
Submitted:
01 July 2023
Posted:
04 July 2023
You are already at the latest version
Abstract
Phytocannabinoids, including the non-addictive cannabis component cannabidivarin (CBDV), have been reported to hold therapeutic potential in several neurodevelopmental disorders (NDDs). Nonetheless, the therapeutic value of phytocannabinoids for treating Fragile X syndrome (FXS), a major NDD, remains unexplored. Here, we characterized the neurobehavioral effects of CBDV at doses of 20 or 100 mg/kg in the Fmr1-knockout (Fmr1-KO) mouse model of FXS using two temporally-different intraperitoneal regimens: subchronic 10-day delivery during adulthood (Study 1: rescue treatment) or chronic 5-week delivery at adolescence (Study 2: preventive treat-ment). Behavioral tests assessing FXS-like abnormalities included anxiety, locomotor, cognitive, social and sensory alterations. Expression of inflammatory and plasticity markers was investigated in the hippocampus and prefrontal cortex. When administered during adulthood (Study 1), the effects of CBDV were marginal, rescuing at the lower dose only the acoustic hyper-responsiveness of Fmr1-KO mice and at both doses, their altered hippocampal expression of neurotrophins. When administered during adolescence (Study 2), CBDV at both doses prevented the cognitive, social and acoustic alterations of adult Fmr1-KO mice and modified the expression of several inflammatory brain markers in both wild-type littermates and mutants. These findings warrant the therapeutic potential of CBDV for preventing neurobehavioral alterations associated with FXS, highlighting the relevance of its early administration.
Keywords:
Phytocannabinoids
; Fmr1
; mouse behavior
; neurodevelopmental disorders
; interleukins
; neurotrophins
1. Introduction
Recent lines of evidence have highlighted the relevance of the endocannabinoid system (ECS) as an important modulator of neuronal functions and a promising therapeutic target for treating a variety of psychiatric disorders [1]. In the brain, the ECS comprises CB1 and CB2 cannabinoid receptors, their endogenous lipid ligands referred to as endocannabinoids (ECBs) and the enzymatic machinery involved in the synthesis and degradation of ECBs. The neurofunctional relevance of this signaling system is well acknowledged, as demonstrated by the abundance of CB1 receptors throughout the brain relative to CB2 receptors whose lower expression is mainly restricted to microglia and vascular components [2]. Furthermore, CB1 receptors, generally predominantly expressed presynaptically, modulate brain functions via the direct control of the release of several neurotransmitters including GABA, glutamate, and serotonin. The ECS also regulates synaptogenesis and neuronal interconnectivity during development [3,4,5], two processes whose defects have been reported as key determinants in major neurodevelopmental disorders (NDDs). Hence, the role of the ECS in NDDs has attracted considerable interest (e.g., [6,7,8]), particularly as concerns its implication in the pathophysiology of Fragile X Syndrome (FXS) [9,10,11,12].
FXS is a rare NDD due to an unstable expansion of CGG repeats in the X-linked gene FMR1, producing loss of FMRP, a synaptically expressed RNA-binding protein regulating protein synthesis [13,14]. FXS is considered the most common monogenic cause of inherited intellectual disability and autism. Building upon the widely used animal model of FXS, i.e., the Fmr1-knockout (Fmr1-KO) mouse, several studies have suggested a major role of the ECS in the pathogenesis of this disease. First, FXS is characterized by an uncontrolled activity of the metabotropic glutamate receptor 5 [15,16] and abnormal mTOR signaling [17,18,19], two pathways that are heavily involved in ECS functionality [20,21,22,23]. Second, previous findings [9,10,11,12], including our own [24], have revealed an aberrant hyperfunctionality of the ECS in Fmr1-KO mice, highlighting the possibility that behavioral deficits may result from dysfunctional CB1-mediated signaling in this mouse model. Accordingly, downregulating the ECS as a therapeutic strategy in Fmr1-KO mice has yielded encouraging results [9,25]. Within this pharmacological framework, cannabidiol (CBD) and its analogs can modulate the functionality of the ECS by inhibiting the enzymes involved in the synthesis of endocannabinoids [26]. Importantly, these phytocannabinoids do not directly act on the major cannabinoid receptor CB1, thus avoiding the heavy undesirable side effects of CB1 antagonists, for instance rimonabant, as well as the noxious psychotropic and addictive effects of other cannabinoids, such as the most naturally abundant and best known Delta9-tetrahydrocannabinol (THC). Hence, these non-psychoactive phytocannabinoids hold promises for treating FXS as they could attenuate the ECS hyperfunctionality observed in FXS without the burden of major side effects. The therapeutic applications of these compounds have already been explored in a variety of pathologies [27,28,29,30,31,32]. In particular, pre-clinical studies in rodents have shown that cannabidivarin (CBDV), a propyl analog of CBD, can induce anti-convulsant [33] and anxiolytic effects [34], together with powerful anti-inflammatory properties [29]. CBDV has also been reported to rescue some of the neurobehavioral alterations found in animal models of Rett syndrome [6,7] and autism spectrum disorder (ASD; [35]). However, the therapeutic potential of CBDV for treating FXS has not been investigated yet.
Here, we aimed at evaluating the therapeutic impact of CBDV in the Fmr1-KO mouse model of FXS by combining two experimental approaches (as schematized in Figure 1). In the first study (Study 1), we evaluated whether FXS-like neurobehavioral phenotypes could be rescued by a sub-chronic (10-day) CBDV treatment that started at adulthood, i.e., once the pathology is fully expressed in the Fmr1-KO mouse model [36]. The goal of the second study (Study 2) was instead to investigate whether the therapeutic effects of CBDV could somehow be enhanced by administering the CBDV treatment preventively at weaning, a developmental stage with high levels of neurobehavioral plasticity [37]. Our hypothesis was that juvenile chronic (during 5 weeks) CBDV administration would be more efficacious than an adult sub-chronic treatment and could prevent the expression of the neurobehavioral abnormalities typically observed in adult Fmr1-KO mice. Both studies used the doses of 20 and 100 mg/kg, as in previous research using CBDV in animal models of other NDDs, i.e., Rett syndrome [6,7] and ASD [35]. In the two studies, Fmr1-KO mice and their littermates underwent a comprehensive test battery tailored to several behavioral domains that we and others previously reported to be robustly altered in this mouse model of FXS [38,39,40,41,42]. Accordingly, behavioral screening included paradigms taxing motor activity in the open field, object recognition memory, social interest (in the three-compartment and direct social interaction tests) and sensory processing using acoustic startle. Although not consistently described in Fmr1-KO mice, emotional alterations in the elevated plus maze were also evaluated in order to assess potential confounding differences in anxiety-like behavior induced by CBDV treatments. At the end of both studies, brain samples were collected from tested mice in order to investigate the ability of CBDV to reverse (Study 1) or prevent (Study 2) the altered expression patterns of inflammatory and plasticity markers in cortex and hippocampus, two brain regions where the FMRP protein is most abundant [43,44] and in which expression of these markers has been reported to be altered in Fmr1-KO mice [40]. Also, the brain expression of some of these inflammatory markers (e.g., TNFα, CD11b) was previously shown to be modulated by CBDV, at least in other animal models of developmental pathologies [35].
2. Materials and Methods
2.1. Animals
Subjects were male C57BL/6J (B6) Fmr1-KO mice and their wild-type (WT) littermates, bred in our animal facility at Bordeaux University for more than 10 generations. Breeding trios were formed by mating two heterozygous Fmr1 females with a wild-type B6 male purchased from Janvier (Le Genest St Isle, France). After 2 weeks, the sire was removed and females were single caged and left undisturbed until post-natal day (PND) 8 of the pups. On this same day, pups were marked with paw tattoos using a non-toxic ink (Ketchum permanent Tattoo Inks green paste, Ketchum MFG Co, NY) and tail samples were collected for PCR assessment of the genotypes as previously described [45]. Mice were weaned at PND 21 and group-housed with their same-sex littermates (3–5/cage). Only male mice were used for the study, as they are the most commonly employed in mouse studies on FXS due to the higher prevalence of this syndrome in the male sex [36]. Only litters including males of both genotypes (WT and KO) were used for all experiments.
NMRI female mice (12 ± 2 week-old) and juvenile (4 week-old) males purchased from Janvier (Le Genest St Isle, France) were used as social stimuli during the direct social interaction and three-compartment tests, respectively. This strain has been selected for its high level of sociability [46] and was previously employed in several social studies on Fmr1-KO mice [40,41,42]. Mice were group-housed (4-5 cage) and left undisturbed upon arrival for one week before the social interaction test.
All animals were housed in polycarbonate standard cages (33 x 15 x 14 cm in size; Tecniplast, Limonest, France), provided with litter (SAFE, Augy, France) and a stainless steel wired lid. Food (SAFE, Augy, France) and water were provided ad libitum. The animals were maintained in a temperature (22 ± 1°C) and humidity (55 ± 10%) controlled vivarium, under a 12:12 h light–dark cycle (lights on at 7 a.m.). All experimental procedures were in accordance with the European Communities Council Directive of 24 November 1986 (86/609/EEC) and local French legislation (Authorization N° 2017073113175079).
2.2. Experimental procedures
2.2.1. CBDV administration at adulthood (Study 1)
In Study 1, adult (12 ± 1 weeks of age) mice of both genotypes were assigned to one of the three following experimental conditions: vehicle (VEH: Cremophor® EL:Ethanol:saline in a ratio of 1:2:17), CBDV at a dose of 20 mg/kg (CBDV-20) or CBDV at a dose of 100 mg/kg (CBDV-100). CBDV (synthetic; purity by HPLC > 99%) was supplied by GW Research Limited (now part of Jazz Pharmaceuticals, Inc., Cambridge, UK) and stored at approximately -20°C, protected from light. Injectable solutions were prepared fresh each day and were continuously stirred until injection. A total of 46 adult males were used as subjects in Study 1 (n=8 for WT-VEH, KO-VEH, WT-CBDV-20 and KO-CBDV-100 groups; n=7 for Fmr1-KO-CBDV-20 and WT-CBDV-100 groups). The number of subjects per experimental conditions was similar to that used in previous studies showing therapeutic effects of phytocannabinoids in other animal models (e.g., [6,7]).
Fmr1-KO mice and WT littermate controls were injected intraperitoneally (10 ml/kg), once a day around 9.00 a.m., during the entire duration of the study, i.e., for 17 consecutive days. Behavioral tests began after 10 days of injections, according to the timing described in Figure 1a. The order of the tests was based on the need of performing first the tests that are the most sensitive to previous testing experience (such as the elevated plus maze), while leaving for last those involving a higher degree of stressful experience (such as the acoustic startle which requires a short confinement in the startle box). A similar sequence of behavioral tests was employed in several previous studies on Fmr1-KO mice (e.g., [40,41,47,48]). All procedures were performed during the light phase, between 9 a.m. and 5 p.m. Mice assigned to each of the three experimental conditions were injected one hour before the beginning of each testing procedure; after injection each mouse was left undisturbed in a waiting cage containing sawdust bedding, food and water. Mice were habituated to the testing room at least 90 min before the beginning of each behavioral test.
2.2.2. CBDV administration at weaning (Study 2)
In Study 2, the day after weaning (i.e., on PND 22), male mice of both genotypes were assigned to one of the three following experimental conditions, VEH, CBDV-20 or CBDV-100. All solutions were prepared as described for Study 1. A total of 47 mice was subjected to all behavioral tests and brain analysis (n=7 for WT-VEH and WT-CBDV-100 groups; n=8 for WT-CBDV-20, KO-CBDV-20 and KO-CBDV-100 groups; n=9 for the KO-VEH group). Fmr1-KO mice and WT littermates were injected intraperitoneally once a day during the entire duration of the study (Figure 1b). Behavioral tests began after 5 weeks of injections, following the timing described in Figure 1b and using the same test sequence as for Study 1.
All procedures were performed during the light phase, as in Study 1. On testing days, the treatments were given after the end of each behavioral test, approximately 24 hours from behavioral and brain assessment to minimize acute effects of CBDV. The order of testing was counterbalanced across experimental groups and mice were habituated to the testing room before behavioral testing as described for Study 1.
2.2.3. Behavioral assessment (studies 1 and 2)
Elevated plus maze. The apparatus and procedures were described in details elsewhere [47,49]. Briefly, each mouse was gently placed in the center of the elevated plus maze and left free to explore the maze during 5 min. Videos recorded from a digital camera above the maze were analyzed manually by an observer blind to the experimental condition of the animals using Observer XT (Version 7, Noldus Technology, the Netherlands). Anxiety-like behaviors were measured as follows: percent time in open arms = time in open arms / time in all arms × 100. Locomotor activity was assessed by scoring the total number of entries into the arms of the maze.
Open field (habituation phase of object recognition test). The apparatus consisted of 2 identical plastic 24 x 30 cm rectangular arenas surrounded by 22 cm-high walls. The arenas were located in a testing room under diffused dim lighting (30 lux in the arena centre). A digital camera was mounted directly above the arenas, capturing images at 5 Hz that were transmitted to a PC running the Ethovision tracking system (version 11, Noldus, the Netherlands). Each mouse was gently placed in the centre of the appropriate arena and allowed to explore it undisturbed for 20 min. The choice of the arena was counterbalanced across experimental groups. Locomotor activity was indexed by the total distance travelled. Anxiety was assessed by calculating the percentage of time spent in the center of the arena.
Object recognition. The open field test served as the habituation phase for the object recognition test that was described in details elsewhere [40]. Briefly, at the end of the open field session, two identical objects were placed in two opposite corners and the mouse was introduced in the center of the arena for a 5-min sample phase. Twenty-four hours later, the mouse was returned to the arena for a 5-min test phase, where one of the objects was replaced by a novel one of different shape and material. Both the type of object used for the sample phase and the position of the novel object during the test phase were counterbalanced across experimental groups. During the training and test phases, the time spent sniffing each object was manually scored by an observer unaware of the experimental conditions of the animals using Observer XT (version 7, Noldus, the Netherlands). To measure object recognition during the test phase, a percent recognition index was calculated using the following formula: Tnovel object/(Tnovel object + Tfamiliar object) x 100 (T: time). Lack of novel object recognition was set at 50%. At the end of the sample and test phases, the apparatus as well as the objects were cleansed with a 30% ethanol solution and dried.
Three compartment test. The apparatus (described in details elsewhere [41]) consisted of a central chamber connected on each side to another compartment containing a perforated stimulus cage (8 x 8 x15 cm) to allow the test mouse to interact with the mouse or the object inside the stimulus cage. The object employed for the test was a plastic black cylinder and the stimulus mice were NMRI juvenile males, in order to minimize aggressive tendencies and exclude sexual interest. Each experimental subject was introduced in the middle of the central compartment and allowed to explore the apparatus for 3 trials of 5 min each. In trial 1 habituation to the apparatus containing empty stimulus cages was evaluated, while in trial 2, the preferential exploration of the social (a juvenile male mouse) versus the non-social (an object) novel stimulus was measured. In trial 3, the preferential exploration of a novel versus familiar social stimulus was assessed by replacing the object with a novel stimulus mouse.
In all trials, the total distance travelled as well as the time spent in each contact area (20 x 22 cm) containing the stimulus cages was computed using the Ethovision tracking system (version 11, Noldus, the Netherlands). A percentage score was also computed for the last two trials as follows:
On trial 2: Sociability index = Tsocial stimulus/(Tsocial stimulus + Tnon-social stimulus) x 100.
On trial 3: Social novelty preference index = Tnovel social stimulus/(Tnovel social stimulus + Tfamiliar social stimulus) x 100.
At the end of each trial, the experimental animal was confined for 30 sec in the central compartment by means of two Plexiglas magnetic doors. At the end of the third trial, the apparatus as well as the object and the stimulus cages were cleansed with a 30% ethanol solution and dried.
Direct social interaction. Direct social interaction was assessed as described in detail elsewhere [41]. Briefly, an unfamiliar adult NMRI female mouse was introduced into a testing cage (32 x 14 x 12.5 cm, with a flat metal grid as cover and approximately 3 cm of clean sawdust bedding) to which experimental subjects were habituated for one hour. Six min testing sessions were recorded and videos analyzed with Observer XT (version 7, Noldus, the Netherlands). One observer who was unaware of the experimental conditions of the animals scored the time spent performing affiliative behaviors, i.e., social investigation (nose, body and anogenital sniffing) and contacts. At the beginning of the testing day, the estrous cycle of the stimulus females was assessed through the analysis of the vaginal smear, so that only females in the non-estrous phase were used for social interaction sessions.
Sensory responsiveness (acoustic startle test). The apparatus consisted of four acoustic startle chambers for mice (SR-LAB, San Diego Instruments, San Diego, CA, USA) described in details elsewhere [42,47]. Twenty-four hours before testing, mice were placed in the startle chamber for 5 min without being exposed to any stimuli in order to habituate them to the confinement and reduce the related stress. On the test day, after a 5 min habituation period with continuous white noise of 66 dB (background), mice were presented with pulses of 20 ms duration and of varying intensity: +6, +12 +18 and +24 dB over background levels (namely 72, 78, 84 and 90 dB), as previously described in details [42,47]. Vibrations of the Plexiglas enclosure caused by the whole-body startle response of the animal were converted into analogue signals, digitized and stored by a computer.
2.2.4. Brain assessment of inflammatory and plasticity markers through RT-PCR (Studies 1 and 2)
Mice were sacrificed by cervical dislocation. Brains were immediately extracted and cut in two hemispheres that were separately frozen using dry ice. Only one hemisphere underwent RNA extraction while the other was stored at -80°C as backup. Whole brain tissue was sectioned using a Leica cryostat to generate 50 μm-thick sections. Sections were collected on polyethylene-naphthalate membrane 1 mm glass slides (series of 8-10 sections per slide)that were pretreated with heat to inactivate RNases. Series corresponding to the prefrontal cortex (PFC) (infralimbic and prelimbic areas) were collected from bregma +1.98 mm to +1.54 mm according to the Paxinos mouse stereotaxic atlas. Series corresponding to Cornu Ammonis 1 (CA1), cornu Ammonis 3 (CA3) and dentate gyrus (DG) of the hippocampus were collected from bregma -1.22 mm to -2.80 mm. To enable cell identification, sections were then stained with cresyl violet using a protocol compatible with subsequent RNA isolation. RNase-free water and solutions were used for all steps.Sections were fixed for 30 seconds with 95% ethanol, followed by 75% ethanol for 30 seconds and by 50% ethanol for 30 seconds to remove the OCT tissue freezing medium compound used during cryostat sectioning to provide a specimen matrix. Sections were subsequently stained with 1% cresyl violet in 50% ethanol for 30 seconds and dehydrated in 50%, 75% and 95% ethanol for 30 seconds each, and 2 times in 100% ethanol for 30 seconds. Stained slides were stored at -80°C until laser capture microdissection.
