858 tissue samples were tested and reported centrally [28]. Considering all samples examined by Next Generation Sequencing (NGS), 83% were primary tumor samples (96% were archival and 4% newly obtained) and 17% were metastatic tumor samples (60% were archival and 33% newly obtained). NGS results were generated more frequently from newly obtained compared with archival samples (63.9% vs. 56.9%) and metastatic compared with primary samples (63.9% vs. 56.2%). Although generation of an NGS result declined with increasing sample age, approximately 50% of samples >10 years generated results.
Somatic analysis of HRR mutation in PC should be considered first, compared to germline analysis. When mutations of HRR genes are acquired during the progression of the disease, biopsy of the metastatic tumor represents the ideal approach to identify molecular alterations. However, biopsies of metastatic lesions can be challenging or not feasible, and at the same time, a single biopsy may not reveal tumor heterogeneity among metastases. The analysis of free circulating DNA (cDNA) is a promising approach as it could overcome the difficulty in obtaining tissue in many cases where this is not possible; currently there is no data that allows this test to be used reliably. The first study that analyzed cDNA in this field is GHALAND study [29] where the treatment efficacy analysis was performed on the amount of circulating tumor cells present from the eighth week of treatment. Best results were obtained in BRCA cohort than non-BRCA cohort, with a 24% of CTC (circulating tumor cell) response. However, incongruences with different commercial tests currently available for cDNA analysis are as high as 40%, with the risk of patients receiving inappropriate or no treatment.
3. Clinical trials and actual recommendations for PARP inhibitors in second line mCRPC
The latest guidelines of the European Urological Association (EAU) recommend PARPi in pre-treated mCRPC patients with relevant DNA repair gene mutations.
So far, only two PARPi, olaparib and rucaparib, are licensed by the FDA (EMA only approved olaparib), but other drugs of the same class are under evaluation (e.g., niraparib, talazoparib). The actual recommendations are supported by prospective trials analyzing a tailored therapy with PARPi for the treatment of mCRPC progressing after first-line treatment with ARSI and taxanes.
In the second line mCRPC setting, PARPi were analyzed as monotherapies in patients with HRR mutations. This type of analysis also determined a ranking in the HRR PV, withBRCA2 mutation associated with the most significant benefits from all types of PARP therapy.
Olaparib was first evaluated in the TOPARP-B trial [30], a multicenter, open label, randomised phase II trial. Eligible patients had a mCRPC and a putatively pathogenic mutation or homozygous deletion in a DDR gene that could be associated with sensitivity to PARP inhibition as identified by NGS. Patients were required to have previously received at least one but no more than two taxane based chemotherapy regimens (regardless of prior exposure to novel hormonal drugs). The 98-eligible-patients with DDR gene aberrations were divided into two dose cohort, randomly assigned to each one to receive 400mg or 300mg olaparib twice daily. Only the cohort receiving400mg twice daily met the predefined criteria for success, showing better percentages in all primary endpoints: radiological objective response, 50% decrease in PSA from baseline, conversion of circulating tumour cell count. Even though the results observed varied considerably for different HRR gene aberrations, the greatest antitumor activity was seen in the subgroup with BRCA1/2 mutations.
The Profound trial [31] can be considered the most relevant study in this field. A prospective, randomized, open-label, phase III trial was carried out at 206 sites in 20 countries and evaluated the safety and efficacy of olaparib in men with mCRPC who progressed while receiving a new hormonal agent (enzalutamide or abiraterone).
Patients were divided into two cohorts depending on their qualifying gene alteration selected for their direct or indirect role in HRR: patients with at least one alteration in BRCA1, BRCA2, or ATM were assigned to cohort A, patients with alterations in any of the other 12 genes were assigned to cohort B. In each cohort patients were randomly assigned to receive olaparib (300mg twice daily) or the physician’s choice of either enzalutamide (160mg once daily) or abiraterone (1000mg once daily) plus prednisone (control group).
Overall, 387 patients met all eligibility criteria and thus underwent randomization from April 2017 through November 2018. The primary endpoint was radiological progression-free survival (PFS), which was significantly longer in the olaparib arm (7.4 months vs 3.6 months in the control group) [HR for progression or death 0.34; 95% CI, 0.25–0.47; P < 0.001], especially in cohort A patients (which have a 66% lower risk of disease progression or death).
In cohort A, as secondary endpoints, the confirmed Objective Response Rate (ORR) was (olaparib vs. control group) 33% vs. 2%, a 50% reduction in PSA was seen in 43% vs. 8% and the median Overall Survival (OS) was 19.1 months vs. 14.7 months (HR 0.69, 95% CI 0.50–0.97, P = 0.0175).
The common adverse events of any grade in the olaparib group were anemia (46% vs. 15%) followed by nausea and fatigue and the overall incidence of grade ≥ 3 adverse events was higher in the olaparib group (51% vs 38%).
The other FDA-approved PARPi, rucaparib, was evaluated in the TRITON-2 trial [32], an open-label, phase II study. The trial enrolled 115 patients who progressed after one or two lines of next-generation AR-directed therapy and one taxane-based chemotherapy for mCRPC and who had a deleterious germline or somatic alteration in one HRR gene that may confer sensitivity to PARP inhibition. Patients received a starting dose of 600mg twice daily. As primary endpoint, ORR was 43.5% (95% CI, 31.0% to 56.7%;) per independent radiology review and 50.8% (95% CI, 38.1% to 63.4%) per investigator assessment. ORRs were similar for patients with a germline or somatic BRCA alteration and for patients with a BRCA1 or BRCA2 alteration, while a higher PSA response rate was observed in patients with a BRCA2 alteration. PSA responses seemed smaller in the BRCA1 (15.4%) or mono-allelic patients (11.1%) compared to BRCA2 (59.8%) or biallelic patients (75.0%). The PSA response rate was 4.1%, 6.7% and 16.7% in the ATM group (49 patients), CDK12 cohort (15 patients) and CHEK2 group (12 patients), respectively. The ORR was 10.5% in the ATM group, 0% in the CDK12 cohort, and 11.1% in the CHEK2 group. The most frequent treatment-emergent adverse events (TEAEs) of any grade were asthenia (61.7%), nausea (52.2%), and anemia (43.5%).
