
Submitted:
02 July 2023
Posted:
03 July 2023
You are already at the latest version
Abstract
Gastrodia elata Bl., one of the well-known edible plants in China, belongs to the family of Orchidaceae. In recent years, the consumption rate of this plant as food supplements or health products is gradually rising in China. However, the traditional industrialization processing procedures (TP) for this plant was found to be too complicated and not friendly to the environment with time and energy consuming. Hence, we investigated whether the processing procedures could be simplified without affecting its overall chemical profile and anti-oxidation activity. In this experiment, a new processing procedure (NP) was designed for processing this plant, and the chemical compositions of the extract for Gastrodia elata produced by two different processing procedures were identified by UPLC-Q-TOF/MS. It was found that phenols and parisins were the most abundant chemical components for this extract. Among all the identified components, the contents of gastrodin and its citric acid derivatives were obviously higher than that of other components. Therefore, the contents of six gastrodin derivatives and ethanol soluble extract rate for Gastrodia elata produced by two different processing procedures, traditional and newly designed processing procedures, were further comparatively investigated for the validation of the applicability. The DPPH radical scavenging activities of the extract from Gastrodia elata produced by two different processing procedures were also investigated. The results demonstrated that the contents of gastrodin and p-hydroxybenzyl alcohol in all samples were slightly decreased in NP samples compared with that of TP samples (except batches of S3 and S6 samples), but no significant difference was observed between these two groups (p = 0.8273 for gastrodin and p= 0.2320 for p-hydroxybenzyl alcohol). Additionally, the total contents of parisins were also in a decreased trend in NP samples but with no significant difference when comparing with that in TP samples (p = 0.1721). The results for ethanol soluble extract rate also had the same situation (p = 0.1094). Taken together, it was suggested that the chemical profiles and contents of gastrodin derivatives and ethanol soluble extract rate for NP samples was not significant different to that of TP samples. The DPPH radical scavenging activity of the extract of Gastrodia elata produced with NP method was not significant different from that of TP samples. As a result, the industrial processing procedures for the preservation of this material plant could be simplified to a more time and energy saving procedure without changing the quality of the products, which is also consistent with the concept of green and sustainable industrial production.
Keywords:
Gastrodia elata
; industrial production
; gastrodin
; parishin
; edible plants
; processing procedure
1. Introduction
In China, Gastrodia elata Bl. (G. elata) is a famous edible plant that is often consumed as a food supplement for daily health care. To date, the consumption of G. elata as a food supplement is gradually increasing in China, and G. elata has been involved in approximately 140 kinds of health food products in China produced by several manufacturers. It is also commonly used in other Asian countries, and it serves as a functional food for the improvement of sexual potency and vision to prevent abortion. In China, this edible plant is often consumed as a functional food for the improvement of hypertension, antioxidation, regulation of the immune system and alleviation of nervous system discomfort. Certain cosmetic products produced in China also use the extract of G. elata tubers as the active ingredient.
To date, more than 81 compounds from G. elata have been isolated and identified, including phenolics, polysaccharides, organic acids, sterols and organic acids [1]. Gastrodin (GA) and its derivatives are commonly considered bioactive ingredients in Gastrodia elata [2]. Among them, GA and p-hydroxybenzyl alcohol (HA) have potent neuroprotective effects, such as hypnotic sedation, anticonvulsant, antidizziness, and memory improvement [3]. The parishins formed by the condensation of GA and citric acid are also considered as important bioactive ingredients in G. elata [4,5,6] since they can be degraded into GA in vivo [7,8,9]. Polysaccharides from G. elata have also been reported to be active against dengue virus, inhibit GABA transaminase and angiotensin-I converting enzyme (ACE) activities and promote angiogenesis [10,11,12,13,14].
Approximately 5 variants are mainly considered Chinese domestic G. elata, and they are identified as G. elata f. elata (Hong Tianma in Chinese), G. elata f. glauca (Wu Tianma in Chinese), G. elata f. viridis (Lv Tianma in Chinese), G. elata f. flavid (Huang Tianma in Chinese) and G. elata f. alba (Song Tianma in Chinese) by the characteristics and shapes of their tuber, flower stem and flower color [15]. Among them, the seeds of G. elata f. elata (Hong Tianma in Chinese) carry the characteristics of a high germination rate and yield, strong adaptability and cold resistance[15]. For G. elata f. glauca (Wu Tianma in Chinese), the tubers of this strain are low in water content and good quality for dry products. In China, both variants and their hybrid variants were selected as novel strains for wide cultivation [16]. Different strains and origins of G. elata have a great impact on the quality of medicinal materials and therefore on their bioactivities. Reports demonstrate that the anticonvulsant and neuroprotective effects of G. elata derived from different origins or strains are significantly different [17], and hence, samples of the representative variants of G. elata, including G. elata f. glauca (Wu Tianma in Chinese), G. elata f. elata (Hong Tianma in Chinese) and hybrid variants of G. elata f. elata and G. elata f. glauca (breeding by our group), planted in different regions were selected for investigation in this experiment.
Processing is one of the key procedures for the preservation of freshly collected G. elata tubers either for edible or medicinal applications. In traditional processing procedures, newly harvested G. elata tubers are often processed by several steps, including steaming for termination, drying for steamed tubers, wetting and softening for dried tubers, slicing into pieces and drying, and then being used for edible or medicinal use. We think that the processing procedures for G. elata are too complex to be energy efficient and environmentally friendly; hence, it is hypothesized that another simple and alternative processing procedure for G. elata could be designed and compared with the traditional processing procedure in this study. To fully understand the chemical profiles of the extract of G. elata, LC-Q-TOF/MS was used to identify as many chemical constituents as possible before the experiments. Then, the content of the chemical constituents with the most abundance in G. elata processed with different procedures was evaluated. The quality evaluation standard for processed G. elata was briefly summarized as follows: the content of GA and HA was not less than 0.25%, and the alcohol soluble extract rate was not less than 15%. In this work, the chemical information of the extract of G. elata was fully isolated and identified by LC-Q-TOF/MS, and the contents of GA and HA as well as the alcohol soluble extract rate were calculated and compared in G. elata tubers processed by different process procedures. Taking the potential activities of other major GA derivatives into consideration, the content of parisonsides was also comparatively studied.