Laser Pressure Catapulting microdissection (LPC) of samples was performed using a PALM MicroBeam microdissection system version 4.6 equipped with the PALM RoboSoftware (P.A.L.M. Microlaser Technologies AG, Bernried, Germany). Laser power and duration were adjusted to optimize capture efficiency. Microdissection was performed at 5X magnification. The microdissection of pure brain structures were collected in adhesives caps and re-suspended in 250 µl guanidine isothiocyanate-containing buffer (BL buffer from ReliaPrep™ RNA Cell Miniprep System, Promega, Wisconsin,USA) with 10 µl 1-Thioglycerol, and stored at −80°C until extraction was done. Total RNA was extracted from microdissected tissues using the ReliaPrep™ RNA Cell Miniprep System (Promega, Wisconsin,USA) according to the manufacturer’s protocol. The integrity of the RNA was checked by capillary electrophoresis using the RNA 6000 Pico Labchip kit and the Bioanalyser 2100 (Agilent Technologies, Massy, France), and quantity was estimated using a Nanodrop 1000 (Thermo Scientific, Waltham, USA). RNA integrity numbers (RIN) were above 7/8.
RNA was processed and analyzed according to an adaptation of published methods [50]. Briefly, cDNA was synthesized from 140 ng of total RNA for each structure by using qSriptTM cDNA SuperMix (Quanta Biosciences). qPCR was performed with a LightCycler® 480 Real-Time PCR System (Roche, Meylan, France). qPCR reactions were done in duplicate for each sample by using LightCycler 480 SYBR Green I Master (Roche) in a final volume of 10 μl. The qPCR data were exported and analyzed in an informatics tool (Gene Expression Analysis Software Environment) developed at the University of Bordeaux. The Genorm method was used to determine the reference gene [50]. Relative expression analysis was normalized against two reference genes. Succinate dehydrogenase complex subunit (Sdha) and tubulin alpha 4a (Tuba4a) were used as reference genes for PFC. Succinate dehydrogenase complex subunit (Sdha) and tyrosine 3 mono-oxygenase tryptophan 5 monooxygenase (Ywhaz) were used as reference genes for CA1. Tuba4a and glyceraldehyde-3-phosphate dehydrogenase (Gapdh) were used as reference genes for CA3. Tuba4a and non-POU-domain-containing octamer binding protein (Nono) were used as reference genes for DG. The relative level of expression was calculated with the comparative (2−ΔΔCT) method [51]. Primer sequences are reported in Supplementary Table S1.
2.3. Statistical analysis
Behavioral and brain marker data were inspected for the identification of possible outliers using the Grubbs’ test or the extreme studentized deviate ESD method. Identified outliers for a given variable were excluded from the statistical analysis. This explains the slight differences in the total number of animals per group among certain statistical comparisons. For each variable analyzed, the exact sample size is indicated in the corresponding figure or in its legend.
Normality was assessed through the Shapiro-Wilks test for each experimental group and each variable of interest. Data from startle reactivity did not show a normal distribution at all stimulus intensities and were therefore subjected to natural logarithmic (ln) transformation in order to meet the normality requirements of ANOVA. For all other variables, data distribution was found to be normal and a parametric 2 x 3 ANOVA with genotype and treatment as the between-subject factors was applied. Within-subject factors were included according to the specific test and used as repeated measures in the ANOVA. These included for example, 5-min bins for the total distance travelled in the open field, 3-min-time bins for the social interaction test and the stimulus intensity for the acoustic startle assessment.
Post-hoc comparisons were conducted using the Tuckey’s HSD test when a significant interaction was found. Otherwise, separate one-way ANOVAs for each treatment group with genotype as the between subject factor were conducted, if appropriate. For the object recognition index, sociability and social novelty scores in the three compartment test, a one-sample t-test was used for comparison with chance level/lack of preference (i.e., 50%), instead of the ANOVA, as done in previous behavioral studies (see for example, [39,52]). All analyses were carried out using Statview (SAS Institute Inc.) and PASW Statistics 18 (SPSS Inc.) softwares.
3. Results
3.1. Study 1: Effects of subchronic administration of CBDV at adulthood
3.1.1. Behavioral profiling
- Elevated plus maze
We examined the percent time spent in the open arms of the maze as an index of anxiety-like behavior. A two-way ANOVA revealed no significant effect of genotype (F1,24 = 2.77; NS] or CBDV treatment (F1,24 = 4.00; NS). Fmr1-KO mice and their WT littermates spent a similar amount of time in the open arms, a pattern that was unaffected by CBDV, regardless of the dose (genotype x treatment interaction: F1,24 = 2.54; NS). Thus, anxiety levels were similar across groups as shown in Figure S1a. Likewise, locomotor activity did not differ among experimental groups, as demonstrated by a similar number of total arm entries across groups [no effect of genotype (F1,24 = 0.96; NS), CBDV treatment (F1,24 = 0.22; NS) or their interaction (F1,24 = 0.89; NS) (Figure S1b).
- Object recognition test
Habituation phase. We took advantage of the open field arena used for the habituation phase of the object recognition paradigm to first assess hyperactivity, which is considered a robust end point for Fmr1-KO mice. As illustrated in Figure 2a, Fmr1-KO mice were indeed more active than their WT littermates in the empty arena (genotype effect: F1,39 = 15.65, p = 0.0003], but this phenotype was not attenuated by CBDV regardless of the dose (genotype x treatment interaction: F2,39 = 2.23; NS]. Although there was a main effect of CBDV treatment (F2,39 = 11.29, p = 0.0001), this was explained by CBDV-induced hyperactivity at the dose of 20 mg/kg in both WT and KO mice (Figure 2a). To assess locomotor habituation, we plotted the total distance traveled in 5-min bins over the 20 min habituation session (Figure S2). This more detailed analysis yielded similar results, with an overall time-dependent reduction in locomotion across all experimental groups (5-min bin effect: F3,117 = 156.08, p < 0.0001), independently of the genotype (F3,117 = 0.69; NS) or treatment (F6,117 = 0.73; NS)(Figure S2, left and right).
We next examined the percentage of time spent in the center of the arena as an index of anxiety (Figure 2b). While similar between WT and Fmr1-KO mice controls injected with vehicle, this parameter was increased only in Fmr1-KO mice injected with CBDV at the highest dose of 100 mg/kg (genotype x treatment interaction: F2,40 = 3.54, p = 0.04; Figure 2b), suggesting the occurrence of an anxiolytic phenotype in Fmr1-KO mice when endocannabinoid signaling is highly stimulated.
Sample phase. During the sample phase, all mice explored similarly the two sample objects irrespective of their position (data not shown). Although not significant, noteworthy is the tendency towards an increased exploration of the two objects by vehicle-injected Fmr1-KO mice (Figure 2c) compared to WT controls, consistent with their hyperactive phenotype reported in Figure 2a. While without effect in WT mice, CBDV treatment decreased the object exploration time in Fmr1-KO animals which resulted in an almost significant genotype x treatment interaction (F2,40 = 3.21, p = 0.051; Figure 2c].
Test phase. During the test phase (Figure 2d), WT control but not Fmr1-KO mice injected with vehicle showed a recognition index that was significantly above chance level [one sample t-test versus 50%: WT-VEH, t(7) = 3.27; p = 0.01, KO-VEH, t(7) = 1.21; NS]. CBDV treatment at both doses did not rescue the memory deficits shown by Fmr1-KO and impaired object recognition in WT mice (Figure 2d). Performance of the two CBDV-treated WT and KO groups was indeed not different from chance level [one sample t-test versus 50% chance level: WT-20: t(7) = 1.02; NS; WT-100: t(6) = 0.69; NS; KO-20: t(6) = -0.64; NS; KO-100: t(7) = 1.91; NS; Figure 2d].
- Three-compartment test for sociability and social novelty
On trial 1 (habituation phase), all vehicle- and CBDV-treated mutant and WT mice travelled the same distance in the apparatus [genotype (F1,39 = 0.24; NS), treatment (F2,39 = 0.88; NS), genotype x treatment (F2,39 = 0.64; NS); Figure 3a] and equally explored the two stimulus cages (genotype x treatment interaction: F2,40 = 1.31; NS), indicating no bias for any of the two side compartments (Figure 3b).
On trial 2 (sociability) shown in Figure 3c, again no difference was found on locomotion [genotype (F1,40 = 1.41; NS), treatment (F2,40 = 1.57; NS), genotype x treatment (F2,40 = 2.27; NS)]. All groups preferentially explored the social stimulus compared to the inanimate stimulus, as demonstrated by the mean percentage time spent in the area containing the juvenile male mouse that was significantly above 50% (one sample t-test versus 50%: p < 0.05 for all groups; Figure 3d). The absence of sociability deficit in KO-VEH mice was expected, based on previous data from our group and other studies (reviewed in [36]).
On trial 3 (novelty), locomotor activity did not differ significantly between experimental groups [genotype (F1,40 = 0.97; NS), treatment (F2,40 = 2.57; NS), genotype x treatment interaction (F2,40 = 1.84; NS; Figure 3e]. In contrast to trial 2, only the WT-VEH control group showed a preference for the novel social stimulus (one sample t-test versus 50% chance level: t(7) = 2.63; p = 0.03; Figure 3f), indicating a deficit in Fmr1-KO mice that was, however, not rescued by the CBDV treatment, regardless of the dose (one sample t-test versus 50% chance level: KO-VEH, t(7) = -1.14, NS; KO-CBDV-20, t(6) = 1.39, NS; KO-CBDV-100: t(7) = 1.19, NS; Figure 3f)). Preference for the novel social stimulus was abolished in WT mice treated with both doses of CBDV (20 mg/kg dose: one sample t-test versus 50% chance level: t(7) = 0.47, NS; 100 mg/kg dose: t(6) = 1.04, NS; Figure 3f).
- Direct social interaction with an adult female
The 6-min interaction session was analyzed using two consecutive bins of 3 minutes in order to assess habituation to the social stimulus (Figure 4a). Most social interactions, as measured by the amount of time spent in affiliative behaviors, were displayed during the first 3 min of the interaction session and decreased afterwards independently of genotype or CBDV treatment (bin-effect: F1,37 = 29.91, p < 0.0001). When restricted to the first bin of 3 minutes, a significant genotype x treatment interaction (F2,37 = 10.16, p = 0.0003] emerged with Fmr1-KO-VEH mice exhibiting a significant decrease in the affiliation time compared to WT-VEH mice (Figure 4a, left). However, despite a trend, both doses of CBDV failed in rescuing this impaired social phenotype. In contrast, in WT mice, the CBDV treatment at the dose of 100 mg/kg decreased the amount of affiliative behavior (Figure 4a, left). During the last bin of 3 min, no between group differences were observed, all comparisons being non-significant.
- Acoustic startle
As expected, body startle response shown in Figure 4b increased with noise intensity in all groups (intensity effect: F3,120 = 21.13, p < 0.0001). Fmr1-KO mice injected with vehicle showed an overall startle hyper-responsiveness compared to WT-vehicle controls which was attenuated only in Fmr1-KO mice treated with the 20 mg/kg dose of CBDV (Figure 4b), as demonstrated by separate ANOVAs yielding genotype effects in VEH (F1,14 = 10.84, p = 0.005) and CBDV-100 (F1,13 = 8.29, p = 0.01) groups, but not in the CBDV-20 group (F1,13 = 0.60; NS).
3.1.2. Brain analyses
- Hippocampus
The effects of CBDV administration on the expression patterns of inflammatory (TNF-α, IL1b, IL-6, IL-10, CD45 or CD11b) and plasticity (BDNF) markers were analyzed in the CA1, CA3 and dentate gyrus of hippocampus of WT and Fmr1-KO mice (Figures S3, 5 and 6). Neither the Fmr1 mutation nor CBDV significantly affected RNA levels of these genes in the CA1 area (Figure S3). In contrast, in the CA3 area, RNA expression of the pro-inflammatory cytokine gene TNF-α was decreased in Fmr1-KO mice compared to WT mice (genotype effect: F1,38 = 7.76, p = 0.008; Figure 5h). However, despite a trend, this effect was not rescued by the CBDV treatment, regardless of the dose (genotype x treatment interaction: F2,38 = 2.55; NS).
Despite the lack of genotype effect (F2,39 = 0.39; NS), a significant effect of CBDV treatment was observed on the pro-inflammatory cytokine gene IL-1b (F2,39 = 5.13, p = 0.01; Figure 5d) whose RNA expression was increased in the CA3 of WT, but not of Fmr1-KO, mice injected at the dose of 100 mg/kg compared to vehicle-injected WT mice (genotype x treatment effect close to reaching significance: F2,39 = 2.99, p = 0.06; treatment effect from separate ANOVAs in WTs: F2,19 = 6.87, p = 0.006, in KOs: F2,20 = 0.17, NS, Figure 5d). CBDV also affected the CD-45 gene involved in cytokine production and proliferation of T cells [treatment effect: F2,39 = 7.0, p = 0.002; Figure 5g]. RNA expression of CD-45 increased in a dose-dependent manner, an effect that seemed more marked in Fmr1-KO mice. However, there was no significant genotype effect (F1,39 = 1.49; NS) or genotype x treatment interaction (F2,39 = 2.5, NS). There were no significant between-group changes in the RNA levels of the neurotrophic factor BDNF, the anti-inflammatory cytokines IL-6 and IL-10 or the microglia marker CD11b (Figure 5b, c, e and f).
In the dentate gyrus, RNA levels of BDNF were increased in Fmr1-KO mice compared to WT animals injected with vehicle (genotype x treatment interaction: F3,40 = 3.27; p = 0.048; Figure 6b), but they were not significantly affected by CBDV treatment (F2,40 = 0.30, NS). IL-1b RNA levels were overall increased in Fmr1-KO mice compared to their WT littermates [genotype effect: F1,38 = 4.75, p =0.036, Figure 6d], without any significant effect of CBDV treatment [genotype x treatment: F2,38 = 3.05, NS). As to the other neuroinflammatory markers, neither the Fmr1 mutation nor the CBDV treatment affected IL10, IL6, CD11b, CD45 or TNFα RNA levels (Figure 6c, e, f, g and h).
- Prefrontal cortex
In the prefrontal cortex, none of the brain markers examined were significantly affected by either the Fmr1 mutation or the CBDV treatments (Figure S4).
3.2. Study 2: Effects of chronic administration of CBDV at weaning
3.2.1. Behavioral profiling
- Elevated plus maze
The effects of juvenile chronic administration of CBDV on anxiety-like behavior in the elevated plus maze are presented in Figure S5. Anxiety levels, assessed by the percent time spent in the open arms, were reduced in Fmr1-KO mice independently of CBDV treatment (genotype effect: F1,39 = 13.09, p = 0.0008; genotype x treatment interaction: F2,39 = 1.75, NS; Figure S5a). Locomotor activity, indexed by the number of total arm entries, was overall enhanced in KO mice compared to their WT littermates and this hyperactive phenotype was unaffected by CBDV treatment (genotype effect: F1,39 = 14.97, p = 0.0004; genotype x treatment interaction: F2,39 = 1.55, NS; Figure S5b].
- Object recognition test
Habituation phase. As expected, Fmr1-KO mice were more active than their WT littermates (genotype effect: F1,41 = 17.65, p = 0.0001; Figure 7a]. However, CBDV treatment, regardless of the dose, did not normalize this locomotor phenotype (genotype x treatment interaction: F2,41 = 0.37; NS). All experimental groups displayed locomotor habituation as shown by a time-dependent reduction in locomotion (5 min-bin effect: F3,123 = 117.44, p < 0.0001; Fig.S6) independently of the genotype (genotype x 5 min-bin interaction: F3,123 = 1.64; NS) or treatment (treatment x 5 min-bin interaction: F3,123 = 1.68; NS).
Anxiety levels appeared slightly reduced in Fmr1-KO mice compared to their WT littermates as shown by the increased time spent in the center of the arena (genotype effect: F1, 41 = 9.42, p = 0.004; Figure 7b). However, this anxiolytic phenotype in the open field was not modified by CBDV treatments (genotype x treatment interaction: F2,41 = 0.09; NS).
Sample phase. All mice explored similarly the two sample objects irrespective of their position, indicating the absence of any spatial bias (data not shown). There was no significant difference in object exploration across experimental groups as shown by the absence of genotype or CBDV treatment effects (F1,41 = 0.56, NS and F2,41 = 0.24, NS) or a genotype x treatment interaction (F2,41 = 0.03; NS; Figure 7c].
Test phase. During the test phase, as already observed in Study 1, a clear object recognition deficit was detected in Fmr1-KO-VEH [one sample t-test versus 50%: t(8) = -0.53, NS] but not inWT-VEH mice that preferentially explored the novel object [t(5) = 6.04, p = 0.002]. This deficit was eliminated by both doses of CBDV (one sample t-test versus 50%: Fmr1-KO CBDV-20, t(7) = 259x, p = 0.04; Fmr1-KO-CBDV-100, t(7) = 2.52, p = 0.03; Figure 7d). In contrast, CBDV treatments impaired recognition memory of WT mice that was not significantly different from chance level [one sample t-test versus 50%: t(7) = 2.59, p = 0.036 for Fmr1-KO-CBDV-20 mice and t(7) = 2.69, p = 0.034 for Fmr1-KO-CBDV-100 mice; Figure 7d].
- Three-compartment test for sociability and social novelty
On trial 1 (habituation phase), no main effects of genotype (F1,41 = 0.83, NS), treatment (F2,41 = 0.86, NS) or genotype x treatment interaction (F2,41 = 0.09, NS) were found on locomotor activity (Figure 8a). All vehicle- and CBDV-treated mice explored similarly the two side compartments (one sample t-test versus 50%: NS for all groups; Figure 8b), indicating no particular spatial bias.