The anti-tumour activity and safety of the PARPi niraparib was evaluated in the GALAHAD trial [29], a multicenter, open-label, single-arm, phase II study. Patients with germline pathogenic or somatic biallelic pathogenic alterations in BRCA1 or BRCA2 (BRCA cohort, n = 142) or biallelic alterations in other prespecified HRR (non-BRCA cohort; n = 81) were included. Enrolled patients received niraparib 300mg orally once daily until treatment discontinuation, death, or study termination. At final analysis, with a median follow-up of 10 months, the ORR in the “BRCA group” was 41% (95% CI 23.5–61.1%) compared to 9% (95% CI 1.1–29.2%) in the “non-BRCA group”; and the CRR was 63% (95% CI 47.6–76.8%) compared to 17% (95% CI 6.6–33.7%), respectively. Median PFS and OS in the BRCA cohort were 8.2 months (95% CI 5.2–11.1 months) and 12.6 months (95% CI 9.2–15.7 months), respectively, versus 5.3 months (95% CI 1.9–5.7 months) and 14.0 months (95% CI 5.3–20.1 months) in the non-BRCA group.
The most common TEAEs of any grade were nausea (58%), anaemia (54%) and vomiting (38%); the most common grade ≥ 3 events were anaemia, thrombocytopenia and neutropenia.
The last PARPi under investigation, talazoparib, was assessed in the TALAPRO-1 trial [33] (open-label, phase II). The inclusion criteria were the same of the GALAHAD trial and eligible patients were given oral talazoparib (1mg per day or 0.75mg per day in patients with moderate renal impairment). Finally, 127 cases received at least one dose of the study drug (safety population) and 104 had HRR-deficient measurable disease (antitumor activity population). The primary endpoint was confirmed ORR, whereas secondary endpoints were time to objective response, duration of objective response, proportion of patients with a decrease in PSA ≥ 50% from baseline, time to PSA progression, radiological PFS, OS, safety. After a median follow-up of 16.4 months, the ORR was 29.8% (95% CI; 21.2–39.6), 46% in cases with BRCA1/2 mutations with a radiological PFS of 11.2 months, 12% in cases with ATM mutations. Talazoparib produced a 50%-PSA response in 66%, 7%, 6%, and a CTC conversion in 81%, 50%, 20%, in cases with BRCA 1/2, ATM and other mutations respectively.
The most common grade ≥ 3 TEAEs in the overall population were: anaemia (31%), thrombocytopenia (9%), and neutropenia (8%).
4. The prognostic role of HRR PV in non-metastatic and mHSPC as indicators of anticipation tailored treatment
It is widely accepted that BRCA2 gene PV increase the chance of developing PC and are linked to an earlier onset, higher rates of lymph node involvement and distant metastasis at the time of diagnosis [34,35]. In this context, the IMPACT study analyzed the role of PSA screening in BRCA PV carriers compared to controls after 3 years of follow-up [36]. A total of 2932 patients were recruited, 919 were BRCA1 carriers, 709 BRCA1 noncarriers, 902 BRCA2 carriers and 497 BRCA2 noncarriers. The incidence of PC at biopsy was 5.2% in BRCA2 carriers and 3.0% in non-carriers. Moreover, BRCA2 gene PV were associated with a younger age at diagnosis and a higher incidence of intermediate-risk or high-risk disease (77% in BRCA2 carriers vs 40% in non-carriers). These data suggested the possibility of a tailored PV screening strategy for this more vulnerable population with a higher chance of high-risk disease.
Active surveillance (AS) is a well-established option for men diagnosed with favorable-risk and intermediate-risk PC. A study by Carter H.B. et al. [21] evaluated whether BRCA 1/2 and ATM PV were associated with grade reclassification (GR) in patients undergoing AS and thus with the need of a more aggressive choice of treatment from diagnosis. Considering all 1211 participants, the rate of PV carriers was significantly higher in those reclassified both for the three-gene panel and for BRCA2 alone (3.8% and 2.1%, respectively) than those not reclassified (1.6% and 0.5%, respectively). However, the retrospective nature of this study and the absence of multiparametric magnetic resonance targeted biopsies demand for further evaluation of the role of AS in DDR PV carriers through prospective studies [37].
Castro E. et al. [22] evaluated the effect of BRCA PV on metastatic relapse and cause-specific survival after radical treatment (surgery and radiotherapy) for localized disease. A total of 1302 patients were included in the study, of which 67 carried a germline BRCA PV (18 BRCA1 and 49 BRCA2). At 3, 5, and 10 years after treatment, 97%, 94%, and 84% of noncarriers vs 90%, 72%, and 50% of carriers were metastasis-free. The 3-, 5-, and 10-year cancer specific survival (CSS) rates from radical treatment were significantly better in the noncarriers (99%, 97%, and 85%, respectively) than in the carrier cohort (96%, 76%, and 61%, respectively; p < 0.001). In line with these results, Martinez Chanza M. et al. [38] concluded that BRCA PV were associated with a greater relapse risk in a retrospective series of 380 patients with localized and metastatic HSPC. Data from these studies supports closer follow-up of these patients and the need for randomized prospective clinical trials to standardize the most appropriate management strategies of localized PC in HRR PV carriers.
Different studies have analyzed the impact of BRCA2 and other HRR PV in metastatic PC. In a recent observational study, Antonarakis et al. [39] investigated the clinical impact of germline HRR PV on the efficacy of first-line ARSI among 172 mCRPC patients. Clinical/radiologic PFS, the primary endpoint, was longer both in patients with versus without any germline DNA-repair PV (median 13.3 vs 10.3 months) and in those with versus without BRCA/ATM PV specifically (median 15.2 vs 10.8 months). These results appear to be in conflict with a retrospective study of 319 patients with mCRPC by Annala M. et al. [40], which showed that HRR PV carriers had a significant shorter PFS rate compared to non-carriers (3.3 vs. 6.2 months, p = 0.01). Despite that, another recent study [41], in accordance with Antonarakis et al. [39], suggested a better response to first-line abiraterone treatment in germline or somatic HRR PV carriers compared to non-carriers, supporting the idea of a “synthetic lethality” of treatment with more effective AR-targeted therapies in patients harboring germline HRR PV.