2. Material and Methods
2.1. Materials and Chemicals
GA (lot No. MUST-13101011, purity 99.99%) was purchased from Chengdu Must Biotechnology Co., Ltd. HA (lot No. BCBT1560, purity 99%) was purchased from Sigma‒Aldrich Corporation. Parishin E (PE, lot No. HS20901B1, purity 98%), Parishin B (PB, lot No. HR2098B1, purity 98%), Parishin C (PC, lot No. HS20905B1, purity 98%) and Parishin (PA, lot No. HS20904B3, purity 98%) were purchased from Baoji Herbest Bio-Tech Co., Ltd. DDPH (1,1-Diphenyl-2-picrylhydrazyl radical) scavenging activity test kits (A153-1-1) were purchased from Nanjing Jiancheng Bioengineering Institute (Nanjing, China). Acetonitrile (HPLC grade) was supplied by TEDIA Company. Formic acid (HPLC grade) was purchased from Aladdin Reagent Company. The ultrapure water used in this study was prepared by a Milli-Q Integral-3 Ultrapure Water System (Darmstadt, Germany). The medicinal material samples of Gastrodia elata were mainly collected from Sichuan, Shanxi, Gansu, Chongqing and Yunnan provinces from November 2020 to March 2021. All the samples were identified as the tubers of G. elata by Professor Hao Zhang from Sichuan University. A voucher specimen Tianma-20102103 is now planted at the laboratory center of the School of Preclinical Medicine of Chengdu University. Other chemicals used in this study were both AR grade.
2.2. Processing Procedures for Material Samples of G. elata
All the collected fresh medicinal material samples of G. elata were screened by their sizes and subjected to steam for termination for 60 min after being well washed. The information for the sample collection is listed in Table 1. For samples processed in the traditional process manner (TP), the steamed tubers of G. elata were dried at 65 °C and sealed for storage. These samples were wetted and softened again with water, sliced into pieces approximately 4 mm thick and dried before edible or medicinal use. For samples processed in the newly designed procedures referred to in this article (NP), the steamed tubers of G. elata were directly sliced into pieces approximately 4 mm thick, which were subjected to drying at 65 °C for approximately 12 h, and these samples were directly used for edible or medicinal use. The scheme of processing procedures for the material samples of G. elata is presented in Figure 1A. Each sample was prepared in triplicate.
2.3. UHPLC-Q-TOF/MS Analysis
UHPLC-Q-TOF/MS analysis was performed on an Agilent 1290 Infinity LC system (Santa Clara, CA) using a Waters ACQUITY UPLC® BEH C18 analytical column (2.1 × 100 mm, 1.7 μm; Milford, MA, USA). Mass spectrometry was conducted on an Agilent 6545 UHD accurate-mass Q-TOF/MS system with a dual jet stream electrospray ion source. Mobile phases A and B were 0.1% (v/v) formic acid in water and acetonitrile, respectively. The optimized elution progress at a flow rate of 0.25 mL/min was set as follows: 0-0.5 min, 2% B; 0.5-3 min, 2-3.5% B; 3-10 min, 3.5-9% B; 10-16 min, 9-20% B; 16-18 min, 20-50% B; 18-20 min, 50-70% B; 20-24 min, 70-95% B; 24-26.9 min, 95% B; 26.9-27 min, 95-2% B with a post time of 2 min. The injection volume was 5 μL, and the column temperature was maintained at 35 °C. The electrospray ionization (ESI) source was operated in negative ion mode with the following parameters: drying gas temperature of 325 °C flowing at 11 L/min, sheath gas temperature of 350 °C flowing at 11 L/min, capillary voltage of 4000 V, nozzle voltage of 1500 V, fragmentary voltage of 175 V, nebulizer pressure of 35 psig, etc. The mass scan range was acquired from m/z 50 to m/z 1500. Accurate mass measurements were calibrated by a low flow of TOF reference mixture containing the internal reference C18H18F24N3O6P3 (m/z 922.0098). Automated MS/MS acquisition was applied at collision energies of 10, 20, 30, 40, 50 and 60 eV for comprehensive collection of fragments.
2.4. UHPLC Chromatographic Conditions
UPLC analysis was performed on an ACQUITY UPLC I-Class PLUS system containing a high-pressure binary pump, automatic sampler, column temperature control chamber and PDA detector (Waters Corporation, MA, USA) with a Waters BEH C18 analytical column (50*2.1 mm, 1.7 μm; Waters Corporation, MA, USA), which was maintained at 35 °C. The injected volume for each sample was 0.3 μL. The mobile phase was composed of water (containing 0.1% formic acid, v/v) (A) and acetonitrile (B), and the flow rate was set at 0.4 mL/min. Data acquisition was performed using an Empower workstation. The analytes were eluted by a gradient program as follows: 0~1 min, 3% B; 1~5 min, 3~20% B; 5~6 min, 20~90% B. The detection wavelength was set as 220 nm.
2.5. Preparation of Stock Solution and Calibration Curve
Stock solutions of GA, HA, PE, PB, PC and PA at a concentration of 1.0 mg/mL were prepared by dissolving the accurately weighed substances in 50% methanol separately. The standard mixture working solutions containing all the analytes were prepared by dilution of the stock solutions of GA, HA, PE, PB, PC and PA with 50% methanol. The concentrations of GA, HA, PE, PB, PC and PA in the working solutions were in the ranges of 3.91-250, 3.10-250, 31.3-1000, 31.3-1000, 7.81-250 and 39.1-1250 μg/mL, respectively. The stock and working solutions were all stored at 4 °C before use.
2.6. Extraction and Preparation of Samples
All the processed G. elata samples produced in a traditional or optimized manner were crushed and passed through a 60 mesh screen. Approximately 1.0 g powder of G. elata processed samples was accurately weighed and ultrasonically extracted (SB25-12 DTD ultrasonic cleaner, Ningbo Scinentz Biotechnology CO., Ltd, Ningbo, China). The parameters were set as follows: power 180 W, frequency: 40 kHz) by 20 mL 50% methanol in a stoppered conical flask for 30 min. Then, an additional accurate volume of 50% methanol was added to supplement the lost weight of solution after keeping at room temperature for 30 min. After well shaking, the solution was filtered through a 0.22 μm membrane, and the subsequent filtration was collected for analysis.
2.7. Linearity and Sample Quantification
The linearity for sample quantification was prepared by analysis of a series of different concentrations of mixed reference solutions diluted with 50% methanol. The standard calibration curves for different references were achieved by taking the sample concentration as the independent variable and the corresponding peak area as the dependent variable. The value of signal-to-noise (S/N) ~10 was considered the lowest limit of quantification (LLOQ) for the references.
2.8. Precision and Accuracy
The precision was assessed by analysis of GA, HA, PE, PB, PC and PA at concentrations of 18.75, 18.75, 75.00, 75, 18.75 and 150.00 μg/mL with six replicates respectively, and the RSD values of the corresponding peak area for each compound were calculated. The method for preparing samples for the accuracy test was summarized as follows: approximately 0.5 g powder of G. elata sample (Batch S5 TP) was accurately weighed into a flask, and the equivalent to 100% of the known content for each compound was added, which was subsequently subjected to ultrasonic extraction by 50% methanol for 30 min. Then, an additional accurate volume of 50% methanol was added to supplement the lost weight of solution after keeping at room temperature for 30 min. After well shaking, the solution was filtered through a 0.22 μm membrane, and the subsequent filtration was collected for analysis. The accuracy was confirmed by detection of the relative recovery of the added content.