On trial 2 (sociability), no difference in the distance travelled across groups was observed (genotype and treatment effects: F1,41 = 0.08, F2,41 = 1.45, NS; genotype x treatment interaction: F2,41 = 0.62, NS; Figure 8c). All mice preferred to explore the social versus the inanimate stimulus, as shown by the mean sociability index that was significantly above 50% in all experimental groups (one sample t-test versus 50%: p < 0.05 for all groups; Figure 8d).
On trial 3 (novelty), locomotor activity did not differ between experimental groups (genotype and treatment effects: F1,40 = 1.71, NS; F2,40 = 2.52, NS; genotype x treatment interaction: F2,40 = 0.69, NS; Figure 8e). As opposed to WT-VEH control mice which showed a preference for the novel social stimulus (one sample t-test versus 50%: t(5) = 4.17, p = 0.01), the Fmr1-KO-VEH group exhibited a clear and expected lack of preference for social novelty (t(8) = -1.31; NS). This deficit was prevented in mutant mice by CBDV chronic administration at both 20 mg/kg and 100 mg/kg doses (one sample t-test versus 50% in Fmr1-KO-20 mice: t(7) = 2.50; p = 0.04; in Fmr1-KO-100 mice: t(7) = 2.88; p = 0.02; Figure 8f). In contrast, the CBDV treatment was deleterious in WT mice which failed to exhibit a significant preference for the novel social stimulus at both 20 mg/kg and 100 mg/kg doses. Performance of WT mice treated with CBDV was not different from chance level (one sample t-test versus 50%: WT-20 group, t(7) = -0.79, NS; WT-100 group: t(6) = -0.93, NS; Figure 8f).
- Direct social interaction with an adult female
Social habituation occurred over a 6-min interaction session that was separated into two time bins of 3 minutes each. There was a main effect of time bin (F1,41 = 225.45, p < 0.0001; Figure 9a), confirming that the highest levels of social affiliative behaviors are displayed during the first 3 min of interaction (Figure 9a, left). As for study 1, these behaviors decreased during the second time bin independently of genotype or CBDV treatment (Figure 9a, right). The ANOVA revealed group differences on social interaction only during the first time bin (interaction genotype x treatment x 3-min bin: F2,41 = 16.12, p < 0.0001) with Fmr1-KO-VEH mice displaying a significant reduction of their affiliation time compared to WT-VEH mice (Figure 9a, left). This impairment during the first 3 min was attenuated following CBDV treatments at both doses. However, CBDV was not beneficial for WT mice which exhibited a reduced affiliation time for both low and high doses of CBDV [genotype x treatment interaction on the first 3 min-bin: F2,41 = 15.95, p < 0.0001; Figure 9a, left). During the last bin of 3 min, no between-group differences were observed, all comparisons being non-significant.
- Acoustic startle
All groups exhibited a body startle response which increased with stimulus intensity (intensity effect: F3,123 = 11.65, p < 0.0001; Figure 9b]. Fmr1-KO mice showed an overall startle hyper-responsiveness (genotype effect: F1,41 = 12.41, p = 0.001). This phenotype was abolished by CBDV administration (genotype x treatment interaction: F2,41 = 7.68, p = 0.002; Figure 9b), with both the 20 mg/kg and 100 mg/kg doses being equally efficacious. CBDV treatments did not affect body startle response in WT-mice, indicating no side effects.
3.2.2. Brain analyses
- Hippocampus
Expression patterns of inflammatory (TNF-α, IL1b, IL-6, IL-10, CD45 or CD11b) and plasticity (BDNF) markers were marginally affected in the CA1 area of the hippocampus of WT and Fmr1-KO mice (Figure S7). The only statistically significant difference concerned RNA expression of the IL-6 gene (interaction genotype x treatment: F2,36 = 2.27, p = 0.049; Figure S7e). However, all post-hoc comparisons failed to reach significance. In the CA3 area, amongst all brain markers analyzed (Figure 10), the only significant differences were observed for the pro-inflammatory cytokine gene CD11b (Figure 10f). Its RNA expression was not altered in Fmr1-KO compared to WT mice (genotype: F1,41= 0.22, NS). However, CBDV increased CD11b expression in both WT and KO mice (treatment effect: F2,41 = 15.23, p < 0.0001 ; Figure 10f), the dose of 20 mg/kg being the most effective.
In the dentate gyrus, RNA levels of the CD45 gene were increased only by the 100 mg/kg dose of CBDV in mice of both genotypes (treatment effect: F2,40 = 5.13, p = 0.01; Figure 11g). Likewise, the highest dose of CBDV increased CD11b RNA expression in all mice (treatment effect: F2,40 = 3.84, p = 0.03 Figure 11f). There was however no genotype effect for these two genes (Figure 11f and 11g). For the IL-10 gene, there was a genotype x treatment interaction (F2,37 = 3.67, p =0.03; Figure 11c), but all post-hoc comparisons failed to reach statistical significance. Neither the Fmr1 mutation nor CBDV significantly affected RNA levels of all the other brain markers (Figure 11b, d, e, h).
- Prefrontal cortex
RNA levels of BDNF were similar in Fmr1-KO and WT mice (genotype effect: F1,38 = 0.10, NS). CBDV altered RNA expression of the BDNF gene in both WT and Fmr1-KO mice in a dose-dependent manner (Figure S8b). While increased by the 20 mg/kg dose, RNA levels of BDNF were decreased by the 100 mg/kg dose (treatment effect: F2,38 = 3.32, p = 0.047). Neither the Fmr1 mutation nor CBDV significantly affected RNA levels of all the other brain markers (Figure S8, c-h).
4. Discussion
Our findings provide for the first time a full characterization of the neurobehavioral effects of CBDV administration in the Fmr1-KO mouse model of FXS. In the first study (Study 1) which consisted in administering CBDV during 10 days starting at adult age (3 months), we show a rescue effect of CBDV at the 20 mg/kg dose only on the startle hyper-responsiveness of Fmr1-KO mice, and at both doses on their altered BDNF levels in the dentate gyrus. In the second study (Study 2), when treatments started at weaning (PND 21) and were given chronically for 5 weeks, we instead demonstrated a larger array of beneficial effects of CBDV in Fmr1-KO mice. All FXS-like behavioral phenotypes in these mutants were prevented, except hyperactivity. Furthermore, in Study 2, CBDV modified the expression of several inflammatory brain markers in mice of both genotypes. Overall, our results indicate the emergence of genotype and main treatment effects, as well as interactions, for selected behavioral and brain measures. These effects are summarized in Table 1 and Table 2.
Concerning the main behavioral effects of genotype (Table 1), we confirmed in both studies the most robust behavioral phenotypes of the Fmr1-KO mouse model, as previously reported by us and others (for a review, see [53]). These included hyperactivity in the open field [39,40,54,55,56,57,58,59,60,61,62,63,64,65,66,67], reduced direct social interaction [36,39,41,55,64,68], lack of preference for social novelty [38,41,54,55,59,64,69,70], sensory hyper-responsiveness in the acoustic startle paradigm [42,71] and deficits in novel object memory [40,54,55,72]. We also found anxiety-like behavior to be an inconsistent phenotype of this mouse model. Indeed, whereas we did not observe any genotype difference in Study 1, in accordance with previous studies by us [38] and others (e.g., [57,60,64,75,77]), reduced anxiety-like behaviors of Fmr1-KO mice were detected in the elevated plus maze and open field test in Study 2, as previously reported as well [10,58,64,65,74]. Our data therefore further underline the inconsistency of the emotional phenotype of Fmr1-KO mice [53], in contrast with what is typically observed in FXS patients who exhibit a robust increase in anxiety levels. As for sociability (on trial 2 of the 3-compartment test), the lack of a Fmr1-KO phenotype was confirmed in both studies, as previously demonstrated by us [38,41] and others [54,55,59,64,69,70]. It should be noted that the hyperactivity exhibited by Fmr1-KO mice in the open field of both Study 1 and Study 2 was less marked than what was previously reported in the literature, including by us (for a review see [36]), as this parameter emerged only as an overall genotype difference, but failed to reach significance when WT-VEH and Fmr1-KO-VEH mice were compared. This finding may be explained, at least in part, by the confounding stressful effects of the repeated intraperitoneal injections performed in both studies.
The behavioral phenotypes of Fmr1-KO mice were normalized by CBDV depending on the treatment schedule (Table 1). While the efficacy of subchronic CBDV treatment at adulthood was limited to acoustic startle and at the lowest dose (Study 1), chronic administration of both doses of CBDV from weaning (Study 2) eliminated all the behavioral alterations of Fmr1-KO mice, except hyperactivity and reduced anxiety, thus preventing the most relevant symptoms associated with the FXS. These results therefore confirm our hypothesis that juvenile chronic CBDV administration is more efficacious than a subchronic treatment delivered at the adult stage. They further corroborate the view that early timing of intervention is critical for alleviating the behavioral alterations induced by the Fmr1 mutation [39,40] and identify the juvenile/adolescence phase as a sensitive period for pharmacological targeting in preclinical models of NDDs. Although such a differential effect could be anticipated based on the high levels of neurobehavioral plasticity characterizing the adolescent phase in mice as in humans [78,79], it does not represent an obvious finding. Indeed, previous studies have described the efficacy of 10-day pharmacological treatments other than phytocannabinoids (e.g., metformin or potassium-channel agonists; [80,81]) in reversing multiple neurobehavioral phenotypes of Fmr1-KO mice when administered at adulthood. Interestingly, these treatments were also able to rescue the alterations in hippocampal spine density and morphology shown by Fmr1-KO mice at adulthood, a brain phenotype that we did not assess here and which may represent a key mechanism for the full rescue of Fmr1-KO behavioral phenotypes, as previously suggested [39,80,81].
While CBDV had marginal effects in terms of the behavioral rescue of mutant mice when administered subchronically at adulthood (Study 1), it also exerted several behavioral effects in WT mice (Table 1). Interestingly, these effects were highly comparable to those observed in Study 2 (Table 1), thus suggesting that the timing of delivery does not represent a critical determinant for the behavioral effects of CBDV in WT animals, in contrast to its effects in Fmr1 mutants. In both studies, cognitive and social deficits occurred in WT mice treated with both doses of CBDV in the novel object recognition and 3-chamber tests, as well as in the direct social interaction paradigm, while anxiety was unaltered (Table 1). It can be hypothesized that CBDV may induce beneficial effects in Fmr1-KO mice by normalizing the ECS hyper-functionality of these animals, while opposite detrimental effects are induced when a “normally” functioning ECS is modulated, as in the case of WT subjects. This hypothesis could be tested by assessing the differential effects of CBDV in our Fmr1-KO and WT mice on ECS functionality, e.g., assessing mTOR activation, since this signaling pathway is altered in Fmr1-KO mice [9]. To the best of our knowledge, this is the first study reporting clear detrimental behavioral outcomes of phytocannabinoids in WT mice, as no or minor effects were described in previous preclinical studies that have examined the consequences of CBD [82] and CBDV [6] treatments in other mouse models of NDDs.Close inspection of the findings obtained using the mouse model of Rett syndrome indicates that CBDV at doses of 20 and 100 mg/kg induced a lack of social preference (close to chance level) in the three compartment test (Figure 2d in [6]). Differences in the genetic background (as only our mice are on a pure B6) could contribute to explain the discrepancies with previous studies. Further experiments using WT mice bred from WT breeders would be necessary to generalize and extend our results to B6 mice, as our WT controls here were bred from Fmr1-HET females and could perhaps be oversensitive to the effects of CBDV. Previous studies have indeed shown that WT mice of the Fmr1 line present several behavioral differences compared to WT animals of the same background obtained from WT breeders [83].
In contrast to the behavioral phenotypes, the brain alterations displayed by Fmr1-KO mice injected with vehicle were less robust and variable between our two studies (Table 2). In Study 1, Fmr1-KO animals displayed reduced expression of TNF-α in the CA3, higher levels of Il1b and BDNF in the DG, while no difference emerged in Study 2. The discrepancy between the two studies may be due to differences in experimental conditions, such that the chronic versus subchronic injection regimen, and support the weakness and high variability of the brain inflammatory phenotype of the Fmr1-KO model. Previous results indeed suggested a subtle imbalance in hippocampal and cortical inflammatory markers in these mutants, with a decrease in some specific regions, and an increase in others [40]. Similarly, the increase in BDNF expression restricted to the DG of Fmr1 mutants in Study 1 was in disagreement with previous studies showing reduced BDNF levels in both CA1 and PFC in untreated Fmr1-KO mice [40], though more in line with other findings from a mouse model of another developmental disorder, the Rett syndrome [6].
CBDV was able to affect several brain markers in both WT and KO mice, as illustrated in Table 2. These pharmacological effects were detected in both studies and appeared slightly more pronounced in Study 2, where they emerged across more brain areas and affected the RNA expression of both inflammatory and plasticity markers. Hence, these findings corroborate the higher efficacy of CBDV in preventing behavioral impairments when delivered chronically during the juvenile period. Although slight region- and dose-specific differences exist between our two studies, the effects of CBDV seemed to consistently increase the expression of CD11b and CD45 (Table 2). Although an opposite effect of CBDV was previously described in a mouse model of ASD [35], a similar effect on the same inflammatory markers was previously induced by the early administration of omega-3-enrichment, a non-pharmacological treatment that induced beneficial behavioral effects in the Fmr1-KO mouse model [40]. Our study examined mRNA expression levels which correlate only partially with functional protein levels due to post-transcriptional regulation of gene expression. Caution is therefore required when relating changes in brain marker expression to behavioral outcomes. This may explain, at least in part, some discrepancies between studies on the effects of endocannabinoids on cerebral expression levels of neuroinflammatory and plasticity markers.
The marginal between-group differences emerging on the brain markers that we evaluated in the present study suggest that other complementary mechanisms may underlie the behavioral effects of CBDV. Like its phytocannabinoid analog cannabidiol, CBDV is thought to exhibit neuroprotective anti-inflammatory properties [29] that may act in concert with the modulation of ECS functionality to support the beneficial effects of CBDV, especially its greater efficacy during the early stage of the FXS. Loss of FMRP in Fmr1 mutants is thought to disrupt ECS signaling [9,10,11,12], leading to neuronal hyper-excitability and the FXS- and ASD-like behavioral phenotypes of KO mice. It is therefore likely that CBDV may rescue the altered endocannabinoid tone in the brain of Fmr1 mutants, for instance by normalizing the endocannabinoid-degrading enzymes, fatty acid amide hydrolase (FAAH) and monoacylglycerol lipase (MAGL), as already shown in the valproate rat model of ASD [35].
5. Conclusions
Overall, these data demonstrate that CBDV, when administered chronically and starting at juvenile age, holds a solid therapeutic potential for FXS as it prevented the most relevant behavioral alterations shown by Fmr1-KO mice. Early timing of treatment appears as a critical determinant to ensure the beneficial effects of CBDV. These results thus encourage future clinical studies using this phytocannabinoid for treating not only FXS but also other neurodevelopmental disorders.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Table S1: Primer Sequences used in RT-PCR; Figure S1: Effects of adult subchronic CBDV treatment (20 or 100 mg/Kg) in the elevated plus maze (Study 1); Figure S2: Effects of adult subchronic CBDV treatment on locomotor habituation in the open field (Study 1); Figure S3: Effects of adult subchronic CBDV treatment on the expression of BDNF and inflammatory markers in CA1 (Study 1); Figure S4: Effects of subchronic adult CBDV treatment on the expression of BDNF and inflammatory markers in the prefrontal cortex (Study 1); Figure S5: Effects of juvenile chronic CBDV treatment (20 or 100 mg/Kg) in the elevated plus maze (Study 2); Figure S6: Effects of juvenile chronic CBDV treatment on locomotor habituation in the open field (Study 2); Figure S7: Effects of juvenile chronic CBDV treatment on the expression of BDNF and inflammatory markers in CA1 (Study 2); Figure S8: Effects of juvenile chronic CBDV treatment on the expression of BDNF and inflammatory markers in the prefrontal cortex (Study 2).
Author Contributions
Conceptualization, S.P. and M.W-R.; methodology, S.P. and M.W-R.; experimental investigation and data collection, M.P., W.F., L.B., V.L.; data curation, S.P., M.W-R. and B.B.; writing—original draft preparation, S.P. and B.B.; writing—review and editing, S.P. and B.B.; supervision and project administration, S.P.; funding acquisition, S.P. All authors have read and agreed to the published version of the manuscript.
Funding
The work was funded by GW Pharmaceutical (Cambridge, UK). V. Lemaire, B. Bontempi and S. Pietropaolo received additional funding from CNRS and the University of Bordeaux. W. Fyke received a PhD fellowship from SUNY Downstate University.
Institutional Review Board Statement
The studies presented here were approved by the Institutional Ethics Committee (“Comité d’Ethique pour l’experimentation animale de Bordeaux”, CE 50) and the French Ministry (“Ministere de l’enseignement superieur de la recherché et de l’innovation”; protocol code APAFIS#10873-2017073113175079 v12)”.
Data Availability Statement
All original data are available upon reasonable request.
Acknowledgments
The authors thank M. Maitre and H. Doat of the NeuroCentre Magendie Inserm U1215 for their work at the Laser Microdissection capture facility funded by Inserm, LabEX BRAIN ANR-10-LABX-43 and FRM DGE20061007758. This work benefited from the support of the Transcriptomic facility funded by Inserm and LabEX BRAIN ANR-10-LABX-43; thanks to T. Leste-Lasserre and the personnel of the Transcriptomic platform of the NeuroCentre Magendie Inserm U1215. We thank Delphine Gonzales and the genotyping facility of Neurocentre Magendie, funded by Inserm and LabEX BRAIN ANR-10-LABEX-43, for animal genotyping. The authors thank Elodie Poinama for the expert animal care, Thierry Lafon for technical assistance, Marie-Laure Rousseau for administrative support.