Prorepair-B was the first prospective trial to analyze the prognostic impact of BRCA1–2 and other HRR genes on CSS in mCRPC patients [42]. There was no significant difference in CSS rates when considering all HRR mutations together between carriers and non-carriers (HRR PV carriers, 23.3 months vs. non-carriers 33.2 months; p = 0.264). Surprisingly, when only BRCA2 was taken into account, the authors found statistical difference in CSS (BRCA2 PV carriers, 17.4 months vs non-carriers, 33.2 months; p = 0.027). Moreover, a subgroup analyses to determine whether the treatment sequence adopted had any effect on the carrier status' impact on CSS was carried out. BRCA2 carriers were found to have worse outcomes compared to non-carriers when treated with the sequence docetaxel-ARSI (median, 10.7 vs 28.4 months; p < 0.001) but not when treated with the sequence ARSI-docetaxel (median, 24.0 vs 31.2, p = 0.901). Despite the need for validation in larger series, these results suggest that the prognostic impact of BRCA2 mutations may be influenced by the choice of first-line therapy.
5. PARP-AR crosstalk and current clinical trials with PARPi in anticipated first line mCRPC
An actual point of great interest is the crosstalk between androgen receptors (AR) and DNA repair. PARP system is involved in androgen-dependent transcription and PARPi can impair it. On the other hand, AR pathway regulates transcription of DNA repair genes and androgen depletion impairs HRR which renders the tumor susceptible to PARPi [9,10]. Experiments showed that ARSI are able to downregulate DDR genes expression in CRPC xenografts, [43] resulting in decreased DNA repair in cells and increased double stranded DNA breaks. This experimental evidence opens the possibility of new combination strategies with ARSI and PARPi. It is interesting to highlight how, in this case, the development of this new combination therapy relates to the attempt to anticipate the use of PARPi and to enlarge their indication independently of HRR mutations in PC patients. Currently, the major clinical trials underway analyzing this combination are anticipating its use as first-line therapy in mCRPC. However, other trials are being defined to extend the anticipation also in the mHSPC setting. There are currently five major clinical trials evaluating the combination of PARPi and ARSI in an anticipated phase of mCRPC. These trials, all phase 3 randomized placebo-controlled trials, are at different level of evolution. The primary endpoint is radiologic PFS and different approaches in terms of HRR mutations are used (Table 1).
5.1. PROPEL trial: abiraterone + Olaparib
The PROPEL trial (clinicaltrials.gov ID:NCT03732820) has been recently concluded and published [44]. This double blind, placebo-controlled, phase 3 trial enrolled 796 first-line mCRPC randomized to abiraterone plus Olaparib or placebo. Assignment in the two groups was stratified by distant metastasis type (bone, visceral, others) and by docetaxel treatment at mHSPC stage. Enrollment was not based on HRR status, however tissue and blood samples at baseline were collected and results were also analyzed in terms of HRR PV. The primary end-point was radiological PFS and OS was considered as secondary end-point. Based on HRR PV analysis, 28.4% of patients were included in the HRR mutated subgroup. The radiological PFS was significantly longer in the abiraterone + Olaparib than in the abiraterone + placebo arm (24.8 vs 16.6 months; HR 0.66; 95%CI 0.54-0.81; p<0.001). The advantage of the abiraterone + Olaparib combination was observed across all subgroups (type of distant metastases, previous docetaxel) and irrespectively of HRR status (HRR mutated subgroup: HR 0.50; 95%CI 0.34-0.73; HRR non-mutated subgroup: HR 0.76; 95%CI 0.60-0.97). Considering only BRCA PV, the advantage of the combination therapy over abiraterone alone was strongly higher in the BRCA mutated (HR 0.23; 95%CI 0.12-0.43) than in the non-mutated subgroup (HR 0.76; 95%CI 0.61-0.94). These data confirmed previous evidence from phase 2 studies on abiraterone + Olaparib [45,46]. The most common side effects in the combination group were anemia (grade 3 in 15.1% in the abiraterone + Olaparib and 3.3% in the abiraterone and placebo arm) and fatigue. The rate of cardiovascular events was similar between the two arms. OS data were immature at the first analysis, however at the ASCO GU 2023 a final analysis on OS for PROPEL patients [47] showed a 7-month advantage for the abiraterone + Olaparib (42.1 months) when compared to the abiraterone + placebo (34.7 months) arm (HR 0.81; 95%CI 0.67-1.00; p=0.0544). Also for OS, the advantage of the combination therapy was observed in all subgroups and irrespectively of HRR PV (HRR mutated subgroup: HR 0.66; 95%CI 0.45-0.95; HRR non-mutated subgroup: HR 0.89; 95%CI 0.70-1.14). 374 patients had a subsequent therapy, most commonly chemotherapy, but for now data are immature to calculate PFS2 (HR 0.76; 95%CI 0.59-0.99).
5.2. MAGNITUDE: abiraterone + Niraparib
The MAGNITUDE trial (Clinicaltrials.gov ID: NCT03748641) has been recently published [48]. This double blind, placebo-controlled, phase 3 trial enrolled 423 first-line mCRPC randomized to abiraterone plus Niraparib or placebo. Assignment in the two groups was stratified by distant metastasis type (bone, visceral, others). Tissue and blood samples at baseline were collected and results were also analyzed in terms of HRR PV. The primary endpoint was radiological PFS and OS was considered as secondary end-point. In the BRCA1/2 mutated subgroup, median radiological PFS was significantly longer in the abiraterone + niraparib than in the abiraterone + placebo arm (16.6 vs 10.9 months; HR 0.53;95%CI 0.36-0.79; p=0.001). Similarly, considering all HRR mutated subgroup, niraparib + abiraterone was associated with a longer radiological PFS (16.5 vs 13.7 months; HR 0.73; 95%CI 0.56-0.96; p=0.022). This result remained significant irrespectively of the site of metastases. In the HRR non-mutant cohort, no significant differences in terms of radiological PFS were found between the two treatment arms (HR 1.09; 95%CI 0.75-1.57; p=0.66) and the cohort was closed to further enrollment. Results in terms of OS in the BRCA+ cohort are still immature (HR 0.88; 95%CI 0.58-1.34; p=0.5505). The most common grade 3 adverse events were anemia (28.3% vs 7.6%) and hypertension (14.6% vs 12.3%) with abiraterone + niraparib versus abiraterone + placebo.