2.9. Reproducibility Test
The method for preparing samples for the reproducibility test was summarized as follows: approximately 1.0000 g powder of G. elata sample (Batch S5 TP) with 6 replicates was accurately weighed into a flask and ultrasonically extracted (SB25-12 DTD ultrasonic cleaner, power 180 W, frequency: 40 kHz) by 20 mL 50% methanol in a stoppered conical flask for 30 min. Then, an additional accurate volume of 50% methanol was added to supplement the lost weight of the solution after being kept at room temperature for 30 min. After well shaking, the solution was filtered through a 0.22 μm membrane, and the subsequent filtration was collected for analysis. All the prepared samples were subsequently subjected to analysis. The average contents of GA, HA, PE, PB, PC and PA were calculated. The reproducibility was confirmed by calculating the RSD of the content of each compound.
2.10. Recovery Test
Approximately 0.5000 g powder of G. elata sample (Batch S5 TP) with six replicates was accurately weighed into a flask, and then a known amount of mixed standard solution (equal 100% to the original content) was accurately added and subsequently subjected to ultrasonic extraction, which followed the sample preparation procedures described in the ‘Extraction and preparation of samples’ section. The recoveries for the analytes were assessed by the following formula: Recovery (%) = (detected content-original content/spiked content) *100%.
2.11. Stability Test of Samples
The stability test for samples was performed by detection of the peak area of GA, HA, PE, PB, PC and PA in samples of Batch No. 5 TP at 0, 1, 2, 4, 6 and 8 h after being prepared, and the RSD values of peak areas for each compound were calculated.
2.12. Data Mining Strategy
The method for analysis of the data from UPLC-Q-TOF/MS was mainly described in the references [18,19]. For the processing of data acquired with LC‒MS, the combination of MS-DIAL and MS-FINDER was used. The raw data (.D) from UHPLC‒MS were first converted into.abf file format using Analysis Base File Converter. The parameters of data collection were 0.01 Da for MS1 tolerance and 0.025 Da for MS2 tolerance. Peak detection was applied with 1000 amplitude for minimum peak height and 0.1 Da for the width of the mass slice. The data deconvolution was performed using MS-DIAL software (ver. 4.7), and the corresponding parameters included a sigma window value of 0.1, MS/MS abundance cutoff of five amplitudes, and retention time tolerance of 0.1 min. The molecular spectra network was established using MS/MS similarity, ontology similarity, and a formula-based bioreaction program MS-DIAL. The formula finder was exclusively processed with C, H, O, and N atoms. The potential candidates for each constituent were obtained by consulting the databases (MassBank, GNPS, KNApSAcK, PubChem, COCONUT, UNPD) in MS-FINDER and then ranked according to their similarity scores, which were calculated based on the comparison between the experimental MS/MS spectra and those in the databases. The top-ranked candidates were usually selected; however, if the key fragments could not be explained well or the scores were the same, the most likely structure among these candidates was manually screened.
2.13. Ethanol-Soluble Extract Determination
The determination of ethanol-soluble extract for collected samples was recorded in the Chinese Pharmacopeia Year 2020 version (General rule 2201). The hot ethanol extract method was used for determination of ethanol-soluble extracts for collected G. elata samples. In detail, approximately 2.0000 g powder of G. elata sample was accurately weighed and extracted with 100 mL hot ethanol for 1 h. Then, an additional accurate volume of ethanol was added to supplement the lost weight of solution after keeping at room temperature for 30 min. Subsequently, the solution was well mixed and filtered, and 25 mL filtration was evaporated in a water bath and dried to constant weight at 105 °C for 3 h. Then, the amount of the extract was quickly and accurately weighed after being cooled in a desiccator for 30 minutes. The content (%) of ethanol-soluble extract for the sample was calculated based on the dry product.
2.14. Determination of The Content of Constituents in Samples
The sample solutions were injected into the UPLC system for analysis, and the contents of GA, HA, PE, PB, PC and PA in the samples were calculated by the corresponding calibration curves.
2.15. DPPH Radical Scavenging Activity Test
Approximately 0.5 g powder of G. elata processed samples were accurately weighed and ultrasonically extracted (SB25-12 DTD ultrasonic cleaner, Ningbo Scinentz Biotechnology CO., Ltd, Ningbo, China). The parameters were set as follows: power 180 W, frequency: 40 kHz) by 20 mL 50% methanol in a stoppered conical flask for 30 min. Then, an additional accurate volume of 50% methanol was added to supplement the lost weight of solution after keeping at room temperature for 30 min. Subsequently, the solutions were centrifuged at 3500 rpm, and 400 μL of solution was subjected to the DPPH radical scavenging activity assay according to the manufacturer’s instructions. The DPPH radical scavenging activity was expressed as μg Trolox/mL.
2.16. Data Analysis
Statistical analysis and data representation were performed with SPSS 13.0 software (SPSS Inc., Chicago, IL, USA). Data are given as the mean values ± standard deviations (SD) unless otherwise indicated. Paired Student’s t test or the Wilcoxon matched pairs test was used for comparison of groups, and a p value of less than 0.05 represented a significant difference.
3. Results and Discussion
3.1. UHPLC-Q-TOF/MS Analysis
To better understand the chemical profiles of the extract from G. elata, UHPLC-Q-TOF/MS was used for identification of the chemical constituents, and the combination of MS-DIAL and MS-FINDER was used for analysis of the chemical structures of the detected components. A total of 237 compounds were found in the extract, and among all the isolates, phenols and citric acid derivatives of GA accounted for the highest proportion. The representative and typical fragment ions of GA and its citric acid derivatives are presented in Figure 2. As shown in Figure 2A, the citric acid derivatives of GA accounted for the most abundance among all the isolates. For Parishin G and E (C19H24O13, MW: 460.1217), in negative ion mode, the common fragment ions for these two compounds were m/z=173.0101 and 111.009. However, the relative abundances of the ions at m/z=173.0101 and 111.009 were significantly different. Additionally, for Parishin E, the ions of m/z=129.0201 and 87.0097 were also found in the MS/MS data. The compounds of citrate (gastroxyl/0/4-HB), Parishin B and C share the same MW of 728.2164, and are both composed by one molecule of citric acid and two molecules of GA. The relative abundance of the fragment ion of m/z 441.1047 for citrate (gastroxyl/0/4-HB) was significantly higher than that of the other two compounds. However, the fragment ions for Parishin B and C share almost the same MS profile. For Parishin A, composed of one molecule of citric acid and three molecules of GA, the fragment ion of m/z=727.2114 accounted for the most abundance. In our experiments, the analog of GA, isogastrodin, was also detected and identified in the extract. The different linkage sites for glucose and the hydroxyl group on the benzene ring resulted in different fragment ions for these two compounds. As shown in Figure 2B, fragment ions of m/z=179.0626 and 161.0094 were detected for isogastrodin and GA, respectively. It was also found that the fragment ion of m/z=123.0454 accounted for the highest abundance among all the ions for GA, which was quite different from that of isogastrodin.