Conflicts of Interest
Marie Woolley-Roberts was a full-time employee of Jazz Pharmaceuticals who, in the course of employment, received stock options exercisable for, and other stock awards of, ordinary shares of Jazz Pharmaceuticals, plc. Funding for the study was provided by GW Research Limited, now part of Jazz Pharmaceuticals, Inc (Cambridge, UK). All other authors declare no conflict of interest.
References
- Parolaro, D., N. Realini, D. Vigano, C. Guidali, and T. Rubino. “The Endocannabinoid System and Psychiatric Disorders.” Exp Neurol 224, no. 1 (2010): 3-14. [CrossRef]
- Mackie, K. “Distribution of Cannabinoid Receptors in the Central and Peripheral Nervous System.” Handb Exp Pharmacol, no. 168 (2005): 299-325. [CrossRef]
- Basavarajappa, B. S., R. A. Nixon, and O. Arancio. “Endocannabinoid System: Emerging Role from Neurodevelopment to Neurodegeneration.” Mini Rev Med Chem 9, no. 4 (2009): 448-62. [CrossRef]
- Berghuis, P., A. M. Rajnicek, Y. M. Morozov, R. A. Ross, J. Mulder, G. M. Urban, K. Monory, G. Marsicano, M. Matteoli, A. Canty, A. J. Irving, I. Katona, Y. Yanagawa, P. Rakic, B. Lutz, K. Mackie, and T. Harkany. “Hardwiring the Brain: Endocannabinoids Shape Neuronal Connectivity.” Science 316, no. 5828 (2007): 1212-6. [CrossRef]
- Monory, K., F. Massa, M. Egertova, M. Eder, H. Blaudzun, R. Westenbroek, W. Kelsch, W. Jacob, R. Marsch, M. Ekker, J. Long, J. L. Rubenstein, S. Goebbels, K. A. Nave, M. During, M. Klugmann, B. Wolfel, H. U. Dodt, W. Zieglgansberger, C. T. Wotjak, K. Mackie, M. R. Elphick, G. Marsicano, and B. Lutz. “The Endocannabinoid System Controls Key Epileptogenic Circuits in the Hippocampus.” Neuron 51, no. 4 (2006): 455-66. [CrossRef]
- Vigli, D., L. Cosentino, C. Raggi, G. Laviola, M. Woolley-Roberts, and B. De Filippis. “Chronic Treatment with the Phytocannabinoid Cannabidivarin (Cbdv) Rescues Behavioural Alterations and Brain Atrophy in a Mouse Model of Rett Syndrome.” Neuropharmacology 140 (2018): 121-29. [CrossRef]
- Zamberletti, E., M. Gabaglio, F. Piscitelli, J. S. Brodie, M. Woolley-Roberts, I. Barbiero, M. Tramarin, G. Binelli, N. Landsberger, C. Kilstrup-Nielsen, T. Rubino, V. Di Marzo, and D. Parolaro. “Cannabidivarin Completely Rescues Cognitive Deficits and Delays Neurological and Motor Defects in Male Mecp2 Mutant Mice.” J Psychopharmacol 33, no. 7 (2019): 894-907. [CrossRef]
- Navarro-Romero, A., L. Galera-Lopez, P. Ortiz-Romero, A. Llorente-Ovejero, L. de Los Reyes-Ramirez, I. Bengoetxea de Tena, A. Garcia-Elias, A. Mas-Stachurska, M. Reixachs-Sole, A. Pastor, R. de la Torre, R. Maldonado, B. Benito, E. Eyras, R. Rodriguez-Puertas, V. Campuzano, and A. Ozaita. “Cannabinoid Signaling Modulation through Jzl184 Restores Key Phenotypes of a Mouse Model for Williams-Beuren Syndrome.” Elife 11 (2022). [CrossRef]
- Busquets-Garcia, A., M. Gomis-Gonzalez, T. Guegan, C. Agustin-Pavon, A. Pastor, S. Mato, A. Perez-Samartin, C. Matute, R. de la Torre, M. Dierssen, R. Maldonado, and A. Ozaita. “Targeting the Endocannabinoid System in the Treatment of Fragile X Syndrome.” Nat Med 19, no. 5 (2013): 603-7. [CrossRef]
- Jung, K. M., M. Sepers, C. M. Henstridge, O. Lassalle, D. Neuhofer, H. Martin, M. Ginger, A. Frick, N. V. DiPatrizio, K. Mackie, I. Katona, D. Piomelli, and O. J. Manzoni. “Uncoupling of the Endocannabinoid Signalling Complex in a Mouse Model of Fragile X Syndrome.” Nat Commun 3 (2012): 1080. [CrossRef]
- Maccarrone, M., S. Rossi, M. Bari, V. De Chiara, C. Rapino, A. Musella, G. Bernardi, C. Bagni, and D. Centonze. “Abnormal Mglu 5 Receptor/Endocannabinoid Coupling in Mice Lacking Fmrp and Bc1 Rna.” Neuropsychopharmacology 35, no. 7 (2010): 1500-9. [CrossRef]
- Zhang, L. , and B. E. Alger. “Enhanced Endocannabinoid Signaling Elevates Neuronal Excitability in Fragile X Syndrome.” J Neurosci 30, no. 16 (2010): 5724-9. [CrossRef]
- Pieretti, M., F. P. Zhang, Y. H. Fu, S. T. Warren, B. A. Oostra, C. T. Caskey, and D. L. Nelson. “Absence of Expression of the Fmr-1 Gene in Fragile X Syndrome.” Cell 66, no. 4 (1991): 817-22. [CrossRef]
- Verkerk, A. J., M. Pieretti, J. S. Sutcliffe, Y. H. Fu, D. P. Kuhl, A. Pizzuti, O. Reiner, S. Richards, M. F. Victoria, F. P. Zhang, and et al. “Identification of a Gene (Fmr-1) Containing a Cgg Repeat Coincident with a Breakpoint Cluster Region Exhibiting Length Variation in Fragile X Syndrome.” Cell 65, no. 5 (1991): 905-14. [CrossRef]
- Bear, M. F., K. M. Huber, and S. T. Warren. “The Mglur Theory of Fragile X Mental Retardation.” Trends Neurosci 27, no. 7 (2004): 370-7. [CrossRef]
- Michalon, A., A. Bruns, C. Risterucci, M. Honer, T. M. Ballard, L. Ozmen, G. Jaeschke, J. G. Wettstein, M. von Kienlin, B. Kunnecke, and L. Lindemann. “Chronic Metabotropic Glutamate Receptor 5 Inhibition Corrects Local Alterations of Brain Activity and Improves Cognitive Performance in Fragile X Mice.” Biol Psychiatry 75, no. 3 (2014): 189-97. [CrossRef]
- Hoeffer, C. A., E. Sanchez, R. J. Hagerman, Y. Mu, D. V. Nguyen, H. Wong, A. M. Whelan, R. S. Zukin, E. Klann, and F. Tassone. “Altered Mtor Signaling and Enhanced Cyfip2 Expression Levels in Subjects with Fragile X Syndrome.” Genes Brain Behav 11, no. 3 (2012): 332-41. [CrossRef]
- Price, T. J., M. H. Rashid, M. Millecamps, R. Sanoja, J. M. Entrena, and F. Cervero. “Decreased Nociceptive Sensitization in Mice Lacking the Fragile X Mental Retardation Protein: Role of Mglur1/5 and Mtor.” J Neurosci 27, no. 51 (2007): 13958-67.
- Sharma, A., C. A. Hoeffer, Y. Takayasu, T. Miyawaki, S. M. McBride, E. Klann, and R. S. Zukin. “Dysregulation of Mtor Signaling in Fragile X Syndrome.” J Neurosci 30, no. 2 (2010): 694-702. [CrossRef]
- Kano, M., T. Ohno-Shosaku, Y. Hashimotodani, M. Uchigashima, and M. Watanabe. “Endocannabinoid-Mediated Control of Synaptic Transmission.” Physiol Rev 89, no. 1 (2009): 309-80. [CrossRef]
- Varma, N., G. C. Carlson, C. Ledent, and B. E. Alger. “Metabotropic Glutamate Receptors Drive the Endocannabinoid System in Hippocampus.” J Neurosci 21, no. 24 (2001): RC188. [CrossRef]
- Puighermanal, E., A. Busquets-Garcia, R. Maldonado, and A. Ozaita. “Cellular and Intracellular Mechanisms Involved in the Cognitive Impairment of Cannabinoids.” Philos Trans R Soc Lond B Biol Sci 367, no. 1607 (2012): 3254-63. [CrossRef]
- Puighermanal, E., G. Marsicano, A. Busquets-Garcia, B. Lutz, R. Maldonado, and A. Ozaita. “Cannabinoid Modulation of Hippocampal Long-Term Memory Is Mediated by Mtor Signaling.” Nat Neurosci 12, no. 9 (2009): 1152-8. [CrossRef]
- Pietropaolo S., L. Bellocchio, Oddi D., D’Amato F.R., G. Marsicano, and Crusio W.E. “The Fmr1 Mouse Line as a Model for Autism: The Relevance of the Genetic Background and of the Endocannabinoid System.” Paper presented at the FENS Annual Meeting, Amsterdam, The Netherlands,, 3-7 July 2010 2010.
- Busquets-Garcia, A., R. Maldonado, and A. Ozaita. “New Insights into the Molecular Pathophysiology of Fragile X Syndrome and Therapeutic Perspectives from the Animal Model.” Int J Biochem Cell Biol 53 (2014): 121-6. [CrossRef]
- Bisogno, T., F. Howell, G. Williams, A. Minassi, M. G. Cascio, A. Ligresti, I. Matias, A. Schiano-Moriello, P. Paul, E. J. Williams, U. Gangadharan, C. Hobbs, V. Di Marzo, and P. Doherty. “Cloning of the First Sn1-Dag Lipases Points to the Spatial and Temporal Regulation of Endocannabinoid Signaling in the Brain.” J Cell Biol 163, no. 3 (2003): 463-8. [CrossRef]
- Hill, A. J., C. M. Williams, B. J. Whalley, and G. J. Stephens. “Phytocannabinoids as Novel Therapeutic Agents in Cns Disorders.” Pharmacol Ther 133, no. 1 (2012): 79-97. [CrossRef]
- Blessing, E. M., M. M. Steenkamp, J. Manzanares, and C. R. Marmar. “Cannabidiol as a Potential Treatment for Anxiety Disorders.” Neurotherapeutics 12, no. 4 (2015): 825-36. [CrossRef]
- Burstein, S. “Cannabidiol (Cbd) and Its Analogs: A Review of Their Effects on Inflammation.” Bioorg Med Chem 23, no. 7 (2015): 1377-85. [CrossRef]
- Campos, A. C., M. V. Fogaca, A. B. Sonego, and F. S. Guimaraes. “Cannabidiol, Neuroprotection and Neuropsychiatric Disorders.” Pharmacol Res (2016). [CrossRef]
- Chakravarti, B., J. Ravi, and R. K. Ganju. “Cannabinoids as Therapeutic Agents in Cancer: Current Status and Future Implications.” Oncotarget 5, no. 15 (2014): 5852-72. [CrossRef]
- Fasinu, P. S., S. Phillips, M. A. ElSohly, and L. A. Walker. “Current Status and Prospects for Cannabidiol Preparations as New Therapeutic Agents.” Pharmacotherapy 36, no. 7 (2016): 781-96. [CrossRef]
- Hill, A. J., M. S. Mercier, T. D. Hill, S. E. Glyn, N. A. Jones, Y. Yamasaki, T. Futamura, M. Duncan, C. G. Stott, G. J. Stephens, C. M. Williams, and B. J. Whalley. “Cannabidivarin Is Anticonvulsant in Mouse and Rat.” Br J Pharmacol 167, no. 8 (2012): 1629-42. [CrossRef]
- Deiana, S., A. Watanabe, Y. Yamasaki, N. Amada, M. Arthur, S. Fleming, H. Woodcock, P. Dorward, B. Pigliacampo, S. Close, B. Platt, and G. Riedel. “Plasma and Brain Pharmacokinetic Profile of Cannabidiol (Cbd), Cannabidivarine (Cbdv), Delta(9)-Tetrahydrocannabivarin (Thcv) and Cannabigerol (Cbg) in Rats and Mice Following Oral and Intraperitoneal Administration and Cbd Action on Obsessive-Compulsive Behaviour.” Psychopharmacology (Berl) 219, no. 3 (2012): 859-73.
- Zamberletti, E., M. Gabaglio, M. Woolley-Roberts, S. Bingham, T. Rubino, and D. Parolaro. “Cannabidivarin Treatment Ameliorates Autism-Like Behaviors and Restores Hippocampal Endocannabinoid System and Glia Alterations Induced by Prenatal Valproic Acid Exposure in Rats.” Front Cell Neurosci 13 (2019): 367. [CrossRef]
- Pietropaolo, S. , and E. Subashi. “Mouse Models of Fragile X Syndrome.” In Behavioral Genetics of the Mouse, edited by S. Pietropaolo, F. Sluyter and W.E. Crusio, 146-63. Cambridge: Cambridge University Press, 2014.
- Spear, L. P. “The Adolescent Brain and Age-Related Behavioral Manifestations.” Neurosci Biobehav Rev 24, no. 4 (2000): 417-63. [CrossRef]
- Hebert, B., S. Pietropaolo, S. Meme, B. Laudier, A. Laugeray, N. Doisne, A. Quartier, S. Lefeuvre, L. Got, D. Cahard, F. Laumonnier, W. E. Crusio, J. Pichon, A. Menuet, O. Perche, and S. Briault. “Rescue of Fragile X Syndrome Phenotypes in Fmr1 Ko Mice by a Bkca Channel Opener Molecule.” Orphanet J Rare Dis 9 (2014): 124. [CrossRef]
- Oddi, D., E. Subashi, S. Middei, L. Bellocchio, V. Lemaire-Mayo, M. Guzman, W. E. Crusio, F. R. D’Amato, and S. Pietropaolo. “Early Social Enrichment Rescues Adult Behavioral and Brain Abnormalities in a Mouse Model of Fragile X Syndrome.” Neuropsychopharmacology 40, no. 5 (2015): 1113-22.
- Pietropaolo, S., M. G. Goubran, C. Joffre, A. Aubert, V. Lemaire-Mayo, W. E. Crusio, and S. Laye. “Dietary Supplementation of Omega-3 Fatty Acids Rescues Fragile X Phenotypes in Fmr1-Ko Mice.” Psychoneuroendocrinology 49 (2014): 119-29. [CrossRef]
- Pietropaolo, S., A. Guilleminot, B. Martin, F. R. D’Amato, and W. E. Crusio. “Genetic-Background Modulation of Core and Variable Autistic-Like Symptoms in Fmr1 Knock-out Mice.” PLoS ONE 6, no. 2 (2011): e17073.
- Zhang, Y., A. Bonnan, G. Bony, I. Ferezou, S. Pietropaolo, M. Ginger, N. Sans, J. Rossier, B. Oostra, G. LeMasson, and A. Frick. “Dendritic Channelopathies Contribute to Neocortical and Sensory Hyperexcitability in Fmr1(-/Y) Mice.” Nat Neurosci 17, no. 12 (2014): 1701-9.
- Khandjian, E. W. “Biology of the Fragile X Mental Retardation Protein, an Rna-Binding Protein.” Biochem Cell Biol 77, no. 4 (1999): 331-42.
- Bakker, C. E., Y. de Diego Otero, C. Bontekoe, P. Raghoe, T. Luteijn, A. T. Hoogeveen, B. A. Oostra, and R. Willemsen. “Immunocytochemical and Biochemical Characterization of Fmrp, Fxr1p, and Fxr2p in the Mouse.” Exp Cell Res 258, no. 1 (2000): 162-70.
- Dutch-Belgian Fragile X Consortium. “Fmr1 Knockout Mice: A Model to Study Fragile X Mental Retardation.” Cell 78, no. 1 (1994): 23-33.
- Moles, A. , and R. D’Amato F. “Ultrasonic Vocalization by Female Mice in the Presence of a Conspecific Carrying Food Cues.” Anim Behav 60, no. 5 (2000): 689-94.
- Gaudissard, J., M. Ginger, M. Premoli, M. Memo, A. Frick, and S. Pietropaolo. “Behavioral Abnormalities in the Fmr1-Ko2 Mouse Model of Fragile X Syndrome: The Relevance of Early Life Phases.” Autism Res 10, no. 10 (2017): 1584-96. [CrossRef]
- Gauducheau, M., V. Lemaire-Mayo, F. R. D’Amato, D. Oddi, W. E. Crusio, and S. Pietropaolo. “Age-Specific Autistic-Like Behaviors in Heterozygous Fmr1-Ko Female Mice.” Autism Res 10, no. 6 (2017): 1067-78.
- Pietropaolo, S. , and W. E. Crusio. “Strain-Dependent Changes in Acoustic Startle Response and Its Plasticity across Adolescence in Mice.” Behav Genet (2009). [CrossRef]
- Bustin, S. A., V. Benes, J. A. Garson, J. Hellemans, J. Huggett, M. Kubista, R. Mueller, T. Nolan, M. W. Pfaffl, G. L. Shipley, J. Vandesompele, and C. T. Wittwer. “The Miqe Guidelines: Minimum Information for Publication of Quantitative Real-Time Pcr Experiments.” Clin Chem 55, no. 4 (2009): 611-22. [CrossRef]
- Livak, K. J. , and T. D. Schmittgen. “Analysis of Relative Gene Expression Data Using Real-Time Quantitative Pcr and the 2(-Delta Delta C(T)) Method.” Methods 25, no. 4 (2001): 402-8.
- Vandesquille, M., M. Baudonnat, L. Decorte, C. Louis, P. Lestage, and D. Beracochea. “Working Memory Deficits and Related Disinhibition of the Camp/Pka/Creb Are Alleviated by Prefrontal Alpha4beta2*-Nachrs Stimulation in Aged Mice.” Neurobiol Aging 34, no. 6 (2013): 1599-609.