5.3. AMPLITUDE trial: Abiraterone + niraparib
AMPLITUDE trial (Clinicaltrial.ov ID: NCT04497844) is an ongoing randomized placebo-controlled study in first-line mCRPC with HRR PV (somatic or germline), comparing abiraterone + niraparib versus abiraterone + placebo [49]. The primary endpoint is radiological PFS and OS is considered as secondary endpoint. Expected enrollment to be reached in November 2024 is 790 patients.
5.4. CASPAR trial: enzalutamide + rucaparib
CASPAR trial (A031902; Clinicaltrials.gov ID: NCT04455750) is an ongoing randomized phase 3 study in which 984 first line mCRPC irrespectively of HRR PV will be randomized on a 1:1 basis to enzalutamide + rucaparib versus enzalutamide + placebo[50,51]. Co-primary endpoints are radiological PFS and OS. The OS analysis will be undertaken as a primary endpoint if the radiological PFS endpoint is met. Endpoints will be verified in subgroups with vs without pathogenetic BRCA1, BRCA2 mutations.
5.5. TALAPRO-2: enzalutamide + talazoparib
TALAPRO-2 trial (Clinicaltrials.gov ID: NCT03395197) is an ongoing phase 3 placebo- controlled study in first-line mCRPC (with and without HRR alterations) randomized to enzalutamide + talazoparib vs enzalutamide + placebo [52]. The enrollment goal is 1037 patients in two cohorts: an all-comers cohort where patients are not required to have HRR alterations and an HRR PV cohort. The primary endpoint in both cohorts is radiological PFS and OS is considered as secondary endpoint.
6. Conclusions
Therapeutic strategies for the treatment of metastatic PC have undergone radical changes in recent years. These changes can be summarized in the search for an anticipation of cure, intensification through the combination with therapeutic doublets or triplets and the search for a personalized medicine of precision. PARPi are also being affected by these strategies: starting from a recommendation in the late phase of mCRPC, the concept of anticipation has produced evidence in the first-line of mCRPC treatment and will probably soon be able to find results also in the mHSPC phase. In parallel, the concept of intensification of care has led to the combinations of PARPi with abiraterone and enzalutamide. Unlike previous strategies, PARPi represent the first evidence of a precision medicine in PC linked to the presence of PV in HRR genes. The link between PARP and genetic analysis enhances the therapeutic indication and paves the way for a characterization of the genetic risk, anticipated from the metastatic to the non-metastatic phase of PC.
Contrary to this strategy, the new message emerging is that the combination of PARPi with ARSI may work independently of genetic analysis and may be indicated as first-line therapy regardless of HRR status. Most of the recent trials analyzing PARP inhibitors plus abiraterone or enzalutamide enrolled first-line mCRPC irrespectively of HRR mutations. In both PROPEL and MAGNITUDE trials, the combination therapy showed a significant advantage in terms of radiological PFS when compared to abiraterone alone. Despite the highest advantage reported in the BRCA 1/2 mutated subgroup, the PROPEL trial concluded that the benefit of the combination therapy was independent of HRR status. The MAGNITUDE trial, however, showed a significant advantage only in the DDR mutated subgroup and the DDR non-mutated cohort was closed for further enrollment. Moreover, radiological PFS is a relevant end-point but we are waiting for mature data in terms of overall survival. It is not unrealistic that considering this major endpoint, the influence of HRR mutations could become stronger. The combination of PARPi with ARSI represents a significant strategy in the concept of anticipation and intensification of care in metastatic PC. However, it should not nullify the advantages of precision medicine linked to the genetic analysis of HRR.
Author Contributions
conceptualization, Alessandro Sciarra, Stefano Salciccia, Flavio Forte, Paolo Casale; formal analysis and investigation, Valerio Santarelli, Lorenzo Santodirocco, Marco Frisenda, Martina Moriconi, Susanna Cattarino; data curation, Giulio Bevilacqua, Alessandro Gentilucci, Beatrice Sciarra, Gianna Mariotti; writing— draft preparation, Alessandro Sciarra, Valerio Santarelli, Loreonzo Santodirocco, Martina Moriconi, Stefano Salciccia ; All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding. It was commissioned from Current Oncology Editorial Office for a Special Issue at 0 APC.
Institutional Review Board Statement
Not required and not applicable for a review article.
Informed Consent Statement
Not required and not applicable for a review article.
Data Availability Statement
not applicable.