3.2. Linearity and LLOQ Results
The standard calibration curves were constructed within the ranges 3.91-250.00 μg/mL for GA, 3.10~250.00 μg/mL for HA, 31.30~1000.00 μg/mL for PE, 31.30~1000.00 μg/mL for PB, 7.81~250.00 μg/mL for PC, and 39.10~1250.00 μg/mL for PA. Typical regression equations for GA, HA, PE, PB, PC and PA were y = 1763 x + 1922 (r = 0.9999), y = 2937x + 2070 (r = 0.9999), y = 991x + 2399 (r = 0.9999), y = 1276 x + 10722 (r = 0.9999), y = 1071x + 937 (r = 0.9999) and y = 1256x + 1922 (r = 0.9999), respectively (where x is the concentration of the solution and y is the corresponding peak area). The LLOQ values were 1.96 μg/mL for GA, 1.55 μg/mL for HA, and 3.13, 3.13, 3.90 and 3.90 μg/mL for PE, PB, PC and PA, respectively.
3.3. Precision and Accuracy Test Results
The six compounds were evaluated for precision and accuracy in three daily runs. The intraday precision (RSD) values ranged from 1.01 to 1.12% for GA, 1.07 to 1.18% for HA, 1.14 to 1.45% for PE, 1.17 to 1.29% for PB, 0.97 to 1.13% for PC and 0.92 to 0.99% for PA, and the interday precision (RSD) values ranged from 0.86 to 1.23% for GA, 1.35 to 1.56% for HA, 1.64 to 2.18% for PE, 1.28 to 1.72% for PB, 1.48 to 1.98% for PC and 1.39 to 2.13% for PA. The RSD conformed to satisfy the criteria.
3.4. Reproducibility Test Results
The RSD values for the content of the compounds in samples were 1.20% for GA, 1.24% for HA, 1.82% for PE, 1.56% for PB, 1.65% for PC, and 1.67% for PA. The results were confirmed to have good reproducibility and satisfied the criteria for analysis.
3.5. Recovery Test Results
The average recoveries for GA, HA, PE, PB, PC and PA were 95.90 ± 2.04%, 97.70 ± 1.34%, 99.50 ± 1.21%, 94.30 ± 0.93%, 104.00 ± 1.16% and 93.8 ± 2.4%, respectively. The results were confirmed to satisfy the criteria for analysis.
3.6. Stability Test Results
The peak areas for each compound in the samples were calculated for the stability test. The RSD values for GA, HA, PE, PB, PC and PA were 1.01%, 0.48%, 0.92%, 0.66%, 1.06% and 0.61%, respectively. The results demonstrated that the tested compounds in the samples were all stable for 8 h.
3.7. Determination of The Contents of GA, HA, PE, PB, PC and PA in TP and NP Samples
Through systematic evaluation of the analysis method for the determination of GA, HA, PE, PB, PC and PA in samples, the contents of these constituents in samples processed by different procedures (TP and NP) were tested under chromatographic conditions. As shown in Figure 2A, all six compounds were well separated on the column. The retention times for GA, HA, PE, PB, PC and PA were 0.79 min, 1.38 min, 2.72 min, 3.83 min, 4.10 min and 4.71 min, respectively. To further identify the compounds in the processed samples, the spectral profiles of the peaks from processed samples were obtained and compared with those of GA, HA, PE, PB, PC and PA standards. As shown in Figure 2B, all the analytes shared the same spectral profiles with the corresponding standards. The maximum absorption wavelength (λmax) of all the analytes ranged from 221.4~222.6 nm. Hence, the detection wavelength for the analytes in the following experiments was set as 220 nm. Additionally, the second absorption wavelengths for PE, PB, PC and PA were all approximately 269 nm since they share almost the same chemical structure.
Seven batches of G. elata collected from different regions were subjected to processing by TP and NP. Quantitative analysis of the contents of GA, HA, PE, PB, PC and PA in all the processed samples was subsequently performed. As shown in Figure 3, the results demonstrated that the content of GA in the NP and TP samples ranged from 0.164~0.424% and 0.074~0.479%, respectively. The content of HA in the NP and TP samples ranged from 0.126~0.257% and 0.009~0.267%, respectively. Paired t test difference analysis was performed to comparatively investigate the contents of GA and HA in the S1 ~ S7 NP and TP samples. The contents of GA and HA in the S1 ~ S7 NP samples were not significantly different from those in the TP groups. The p values for GA and HA were 0.827 and 0.232, respectively.
The quality control for the processed product of this plant recorded in Chinese pharmacopeia is specified as the total contents of GA and HA should be no less than 0.25%. Subsequently, the total contents of GA and HA in the NP and TP samples were calculated. As shown in Figure 3, the total contents of GA and HA in the NP and TP samples ranged from 0.290~0.629% and 0.083~0.715%, respectively. The total contents of GA and HA in all NP samples were all more than 0.25%. However, the results were quite different from those of the TP samples. The total contents of GA and HA in the S2, S5 and S6 TP samples were less than 0.25%, especially for the S6 TP sample, and the total content of these two constituents decreased by approximately 80% compared with that of the S6 NP sample. Paired t test difference analysis was performed to comparatively investigate the total content of these two constituents in the samples, and the results showed that the total contents of GA and HA in the S1 ~ S7 NP samples were not significantly different from those in the TP groups, with a p value of 0.639.
As derivatives of GA, parishins, including PE, PB, PC and PA, are rich in the tubers of G. elata, and these constituents are also considered potent bioactive constituents. In this experiment, the contents of these derivatives were simultaneously detected, and the results are shown in Figure 3. The contents of PE, PB, PC and PA in NP samples ranged from 0.518~1.199%, 0.540~0.791%, 0.121% ~ 0.167% and 0.799% ~ 1.966%, respectively, and the contents of PE, PB, PC and PA in TP samples ranged from 0.509~1.242%, 0.499~1.049%, 0.121~0.226% and 0.719~2.773%, respectively. Among all the parishins, PA was one of the most abundant constituents in the processed samples. The total contents of the parishins are shown in Figure 3, and the total contents of parishins in the TP and NP samples were in the ranges of 2.978~4.420% and 1.992~5.826%, respectively. Statistical analysis for the total content of parishins in TP and NP samples demonstrated no significant difference between these two groups with a p value of 0.297. Taken together, the contents of the detected constituents in NP samples were not significantly different from those in TP samples.