- Kat, R., M. Arroyo-Araujo, R. B. M. de Vries, M. A. Koopmans, S. F. de Boer, and M. J. H. Kas. “Translational Validity and Methodological Underreporting in Animal Research: A Systematic Review and Meta-Analysis of the Fragile X Syndrome (Fmr1 Ko) Rodent Model.” Neurosci Biobehav Rev 139 (2022): 104722. [CrossRef]
- Bhattacharya, A., H. Kaphzan, A. C. Alvarez-Dieppa, J. P. Murphy, P. Pierre, and E. Klann. “Genetic Removal of P70 S6 Kinase 1 Corrects Molecular, Synaptic, and Behavioral Phenotypes in Fragile X Syndrome Mice.” Neuron 76, no. 2 (2012): 325-37.
- Dahlhaus, R. , and A. El-Husseini. “Altered Neuroligin Expression Is Involved in Social Deficits in a Mouse Model of the Fragile X Syndrome.” Behav Brain Res 208, no. 1 (2010): 96-105. [CrossRef]
- de Diego-Otero, Y., Y. Romero-Zerbo, R. el Bekay, J. Decara, L. Sanchez, F. Rodriguez-de Fonseca, and I. del Arco-Herrera. “Alpha-Tocopherol Protects against Oxidative Stress in the Fragile X Knockout Mouse: An Experimental Therapeutic Approach for the Fmr1 Deficiency.” Neuropsychopharmacology 34, no. 4 (2009): 1011-26.
- Eadie, B. D., W. N. Zhang, F. Boehme, J. Gil-Mohapel, L. Kainer, J. M. Simpson, and B. R. Christie. “Fmr1 Knockout Mice Show Reduced Anxiety and Alterations in Neurogenesis That Are Specific to the Ventral Dentate Gyrus.” Neurobiol Dis 36, no. 2 (2009): 361-73. [CrossRef]
- Hayashi, M. L., B. S. Rao, J. S. Seo, H. S. Choi, B. M. Dolan, S. Y. Choi, S. Chattarji, and S. Tonegawa. “Inhibition of P21-Activated Kinase Rescues Symptoms of Fragile X Syndrome in Mice.” Proc Natl Acad Sci U S A 104, no. 27 (2007): 11489-94. [CrossRef]
- Liu, Z. H., D. M. Chuang, and C. B. Smith. “Lithium Ameliorates Phenotypic Deficits in a Mouse Model of Fragile X Syndrome.” Int J Neuropsychopharmacol (2011): 1-13. [CrossRef]
- Mineur, Y. S., F. Sluyter, S. de Wit, B. A. Oostra, and W. E. Crusio. “Behavioral and Neuroanatomical Characterization of the Fmr1 Knockout Mouse.” Hippocampus 12, no. 1 (2002): 39-46.
- Olmos-Serrano, J. L., J. G. Corbin, and M. P. Burns. “The Gaba(a) Receptor Agonist Thip Ameliorates Specific Behavioral Deficits in the Mouse Model of Fragile X Syndrome.” Dev Neurosci 33, no. 5 (2011): 395-403.
- Peier, A. M., K. L. McIlwain, A. Kenneson, S. T. Warren, R. Paylor, and D. L. Nelson. “(over)Correction of Fmr1 Deficiency with Yac Transgenics: Behavioral and Physical Features.” Hum Mol Genet 9, no. 8 (2000): 1145-59. [CrossRef]
- Restivo, L., F. Ferrari, E. Passino, C. Sgobio, J. Bock, B. A. Oostra, C. Bagni, and M. Ammassari-Teule. “Enriched Environment Promotes Behavioral and Morphological Recovery in a Mouse Model for the Fragile X Syndrome.” Proc Natl Acad Sci U S A 102, no. 32 (2005): 11557-62. [CrossRef]
- Spencer, C. M., O. Alekseyenko, S. M. Hamilton, A. M. Thomas, E. Serysheva, L. A. Yuva-Paylor, and R. Paylor. “Modifying Behavioral Phenotypes in Fmr1ko Mice: Genetic Background Differences Reveal Autistic-Like Responses.” Autism Res 4, no. 1 (2011): 40-56. [CrossRef]
- Spencer, C. M., O. Alekseyenko, E. Serysheva, L. A. Yuva-Paylor, and R. Paylor. “Altered Anxiety-Related and Social Behaviors in the Fmr1 Knockout Mouse Model of Fragile X Syndrome.” Genes Brain Behav 4, no. 7 (2005): 420-30. [CrossRef]
- Thomas, A. M., N. Bui, D. Graham, J. R. Perkins, L. A. Yuva-Paylor, and R. Paylor. “Genetic Reduction of Group 1 Metabotropic Glutamate Receptors Alters Select Behaviors in a Mouse Model for Fragile X Syndrome.” Behav Brain Res 223, no. 2 (2011): 310-21. [CrossRef]
- Uutela, M., J. Lindholm, V. Louhivuori, H. Wei, L. M. Louhivuori, A. Pertovaara, K. Akerman, E. Castren, and M. L. Castren. “Reduction of Bdnf Expression in Fmr1 Knockout Mice Worsens Cognitive Deficits but Improves Hyperactivity and Sensorimotor Deficits.” Genes Brain Behav 11, no. 5 (2012): 513-23. [CrossRef]
- Mineur, Y. S., L. X. Huynh, and W. E. Crusio. “Social Behavior Deficits in the Fmr1 Mutant Mouse.” Behav Brain Res 168, no. 1 (2006): 172-5. [CrossRef]
- Heitzer, A. M., A. K. Roth, L. Nawrocki, C. C. Wrenn, and M. G. Valdovinos. “Brief Report: Altered Social Behavior in Isolation-Reared Fmr1 Knockout Mice.” J Autism Dev Disord (2012). [CrossRef]
- Mines, M. A., C. J. Yuskaitis, M. K. King, E. Beurel, and R. S. Jope. “Gsk3 Influences Social Preference and Anxiety-Related Behaviors During Social Interaction in a Mouse Model of Fragile X Syndrome and Autism.” PLoS One 5, no. 3 (2010): e9706. [CrossRef]
- Michalon, A., M. Sidorov, T. M. Ballard, L. Ozmen, W. Spooren, J. G. Wettstein, G. Jaeschke, M. F. Bear, and L. Lindemann. “Chronic Pharmacological Mglu5 Inhibition Corrects Fragile X in Adult Mice.” Neuron 74, no. 1 (2012): 49-56. [CrossRef]
- Ventura, R., T. Pascucci, M. V. Catania, S. A. Musumeci, and S. Puglisi-Allegra. “Object Recognition Impairment in Fmr1 Knockout Mice Is Reversed by Amphetamine: Involvement of Dopamine in the Medial Prefrontal Cortex.” Behav Pharmacol 15, no. 5-6 (2004): 433-42.
- Bilousova, T. V., L. Dansie, M. Ngo, J. Aye, J. R. Charles, D. W. Ethell, and I. M. Ethell. “Minocycline Promotes Dendritic Spine Maturation and Improves Behavioural Performance in the Fragile X Mouse Model.” J Med Genet 46, no. 2 (2009): 94-102. [CrossRef]
- Heulens, I., C. D’Hulst, D. Van Dam, P. P. De Deyn, and R. F. Kooy. “Pharmacological Treatment of Fragile X Syndrome with Gabaergic Drugs in a Knockout Mouse Model.” Behav Brain Res 229, no. 1 (2012): 244-9. [CrossRef]
- Nielsen, D. M., W. J. Derber, D. A. McClellan, and L. S. Crnic. “Alterations in the Auditory Startle Response in Fmr1 Targeted Mutant Mouse Models of Fragile X Syndrome.” Brain Res 927, no. 1 (2002): 8-17. [CrossRef]
- Qin, M., Z. Xia, T. Huang, and C. B. Smith. “Effects of Chronic Immobilization Stress on Anxiety-Like Behavior and Basolateral Amygdala Morphology in Fmr1 Knockout Mice.” Neuroscience 194 (2011): 282-90. [CrossRef]
- Veeraragavan, S., D. Graham, N. Bui, L. A. Yuva-Paylor, J. Wess, and R. Paylor. “Genetic Reduction of Muscarinic M4 Receptor Modulates Analgesic Response and Acoustic Startle Response in a Mouse Model of Fragile X Syndrome (Fxs).” Behav Brain Res 228, no. 1 (2011): 1-8. [CrossRef]
- Spear, L. P. “Adolescent Brain Development and Animal Models.” Ann N Y Acad Sci 1021 (2004): 23-6. [CrossRef]
- Spear, L. P. , and S. C. Brake. “Periadolescence: Age-Dependent Behavior and Psychopharmacological Responsivity in Rats.” Dev Psychobiol 16, no. 2 (1983): 83-109. [CrossRef]
- Gantois, I., A. Khoutorsky, J. Popic, A. Aguilar-Valles, E. Freemantle, R. Cao, V. Sharma, T. Pooters, A. Nagpal, A. Skalecka, V. T. Truong, S. Wiebe, I. A. Groves, S. M. Jafarnejad, C. Chapat, E. A. McCullagh, K. Gamache, K. Nader, J. C. Lacaille, C. G. Gkogkas, and N. Sonenberg. “Metformin Ameliorates Core Deficits in a Mouse Model of Fragile X Syndrome.” Nat Med 23, no. 6 (2017): 674-77. [CrossRef]
- Lemaire-Mayo, Valerie, Marion Piquemal, Wim E. Crusio, Eric Louette, and Susanna Pietropaolo. “Therapeutic Effects of Chlorzoxazone, a Bkca Channel Agonist, in a Mouse Model of Fragile X Syndrome.” bioRxiv (2020): 2020.12.11.389569.
- Cheng, D., J. K. Low, W. Logge, B. Garner, and T. Karl. “Chronic Cannabidiol Treatment Improves Social and Object Recognition in Double Transgenic Appswe/Ps1e9 Mice.” Psychopharmacology (Berl) 231, no. 15 (2014): 3009-17. [CrossRef]
- Zupan, B. , and M. Toth. “Wild-Type Male Offspring of Fmr-1+/- Mothers Exhibit Characteristics of the Fragile X Phenotype.” Neuropsychopharmacology 33, no. 11 (2008): 2667-75.
Figure 1.
Experimental timeline and procedures used in Study 1 and Study 2. For study 1 (a), treatment administration started at adulthood (i.e., approximately 3 months of age) and 10 days before the beginning of behavioral testing. Daily intraperitoneal injections of vehicle (Veh) or cannabidivarin (CBDV) solutions (20 or 100 mg/kg) were given during the entire experimental period, including the days of behavioral testing and brain sampling (one hour before their beginning). For study 2 (b), daily intraperitoneal injections started the day after weaning (i.e., at 3 weeks of age) and 5 weeks before the beginning of behavioral tests. Injections were continued during the entire experimental period, including the days of behavioral testing during which they were administered after completion of each testing procedure. EPM = elevated plus maze, OF = open field, OR = object recognition, 3-COMP = three-compartment test for sociability, SI = social interaction, AS = acoustic startle. PND = post-natal day.
Figure 1.
Experimental timeline and procedures used in Study 1 and Study 2. For study 1 (a), treatment administration started at adulthood (i.e., approximately 3 months of age) and 10 days before the beginning of behavioral testing. Daily intraperitoneal injections of vehicle (Veh) or cannabidivarin (CBDV) solutions (20 or 100 mg/kg) were given during the entire experimental period, including the days of behavioral testing and brain sampling (one hour before their beginning). For study 2 (b), daily intraperitoneal injections started the day after weaning (i.e., at 3 weeks of age) and 5 weeks before the beginning of behavioral tests. Injections were continued during the entire experimental period, including the days of behavioral testing during which they were administered after completion of each testing procedure. EPM = elevated plus maze, OF = open field, OR = object recognition, 3-COMP = three-compartment test for sociability, SI = social interaction, AS = acoustic startle. PND = post-natal day.
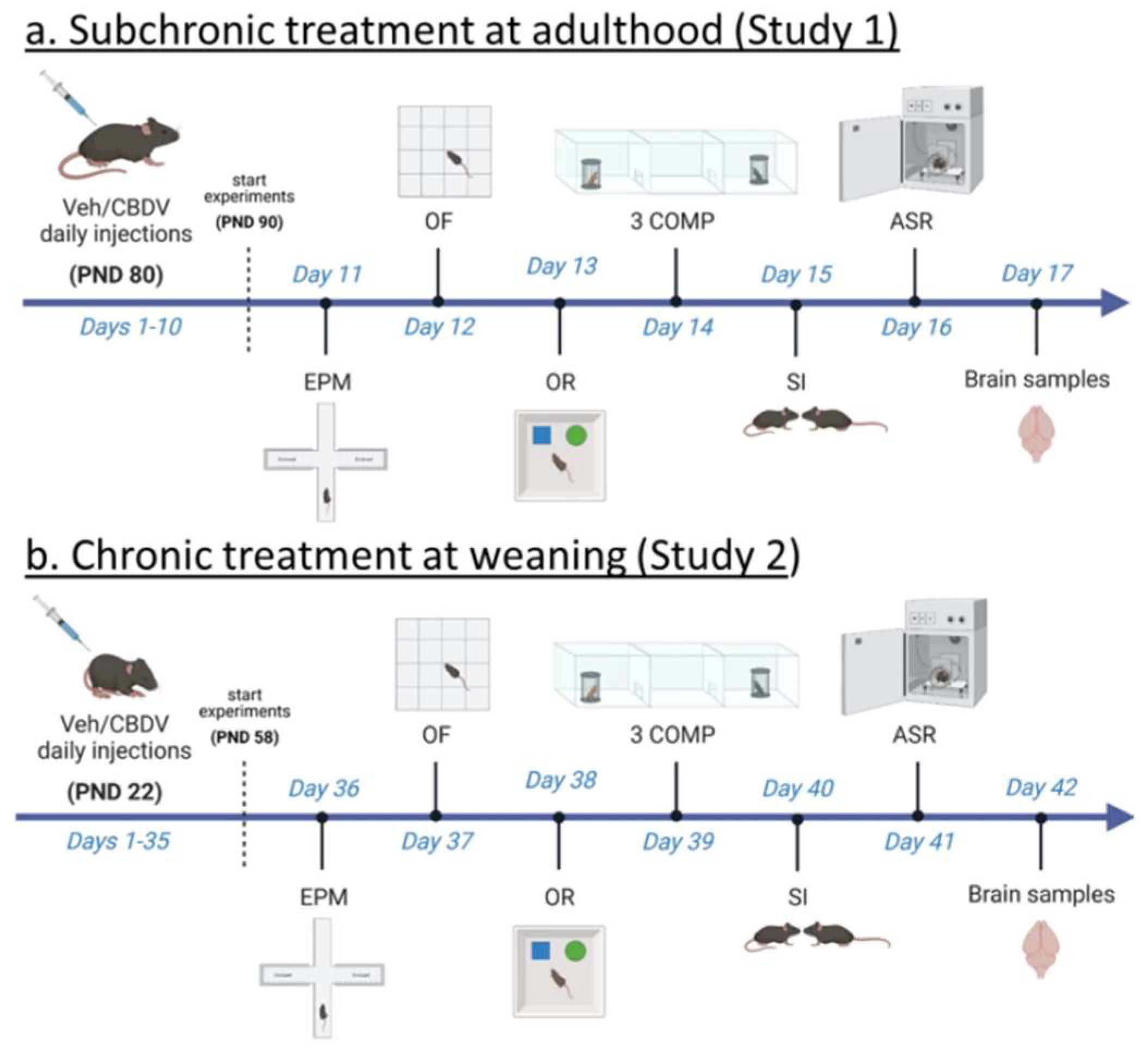
Figure 2.
Effects of adult subchronic CBDV administration on object recognition memory (Study 1). Locomotor activity (a) and anxiety-like behavior (b) were measured during the 20-min habituation session in the empty open field. The time spent exploring both objects (c) and the novel object recognition index (d) were computed during the 5-min sample and test phases, respectively. All behavioral parameters were measured in WT and Fmr1-KO mice treated with vehicle solution, CBDV at the dose of 20 mg/kg (CBDV-20) or 100 mg/kg (CBDV-100). Data are expressed as mean ± SEM. Numbers in histograms indicate sample size for each group. *p < 0.05; § versus chance level (50%, red dotted line).
Figure 2.
Effects of adult subchronic CBDV administration on object recognition memory (Study 1). Locomotor activity (a) and anxiety-like behavior (b) were measured during the 20-min habituation session in the empty open field. The time spent exploring both objects (c) and the novel object recognition index (d) were computed during the 5-min sample and test phases, respectively. All behavioral parameters were measured in WT and Fmr1-KO mice treated with vehicle solution, CBDV at the dose of 20 mg/kg (CBDV-20) or 100 mg/kg (CBDV-100). Data are expressed as mean ± SEM. Numbers in histograms indicate sample size for each group. *p < 0.05; § versus chance level (50%, red dotted line).