Acknowledgments
no acknowledgments.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Poon DM, Chan K, Lee SH, Chan TW, Sze H, Lee EK, Lam D, Chan MF. Abirateroneacetate in metastatic castration-resistant prostate cancer - the unanticipated real-world clinical experience. BMC Urol. 2016 Mar 22;16:12-18. [CrossRef]
- Beer, T.M.; Armstrong, A.J.; Rathkopf, D.; Loriot, Y.; Sternberg, C.N.; Higano, C.S.; Iversen, P.; Evans, C.P.; Kim, C.-S.; Kimura, G.; et al. Enzalutamide in Men with Chemotherapy-naïve Metastatic Castration-resistant Prostate Cancer: Extended Analysis of the Phase 3 PREVAIL Study. Eur. Urol. 2016, 71, 151–154. [Google Scholar] [CrossRef] [PubMed]
- Armstrong AJ, Szmulewitz RZ, Petrylak DP, Holzbeierlein J, Villers A, Azad A,Alcaraz A, Alekseev B, Iguchi T, Shore ND, Rosbrook B, Sugg J, Baron B, Chen L,Stenzl A. ARCHES: A Randomized, Phase III Study of Androgen Deprivation Therapy With Enzalutamide or Placebo in Men With Metastatic Hormone-Sensitive ProstateCancer. J Clin Oncol. 2019 Nov 10;37(32):2974-2986. [CrossRef]
- Chi, K.N.; Chowdhury, S.; Bjartell, A.; Chung, B.H.; Gomes, A.J.P.d.S.; Given, R.; Juárez, A.; Merseburger, A.S.; Özgüroğlu, M.; Uemura, H.; et al. Apalutamide in Patients With Metastatic Castration-Sensitive Prostate Cancer: Final Survival Analysis of the Randomized, Double-Blind, Phase III TITAN Study. J. Clin. Oncol. 2021, 39, 2294–2303. [Google Scholar] [CrossRef] [PubMed]
- Fizazi, K.; Tran, N.; Fein, L.; Matsubara, N.; Rodriguez-Antolin, A.; Alekseev, B.Y.; Özgüroğlu, M.; Ye, D.; Feyerabend, S.; Protheroe, A.; et al. Abiraterone acetate plus prednisone in patients with newly diagnosed high-risk metastatic castration-sensitive prostate cancer (LATITUDE): final overall survival analysis of a randomised, double-blind, phase 3 trial. Lancet Oncol. 2019, 20, 686–700. [Google Scholar] [CrossRef] [PubMed]
- Kyriakopoulos, C.E.; Chen, Y.-H.; Carducci, M.A.; Liu, G.; Jarrard, D.F.; Hahn, N.M.; Shevrin, D.H.; Dreicer, R.; Hussain, M.; Eisenberger, M.; et al. Chemohormonal Therapy in Metastatic Hormone-Sensitive Prostate Cancer: Long-Term Survival Analysis of the Randomized Phase III E3805 CHAARTED Trial. J. Clin. Oncol. 2018, 36, 1080–1087. [Google Scholar] [CrossRef] [PubMed]
- Fizazi, K.; Foulon, S.; Carles, J.; Roubaud, G.; McDermott, R.; Fléchon, A.; Tombal, B.; Supiot, S.; Berthold, D.; Ronchin, P.; et al. Abiraterone plus prednisone added to androgen deprivation therapy and docetaxel in de novo metastatic castration-sensitive prostate cancer (PEACE-1): a multicentre, open-label, randomised, phase 3 study with a 2 × 2 factorial design. Lancet 2022, 399, 1695–1707. [Google Scholar] [CrossRef] [PubMed]
- Smith MR, Hussain M, Saad F, Fizazi K, Sternberg CN, Crawford ED, KopyltsovE, Park CH, Alekseev B, Montesa-Pino Á, Ye D, Parnis F, Cruz F, Tammela TLJ,Suzuki H, Utriainen T, Fu C, Uemura M, Méndez-Vidal MJ, Maughan BL, Joensuu H,Thiele S, Li R, Kuss I, Tombal B; ARASENS Trial Investigators. Darolutamide andSurvival in Metastatic, Hormone-Sensitive Prostate Cancer. N Engl J Med. 2022 Mar 24;386(12):1132-1142. [CrossRef]
- Sciarra, A.; Frisenda, M.; Bevilacqua, G.; Gentilucci, A.; Cattarino, S.; Mariotti, G.; Del Giudice, F.; Di Pierro, G.B.; Viscuso, P.; Casale, P.; et al. How the Analysis of the Pathogenetic Variants of DDR Genes Will Change the Management of Prostate Cancer Patients. Int. J. Mol. Sci. 2022, 24, 674. [Google Scholar] [CrossRef]
- Sciarra A, Fiori C, Del Giudice F, DI Pierro G, Bevilacqua G, Gentilucci A,Cattarino S, Mariotti G, Salciccia S. DDR genes analysis and PARP-inhibitors therapy as tailored management in metastatic prostate cancer: achieved answers, open questions and future perspectives. Minerva Urol Nephrol. 2022 Dec;74(6):649-652. [CrossRef]
- Giglia-Mari G, Zotter A, Vermeulen W. DNA damage response. Cold Spring Harb Perspect Biol. 2011;3(1):a000745. [CrossRef]
- Caldecott, K.W. Mammalian single-strand break repair: Mechanisms and links with chromatin. DNA Repair 2007, 6, 443–453. [Google Scholar] [CrossRef]
- Rouleau, M.; Patel, A.; Hendzel, M.J.; Kaufmann, S.H.; Poirier, G.G. PARP inhibition: PARP1 and beyond. Nat. Rev. Cancer 2010, 10, 293–301. [Google Scholar] [CrossRef]
- Liu, C.; Vyas, A.; Kassab, M.A.; Singh, A.K.; Yu, X. The role of poly ADP-ribosylation in the first wave of DNA damage response. Nucleic Acids Res. 2017, 45, 8129–8141. [Google Scholar] [CrossRef]
- Ceccaldi, R.; Liu, J.C.; Amunugama, R.; Hajdu, I.; Primack, B.; Petalcorin, M.I.R.; O’connor, K.W.; Konstantinopoulos, P.A.; Elledge, S.J.; Boulton, S.J.; et al. Homologous-recombination-deficient tumours are dependent on Polθ-mediated repair. Nature 2015, 518, 258–262. [Google Scholar] [CrossRef]
- Hegan, D.C.; Lu, Y.; Stachelek, G.C.; Crosby, M.E.; Bindra, R.S.; Glazer, P.M. Inhibition of poly(ADP-ribose) polymerase down-regulates BRCA1 and RAD51 in a pathway mediated by E2F4 and p130. Proc. Natl. Acad. Sci. 2010, 107, 2201–2206. [Google Scholar] [CrossRef] [PubMed]