3.8. Ethanol Soluble Extract Rate Determination Results
In this experiment, the extraction rate for ethanol-soluble ingredients was also detected for NP and TP samples according to the rules in Chinese pharmacopeia. The extraction rate for ethanol-soluble ingredients for the processed G. elata samples should be less than 15%. The results are shown in Figure 3C. The extraction rate for ethanol-soluble ingredients for all seven batches of processed samples met the regulations. It was also found that the extraction rate for ethanol-soluble ingredients for NP samples was higher than that for TP samples except for the S3 sample. The extraction rates for ethanol-soluble ingredients for NP and TP samples were in the range of 18.80~31.60% and 19.20~30.90%, respectively. Statistical analysis results demonstrated that the extraction rate for ethanol-soluble ingredients for NP and TP samples was also not significantly different between these two groups, with a p value of 0.076. It was suggested that the extraction rate for ethanol-soluble ingredients for NP samples showed an increasing trend (S3 was excluded) but with no significant difference from that for TP samples.
3.9. DPPH Radical Scavenging Activity Test Results
To further investigate the antioxidation activity of the extract of G. elata processed by different procedures, the DPPH radical scavenging activity test was performed. As shown in Figure 4A, the results demonstrated that the antioxidation activity of the extract of G. elata samples from the NP group was not significant compared to that of the TP group, with a p value of 0.469. The antioxidation activity of G. elata processed by NP processing procedures was not significantly changed. In this experiment, it was also found that the DPPH radical scavenging activity of some batches of samples from the NP group was upregulated compared with the corresponding samples from the TP group, such as S1, S3, S4 and S5. To further ensure whether the DPPH radical scavenging activity of the extract of G. elata was correlated to the contents of the gastrodin derivatives, the correlations between the content of parsins, GA and HA, as well as all six derivatives, and DPPH radical scavenging activity were statistically analyzed. All the investigated indices were weakly correlated with the DPPH radical scavenging activity (shown in Figure 4B).
Processing is one of the key procedures for the storage and preservation of freshly collected G. elata tubers either for edible or medicinal applications. In the process of industrial production of G. elata products, we find that the traditional procedures for processing G. elata tubers are too complicated with many repeated processes. Hence, in this experiment, we designed a new processing procedure for processing G. elata tubers; that is, the steamed tubers are directly sliced into pieces and subsequently dried for storage (NP) (as shown in Figure 1). Quantitative analysis of the gastrodin derivatives in processed samples (both TP and NP samples) was performed to comparatively investigate the effects of processing procedures on the quality of the products. The results demonstrated that the content of gastrodin derivatives in the NP samples was not significantly different from that in the TP samples (p>0.05). By comprehensively observing all the contents of gastrodin derivatives in NP samples, it was found that the contents of the detected ingredients decreased but were not significantly different from those in TP samples. We deduced that the juice of the steamed tubers, which was directly sliced into pieces and subsequently dried for storage (NP samples), might be lost during the processing procedures and lead to the decreased content of the gastrodin derivatives in NP samples. It was also suggested that the processing procedures for NP samples might be improved by reducing the loss of juice in steamed tubers in the process of cutting into slices and drying. The quality of NP samples in terms of the content of gastrodin derivatives produced by the present newly designed processing procedures was not significantly different from that of TP samples, and they all met the standard of G. elata recorded in the Chinese Pharmacopeia. The extraction ratio of ethanol-soluble extract for NP samples was also not significantly changed, and the extraction ratio for the ethanol-soluble ingredients showed an increasing trend (except for the S3 sample).
Apart from the above findings, the newly designed processing procedures were also found to be potent with some advantages. From the viewpoint of energy savings, environmental friendliness and simplifying processing procedures, the newly designed procedures with optimization for freshly collected G. elata tubers in this experiment might be recommended for industrial production. First, the drying time and cycle for products were largely shortened for the newly designed procedures compared with those of the traditional processing procedures, and hence, much energy was saved. Additionally, following the traditional processing procedures, G. elata tubers will go through drying, wetting for soft, slicing into pieces and drying before edible or medicinal application. Taking these factors together, we believe the newly designed procedures with optimization for freshly collected G. elata tubers in this experiment might be further promoted and applied in the industrial production of this plant.
In this experiment, the contents of gastrodin derivatives and ethanol-soluble extracts in G. elata processed by different procedures were mainly investigated. However, apart from the investigated constituents, other bioactive ingredients in the processed samples, such as polysaccharides, sterols and organic acids, were not studied [10,12,20]. In future work, more indices will be included for the quality assessment of the processed products produced by these new processing procedures.
4. Conclusions
Briefly, UPLC-Q-TOF/MS analysis demonstrated that GA and its citric acid derivatives were the most abundant in the extract of G. elata in the NP and TP samples, and the contents of gastrodin derivatives, ethanol soluble extract rate and antioxidation activity of the NP samples were not significantly different from those of the TP samples. It was suggested that the processing procedures for this material plant might be simplified to a more time- and energy-efficient procedure, which is also consistent with the concept of green industry for industrialization research and development. The newly designed procedures for freshly collected G. elata tubers in this experiment might be further optimized for promotion and application in the industrial production of this plant.
Author Contributions
Anqi Wang, Ying Liu and Leilei Du designed the experiments. Ying Liu and Leilei Du performed the experiments, collected and analyzed the data. Anqi Wang and Ying Liu prepared tables and figures. Anqi Wang and Ying Liu drafted the manuscript. Anqi Wang, Ying Liu, Wei Li, Chu Chen, Dingtao Wu, Liang Zou, Jiaqian Chen, Chi Teng Vong and Leilei Du critically revised the manuscript. All authors read and approved the final version of the manuscript.
Funding
This work was financially supported by Sichuan Science and Technology Program (2021YFH0169, 2022YFS0581), Sichuan Science and Technology Plan Key Project (2020YFN0152) and Key R&D Program in Sichuan Province (2021YFS0042).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data will be made available on request.