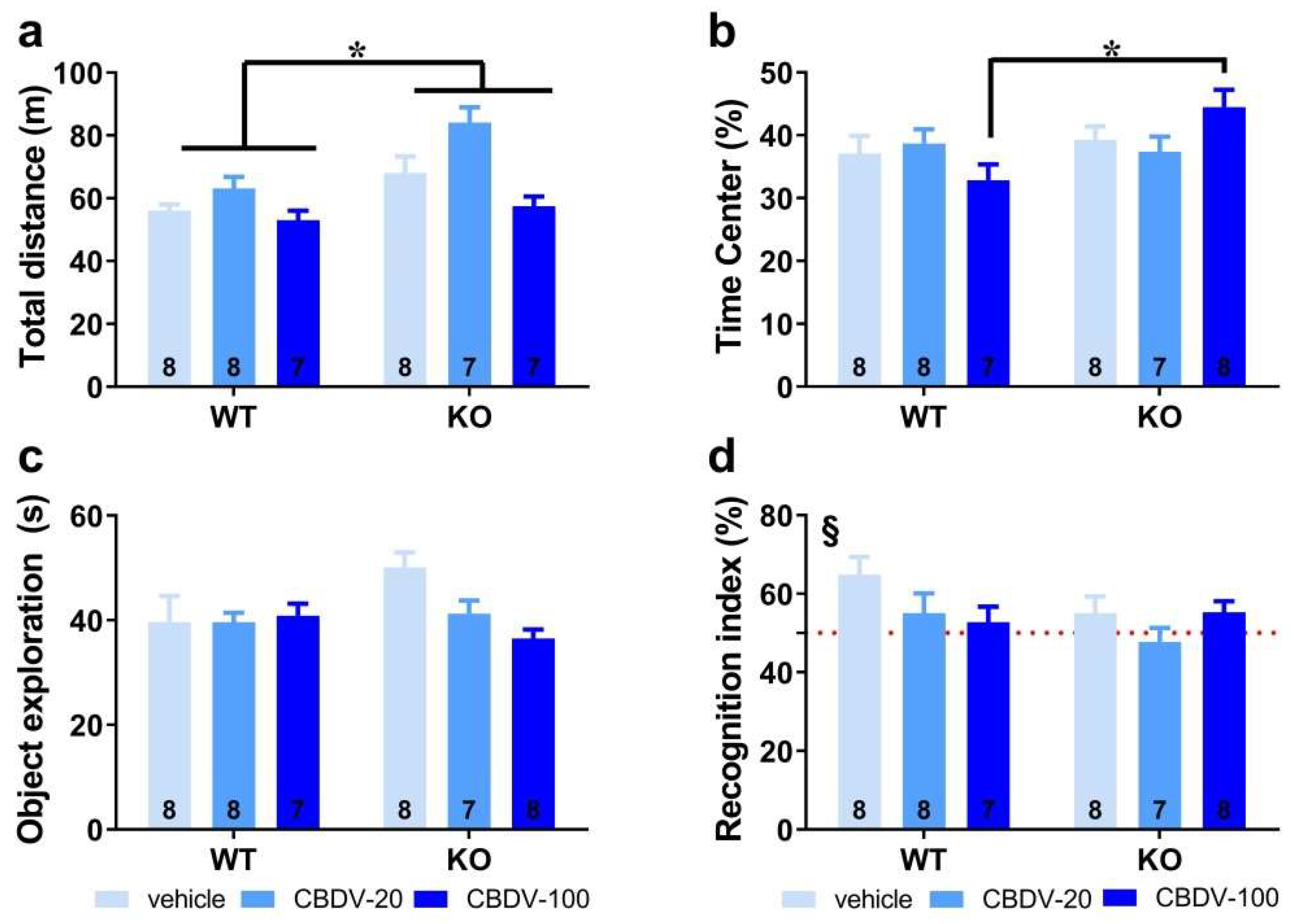
Figure 3.
Effects of adult subchronic CBDV administration on sociability and social novelty preference in the three-compartment paradigm (Study 1). Locomotion (a, c and e), percent sociability (b, d) and social novelty recognition (f) scores are shown for each of the three 5-min trials of the test. These included a first trial of habituation to the apparatus containing the empty stimulus cages (a, b), a second trial of sociability (c, d) aimed at assessing the percent preference for a social versus non-social novel stimulus (juvenile male mouse versus inanimate object), and a third trial of social novelty preference (e, f) assessing the percent preference for a novel versus familiar stimulus mouse. Data are expressed as mean ± SEM. Numbers in histograms indicate sample size for each group. § versus chance level (50%, red dotted line).
Figure 3.
Effects of adult subchronic CBDV administration on sociability and social novelty preference in the three-compartment paradigm (Study 1). Locomotion (a, c and e), percent sociability (b, d) and social novelty recognition (f) scores are shown for each of the three 5-min trials of the test. These included a first trial of habituation to the apparatus containing the empty stimulus cages (a, b), a second trial of sociability (c, d) aimed at assessing the percent preference for a social versus non-social novel stimulus (juvenile male mouse versus inanimate object), and a third trial of social novelty preference (e, f) assessing the percent preference for a novel versus familiar stimulus mouse. Data are expressed as mean ± SEM. Numbers in histograms indicate sample size for each group. § versus chance level (50%, red dotted line).
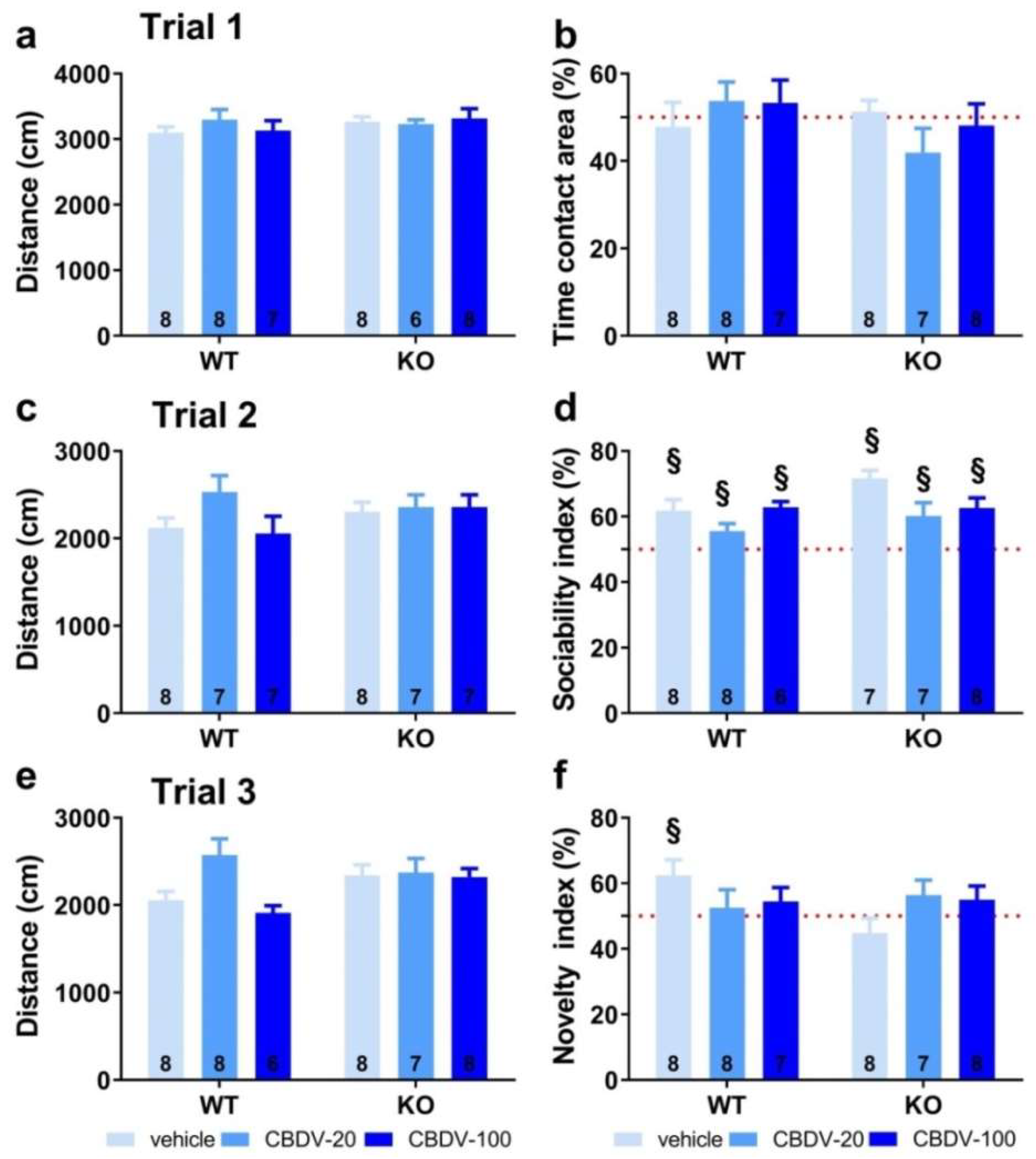
Figure 4.
Effects of subchronic adult CBDV treatment on social interaction and acoustic startle response (Study 1). Time spent performing direct affiliative behaviors (including sniffing and contact) towards an unfamiliar adult NMRI female mouse was assessed during the first and second 3-min time bins of a 6-min interaction session (a). Body startle (ln-transformed to meet the normality assumptions of parametric ANOVA) was measured in response to acoustic stimuli of 6, 12, 18 and 24 dB over a background of 66 dB (b). Data are expressed as mean ± SEM. Numbers in histograms indicate sample size for each group. *p < 0.05 from post-hoc comparisons following a significant interaction (a) or from separate ANOVAs in each treated group (b).
Figure 4.
Effects of subchronic adult CBDV treatment on social interaction and acoustic startle response (Study 1). Time spent performing direct affiliative behaviors (including sniffing and contact) towards an unfamiliar adult NMRI female mouse was assessed during the first and second 3-min time bins of a 6-min interaction session (a). Body startle (ln-transformed to meet the normality assumptions of parametric ANOVA) was measured in response to acoustic stimuli of 6, 12, 18 and 24 dB over a background of 66 dB (b). Data are expressed as mean ± SEM. Numbers in histograms indicate sample size for each group. *p < 0.05 from post-hoc comparisons following a significant interaction (a) or from separate ANOVAs in each treated group (b).
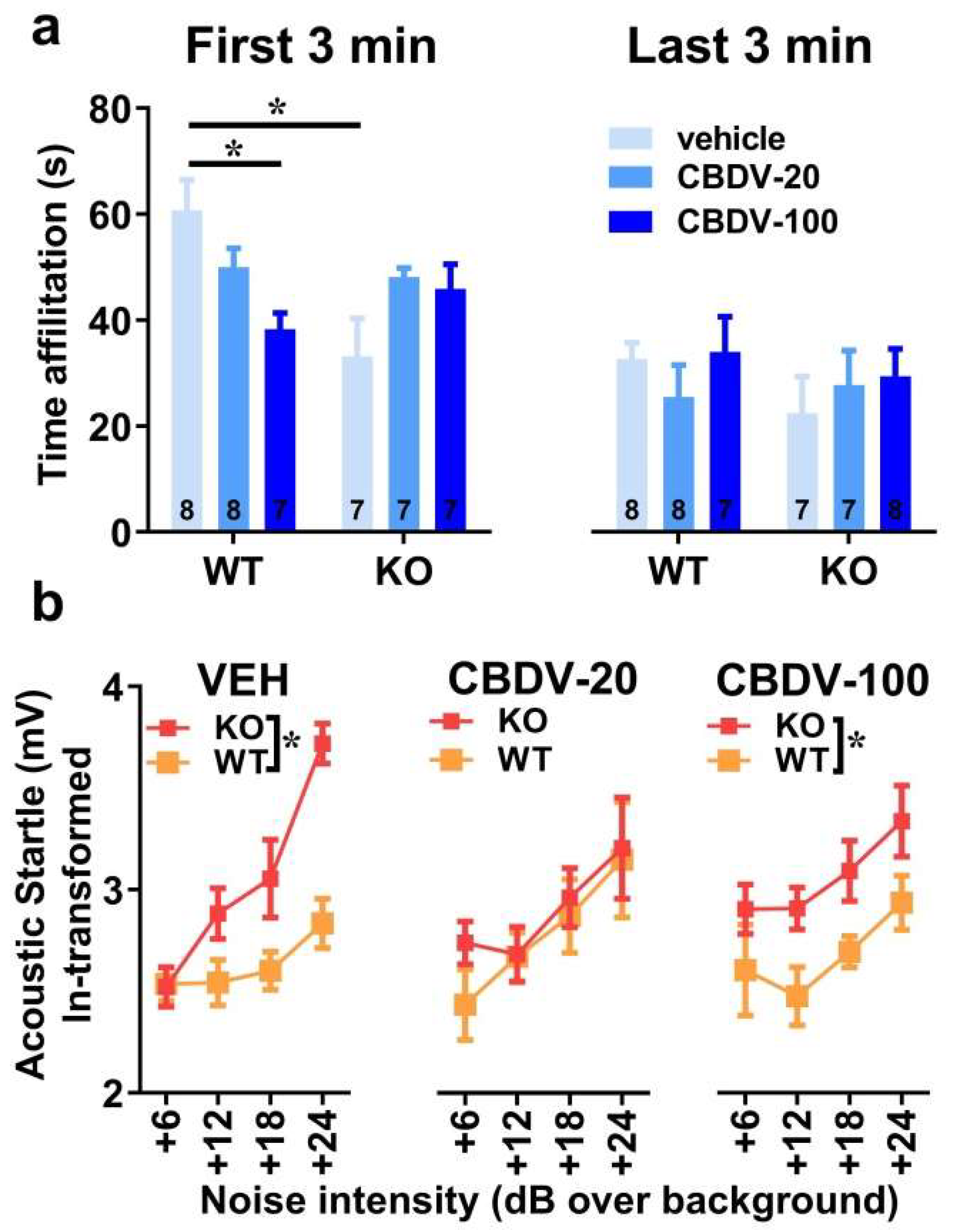
Figure 5.
Effects of subchronic adult CBDV administration on the expression of BDNF and inflammatory markers in the CA3 subfield of the hippocampus (Study 1). Representative image of CA3 obtained by laser microdissection (a) and expression levels of plasticity (b) and inflammatory markers (c-h). Data are expressed as mean ± SEM. Numbers in histograms indicate sample size for each group.
Figure 5.
Effects of subchronic adult CBDV administration on the expression of BDNF and inflammatory markers in the CA3 subfield of the hippocampus (Study 1). Representative image of CA3 obtained by laser microdissection (a) and expression levels of plasticity (b) and inflammatory markers (c-h). Data are expressed as mean ± SEM. Numbers in histograms indicate sample size for each group.
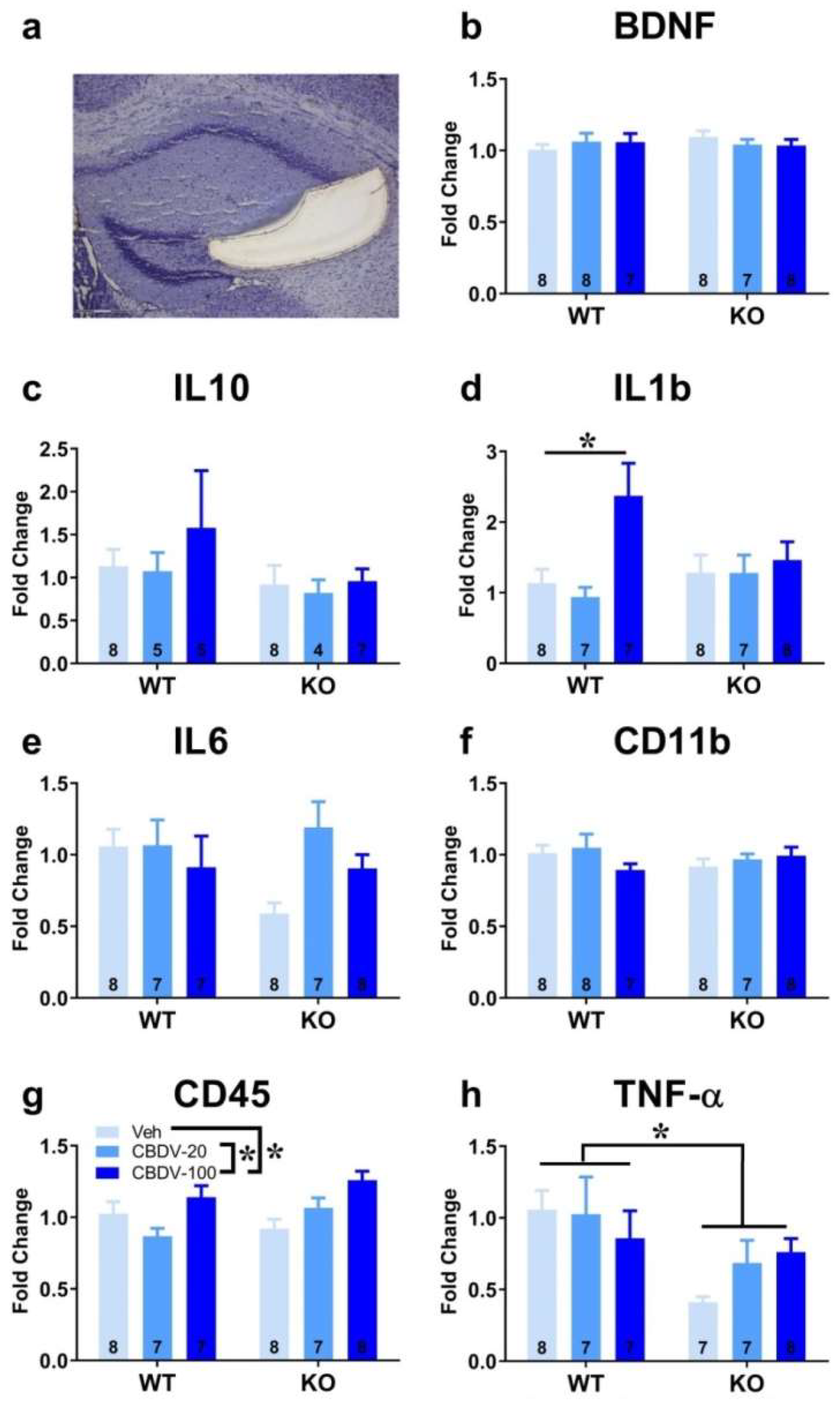
Figure 6.
Effects of subchronic adult CBDV administration on the expression of BDNF and inflammatory markers in the dentate gyrus of the hippocampus (Study 1). Representative image of the dentate gyrus obtained by laser microdissection (a) and expression levels of plasticity (b) and inflammatory markers (c-h). Data are expressed as mean ± SEM. Numbers in histograms indicate sample size for each group.
Figure 6.
Effects of subchronic adult CBDV administration on the expression of BDNF and inflammatory markers in the dentate gyrus of the hippocampus (Study 1). Representative image of the dentate gyrus obtained by laser microdissection (a) and expression levels of plasticity (b) and inflammatory markers (c-h). Data are expressed as mean ± SEM. Numbers in histograms indicate sample size for each group.