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.J.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef] [PubMed]
- Bryant, H.E.; Schultz, N.; Thomas, H.D.; Parker, K.M.; Flower, D.; Lopez, E.; Kyle, S.; Meuth, M.; Curtin, N.J.; Helleday, T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005, 434, 913–917. [Google Scholar] [CrossRef] [PubMed]
- Domchek SM, Aghajanian C, Shapira-Frommer R. Efficacy and safety of olaparib monotherapy in germline BRCA1/2 mutation carriers with advanced ovarian cancer and three or more lines of prior therapy. Gynecol Oncol. 2016;140(2):199-203. [CrossRef]
- Castro E, Goh C, Olmos D. Germline BRCA mutations are associated with higher risk of nodal involvement, distant metastasis, and poor survival outcomes in prostate cancer. J Clin Oncol. 2013;31(14):1748-1757. [CrossRef]
- Carter HB, Helfand B, Mamawala M. Germline Mutations in ATM and BRCA1/2 Are Associated with Grade Reclassification in Men on Active Surveillance for Prostate Cancer. Eur Urol. 2019;75(5):743-749. [CrossRef]
- Castro E, Goh C, Leongamornlert D. Effect of BRCA Mutations on Metastatic Relapse and Cause-specific Survival After Radical Treatment for Localised Prostate Cancer. Eur Urol. 2015;68(2):186-193. [CrossRef]
- Lang, S.H.; Swift, S.L.; White, H.; Misso, K.; Kleijnen, J.; Quek, R.G. A systematic review of the prevalence of DNA damage response gene mutations in prostate cancer. Int. J. Oncol. 2019, 55, 597–616. [Google Scholar] [CrossRef] [PubMed]
- ICGC/TCGA Pan-Cancer Analysis of Whole Genomes Consortium. Pan-cancer analysis of whole genomes Nature. 2020;578(7793):82-93. [CrossRef]
- Eeles, R.; Goh, C.; Castro, E.; Bancroft, E.; Guy, M.; Al Olama, A.A.; Easton, D.; Kote-Jarai, Z. The genetic epidemiology of prostate cancer and its clinical implications. Nat. Rev. Urol. 2013, 11, 18–31. [Google Scholar] [CrossRef] [PubMed]
- Kim IE Jr, Kim S, Srivastava A. Similar incidence of DNA damage response pathway alterations between clinically localized and metastatic prostate cancer. BMC Urol. 2019;19(1):33. [CrossRef]
- Marshall, C.H.; Fu, W.; Wang, H.; Baras, A.S.; Lotan, T.L.; Antonarakis, E.S. Prevalence of DNA repair gene mutations in localized prostate cancer according to clinical and pathologic features: association of Gleason score and tumor stage. Prostate Cancer Prostatic Dis. 2018, 22, 59–65. [Google Scholar] [CrossRef]
- Hussain, M.; Corcoran, C.; Sibilla, C.; Fizazi, K.; Saad, F.; Shore, N.; Sandhu, S.; Mateo, J.; Olmos, D.; Mehra, N.; et al. Tumor Genomic Testing for >4,000 Men with Metastatic Castration-resistant Prostate Cancer in the Phase III Trial PROfound (Olaparib). Clin. Cancer Res. 2022, 28, 1518–1530. [Google Scholar] [CrossRef]
- I Scher, H.; Sandhu, S.; Efstathiou, E.; Lara, P.N.; Yu, E.Y.; Saad, F.; Ståhl, O.; Olmos, D.; E Mason, G.; Espina, B.M.; et al. Niraparib in patients with metastatic castration-resistant prostate cancer and DNA repair gene defects (GALAHAD): a multicentre, open-label, phase 2 trial. Lancet Oncol. 2022, 23, 362–373. [Google Scholar] [CrossRef]
- Mateo, J.; Porta, N.; Bianchini, D.; McGovern, U.; Elliott, T.; Jones, R.; Syndikus, I.; Ralph, C.; Jain, S.; Varughese, M.; et al. Olaparib in patients with metastatic castration-resistant prostate cancer with DNA repair gene aberrations (TOPARP-B): a multicentre, open-label, randomised, phase 2 trial. Lancet Oncol. 2019, 21, 162–174. [Google Scholar] [CrossRef]
- De Bono, J.; Mateo, J.; Fizazi, K.; Saad, F.; Shore, N.; Sandhu, S.; Chi, K.N.; Sartor, O.; Agarwal, N.; Olmos, D.; et al. Olaparib for Metastatic Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2020, 382, 2091–2102. [Google Scholar] [CrossRef]
- Abida, W.; Patnaik, A.; Campbell, D.; Shapiro, J.; Bryce, A.H.; McDermott, R.; Sautois, B.; Vogelzang, N.J.; Bambury, R.M.; Voog, E.; et al. Rucaparib in Men With Metastatic Castration-Resistant Prostate Cancer Harboring a BRCA1 or BRCA2 Gene Alteration. J. Clin. Oncol. 2020, 38, 3763–3772. [Google Scholar] [CrossRef] [PubMed]
- de Bono, J.S.; Mehra, N.; Scagliotti, G.V.; Castro, E.; Dorff, T.; Stirling, A.; Stenzl, A.; Fleming, M.T.; Higano, C.S.; Saad, F.; et al. Talazoparib monotherapy in metastatic castration-resistant prostate cancer with DNA repair alterations (TALAPRO-1): an open-label, phase 2 trial. Lancet Oncol. 2021, 22, 1250–1264. [Google Scholar] [CrossRef] [PubMed]
- Mitra, A.; Collaborators, T.I.A.E.; Fisher, C.; Foster, C.S.; Jameson, C.; Barbachanno, Y.; Bartlett, J.; Bancroft, E.; Doherty, R.; Kote-Jarai, Z.; et al. Prostate cancer in male BRCA1 and BRCA2 mutation carriers has a more aggressive phenotype. Br. J. Cancer 2008, 98, 502–507. [Google Scholar] [CrossRef]
- Thorne, H.; Willems, A.J.; Niedermayr, E.; Hoh, I.M.Y.; Li, J.; Clouston, D.; Mitchell, G.; Fox, S.; Hopper, J.L.; Bolton, D.; et al. Decreased Prostate Cancer-Specific Survival of Men with BRCA2 Mutations from Multiple Breast Cancer Families. Cancer Prev. Res. 2011, 4, 1002–1010. [Google Scholar] [CrossRef] [PubMed]