Conflicts of Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Ye, X.; Wang, Y.; Zhao, J.; Wang, M.; Avula, B.; Peng, Q.; Ouyang, H.; Lingyun, Z.; Zhang, J.; Khan, I.A. Identification and Characterization of Key Chemical Constituents in Processed Gastrodia elata Using UHPLC-MS/MS and Chemometric Methods. J. Anal. Methods Chem. 2019, 2019, 1–10, . [CrossRef]
- Liu, Y.; Gao, J.; Peng, M.; Meng, H.; Ma, H.; Cai, P.; Xu, Y.; Zhao, Q.; Si, G. A Review on Central Nervous System Effects of Gastrodin. Front. Pharmacol. 2018, 9, 24, . [CrossRef]
- Xiao, H., Jiang, Q., Qiu, H., Wu, K., Ma, X., Yang, J., and Cheng, O. Gastrodin promotes hippocampal neurogenesis via PDE9-cGMP-PKG pathway in mice following cerebral ischemia. Neurochemistry international 2021,150, Article ID 105171. [CrossRef]
- Zhao, X.; Zhou, S.; Yan, R.; Gong, C.; Gui, Q.; Zhang, Q.; Xiang, L.; Chen, L.; Wang, P.; Li, S.; et al. Parishin From Gastrodia Elata Ameliorates Aging Phenotype in Mice in a Gut Microbiota-Related Manner. Front. Microbiol. 2022, 13, 877099, . [CrossRef]
- Wang, T.; Chen, /.H.; Xia, /.S.; Chen, X.; Sun, H.; Xu, Z. Ameliorative Effect of Parishin C Against Cerebral Ischemia-Induced Brain Tissue Injury by Reducing Oxidative Stress and Inflammatory Responses in Rat Model. Neuropsychiatr. Dis. Treat. 2021, ume 17, 1811–1823, . [CrossRef]
- Lin, Y. F., Sun, Y. J., Weng, Y. F., Matsuura, A., Xiang, L., and Qi, J. H. Parishin from Gastrodia elata Extends the Lifespan of Yeast via Regulation of Sir2/Uth1/TOR Signaling Pathway. Oxid. Med. Cell. Longev. 2016,2016, Article ID 4074690. [CrossRef]
- Tang, C.; Wang, L.; Liu, X.; Cheng, M.; Xiao, H. Pharmacokinetic study of Gastrodia elata in rats. Anal. Bioanal. Chem. 2015, 407, 8903–8910, . [CrossRef]
- Dong, J.; Ji, D.; Su, L.; Zhang, F.; Tong, H.; Mao, C.; Lu, T. A simplified LC−MS/MS approach for simultaneous quantification and pharmacokinetics of five compounds in rats following oral administration of Gastrodia elata extract. J. Anal. Sci. Technol. 2020, 11, 1–9, . [CrossRef]
- He, J., Li, X., Yang, S., Li, Y., Lin, X., Xiu, M., Li, X., and Liu, Y. Gastrodin extends the lifespan and protects against neurodegeneration in the Drosophila PINK1 model of Parkinson's disease. Food Funct. 2021,12, 7816-7824.
- Zhu, Z.-Y.; Chen, C.-J.; Sun, H.-Q.; Chen, L.-J. Structural characterisation and ACE-inhibitory activities of polysaccharide from Gastrodia elata Blume. Nat. Prod. Res. 2019, 33, 1721–1726, . [CrossRef]
- Xie, H.; Chen, Y.; Wu, W.; Feng, X.; Du, K. Gastrodia elata Blume Polysaccharides Attenuate Vincristine-Evoked Neuropathic Pain through the Inhibition of Neuroinflammation. Mediat. Inflamm. 2021, 2021, 1–10, . [CrossRef]
- Liu, Y.; Huang, G. The Chemical Composition, Pharmacological Effects, Clinical Applications and Market Analysis of Gastrodia Elata. Pharm. Chem. J. 2017, 51, 211–215, . [CrossRef]
- Lee, O.-H.; Kim, K.-I.; Han, C.-K.; Kim, Y.-C.; Hong, H.-D. Effects of Acidic Polysaccharides from Gastrodia Rhizome on Systolic Blood Pressure and Serum Lipid Concentrations in Spontaneously Hypertensive Rats Fed a High-Fat Diet. Int. J. Mol. Sci. 2012, 13, 698–709, . [CrossRef]
- Ji, N.; Liu, P.; Zhang, N.; Yang, S.; Zhang, M. Comparison on Bioactivities and Characteristics of Polysaccharides From Four Varieties of Gastrodia elata Blume. Front. Chem. 2022, 10, 956724, . [CrossRef]
- Hu, Q., Wang, Y., XU, X., Li, M., and Wang, T. Progress on cultivation technique and variety of Gastrodia elata BL. Chin. Pharm. J. 2021,56, 868-894.
- Wang, W., Wen, H., Zhang, D., Wang, Q., Peng, C., and Gao, J. Quality comparison of different types of Gastrodia elata Bl. Food Ind. 2018,39, 305-307.
- Huang, Y., Tang, J., Tian, X., Lu, L., Pan, M., Chai, Y., Xie, Y., Ma, S., Liu, C., Niu, J., and Zhang, L. Study on the anticonvulsant and neuroprotective effects of Gastrodia elata freeze-dried powder from five different strains or origins. Lishizhen Med. Mater. Med. Res. 2021,32, 1996-1999.
- Wang, X.; Li, N.; Chen, S.; Ge, Y.-H.; Xiao, Y.; Zhao, M.; Wu, J.-L. MS-FINDER Assisted in Understanding the Profile of Flavonoids in Temporal Dimension during the Fermentation of Pu-erh Tea. J. Agric. Food Chem. 2022, 70, 7085–7094, . [CrossRef]
- Miao, W.; Liu, X.; Li, N.; Bian, X.; Zhao, Y.; He, J.; Zhou, T.; Wu, J.-L. Polarity-extended composition profiling via LC-MS-based metabolomics approaches: A key to functional investigation of Citrus aurantium L. Food Chem. 2023, 405, 134988, . [CrossRef]
- Zhan, H.-D.; Zhou, H.-Y.; Sui, Y.-P.; Du, X.-L.; Wang, W.-H.; Dai, L.; Sui, F.; Huo, H.-R.; Jiang, T.-L. The rhizome of Gastrodia elata Blume – An ethnopharmacological review. J. Ethnopharmacol. 2016, 189, 361–385, . [CrossRef]
Figure 1.
Flow chart of investigation on the chemical profile and anti-oxidation activity of the extract of G. elata tubers produced by new optimized and traditional processing procedures.
Figure 1.
Flow chart of investigation on the chemical profile and anti-oxidation activity of the extract of G. elata tubers produced by new optimized and traditional processing procedures.
Figure 2.
The representative MS/MS data of gastordin and its citric acid derivatives identified in the extract of G. elata tubers produced by new optimized and traditional processing procedures. A, the representative MS/MS data of citric acid derivatives of GA acquired in negative ion mode; B, the representative MS/MS data of Isogastrodin and GA acquired in negative ion mode.
Figure 2.
The representative MS/MS data of gastordin and its citric acid derivatives identified in the extract of G. elata tubers produced by new optimized and traditional processing procedures. A, the representative MS/MS data of citric acid derivatives of GA acquired in negative ion mode; B, the representative MS/MS data of Isogastrodin and GA acquired in negative ion mode.