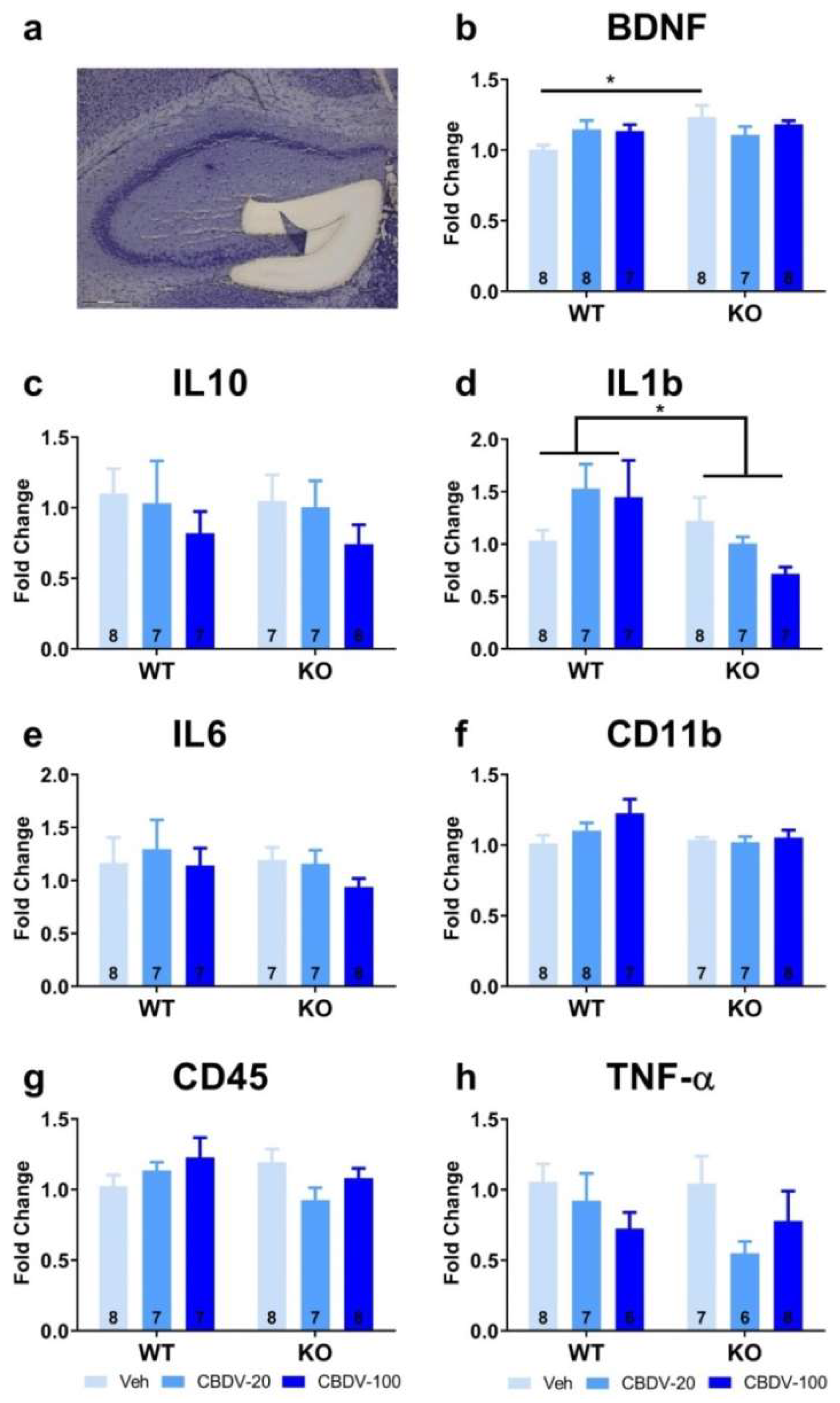
Figure 7.
Effects of juvenile chronic CBDV administration on object recognition memory (Study 2). Locomotor activity (a) and anxiety-like behavior (b) were measured during the 20-min habituation session in the empty open field. The time spent exploring both objects (c) and the novel object recognition index (d) were computed during the 5-min sample and test phases, respectively. All behavioral parameters were measured in WT and Fmr1-KO mice treated with vehicle solution, CBDV at the dose of 20 mg/kg (CBDV-20) or 100 mg/kg (CBDV-100). Data are expressed as mean ± SEM. Numbers in histograms indicate sample size for each group. *p < 0.05; § versus chance level (50%, red dotted line).
Figure 7.
Effects of juvenile chronic CBDV administration on object recognition memory (Study 2). Locomotor activity (a) and anxiety-like behavior (b) were measured during the 20-min habituation session in the empty open field. The time spent exploring both objects (c) and the novel object recognition index (d) were computed during the 5-min sample and test phases, respectively. All behavioral parameters were measured in WT and Fmr1-KO mice treated with vehicle solution, CBDV at the dose of 20 mg/kg (CBDV-20) or 100 mg/kg (CBDV-100). Data are expressed as mean ± SEM. Numbers in histograms indicate sample size for each group. *p < 0.05; § versus chance level (50%, red dotted line).
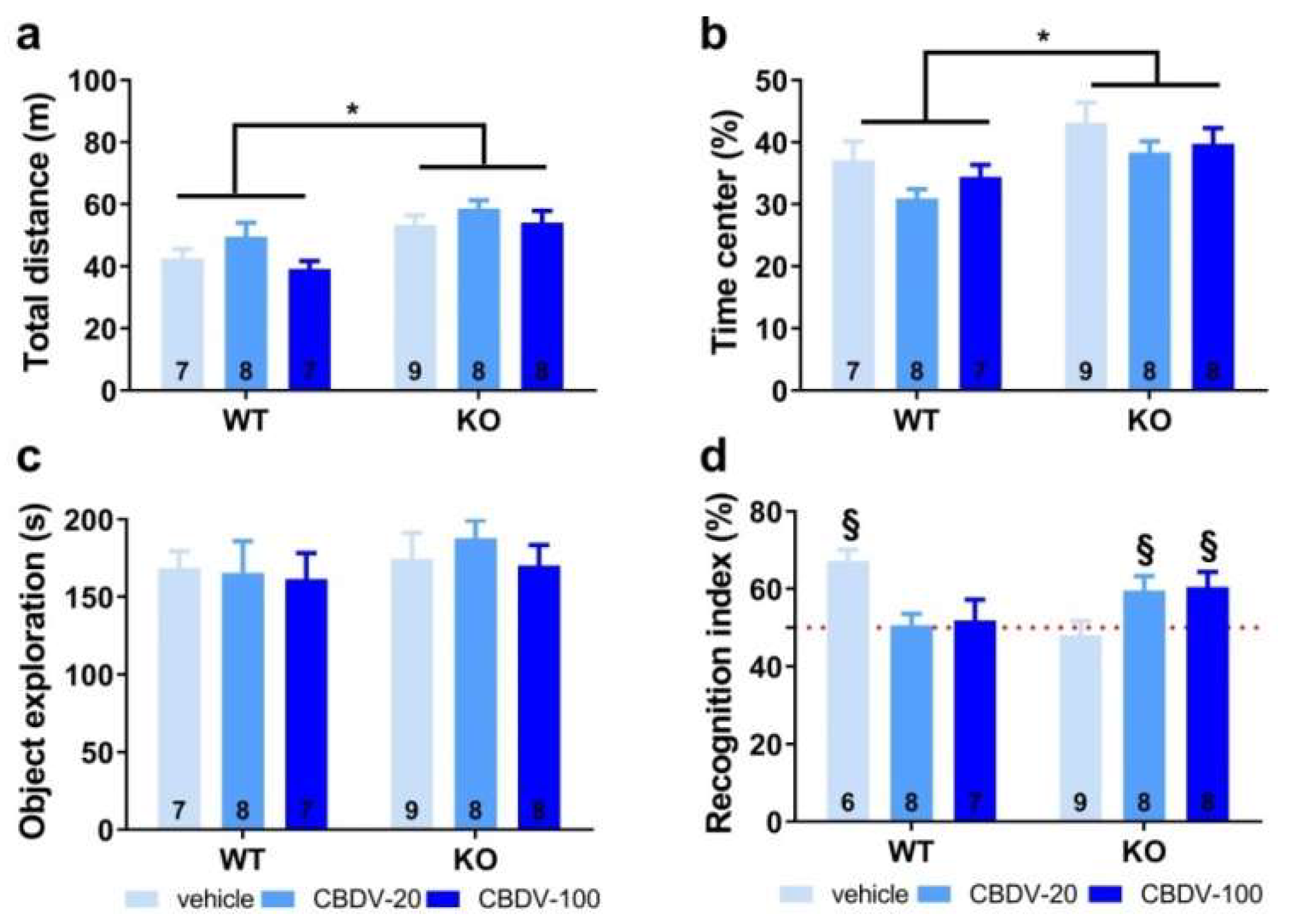
Figure 8.
Effects of juvenile chronic CBDV administration on sociability and social novelty preference in the three-compartment paradigm (Study 2). Locomotion (a, c and e), percent sociability (b, d) and social novelty recognition (f) scores are shown for each of the three 5-min trials of the test. These included a first trial of habituation to the apparatus containing the empty stimulus cages (a, b), a second trial of sociability (c, d) aimed at assessing the percent preference for a social versus non-social novel stimulus (juvenile male mouse versus inanimate object), and a third trial of social novelty preference (e, f) assessing the percent preference for a novel versus familiar stimulus mouse. Data are expressed as mean ± SEM. Numbers in histograms indicate sample size for each group. § versus chance level (50%, red dotted line).
Figure 8.
Effects of juvenile chronic CBDV administration on sociability and social novelty preference in the three-compartment paradigm (Study 2). Locomotion (a, c and e), percent sociability (b, d) and social novelty recognition (f) scores are shown for each of the three 5-min trials of the test. These included a first trial of habituation to the apparatus containing the empty stimulus cages (a, b), a second trial of sociability (c, d) aimed at assessing the percent preference for a social versus non-social novel stimulus (juvenile male mouse versus inanimate object), and a third trial of social novelty preference (e, f) assessing the percent preference for a novel versus familiar stimulus mouse. Data are expressed as mean ± SEM. Numbers in histograms indicate sample size for each group. § versus chance level (50%, red dotted line).
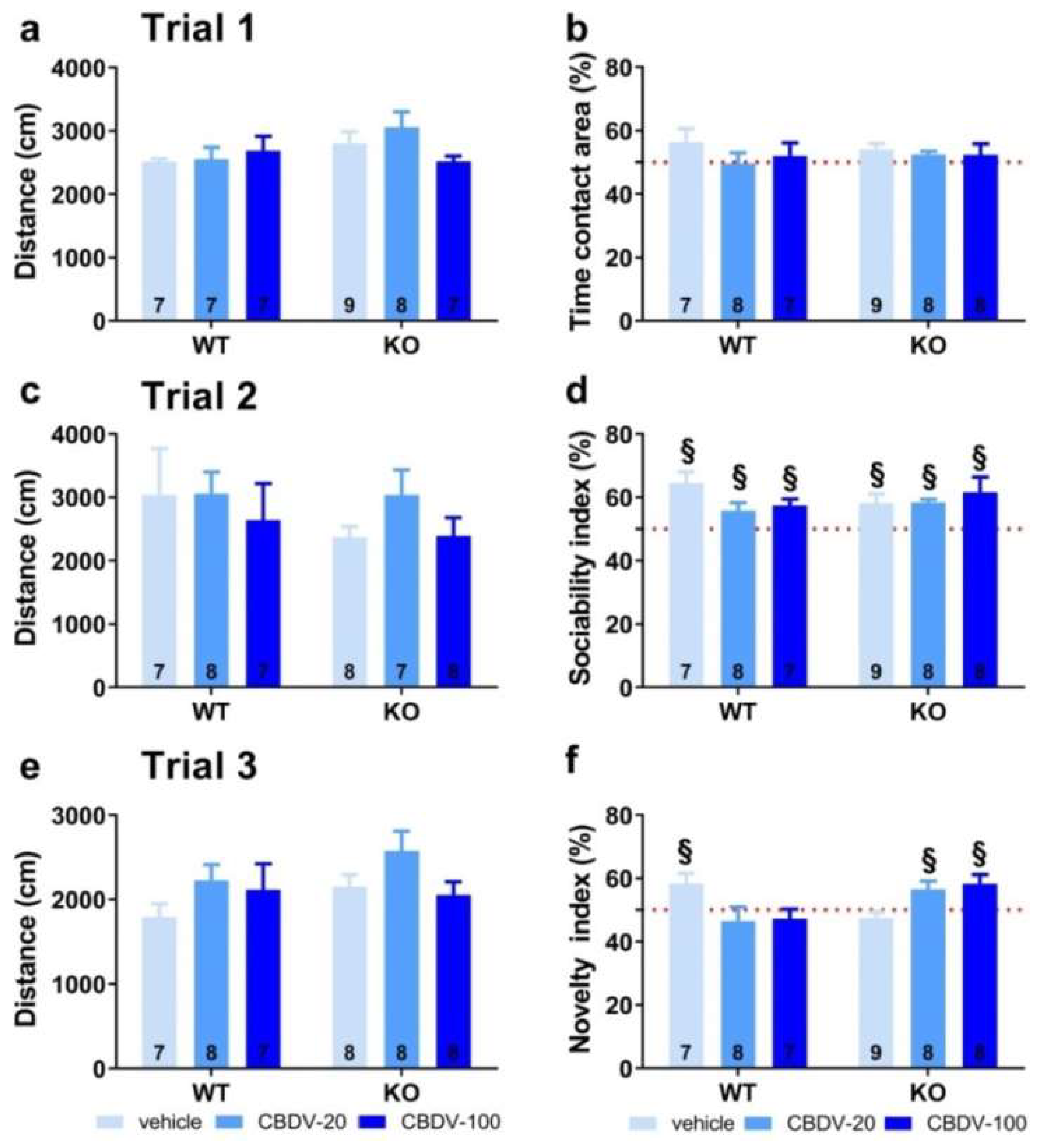
Figure 9.
Effects of juvenile chronic CBDV treatment on social interaction and acoustic startle response (Study 2). Time spent performing direct affiliative behaviors (including sniffing and contact) towards an unfamiliar adult NMRI female mouse was assessed during the first and second 3-min time bins of a 6-min interaction session (a). Body startle (ln-transformed to meet the normality assumptions of parametric ANOVA) was measured in response to acoustic stimuli of 6, 12, 18 and 24 dB over a background of 66 dB (b). Data are expressed as mean ± SEM. Numbers in histograms indicate sample size for each group. *p < 0.05 from post-hoc comparisons following a significant interaction (a) or from separate ANOVAs in each treatedgroup (b).
Figure 9.
Effects of juvenile chronic CBDV treatment on social interaction and acoustic startle response (Study 2). Time spent performing direct affiliative behaviors (including sniffing and contact) towards an unfamiliar adult NMRI female mouse was assessed during the first and second 3-min time bins of a 6-min interaction session (a). Body startle (ln-transformed to meet the normality assumptions of parametric ANOVA) was measured in response to acoustic stimuli of 6, 12, 18 and 24 dB over a background of 66 dB (b). Data are expressed as mean ± SEM. Numbers in histograms indicate sample size for each group. *p < 0.05 from post-hoc comparisons following a significant interaction (a) or from separate ANOVAs in each treatedgroup (b).
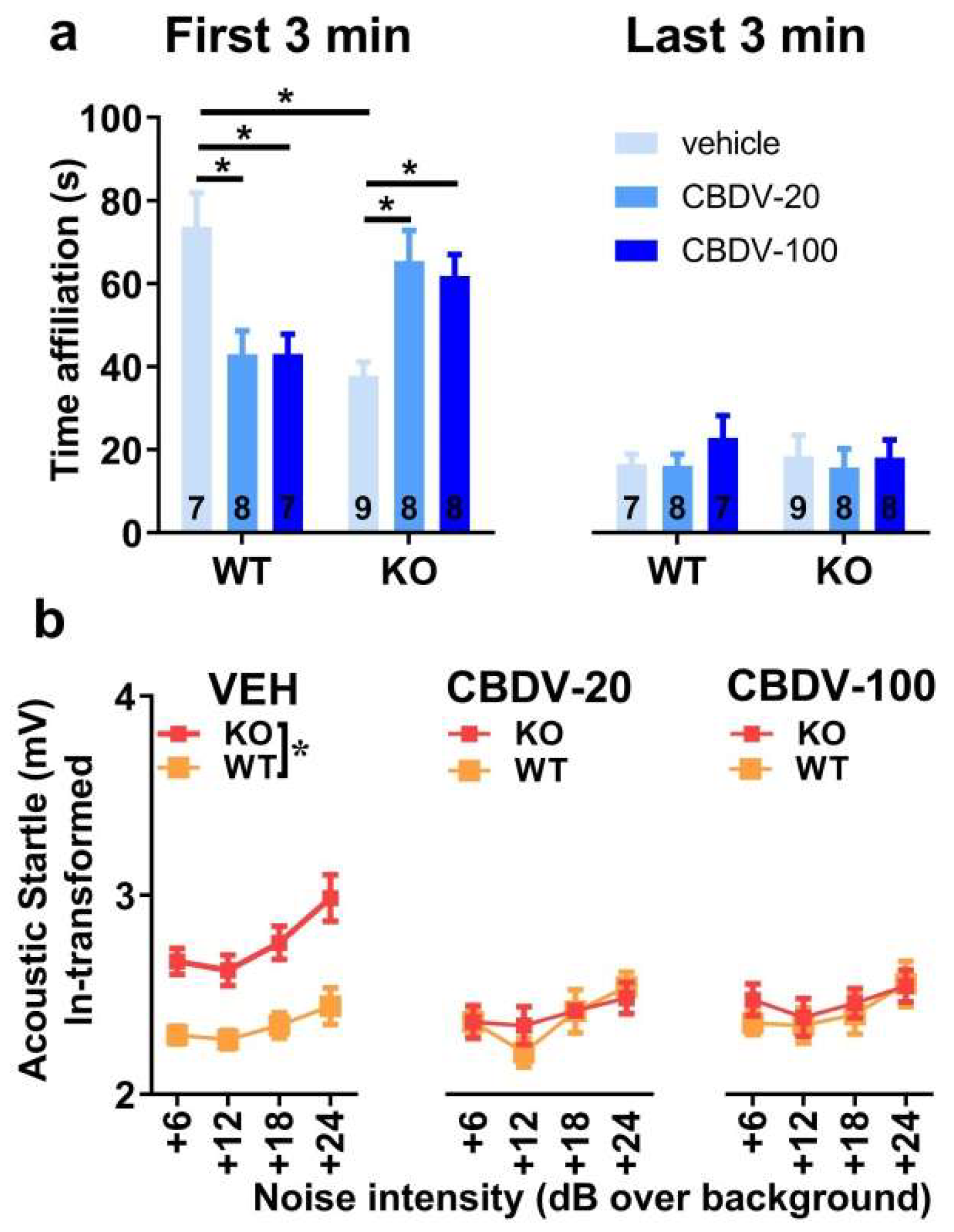
Figure 10.