- Page, E.C.; Bancroft, E.K.; Brook, M.N.; Assel, M.; Hassan Al Battat, M.; Thomas, S.; Taylor, N.; Chamberlain, A.; Pope, J.; Ni Raghallaigh, H.; et al. Interim Results from the IMPACT Study: Evidence for Prostate-specific Antigen Screening in BRCA2 Mutation Carriers. Eur. Urol. 2019, 76, 831–842. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.C.; Rumble, R.B.; Loblaw, D.A.; Finelli, A.; Ehdaie, B.; Cooperberg, M.R.; Morgan, S.C.; Tyldesley, S.; Haluschak, J.J.; Tan, W.; et al. Active Surveillance for the Management of Localized Prostate Cancer (Cancer Care Ontario Guideline): American Society of Clinical Oncology Clinical Practice Guideline Endorsement. J. Clin. Oncol. 2016, 34, 2182–2190. [Google Scholar] [CrossRef] [PubMed]
- Chanza, N.M.; Bernard, B.; Barthelemy, P.; Accarain, A.; Paesmans, M.; Desmyter, L.; de Roodenbeke, D.T.; Gil, T.; Sideris, S.; Roumeguere, T.; et al. Prevalence and clinical impact of tumor BRCA1 and BRCA2 mutations in patients presenting with localized or metastatic hormone-sensitive prostate cancer. Prostate Cancer Prostatic Dis. 2021, 25, 199–207. [Google Scholar] [CrossRef]
- Antonarakis ES, Lu C, Luber B, Liang C, Wang H, Chen Y, Silberstein JL, Piana D, Lai Z, Chen Y, Isaacs WB, Luo J. Germline DNA-repair Gene Mutations and Outcomes in Men with Metastatic Castration-resistant Prostate Cancer Receiving First-line Abiraterone and Enzalutamide. Eur Urol. 2018 Aug;74(2):218-225. [CrossRef]
- Annala M, Struss WJ, Warner EW, Beja K, Vandekerkhove G, Wong A, Khalaf D, Seppälä IL, So A, Lo G, Aggarwal R, Small EJ, Nykter M, Gleave ME, Chi KN, Wyatt AW. Treatment Outcomes and Tumor Loss of Heterozygosity in Germline DNA Repair-deficient Prostate Cancer. Eur Urol. 2017 Jul;72(1):34-42. [CrossRef]
- Hussain M, Daignault-Newton S, Twardowski PW. Targeting androgen receptor and DNA repair in metastatic castration-resistant prostate cancer: results from NCI 9012. J Clin Oncol 2018, 36, 991–9. [CrossRef]
- Castro E, Romero-Laorden N, Del Pozo A, Lozano R, Medina A, Puente J, Piulats JM, Lorente D, Saez MI, Morales-Barrera R, Gonzalez-Billalabeitia E, Cendón Y, García-Carbonero I, Borrega P, Mendez Vidal MJ, Montesa A, Nombela P, Fernández-Parra E, Gonzalez Del Alba A, Villa-Guzmán JC, Ibáñez K, Rodriguez-Vida A, Magraner-Pardo L, Perez-Valderrama B, Vallespín E, Gallardo E, Vazquez S, Pritchard CC, Lapunzina P, Olmos D. PROREPAIR-B: A Prospective Cohort Study of the Impact of Germline DNA Repair Mutations on the Outcomes of Patients With Metastatic Castration-Resistant Prostate Cancer. J Clin Oncol. 2019 Feb 20;37(6):490-503. [CrossRef]
- Rao A, Moka N, Hamstra DA, Ryan CJ. Co-inhibition of androgen receptor and PARP as a novel treatment paradigm in prostate cancer: where are we now? Cancer 2022;14;801:1-23. [CrossRef]
- Clarke NW, Armstrong AJ, Oya M, Shore N, Loredo E, Procopio G, Girotto G, for the Propel investigators. Abiraterone and Olaparib for metastatic castration resistant prostate cancer. NEJM Evidence,; 1 (9): 1-16. 3 June. [CrossRef]
- Carr, T.H.; Adelman, C.; Barnicle, A.; Kozarewa, I.; Luke, S.; Lai, Z.; Hollis, S.; Dougherty, B.; Harrington, E.A.; Kang, J.; et al. Homologous Recombination Repair Gene Mutation Characterization by Liquid Biopsy: A Phase II Trial of Olaparib and Abiraterone in Metastatic Castrate-Resistant Prostate Cancer. Cancers 2021, 13, 5830. [Google Scholar] [CrossRef]
- Saad, F.; Thiery-Vuillemin, A.; Wiechno, P.; Alekseev, B.; Sala, N.; Jones, R.; Kocak, I.; Chiuri, V.E.; Jassem, J.; Fléchon, A.; et al. Patient-reported outcomes with olaparib plus abiraterone versus placebo plus abiraterone for metastatic castration-resistant prostate cancer: a randomised, double-blind, phase 2 trial. Lancet Oncol. 2022, 23, 1297–1307. [Google Scholar] [CrossRef]
- Clarke N. Final overall survival in PROpel: abiraterone and Olaparib versus abiraterone and placebo as first-line therapy for metastatic castration-resistant prostate cancer. ASCO GU 2023. Available online: https://meetings.asco.org/abstracts-presentations/217650.
- Chi, K.N.; Rathkopf, D.; Smith, M.R.; Efstathiou, E.; Attard, G.; Olmos, D.; Lee, J.Y.; Small, E.J.; Gomes, A.J.P.d.S.; Roubaud, G.; et al. Niraparib and Abiraterone Acetate for Metastatic Castration-Resistant Prostate Cancer. J. Clin. Oncol. 2023, 41, 3339–3351. [Google Scholar] [CrossRef] [PubMed]
- Rathkopf, D. A Phase 3 Randomized, Placebo-controlled, Double-blind Study of Niraparib in Combination With Abiraterone Acetate and Prednisone Versus Abiraterone Acetate and Prednisone for the Treatment of Participants With Deleterious Germline or Somatic Homologous Recombination Repair (HRR) Gene-Mutated Metastatic Castration-Sensitive Prostate Cancer (mCSPC).2023. Available online: https://clinicaltrials.gov/ct2/show/NCT04497844.