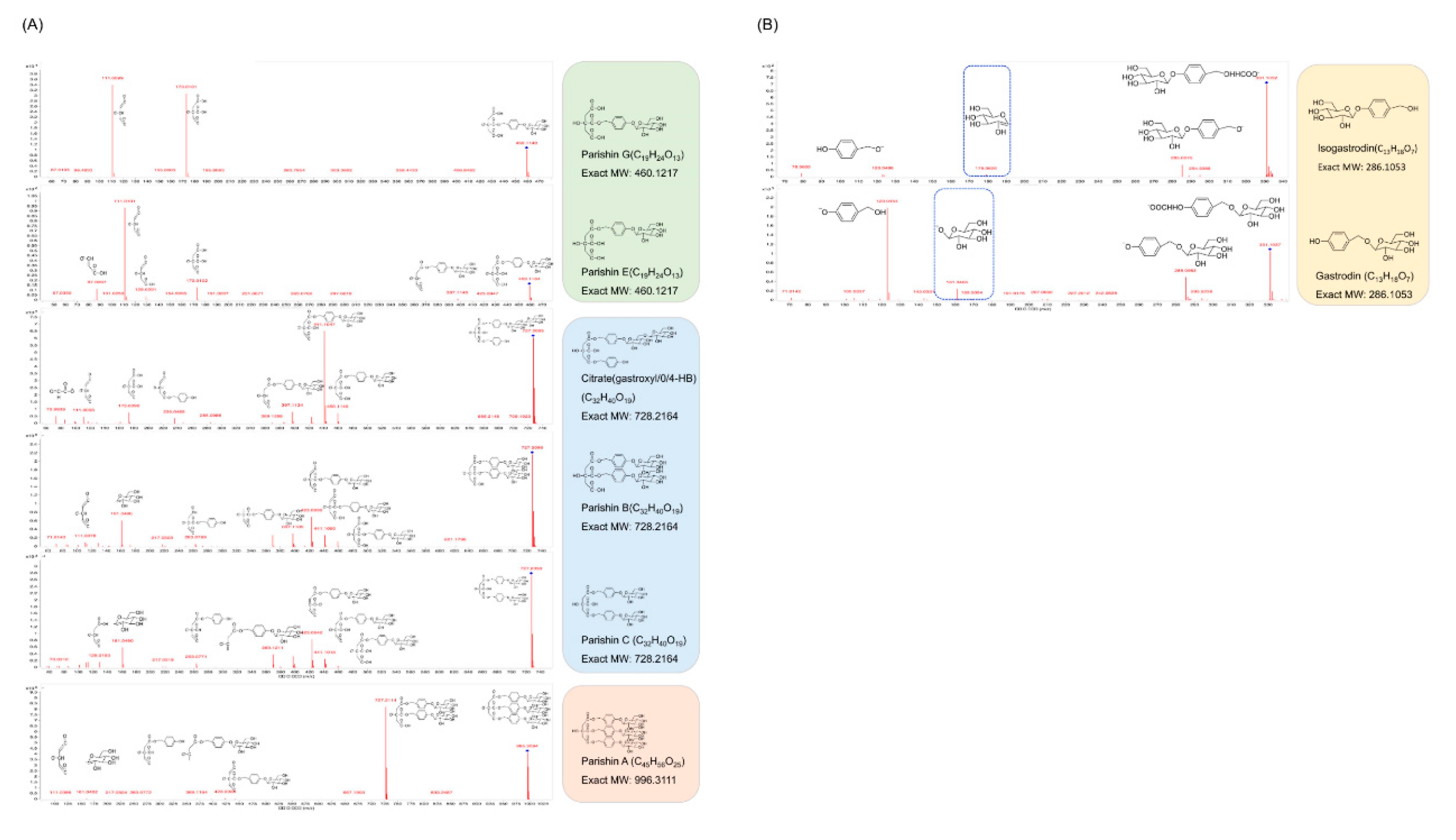
Figure 3.
The content of gastrodin and its derivatives in 7 batches of Gastrodia elata produced by different processing procedures. A, the content of gastrodin, p-hydroxybenzyl alcohol, Parishin E, B, C and Parishin in batches of Gastrodia elata produced by different processing procedures; B, the content of gastrodin and p-hydroxybenzyl alcohol (Total of GA and HA), Parishin E, B, C and Parishin (Total of Parishins), and all the derivatives in 7 batches of G. elata produced by different processing procedures (Total); C, the extraction rate of ethanol-soluble extract for 7 batches of G. elata produced by different processing procedures (n=3).
Figure 3.
The content of gastrodin and its derivatives in 7 batches of Gastrodia elata produced by different processing procedures. A, the content of gastrodin, p-hydroxybenzyl alcohol, Parishin E, B, C and Parishin in batches of Gastrodia elata produced by different processing procedures; B, the content of gastrodin and p-hydroxybenzyl alcohol (Total of GA and HA), Parishin E, B, C and Parishin (Total of Parishins), and all the derivatives in 7 batches of G. elata produced by different processing procedures (Total); C, the extraction rate of ethanol-soluble extract for 7 batches of G. elata produced by different processing procedures (n=3).
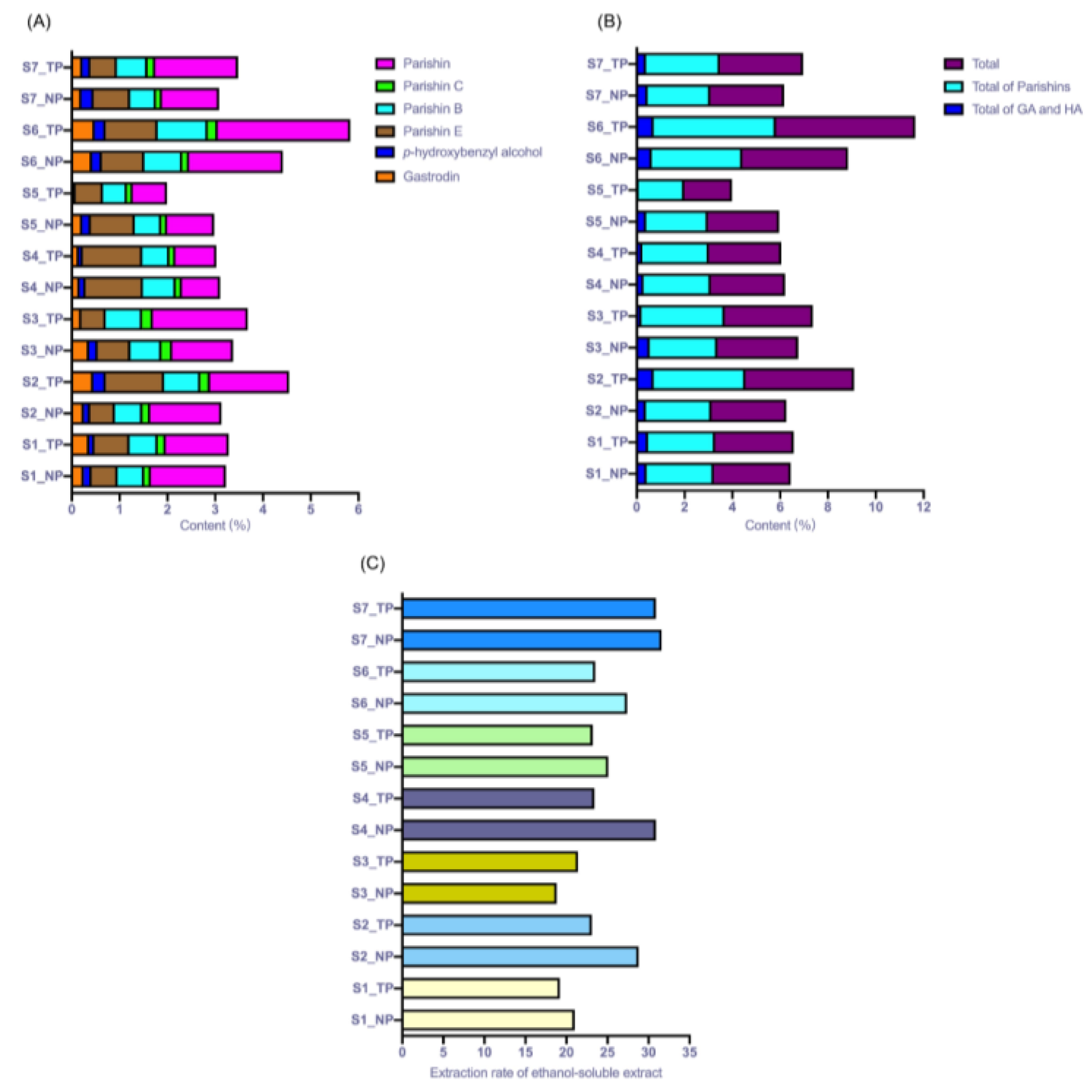
Figure 4.