Effects of juvenile chronic adult CBDV administration on the expression of BDNF and inflammatory markers in the CA3 subfield of the hippocampus (Study 2). Representative image of CA3 obtained by laser microdissection (a) and expression levels of plasticity (b) and inflammatory markers (c-h). Data are expressed as mean ± SEM. Numbers in histograms indicate sample size for each group.
Figure 10.
Effects of juvenile chronic adult CBDV administration on the expression of BDNF and inflammatory markers in the CA3 subfield of the hippocampus (Study 2). Representative image of CA3 obtained by laser microdissection (a) and expression levels of plasticity (b) and inflammatory markers (c-h). Data are expressed as mean ± SEM. Numbers in histograms indicate sample size for each group.
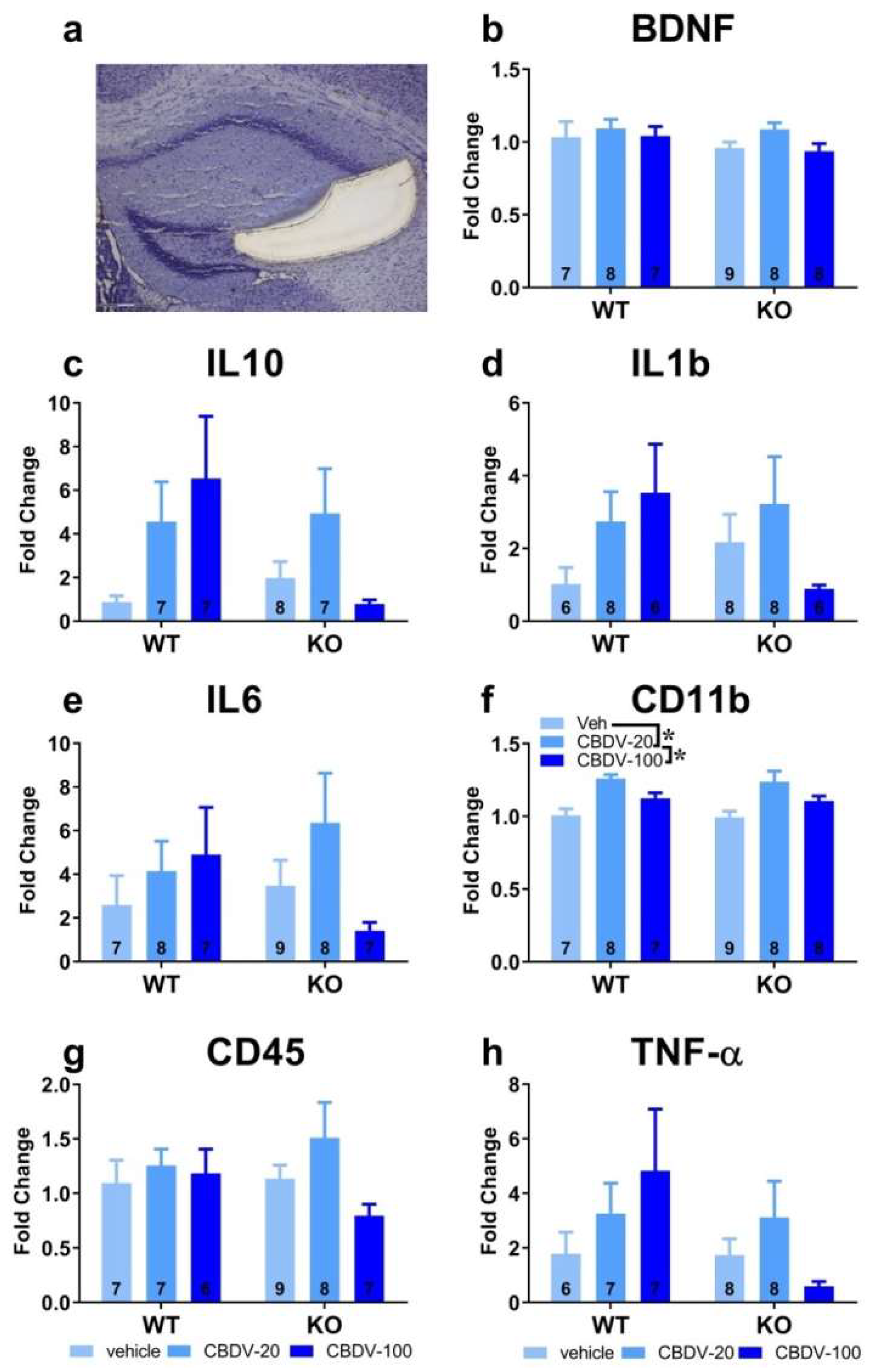
Figure 11.
Effects of juvenile chronic adult CBDV administration on the expression of BDNF and inflammatory markers in dentate gyrus of the hippocampus (Study 2). Representative image of dentate gyrus obtained by laser microdissection (a) and expression levels of plasticity (b) and inflammatory markers (c-h). Data are expressed as mean ± SEM. Numbers in histograms indicate sample size for each group.
Figure 11.
Effects of juvenile chronic adult CBDV administration on the expression of BDNF and inflammatory markers in dentate gyrus of the hippocampus (Study 2). Representative image of dentate gyrus obtained by laser microdissection (a) and expression levels of plasticity (b) and inflammatory markers (c-h). Data are expressed as mean ± SEM. Numbers in histograms indicate sample size for each group.
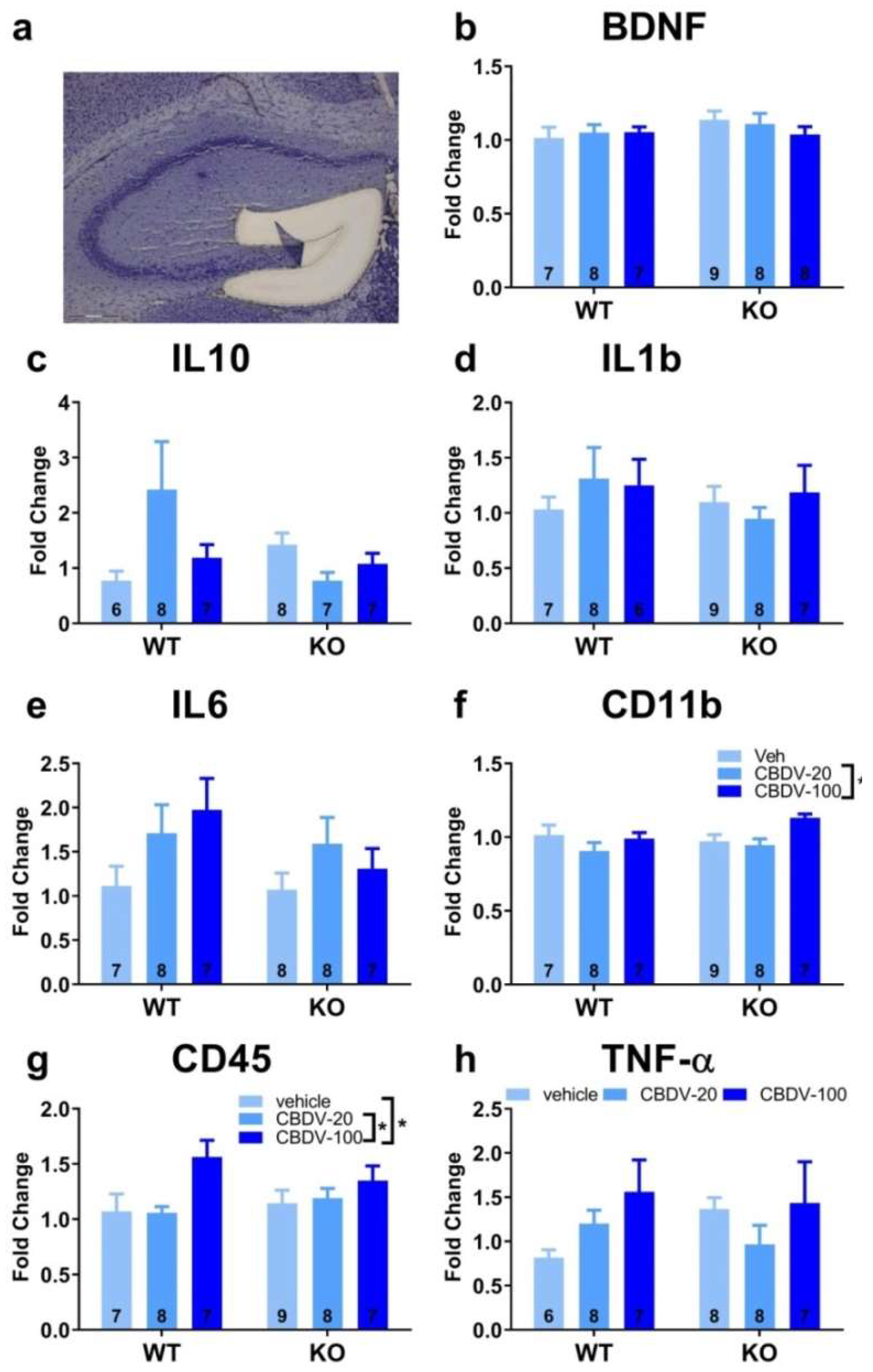

Table 1.
Summary of the behavioral effects of the Fmr1-KO genotype and CBDV treatments. Significant genotype x treatment interactions are highlighted in blue. EPM = elevated plus maze; OF = open field; OR = object recognition; 3-COMP = 3-compartment test for sociability and social novelty; SI = social interaction; AS = acoustic startle.
Table 1.
Summary of the behavioral effects of the Fmr1-KO genotype and CBDV treatments. Significant genotype x treatment interactions are highlighted in blue. EPM = elevated plus maze; OF = open field; OR = object recognition; 3-COMP = 3-compartment test for sociability and social novelty; SI = social interaction; AS = acoustic startle.
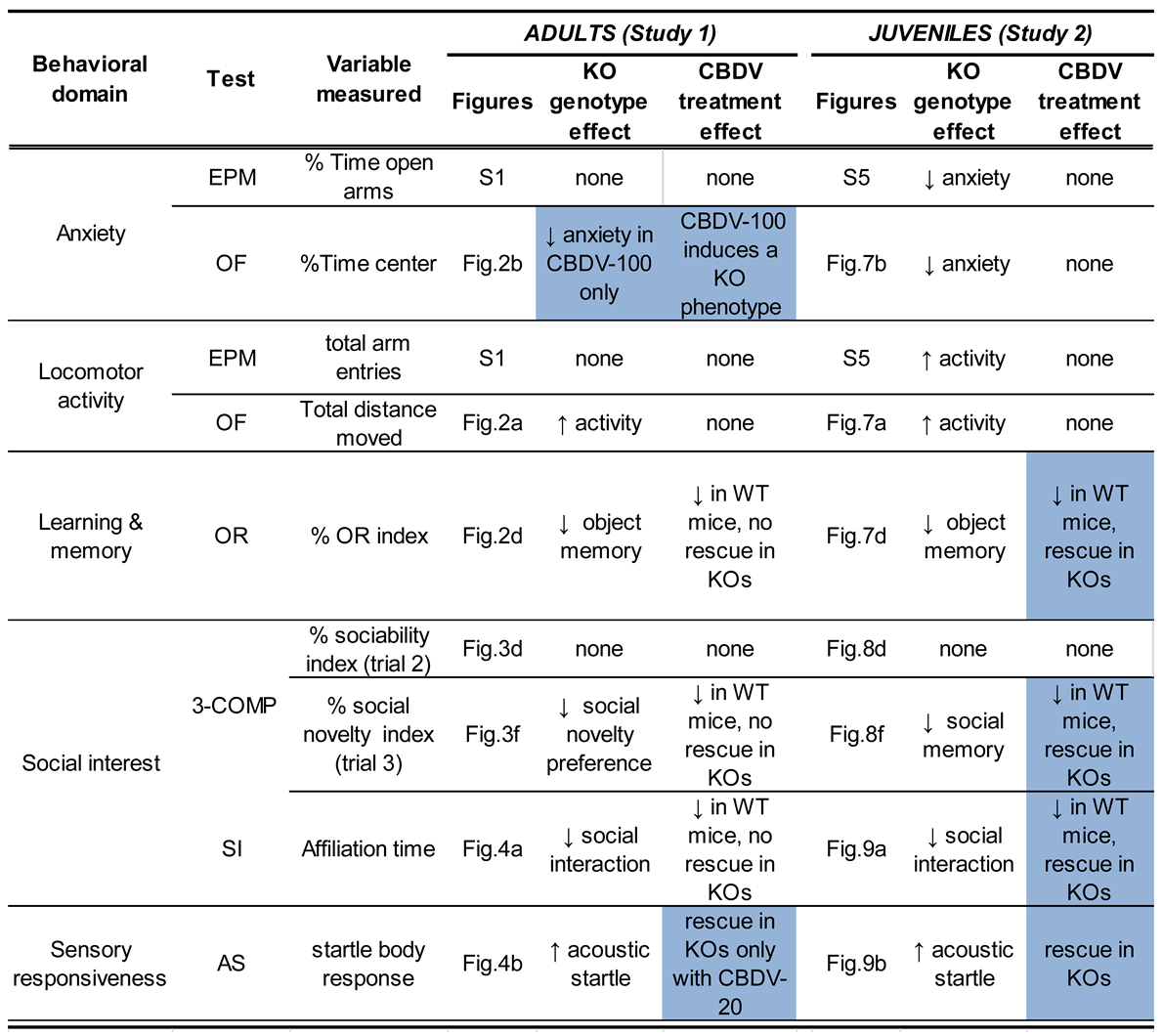 |
Table 2.
Summary of the effects of the Fmr1-KO genotype and CBDV treatments on the expression of brain markers. Significant genotype x treatment interactions are highlighted in blue. DG= dentate gyrus; PFC= prefrontal cortex.
Table 2.
Summary of the effects of the Fmr1-KO genotype and CBDV treatments on the expression of brain markers. Significant genotype x treatment interactions are highlighted in blue. DG= dentate gyrus; PFC= prefrontal cortex.
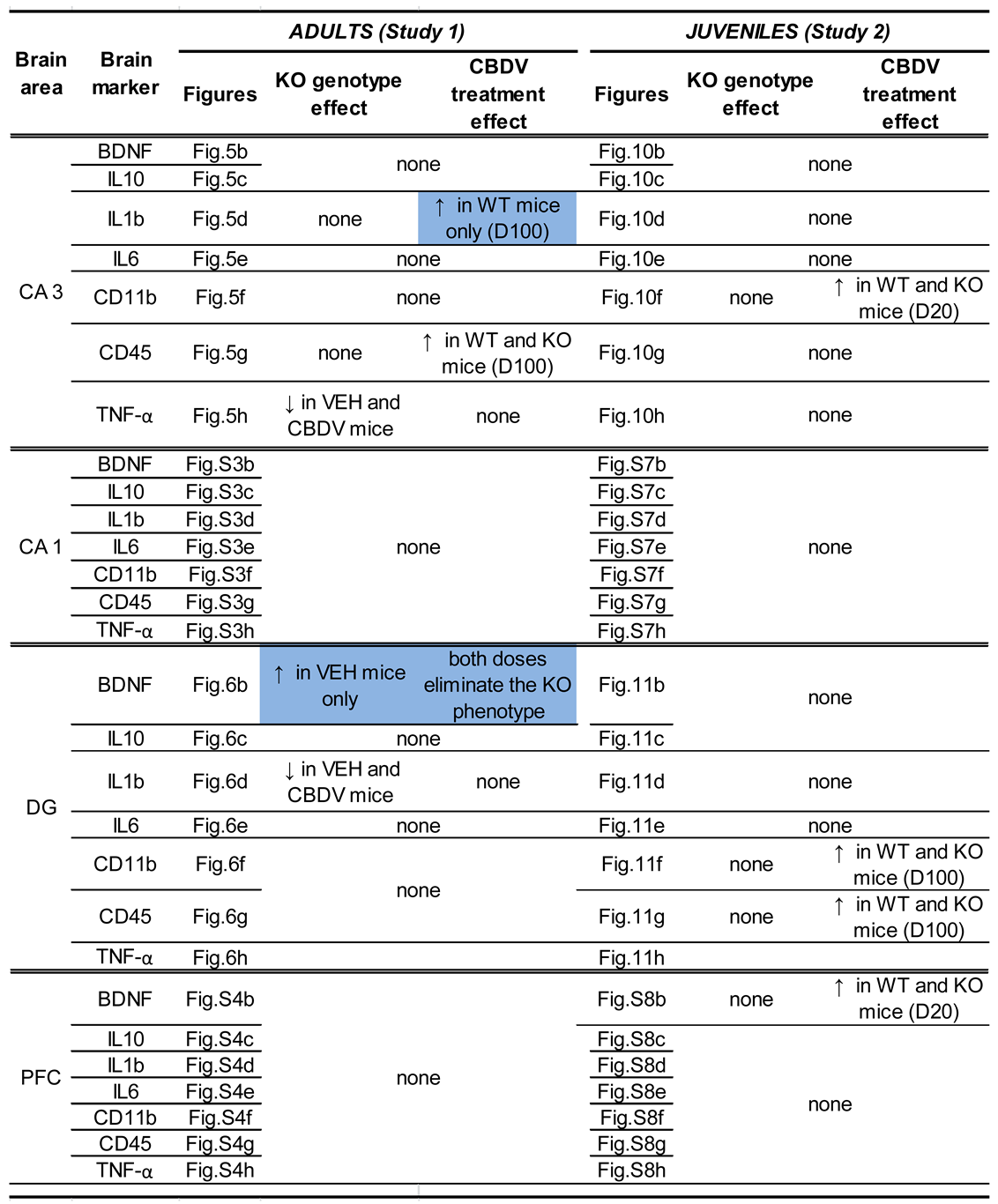 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