- Rao A, Ryan CJ, VanderWeele DJ, Heller G, Lewis LD, Watt C, Chen RC, Grubb R, Hahn OM, Beltran H. CASPAR (Alliance A031902): A Randomized, Phase III Trial of Enzalutamide (ENZ) with Rucaparib (RUCA)/Placebo (PBO) as aNovel Therapy in First-Line Metastatic Castration-Resistant Prostate Cancer (MCRPC). J. Clin. Oncol. 2021, 39, TPS181.
- ClinicalTrials.gov. A Clinical Study Evaluating The Benefit of Adding Rucaparib to Enzalutamide for Men With Metastatic Prostate Cancer That Has Become Resistant To Testosterone-Deprivation Therapy. Available online: https://clinicaltrials.gov/ct2/show/NCT04455750.
- Agarwal, N.; Azad, A.; Shore, N.D.; Carles, J.; Fay, A.P.; Dunshee, C.; Karsh, L.I.; Paccagnella, M.L.; Di Santo, N.; Elmeliegy, M.; et al. Talazoparib plus enzalutamide in metastatic castration-resistant prostate cancer: TALAPRO-2 phase III study design. Futur. Oncol. 2022, 18, 425–436. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
The concept of anticipation and intensification of care in the systemic therapy of metastatic PC. How also PARP inhibitors and HRR genetic analysis are involved. (mCRPC= metastatic castration resistant prostate cancer; mHSPC= metastatic hormone sensitive prostate cancer; nmCRPC= non metastatic castration resistant prostate cancer; PFS= progression free survival; HR= hazard ratio; HRR= homologous recombinant repair; ABI= abiraterone; ENZA=enzalutamide; PARPi= Poly ADP ribose polymerase inhibitors).
Figure 1.
The concept of anticipation and intensification of care in the systemic therapy of metastatic PC. How also PARP inhibitors and HRR genetic analysis are involved. (mCRPC= metastatic castration resistant prostate cancer; mHSPC= metastatic hormone sensitive prostate cancer; nmCRPC= non metastatic castration resistant prostate cancer; PFS= progression free survival; HR= hazard ratio; HRR= homologous recombinant repair; ABI= abiraterone; ENZA=enzalutamide; PARPi= Poly ADP ribose polymerase inhibitors).

Table 1.
Characteristics of 5 clinical trials analyzing the combination of ARSI and PARP inhibitors in an anticipated first line mCRPC setting. (mCRPC= metastatic castration resistant prostate cancer; mHSPC= metastatic hormone sensitive prostate cancer; nmCRPC= non metastatic castration resistant prostate cancer; PFS= progression free survival; HR= hazard ratio; HRR= homologous recombinant repair; PARPi= Poly ADP ribose polymerase inhibitors).
Table 1.
Characteristics of 5 clinical trials analyzing the combination of ARSI and PARP inhibitors in an anticipated first line mCRPC setting. (mCRPC= metastatic castration resistant prostate cancer; mHSPC= metastatic hormone sensitive prostate cancer; nmCRPC= non metastatic castration resistant prostate cancer; PFS= progression free survival; HR= hazard ratio; HRR= homologous recombinant repair; PARPi= Poly ADP ribose polymerase inhibitors).
| Study | Design | Treatments | Patient Selection | Primary Endpoint | Main Results |
|---|---|---|---|---|---|
| PROPEL [44] | Phase 3, randomized, placebo-controlled, double-blinded |
Abiraterone + Olaparib (360) Abiraterone + Placebo (360) |
First line mCRPC Prior ARSI not allowed Prior docetaxel allowed for mHSPC HRR mutation not required (but analyzed as subgroup) |
Radiographic PFS | Radiological PFS 24.8 vs 16.6 months; HR 0.66; 95%CI 0.54-0.81; p<0.001, irrespectively to HRR status (HRR mutated subgroup: HR 0.50; 95%CI 0.34-0.73; HRR non-mutated subgroup: HR 0.76; 95%CI 0.60-0.97). Overall survival 42.1 vs 34.7 months; HR 0.81; 95%CI 0.67-1.00; p=0.0544, irrespectively to HRR mutation (HRR mutated subgroup: HR 0.66; 95%CI 0.45-0.95; HRR non-mutated subgroup: HR 0.89; 95%CI 0.70-1.14) |
| CASPAR [50] | Phase 3, randomized, placebo-controlled, double-blinded |
Enzalutamide + Rucaparib (496) Enzalutamide + Placebo (496) |
First line mCRPC Prior ARSI allowed For mHSPC and nmCRPC Prior docetaxel allowed for mHSPC HRR mutation not required (but analyzed as subgroups) |
Radiographic PFS and overall survival |
ongoing |
| MAGNITUDE [48] | Phase 3, randomized, placebo-controlled, double-blinded |
HRR-mutant cohort Abiraterone + Niraparib (200) Abiraterone + Placebo (200) -------------------------- HRR- no mutated cohort Abiraterone + Niraparib (300) Abiraterone + Placebo (300) |
First line mCRPC Prior ARSI not allowed Prior docetaxel allowed for mHSPC ------------------------------ First line mCRPC Prior ARSI not allowed Prior docetaxel allowed for mHSPC |
Radiographic PFS ----------------------- Radiographic PFS |
In HRR- mutant : PFS 16.5 vs 13.7 months; HR 0.73; 95%CI 0.56-0.96;p=0.022. In HRR- no mutant: HR 1.09; 95%CI 0.75-1.57;p=0.66 and the cohort was closed to further enrollment. |
| TALAPRO-2 [52] | Phase 3, randomized, placebo-controlled, double-blinded |
Enzalutamide + Talazoparib (509) Enzalutamide + Placebo (509) |
First line mCRPC Prior abiraterone allowed (no novel AR inhibitors) for mHSPC and nmCRPC Prior docetaxel allowed for mHSPC HRR mutation not required but analyzed as subgroups |
Radiographic PFS | ongoing |
| AMPLITUDE [49] | Phase 3, randomized, placebo-controlled, double-blinded |
Abiraterone+ niraparib (395) Abiraterone + Placebo (395) |
First line mCRPC with HRR pathogenetic variants Prior ARSI not allowed |
ongoing |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions, or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