The DPPH radical scavenging activity of the extract of Gastrodia elata produced by different processing procedures (A) and the correlations between the content of parsins, GA and HA, as well as all the six derivatives and DPPH radical scavenging activity (B).
Figure 4.
The DPPH radical scavenging activity of the extract of Gastrodia elata produced by different processing procedures (A) and the correlations between the content of parsins, GA and HA, as well as all the six derivatives and DPPH radical scavenging activity (B).
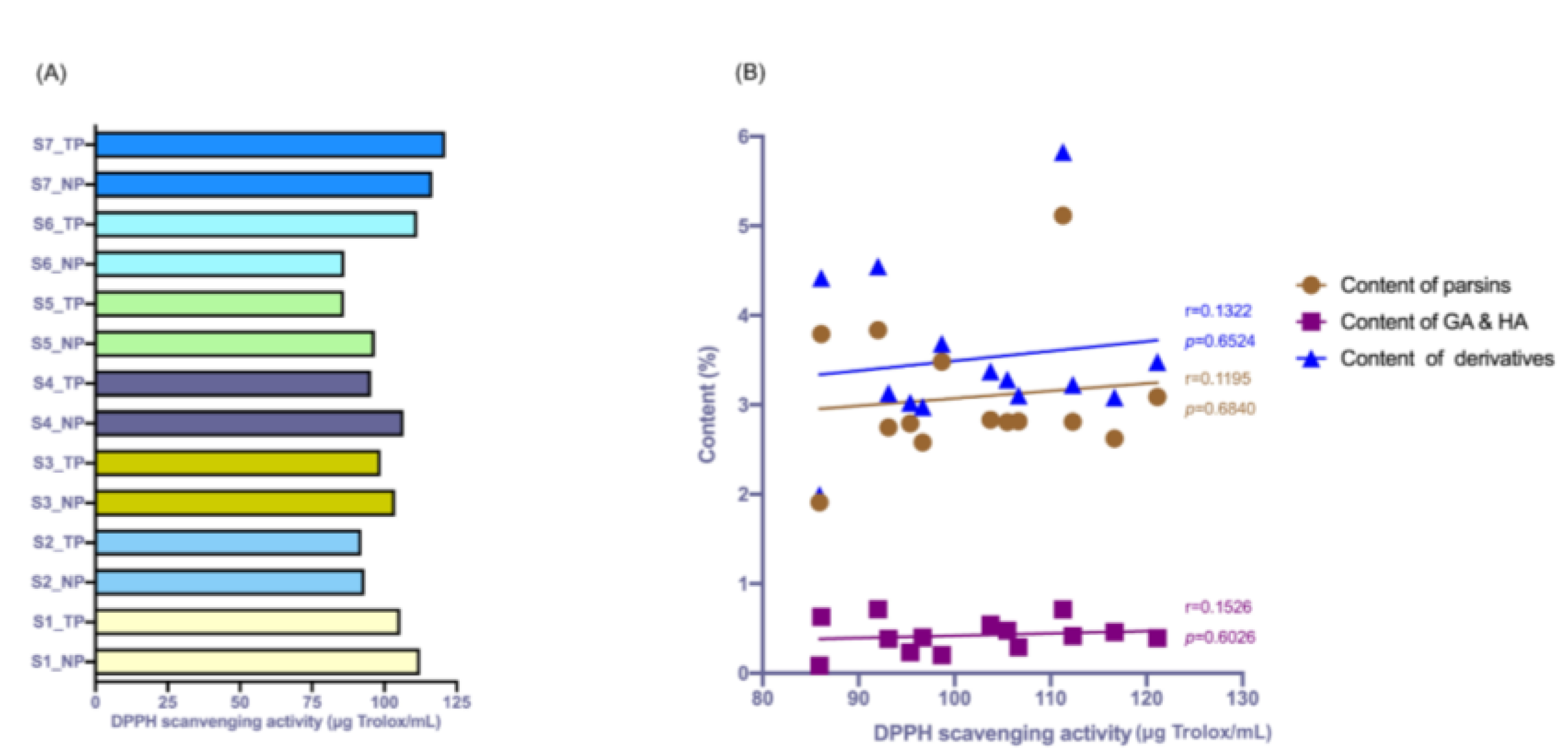

Table 1.
Information of Gastrodia elata collected from different regions used in the present study.
| Sample NO | Province | City | Variants of Gastrodia elata |
|---|---|---|---|
| S1 | Sichuan | Guangyuan | G. elata f. elata (Hong Tianma in Chinese) |
| S2 | Sichuan | Guangyuan | G. elata f. glauca (Wu Tianma in Chinese) |
| S3 | Gansu | Longnan | G. elata f. elata (Hong Tianma in Chinese) |
| S4 | Sichuan | Guangyuan | Hybrid variants of G. elata f. elata and G. elata f. glauca |
| S5 | Chongqing | Yunyang | G. elata f. glauca (Wu Tianma in Chinese) |
| S6 | Yunnan | Zhaotong | G. elata f. glauca (Wu Tianma in Chinese) |
| S7 | Sichuan | Mianyang | G. elata f. glauca (Wu Tianma in Chinese) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



